
Microhydration of small protonated polyaromatic hydrocarbons: a first principles study†
Muthuramalingam
Prakash
 *a,
K.
Rudharachari Maiyelvaganan‡
a,
N.
Giri Lakshman‡
a,
C.
Gopalakrishnan‡
a and
Majdi
Hochlaf
*b
*a,
K.
Rudharachari Maiyelvaganan‡
a,
N.
Giri Lakshman‡
a,
C.
Gopalakrishnan‡
a and
Majdi
Hochlaf
*b
aComputational Chemistry Research Laboratory (CCRL), Department of Chemistry, SRM Institute of Science and Technology, Kattankulathur-603 203, Chengalpattu, Tamil Nadu, India. E-mail: prakashspm@gmail.com; prakashm4@srmist.edu.in; Tel: +91 44 2741 6230
bUniversité Gustave Eiffel, COSYS/IMSE, 5 Bd Descartes, 77454, Champs Sur Marne, France. E-mail: majdi.hochlaf@univ-eiffel.fr
First published on 13th May 2024
Abstract
Using first principles methodology, we investigate the microsolvation of protonated benzene (BzH+), protonated coronene (CorH+) and protonated dodecabenzocoronene (DbcH+). Gas phase complexes of these small protonated polyaromatic hydrocarbons (H+PAHs) with mono-, di-, and tri-hydrated water molecules are considered. Their most stable forms are presented, where we discuss their structural, energetic aromaticity and IR and UV spectral features. In particular, we focus on the analysis of the bonding and various non-bonded interactions between these protonated aromatics and water clusters. The strength of non-bonded interactions is quantified and correlated with their electron density profiles. Furthermore, insights into the interfacial interactions and stability of these complexes were obtained through non-covalent index and symmetry-adapted perturbation theory (SAPT0) analyses. We also discuss the effects of the extension of the π aromatic cloud on the water solvation of these protonated aromatics. In particular, we extended our predictions for the S0 → S1 and S0 → T1 wavelength transitions of micro hydrated H+PAHs to deduce those of these species solvated in aqueous solution. The present findings should be useful for understanding, at the microscopic level, the effects of water interacting with H+PAHs, which are relevant for organic chemistry, astrochemistry, atmospheric chemistry, combustion and materials science.
I. Introduction
Polyaromatic hydrocarbons (PAHs) are aromatic organic compounds of great importance in atmospheric chemistry,1,2 materials science3,4 and combustion.5–9 For instance, they can be formed during the incomplete combustion of organic compounds and be found in various environmental and atmospheric media such in air, food, soil and water with serious drawbacks for the health of humans, animals and plants because of their wide structural distribution and potential toxicity.10–12 Besides, PAHs are very important in astrochemistry.13–15 Indeed, they are highly suspected to be present in interstellar media (ISM) and on the origin of the unidentified infrared emission bands (UIR). Although the characteristics of some of the vibrations of PAHs are closely similar to those recorded in surveys, several features remain unassigned. In this context, several PAH (ionized or nitrogen or silicon containing) derivatives and clusters were proposed instead. Indeed, the recent detections of small aromatics (e.g., benzonitrile,16 1-cyanonaphthalene and 2-cyanonaphthalene,17 1-cyano-cyclopentadiene,18 cyclopentadiene and indene19) and of the commonly admitted chemical intermediates for their formation in the ISM (e.g., 1-ethynyl-cyclopentadiene and 2-ethynyl-cyclopentadiene20 and fulvenallene21) are very encouraging towards their definitive identification and for the understanding of their physical chemistry there. In particular, protonated PAHs (H+PAHs) were proposed as important intermediates in the chemical models leading to PAHs, since some protonated compounds were also detected in the ISM.22 Therefore, these results motivated extensive spectroscopic laboratory investigations of protonated aromatics targeting their detection in astrophysical media.23–31PAHs are formed by fused aromatic rings with wide π-electron delocalization. A priori, several protonation sites of these aromatics are possible resulting in: (i) a σ complex where the proton is attached to an external carbon of the aromatic compound (i.e., aliphatic sp3 C–H+); (ii) a π complex where the proton interacts with the π orbital of these aromatics; and (iii) a σ complex where the proton is attached to an internal carbon of PAHs possessing more than one aromatic ring. The identification of the protonation site, the relative stability of the corresponding protonated aromatic compound and the nature of their mutual interactions motivated intensive theoretical and experimental investigations during the two last decades.
Back to 2001, the computational study of Frash et al.32 showed that the proton attaches preferentially to one carbon atom (either external or internal) of corannulene, forming a σ-complex. Then, Solcà and Dopfer29 and Jones et al.33 showed that a σ-complex (case (i)) is obtained upon attaching a proton to benzene forming a benzenium ion through the analysis of the corresponding IR spectrum. For the benzenium ion, Duncan and co-workers34–36 confirmed these findings using infrared and photodissociation spectroscopy (IRPD) combined with ab initio and DFT computations. Later on, Knorke et al.28 and Lorenz et al.37 extended such investigations to gas phase protonated naphthalene, anthracene, tetracene, pentacene, and coronene. Indeed, these authors produced various protonated forms of such polyaromatic ring compounds. They characterized them by IR multiple-photon dissociation (IRMPD) spectroscopy. Through comparison to their computed DFT IR spectra, these authors confirmed the formation of case (i) ions. For protonated naphthalene and protonated coronene, Bahou et al.30,38 recorded highly resolved IR spectra of these molecular cations trapped in cold solid para-hydrogen matrices. Through the analysis of these spectra, these authors came to the conclusion that H+PAHs might be among the carriers of UIR bands in the ISM.
Besides, UV-vis absorption and fluorescence spectroscopies were used to give insights into the pattern of the electronic excited states of H+PAHs and on their photophysics. Experimentally, vibrationally resolved electronic spectra of protonated species of benzene, toluene, naphthalene, anthracene, and tetracene were recorded.39–44 Maier and co-workers45–49 extended these studies to larger H+PAHs, such as protonated pyrene and protonated coronene in addition to protonated naphthalene. These cations were trapped in cold solid neon matrices or in ion traps combined with collisional cooling. Such experimentally very cold media (T ∼ 15 K) mimic the ISM temperature allowing stating, nevertheless, that these cations are not possible carriers of any known diffuse interstellar band. Moreover, these works showed that protonation strongly affects the electronic structure of the respective PAHs resulting in an unusually large red shift with respect to the corresponding neutral bands. The theoretical investigations50–56 of the S0 → S1 transition of such protonated PAHs showed that this transition exhibits a charge transfer character as observed experimentally. Complex dynamics on the electronic states of H+PAHs was noticed, such as reversible photoisomerization of protonated naphthalenes. Very recently, Laws et al.57 recorded highly resolved excitation spectra of very cold protonated anthracene cations obtained by triple-resonance dissociation spectroscopy. These spectra present the rich structure signature of a symmetry-breaking intramolecular charge-transfer process through active Herzberg–Teller progressions. In fact, these authors revealed that the electronic structure and photophysics of H+PAHs, either isolated or interacting with inert ligands, are still challenging and worth investigating in-depth.
Although H+PAHs were extensively studied, this is not the case, however, of their complexes with ligands such as water or rare gases. Back to 2003, Solcà and Dopfer58 detected protonated benzene – complexed with various inert ligands, including water (W). They identified the protonated benzene–water dimer (BzH+–W) by IRPD spectroscopy complemented by MP2/6-311G(2df,2pd) computations for assignment. In 2012, the structures, bonding, and IR spectra of protonated benzene (BzH+)n–Wm (with n and/or m up to 3) clusters were studied by Duncan and co-workers59,60 using IRPD spectroscopy and quantum chemical approaches. They found that specific induced shifts on IR bands compared to bare protonated benzene allow identification of the corresponding cluster. Also, Jouvet and co-workers61,62 treated experimentally the effects on the electronic spectra of protonated naphthalene caused by microhydration or its clustering with Ar. Complex vibrationally resolved photodissociation spectral features were recorded, for which assignments were achieved using quantum chemical calculations and Franck–Condon simulations of the vibrational bands. More recently, Chatterjee and Dopfer63 used IRPD and B3LYP-D3/aug-cc-pVTZ computations, in the C–H and O–H stretch spectral range (2750–3800 cm−1), to probe clustering induced shifts on bare and Ar-tagged protonated naphthalene–Wn complexes (n ≤ 3). Interestingly, they found a solvation-dependent intracluster proton transfer where a proton is attached to naphthalene for n = 1, whereas the proton binds preferentially to water clusters for n ≥ 2.
Previous theoretical and experimental studies proved that the most stable protonated aromatic species correspond to case (i) cations. They showed also that IR and UV-vis spectroscopies combined with first principles computations represent powerful tools and a critical probe of the structure, the bonding and the electronic, vibrational and rotational spectra of organic/aromatic–Wn complexes. The present study uses such first principles methodologies to investigate the structural stability, IR and UV-vis spectra and energetics of water cluster networks (Wn, where n = 1 to 3) interacting with three different protonated aromatic carbon compounds of increasing size (i.e. benzene (BzH+), coronene (CorH+) and dodecabenzocoronene (DbcH+). We thus aim at understanding the effect of the aromatic π-extension from benzene to coronene to dodecabenzocoronene. These extended models are considered to mimic the effect of large, protonated PAH–water cluster interactions. Although computations and experiments on the latter are challenging, recent investigations showed, indeed, that studying the series of small PAHs with increasing size gives several insights on the larger ones.64–66 Besides, we target shedding light on the microsolvation induced effect of these H+PAHs targeting their physical chemical properties in aqueous solutions.
II. Computational methods
We focus on the structures, stabilities, energetics, electron density profiles, and nature of the interfacial interactions within BzH+–Wn, CorH+–Wn and DbcH+–Wn (n = 1–3) clusters, and on their clustering induced IR and UV-vis spectral features. For these clusters, we thus perform geometry optimizations, frequency calculations, bonding and intermonomer analyses, binding energy calculations and IR and UV spectra determinations. For electronic structure calculations and spectroscopic investigations, we use the Gaussian 1667 suite of programs. For all species, we carry out computations at the PBE0/6-311++G** and PBE0-D3/6-311++G** levels, where D3 corresponds to the Grimme's dispersion correction to account for dispersion.68 As established in the literature,66,69 the PBE0-D3/6-311++G** level of theory is accurate enough to describe such protonated aromatic–water clusters since benchmarks revealed that PBE0-D3/6-311++G** competes with MP2/CBS and CCSD(T)/CBS, whereas a strong reduction in computational costs is noticed. In particular, we carried out additional MP2/6-311++G** calculations for BzH+–Wn (n = 1–3) clusters. These results are provided in Fig. S1 and Table S1 of the ESI.† These MP2 computations show that the O–H+⋯π mode of interaction is more favorable than the C–H+⋯O interaction. Indeed, all PBE0-D3 optimized CH+⋯O isomers convert into O–H+⋯π isomers. This is due to the strong inductive effect that stabilizes the Cation+⋯π complex (see ref. 83 for more details). Nevertheless, PBE0-D3 describes well σ C–H+⋯O interaction with reduced computational cost. Since this is the focus of the present work, we will adopt PBE0-D3 for all electronic computations of BzH+–Wn, CorH+–Wn and DbcH+–Wn (n = 1–3) complexes.For visualization, we use Gauss View 6.0 with Chemcraft software.70,71 We also do an energy decomposition analysis (EDA) at the SAPT0/aug-cc-pVDZ level. For these symmetry-adapted perturbation theory (SAPT) computations, we use PSI-4 software.72,73
For cluster optimizations, several initial configurations are considered. Using the Conformer–Rotamer ensemble sampling tool (CREST) technique, which is based on the semiempirical tight-binding quantum chemistry methods (GFN1-xTB and GFN2-xTB), the conformational space for clusters is sampled.74–76 Within the framework of meta-dynamics (MTD), the semiempirical tight-binding based quantum chemistry approach GFN-xTB is utilized to explore the different chemical compounds, conformers, and reaction space. First, geometries are pre-optimized with the semi-empirical tight-binding XTB2 method.77 The corresponding results are listed in Table S2 of the ESI.†
We set very low convergence criteria since the intermonomer interactions are weak and these clusters are floppy resulting in flat potentials close to equilibria. We check on the minimal nature of the optimized stationary points by having all positive vibrational frequencies. Afterwards, only the most stable minimum structures of BzH+–Wn, CorH+–Wn and DbcH+–Wn (n = 1–3)-type clusters are retained for further treatment. Thus, we deduce their anharmonic frequencies by scaling the harmonic ones by a factor of 0.959378 and compute their IR spectra and their UV-vis absorption spectra. In addition, we perform full anharmonic frequency calculations for BzH+–Wn (n = 1–3) and CorH+–Wn (n = 0–2) species using perturbation theory as implemented in Gaussian 16. For electronic excited states and UV-vis spectra calculations, we use the time-dependent density functional theory (TD-DFT) technique as implemented in Gaussian 16. All TD-DFT calculations are carried out on the PBE0/6-311++G** optimized geometries of the clusters, where the TD = (Nstates = 30, 50–50) keywords for the singlet and triplet excited states are used. Furthermore, to study the aromatic character of the protonated species after incorporation of water molecules, nucleus independent chemical shift (NICS) values are computed at the center of the ring (NICS (0 Å)zz) and at 1 Å above (NICS (1 Å)zz) for all the clusters.
To characterize the bonding and the intermonomer interactions within these complexes, we carry out atoms-in-molecules (AIM) electron density and non-covalent interaction (NCI) index analyses using AIM 200079 and Multiwfn80 software, respectively. We also compute their binding energies (BE(AB)) at the PBE0(-D3)/6-311++G** level. For BzH+-containing clusters, we derive also their BEs using MP2/6-311++G**. BE(AB) is calculated within the supramolecular approach and corrected for basis set superposition error using the counterpoise method:81
| BE(AB) = [E(AB) − (E(A) + E(B))] | (1) |
III. Results
1. Most stable forms of BzH+–Wn, CorH+–Wn and DbcH+–Wn (where n = 1–3) clusters
Fig. 1 shows the PBE0-D3/6-311++G** optimized geometries of BzH+, CorH+, DbcH+ and of their clusters with water (i.e., BzH+–Wn, CorH+–Wn and DbcH+–Wn (n = 1–3). The coordinates obtained from the PBE0-D3/6-311++G** method are provided in Table S3 of the ESI,† where we also list those obtained without D3 correction (Table S4, ESI†) and the MP2/6-311++G** (Table S5, ESI†) methods. Only in case (i), protonated aromatics are considered as interacting with water molecules since their monohydrates are the most stable ones. Indeed, this was already confirmed for BzH+ hydrates and we show in Tables S4 and S5 (ESI†) that this is also the case for CorH+ and for CorH+–W1. Strictly speaking, upon protonation, BzH+ species are no more aromatic; however, those containing CorH+ and DbcH+ are still aromatic.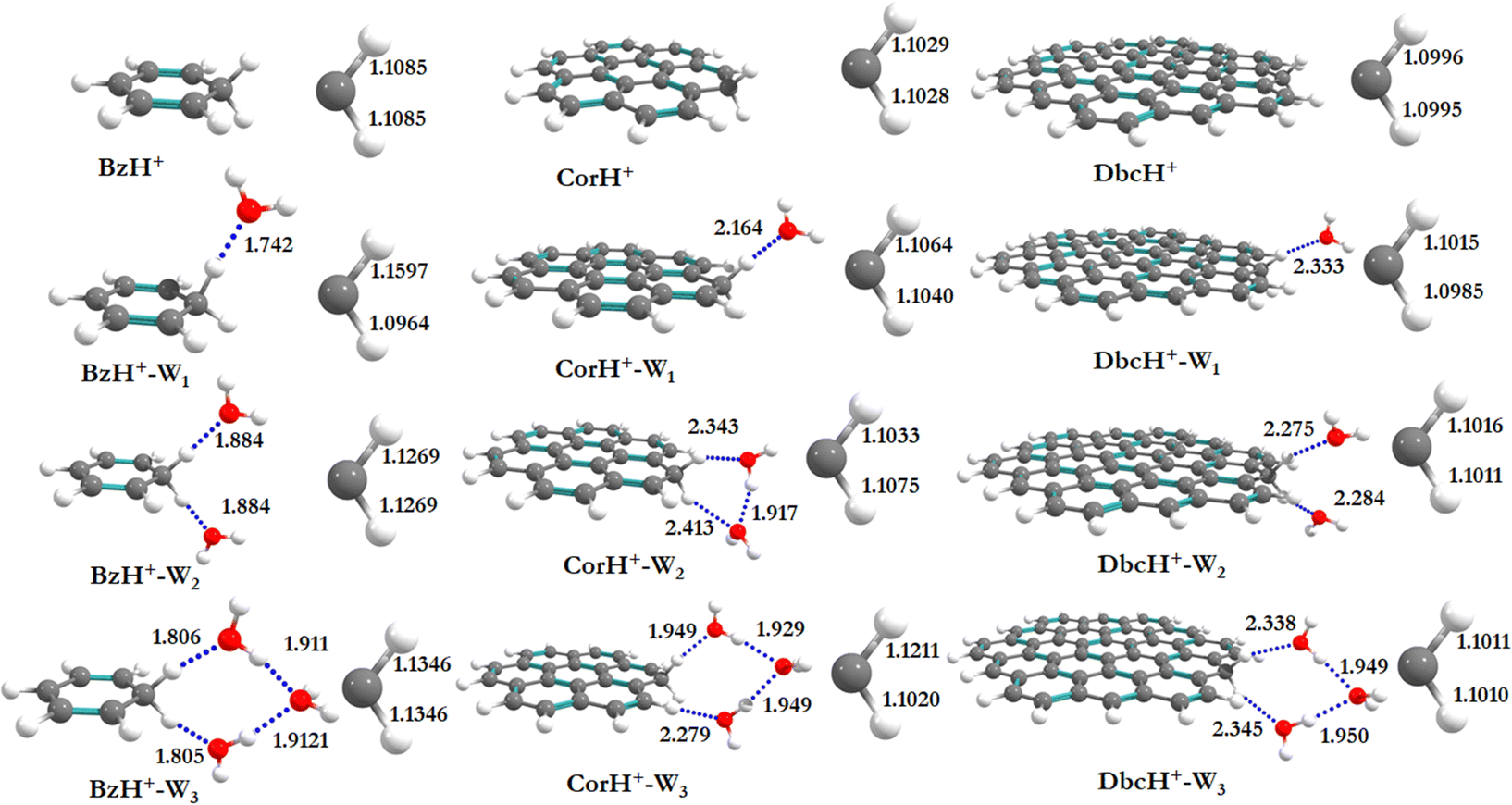 | ||
| Fig. 1 PBE0-D3/6-311++G** optimized equilibrium structures of BzH+, CorH+ and DbcH+ and of the most stable forms of the BzH+–Wn, CorH+–Wn and DbcH+–Wn (n = 1–3) complexes. Close views of the sp3 C–H bond distances (before and after hydration) and the intermonomer distances (in Å) are given. | ||
Fig. 1 reveals that, after optimization, the proton remains attached to the aromatic, where the water molecules form an H-bonded network in its vicinity. For instance, we have an H-bond between the excess proton (i.e., sp3 C–H+ site) and the oxygen of a water molecule for the BzH+–W1, CorH+–W1 and DbcH+–W1 complexes. We have also the establishment of H-bonds between the hydrogens attached to the sp3 carbon of BzH+, CorH+ and DbcH+ and an oxygen of the water molecule for BzH+–W2, CorH+–W2 and DbcH+–W2, without interaction between the two water molecules. Nevertheless, a H2O–H2O–H2O network is formed while attaching three water molecules to BzH+, CorH+ and DbcH+ to form BzH+–W3, CorH+–W3 and DbcH+–W3, where an H-bond network involving the hydrogens of the sp3 carbon of BzH+, CorH+ and DbcH+ and H/O of the water molecules is observed.
Specifically, the optimized geometries of BzH+–Wn (n = 1–3) clusters (Fig. 1) reveal that the excess proton is always localized/attached to Bz establishing H-bond interactions with the neighbouring water molecules. The corresponding H-bond distances vary from 1.742 (1.776) Å, to 1.884 (1.905) Å, to 1.805 (1.825) Å for BzH+–W1, BzH+–W2, and BzH+–W3, respectively, where parenthesised values are obtained without D3 correction. The corresponding distances calculated using the MP2/6-311++G** method are 1.674 Å, 1.941 Å, and 1.849 Å (Fig. S1, ESI†). The comparison of these distances reveals that the incorporation of D3 correction significantly strengthens the H-bonds. The MP2 distances exhibit a similar trend upon attaching water molecules and remain close to those deduced using PBE0-D3/6-311++G**.
For BzH+–Wn (n = 1–3), the calculated sp3 C–H bond distances of bare BzH+ are ∼1.11 Å, whereas hydrated BzH+ C–H bond distances vary from 1.096 to 1.160 Å. This is due to the formation of a H-bonded network with water clusters. The mono-hydrated protonated Bz has the shortest distance with water when compared to di-hydrated (i.e., bifurcated) ones. When n = 3, water molecules form a cyclic H-bonded network, which has a close resemblance with the tetrameric form of such water clusters with different charged species.82,83 While considering CorH+–Wn and DbcH+–Wn (n = 1–3) clusters, we have similar findings. Indeed, monohydrated CorH+ and DbcH+ are stabilised by an intermonomer H-bond. Di-hydrated CorH+ and DbcH+ consist of two water molecules establishing H-bonds, each with an aliphatic C–H of CorH+ or DbcH+. For CorH+–W3 and DbcH+–W3, we have a network of three water molecules bonded by four H-bonds. In addition, Fig. 1 shows that extending the aromatic π-clouds affects the H-bond distances and geometrical parameters. For example, the primary shell H-bond distance in BzH+–W1 is ∼1.742 Å, which is lengthened, due to the H-bond strength weakening, to 2.164 Å and 2.333 Å in CorH+–W and in DbcH+–W, respectively. A similar trend is observed for CorH+–W2, CorH+–W3, DbcH+–W2 and DbcH+–W3 clusters. In sum, the extension of the π-aromatic cloud significantly influences the strength of C–H+⋯O and O–H⋯O interactions, i.e. it reduces the strength of the hydration/solvation structure. In contrast, the proton transfer process enhances the H-bonding strength as well as shortens the water–water interactions in protonated water PAH clusters.66
2. Binding energies of BzH+–Wn, CorH+–Wn and DbcH+–Wn (where n = 1–3) complexes
Table 1 lists the BEs of BzH+–Wn, CorH+–Wn and DbcH+–Wn (n = 1–3) complexes as computed at the PBE0/6-311++G** and PBE0-D3/6-311++G** levels i.e., without and with considering the D3 dispersion correction. This table shows that dispersion correction leads to 5–10% larger BEs (in absolute values). This is due to the better consideration of the long-range interactions within the complexes. This table shows that MP2 leads to less deep potential wells compared to PBE0-D3, whereas the differences remain small. Hereafter, we will refer to the PBE0-D3/6-311++G** data.| N | BzH+–Wn | CorH+–Wn | DbcH+–Wn |
|---|---|---|---|
| a Taken from ref. 66 and 69. b Using the aug-cc-pVTZ basis set. Taken from ref. 69. c This geometry is from this work for the cyclic H-bonded water cluster model. | |||
| 1 | −12.76a (−11.92)b | −7.09 | −5.40 |
| −13.97 | −8.17 | −6.55 | |
| −13.15b | — | — | |
| 2 | −21.49a (−19.84)b | −13.40 | −9.92 |
| −24.10 | −13.46 | −13.03 | |
| −19.44 (−20.07)b | — | — | |
| 3 | −36.40a (−34.17)b | −25.24 | −20.91 |
| −32.69 | −21.2 | −17.1 | |
| −28.16c | — | — | |
For BzH+–Wn, CorH+–Wn and DbcH+–Wn (n = 1–3) clusters, Table 1 shows that, for a given number of water molecules, the C–H+⋯O H-bond interaction energies decrease, in absolute values, while increasing the size of the aromatic compound. For instance, we compute BEs of −13.97, −8.17, and −6.55 in kcal mol−1 for BzH+–W1, CorH+–W1 and DbcH+–W1, respectively. Thus, the reduction in the strength reaches up to 40 to 50% upon extending the π-cloud. In fact, the extension of the aromatic surface leads to a decrease of the interaction strength irrespective of the nature of the water clusters signature of the decrease of the hydration energy of the proton when the excess proton is attached to such an organic surface model.
In contrast, the |BEs| increase while increasing the number of water molecules surrounding protonated BzH+, CorH+ or DbcH+, where we observe an increase of 3–4 times when going from the mono- to di- to tri-hydrated respective clusters. Interestingly, our work shows that we can modulate, via the extension of the size of the aromatic, the interaction, at the microscopic level, between the protonated sp3 carbon of the aromatic compounds, and the neighboring network of water molecules participating in the solvation of these species. This may result in the activation of the solvated compound as we noticed for CO2 interacting, via σ H-bond or π stacking interactions, with organic molecules, metal surfaces or ionic liquids.84–86 We expect applications of these findings at least in organic chemistry where protonated aromatics are important intermediates, for instance, in electrophilic substitutions.
a. AIM electron density topography. To get some insights into the interfacial interactions, the AIM approach is used. This allows description of the non-covalent interactions between BzH+, CorH+ and DbcH+ and water clusters, where topological parameters at bond critical points (BCPs) are determined. Indeed, the strength and the nature of non-covalent interactions are connected to the electron density (ρ(rc)) and its Laplacian (∇2ρ(rc)) values at BCPs. Fig. 2 shows the electron density analysis at H-bonded critical points (HBCPs) and non-bonded critical points (NBCPs) for BzH+–Wn, CorH+–Wn and DbcH+–Wn (n = 1–3). This figure reveals that there are several kinds of non-covalent interactions stabilizing these complexes, where various bond paths are formed between protonated aromatic compounds and water clusters.
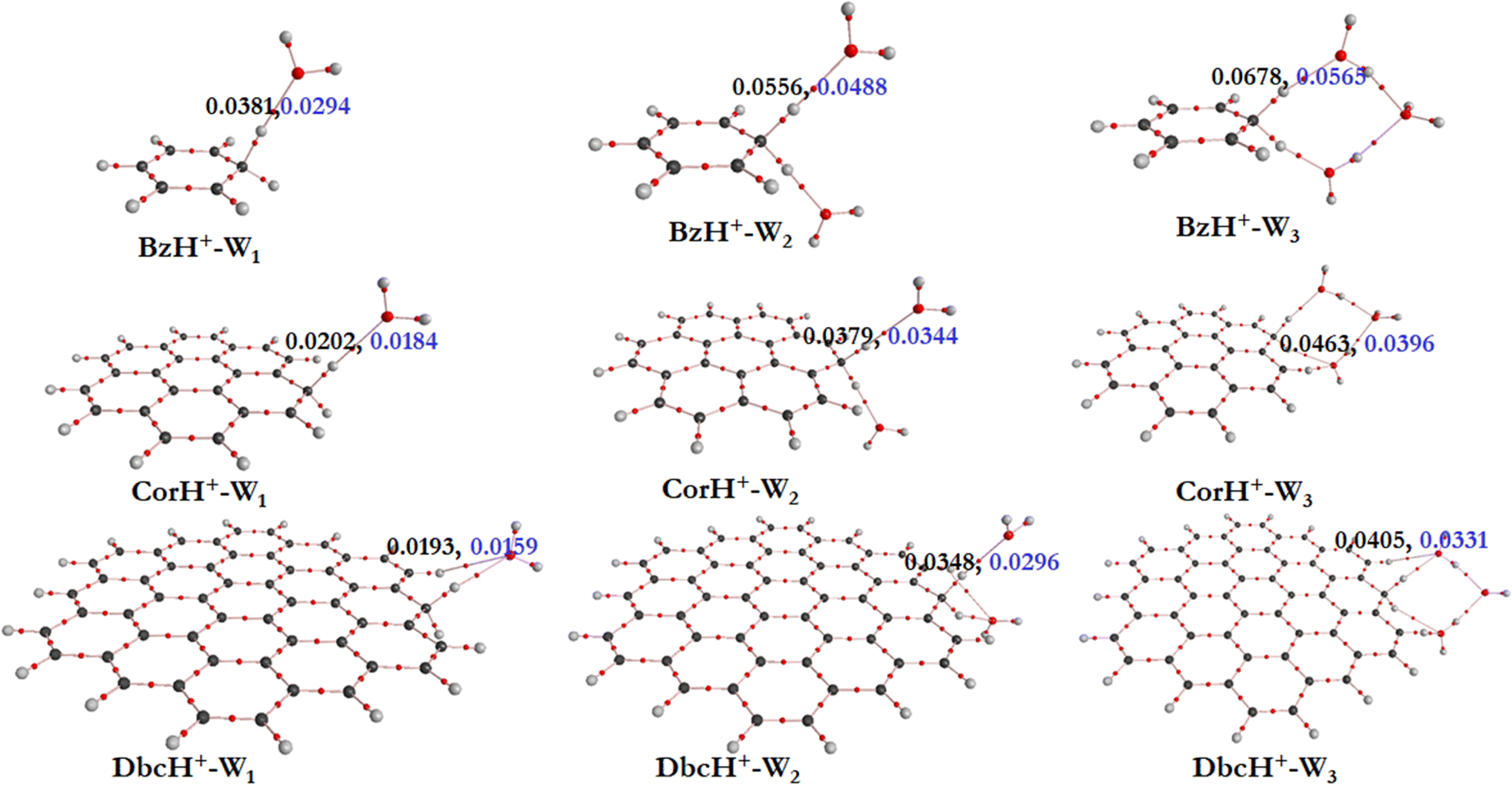 | ||
| Fig. 2 PBE0/6-311++G** molecular topography analyses of BzH+–Wn, CorH+–Wn and DbcH+–Wn (n = 1–3) clusters as determined from the corresponding total electron densities. Red dots correspond to bond critical points (BCPs). We give also their total electron density ρ(rc) (in black color) and ∇2ρ(rc) (in blue color). These values are shown in a.u. | ||
For BzH+–W1, the calculated ρ(rc) and ∇2ρ(rc) values at the HBCP of C–H+⋯O interaction are 0.0381 and 0.0294 a.u., respectively. In the case of BzH+–W2 and BzH+–W3 clusters, the ρ(rc) (∇2ρ(rc)) values at the HBCP are 0.0556 (0.0488) and 0.0678 (0.0565) a.u. It is worth noticing that the electron density increases while increasing the hydration number. This reveals that there is no proton migration to water clusters i.e., the excess proton remains localized at the aromatic organic side as discussed above. Also, it is interesting to note that when the hydration number increases from 1 to 3, the H-bond strength gradually decreases. This is due to the charge distribution over the whole cluster. This is in line with the lengthening of the H-bond(s) between BzH+ upon increasing the number of water molecules attached to BzH+. Similar effects are found for the protonated CorH+/DbcH+ hydrates.
b. Non-covalent interaction indexes. Non-covalent interaction indexes allow the quantitative assessment of the non-covalent interactions established between water clusters and protonated aromatics. Fig. 3 shows the NCI plots of BzH+–Wn, CorH+–Wn and DbcH+–Wn (n = 1–3) clusters. We observe the existence of strong interaction regions (in blue) between water clusters and these aromatics. In addition, we notice the further stabilization of some water clusters via weak van der Waals interactions (in green). The relative contribution of both kinds of interactions depends on the cluster type.
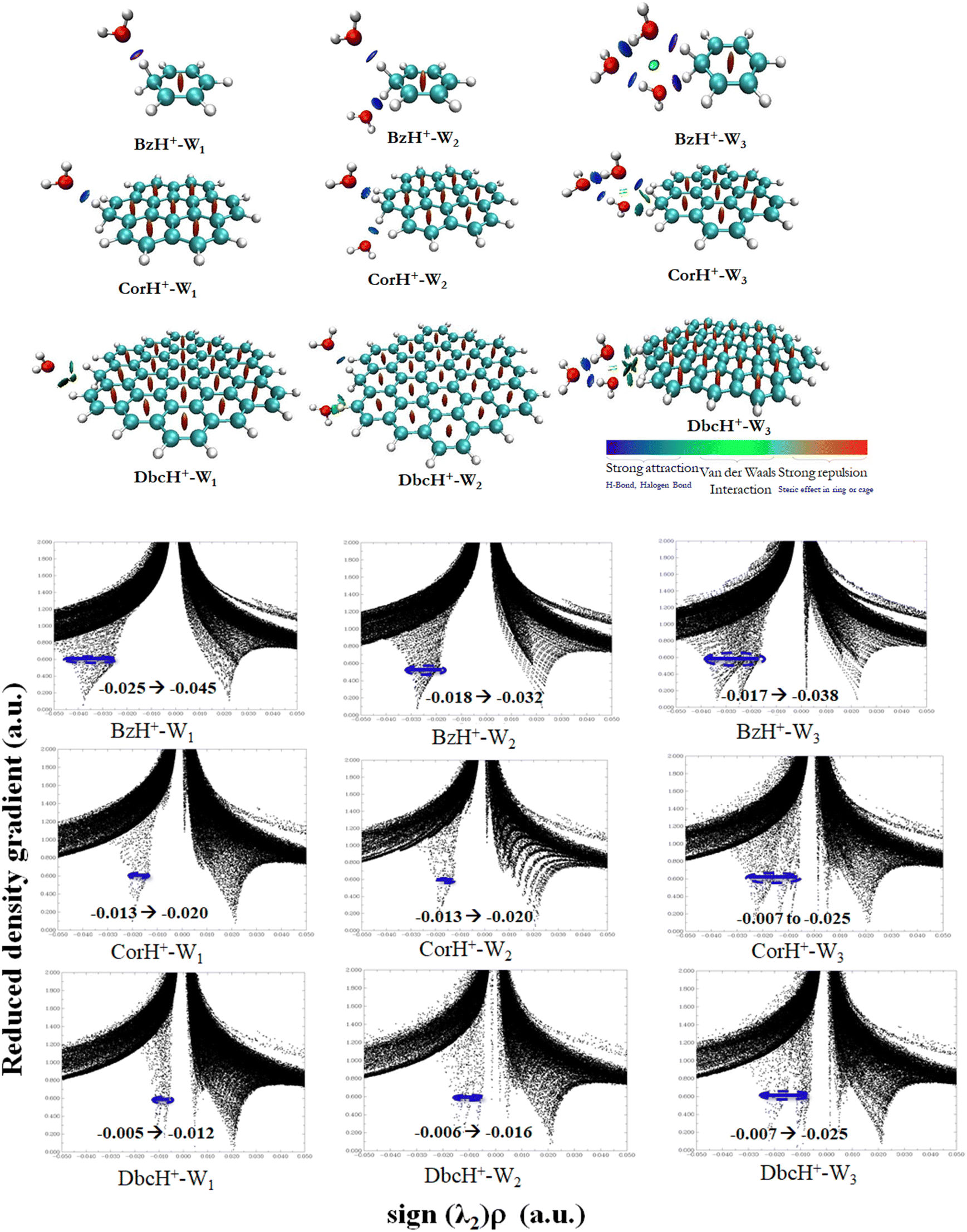 | ||
| Fig. 3 Top: NCI indexes of the BzH+–Wn, CorH+–Wn and DbcH+–Wn (n = 1–3) clusters with an isosurface of 0.59 a.u. We show the strong and weak interaction regions in blue and green, respectively. Bottom: Bi-dimensional plots of reduced density gradient RDG versus sign (λ2)*ρ of BzH+–Wn, CorH+–Wn and DbcH+–Wn (n = 1–3) clusters. | ||
Mainly, strong interactions dominate within BzH+–Wn, CorH+–Wn and DbcH+–Wn (n = 1–3) clusters. Therefore, similar covalent and non-covalent interactions, with various strengths, are in action for the stabilization of these clusters whatever the extension of the π-aromatic cloud. The bi-dimensional plots of reduced density gradient RDG versus sign (λ2)*ρ (Fig. 3) allow the assessment of quantitative information about the strength of H-bonded interaction within these various clusters. The strong repulsion and attraction in van der Waals interactions are represented in red and green areas, respectively; meanwhile, the H-bonds are represented by blue area.
c. Energy decomposition analysis (EDA). As revealed by the NCI analyses, various terms contribute to the intermonomer interactions within BzH+–Wn, CorH+–Wn and DbcH+–Wn (n = 1–3). Such effects can be quantified via energy decomposition analyses, which allow an understanding of the different energy contributions. At present, these are done at the SAPT0/aug-cc-pVDZ level as implemented in PSI-4. Within this approach, the total interaction energy (IEtotal) contains the following contributions: electrostatic (E(electrostatic)), exchange-repulsion (E(exchange-repulsion)), induction (E(induction)), and dispersion (E(dispersion)) energies:
| IEtotal = E(electrostatic) + E(exchange-repulsion) + E(induction) + E(dispersion) | (2) |
Fig. 4 plots the individual contributions for BzH+–Wn, CorH+–Wn and DbcH+–Wn (n = 1–3) complexes. This figure shows that the E(electrostatic), E(induction) and E(dispersion) energies are negative, favouring thus the stability of these weakly bound complexes. The dominant stabilising contributions are of electrostatic nature since |E(electrostatic)| is the largest. E(exchange-repulsion) energies are, however, positive and thus contribute to the non-stability of these molecular species. Nevertheless, the sum of E(electrostatic), E(induction) and E(dispersion) is larger, in absolute value, than E(exchange-repulsion) leading to negative IEtotal energies, which are in line with the stable character of the clusters under study as certified by their negative BEs and AIM analyses. Moreover, Fig. 4 reveals that the IEtotal energies and their various terms increase, in absolute value, upon increasing the number of water molecules because of the establishment of additional σ-type H-bonded interactions within such species. In contrast, the opposite trend is observed when the π-cloud is extended from Bz to Cor to Dbc.
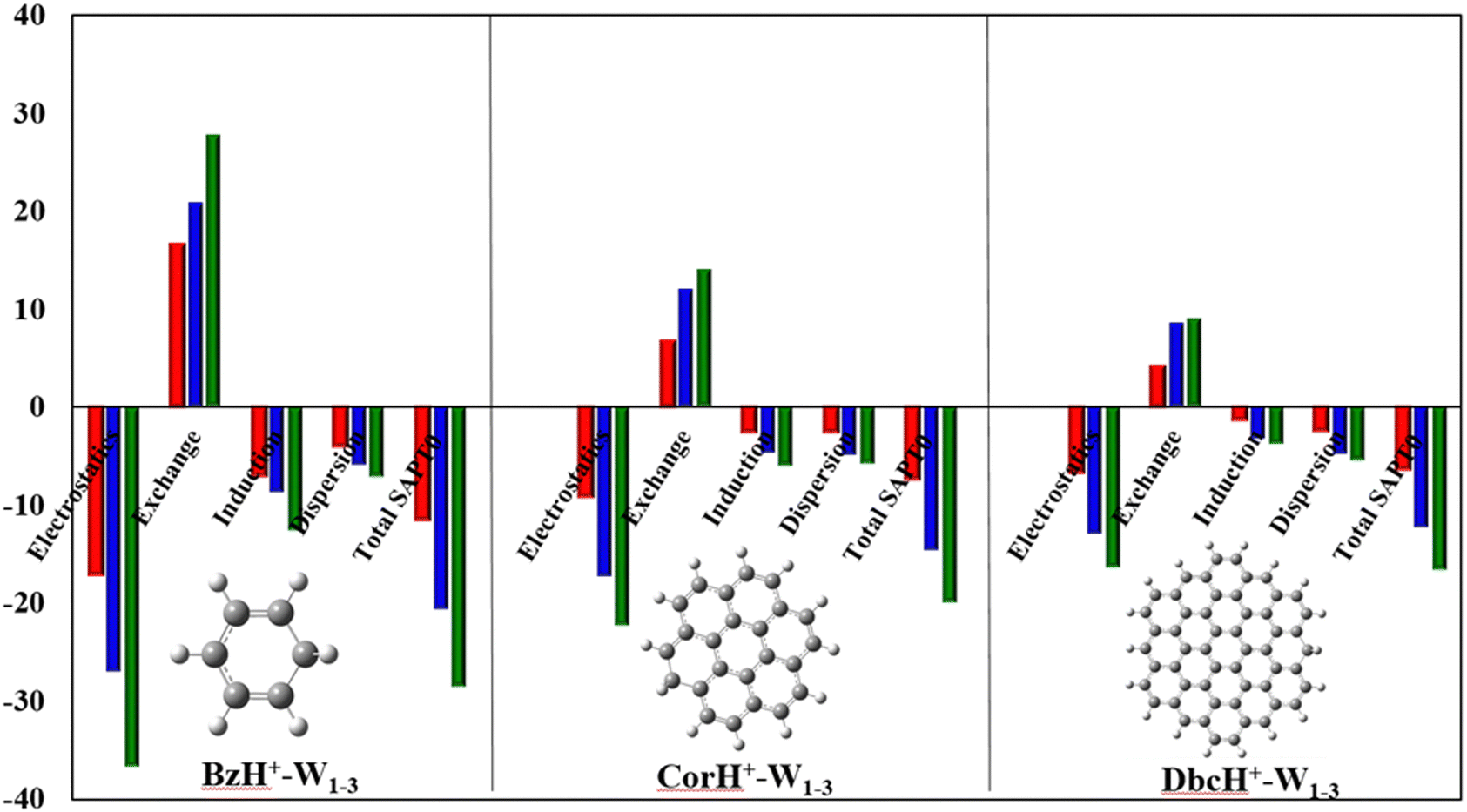 | ||
| Fig. 4 SAPT0/aug-cc-pVDZ energy decomposition analyses of BzH+–Wn, CorH+–Wn and DbcH+–Wn (n = 1–3) complexes. The mono-, di- and tri-hydrated values are given in red, blue and green, respectively. All values are in kcal mol−1. | ||
d. Vibrational analyses and IR spectra. Previous works showed that IR spectroscopy is useful to characterize protonated aromatic compounds in the gas phase and to probe the interfacial interactions between organic compounds and water molecules. We thus computed, at the PBE0(-D3)/6-311++G** level, the vibrational harmonic frequencies of bare BzH+, CorH+ and DbcH+ and those of their complexes with one, two and three water molecules. Afterwards we deduced the corresponding anharmonic frequencies by scaling the harmonic ones. They are listed as νs in Tables 2 and 3. For BzH+–Wn (n = 0–3) and CorH+–Wn (n = 0–1), we have also computed their anharmonic frequencies at the PBE0 + D3/6-311++G** level and using perturbation theory (PT2) as implemented in Gaussian 16. The latter are denoted as νp in Tables 2 and 3. In the following, we will concentrate on the anharmonic frequencies > 2500 cm−1. Mainly, they correspond to the C–H+ (sp3) and the C–H (sp2) stretchings and those arising from the complexation with water molecules. The latter are classified as Awater, AAwater and ADwater for single acceptor, double acceptor and single acceptor-single donor H-bonded stretching in water molecules, respectively. We give also the respective complexation induced shifts. The shifted frequencies were evaluated as the differences between the frequencies of the bare protonated aromatic and water molecule and those of this compound complexed with water clusters. For an isolated water molecule, the experimental and calculated stretching frequencies are 3757 cm−1
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 87 and 3824 cm−1, respectively. Note that free sp3 C–H stretchings have low IR intensities and they will not be discussed further.
87 and 3824 cm−1, respectively. Note that free sp3 C–H stretchings have low IR intensities and they will not be discussed further.
| n | Assignment | PBE0 | PBE0-D3 | |
|---|---|---|---|---|
| ν s | ν s | ν p | ||
| a Experimental IR spectroscopy in the gas phase. Ref. 29. b Experimental IR spectroscopy in the gas phase. Ref. 36. c Experimental IR spectroscopy in the gas phase. Ref. 33. d Experimental IR spectroscopy in the gas phase. Ref. 36. | ||||
| 0 | Aliphatic sp3 C–H+ stretching | 2856 | 2858 | 2977 |
| 2810a | ||||
| 2820b | ||||
| 2700–3200c | ||||
| 2891d | ||||
| Aromatic sp2 C–H stretching | 3080 | 3079 | 3210 | |
| 3110a | ||||
| 3006b | ||||
| 1 | Free sp3 C–H+ stretching mode | 2952 (−96) | 2964 (−106) | 3089 (−112) |
| Free sp2 C–H stretching | 3082 (−2) | 3081 (−2) | 3211 (−1) | |
| H-bonded C–H+ stretching to Awater (νss) | 2323 (533) | 2269 (589) | 2365 (612) | |
| Free O–H stretching in Awater (νas) | 3793 (31) | 3794 (31) | 3955 (35) | |
| 2 | Free sp3 C–H+ stretching | Absence of sp3 C–H+ stretching | ||
| Free sp2 C–H stretching | 3080 (0) | 3079 (0) | 3209 (1) | |
| H-bonded C–H+ stretching to Awater (νas) | 2637 (219) | 2581 (277) | 2690 (287) | |
| Free O–H stretching in Awater (νas) | 3800 (24) | 3801 (26) | 3938 (52) | |
| 3 | Free sp2 C–H stretching | 3070 (10) | 3080 (−1) | 3210 (0) |
| H-bonded C–H+ stretching to ADwater | 2577 (279) | 2447 (411) | 2679 (298) | |
| H-bonded O–H stretching in ADwater (νas) | 3780 (48) | 3780 (47) | 3943 (47) | |
| Free O–H stretching in AAwater (νas) | 3776 (52) | 3778 (49) | 3938 (52) | |
| n | Assignment | CorH+–Wn | DbcH+–Wn | |||
|---|---|---|---|---|---|---|
| PBE0 | PBE0-D3 | PBE0 | PBE0-D3 | |||
| ν s | ν s | ν p | ν s | ν s | ||
| a Experimental IR spectroscopy in the gas phase. Ref. 38. | ||||||
| 0 | Aliphatic sp3 C–H+ stretching | 2898 | 2899 | 3022 | 2926 | 2926 |
| 2813a | ||||||
| Aromatic sp2 C–H stretching | 3085 | 2954 | 3215 | 3082 | 3081 | |
| 1 | Free sp3 C–H+ stretching mode | 2900 (−2) | 2843 (56) | 3003 (19) | 2887 (39) | 2890 (30) |
| Free sp2 C–H stretching | 3084 (1) | 2956 (−2) | 3197 (18) | 3064 (18) | 3080 (−1) | |
| H-bonded C–H+ stretching to Awater (νss) | 2771 (127) | 2841 (58) | 2961 (61) | 2926 (0) | 2928 (−2) | |
| Free O–H stretching in Awater (νas) | 3807 (20) | 3802 (25) | 3963 (27) | 3805 (19) | 3805 (22) | |
| 2 | Free sp3 C–H+ stretching | Absence of sp3 C–H+ stretching | ||||
| Free sp2 C–H stretching | 3083 (2) | 3071 (−117) | 3190 (25) | 3081 (1) | 3080 (−154) | |
| H-bonded C–H+ stretching to Awater (νss) | 2791 (107) | 2840 (100) | 2962 (60) | 2846 (80) | 2883 (43) | |
| Free O–H stretching in Awater (νas) | 3807 (20) | 3786 (41) | 3946 (44) | 3807 (20) | 3807 (20) | |
| 3 | Free sp2 C–H stretching | 3068 (17) | 3071 (−117) | — | 3064 (18) | 3081 (0) |
| H-bonded C–H+ stretching to ADwater | 2693 (205) | 2668 (72) | — | 2891 (35) | 2905 (21) | |
| H-bonded O–H stretching in ADwater (νas) | 3780 (47) | 3781 (46) | — | 3782 (45) | 3782 (45) | |
| Free O–H stretching in AAwater (νas) | 3785 (42) | 3786 (41) | — | 3790 (37) | 3791 (36) | |
Tables 2 and 3 show that the PBE0 and PBE0-D3 νs values are very close, whereas the corresponding data are quite different from the respective νp, questioning thus the validity of the scaling factor used and of the applicability of perturbation theory to deduce such a quantity. Indeed, the scaling factor was derived ∼20 years ago and it should be updated. Besides, perturbation theory may not be well suited for such weakly bound complexes, which exhibit low frequency modes, possibly inducing some divergences upon determination of the derivatives needed for anharmonicity effect evaluations, even for modes > 2500 cm−1. One needs highly resolved IR spectra for validation. Interestingly, the complexation induced shifts are not affected, where we have similar shifts either including D3 or not and either scaled or PT2 anharmonic frequencies.
For the benzenium cation, the PBE0 and PBE0-D3 C–H+ (sp3) and the C–H (sp2) stretching νs are computed as ∼2856 and ∼3080 cm−1, respectively. These values are close to the experimental ones. For instance, the C–H+ (sp3) stretching was measured as 2810, 2820, 2700–3200, and 2891 cm−129,33,36 and the C–H (sp2) stretching was determined as 3110 and 3006 cm−1.29,36 Our computed values are also close to those determined using the B3LYP/6-311+G(d,p) method (i.e. the C–H+ (sp3) stretching = 2887 cm−1 and the C–H (sp2) stretching = 3114 cm−1).36 In contrast, the corresponding νp values are relatively high (= 2977 and 3210 cm−1, respectively). A priori, this validates the use of the scaling factor and not of PT2 for BzH+.
Table 3 gives the νs values of CorH+–Wn and DbcH+–Wn clusters. In the following we will quote those computed at the PBE0-D3/6-311++G** level. For CorH+, we calculate 2899 cm−1 and 2954 cm−1 for the C–H+ (sp3) and C–H (sp2) stretchings, which compare favorably with the experimental value of aliphatic sp3 C–H+ stretching (2813 cm−1).38 For bare DbcH+, our predictions for these modes are 2926 cm−1 and 3081 cm−1, respectively. The existence of H-bonded interactions between aliphatic C–H+ and water molecule(s) may influence the C–H stretching modes. The conventional H-bonded interactions induce a shift in frequency. For monohydrates of BzH+, CorH+ and DbcH+, the H-bonded aliphatic C–H+ stretchings are reduced upon increasing the aromatic π-clouds. Indeed, we compute a redshift of 589 cm−1 for BzH+–W1 that drops to 58 cm−1 for CorH+–W1 and vanishes for DbcH+–W1. Besides, there is a blue shift in free aliphatic C–H stretching in BzH+–W1. This stretching mode in the extended π-cloud in CorH+–W1 and DbcH+–W1 is predicted to be 2843 and 2890 cm−1. Interestingly, the free aliphatic sp3 C–H+ stretching mode in BzH+ is blue shifted, whereas in DbcH+ it is redshifted (by ∼30 cm−1). This important finding shows how the extension of the π-cloud influences the non-H-bonded C–H stretching modes via the perturbation of the electronic structure of the aromatic rings.
For dihydrates of BzH+, CorH+ and DbcH+, both aliphatic sp3 C–H+ modes are directly involved in the H-bonded stretching to water molecules. For these clusters, Tables 2 and 3 show that the water stretching redshift values are decreasing, while increasing the aromatic π-cloud. Indeed, the corresponding redshift values are 277 cm−1, 100 cm−1 and 43 cm−1 for BzH+–W2, CorH+–W2 and DbcH+–W2, respectively. In trihydrated clusters of BzH+, CorH+ and DbcH+, the H-bond to water is observed in both the hydrogens present in sp3 carbon and stretching redshift values are affected due to the third AA (double acceptor)-water present in between them, where the water molecules form a cyclic structure with the hydrogen atom. The redshifts for BzH+–W3, CorH+–W3 and DbcH+–W3 are 411 cm−1, 72 cm−1 and 21 cm−1, respectively. As expected, the free sp2 C–H stretchings are slightly affected by microhydration of BzH+, CorH+ and DbcH+.
Fig. 5 displays the IR spectra of bare BzH+, CorH+ and DbcH+, and of their mono-, di and trihydrates as computed using the PBE0-D3/6-311++G** method. For the clusters not probed yet by IR, these spectra may be used for the assignment of the corresponding spectra whenever measured. In particular, they can help to disentangle the IR spectra of protonated aromatics interacting with water clusters and those of aromatics interacting with protonated water clusters in a mixture using the presently computed IR spectra and those of Bz–H+Wn, Cor–H+Wn and Dbc–H+Wn (n = 1–3) as given in ref. 66. Indeed, different spectral shapes are computed for each set of complexes.
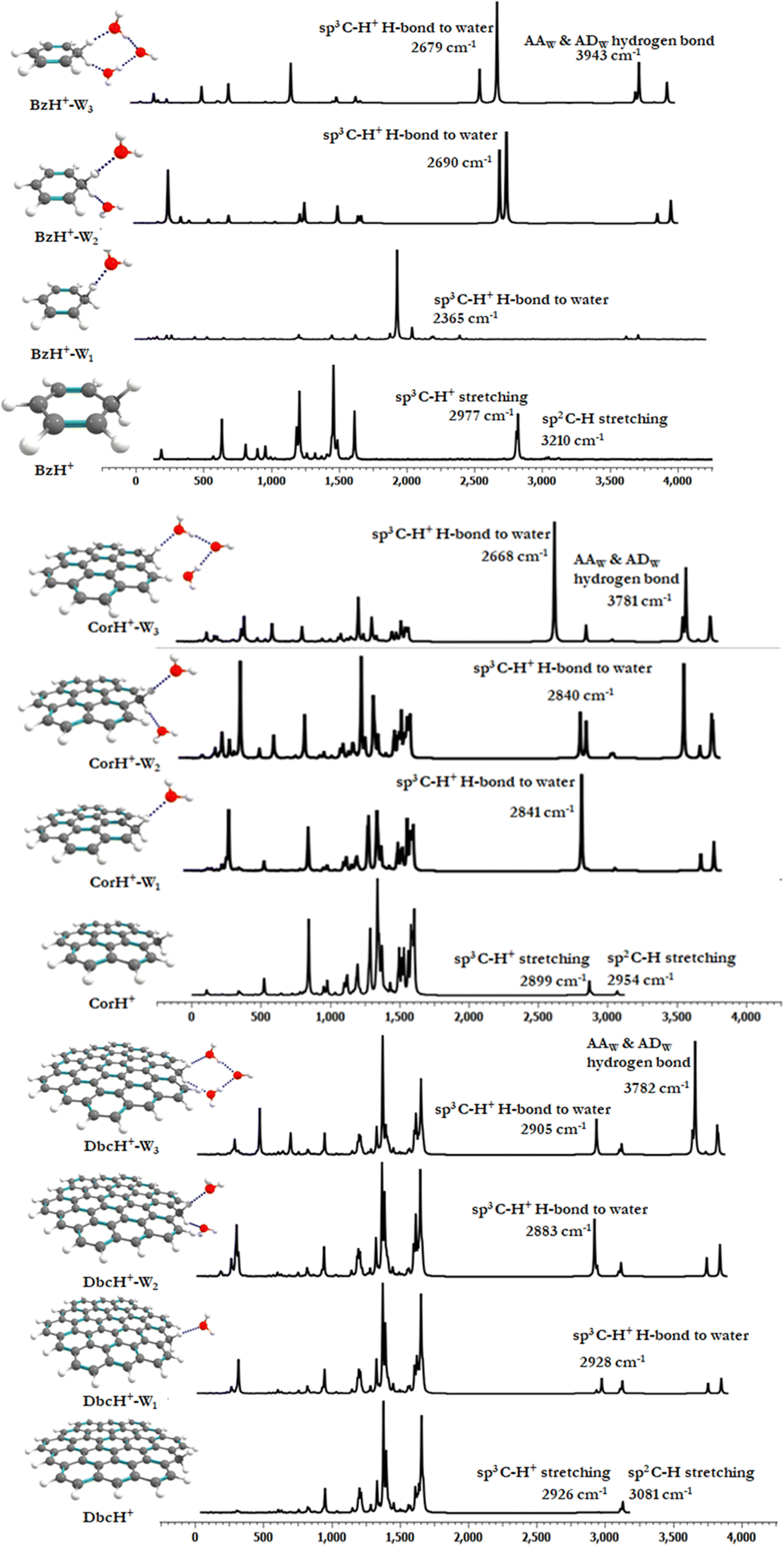 | ||
| Fig. 5 IR spectra of bare BzH+, CorH+ and DbcH+ and of their clusters with water as computed at the PBE0-D3/6-311++G** level. | ||
e. UV-vis spectra. The full set of singlet and triplet electronic states of BzH+, CorH+ and DbcH+, and of their clusters with water (i.e., BzH+–Wn, CorH+–Wn and DbcH+–Wn (n = 1–3)) is listed in Tables S6 and S7 (ESI†) for transitions with λ > 200 nm. These data are obtained at the TD-DFT PBE0/6-311++G** level. We give also their tentative assignments. Mostly, they correspond to the promotion of one electron from the occupied highest molecular orbital (MO) to the lowest unoccupied MO. These MOs are depicted in Fig. S2–S4 (ESI†), which show that they are mainly located on the organic part with some contributions on the water clusters. Although, the lowest singlets of BzH+ and of CorH+ were studied both theoretically and experimentally (Table 4),39–41,44,46 the singlet states of DbcH+ and those of their mono-, di- and trihydrates are treated here for the first time. Also, the results on the triplets represent predictions.
| n | Transition | E | λ | f |
|---|---|---|---|---|
| a Adiabatic electronic excitation transition. | ||||
| BzH+ | ||||
| 0 | S0 → T1 | 2.21 | 560 | |
| S0 → S1 | 4.24 | 292 | 0.12 | |
| (330)40,41 | ||||
| (325)39 | ||||
| (325)44 | (0.14)44 | |||
| 1 | S0 → T1 | 2.51 | 493 | |
| S0 → S1 | 4.40 | 281 | 0.11 | |
| 2 | S0 → T1 | 2.52 | 492 | |
| S0 → S1 | 4.38 | 282 | 0.11 | |
| 3 | S0 → T1 | 2.65 | 467 | |
| S0 → S1 | 4.45 | 278 | 0.10 | |
| +∞ | S0 → T1 | 464 ± 50 | ||
| 41389 | ||||
| S0 → S1 | 277 ± 15 | |||
| 28488 | ||||
| CorH+ | ||||
| 0 | S0 → T1 | 1.20 | 1031 | |
| S0 → S1 | 2.08 | 595 | 0.08 | |
| (1.78)46a |
(695.6)46a |
|||
| 1 | S0 → T1 | 1.28 | 961 | |
| S0 → S1 | 2.16 | 573 | 0.08 | |
| 2 | S0 → T1 | 1.36 | 905 | |
| S0 → S1 | 2.23 | 555 | 0.08 | |
| 3 | S0 → T1 | 1.39 | 889 | |
| S0 → S1 | 2.25 | 549 | 0.08 | |
| +∞ | S0 → T1 | 849 ± 50 | ||
| S0 → S1 | 534 ± 15 | |||
| DbcH+ | ||||
| 0 | S0 → T1 | 0.87 | 1411 | |
| S0 → S1 | 1.48 | 836 | 0.14 | |
| 1 | S0 → T1 | 0.91 | 1356 | |
| S0 → S1 | 1.51 | 816 | 0.14 | |
| 2 | S0 → T1 | 0.96 | 1287 | |
| S0 → S1 | 1.56 | 791 | 0.15 | |
| 3 | S0 → T1 | 0.97 | 1273 | |
| S0 → S1 | 1.57 | 785 | 0.15 | |
| +∞ | S0 → T1 | 1196 ± 50 | ||
| S0 → S1 | 754 ± 15 | |||
The UV-vis absorption spectra of BzH+–Wn, CorH+–Wn and DbcH+–Wn (n = 1–3) are shown in Fig. 6. This figure reveals that different UV-vis spectra are computed for each molecular species, depending on the extension of the aromatic compound and on the number of water molecules clustered with them. For instance, Fig. 6 shows that the characteristic peaks of BzH+–W1, CorH+–W1 and DbcH+–W1 clusters have different energy positions from those of BzH+, of CorH+, and of DbcH+, where shorter wavelengths are computed upon attaching water molecules to these H+PAHs. This trend is found for BzH+–Wn, CorH+–Wn and DbcH+–Wn clusters upon increasing n. This is further confirmed by the computed vertical transition energies/wavelengths given in Table 4. Indeed, we compute S0 → S1 transitions at 292, 281, 282 and 278, in nm, for BzH+, BzH+–W1, BzH+–W2 and BzH+–W3, respectively. For CorH+–Wn clusters, we also have such a decrease of wavelengths upon increasing the number of water molecules interacting with Cor+, where we compute S0 → S1 transitions at 595, 573, 555 and 549 nm for n = 0, 1, 2 and 3, respectively. The S0 → S1 transitions for DbcH+–Wn (n = 0, 1, 2 and 3) are predicted at 836, 816, 791 and 785 nm.
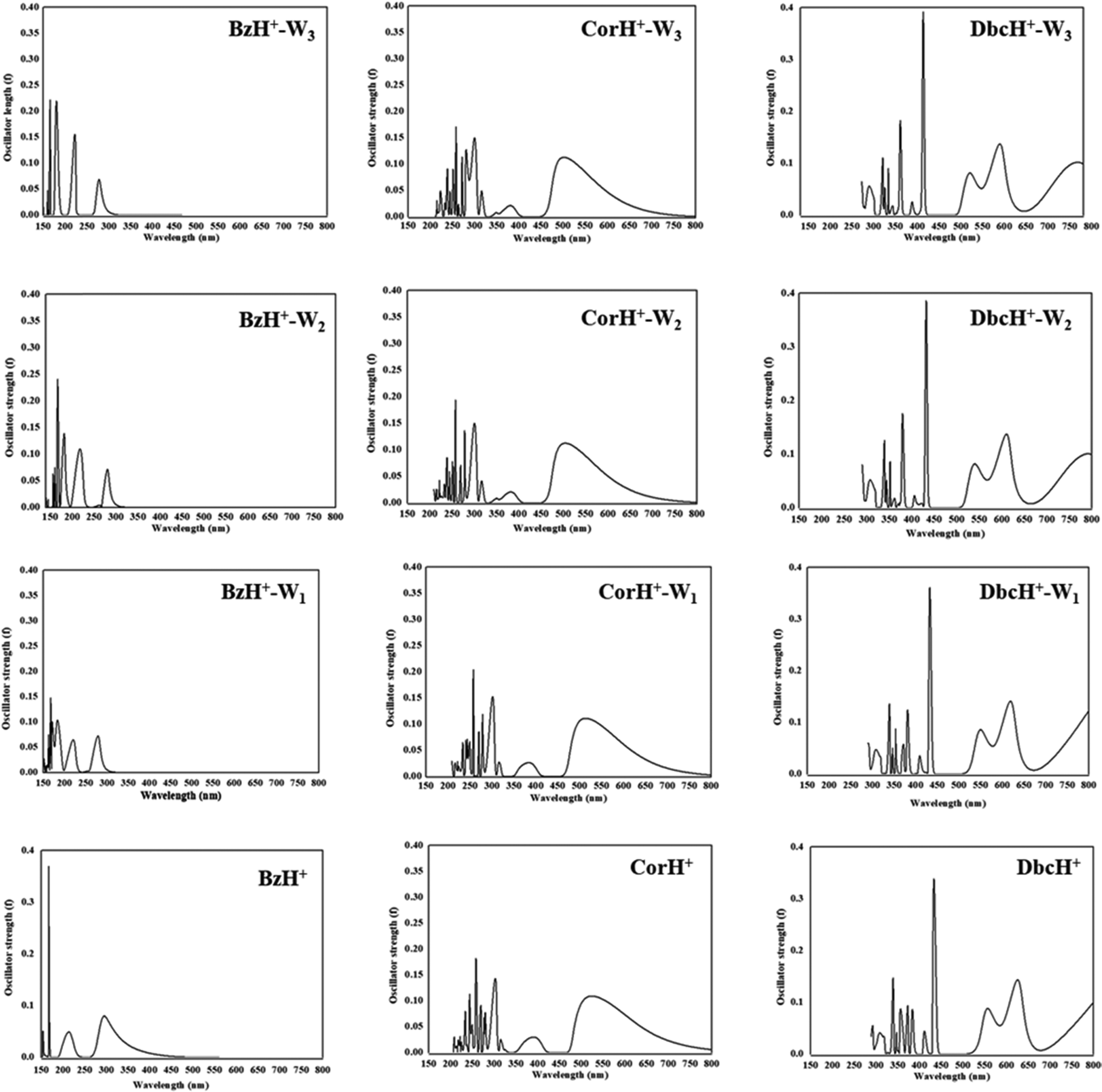 | ||
| Fig. 6 UV-vis spectra of bare BzH+, CorH+ and DbcH+, and of their clusters with water BzH+–Wn, CorH+–Wn and DbcH+–Wn (where n = 1–3) as computed at the TD-DFT PBE0/6-311++G** level. | ||
Fig. 6 shows that bands appear in the visible domain upon extending the π-aromatic cloud from BzH+ to CorH+ to DbcH+ as already noticed experimentally for the extension from BzH+ to CorH+44,46 and for other isolated H+PAHs such as protonated naphthalene, protonated anthracene, protonated tetracene, and protonated pyrene.42,43,46 We have a similar trend for these protonated species complexed with water molecules. For illustration, Table 4 shows that the S0 → T1 and S0 → S1 transition wavelengths increase upon extending the π-aromatic cloud for these bare H+PAHs. For the S0 → S1 transitions, they are predicted at 292 nm, 595 nm and 836 nm for BzH+, CorH+ and DbcH+, respectively. Also, Table 4 shows that this trend is observed for the S0 → T1 transitions of all species and for the S0 → S1 transitions of BzH+–Wn, CorH+–Wn and DbcH+–Wn (n = 1–3). For instance, the computed λ values are 493 nm, 961 nm and 1356 nm for the S0 → T1 BzH+–W1, CorH+–W1 and DbcH+–W1 transitions, respectively. Note that our calculated S0 → S1 wavelengths for BzH+ and CorH+ are slightly lower than the measured ones. The differences may come from perturbations of the singlets states by vibronic or Herzberg–Teller couplings as pointed out in ref. 39–41, 44 and 46 not accounted for in the present computations.
The trends upon the extension of the π-aromatic cloud or upon adding water molecules can be explained by examining the frontier MOs given in Fig. S2–S4 (ESI†). Indeed, the S0 → T1 and S0 → S1 transitions are associated with the promotion of an electron from the HOMO to the LUMO (Tables S6 and S7, ESI†). Fig. S2–S4 (ESI†) show that the HOMOs of BzH+–Wn, CorH+–Wn and DbcH+–Wn (n = 1–3) species are destabilized by the anti-bounding nature of the interaction between the organic and the water molecules’ MOs. This results in the increase of the energies of these HOMOs, whereas we observe a favorable bonding between both entities in the LUMOs. In total, we have reduction of their HOMO–LUMO gaps and the increase of the corresponding wavelength transitions.
Besides, the S0 → T1 and S0 → S1 transition wavelengths decrease by increasing the number of water molecules attached to the protonated aromatics. For instance, the S0 → S1 transition wavelengths change from 292 nm to 278 nm for BzH+–Wn (n = 0–3), from 595 nm to 549 nm for CorH+–Wn (n = 0–3) and from 836 nm to 785 nm for DbcH+–Wn (n = 0–3). For the S0 → T1 transitions, these values change from 560 nm to 467 nm, from 1031 nm to 889 nm and from 1411 nm to 1273 nm in the same order. These evolutions are non-monotonic and they can be fitted by A + B × exp(−C × n), where n is the number of water molecules attached to these protonated aromatics. In this formula, the A parameter corresponds to the λ+∞ wavelength transition for n → +∞ and thus could be assumed to be the value of the wavelength transition for these H+PAHs in aqueous media. Table 4 gives the values of λ+∞ for the S0 → S1 and S0 → T1 transitions. We show also the λ values of BzH+ dissolved in liquid hydrogen fluoride containing boron trifluoride and of BzH+MX4− (MX4− = BF4−, AlCl4−, AlBr4−, GaCl4−, GaBr4−) ion pair in an organic solvent solution as measured by Reid88 and by Perkampus and Baumgarten,89 respectively. Although relatively old, these data correspond to the unique data available for BzH+ in the condensed phase to compare with.
For BzH+, the extrapolated S0 → S1 wavelength transition is λ+∞ = 277 ± 15 nm, which agrees quite well with the 284 nm wavelength measured for BzH+ ions dissolved in liquid hydrogen fluoride containing boron trifluoride.88 However, the 413 nm value measured for the BzH+MX4− (MX4− = BF4−, AlCl4−, AlBr4−, GaCl4−, GaBr4−) ion pair in organic solvents89 is obviously far from the present λ+∞ and that of ref. 88. Thus, it cannot be assigned to the S0 → S1 transition. Instead, our extrapolated value for the S0 → T1 transition (λ+∞ = 464 ± 50 nm) is closer and falls within both experimental and theoretical error bars. Accordingly, we suggest to reassign this band to the BzH+ S0 → T1 transition, although it is spin-forbidden. Anyway, such transitions become slightly allowed in emission and responsible for the phosphorescence spectra of molecules. Back to the 1940's, Lewis and Kasha90,91 proved that such S–T absorption transitions can be viewed as a reverse process of phosphorescence and become slightly allowed under spin–orbital perturbations, for instance, by surrounding compounds92 such as solvents or the MX4− anion within the BzH+MX4− ion pair. Since then, such S–T transitions were detected for several compounds93 and even for some neutral PAHs such as anthracene and its derivatives.93 For CorH+ in aqueous solution, we predict S0 → T1 and S0 → S1λ+∞'s at 849 ± 50 nm and 534 ± 15 nm, respectively. These values decrease to λ+∞ = 1196 ± 50 and λ+∞ = 754 ± 15 nm for DbcH+ in a water solvent. Generally, the wavelength transitions in the condensed phase are quite different from those of gas phase species, which illustrates the drastic impact of the surroundings on these H+PAH optical properties. These predictions are worth investigating experimentally. Such experiments are challenging since the extinction coefficients for the singlet–triplet transitions are several orders of magnitude smaller than the values for the singlet–singlet transitions.93
f. Nucleus-independent chemical shift (NICS) analysis. In ref. 94–96 a methodology was proposed to quantify the nature of aromaticity for any molecular system using nucleus independent chemical shifts (NICS) as the negative of the absolute magnetic shielding. The latter is here calculated at the center of the ring (0 Å) and 1 Å above resulting in NICS (0 Å)zz and NICS (1 Å)zz, respectively. The calculated NICS isotropic terms are shown in Table 5. Calculations suggest that the protonation of the PAH significantly reduces the aromatic character at both 0 and 1 Å. In contrast, incorporation of water molecules to the protonated PAH moiety leads to an enhancement of the NICS values (more negative terms mean more aromatic character). Overall, the aromatic character at 1 Å is always higher than at the center of the ring irrespective of the nature of the considered H+PAH and size of the water cluster. In particular, the binding of excess protons on the Bz ring affects NICS (0 Å)zz, whereas the NICS (1 Å)zz terms are slightly affected. At the same time, the extension of the PAH with H-bonded water clusters enhances the aromaticity, and this will help in the stability of the molecular clusters. It is interesting to note that the NICS (0 Å)zz and NICS (1 Å)zz values of bare Cor are significantly lower, in absolute value, at the center of the ring than in the outer ring, where we compute values of −9.6 ppm and −11.3 ppm, respectively. In fact, Cor systems are slightly more aromatic than Bz systems and less aromatic than Dbc ones. This effect is due to the dual aromaticity that exists in the Cor surface. It is also the case in the presence of proton and water clusters attached at the Cor surface.
| PAH | Bz | Cora | Dbc | |||
|---|---|---|---|---|---|---|
| NICS (0 Å)zz | NICS(1 Å)zz | NICS (0 Å)zz | NICS (1 Å)zz | NICS (0 Å)zz | NICS (1 Å)zz | |
| a For Cor, two types of aromaticity exist in the inner and outer rings. The calculated NICS (0 Å)zz and NICS (1 Å)zz for the outer ring of COR are provided in parenthesis. | ||||||
| Bare PAH | −8.2 | −10.4 | −0.1 (−9.6) | −4.6 (−11.3) | −12.9 | −15.6 |
| H+PAH | −0.5 | −5.9 | −1.6 (−8.4) | −5.7 (−10.9) | −2.9 | −6.9 |
| H+PAH-W1 | −2.0 | −6.9 | −1.5 (−8.7) | −5.9 (−12.0) | −3.8 | −7.7 |
| H+PAH-W2 | −1.9 | −6.9 | −1.4 (−8.9) | −5.6 (−11.5) | −4.6 | −8.3 |
| H+PAH-W3 | −2.5 | −7.2 | −1.3 (−7.3) | −5.7 (−10.5) | −4.6 | −8.4 |
IV. Conclusions
The structure, energetic stability, aromaticity, IR and UV-vis features of various H+PAHs with Wn clusters (where n = 1–3) have been computed. It is interesting to note that the extension of the π-cloud directly influences the structure, energetic stability and spectral features of hydrated H+PAH cations. Also, we show that there is a substantial amount of reduction in the energetics, which does not disturb the hydration pattern irrespective of the size of the H+PAH cations. For explanation, we found that this is due to noncovalent interactions that are dominating the microsolvation of H+PAH cations. Furthermore, NICS(0 Å)zz and NICS(1 Å)zz calculations confirm the aromatic character of the Cor and Dbc bearing clusters.The present work shows that the mode of interaction plays a vital role in understanding the strength and stability of the proton at the interface and different vibrational shifts were computed for the complexes under study. The calculated induced complexation shifts are large enough to be probed by IR. Also, computations reveal that the UV-vis spectral fingerprints are specific for each compound. Indeed, different UV-vis spectra are computed depending on the extended aromatic compound and on the number of water molecules interacting with these protonated aromatics. In particular, we show that UV-vis spectroscopy can be used to probe complexation induced effects of H+PAHs upon microhydration. Besides, this spectroscopy can be used to identify the excess proton localized in either the carbon surface or water clusters after comparison of the present spectra to those predicted recently for Bz–H+Wn, Cor–H+Wn and Dbc–H+Wn (n = 1–3) clusters.66 Indeed, the presently computed UV-vis band wavelengths are significantly lower than those when the excess proton is localized at the water clusters.
Finally, our work allows filling the gap between the gas phase H+PAH properties and those of these species in aqueous solution. In particular, we characterized the S0–S1 and S0–T1 transitions of BzH+–Wn, CorH+–Wn and DbcH+–Wn (n = 1–3) in both gas and condensed phases. Although the intensities of the latter are expected to be weak, the direct population of triplet states is important for applications such as designing materials targeting visible-light excitable organic afterglow and for the development of triplet-state involved organic optoelectronic technologies.97
Conflicts of interest
There are no conflicts to declare.Acknowledgements
K. R. M., N. G. L., and C. G. acknowledge the SRM Institute of Science and Technology (SRM-IST) Research Fellowship for their research work. M. P. thanks the Department of Science and Technology, Science and Engineering Research Board (DST-SERB) of India for financial support from the CRG scheme. The authors also thank SRM Supercomputer Centre (HPCC), SRM Institute of Science and Technology for providing the computational facility and financial support.References
- K.-H. Kim, S. A. Jahan, E. Kabir and R. J. Brown, Environ. Int., 2013, 60, 71–80 CrossRef CAS PubMed.
- I. J. Keyte, R. M. Harrison and G. Lammel, Chem. Soc. Rev., 2013, 42, 9333–9391 RSC.
- J. E. Anthony, Angew. Chem., Int. Ed., 2008, 47, 452–483 CrossRef CAS PubMed.
- J. E. Anthony, Chem. Rev., 2006, 106, 5028–5048 CrossRef CAS PubMed.
- M. Frenklach, Phys. Chem. Chem. Phys., 2002, 4, 2028–2037 RSC.
- K.-H. Homann, Angew. Chem., Int. Ed., 1998, 37, 2434–2451 CrossRef PubMed.
- H. Wang, Proc. Combust. Inst., 2011, 33, 41–67 CrossRef CAS.
- H. Richter and J. B. Howard, Prog. Energy Combust. Sci., 2000, 26, 565–608 CrossRef CAS.
- K. O. Johansson, M. P. Head-Gordon, P. E. Schrader, K. R. Wilson and H. A. Michelsen, Science, 2018, 361, 997–1000 CrossRef CAS PubMed.
- G. R. Sampaio, G. M. Guizellini, S. A. da Silva, A. P. de Almeida, A. C. C. Pinaffi-Langley, M. M. Rogero, A. C. de Camargo and E. A. F. S. Torres, Int. J. Mol. Sci., 2021, 22, 6010 CrossRef CAS PubMed.
- O. Idowu, K. T. Semple, K. Ramadass, W. O’Connor, P. Hansbro and P. Thavamani, Environ. Int., 2019, 123, 543–557 CrossRef CAS PubMed.
- N. Premnath, K. Mohanrasu, R. Guru Raj Rao, G. H. Dinesh, G. S. Prakash, V. Ananthi, K. Ponnuchamy, G. Muthusamy and A. A. Arun, Chemosphere, 2021, 280, 130608 CrossRef CAS PubMed.
- C. Joblin and A. G. G. M. Tielens, PAHs and the Universe, EDP Sciences, 2021, ISBN: 978-2-7598-0624-9 Search PubMed.
- D. S. N. Parker and R. I. Kaiser, Chem. Soc. Rev., 2017, 46, 452–463 RSC.
- R. I. Kaiser, D. S. Parker and A. M. Mebel, Annu. Rev. Phys. Chem., 2015, 66, 43–67 CrossRef CAS PubMed.
- B. A. McGuire, A. M. Burkhardt, S. Kalenskii, C. N. Shingledecker, A. J. Remijan, E. Herbst and M. C. McCarthy, Science, 2018, 359, 202–205 CrossRef CAS PubMed.
- B. A. McGuire, R. A. Loomis, A. M. Burkhardt, K. L. K. Lee, C. N. Shingledecker, S. B. Charnley, I. R. Cooke, M. A. Cordiner, E. Herbst, S. Kalenskii, M. A. Siebert, E. R. Willis, C. Xue, A. J. Remijan and M. C. McCarthy, Science, 2021, 371, 1265–1269 CrossRef CAS PubMed.
- M. C. McCarthy, K. L. K. Lee, R. A. Loomis, A. M. Burkhardt, C. N. Shingledecker, S. B. Charnley, M. A. Cordiner, E. Herbst, S. Kalenskii, E. R. Willis, C. Xue, A. J. Remijan and B. A. McGuire, Nat. Astron., 2021, 5, 176–180 CrossRef.
- J. Cernicharo, M. Agúndez, C. Cabezas, B. Tercero, N. Marcelino, J. R. Pardo and P. de Vicente, Astron. Astrophys., 2021, 649, L12 CrossRef PubMed.
- J. Cernicharo, M. Agúndez, R. I. Kaiser, C. Cabezas, B. Tercero, N. Marcelino, J. R. Pardo and P. de Vicente, Astron. Astrophys., 2021, 655, L1 CrossRef CAS.
- J. Cernicharo, R. Fuentetaja, M. Agúndez, R. I. Kaiser, C. Cabezas, N. Marcelino, B. Tercero, J. R. Pardo and P. de Vicente, Astron. Astrophys., 2022, 663, L9 CrossRef CAS.
- https://www.astrochymist.org/astrochymist_ism.html .
- A. G. G. M. Tielens, Annu. Rev. Astron., 2008, 46, 337 Search PubMed.
- D. M. Hudgins, C. W. Bauschlicher and L. J. Allamandola, Spectrochim. Acta, Part A, 2001, 57, 907 CrossRef CAS PubMed.
- O. Dopfer, Laboratory Spectroscopy of Protonated PAH Molecules Relevant For Interstellar Chemistry in PAHs and the Universe, ed C. Joblin and A. G. G. M. Tielens, EAS Publications Series, 2011, vol. 46, pp. 103–108 Search PubMed.
- M. Hammonds, A. Pathak, A. Candian and P. J. Sarre, Spectroscopy of Protonated and Deprotonated PAHs in PAHs and the Universe, ed C. Joblin and A. G. G. M. Tielens, EAS Publications Series, 2011, vol. 46, pp. 373–379 Search PubMed.
- A. L. F. de Barros, A. L. Mattioda, A. Ricca, G. A. Cruz-Diaz and L. J. Allamandola, Astrophys. J., 2017, 848, 112 CrossRef PubMed.
- H. Knorke, J. Langer, J. Oomens and O. Dopfer, Astrophys. J., 2009, 706, L66–L70 CrossRef CAS.
- N. Solcà and O. Dopfer, Angew. Chem., Int. Ed., 2002, 41, 3628–3631 CrossRef.
- M. Bahou, Y. J. Wu and Y. P. Lee, Angew. Chem., Int. Ed., 2014, 53, 1021–1024 CrossRef CAS PubMed.
- V. A. Kapinus, Photophysical Properties of Protonated Aromatic Hydrocarbons, PhD, California Institute of Technology Pasadena, California, 2005 (Defended December 14, 2004) Search PubMed.
- M. V. Frash, A. C. Hopkinson and D. K. Bohme, J. Am. Chem. Soc., 2001, 123(27), 6687–6695 CrossRef CAS PubMed.
- W. Jones, P. Boissel, B. Chiavarino, M. E. Crestoni, S. Fornarini, J. Lemaire and P. Maitre, Angew. Chem., Int. Ed., 2003, 42, 2057–2059 CrossRef CAS PubMed.
- Michael A. Duncan, J. Phys. Chem. A, 2012, 116, 11477–11491 CrossRef CAS PubMed.
- T. C. Cheng, B. Bandyopadhyay, J. D. Mosley and M. A. Duncan, J. Am. Chem. Soc., 2012, 134, 13046–13055 CrossRef CAS PubMed.
- G. E. Douberly, A. M. Ricks, P. V. R. Schleyer and M. A. Duncan, J. Phys. Chem. A, 2008, 112, 4869–4874 CrossRef CAS PubMed.
- U. J. Lorenz, N. Solcà, J. Lemaire, P. Maître and O. Dopfer, Angew. Chem., Int. Ed., 2007, 46, 6714–6716 CrossRef CAS PubMed.
- M. Bahou, Y. J. Wu and Y. P. Lee, Phys. Chem. Chem. Phys., 2013, 15, 1907–1917 RSC.
- I. Garkusha, J. Fulara, A. Nagy and J. P. Maier, J. Am. Chem. Soc., 2010, 132, 14979 CrossRef CAS PubMed.
- B. S. Freiser and J. L. Beauchamp, J. Am. Chem. Soc., 1976, 98, 3136 CrossRef CAS.
- B. S. Freiser and J. L. Beauchamp, J. Am. Chem. Soc., 1977, 99, 3214 CrossRef CAS.
- I. Alata, C. Dedonder, M. Broquier, E. Marceca and C. Jouvet, J. Am. Chem. Soc., 2010, 132, 17483–17489 CrossRef CAS PubMed.
- I. Alata, R. Omidyan, M. Broquier, C. Dedonder, O. Dopfer and C. Jouvet, Phys. Chem. Chem. Phys., 2010, 12, 14456–14458 RSC.
- N. E.-López, C. D.-Lardeux and C. Jouvet, J. Chem. Phys., 2015, 143, 074303 CrossRef PubMed.
- F.-X. Hardy, O. Gause, C. A. Rice and J. P. Maier, Astrophys. J., Lett., 2013, 778, L30 CrossRef.
- I. Garkusha, J. Fulara, P. J. Sarre and J. P. Maier, J. Phys. Chem. A, 2011, 115, 10972–10978 CrossRef CAS PubMed.
- I. Garkusha, A. Nagy, J. Fulara, M. F. Rode, A. L. Sobolewski and J. P. Maier, J. Phys. Chem. A, 2013, 117, 351–360 CrossRef CAS PubMed.
- I. Garkusha, J. Fulara and J. P. Maier, J. Mol. Struc., 2012, 1025, 147–150 CrossRef CAS.
- C. A. Rice, F. X. Hardy, O. Gause and J. P. Maier, J. Phys. Chem. Lett., 2014, 5, 942–945 CrossRef CAS PubMed.
- B. Saed and R. Omidyan, J. Phys. Chem. A, 2013, 117, 2499–2507 CrossRef CAS PubMed.
- R. Omidyan, Chem. Phys. Lett., 2011, 518, 15–20 CrossRef CAS.
- J. F. Li, G. Tian, Y. Luo and Z. Cao, Chem. Phys. Lett., 2015, 641, 57–61 CrossRef CAS.
- J. Li, Y. Luo and J. Zhang, Spectrochim. Acta, Part A, 2018, 205, 520–527 CrossRef CAS PubMed.
- J. Li and Y. Luo, Spectrochim. Acta, Part A, 2021, 244, 118832 CrossRef CAS PubMed.
- J. Li and N. Li, Comput. Theor. Chem., 2023, 1221, 114028 CrossRef CAS.
- C. H. Chin and S. H. Lin, Phys. Chem. Chem. Phys., 2016, 18, 14569–14579 RSC.
- B. A. Laws, O. Krechkivska, K. Nauta, S. H. Kable and T. W. Schmidt, Phys. Chem. Chem. Phys., 2023, 25, 19524–19531 RSC.
- N. Solcà and O. Dopfer, Chem. – Eur. J., 2003, 9, 3154–3163 Search PubMed.
- M. A. Duncan, J. Phys. Chem. A, 2012, 116, 11477–11491 CrossRef CAS PubMed.
- T. C. Cheng, B. Bandyopadhyay, J. D. Mosley and M. A. Duncan, J. Am. Chem. Soc., 2012, 134, 13046–13055 CrossRef CAS PubMed.
- I. Alata, M. Broquier, C. Dedonder-Lardeux, C. Jouvet, M. Kim, W. Y. Sohn, S. Kim, H. Kang, M. Schütz, A. Patzer and O. Dopfer, J. Chem. Phys., 2011, 134, 074307 CrossRef PubMed.
- A. Patzer, M. Schütz, C. Jouvet and O. Dopfer, J. Phys. Chem. A, 2013, 117, 9785–9793 CrossRef CAS PubMed.
- K. Chatterjee and O. Dopfer, J. Phys. Chem. A, 2020, 124, 1134–1151 CrossRef CAS PubMed.
- A. M. Santa Daría, L. González-Sánchez and S. Gómez, Phys. Chem. Chem. Phys., 2024, 26, 174–184 RSC.
- J. Bourgalais, X. Mercier, M. Mogren Al-Mogren and M. Hochlaf, J. Phys. Chem. A, 2023, 127, 8447–8458 CrossRef CAS PubMed.
- M. Prakash, K. R. Maiyelvaganan, N. G. Lakshman, M. Mogren Al-Mogren and M. Hochlaf, ChemPhysChem, 2023, 24, e202300267 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, N. R. J. Gao, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Wallingford CT, 2016 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- K. R. Maiyelvaganan, M. K. Ravva, M. Prakash and V. Subramanian, Theor. Chem. Acc., 2020, 139, 1–14 Search PubMed.
- R. Dennington, T. A. Keith and J. M. Millam, GaussView 6.0.16, Shawnee Mission, Semichem Inc, 2016 Search PubMed.
- G. A. Zhurko and D. A. Zhurko, Chemcraft Program, academic version 1.8, 2015 Search PubMed.
- B. P. Pritchard, A. Kumar, A. C. Simmonett, F. A. Evangelista, K. Patkowski, A. E. DePrince, J. M. Turney, M. Saitow, R. A. King, R. Di Remigio, D. G. A. Smith, U. Bozkaya, P. Verma, A. Y. Sokolov, E. F. Valeev, E. G. Hohenstein, C. D. Sherrill, R. M. Richard, A. M. James, T. D. Crawford, R. M. Parrish, H. F. Schaefer, L. A. Burns, H. R. McAlexander, X. Wang and J. F. Gonthier, J. Chem. Theory Comput., 2017, 13, 3185–3197 CrossRef PubMed.
- K. Morokuma, J. Chem. Phys., 1971, 55, 1236 CrossRef CAS.
- C. Bannwarth, S. Ehlert and S. Grimme, J. Chem. Theory Comput., 2019, 15, 1652–1671 CrossRef CAS PubMed.
- S. Grimme, C. Bannwarth and P. Shushkov, J. Chem. Theory Comput., 2017, 13, 1989–2009 CrossRef CAS PubMed.
- S. Grimme, C. Bannwarth, S. Dohm, A. Hansen, J. Pisarek, P. Pracht, J. Seibert and F. Neese, Angew. Chem., Int. Ed., 2017, 56, 14763–14769 CrossRef CAS PubMed.
- S. Grimme, J. Chem. Theory Comput., 2019, 15, 2847–2862 CrossRef CAS PubMed.
- Y. Tantirungrotechai, K. Phanasanta, S. Roddecha, P. Surawatanawonga, V. Sutthikhum and J. Limtrakul, THEOCHEM, 2006, 760, 189–192 CrossRef CAS.
- F. Biegler-Konig, J. Schonbohm, R. Derdau, D. Bayles and R. F. W. Bader, AIM 2000, Bielefeld, Germany, 2000 Search PubMed.
- T. Lu and F. W. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- F. Bernardi and S. F. Boys, Mol. Phys., 1970, 19, 553–566 CrossRef.
- M. Prakash, K. Gopalsamy and V. Subramanian, J. Chem. Phys., 2011, 135, 214308 CrossRef CAS PubMed.
- K. R. Maiyelvaganan, M. K. Ravva and M. Prakash, J. Phys. Chem. A, 2020, 124, 3364–3373 CrossRef CAS PubMed.
- M. Prakash, K. Mathivon, D. M. Benoit, G. Chambaud and M. Hochlaf, Phys. Chem. Chem. Phys., 2014, 16, 12503–12509 RSC.
- S. Kamalakannan, K. R. Maiyelvaganan, K. Palanisamy, A. Thomas, R. B. Said, M. Prakash and M. Hochlaf, Chemosphere, 2022, 286, 131612 CrossRef CAS PubMed.
- S. Kamalakannan, K. Palanisamy, M. Prakash and M. Hochlaf, Photocatalysis for Environmental Remediation and Energy Production, ed. S. Garg and A. Chandra, Green Chemistry and Sustainable Technology, 2023, p. 329 Search PubMed.
- https://webbook.nist.gov/ .
- C. Reid, J. Am. Chem. Soc., 1954, 76, 3264–3268 CrossRef CAS.
- H. H. Perkampus and E. Baumgarten, Angew. Chem., Int. Ed. Engl., 1964, 3, 776 CrossRef.
- G. N. Lewis and M. Kasha, J. Am. Chem. Soc., 1945, 67, 994 CrossRef CAS.
- G. N. Lewis and M. Kasha, J. Am. Chem. Soc., 1944, 66, 2100 CrossRef CAS.
- S. K. Lower and M. A. El-Sayed, Chem. Rev., 1966, 66, 199 CrossRef CAS.
- M. A. Omary and H. H. Patterson, Organic Luminophores in Encyclopedia of Spectroscopy and Spectrometry, ed J. C. Lindon, G. E. Tranter and D. W. Koppenaal, 3rd edn, 2017, ISBN: 978-0-12-803224-4 Search PubMed.
- C. Corminboeuf, T. Heine, G. Seifert, P. Von Ragué Schleyer and J. Weber, Phys. Chem. Chem. Phys., 2004, 6, 273–276 RSC.
- H. Fallah-Bagher-Shaidaei, C. S. Wannere, C. Corminboeuf, R. Puchta and P. V. R. Schleyer, Org. Lett., 2006, 8, 863–866 CrossRef CAS PubMed.
- R. Báez-Grez, L. Ruiz, R. Pino-Rios and W. Tiznado, RSC Adv., 2018, 8, 13446–13453 RSC.
- J. Yuan, R. Chen, X. Tang, Y. Tao, S. Xu, L. Jin, C. Chen, X. Zhou, C. Zheng and W. Huang, Chem. Sci., 2019, 10, 5031 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp06000d |
| ‡ These authors contributed equally to this work. |
| This journal is © the Owner Societies 2024 |