
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Simulated conformality of atomic layer deposition in lateral channels: the impact of the Knudsen number on the saturation profile characteristics†
Christine
Gonsalves
 *a,
Jorge A.
Velasco
a,
Jihong
Yim
a,
Jänis
Järvilehto
a,
Ville
Vuorinen
b and
Riikka L.
Puurunen
*a
*a,
Jorge A.
Velasco
a,
Jihong
Yim
a,
Jänis
Järvilehto
a,
Ville
Vuorinen
b and
Riikka L.
Puurunen
*a
aSchool of Chemical Engineering, Department of Chemical and Metallurgical Engineering, Aalto University, P.O. Box 16100, FI-00076 AALTO, Finland. E-mail: christine.gonsalves@aalto.fi; riikka.puurunen@aalto.fi
bSchool of Engineering, Aalto University, P.O. Box 16100, FI-00076 AALTO, Finland
First published on 22nd October 2024
Abstract
Atomic layer deposition (ALD) is exceptionally suitable for coating complex three-dimensional structures with conformal thin films. Studies of ALD conformality in high-aspect-ratio (HAR) features typically assume free molecular flow conditions with Knudsen diffusion. However, the free molecular flow assumption might not be valid for real ALD processes. This work maps the evolution of the saturation profile characteristics in lateral high-aspect-ratio (LHAR) channels through simulations using a diffusion–reaction model for various diffusion regimes with a wide range of Knudsen numbers (106 to 10−6), from free molecular flow (Knudsen diffusion) through the transition regime to continuum flow conditions (molecular diffusion). Simulations are run for ALD reactant partial pressures spanning several orders of magnitude with the exposure time kept constant (by varying the total exposure) and with the total exposure kept constant (by varying the exposure time). In a free molecular flow, for a constant total exposure, the saturation profile characteristics are identical regardless of the LHAR channel height and the partial pressure of the reactant. Under transition regime and continuum conditions, the penetration depth decreases and the steepness of the adsorption front increases with decreasing Knudsen number. The effect of varying individual parameters on the saturation profile characteristics in some cases depends on the diffusion regime. An empirical “extended slope method” is proposed to relate the sticking coefficient to the saturation profile's characteristic slope for any Knudsen number.
1 Introduction
Atomic layer deposition (ALD) is a thin film growth technique that delivers uniform thin films with nanoscale precision,1–4 and has applications in diverse fields ranging from microelectronics to catalysts to optical coatings and beyond.4 With the earliest experiments dating to the 1960s and 1970s,5–7 interest in ALD is rapidly growing due to its unparalleled ability to coat complex three-dimensional structures with a conformal film.8 This ability stems from the use of self-terminating gas-solid reactions, and is taken advantage of, for example, in functional layers in logic and memory chips, in multiple patterning,9 in catalysis,10 and in energy storage.11 Recently, uniform coating of silica aerogel structures with a high-aspect-ratio (AR) of >60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000:1 has been demonstrated.12
000:1 has been demonstrated.12
Experimental studies on the conformality of ALD processes typically rely on specifically developed high-aspect-ratio (HAR) test structures.8 These structures consist either of vertical features etched into silicon8,13,14 or of lateral HAR (LHAR) structures prepared with a limiting height and controlled length.8,15–17 Manually assembled macroscopic LHAR structures typically have a limiting channel height in the 100 μm range,15,18 while LHAR structures made with techniques used in microelectromechanical systems (MEMS) can yield microscopic LHAR structures with a limiting channel height on the order of 100 nm.16,17,19 In microscopic LHAR structures, the ARs can reach over 1000:1, making the structures demanding to coat completely, as the required exposure scales with the AR squared.8,13 Incomplete conformality exposes the saturation profile for detailed analysis, as shown in Fig. 1. The saturation profile contains information on the surface reaction kinetics and has been used for analyzing the sticking coefficient in ALD19–21 and the radical recombination probability in plasma-enhanced ALD.22,23
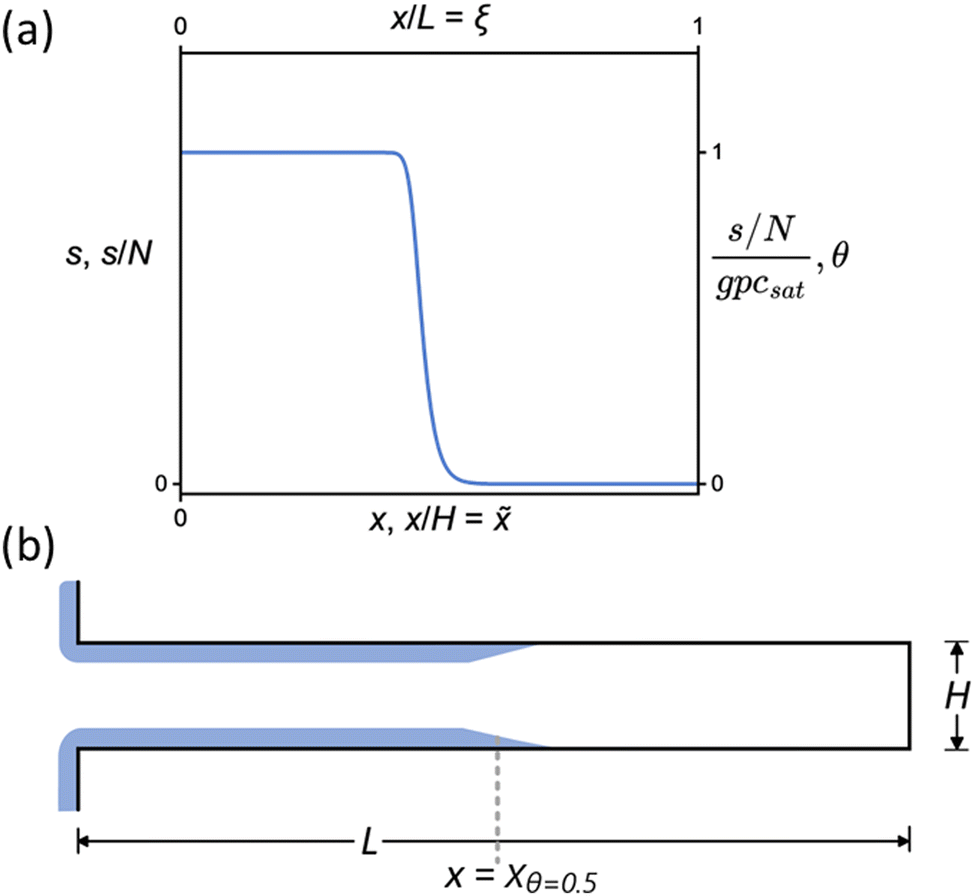 | ||
| Fig. 1 (a) The representative saturation profile as a function of the distance from the channel entrance. (b) The schematic geometry of the LHAR channels used to simulate ALD growth inside the channel for varying Knudsen numbers. | ||
Mass transport in HAR features takes place by diffusion. Whether this diffusion is Knudsen diffusion or molecular diffusion is determined by the dimensionless Knudsen number Kn (−), which gives the ratio of the mean free path λ (m) of the molecules in the gas to the characteristic limiting feature size D (m):8,19,24
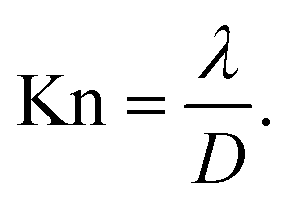 | (1) |
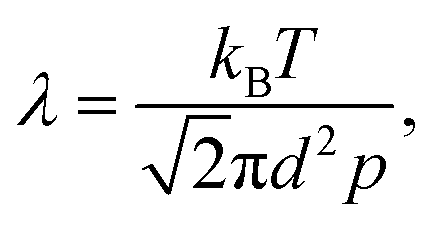 | (2) |
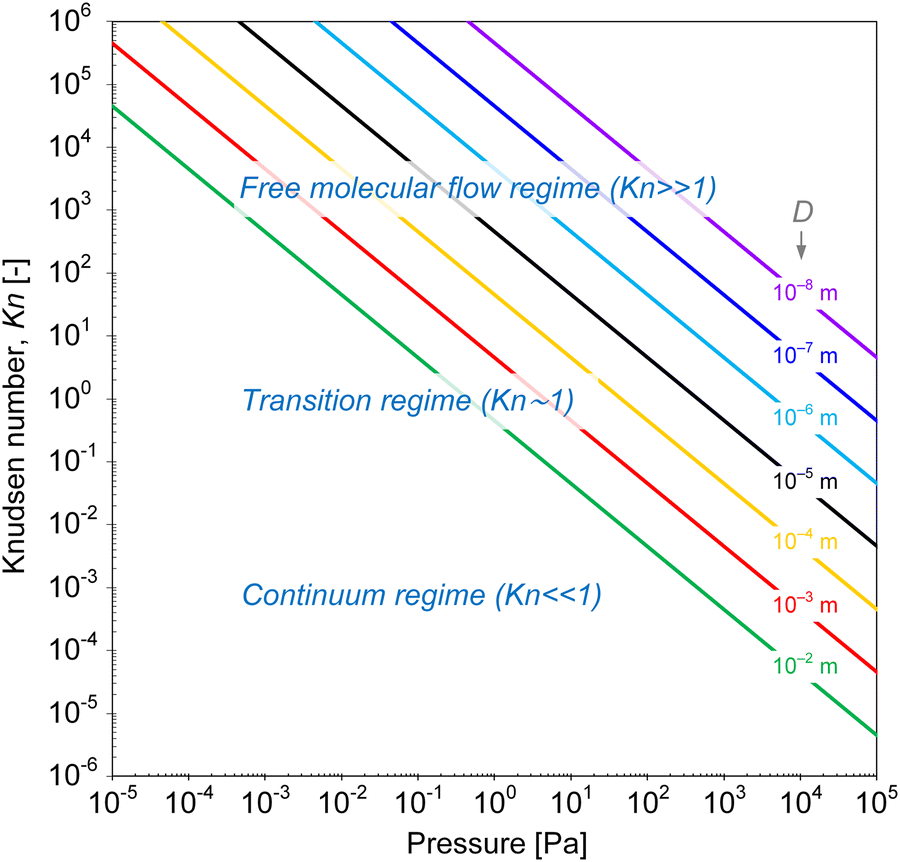 | ||
| Fig. 2 Knudsen number as a function of pressure at 250 °C, for features with different characteristic limiting sizes “D”, calculated from the mean free path (eqn (1) and (2)) for a gas molecule with a hard-sphere diameter of 6 × 10−10 m. | ||
Various models, such as diffusion–reaction models, ballistic transport–reaction models, and Monte Carlo models are used to simulate feature-scale ALD growth in HAR structures.8,26 Diffusion–reaction models and Monte Carlo models can be used at any Knudsen number, while ballistic transport–reaction models are limited to the Knudsen diffusion regime. Computational fluid dynamics simulations, in turn, are useful in the continuum flow regime, with convection and molecular diffusion.27 While all models describe transport and reaction in HAR features, the detailed predictions may differ. This was recently shown in a comparison between a diffusion–reaction model and a ballistic transport–reaction model: the growth penetration was deeper in the ballistic transport–reaction model and the slope of the adsorption front inside the HAR feature was steeper in the diffusion–reaction model.26 In addition, the ballistic transport–reaction model exhibited a “trunk” formed at the feature end, which was absent from the diffusion–reaction model26 (a similar trunk has also been observed in Monte Carlo simulations8). This work continues the series of simulations17,21,26 performed with the Ylilammi et al.19 diffusion–reaction model,21,28 extending its use to the continuum regime (Kn ≪ 1).
Another useful dimensionless number in addition to the Knudsen number is the Thiele modulus hT, which characterizes ALD growth in HAR features. The Thiele modulus has only recently been introduced for ALD,29,30 but it has been in use for decades in the related field of heterogeneous catalysis.29,31 The Thiele modulus is the ratio of the reaction rate to the diffusion rate and can be used to assess the growth-limiting factor (diffusion vs. reaction) in HAR features.29–33 For single-site adsorption on a fresh surface, the Thiele modulus hT can be calculated20,21,29 from:
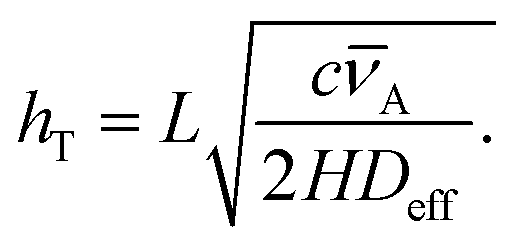 | (3) |
![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) A (m s−1) is the thermal velocity. When the reaction rate is much faster than the diffusion rate (when the Thiele modulus is greater than one), the process is diffusion limited, and inside the HAR features, an adsorption front forms. When the diffusion rate is faster than the reaction rate (the Thiele modulus is lower than one), the process is reaction limited and the thickness inside the HAR features increases uniformly with time. The exact limiting values of the Thiele modulus vary by source; according to Levenspiel,34 in heterogeneous catalysis, hT > 4 corresponds to severe diffusion resistance and hT < 0.4 to the absence of diffusion resistance.8,29,30,33 For diffusion-limited growth in a free molecular flow regime, a simple slope method has been recently developed by Arts et al.20 to extract information on the growth kinetics from the slope of the adsorption front.
A (m s−1) is the thermal velocity. When the reaction rate is much faster than the diffusion rate (when the Thiele modulus is greater than one), the process is diffusion limited, and inside the HAR features, an adsorption front forms. When the diffusion rate is faster than the reaction rate (the Thiele modulus is lower than one), the process is reaction limited and the thickness inside the HAR features increases uniformly with time. The exact limiting values of the Thiele modulus vary by source; according to Levenspiel,34 in heterogeneous catalysis, hT > 4 corresponds to severe diffusion resistance and hT < 0.4 to the absence of diffusion resistance.8,29,30,33 For diffusion-limited growth in a free molecular flow regime, a simple slope method has been recently developed by Arts et al.20 to extract information on the growth kinetics from the slope of the adsorption front.
The goal of this work is to analyze the effect of the Knudsen number on the evolution of conformality in narrow channels under diffusion-limited conditions using the Ylilammi et al.19,21,28 model. Mapping is performed from the free molecular flow governed by Knudsen diffusion (Kn ≫ 1) through the transition regime (Kn ∼ 1) to continuum flow conditions governed by molecular diffusion (Kn ≪ 1) by varying the channel height and pressure. The Knudsen number is varied by 13 orders of magnitude. An extended slope method analogous to the Arts et al.20 slope method is proposed that covers all Knudsen numbers. This work further expands on the trends documented earlier21 for the effect of varying process conditions on the penetration depth and the slope of the saturation profile at various Knudsen numbers. While the numerical results will be somewhat model-specific, the reported trends should be generic. Furthermore, through the (hole-)equivalent aspect ratio (EAR) concept,8 the results can be scaled to HAR geometries other than the narrow channels studied in this work.
2 Methods
2.1 Description of the model
In this work, we used a one-dimensional diffusion–reaction model by Ylilammi et al.19 to simulate the transport of a reactant gas from the channel entrance to the growth surface in a lateral high-aspect-ratio structure (LHAR).21 The diffusion–reaction model used in this work is based on Fick's law of diffusion and assumes Langmuir adsorption. The full model has been previously described in detail.19,21,26 The key equations are written here to guide the reader through the simulations and results of this work.The mean free path of the reactant ‘A’ in a system of two gases (reactant ‘A’ and inert carrier gas ‘I’) can be obtained using the equation8,19,21,35
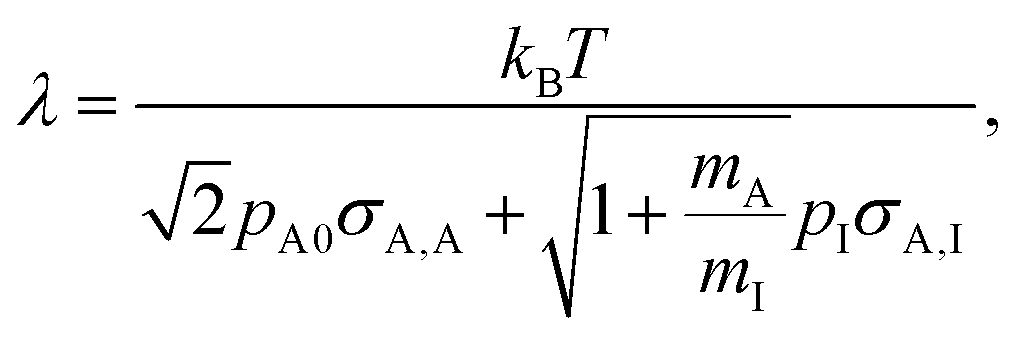 | (4) |
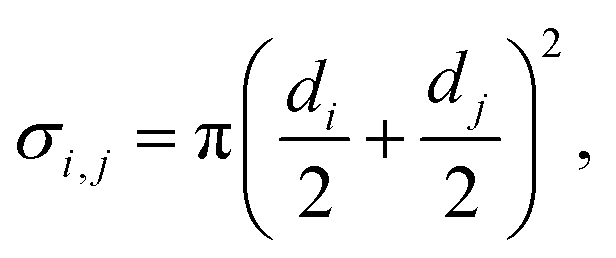 | (5) |
In this model, the ALD surface reactions are described by the Langmuir adsorption model,15,21 which assumes reversible single-site adsorption. Reversible Langmuir adsorption can be expressed by
| A + * ⇌ A*, | (6) |
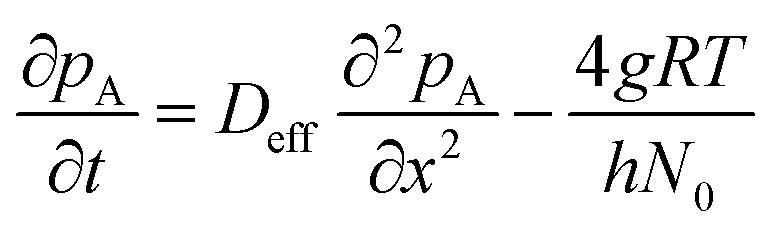 | (7) |
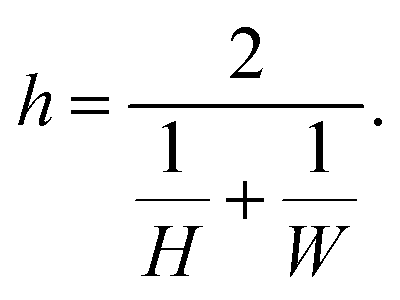 | (8) |
In Langmuir adsorption, a certain number of adsorption sites are occupied by the reactant molecules, and their ratio with respect to the total number of sites is called the surface coverage θ, which has values ranging from 0 to 1. The rate of adsorption fads is proportional to the fraction of unoccupied sites. The g in eqn (7) stands for the net adsorption rate (m−2 s−1), and it is the difference between the rate of adsorption fads and the rate of desorption fdes (m−2 s−1).19,21 The evolution of the fractional surface coverage θ (−) with time from the Langmuir model of adsorption can be given by the rate equation:
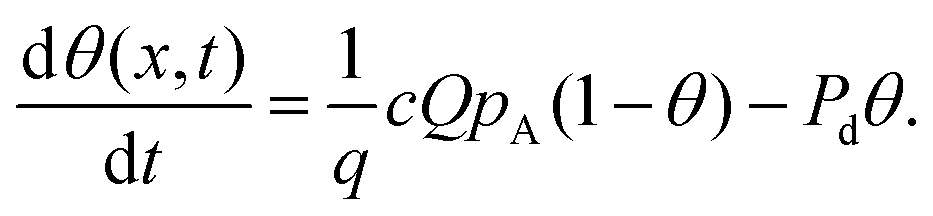 | (9) |
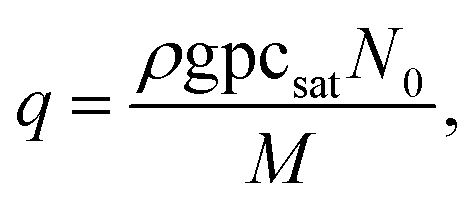 | (10) |
In eqn (9), Q is the collision rate at unit pressure (m−2 s−1 Pa−1), represented as21
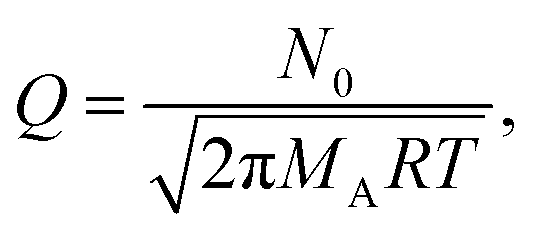 | (11) |
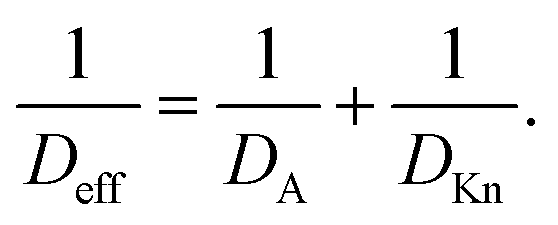 | (12) |
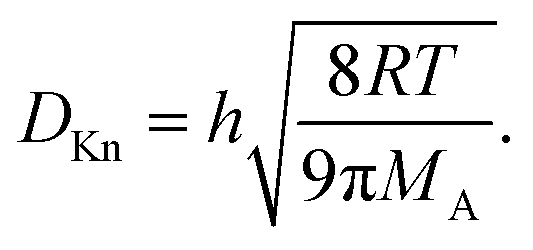 | (13) |
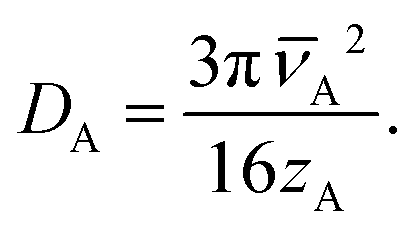 | (14) |
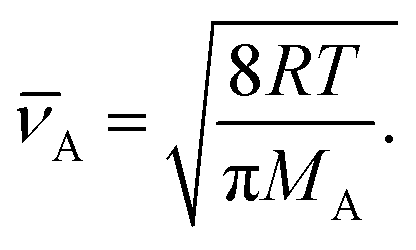 | (15) |
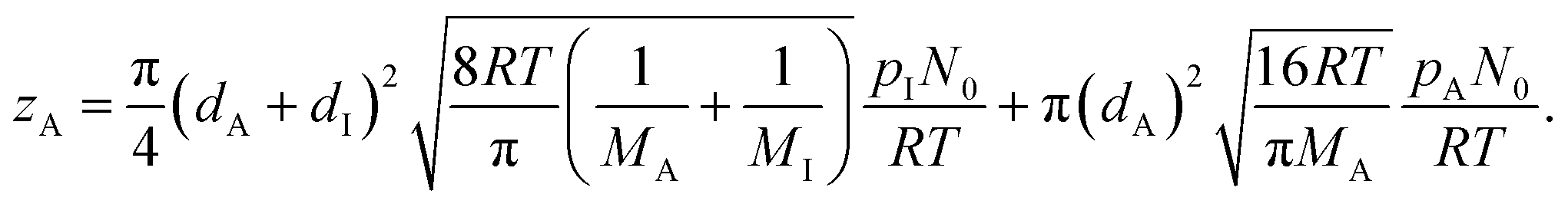 | (16) |
To solve the partial pressure of reactant A along the channel pA(x,t), instead of solving Fick's second law of diffusion, the Ylilammi et al. model19,21 uses an analytical approximation to account for the reactant gas pressure pA along the channel. At the channel entrance, surface saturation is instantaneous (g ≈ 0). The diffusion–reaction equation (eqn (7)) is then simplified to19
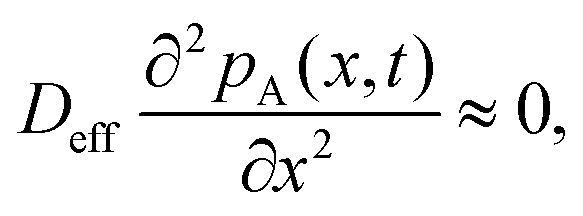 | (17) |
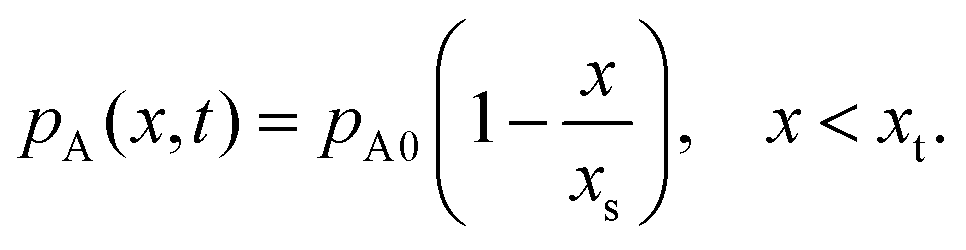 | (18) |
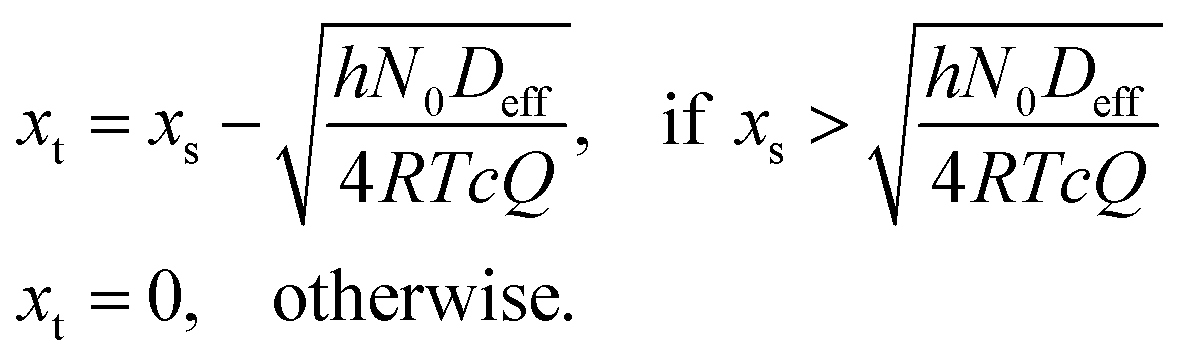 | (19) |
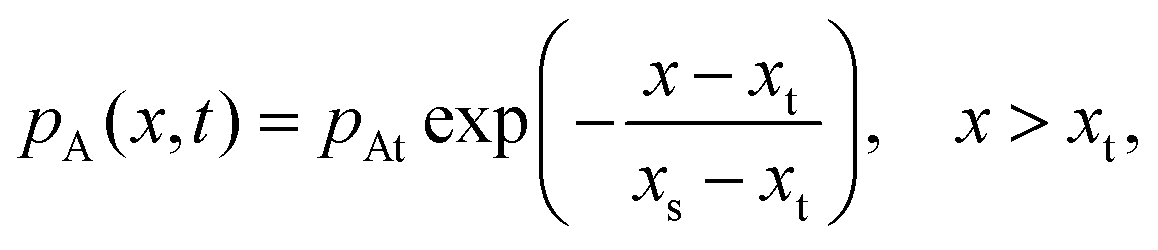 | (20) |
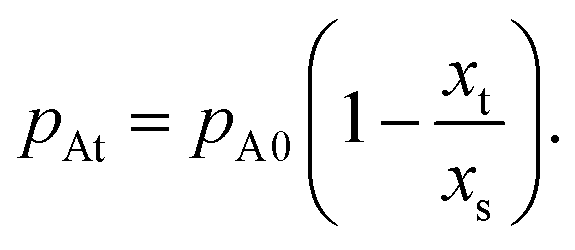 | (21) |
2.2 Simulation details
The equations of the Ylilammi et al. diffusion–reaction model19 were solved using MATLAB®. A detailed description of the implementation of this model in MATLAB was presented previously.21,28 A summary of the parameters used in the simulations in this work is shown in Table 1. These parameters were inspired by the trimethylaluminum and water ALD process.8,19,21 The influence of varying parameters, such as the channel height, reactant partial pressure, and Knudsen number on the saturation profile was studied. All simulations were performed for one ALD reactant pulse, assuming it is the limiting step and represents an ALD cycle. The simulations were carried out using different reactant A partial pressure values (pA0) varying from 10−2 to 104 Pa and the inert gas partial pressure (pI) was nine times this value. Channel heights (H) ranging from 10−8 to 10−2 m were used. To maintain a constant exposure of 10 Pa s (∼7.5 × 104 Langmuirs, where 1 Pa s = 7500 Langmuirs), the time t was varied with the reactant A partial pressure pA0 in the range of 10−3 to 103 s. The dimensionless distance![[x with combining tilde]](https://www.rsc.org/images/entities/i_char_0078_0303.gif) (−) was the ratio of the physical distance x (m) to the channel height H (m) and was used to effectively compare results for channels of varying heights. Simulated surface coverage results were also plotted both as a function of the physical distance x, as well as the dimensionless distance . Fig. 3 shows the Knudsen number and the Thiele modulus calculated for the simulations in this work at different channel heights H and reactant A partial pressure values pA0. Unless otherwise stated, all simulation parameters and conditions are those provided in Table 1.
(−) was the ratio of the physical distance x (m) to the channel height H (m) and was used to effectively compare results for channels of varying heights. Simulated surface coverage results were also plotted both as a function of the physical distance x, as well as the dimensionless distance . Fig. 3 shows the Knudsen number and the Thiele modulus calculated for the simulations in this work at different channel heights H and reactant A partial pressure values pA0. Unless otherwise stated, all simulation parameters and conditions are those provided in Table 1.
| Parameter | Values |
|---|---|
| a Other parameters used: length (L) was varied with height (H) such that L/H = 1000 (minimum L = 10−5 m, maximum L = 10 m); width (W) was varied with height (H) such that W/H ≥ 1000 (minimum W = 10−2 m, maximum W = 10 m); hard-sphere diameter of molecule A (dA) = 6 × 10−10 m; hard-sphere diameter of an inert gas molecule (dI) = 3.4 × 10−10 m; molar mass of reactant A (MA) = 0.0749 kg mol−1; molar mass of the inert gas (MI) = 0.03994 kg mol−1; film mass density (ρ) [kg m−3] = 3500; number of metal atoms per formula unit of film (bfilm) in the Ylilammi model19 [−] = 1; number of metal atoms in reactant molecule bA in the Ylilammi model19 [−] = 1; mass of the film (Mfilm) [kg mol−1] = 0.05. b The partial pressure of inert gas I (pI) was set to nine times the value of the partial pressure of the reactant gas (pA0). c The sticking coefficient used in most of this work was 0.01 unless otherwise stated. | |
| Initial partial pressure of the reactant gas (pA0) [Pa] | 0.01, 0.1, 1, 10, 100, 1000, 10000 |
| Partial pressure of inert gas I (pI)b [Pa] | 0.09, 0.9, 9, 90, 900, 90000 |
| Channel height (H) [m] | 10−8, 10−7, 10−6, 10−5, 10−4, 10−3, 10−2; 5 × 10−7 |
| Time (t) [s] | 0.001, 0.01, 0.1, 1, 10, 100, 1000 |
| Temperature (T) [°C] | 250 |
| Adsorption capacity (q) [m−2] | 4 × 10−18 |
| Desorption probability in unit time (Pd) [s−1] | 0.0001 |
| Sticking coefficient (c) [−] | 0.0001, 0.001, 0.01c, 0.1, 1 |
| Number of ALD cycles N [−] | 1 |
 | ||
| Fig. 3 The calculated values of the Knudsen number and Thiele modulus with a sticking coefficient c of 0.01, for the saturation profile simulations performed in this work using the Ylilammi et al. model.19,21 (a) The Knudsen number as a function of the channel height H for different reactant A partial pressures pA0, (b) the Thiele modulus as a function of the channel height H for different reactant A partial pressures pA0, and (c) the Thiele modulus hT as a function of the Knudsen number for different channel heights. | ||
To follow the penetration depth at half surface coverage θ = 0.5, a linear interpolation was made between the two closest discretization points of the dimensionless distance. The points were chosen such that the difference between the two was less than 1% of the whole range of the y-axis. Furthermore, these two points were used to get the value of the slope at half coverage, i.e., |Δθ/Δ|θ=0.5.
3 Results
3.1 Saturation profiles under increasing total exposure and pressure
A series of simulations were performed in which the reactant partial pressure was varied, keeping the pulse time constant and thus varying the total exposure. Keeping the exposure time t fixed at 0.1 s, the reactant partial pressure pA0 was varied within 10−2 to 104 Pa, giving reactant exposures ranging from 10−3 to 103 Pa s. Simulations were performed for channels with heights from the nanometer to centimeter scale. The detailed process conditions are listed in Table 1. The Knudsen number ranged between 106 and 10−6 for this set of simulations (eqn (1) and Fig. 3). Thus, these simulations cover conditions from free molecular flow (Kn ≫ 1) through the transition regime (Kn ∼ 1) to the continuum regime (Kn ≪ 1).
Fig. 4 shows the saturation profiles for different channel heights H and reactant partial pressures pA0. Moving from panel (a) to panel (g), the channel height increases each time by one order of magnitude, going from 10−8 to 10−2 (10 nm to 1 cm). The last panel (h) corresponds to a channel height H of 500 nm, which is typical for the PillarHall™ test structures.17,37 For a sufficiently large exposure (1 Pa s or larger) with a sticking coefficient c of 0.01, a well-developed saturation profile is seen with full coverage (θ ≈ 1) at the channel entrance and decreasing coverage in an adsorption front deeper in the channel. In Fig. 4, the primary horizontal axis shows the physical distance x and the secondary horizontal axis shows the dimensionless distance = x/H. In terms of the physical distance x, the growth penetrates deeper in larger channels. In terms of the dimensionless distance , the opposite is observed: the growth penetrates deeper in smaller channels. These trends are as expected.21
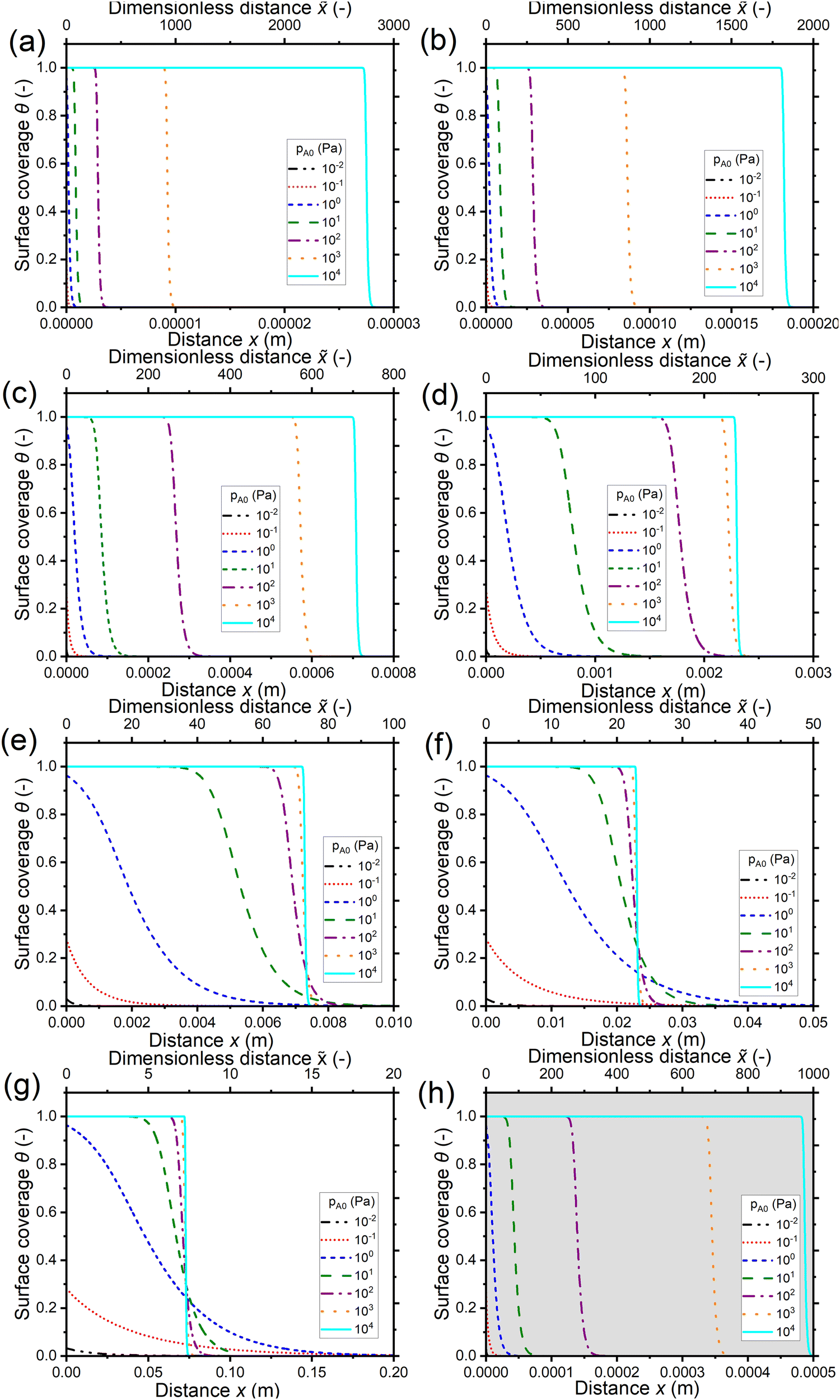 | ||
| Fig. 4 Saturation profiles in wide lateral channels of different heights at varying exposures (Pa s) using a constant reactant pulse time of 0.1 s. Channel heights: (a) 10 nm, (b) 100 nm, (c) 1 μm, (d) 10 μm, (e) 100 μm, (f) 1 mm, (g) 1 cm, and (h) 500 nm (the typical PillarHall™ case17,37). The calculated Knudsen number values are shown in Fig. 3(a). The exposure values are provided in the ESI† (S1). The initial reactant partial pressure pA0 is in the range of 10−2 to 104 Pa. The sticking coefficient c is 0.01. The other simulation conditions are given in Table 1. | ||
Fig. 5(a) shows the penetration depth at half coverage in terms of the dimensionless distance θ=0.5 extracted from the saturation profiles of Fig. 4 as a function of the channel height H. The cases that did not give a well-developed saturation profile (pA0 ≤ 1 Pa) have been excluded from Fig. 5. For a given reactant partial pressure pA0 (and thus a fixed exposure), as the channel height H increases, the penetration depth either decreases or stays constant (Fig. 5(a)). For a given channel height H, as the partial pressure pA0 increases (and thus the exposure increases), the penetration depth either increases or stays constant (Fig. 5(a)). It has been shown in previous literature that with increasing exposure, the penetration depth in ALD increases,13 so the latter trend (no increase) may feel counter-intuitive. The reasons for the observed differences can be understood by examining the trends as a function of the Knudsen number.
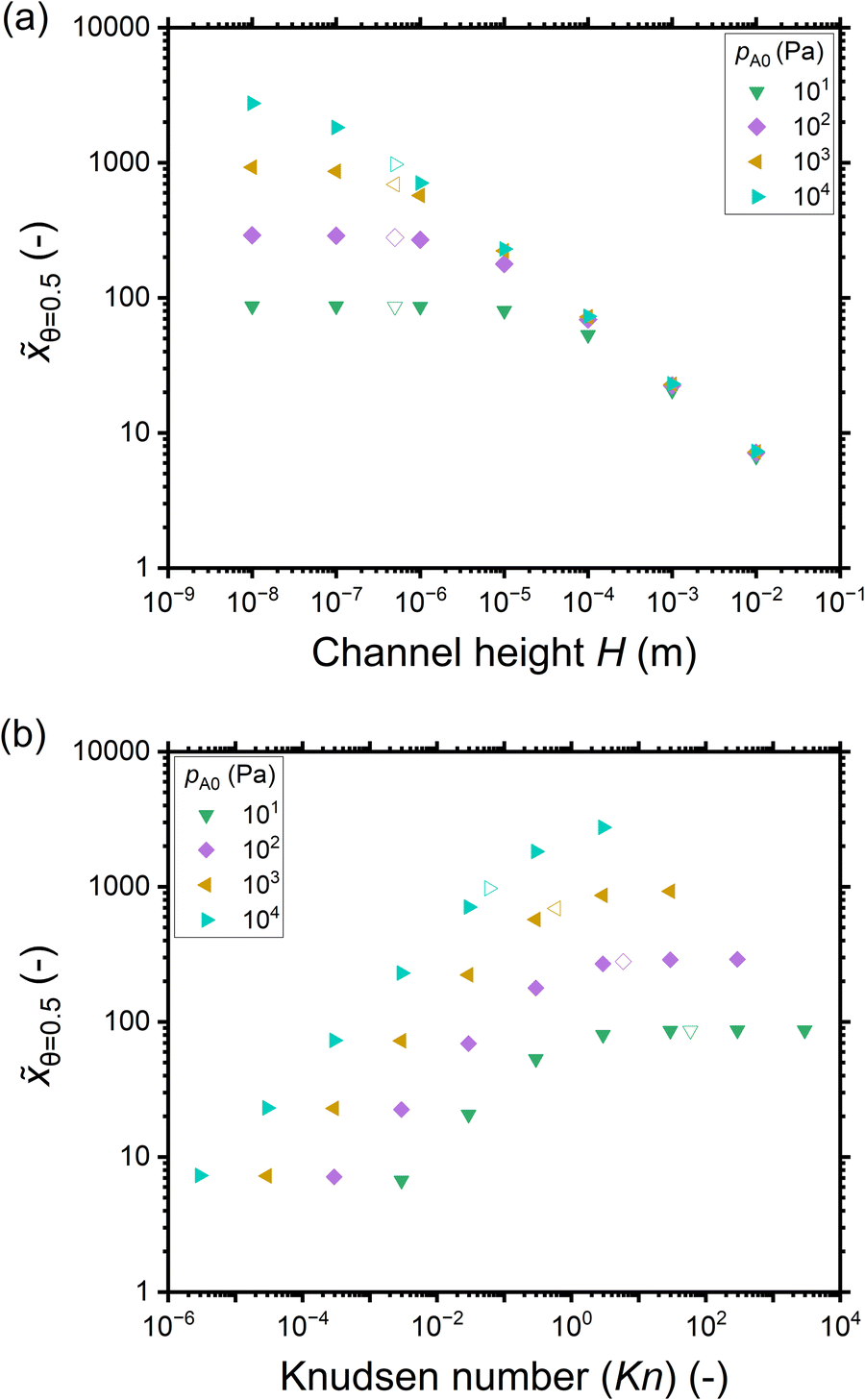 | ||
| Fig. 5 The penetration depth in terms of the dimensionless distance at half coverage θ=0.5 extracted from the saturation profiles shown in Fig. 4 as a function of: (a) the channel height H, and (b) the Knudsen number. Hollow symbols correspond to the typical PillarHall™ case17,37 with a 500 nm channel height. | ||
Fig. 5(b) shows the penetration depth data of Fig. 5(a) further as a function of the Knudsen number. Comparing panels (b) and (a), one can see that the data has been mirrored with respect to the horizontal axis (high H corresponds to low Kn) and higher partial pressure data points are further shifted leftwards. As a result, the low-penetration-depth data points showing no dependence on pA0 and therefore clustered together at the right side of panel (a) are spread out for different Knudsen numbers on the left side of panel (b). With increasing Knudsen number for a given pA0, in the continuum flow (Kn ≪ 1), the penetration depth increases with increasing Knudsen number. In the transition regime (Kn ∼ 1), the penetration depth θ=0.5 continues to increase with increasing Knudsen number but the pace of increase is less, and on reaching free molecular flow (Kn ≫ 1), the penetration depth has settled to a constant value (Fig. 5(b)). Thus, it seems that the increasing effect of exposure on the penetration depth appears counterbalanced by the decreasing effect from the decreasing Knudsen number.
The dependence on the Knudsen number and the effect of pressure on the penetration depth can be explained by the type of diffusion taking place. The values of the diffusion coefficients are shown in Fig. 6 as a function of the Knudsen number: the molecular diffusion coefficient DA describing molecule–molecule interactions (eqn (14)) in panel (a), the Knudsen diffusion coefficient DKn describing molecule–wall interactions (eqn (13)) in panel (b), and the effective diffusion coefficient Deff calculated from DA and DKn through the Bosanquet equation (eqn (12)) in panel (c). The diffusion coefficients correspond to the cases of Fig. 5, while Fig. S2 and S3 in the ESI† represent the diffusion coefficients for all simulation conditions used in this work. Furthermore, in the ESI,† the diffusion coefficients are shown as a function of the partial pressure pA0 and the channel height H (Fig. S4, ESI†). By examining the corresponding equations, one notices that the molecular diffusion coefficient DA has a first-order inverse relationship with the pressure (eqn (14) and (16)), while the Knudsen diffusion coefficient is not impacted by the pressure (eqn (13)). Thus, as the reactant partial pressure pA0 increases, the molecular diffusion coefficient DA decreases (Fig. 6(a) and Fig. S2(a), ESI†), while the change does not affect the Knudsen diffusion coefficient DKn (Fig. 6(b) and Fig. S2(b), ESI†) (to observe the trends with partial pressure, the hollow symbols help to guide the eye). Furthermore, the Knudsen diffusion coefficient DKn has a linear dependence on the channel height H (eqn (13) and Fig. S2(b), ESI†), while the channel height does not influence the molecular diffusion coefficient (eqn (14) and Fig. S2(a), ESI†). The effective diffusion coefficient Deff merges these different trends, with its value being smaller than or equal to the smaller of the two. Hence, in the free molecular flow regime (Kn ≫ 1), in the absence of gas-phase molecule–molecule interactions, the diffusion coefficient does not depend on the pressure, so the penetration depth increases because of the increasing total reactant exposure (Fig. 5). In a continuum flow (Kn ≪ 1), in contrast, the diffusion coefficient Deff decreases linearly with increasing reactant pressure. Consequently, the increasing effect of the reactant partial pressure pA0 on the exposure is counterbalanced by the decreasing effect of increasing pA0 on the effective diffusion coefficient, and the net effect of the increasing partial pressure pA0 on the penetration depth is negligible.
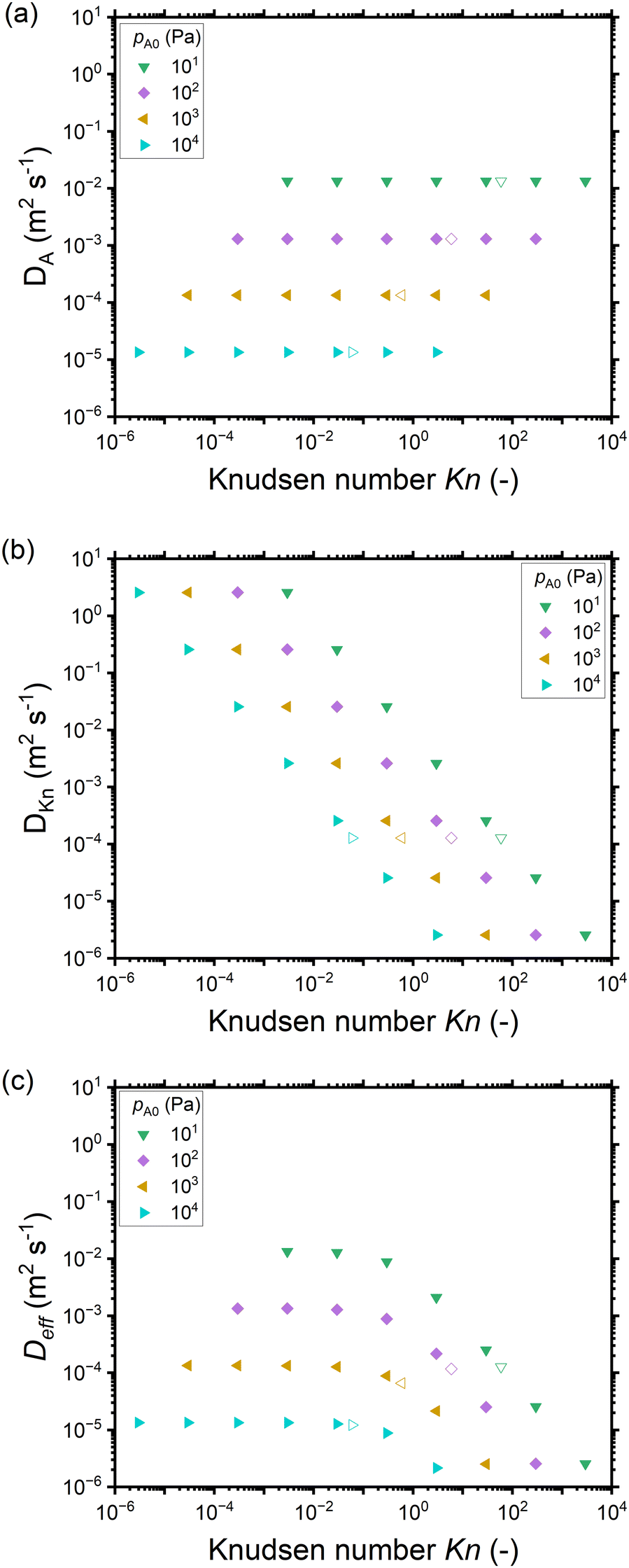 | ||
| Fig. 6 The diffusion coefficients (m2 s−1) as a function of the Knudsen number for different reactant partial pressures pA0 corresponding to Fig. 5: (a) the molecular diffusion coefficient DA, (b) the Knudsen diffusion coefficient DKn, and (c) the effective diffusion coefficient Deff. Hollow symbols represent a 500 nm channel height and correspond to the typical PillarHall™ case.17,37 Diffusion coefficients for the whole range of pA0 are provided in the ESI† (Fig. S2 as a function of Knudsen number and Fig. S3 as function of channel height H). | ||
Examining Fig. 4 further, in addition to trends in the penetration depth, one also observes systematic trends in the shape of the saturation profile. With increasing partial pressure pA0, for all but the most narrow channels, the adsorption front of the saturation profiles becomes shorter, and the slope of the saturation front becomes steeper. In some cases, the changes in shape lead even to a cross-over in the saturation profiles: the leading edge of the adsorption front reaches further for lower partial pressures than for higher pressures. The cross-over can be clearly seen in Fig. 4(f) and (g). The reasons for the change in shape and the cross-over are again differences in the Knudsen number and the effective diffusion coefficient. All cases where cross-over is observed are in the continuum flow regime. The increase in partial pressure pA0 leads to a decrease in the effective diffusion coefficient Deff, the stagnation of the penetration depth, a steeper saturation profile, and hence the cross-over.
3.2 Saturation profiles at a constant reactant exposure
A series of simulations were performed in which the reactant partial pressure pA0 (Pa) and the exposure time t (s) were varied in a way that preserved the total exposure (Pa s). The reactant partial pressure pA0 is directly related to the Knudsen number through the mean free path λ (eqn (1) and (4)). Hence, the evolution of conformality can be studied at a constant reactant exposure for a range of Knudsen numbers, and thus different diffusion regimes. The reactant pressure was varied similarly to in the previous section from 10−2 to 104 Pa, while the exposure was kept constant at 10 Pa s by varying the pulse time between 103 and 10−3 s. Note that the shortest pulse times (10−2–10−3 s) may be physically impractical.
Fig. 7 shows the saturation profiles at constant exposure for different partial pressures of the reactant pA0 and channel heights H. Similarly to in the previous section, the channel heights vary from 10 nm in panel (a) to 1 cm in panel (g) of Fig. 7, with panel (h) representing the 500 nm PillarHall™ case.17,37 The saturation profiles are shown with respect to the dimensionless distance on the primary horizontal axis and the physical distance x on the secondary horizontal axis. In terms of the dimensionless distance , for a given value of the reactant partial pressure pA0, the ALD growth penetrates deeper in smaller channels. In terms of the physical distance x, for the same pA0, the growth is deeper in larger channels.
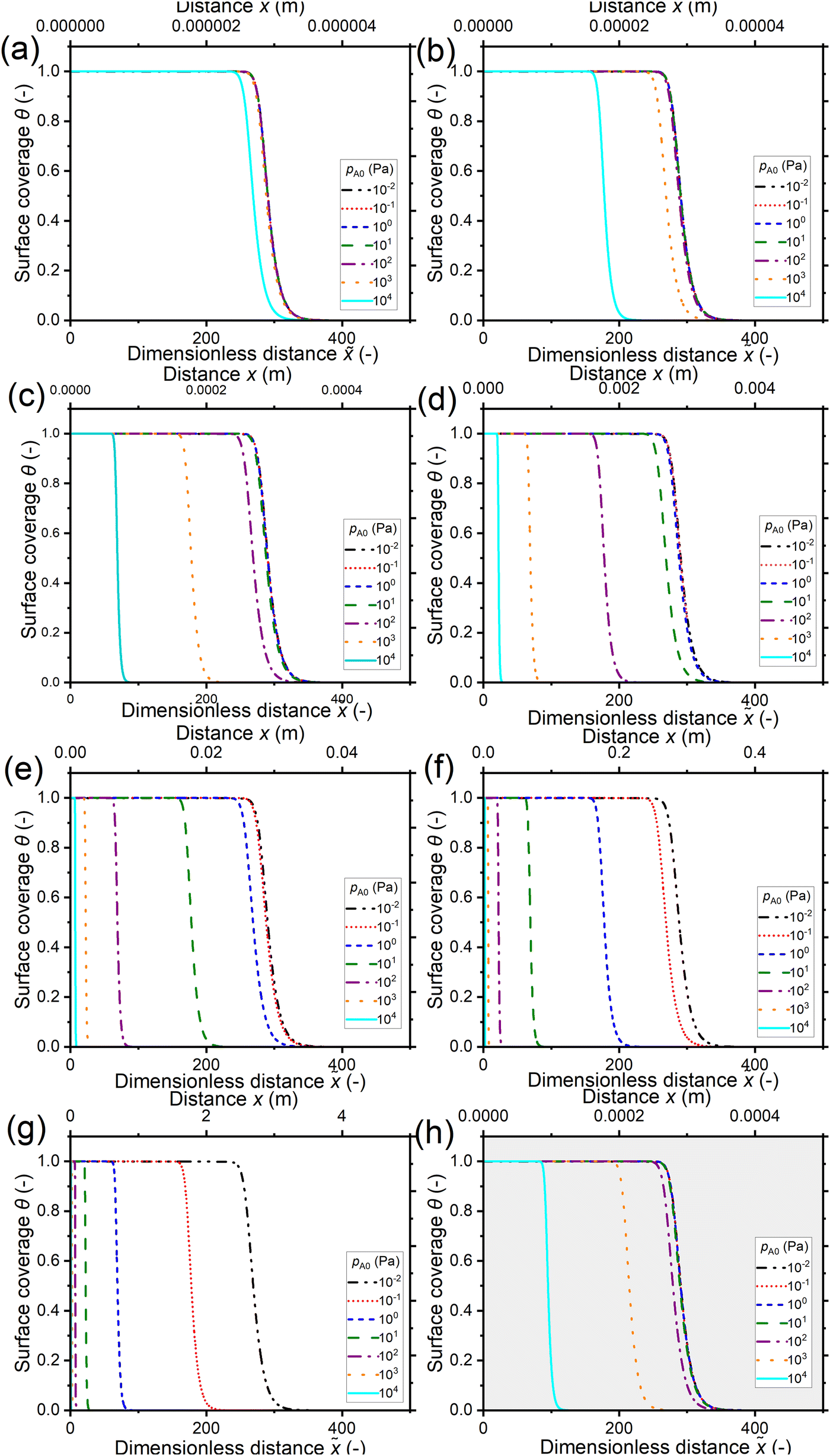 | ||
| Fig. 7 Saturation profiles in wide lateral high-aspect-ratio channels of various heights at a constant reactant exposure of 10 Pa s (300 °C). Channel heights: (a) 10 nm, (b) 100 nm, (c) 1 μm, (d) 10 μm, (e) 100 μm, (f) 1 mm, (g) 1 cm, and (h) 500 nm (the typical PillarHall™ case17,37). The calculated Knudsen number values for the simulated points are shown in Fig. 3(a). To maintain a constant exposure of 10 Pa s, the pulse time was varied in the range of 10−3 to 103 s and the initial reactant partial pressure pA0 was varied in the range of 10−2 to 104 Pa (Section S2 in the ESI†). The other simulation conditions are listed in Table 1. | ||
Fig. 8 shows the penetration depth values in terms of the dimensionless distance extracted from the saturation profiles of Fig. 7. Fig. 8(a) shows that for a given reactant partial pressure pA0, the penetration depth at half coverage θ=0.5 either decreases or remains constant with increasing channel height. For a given channel height H, with an increase in the reactant partial pressure pA0, the penetration depth θ=0.5 either decreases or remains constant. Intuitively, since the exposure is constant, one could expect the penetration depth θ=0.5 to be constant. However, θ=0.5 decreases with increasing channel height and with increasing reactant partial pressure pA0.
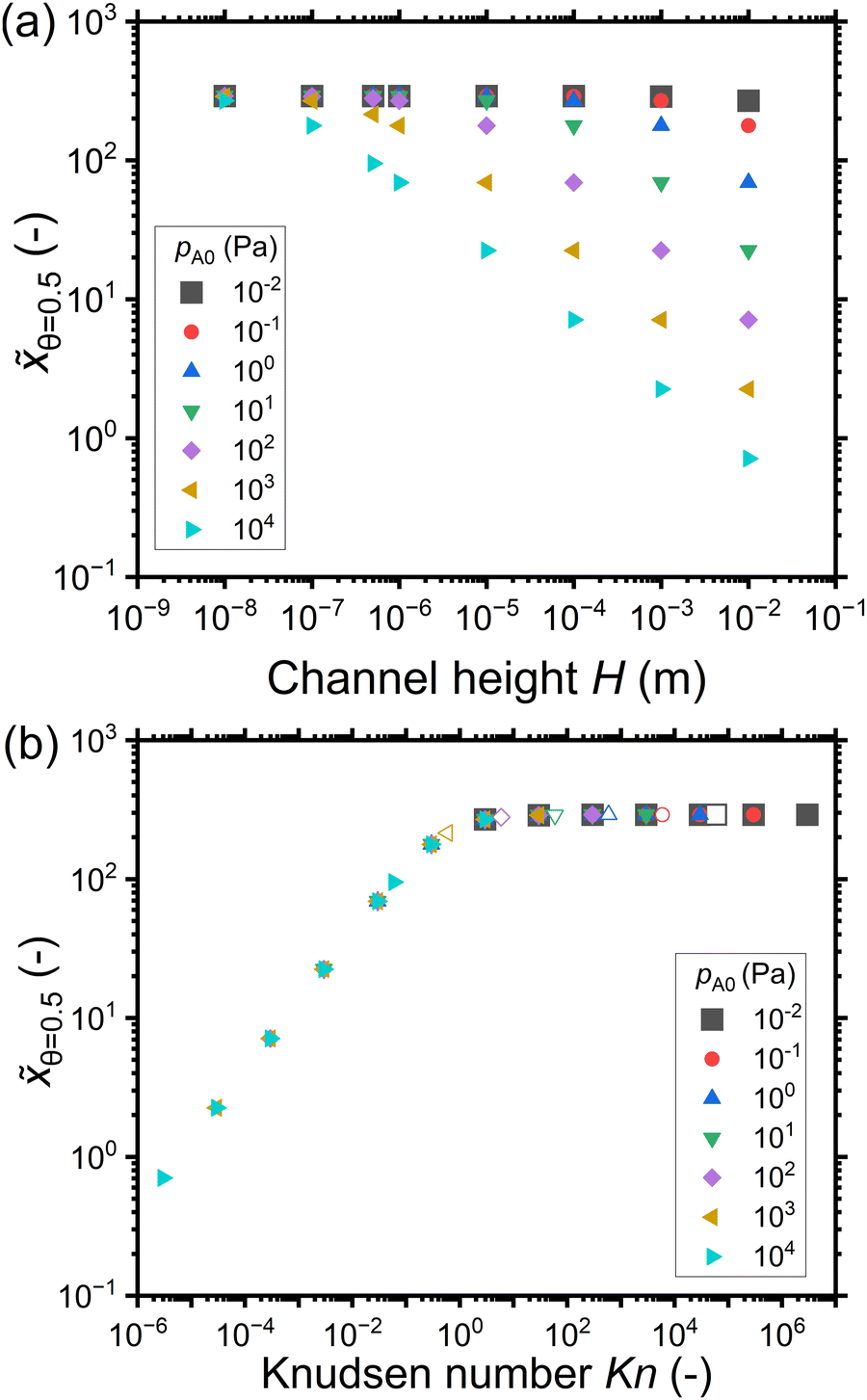 | ||
| Fig. 8 The penetration depth in terms of the dimensionless distance at half coverage θ=0.5 with respect to (a) the channel height H and (b) the Knudsen number. Data was extracted from the saturation profiles shown in Fig. 7. The hollow symbols at 500 nm represent the typical PillarHall™ case.17,37 | ||
When the data of Fig. 8(a) is plotted as function of the Knudsen number, the data points collapse into a single curve in Fig. 8(b). In the continuum flow regime (Kn ≪ 1), as the Knudsen number increases, the penetration depth increases irrespective of the reactant partial pressure. In the transition flow regime (Kn ∼ 1), the increase in the penetration depth levels off. The penetration depth becomes constant with increasing Knudsen number in the free molecular flow regime (Kn ≫ 1). The effect of the Knudsen number on the penetration depth is further illustrated in Fig. 9, where the saturation profiles obtained for various Knudsen numbers are presented together. Larger channel heights correspond to lower Knudsen numbers and hence have a lower penetration depth. Similarly, the higher reactant partial pressures correspond to lower Knudsen numbers and have a lower penetration depth. Once the Knudsen numbers are large enough to be in the free molecular flow regime (Kn ≫ 1), the penetration depth is constant when the total exposure is the same, irrespective of the reactant partial pressure or channel height.
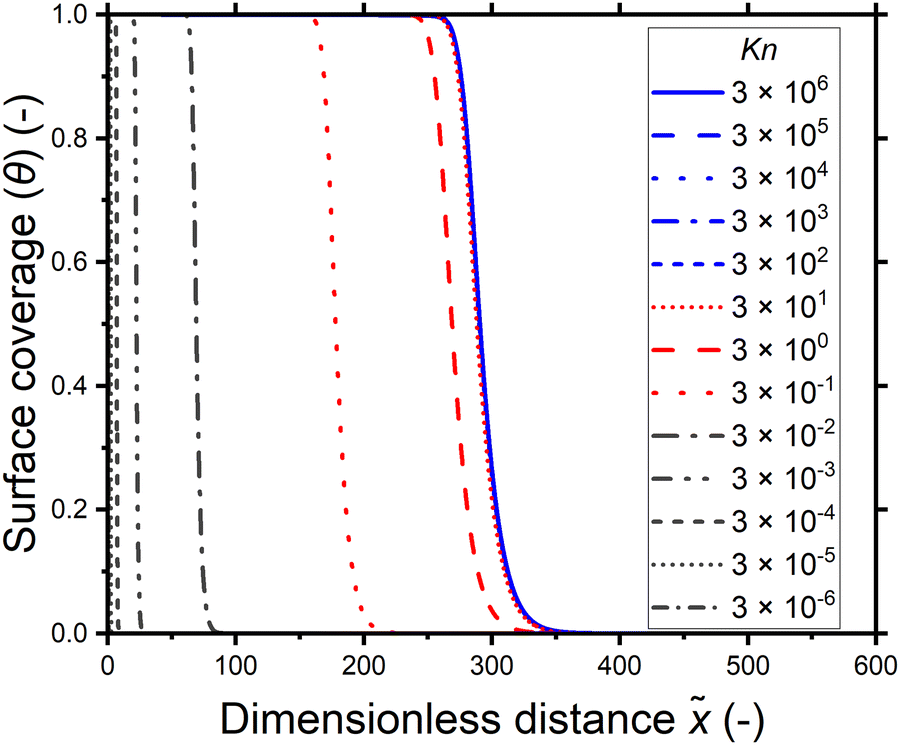 | ||
| Fig. 9 The saturation profiles as a function of the dimensionless distance for different Knudsen numbers at a constant reactant dose of 10 Pa s, collected from Fig. 7 (the data used is specified in the ESI,† Table S3). For Kn ≳ 102, the surface coverage profiles overlap. | ||
To further analyze the effect of the Knudsen number on the saturation profile characteristics, the penetration depth at half coverage and the absolute slope of the adsorption front extracted from the saturation profiles of Fig. 7 are shown in Fig. 10. The penetration depth is lowest in the continuum (Kn ≪ 1), increases with increasing Knudsen number through the transition regime (Kn ∼ 1), and settles to a constant value in the free molecular flow regime (Kn ≫ 1). The absolute value of the slope decreases with increasing Knudsen number in the continuum regime (Kn ≪ 1), the decrease slows down in the transition regime (Kn ∼ 1), and the value settles to constant in the free molecular flow (Kn ≫ 1).
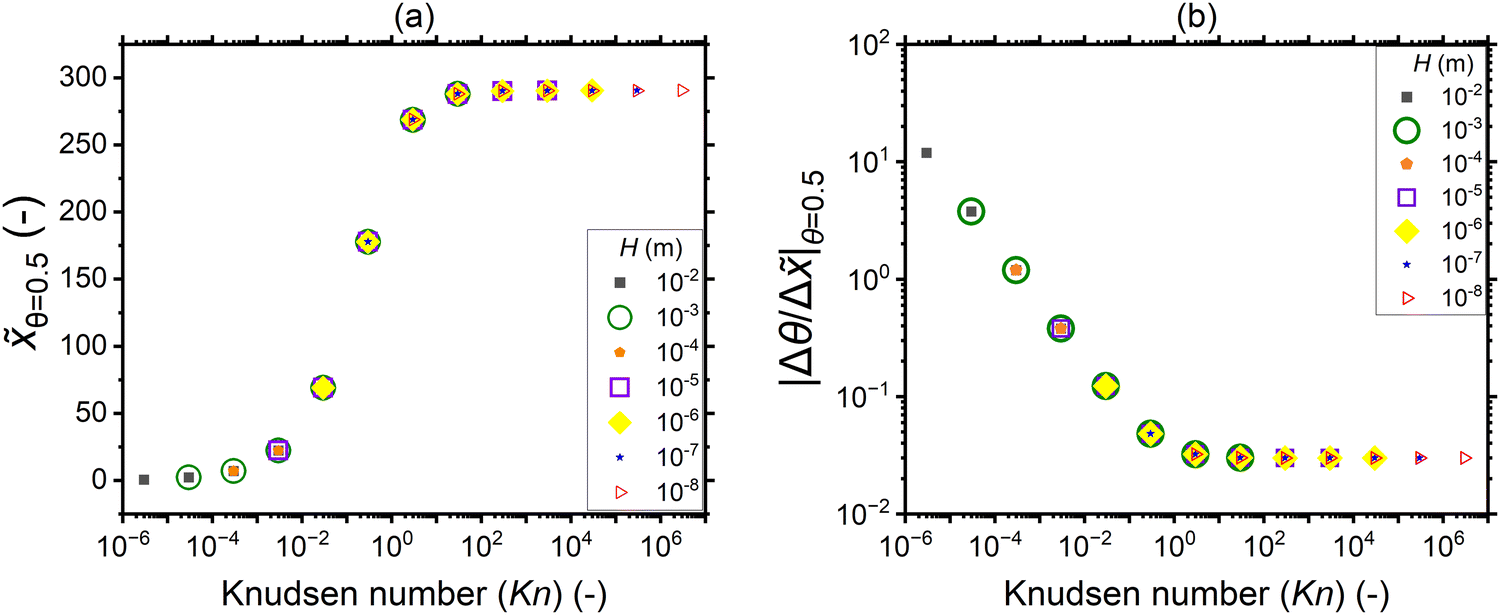 | ||
| Fig. 10 (a) The penetration depth at half coverage θ=0.5 and (b) the absolute value of the slope at half coverage |Δθ/Δ|θ=0.5 plotted as a function of the Knudsen number for a sticking coefficient c of 0.01. The channel heights, reactant partial pressure, and saturation profiles are the same as in Fig. 7. Table 1 lists the other parameters. | ||
3.3 The saturation profile characteristics at constant exposure with a varying sticking coefficient
In this section, the saturation profile characteristics are analyzed for a varied sticking coefficient while keeping the total exposure constant (10 Pa s). Fig. 11 illustrates as a function of the Knudsen number the influence of the sticking coefficient on the penetration depth at half coverage (a) and the slope of the adsorption front at half coverage (b). The corresponding saturation profiles are provided in the ESI† (Fig. S5).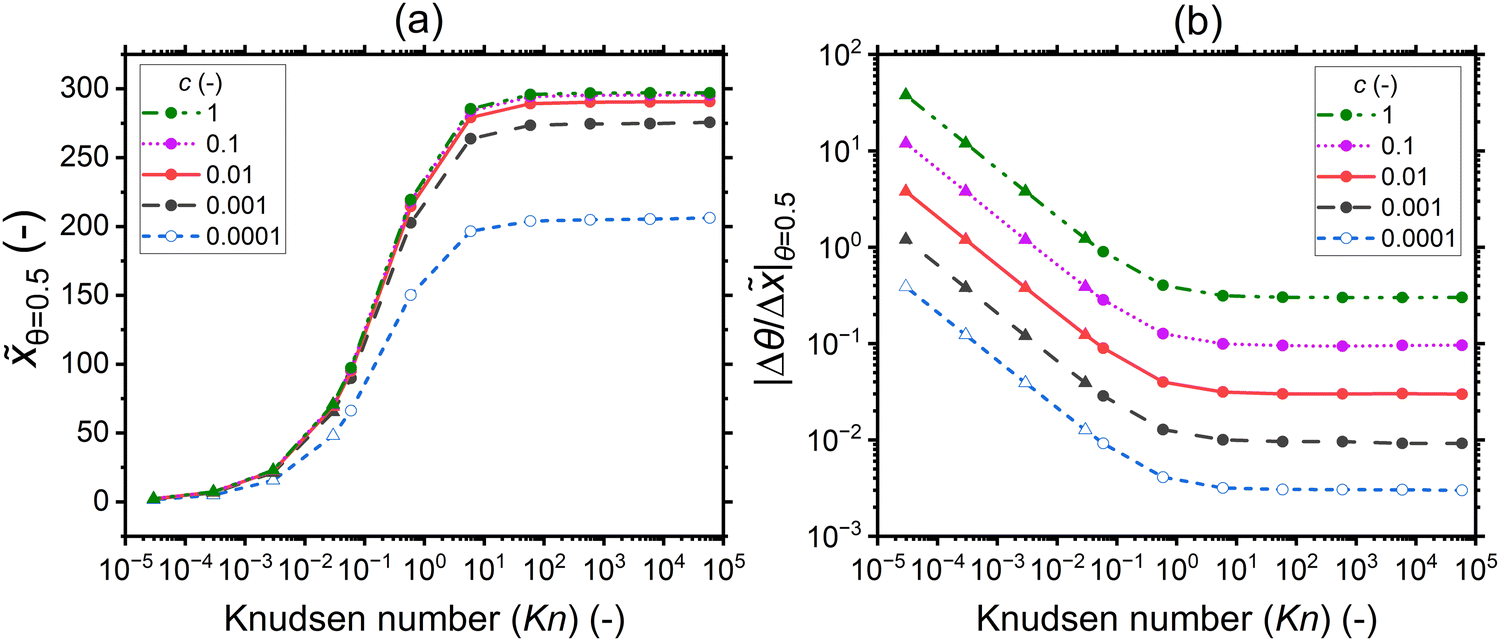 | ||
| Fig. 11 (a) The penetration depth at half coverage θ=0.5 and (b) the absolute value of the slope at half coverage |Δθ/Δ|θ=0.5 as a function of the Knudsen number for varying sticking coefficients: 1, 0.1, 0.01, 0.001, and 0.0001. The channel height used is 500 nm (the typical PillarHall™ case17,37). To obtain the last four points in the lower Knudsen number regime, a channel height of 1 mm is used, as indicated by the solid triangle symbol. The other parameters used for the simulations are provided in Table 1. The hollow symbols used for data of c = 0.0001 signify the fact that the used exposure did not yet saturate the adsorption at the channel entrance (saturation curves in Fig. S7, ESI†). | ||
For all sticking coefficient values, the lowest penetration depth values are obtained in the continuum (Kn ≪ 1), see Fig. 11(a). The values increase with the Knudsen number in the transition regime (Kn ∼ 1), and they settle to a constant value in the free molecular flow (Kn ≫ 1). While the penetration depth at half coverage in general shows not much dependence on the sticking coefficient, especially in the free molecular flow regime, a slight variation in the penetration depth is seen for different sticking coefficients, with a higher sticking coefficient leading to a larger penetration depth at half coverage. The slight variation in the penetration depth at half coverage with the sticking coefficient is, to the authors’ knowledge, related to the simplified approximate way of treating the reactant partial pressure in the Ylilammi et al.,19,21 and not a general feature of all diffusion–reaction models.20,24 Specifically, simulations made with the Ylilammi et al. model19,21 show a pivot point for θ(x,t) with varied sticking coefficient c at θ ≈ 0.3 (see Fig. S7, ESI†), while the full solution of eqn (7) shows a pivot point at about θ ≈ 0.5 (see Fig. 1(b) of ref. 20).20 Would the penetration depth be investigated at θ ≈ 0.3 for simulations made with the Ylilammi et al. model, it would be independent of the sticking coefficient. The case c = 0.0001 differs further from the series, because saturation had not fully taken place even at the channel entrance with the particular simulation parameters used (Fig. S7, ESI†).
The slope of the adsorption front at half coverage depends systematically on the sticking coefficient c, with the specific relation depending on the diffusion regime (Fig. 11(b)). It is meaningful to examine the trends with decreasing Knudsen number. In a free molecular flow (Kn ≫ 1), the slope does not depend on the Knudsen number but depends on the sticking coefficient. The curves are vertically offset, so that a higher sticking coefficient corresponds to a higher absolute value of the slope. This observation was made previously and is the basis of the Arts et al.20 slope method. In the transition regime (Kn ∼ 1), the offset with a higher sticking coefficient corresponding to the higher absolute value of the slope remains, but the Knudsen number starts affecting the result: a lower Knudsen number corresponds to a steeper slope. In the continuum regime (Kn ≪ 1), the offset remains, and the (logarithm of the) slope seems to depend linearly on the (logarithm of the) Knudsen number.
To conclude on the effect of the sticking coefficient on the saturation profile characteristics, the sticking coefficient barely affects the penetration depth, but it strongly affects the slope of the adsorption front at half coverage. The way the sticking coefficient affects the slope depends on the diffusion regime. Further analysis of this dependence will be provided in the Discussion section.
4 Discussion
4.1 Extended slope method for extracting the sticking coefficient from the saturation profile
In Fig. 11(b), it was seen that the slope of the adsorption front of the saturation profile depends on (i) the sticking coefficient and (ii) the Knudsen number. A slope method was previously derived by Arts et al.20 to calculate the sticking coefficient describing the (lumped) kinetics of an ALD reaction in the free molecular flow regime (Kn ≫ 1). In this section, we analyze the simulated trends with the goal of deriving an extended slope method to extract the sticking coefficient from the saturation profile in other regimes (Kn ∼ 1 and Kn ≪ 1).First, the trends are analyzed in the free molecular flow regime (Kn ≫ 1), where the slope of the adsorption front is independent of the Knudsen number (Fig. 11(b)). The least squares fitting of the data shows the following square root dependence of the slope of the adsorption front on the sticking coefficient:
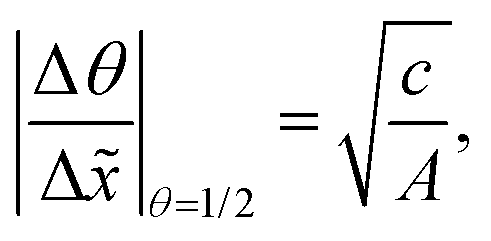 | (22) |
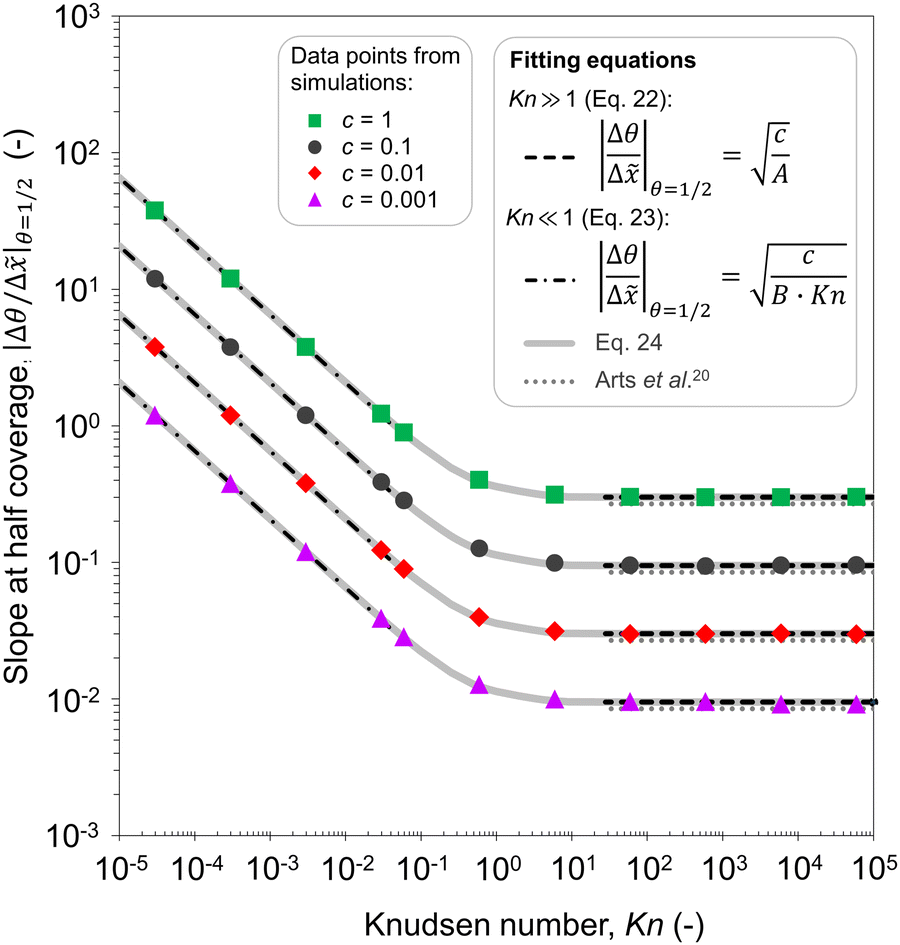 | ||
| Fig. 12 Fitted data of the absolute value of the slope at half coverage as a function of the Knudsen number. Data points for c = 1, 0.1, 0.01, and 0.001 were obtained using the Ylilammi et al.19 model for diffusion-limited conditions. Fitting parameters of the equations: A = 11.1 and B = 23.3. | ||
Second, a similar analysis is performed for the continuum regime (Kn ≪ 1). The least squares fitting with two variables (the sticking coefficient c and Knudsen number Kn) leads to the following equation:
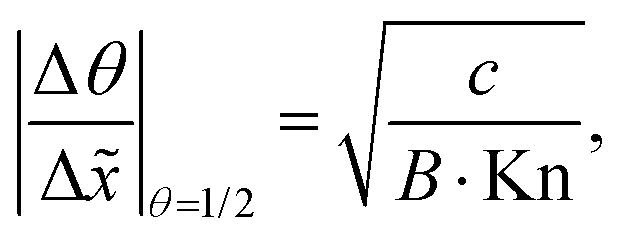 | (23) |
It would be helpful to have one empirical equation to relate the slope of the adsorption front at half coverage to the sticking coefficient for any Knudsen number. The following equation merges eqn (22) and (23) for a free molecular flow and continuum (diffusion-limited conditions), providing an approximate calculation also for the transition regime:
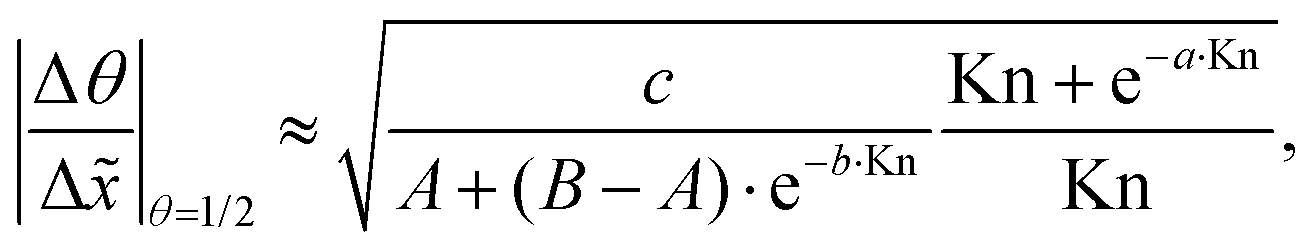 | (24) |
4.2 The effect of varying individual parameters on the saturation profile characteristics
In this section, we discuss the effect of varying a single parameter at a time on the characteristics of the ALD saturation profile. This analysis is carried out using the results obtained from simulations with the Ylilammi et al. diffusion–reaction model19 for a free molecular flow with Knudsen diffusion (Kn ≫ 1), which is the simplest reference case, for the transition regime (Kn ∼ 1), and for a continuum with molecular diffusion (Kn ≪ 1). A similar analysis was previously performed for the free molecular flow and transition regimes;21 this work updates and adds to the previous analysis. As numerical measures of the ALD saturation profile characteristics, we use (i) the penetration depth at half coverageθ=0.5, expressed in terms of the dimensionless distance , (ii) the absolute value of the slope of the adsorption front of the scaled saturation profile (the thickness divided by the number of cycles vs. the dimensionless distance),17 and (iii) the absolute value of the slope of the adsorption front of the Type 1 normalized saturation profile (the normalized thickness vs. dimensionless distance).17 The various ways to plot and interpret the ALD saturation profile (thickness profile) were introduced in ref. 17 and discussed further in ref. 21.
A summary of the interpreted trends is presented in Table 2. The corresponding ALD saturation profiles for the continuum flow regime are presented in the ESI† (Fig. S8). The parameter ranges used in these specific simulations are presented as footnotes in Table 2 and summarized in Table S4 of the ESI.† In the following text, we discuss the effect of varying each parameter individually.
| Simulation parameter (increases) | Kn ≫ 1c | Kn ∼ 1d | Kn ≪ 1e | ||||||
|---|---|---|---|---|---|---|---|---|---|
|
|
|
|
|||||||
| (−) | (nm) | (−) | (−) | (nm) | (−) | (−) | (nm) | (−) | |
|
a The detailed parameters are provide in Table S3 of the ESI.
b The common center point parameters for all flow regimes were: W = 10 mm, N = 1, T = 573.15 K, t1 = 0.1 s, MA = 0.1 kg mol−1, dA = 6 × 10−10 m, MI = 0.028 kg mol−1, dI = 4 × 10−10 m, q = 4 nm−2, ρ = 3500 kg m−3, M = 0.050 kg mol−1, Pd = 0.01 s−1, c = 0.01. These are the same as given by Yim et al.21
c Additional center point parameters for the free molecular regime (Kn ≫ 1): H = 0.05 μm, pA0 = 50 Pa, pI = 250 Pa. These are the same as those of Yim et al.21
d Additional center point parameters for the transition regime (Kn ∼ 1): H = 0.5 μm, pA0 = 500 Pa, pI = 2500 Pa. These are the same as those of Yim et al.21
e Additional center point parameters for the continuum regime (Kn ≪ 1): H = 500 μm, pA0 = 500 Pa, pI = 2500 Pa.
f The initial partial pressure of reactant A pA0 was increased from 1 to 100 Pa with a constant inert gas pressure pI of 250 Pa in the free molecular flow regime and from 100 to 1000 Pa with constant pI of 2500 Pa in the transition and continuum flow regimes as in Yim et al.21
g For Kn ∼ 1, a decrease in the value is seen in the third significant digit. For Kn ≪ 1, a decrease in the value is seen in the second significant digit.
h This is specific to the Ylilammi19 model and other models show no change here.
i The comparison was made for Pd in the range of 10−3 to 10 s−1, as in Yim et al.21 For higher values of the desorption probability (Pd ≥ 10), the absolute value of the slope decreases (Fig. S9, ESI).
j The total pressure p was increased by increasing the partial pressure of the inert gas pI from 0.5 to 250 Pa with a constant pA0 of 50 Pa in the free molecular flow regime and by increasing the pI from 625 to 10000 Pa with a constant pA0 of 500 Pa in the transition and continuum flow regimes.
k In ref. 21, this was indicated as no change. Here it is re-evaluated and shown as a slightly decreasing trend.
|
|||||||||
| Channel height (H) | — | — | — | ↘ | ↗ | ↗ | ↘↘ | ↗↗ | ↗↗ |
| Initial partial pressure of the ALD reactant A (pA0)f | ↗↗ | — | — | ↗↗ | —g | —g | ↗↗ | ↘g | ↘g |
| Reactant pulse time (t1) | ↗↗↗ | — | — | ↗↗↗ | — | — | ↗↗↗ | — | — |
| Sticking coefficient (c) | ↗h | ↗↗↗ | ↗↗↗ | ↗h | ↗↗↗ | ↗↗↗ | ↗h | ↗↗↗ | ↗↗↗ |
| Desorption probability (Pd) | — | — | — | — | — | — | — | — | —i |
| Adsorption capacity (q) | ↘↘↘ | ↗↗ | — | ↘↘↘ | ↗↗ | — | ↘↘↘ | ↗↗ | — |
| Temperature (T) | ↘ | — | — | ↘ | — | — | ↗ | ↘ | ↘ |
| Total pressure (p)j | — | — | — | ↘ | ↗ | ↗ | ↘↘ | ↗↗ | ↗↗ |
| Molar mass of the ALD reactant (MA) | ↘ | — | — | ↘ | ↗ | ↗ | ↘ | ↗ | ↗ |
| Molar mass of the carrier gas (MI) | — | — | — | ↗ | ↘ | ↘ | ↗ | ↘↘ | ↘↘ |
| Size of the reactant molecule (dA) | — | — | — | ↘k | ↗ | ↗ | ↘ | ↗ | ↗ |
| Size of the inert carrier gas molecule (dI) | — | — | — | ↘ | ↗ | ↗ | ↘ | ↗ | ↗ |
| Density of the grown material (ρ) | — | ↘ | — | — | ↘ | — | — | ↘ | — |
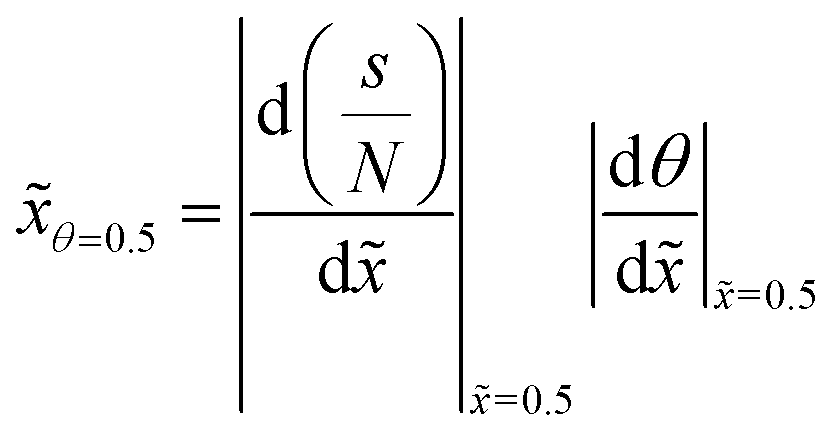
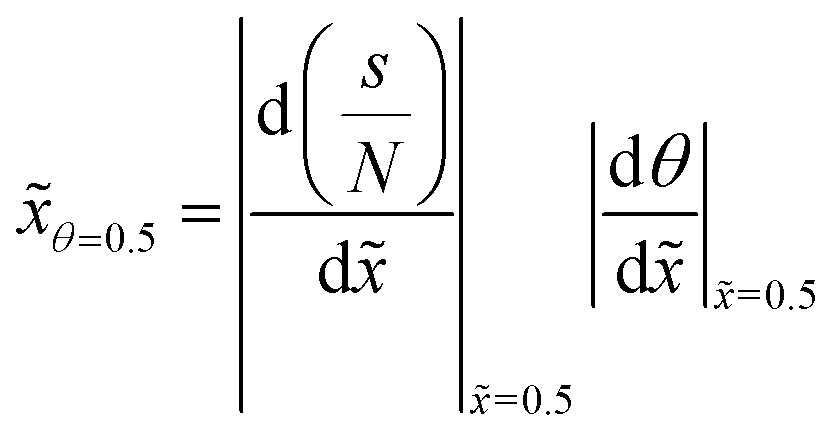
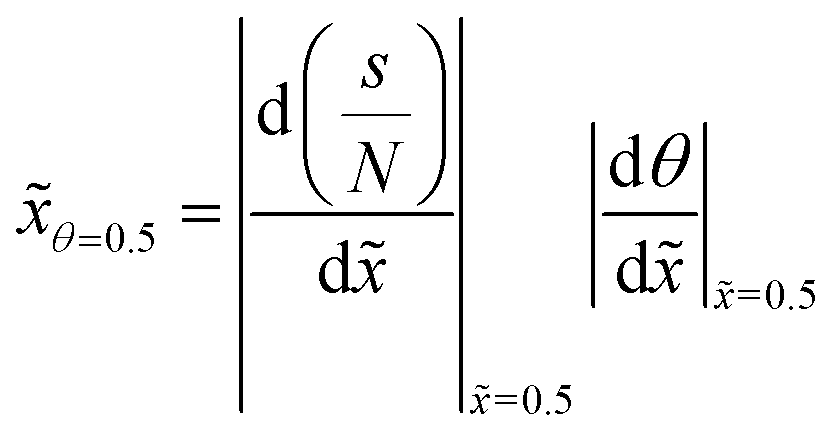
Increasing the channel height H under the reference conditions of the free molecular flow (Kn ≫ 1) does not influence the penetration depth (when expressed as the dimensionless distance ) or the slope of the adsorption front (Table 2). (The penetration depth expressed as physical distance x of course increases with H.) In the transition (Kn ∼ 1) and continuum regimes (Kn ≪ 1), increasing the channel height does have an effect: the penetration depth decreases (more strongly so in the continuum than in the transition flow) and the absolute value of the slope increases (again more strongly so in the continuum than in the transition flow). As discussed in Section 3.2, in the transition and continuum flow regimes, increasing the channel height corresponds to a decrease in the Knudsen number, leading to slower diffusion through more gas-phase molecule–molecule interactions. This leads to a lower penetration depth and a steeper slope of the adsorption front.
Increasing the reactant partial pressure pA0 leads to a higher exposure (pA0·t) and hence an increase in the penetration depth for all the diffusion regimes (free molecular flow (Kn ≫ 1), transition (Kn ∼ 1), continuum (Kn ≪ 1)). (Note that in this simulation series, the inert gas pressure pI was kept constant, meaning that the total pressure increased only slightly.) The slope of the adsorption front is not affected in the free molecular flow (Kn ≫ 1), and there is no effect in the transition regime (Kn ∼ 1) either. In the continuum (Kn ≪ 1), in contrast, the absolute value of the slope of the adsorption front barely noticeably decreases with increasing pA0. If the change was related to molecular diffusion coefficient, which slightly decreases, we would expect to see an increase in the absolute value of the slope, similarly as was the case in results reported in Section 3.1. The origin of the observed trend is clearly different, in this case. While this origin of this trend is currently not fully explained, we speculate that it may be related to the Ylilammi et al. model with the simplified analytic solution for pA(x,t), and not to the full solution of the diffusion equation (eqn (7)).
Increasing the pulse time t1 also leads to a higher exposure (pA0·t) and, consequently, an increase in the penetration depth for all diffusion regimes (free molecular flow (Kn ≫ 1), transition (Kn ∼ 1), continuum (Kn ≪ 1)). The slope of the adsorption front remains unaffected in all cases. In real ALD growth experiments, increasing the exposure time is a typical way to increase the total exposure of a reactant and achieve deeper penetration into a HAR feature, with increasing the partial pressure of the reactant being the other alternative.21 On the basis of this observation, it seems advisable to increase the total exposure in experimental conformality studies preferably by increasing the exposure time, because that does not risk altering the Knudsen number, the diffusion regime, or the slope of the adsorption front, and therefore makes interpretations related to the kinetic parameters more straightforward.
Increasing the sticking coefficient c strongly correlates with an increasing absolute value of the slope of the adsorption front in all three diffusion regimes (the free molecular flow (Kn ≫ 1), transition (Kn ∼ 1), continuum (Kn ≪ 1)). This correlation has been discussed before21 and for the basis of Arts et al.20 slope method as well as the extended slope method proposed in this work. The simulations in this work show a slight positive correlation between the sticking coefficient and the penetration depth for all diffusion regimes. To our best understanding, this correlation is specific to the Ylilammi et al.19 diffusion–reaction model used in this work (and related to the way it treats the partial pressure of the reactant in a simplified analytic way), and it is not expected for all diffusion–reaction models. Indeed, recent simulations, e.g., those by Arts et al.20 with the Yanguas-Gil–Elam model,38,39 showed no correlation between the sticking coefficient and the penetration depth.
The diffusion–reaction model of Ylilammi et al.19 used in this work allows reversible reactions, in contrast to many other ALD models that only allow irreversible reactions. The reversibility is modeled through the desorption probability Pd, or alternatively, the adsorption equilibrium constant K. The two are related through the equation K = cQ/qPd (eqn (20) in ref. 21 and eqn (13) in ref. 19). In the simulations carried out for this summary, varying the desorption probability does not affect the penetration depth or slope in any of the diffusion regimes. However, the values in the current simulations were chosen to be rather low, as we had Pd from 0.001 to 10 s−1. For higher values (Pd ≥ 10), a decreasing effect on the absolute value of the slope is seen, along with a change in the shape of the saturation profile (Fig. S9, ESI†). An example of a significant effect of the desorption probability can be seen in earlier simulations made for the TiCl4–H2O ALD process to grow TiO2.19
Varying the adsorption capacity q, which is a direct measure of the ALD GPC and can be converted into the thickness per cycle through a simple formula (eqn (10)), strongly affects the saturation profile characteristics. The trends are the same regardless of the diffusion regime (Kn ≫ 1, Kn ∼ 1, and Kn ≪ 1). With increasing adsorption capacity q (and thus an increasing GPC), the penetration depth strongly decreases. The decreasing effect of the GPC on the penetration depth was observed earlier in simulations21 and in experimental studies.17,40 When examining the adsorption front for the scaled saturation profile, with increasing adsorption capacity q (increasing GPC) the absolute value of the slope increases. When examining the adsorption front for the Type 1 normalized saturation profile, there is no change to observe in the absolute value of the slope. This case demonstrates how the difference in the two methods used to determine the saturation profile, introduced by Yim et al.,17 is fundamentally important: the scaled saturation profile preserves the information of the core characteristic of the ALD (the GPC), while the Type 1 saturation profile does not show it. While the slope method20 relies on the Type 1 normalized saturation profile, it would be unwise to examine only this normalized saturation profile; the scaled saturation profile is superior in its information content.
Increasing the temperature T of the ALD process influences the characteristics of the saturation profile, with the exact effect depending on the diffusion regime. (Diffusion coefficients are presented in the ESI† as Fig. S11.) In the reference free molecular flow conditions (Kn ≫ 1), the penetration depth decreases slightly with increasing temperature, while the slope of the adsorption front is not affected. The transition regime (Kn ∼ 1) shows the same effect (decrease) as a free molecular flow regime. The continuum regime (Kn ≪ 1) shows the opposite effect: the penetration depth increases slightly with increasing temperature, and at the same time, the slope of the adsorption front gets slightly less steep. These two trends can be understood by considering two effects: (i) the effect of temperature on the density of the gas and (ii) the effect of temperature on the molecular diffusion coefficient. The effect of temperature on the density of the gas is seen through the ideal gas law, pV = nRT. For a given partial pressure of the reactant molecules, the gas is less dense at a higher temperature; that is, the number density of the molecules in the gas (m−3) decreases with increasing temperature (n/V = p/RT). Thus, while the total exposure calculated in the classic way through pA0·t is kept constant in the simulations, the exposure in terms of molecules entering the channel is not constant, because the number density is not constant (gas is less dense at a higher temperature). (Note: to be accurate, the total exposure, calculated from the partial pressure times time, needs temperature as a reference to be accurately defined.) Therefore, the number of molecules entering the channel during the simulation time decreases with temperature, leading to the penetration depth also decreasing under the reference free molecular flow conditions. More detailed analysis of the effect of temperature on the penetration depth in free molecular flow was recently published by Heikkinen et al.41 In the continuum regime, where molecular diffusion and gas-phase interactions (collisions) dominate, the increasing effect of temperature on molecular diffusion (eqn (14)) dominates over the decreasing effect of temperature on the gas density, and the penetration depth thereby increases.
Varying the total pressure p alone has a distinctively different effect on the characteristics of the saturation profile in different diffusion regimes. Note that in this simulation series, the exposure (pA0·t) was kept constant by keeping pA0 constant; the total pressure was varied by varying the pressure of the inert gas pI. In the reference free molecular flow conditions (Kn ≫ 1), the pressure does not influence the saturation profile characteristics – neither the penetration depth nor the slope of the adsorption front. In the transition regime (Kn ∼ 1), the penetration depth slightly decreases and the slope of the adsorption front becomes slightly steeper with increasing total pressure. In the continuum regime (Kn ≪ 1), in contrast, increasing the total pressure leads to a strong decrease in the penetration depth and a significantly steeper slope of the adsorption front. These trends are explained by the decreasing effect of pressure on the molecular diffusion coefficient and were already discussed earlier in Section 3.1.
Increasing the molar mass of reactant A MA causes a lower penetration depth in all diffusion regimes. Under the reference free molecular flow conditions (Kn ≫ 1), increasing the molar mass has no effect on the slope of the adsorption front. In the transition (Kn ∼ 1) and continuum regimes (Kn ≪ 1), a slight increase in the absolute value of the slope (i.e., steepness) of the adsorption front is observed. The decrease in the penetration depth is explained by the slowing down of diffusion through heavier molecules moving more slowly than lighter molecules (eqn (14)–(16)).
Increasing the molar mass of the inert carrier gas molecules MI has no influence on the penetration depth or the slope of the adsorption front under the reference free molecular flow conditions (Kn ≫ 1). Interestingly, in the transition regime (Kn ∼ 1), the penetration depth increases and the absolute value of the slope of the adsorption front decreases with an increasing molar mass of the inert gas. The continuum (Kn ≪ 1) shows similar trends as the transition regime, only stronger. As the molar mass of the inert carrier gas increases, the overall collision frequency decreases (eqn (14) and (16)). This, again, leads to a higher effective diffusion of the reactant gas.
Increasing the hard-sphere diameter of the reactant molecule dA and the inert carrier gas molecule dI has no effect on the characteristics of the saturation profile under the reference free molecular flow conditions (Kn ≫ 1). When entering the transition (Kn ∼ 1) and continuum regimes (Kn ≪ 1), a small effect is seen, where the penetration depth decreases slightly and the steepness of the adsorption front increases slightly with increasing hard-sphere diameters of the reactant molecule and the carrier gas. With increasing hard-sphere diameters, the molecular diffusion coefficient decreases (eqn (14) and (16)). This leads to a lower penetration depth and a steeper adsorption front under the transition and continuum conditions.
The last parameter to vary individually was the density ρ of the material that makes up the thin film being grown by ALD. With all other parameters being constant (including the adsorption capacity q), the density only affects the physical thickness of the film being grown and the GPC, expressed as thickness per cycle. The trends in the saturation profile characteristics are the same regardless of the diffusion regimes (Kn ≫ 1, Kn ∼ 1, and Kn ≪ 1). The penetration depth remains unaffected by the change. With increasing density, the slope calculated from the scaled saturation profile decreases, while the slope calculated from the Type 1 normalized saturation profile remains unaffected. These changes with ρ are as expected and identical to those reported for the free molecular flow and transition regime earlier.21
5 Summary and conclusion
The effect of the Knudsen number on the saturation profile in diffusion–limited ALD in narrow channels was analyzed with a diffusion–reaction model in this work.19,21 Simulations were performed for a large range of realistic channel heights (10−8 to 10−2 m) and ALD reactant partial pressures (10−2 to 104 Pa). The resulting large range of Knudsen numbers (10−6 to 106) covers free molecular flow with Knudsen diffusion (molecule–wall interactions, Kn ≫ 1), the transition regime (Kn ∼ 1), and a continuum with molecular diffusion (molecule–molecule interactions, Kn ≪ 1). A series of simulations were performed (i) while varying the total exposure (the partial pressure of the reactant pA0 times the exposure time t) and (ii) for a constant total exposure (10 Pa s at 523 K).The simulation series with varying total exposure revealed different trends for the saturation profile characteristics with the channel height and partial pressure depending on the Knudsen number. In the free molecular flow (Kn ≫ 1), the penetration depth increased with the reactant partial pressure (i.e., the total exposure), following the well-known square-root-of-exposure trend. In the continuum (Kn ≪ 1), in contrast, the penetration depth in a given channel height stagnated to a constant value with increasing reactant partial pressure. The stagnation was accompanied by a change in the shape of the saturation profile, which led to a cross-over in the simulated saturation profiles: the leading edge of the adsorption front reached further at low reactant partial pressures than at high pressures. While the observed stagnation and cross-over may at first seem counter-intuitive, as discussed in detail in the text, these trends are readily explained by changes in the type of diffusion from Knudsen diffusion (in free molecular flow) to molecular diffusion (in the continuum), and by how increasing pressure inversely affects the diffusion coefficient under continuum conditions.
The simulation series with constant total exposure revealed a constant penetration depth in terms of the dimensionless distance ( = x/H) in a free molecular flow (Kn ≫ 1), irrespective of the specific value of the Knudsen number. (Note: the physical penetration depth x then scales with the channel height H.) When the Knudsen number decreased, in the transition regime (Kn ∼ 1), the dimensionless penetration depth started to decrease, and in the continuum (Kn ≪ 1), it decreased strongly with decreasing Knudsen number. Examining the saturation profile further, it is seen that the slope of the adsorption front has a constant value in a free molecular flow (Kn ≫ 1), starts to increase with decreasing Knudsen number in the transition regime (Kn ∼ 1), and continues to increase with decreasing Knudsen number in the continuum (Kn ≪ 1). An extended slope method was proposed relating the slope of the adsorption front at half coverage to the sticking coefficient at any Knudsen number.
The trends in the saturation profile characteristics for different diffusion regimes (Kn ≫ 1, Kn ∼ 1, Kn ≪ 1) were analyzed while varying individual simulation parameters, extending the earlier analysis for the free molecular flow and transition regime21 to also cover the continuum. The responses to the changes in the individual parameters in the saturation profile characteristics – the penetration depth and slope of the adsorption front – were similar across all diffusion regimes when the following parameters were varied: the pulse time t1, sticking coefficient c, desorption probability Pd, adsorption capacity q, and material density ρ. In contrast, the response depended on the diffusion regime when the following parameters were varied: the channel height H, temperature T, reactant gas pressure pA0, total pressure p, reactant pressure fraction of the total pressure (pA0/p), molar mass of the reactant MA, molar mass of the inert carrier gas MI, diameter of the reactant dA, and diameter of the inert carrier gas dI. Most cases in which the response seen in the saturation profile characteristics depends on the diffusion regime can be explained by the effect of the individual parameter on the diffusion coefficients (Knudsen and molecular diffusion coefficient). Because the total exposure (pA0·t) can be varied by either varying the partial pressure or by varying the pulse time, it is recommended that one should preferably vary the time to make sure one is not affecting the diffusion characteristics at the same time.
This work has shown that the saturation profile characteristics are affected by the diffusion regime, an indicator of which is the Knudsen number. It is recommended that all scientific articles published in the future in the field of ALD should report the pressure range used in the experiments and, especially if kinetic analysis is performed using saturation profiles, the Knudsen number.
List of symbols
| A | Parameter in the extended slope method (eqn (22) and (24)) (−) |
| a | Parameter in the extended slope method (eqn (24)) (−) |
| B | Parameter in the extended slope method (eqn (23) and (24)) (−) |
| b | Parameter in the extended slope method (eqn (24)) (−) |
| b A | Number of metal atoms in a reactant molecule (−) |
| b film | Number of metal atoms per formula unit of film (−) |
| c | Sticking coefficient (−) |
| c ext | Sticking coefficient back-extracted with the slope method20 (−) |
| D | Characteristic feature dimension (m) |
| D A | Molecular diffusion coefficient (m2 s−1) |
| D eff | Effective diffusion coefficient (m2 s−1) |
| D Kn | Knudsen diffusion coefficient (m2 s−1) |
| d A | Hard-sphere diameter of a molecule (m) |
| d A | Hard-sphere diameter of molecule A (m) |
| d I | Hard-sphere diameter of the inert gas molecule (m) |
| f ads | Adsorption rate (m2 s−1) |
| f des | Desorption rate (m2 s−1) |
| g | Net adsorption rate (m2 s−1) |
| gpcsat | Saturation growth per cycle, thickness-based, in the Ylilammi et al.19 model (m) |
| h | Hydraulic diameter of the channel (m) |
| H | Height of the channel (m) |
| h T | Thiele modulus (−) |
| k ads | Adsorption rate constant (m−2 s−1) |
| k des | Desorption rate constant (m−2 s−1) |
| Kn | Knudsen number |
| k B | Boltzmann constant (m2 kg s−2 K−1) |
| λ | Mean free path (m) |
| L | Length of the channel (m) |
| M | Molar mass of the ALD grown film material (kg mol−1) |
| M A | Molar mass of reactant A (kg mol−1) |
| M I | Molar mass of inert gas I (kg mol−1) |
| M film | Mass of the film (kg) |
| N | Number of ALD cycles (−) |
| n A | Particle density of reactant A (m−3) |
| N 0 | Avogadro's constant (mol−1) |
| p | Total pressure pA0 + pI (Pa) |
| p A | Partial pressure of reactant A (Pa) |
| p A0 | Initial partial pressure of reactant A at the beginning of the channel (Pa) |
| p At | Partial pressure of reactant A at xt (Pa) |
| p I | Partial pressure of inert gas I (Pa) |
| P d | Desorption probability in unit time in the Ylilammi et al.19 model (s−1) |
| q | Adsorption density of metal M atoms in the ALD growth of film of the MyZx material (m−2) (i.e., GPC expressed as areal number density) |
| Q | Collision rate of reactant A with the surface at unit pressure in the Ylilammi et al.19 model (m−2 s−1 Pa−1) |
| R | Gas constant (J K−1 mol−1) |
| ρ | Film mass density (kg m3) |
| σ i,j | Collision cross-section between the molecules i and j (m2) |
| θ | Surface coverage of the adsorbed species (−) |
| t | Time (s) |
| T | Temperature (K) |
|
A
| Thermal velocity of molecule A (m s−1) |
| W | Width of the channel (m) |
| x | Physical distance (m) |
|
| Dimensionless distance (−). This is the ratio of the physical distance to the channel height. ( = x/H) |
| x s | Distance where the extrapolated linear part of the reactant pressure is zero in the Ylilammi et al.19 model (m) |
| x t | Distance of the linear part of the reactant pressure distribution in the Ylilammi et al.19 model (m) |
|
θ=0.5
| Penetration depth at half coverage in terms of the dimensionless distance (−) |
![[z with combining tilde]](https://www.rsc.org/images/entities/i_char_007a_0303.gif) A
A
| The collision frequency of reactant A with other gas molecules in a gas mixture of reactant A and inert gas I (s−1) |
Author contributions
The saturation profiles in this work were simulated by C. G. with re-implemented code21,28 based on the Ylilammi et al.19 model. The initial set of saturation profiles was simulated by J. A. V. The final simulation parameters were selected by C. G. and R. L. P. The simulations related to the summary table were carried out by C. G. The extended slope method was mainly derived by J. A. V. The initial version of the manuscript was composed by C. G. and R. L. P. The work was initiated and supervised by R. L. P. All authors discussed and contributed to the final manuscript.Data availability
We aim to publish data of this manuscript in https://Zenodo.org, DOI: 10.5281/zenodo.14016587, to be included in the https://zenodo.org/communities/ald-saturation-profile-open-data/ community. Code used for the simulations is available in GitHub, see DReaM-ALD, https://github.com/Aalto-Puurunen/dream-ald.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Academy of Finland (currently, Research Council of Finland): ALDI consortium (Decision No. 331082 and 333069) and the COOLCAT consortium (Decision No. 329978). Parts of this work were included in an oral presentation at the ALD 2023 conference in Bellevue, Washington, USA,42 in a poster at the ALD 2022 conference in Ghent, Belgium,43 and in an oral presentation at ALD Russia 2021 (online).44 Computational resources were provided by the Aalto Science-IT services.References
- T. Suntola, Mater. Sci. Rep., 1989, 4, 261–312 CrossRef CAS.
- R. L. Puurunen, J. Appl. Phys., 2005, 97, 9 CrossRef.
- S. M. George, Chem. Rev., 2010, 110, 111–131 CrossRef CAS PubMed.
- J. R. van Ommen, A. Goulas and R. L. Puurunen, Chapter “Atomic Layer Deposition”in ”Kirk-Othmer Encyclopedia of Chemical Technology, (Online), 2021, pp. 1–42, https://doi.org/10.1002/0471238961.koe00059.
- A. A. Malygin, V. E. Drozd, A. A. Malkov and V. M. Smirnov, Chem. Vap. Deposition, 2015, 21, 216–240 CrossRef CAS.
- R. L. Puurunen, Chem. Vap. Deposition, 2014, 20, 332–344 CrossRef CAS.
- E. Ahvenniemi, A. R. Akbashev, S. Ali, M. Bechelany, M. Berdova, S. Boyadjiev, D. C. Cameron, R. Chen, M. Chubarov and V. Cremers, et al. , J. Vac. Sci. Technol., A, 2017, 35, 010801 CrossRef.
- V. Cremers, R. L. Puurunen and J. Dendooven, Appl. Phys. Rev., 2019, 6, 021302 Search PubMed.
- A. Mackus, A. Bol and W. Kessels, Nanoscale, 2014, 6, 10941–10960 RSC.
- B. J. ONeill, D. H. Jackson, J. Lee, C. Canlas, P. C. Stair, C. L. Marshall, J. W. Elam, T. F. Kuech, J. A. Dumesic and G. W. Huber, ACS Catal., 2015, 5, 1804–1825 CrossRef CAS.
- Y. Zhao, L. Zhang, J. Liu, K. Adair, F. Zhao, Y. Sun, T. Wu, X. Bi, K. Amine and J. Lu, et al. , Chem. Soc. Rev., 2021, 50, 3889–3956 RSC.
- A. J. Gayle, Z. J. Berquist, Y. Chen, A. J. Hill, J. Y. Hoffman, A. R. Bielinski, A. Lenert and N. P. Dasgupta, Chem. Mater., 2021, 33, 5572–5583 CrossRef CAS.
- R. G. Gordon, D. Hausmann, E. Kim and J. Shepard, Chem. Vap. Deposition, 2003, 9, 73–78 CrossRef CAS.
- M. Rose and J. Bartha, Appl. Surf. Sci., 2009, 255, 6620–6623 CrossRef CAS.
- J. Dendooven, D. Deduytsche, J. Musschoot, R. Vanmeirhaeghe and C. Detavernier, J. Electrochem. Soc., 2009, 156, P63 CrossRef CAS.
- F. Gao, S. Arpiainen and R. L. Puurunen, J. Vac. Sci. Technol., A, 2015, 33, 010601 CrossRef.
- J. Yim, O. M. Ylivaara, M. Ylilammi, V. Korpelainen, E. Haimi, E. Verkama, M. Utriainen and R. L. Puurunen, Phys. Chem. Chem. Phys., 2020, 22, 23107–23120 RSC.
- A. Werbrouck, K. Van de Kerckhove, D. Depla, D. Poelman, P. F. Smet, J. Dendooven and C. Detavernier, J. Vac. Sci. Technol., A, 2021, 39, 062402 CrossRef CAS.
- M. Ylilammi, O. M. Ylivaara and R. L. Puurunen, J. Appl. Phys., 2018, 123, 205301 CrossRef.
- K. Arts, V. Vandalon, R. L. Puurunen, M. Utriainen, F. Gao, W. M. Kessels and H. C. Knoops, J. Vac. Sci. Technol., A, 2019, 37, 030908 CrossRef.
- J. Yim, E. Verkama, J. A. Velasco, K. Arts and R. L. Puurunen, Phys. Chem. Chem. Phys., 2022, 24, 8645–8660 RSC.
- K. Arts, M. Utriainen, R. L. Puurunen, W. M. Kessels and H. C. Knoops, J. Phys. Chem. C, 2019, 123, 27030–27035 CrossRef CAS.
- M. L. van de Poll, H. Jain, J. N. Hilfiker, M. Utriainen, P. Poodt, W. M. Kessels and B. Macco, Appl. Phys. Lett., 2023, 123, 182902 CrossRef CAS.
- A. Yanguas-Gil and J. W. Elam, Chem. Vap. Deposition, 2012, 18, 46–52 CrossRef CAS.
- M. Knudsen, Ann. Phys., 1909, 333, 75–130 CrossRef.
- J. Järvilehto, J. A. Velasco, J. Yim, C. Gonsalves and R. L. Puurunen, Phys. Chem. Chem. Phys., 2023, 25, 22952–22964 RSC.
- G. Ersavas Isitman, D. Izbassarov, R. L. Puurunen and V. Vuorinen, Chem. Eng. Sci., 2023, 277, 118862 CrossRef CAS.
- E. Verkama and R. L. Puurunen, DReaM-ALD (v1.0.0), https://github.com/Aalto-Puurunen/dream-ald, 2023 DOI:10.5281/zenodo.7759195.
- A. Yanguas-Gil, Growth and Transport in Nanostructured Materials: Reactive Transport in PVD, CVD, and ALD, Springer Nature, Cham, 2017 Search PubMed.
- W. Szmyt, C. Guerra-Nuñez, L. Huber, C. Dransfeld and I. Utke, Chem. Mater., 2021, 34, 203–216 CrossRef.
- E. W. Thiele, Ind. Eng. Chem., 1939, 31, 916–920 CrossRef CAS.
- H. S. Fogler and S. H. Fogler, Elements of Chemical Reaction Engineering, Pearson Education, 1999 Search PubMed.
- P. Poodt, A. Mameli, J. Schulpen, W. Kessels and F. Roozeboom, J. Vac. Sci. Technol., A, 2017, 35, 021502 CrossRef.
- O. Levenspiel, Chemical Reaction Engineering, John Wiley & Sons, 3rd edn, 1999 Search PubMed.
- S. Chapman and T. G. Cowling, The Mathematical Theory of Non-uniform Gases, Cambridge University Press, 1990 Search PubMed.
- R. L. Puurunen, Chem. Vap. Deposition, 2003, 9, 249–257 CrossRef CAS.
- PillarHall® – Lateral High Aspect Ratio Test Structures, Accessed May 12, 2023, https://pillarhall.com/.
- A. Yanguas-Gil and J. W. Elam, Machball (0.2.0), https://github.com/aldsim/machball, 2020 Search PubMed.
- A. Yanguas-Gil and J. W. Elam, Theor. Chem. Acc., 2014, 133, 1–13 Search PubMed.
- M. Mattinen, J. Hämäläinen, F. Gao, P. Jalkanen, K. Mizohata, J. Räisänen, R. L. Puurunen, M. Ritala and M. Leskelä, Langmuir, 2016, 32, 10559–10569 CrossRef CAS PubMed.
- N. Heikkinen, J. Lehtonen and R. L. Puurunen, Phys. Chem. Chem. Phys., 2024, 26, 7580–7591 RSC.
- C. Gonsalves, J. A. Velasco, J. Järvilehto, J. Yim, V. Vuorinen and R. L. Puurunen, Oral presentation at AVS 23nd International Conference on Atomic Layer Deposition, Bellevue, Washington, USA, July 23–26, 2023 Search PubMed.
- J. A. Velasco, C. Gonsalves, G. Ersavas Isitman, J. Yim, D. Izabassarov, E. Verkama, V. Vuorinen and R. L. Puurunen, Poster presentation at AVS 22nd International Conference on Atomic Layer Deposition, Ghent, Belgium, June 26–29, 2022 Search PubMed.
- J. A. Velasco, J. Yim, E. Verkama and R. L. Puurunen, Oral online presentation at ALD Russia, September 27–30, 2021 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp00131a |
| This journal is © the Owner Societies 2024 |