
The interaction of size-selected Ru3 clusters with TiO2: depth-profiling of encapsulated clusters†
Liam
Howard-Fabretto
ab,
Timothy J.
Gorey
c,
Guangjing
Li
c,
D. J.
Osborn
d,
Siriluck
Tesana
 ef,
Gregory F.
Metha
d,
Scott L.
Anderson
c and
Gunther G.
Andersson
*ab
ef,
Gregory F.
Metha
d,
Scott L.
Anderson
c and
Gunther G.
Andersson
*ab
aFlinders Institute for Nanoscale Science and Technology, Flinders University, Physical Sciences Building (2111) GPO Box 2100, Adelaide 5001, South Australia 5042, Australia. E-mail: gunther.andersson@flinders.edu.au
bFlinders Microscopy and Microanalysis, College of Science and Engineering, Flinders University, Adelaide, South Australia 5042, Australia
cChemistry Department, University of Utah, 315 S. 1400 E., Salt Lake City, UT 84112, USA
dDepartment of Chemistry, University of Adelaide, Adelaide, South Australia 5005, Australia
eThe MacDiarmid Institute for Advanced Materials and Nanotechnology, School of Physical and Chemical Sciences, University of Canterbury, Christchurch 8041, New Zealand
fNational Isotope Centre, GNS Science, Lower Hutt 5010, New Zealand
First published on 18th June 2024
Abstract
Ru is a metal of interest in catalysis. Monodisperse Ru3 clusters as catalytic sites are relevant for the development of catalysts because clusters use significantly lower amounts of precious materials for forming active sites due to the small size of the cluster. However, retaining the mono-dispersity of the cluster size after deposition is a challenge because surface energy could drive both agglomeration and encapsulation of the clusters. In the present work Ru3 clusters are deposited by chemical vapor deposition (CVD) of Ru3(CO)12 and cluster source depositions of bare Ru3 onto radio frequency sputter-deposited TiO2 (RF-TiO2) substrates, TiO2(100), and SiO2. When supported on RF-TiO2, bare Ru3 is encapsulated by a layer of titania substrate material during deposition with a cluster source. Ligated Ru3(CO)12 is also encapsulated by a layer of titania when deposited onto sputter-treated RF-TiO2, but only through heat treatment which is required to remove most of the ligands. The titania overlayer thickness was determined to be 1–2 monolayers for Ru3(CO)12 clusters on RF-TiO2, which is thin enough for catalytic or photocatalytic reactions to potentially occur even without clusters being part of the very outermost layer. The implication for catalysis of the encapsulation of Ru3 into the RF-TiO2 is discussed. Temperature-dependent X-ray photoelectron spectroscopy (XPS), angle-resolved XPS, and temperature-dependent low energy ion scattering (TD-LEIS) are used to probe how the cluster–surface interaction changes due to heat treatment and scanning transmission electron microscopy (STEM) was used to image the depth of the surface from side-on.
Introduction
Small metal clusters are defined as groups of bound metal atoms with approximately <300 atoms.1–6 Their electronic and catalytic properties depend on the number of atoms forming the cluster.7 Ru clusters are of particular interest as they are among the most efficient catalysts for reactions such as CO and CO2 hydrogenation8–17 and photocatalytic water splitting.18 Clusters deposited on reducible oxides like TiO2 can be strongly affected by the so-called strong metal–support interaction (SMSI), and in some cases may become covered by an overlayer of support material.19,20 This is known as “encapsulation” or “decoration” of the clusters,21,22 and similar occurrences can also be found for scenarios where TiO2 has an overlayer aside from clusters.23 Depending on the combination of materials for the clusters and substrate, varying conditions have been required to induce cluster encapsulation, including sputtering prior to cluster deposition,19,24 and high temperature reduction of the cluster/oxide system in ultra-high vacuum (UHV).19,22,24–32 These changes are measurable using X-ray photoelectron spectroscopy (XPS), where encapsulation increases the concentration of reduced Ti at the surface casing a low binding energy (BE) shoulder in the Ti 2p peak.19,33For catalytic applications, encapsulation of supported metal catalysts by the substrate material is generally not desirable because the catalytic properties can be affected by either the change in properties, or the steric hindrance of reactant molecules being blocked from the catalyst.19,24,31 However, if the overlayer is thin enough some combinations of clusters and overlayers can yield an electronic structure which is suitable for catalysis and/or photocatalysis without direct reactant-cluster contact.34–36 This combination can also be considered as a form of doping, and in these cases there can be other benefits such as increased resistance to cluster agglomeration,34,35 increased reaction selectivity,36 or improving activity by hindering back reactions.35 As an example, the water splitting photocatalytic activity of Au25/BaLa4Ti4O15 is increased 19-fold due to the encapsulation of ∼1 nm diameter clusters by 0.8–0.9 nm thick Cr2O3 (as determined by HRTEM). This system had a dual benefit for catalysis of decreasing the rate of the O2 photoreduction back-reaction, as well as decreasing the level of UV irradiation-induced cluster agglomeration which occurred.35,37
Two commonly utilised approaches for depositing Ru clusters onto substrates under vacuum are depositing gas-phase size-selected, bare clusters using a cluster source (CS), and depositing ligand-stabilised clusters such as Ru3(CO)12 by chemical vapor deposition (CVD).38–48 CS depositions are performed in situ and are typically suited for small scale fundamental research studies, while being difficult to upscale for industrial applications due to the high vacuum required and small cluster deposition areas.49–53 However, upscaling is possible with CVD. Ligated cluster depositions with CVD often requires post-deposition surface treatments such as heating or chemical methods to remove the ligands and expose bare, surface-supported particles.39,40,54,55 The deposition method can affect the resultant cluster properties, however very few studies have directly compared the cluster properties of identically sized clusters resulting from different deposition methods.40,56
TiO2 is a common choice as a substrate for the deposition of clusters,19,24,26,39,40,53,57–65 which is often used due to its photocatalytic activity66 and ease in handling the material. Sputtering TiO2 substrates prior to cluster depositions is a method used to help prevent the agglomeration of clusters;55,67 the anchoring of clusters to defect sites on TiO2 has been demonstrated by experiments55 as well as DFT calculations.68 Rutile TiO2(110) is the most frequently used form of the TiO2 for surface science experiments, however here we use radio frequency (RF) sputter-deposited TiO2 prepared by sputtering a TiO2 wafer over a spinning substrate under high vacuum, which produces a dense, uniform, stoichiometry-controlled layer of TiO269 which is cheaper and more readily available than TiO2(110).
There are various analytical techniques which can be used to depth profile systems of small metal clusters. Angle resolved XPS (ARXPS) is commonly used for non-destructive depth profiling70,71 but is known to be less reliable for samples which have non-monotonic concentration depth profiles or show significant roughness.71 Low energy ion scattering (LEIS) allows for measuring the atomic composition of the topmost layer,25,59,61,72–92 and has been shown to detect cluster encapsulation.25,72,85 Overlayer thickness can be determined by LEIS as described by Brongersma et al.,93 which has previously been applied to determining the overlayer thickness of functionalised Au nanoparticles.94,95
In our previous study, CO temperature programmed desorption (CO-TPD) was performed on Ru3 clusters deposited by CVD (Ru3(CO)12) and CS (bare Ru3) onto sputter-treated RF-TiO2, which provides a measure of the number of surface-exposed Ru atoms.56 These results suggest that the Ru3 clusters became encapsulated by TiOx, due to blocking of the CO sites that would be expected for Ru clusters. For Ru3(CO)12, the number of available Ru–CO binding sites significantly decreased after the first heating run which was attributed to encapsulation of the clusters upon heating and ligand removal. For CS-Ru3, essentially no Ru–CO binding sites are observed even in the first TPD run, suggesting that the sputter-deposited TiO2 encapsulated the Ru3 upon deposition. XPS measurements of the samples after the CO-TPD experiments indicated that the Ru3 clusters are partially oxidised by the sputtered RF-TiO2 substrate after heating to 800 K. However, this work had no direct evidence for cluster encapsulation.
Here, we deposit size-selected Ru clusters onto RF-TiO2 substrates using CVD and CS depositions. TiO2(110) and SiO2 are also studied for comparison with the RF-TiO2 substrates; TiO2(110) is the most commonly used single crystal form of TiO2,96 and SiO2 is a non-reducible oxide that does not normally support encapsulation.66 Clusters are size-selected during depositions as Ru3 but are no longer strictly size-selected Ru3 after the deposition and sample processing. The first aim is to determine whether the Ru3 deposition method has any effect on the resultant surface properties. The second aim is to directly measure the encapsulation of Ru clusters and determine the overlayer depth and temperature at which this occurs. These are all unknown factors for this system which could be of critical importance for practical catalytic applications.
Experimental
Samples
A list of the substrates used, and their abbreviated names are given in Table 1. Separate samples were prepared for each measurement, to ensure that sample damage is minimised prior to analysis. A full list of all 12 samples which were prepared and analysed is presented in the ESI† (Table S1).| Substrate material | Ar+ sputter dose (ions per cm2) | Designated sample name | Abbreviated name |
|---|---|---|---|
| RF-TiO2 | None | Non-sputtered RF-TiO2 | NS-RF-TiO2 |
| RF-TiO2 | 4 × 1013 | Low-dose sputtered RF-TiO2 | LDS-RF-TiO2 |
| RF-TiO2 | 6 × 1014 | High-dose sputtered RF-TiO2 | HDS-RF-TiO2 |
| Rutile TiO2(110) single crystal | 6 × 1014 | TiO2(110) | TiO2(110) |
| SiO2/Si (100) | None | SiO2 | SiO2 |
RF sputter- deposited TiO2 substrates (RF-TiO2) are prepared by RF magnetron-sputtering with a TiO2 target onto an SiO2 wafer. An HHV/Edwards TF500 Sputter Coater was used with a process that has been described in detail in previous publications.56,97 Based on SEM measurements of similarly prepared substrates, the thickness of the RF-TiO2 was approximately 150 nm.97 RF-TiO2 has a nanoparticulate film structure with 25–45 nm spherical-shaped grains distributed across the substrate which enlarge upon heat treatment.69,97–99 RF-TiO2 was treated by heating to 723 K for 10 minutes, then using 3 different pre-deposition Ar+ sputtering treatments: namely, NS-RF-TiO2 (non-sputtered), LDS-RF-TiO2 (low-dose sputtered, 4 × 1013 Ar+ ions per cm2), and HDS-RF-TiO2 (high dose-sputtered, 6 × 1014 Ar+ ions per cm2). LDS-RF-TiO2 was used only in results presented in the ESI.†
For the SiO2 substrate, p-type, boron-doped Si(100) wafers were purchased from MTI Corporation and treated in situ by heating to 700 K for 20 minutes under 7 × 10−6 mbar O2, then 2 minutes under UHV. These substrates are referred to as SiO2 due to the fact that they have an amorphous SiO2 (silica) surface layer.76 A 99.99% pure rutile TiO2(110) single crystal was purchased from MTI Corporation, and sample treatment followed that reported in recent publications59,61 and is detailed further in the ESI† (page 2). The TiO2(110) was dosed with 6 × 1014 Ar+ ions per cm2in situ prior to cluster depositions.
Instrumentation
Cluster depositions and analysis were performed on 3 separate UHV instruments. In situ XPS and LEIS were performed on the University of Utah UHV apparatus and Flinders University UHV apparatus, respectively. ARXPS measurements were performed at the Australian Synchrotron soft X-ray UHV beamline; CVD was performed in situ whereas a CS-Ru3/HDS-RF-TiO2 sample was prepared at the University of Utah and analysed ex situ. All 3 instruments featured their own in situ 2–3 keV Ar+ sputtering systems.Cluster depositions
Ru3 cluster depositions were performed by CVD and CS depositions. CVD was performed in situ at both Flinders University and the Australian Synchrotron, and the samples are referred to as CVD-Ru3(CO)12. The CVD procedure has been described in previous work56 and briefly in the ESI† section on page 2. Ru3(CO)12 was inserted into a loading chamber and allowed. CS depositions were performed by depositing 1.5 × 1014 Ru atoms per cm2 as mass-selected Ru3+, using an in situ laser vaporisation CS which has been described previously.56,73,74,76 The instrument details and deposition procedures are given in the ESI† (pages 2–3). Cluster spots were 2 mm in diameter, defined by an aperture. The deposition energy was set to ∼1 eV per atom to prevent fragmentation of the clusters.100TD-XPS
For TD-XPS measurements, the temperature of a sample is increased in a stepwise manner while XPS is performed at each discrete temperature. Samples are held at each temperature for 10 minutes, then the heating is turned off and XPS is performed as the sample slowly cools. TD-XPS measurements were also performed for substrates with no clusters deposited, which are referred to as “blank samples”. TD-XPS of CVD-Ru3(CO)12 samples were measured at Flinders University, while CS-Ru3 samples were measured at the University of Utah. Details on each experimental setup are provided in the ESI† (page 4). The heat treatment was applied in vacuum and temperature variation for the experiments is reported in the Results section for each of the experiments conducted.101The binding energy (BE) axis was calibrated to C 1s = 285.0 eV for the aliphatic adventitious hydrocarbons on sample surfaces. The absolute uncertainty in reported BEs is ±0.2 eV, however, for comparing BE differences in the same sample before and after treatment the uncertainty is reduced to ±0.1 eV. Based on the XPS peak fitting, atomic concentrations in percentage (At%) were determined and the surface coverage of cluster material was estimated for each cluster deposition. Surface coverages are given in terms of percent of a close-packed monolayer (% ML), where one monolayer (ML) is 1.6 × 1015 Ru atoms per cm2. Further details on the peak fitting and calculations are given in the ESI† (pages 5–7). The At% for TD-XPS results presented are averages over all measured temperatures.
ARXPS
The synchrotron X-ray measurement and ARXPS data analysis procedure are provided in the ESI† (pages 7–9).LEIS
In LEIS, a sample is bombarded with low energy ions which are backscattered and detected.102,103 Backscattered He+ counts were plotted against the ratio E/E0, where E is the backscattered energy and E0 is the incident ion energy. Further details on the LEIS technique and its surface sensitivity, as well as the instrumentation and confirmation of the reproducibility of results are given in the ESI† (pages 9–10). The two LEIS measurement procedures used were “series LEIS” (only reported in the ESI†) and “temperature-dependent LEIS” (TD-LEIS). Series LEIS measurements were performed repeatedly on the same sample area to determine the effects of the He+ beam on the sample. TD-LEIS measurements were performed as the sample temperature was increased stepwise to determine the effects of heating. For each increase in temperature the He+ beam was stopped, and the sample was heated at 3 K s−1 to the next temperature. Heating was stopped as soon as the target temperature was reached, and another LEIS measurement was performed until 900 K. For each TD-LEIS sample, XPS was performed after the TD-LEIS measurements and used to determine Ru surface coverages.STEM
A cross-section of a CVD-Ru3(CO)12/HDS-RF-TiO2 sample was analysed using high resolution scanning transmission electron microscopy (STEM). The experimental details are described in the ESI† section on page 11.Results and discussion
In which way the Ru3 clusters deposited via CVD and CS could contribute to the catalytic activity of the substrate depends strongly on their position relative to the outermost layer after completing the deposition process. We have applied methods with differences in chemical sensitivity and depth resolution to determine the state and depth within the sample of the deposited clusters.TD-XPS
XPS has been applied to determine the chemical state of the clusters after the deposition and ligand removal process and at which depth they are located. A TD version must be applied because the CO ligand removal requires heating.TD-XPS measurements were performed on the 5 substrates listed in Table 1. The resulting spectra and analysis are shown in the ESI† (Fig. S1–S6). Temperature-dependent results are presented for Ru At%, Ru 3d BE, CO/Ru atomic ratio (for ligated clusters), and the ratio of Ti defects (Ti2+ and Ti3+) in the substrate.
It is determined from the Ru 3d BE for CS-Ru3/NS-RF-TiO2 that Ru clusters are partially oxidised due to heating (see Fig. S7 in the ESI† page 17), as indicated by a change in BE which begins at which begins at 500 K and reaches +0.2 ± 0.1 eV by 800 K, agreeing with our previous study.56 The Ru 3d BE is higher in the presence of ligands for both Ru3(CO)12/HDS-RF-TiO2 and Ru3(CO)12/TiO2(110), however after heat treatment to 723–823 K the Ru 3d BE converged to 280.5–280.6 eV for all TiO2-supported samples, indicating that the clusters have similar oxidation states after heating. This suggests that for catalytic purposes the specific deposition process will not likely result in significant differences if the clusters have been heat treated. For the ligated clusters, the initial CO/Ru ratio (Fig. S8, ESI†) is 2.1 ± ∼0.5 for CVD-Ru3(CO)12/HDS-RF-TiO2, and 1.5 ± ∼0.5 for CVD-Ru3(CO)12/TiO2(110). This implies approximate as-deposited cluster chemical formulae of Ru3(CO)6 and Ru3(CO)4.5 respectively, meaning some ligands are lost in the CVD procedures.
The density of Ti surface defects (Fig. S9, low BE shoulder on Ti 2p peak, ESI†) is shown to generally increase with heating for the Ru3(CO)12/TiO2 samples, which can partially be contributed to changes in the substrate. Heating-induced defects for TiO2 under UHV have been reported in previous studies,22,66,104 but in this case the surface defects in cluster-loaded samples increased to a greater level than the blank samples for both HDS-RF-TiO2 and TiO2(110) substrates, suggesting the clusters themselves are involved. Conversely, CS-Ru3/NS-RF-TiO2 did not have an increase in Ti surface defects within the sensitivity of the instrument.
ARXPS
ARXPS measurements were performed on 2 samples; CVD-Ru3(CO)12/HDS-RF-TiO2 (deposited in situ) and CS-Ru3/HDS-RF-TiO2 (deposited ex situ).The ARXPS results for CVD-Ru3(CO)12/HDS-RF-TiO2 and CS-Ru3/HDS-RF-TiO2 are shown in Fig. 1. The fitted Ru 3d and C 1s region spectra are shown in the ESI† (Fig. S10). The lower surface concentrations in Fig. 1b are due to the lower surface coverage used in the CS-Ru3 sample. The Ru surface coverage for both samples is <5% ML, and thus cluster–cluster interactions are considered negligible, and the samples are directly comparable to one another in terms of Ru properties. The estimated Ru surface coverage of the samples is shown in Table 2.
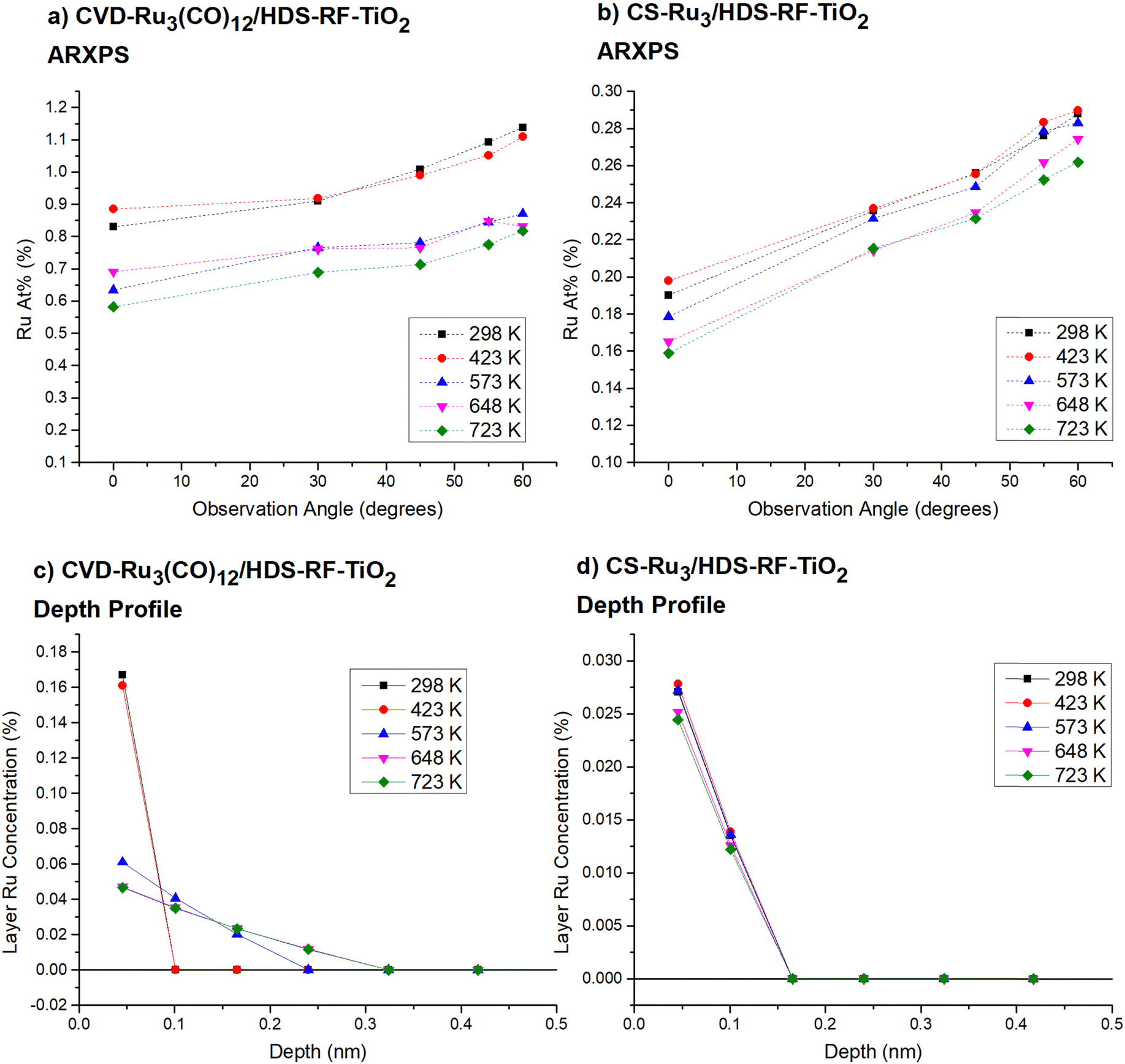 | ||
| Fig. 1 ARXPS data for CVD-Ru3(CO)12/HDS-RF-TiO2 and CS-Ru3/HDS-RF-TiO2. (a) and (b) Show the data for Ru At% vs. observation angle for CVD-Ru3(CO)12 and CS-Ru3, respectively. (c) and (d) Show the ARXPS depth profiles for CVD-Ru3(CO)12 and CS-Ru3, respectively. These show the atomic concentration of Ru per layer for arbitrarily defined layers of the sample, as determined by the ARXPS model. The uncertainty in Ru At% is ±2%, while the uncertainty in the layer concentration is ±20%. | ||
| Deposition | Substrate | Ru At% (%) | Ru surface coverage (% ML) |
|---|---|---|---|
| CVD-Ru3(CO)12 | HDS-RF-TiO2 | 0.77 | 4.0 |
| CS-Ru3 | HDS-RF-TiO2 | 0.18 | 1.0 |
The Ru At% increases with increasing observation angle due to the limited electron mean free path of ejected photoelectrons in the surface, and those Ru cluster that are at or near the surface, have a greater relative signal detected by XPS. We use these results to determine Ru depth profiles for the samples at each temperature (see ESI† pages 8–9 for more details on the data analysis).
Fig. 1c shows that the depth profile of CVD-Ru3(CO)12 clusters on the surface change between 423 K and 573 K. Between 298 K and 423 K all Ru is present on the top surface layer, defined in the calculation as the top 0.6 nm. At 573 K the depth profile changes, suggesting that a substrate overlayer has formed above the clusters. Note that this is equivalent to describing the process as the clusters burrowing deeper into the substrate. The penetration extended deeper again at 648 K to a maximum penetration depth of 0.24 nm ± 0.03 nm, which is close to 1 ML based on the layer estimated thickness of 0.198 nm (based on apical Ti–O bond length of the rutile TiO2 crystal).105 Based on the ARXPS analysis, the depth profile shows that at 723 K there is approximately 60% Ru below the top-most layer, and 40% present on the surface, based on the relative layer concentrations of Ru. The formation of a TiO2 overlayer will also be discussed in the context of the ISS results (vide infra).
Fig. 1d shows the depth profiles for CS-Ru3/HDS-RF-TiO2. The main change observed is a small reduction in the total amount of Ru visible in XPS as the temperature increased. Unlike CVD-Ru3(CO)12, Ru below the top-most layer is present even at 298 K. The ratio of surface to sub-surface Ru is consistent for all temperatures; there is ∼66% on the surface and ∼34% below the top-most layer. The maximum penetration depth is approximately 0.1 nm ± 0.03 nm, which corresponds to approximately 0.5 ML for an overlayer of TiO2. The calculated value being <1 ML may suggest that not all the clusters are covered, or that the clusters are only partially covered by the substrate. The nature of the overlayer for both samples is discussed in further detail in the Encapsulation section below.
Through XPS and ARXPS the chemical state of the Ru3 clusters after their deposition has been revealed and that a very thin overlayer forms over the Ru3 clusters through the deposition process. However, a method with a better depth resolution and surface sensitivity than XPS is required to understand where exactly the clusters are located after the deposition process.
TD-LEIS
LEIS is better suited to investigate where the Ru3 clusters are located due to its excellent sensitivity for the outermost layer. TD-LEIS measurements were performed on 3% ML CS-Ru3/SiO2 (Fig. 2) and 11% ML CVD-Ru3(CO)12/HDS-RF-TiO2 (Fig. 3). For the latter, two identical samples were prepared and analysed (referred to as TD-LEIS A and TD-LEIS B). XPS results are used to estimate the surface coverages of Ru on the samples, as well as to confirm that the level of Ru removed from the surfaces due to the He+ beam in TD-LEIS is sufficiently low to not influence the results (further details provided in ESI,† pages 25–27). These XPS results and the total ion doses used during TD-LEIS measurements are shown in Table S4 (ESI†). Measurements were also performed on CS-Ru3 on RF-TiO2 with 3 different pre-deposition sputtering doses, but the Ru peaks are obscured in the LEIS results. This may indicate the clusters were encapsulated upon deposition; however, this cannot be confirmed because these samples featured a higher level of adventitious carbon which can affect the surface-sensitive LEIS measurements (results shown and discussed in ESI,† pages 28–31, Fig. S15 and S16). Additional details about the encapsulation process is revealed by the ISS data (vide infra).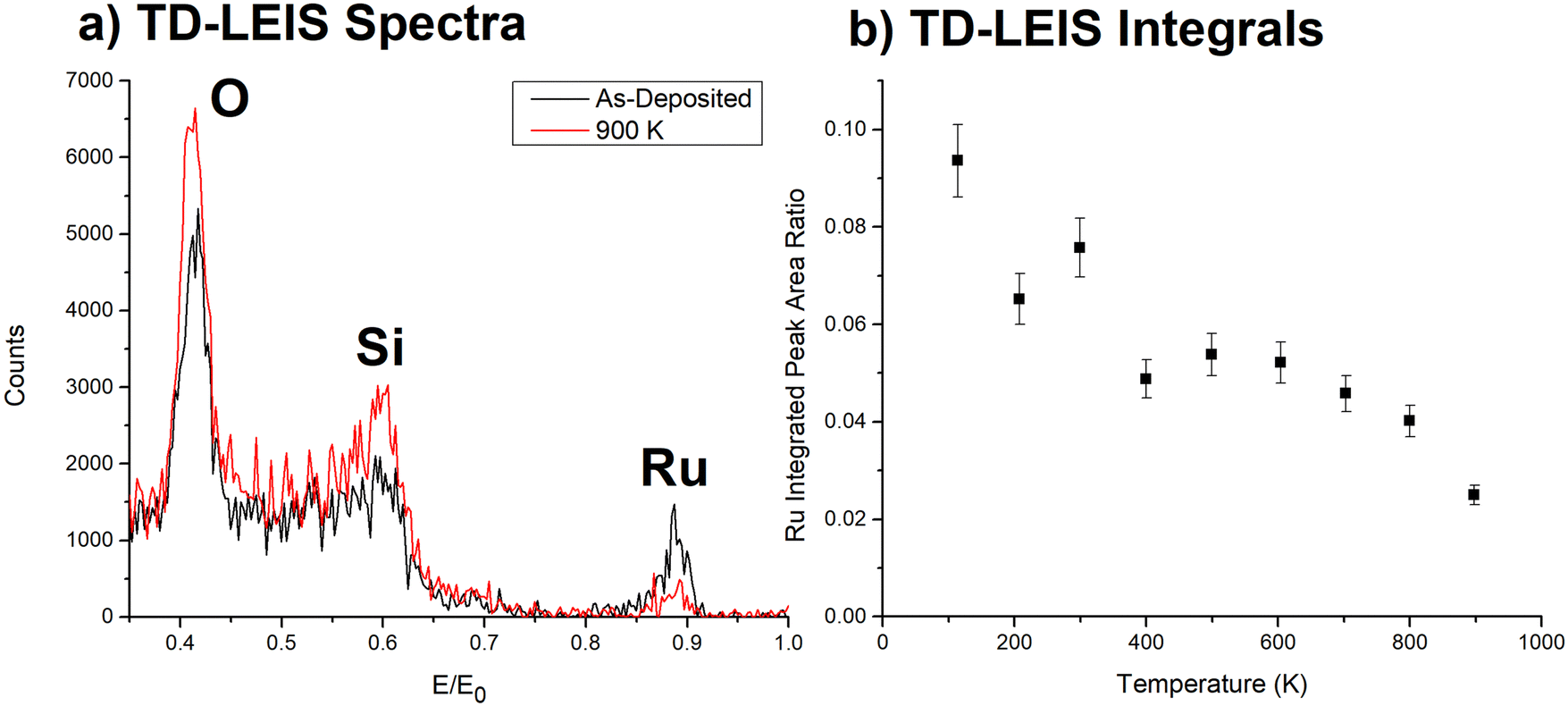 | ||
| Fig. 2 TD-LEIS of CS-Ru3/SiO2. (a) LEIS spectra for initial and final temperature measurements. (b) Integrated Ru/(Si + O) peak ratio vs. temperature. Uncertainties in the Ru peak area ratios are ∼8%. The sputter effects of the He+ on the sample are discussed in the ESI† (see Fig. S14). | ||
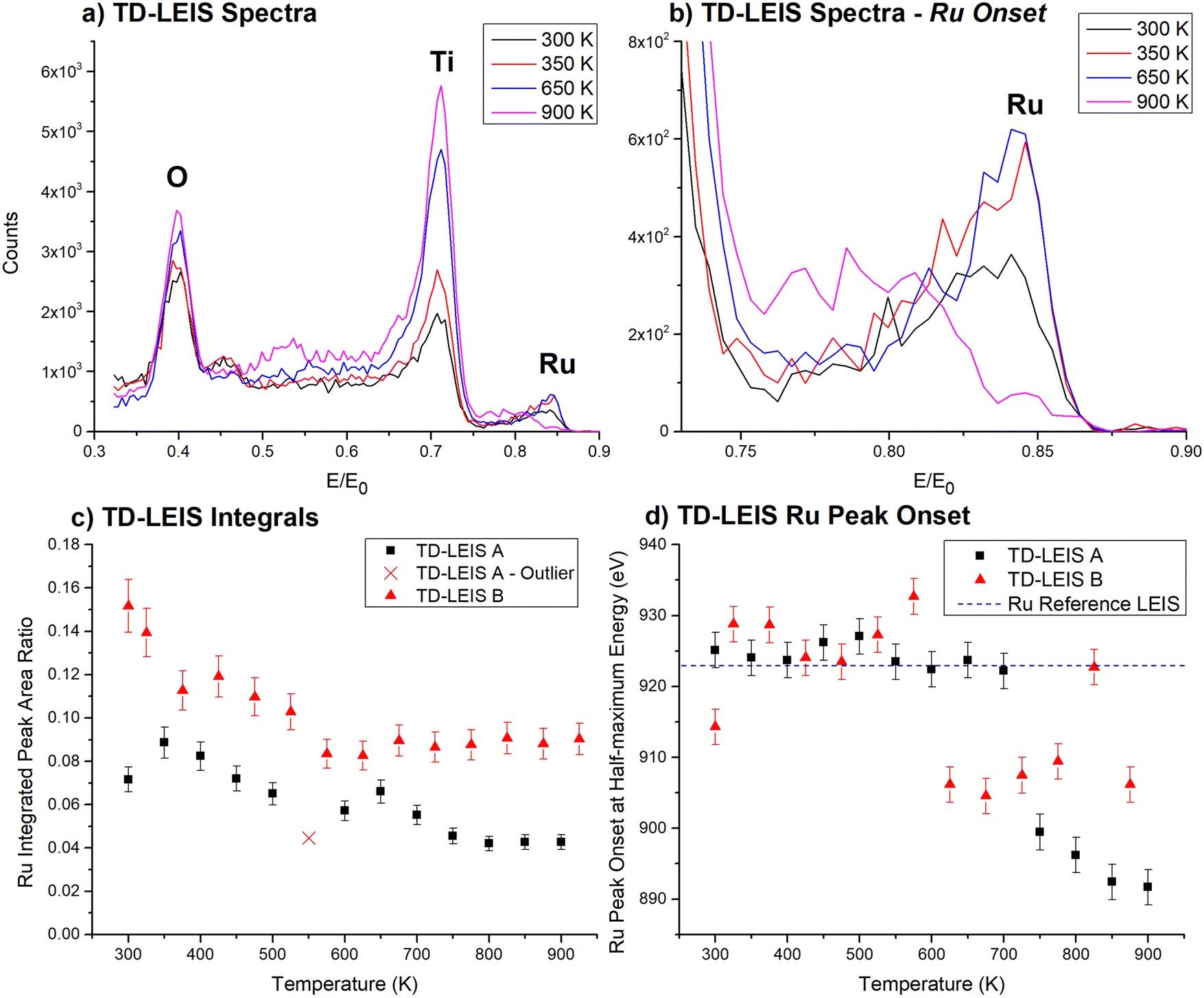 | ||
| Fig. 3 TD-LEIS of CVD-Ru3(CO)12/HDS-RF-TiO2. Separate depositions and TD-LEIS measurements were performed on 2 samples. (a) and (b) Show spectra from TD-LEIS A, while (c) and (d) show collated data from both TD-LEIS measurements. (a) LEIS spectra at selected temperatures. (b) LEIS spectra at selected temperatures, zoomed in to the Ru peak region. (c) Integrated Ru/(Ti + O) peak ratios vs. temperature. Data point × is included for completion but is treated as an outlier. Uncertainties in Ru integrated peak ratios are ∼8%. (d) Half-maximum peak onset for Ru vs. temperature, in terms of backscattered He+ energy. The black dashed line at 923 eV represents the Ru half-maximum onset for a metallic Ru reference sample. The error bars are ±2.5 eV based on the bin width. | ||
In Fig. 2a the intensities of the LEIS peaks from the SiO2 support increase after heating to 900 K, indicating that the fraction of clean substrate increased, attributed to some combination of desorption of adventitious adsorbates and reduction of the fraction of the surface area blocked or shadowed by Ru clusters. Conversely, the Ru peak intensity decreased monotonically with increasing temperature, such that the Ru/(Si + O) intensity ratio (Fig. 2b) dropped by a factor of ∼3 from 300 to 900 K. Such a decrease indicates that the clusters sintered into multi-layer structures where some of the Ru atoms are no longer in the LEIS-accessible surface layer. This is supported by CO-TPD results in our previous work56 where the change in the CS-Ru3/SiO2 CO-TPD spectra with repeated heating cycles provided evidence the clusters are agglomerated when heated to 800 K. It is unlikely that the change is due to an overlayer forming as this is not typically observed for SiO2, and the decrease began at a low temperature when SiO2 should be stable. This conclusion is also consistent with previous studies on other cluster materials, which found that Ptn clusters75,79 and Irn clusters106 deposited on SiO2 undergo sintering when heated over this temperature range, giving rise to similar changes in LEIS signals. The Ru/(Si + O) also decreases at temperatures <300 K. It is unclear why this is occurring. One possibility is that it is due to preferential adsorption from the rest gas in the UHV chamber at temperatures below 300 K. It should also be noted that the effect is not further relevant for the encapsulation study.
Fig. 3a and b show the LEIS spectra at all temperatures measured for CVD-Ru3(CO)12/HDS-RF-TiO2. The signal strength of Ti and O increased with increasing temperature, which is indicative of adventitious adsorbates being desorbed and sputtered away by the He+ beam, most likely being adventitious hydrocarbons. The intensity of signal at the onset of the Ru peak (on the right) decreased at 650–700 K, and decreased further as the temperature is increased to 850–900 K. Starting at 650–700 K, the counts at ∼0.79E/E0 increased while the main peak at ∼0.85 decreased in size, shifting the peak location of Ru to lower E/E0 values showing that the Ru clusters are fully covered with TiO2. In LEIS, peaks shifting to lower E/E0 values in cases such as this can be indicative of the clusters being covered by an overlayer which the He+ needs to penetrate through before and after backscattering. In such cases the He+ projectiles lose energy due to penetrating through the overlayer, which shifts the peak to lower values of E/E0.95,103 Backscattered projectiles have a probability of re-ionisation when leaving the surface which results in a measurable signal of backscattered He+.95,103 It is important to note that the LEIS intensity is lower for backscattering from deeper layers even if the same amount of the specific element – in this case Ru – is present because the probability for reionisation when backscattered from a deeper layer is lower compared to the probability to be backscattered as He+ from the outermost layer.103
The TD-LEIS results are supported further by evidence from our previous study,56 where it was proposed based on CO-TPD results that CVD-Ru3(CO)12/HDS-RF-TiO2 may be encapsulated by the HDS-RF-TiO2 substrate when heated based on the loss of all Ru–CO binding sites after heating the sample to 723 K. In contrast, for CS-Ru3/SiO2, where encapsulation does not occur, the CO binding sites were retained as the sample was heated. These results are consistent with the evidence for formation of a titania overlayer in the TD-LEIS and ARXPS results presented here, resulting in a strong argument that the clusters are encapsulated by the RF-sputtered TiO2 substrate. This is discussed in detail in the Encapsulation section below.
In Fig. 3c the integrated Ru peak ratios are shown vs. temperature, where the integration included both the high and low energy regions of the Ru peak (for surface and sub-surface species). The relative Ru LEIS peak size decreased with increasing temperature. The decrease in intensity is most likely due to the encapsulation of the clusters with the consequence of decrease in reionisation probability of the He+ projectiles when backscattered from a deeper layer as described above. This aligns with the ARXPS results (Fig. 1), the evidence from Fig. 3c where the LEIS peak shifted to lower values,95,103 and our previous CO-TPD results.56 LEIS measurements A and B show similar trends of decreasing integrated Ru intensity and shift in onset energies although TD-LEIS B has a higher Ru/(Ti + O) ratio at all temperatures. The intensity differences are due to TD-LEIS B having a higher background count rate, which is not subtracted for the peak integrations. The data point marked with a “×” had a lower ratio than expected because the sample was left in the vacuum chamber after heating due to an equipment issue, which allowed some adventitious hydrocarbons to adsorb atop the clusters.
Fig. 3d shows the onset half-maximum energy determined from the high E/E0 side of the Ru peaks in each TD-LEIS spectrum. This is the energy where the Ru peak reached half its maximum height. For TD-LEIS A, the energy for the onset of the half-maximum is approximately the same as the metallic Ru value (indicated by the black dotted line) until heating to 750 K, where the energy for the onset of the half-maximum shifted to a lower energy, and after 750 K it decreased slightly further. For TD-LEIS B, the shift to a lower Ru energy for the onset of the half-maximum occurred at 575 K and is otherwise the same as TD-LEIS A within the experimental uncertainty. The large shift in the energy for the onset of the half-maximum observed for both measurements corresponds to the complete loss of the surface Ru peak and is treated as an indicator of the clusters being covered by a substrate overlayer.
The shift in the energy for the onset of the Ru contribution in LEIS allows for determining the thickness of the titania overlayer covering the clusters for the CVD-Ru3(CO)12/HDS-RF-TiO2 sample after heating. A similar method has been used in studies on Au nanoparticles by Belsey et al.95 and Hoffman et al.107 For each TD-LEIS measurement, the onset half-maximum energies are averaged before and after the temperature where the onset shifted, and the energy shift between these is calculated to determine the loss of He+ energy due to the stopping power of the overlayer, ΔE (this did not include the backscattering energy loss). ΔE is determined to be 29.9 eV ± 5.3 eV and 19.7 eV ± 5.2 eV for TD-LEIS A and B respectively, with an average value of 24.8 eV ± 5.3 eV. From this it is estimated (based on apical Ti–O bond length of the rutile TiO2 crystal)105 that the average thickness of the overlayer is 0.35 nm ± 0.08 nm, which is approximately 1.7 ML of titania (calculation details provided in ESI,† pages 27–28).
The TD-LEIS and ARXPS results for overlayer thickness match within the experimental uncertainty. In this case the TD-LEIS results are considered to be more reliable than the AXPS results for overlayer depth because LEIS is known for its extreme surface sensitivity,73 while ARXPS is considered to be unreliable for non-monotonic concentration depth profiles.71 The results are comparable to previously reported results in a study by Fu et al.,33 where 3 samples of Pd nanoparticles on TiO2(110) were analysed using the shift in the Rutherford backscattering high energy edges of Pd and found to have TiOx overlayers with thicknesses of 0.13 nm, 0.14 nm, and 0.27 nm.
STEM
The surface analysis performed so far does not provide information how deep overall the Ru clusters penetrate the sample. This can be achieved through high resolution electron microscopy of cross sections of the samples. High resolution STEM measurements were performed on a CVD-Ru3(CO)12/HDS-RF-TiO2 sample to image the overlayer, determine whether the clusters are still present close to the surface after 723 K heating, and to determine an approximate size distribution for the clusters after the heating and encapsulation. A thin cross-section was generated using FIB to allow measurement of the sample side-on, allowing the imaging of below-surface clusters (see ESI,† Fig. S17 for diagram).Fig. S18 in the ESI† shows that the RF-TiO2 substrate features pores in the surface which contain Ru clusters. Because the CVD process is line-of-sight, this indicates that the clusters are mobile on the substrate surface to some extent and moved into the pores. The TD-XPS results for Ru surface concentration did not change significantly due to heating and ligand removal, so it is most likely that the ligated clusters are mobile before ligand removal. Herein we focus on an image of the clusters at the surface layer because this corresponds to the measured XPS and LEIS results which are surface-sensitive.
Fig. 4a shows a representative image of the sample surface where an overlayer is seen for encapsulated Ru clusters near the surface (clusters have burrowed into the substrate). Fig. 4b shows the particle size distribution of the clusters. An additional STEM surface image and energy dispersive X-ray (EDX) results are shown in the ESI† (Fig. S19–S21) which demonstrate that Fig. 4a is representative of the whole sample, and that the clusters are indeed composed of Ru. Most of the encapsulated clusters are 0.9 nm to 1.1 nm in diameter, but the mean diameter of the clusters is 1.2 nm and some had agglomerated to sizes up to 1.6 nm. This indicates that the Ru particles are likely still in a cluster form rather than nanoparticulate form. However, the theoretical diameter of supported Ru3 is estimated to be 0.265 nm based on the interatomic Ru–Ru distance (bond length),108 which indicates the Ru3 clusters have agglomerated to some extent during the heat treatment. The titania overlayer depth cannot be directly measured using STEM as the sample surface is not consistently at the same height throughout the depth of the slice, so it is difficult to determine the location of the TiO2 surface boundary for each cluster. However, there do appear to be clusters present deeper than the 0.35 nm ± 0.08 nm depth reported based on TD-LEIS, so this value should be treated as the minimum overlayer depth.
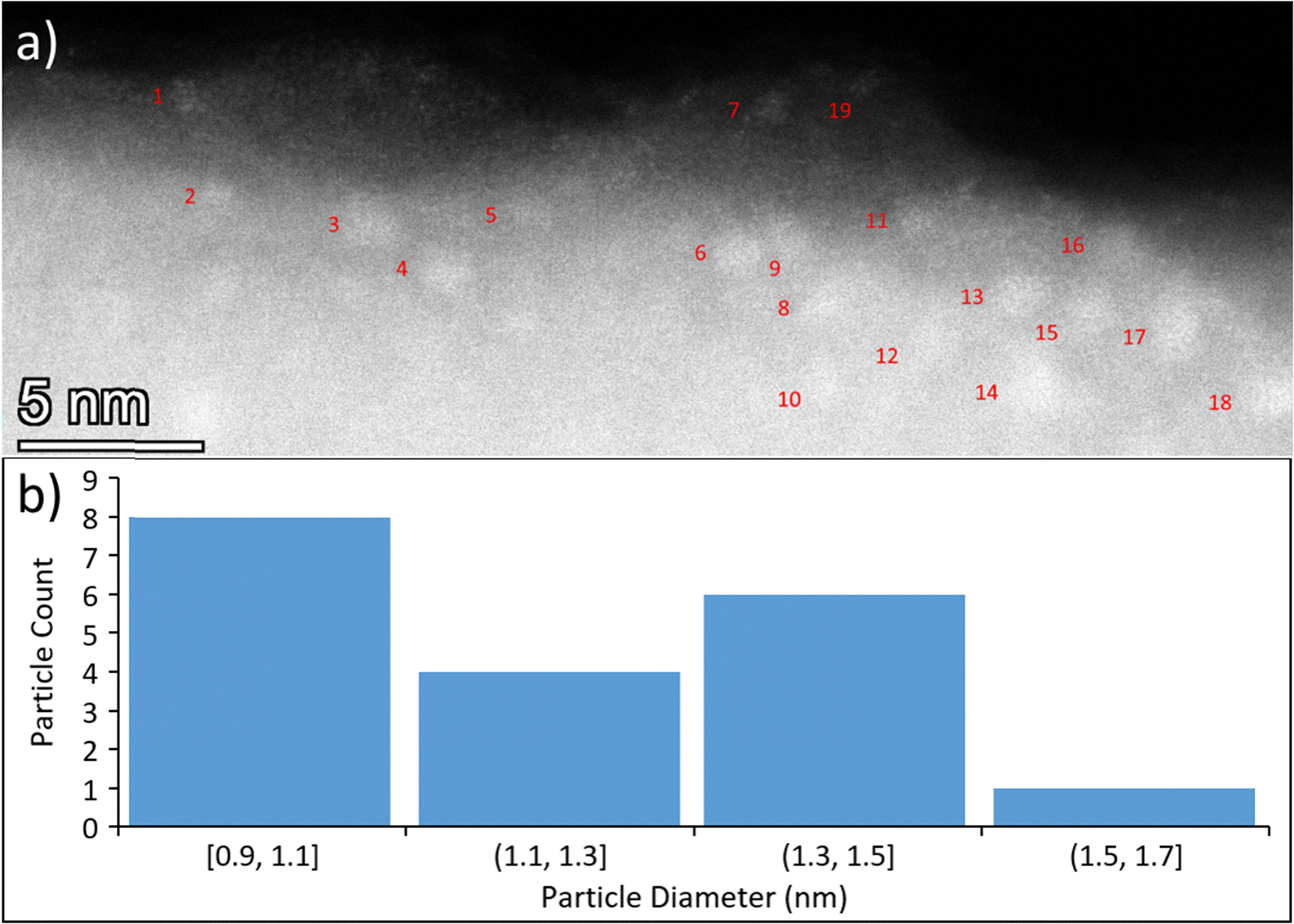 | ||
| Fig. 4 STEM of CVD-Ru3(CO)12/HDS-RF-TiO2, showing side-on perspective of sample. (a) STEM image with numbered, encapsulated Ru clusters (cluster sizes shown individually in Table S5, ESI†). (b) Distribution of particle diameters for encapsulated Ru clusters. | ||
Encapsulation
Having established that the Ru clusters are encapsulated into the TiO2 raises the question what the driving force of the encapsulation is. It is often proposed that for encapsulation to occur there must be a thermodynamic advantage which is given by the tendency of a system to minimize the total surface energy of the system. To drive encapsulation, the surface energy of the metal adsorbate must be greater than the surface energy of the supporting layer.19,22,28,29,31,33 The combination of Ru clusters and TiO2 substrate fits this condition as the surface energy of Ru is 3.409 J m−2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 109 and TiO2(110) is 1.78 J m−2,110,111 meaning it is reasonable to expect encapsulation may occur, especially at higher temperatures.
109 and TiO2(110) is 1.78 J m−2,110,111 meaning it is reasonable to expect encapsulation may occur, especially at higher temperatures.
The ARXPS, TD-LEIS, and STEM results support the postulation that Ru3(CO)12/HDS-RF-TiO2 is encapsulated by an overlayer following heating. The process is schematically shown in Fig. 5. The TD-XPS results for the encapsulated CVD-Ru3(CO)12/HDS-RF-TiO2 showed an increase in the Ti defect ratio beyond the blank sample (Fig. S9, ESI†). In the literature, increases in Ti defects for surfaces of substrates loaded with clusters have previously been associated with clusters being encapsulated by reduced titania for other TiO2-supported systems, including Pd/TiO2,19,31 Pt/TiO2,22,28 and Rh/TiO2.24,29,30 This aligns with the other evidence and suggests that the overlayer is most likely composed of reduced titania, i.e. TiOx, where x < 2.
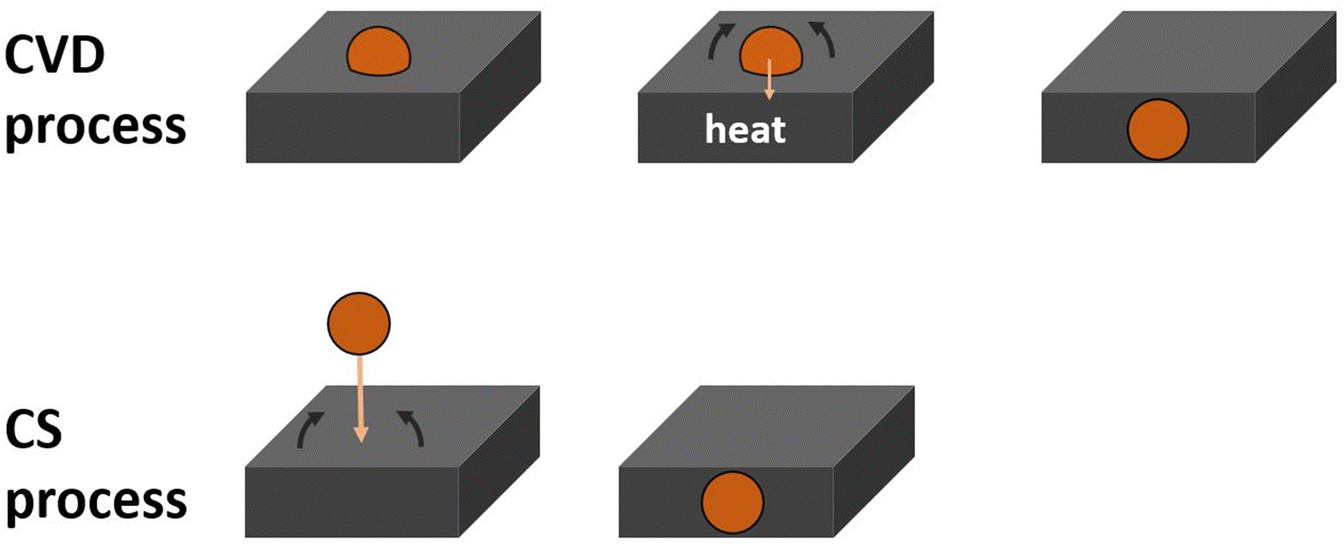 | ||
| Fig. 5 Schematic of the encapsulation of the Ru clusters for the CVD and CS process. The clusters are shown schematically and do not include the ligands of the Ru3(CO)12 clusters and also do not show a schematic for the growth to larger particles. | ||
Regarding CS-deposited clusters, in our previous experiment56 the as-deposited CS-Ru3/HDS-RF-TiO2 did not have any Ru–CO binding sites present on the surface layer before or after heat treatment. This aligns with the ARXPS results in Fig. 1d, showing there is sub-surface Ru present as-deposited (which did not change due to heat treatment). These results suggest that the clusters in this sample are encapsulated as part of the deposition procedure and thus are not affected by heat treatment as shown schematically in Fig. 5. In contrast, heat treatment is required for encapsulation in CVD-Ru3(CO)12-HDS-RF-TiO2. Conversely, there is no evidence for cluster encapsulation occurring for the CS-Ru3/SiO2 sample before or after heat treatment, but there is evidence that the clusters sinter upon heat treatment (Fig. 2). A summary of the encapsulation results is given in Table 3. It is worth noting that there was no evidence found for encapsulation of Pd, Ir or Ni clusters soft landed onto TiO2(110),110,112,113 suggesting that the encapsulation observed for Ru3 soft landed on RF-sputtered TiO2 is a function of the highly defective nature of the sputtered surface. The encapsulation process also might have a potential barrier which could be low enough for some substrates for the encapsulation to occur at room temperature while for other substrates elevated temperatures are required. The defect rich sputter deposited TiO2 is then an example of the former and a single crystal substrate is an example of the latter.
| Sample | As-deposited | After heat treatment |
|---|---|---|
| CVD-Ru3(CO)12/HDS-RF-TiO2 | Clusters on top-most layer | Encapsulated clusters |
| CS-Ru3/HDS-RF-TiO2 | Encapsulated clusters | Encapsulated clusters |
| CS-Ru3/SiO2 | Clusters on top-most layer | Agglomerated clusters, with no encapsulation |
There is an apparent inconsistency between the ARXPS results and both the TD-LEIS results and CO-TPD results from our previous study.56 The ARXPS results (Fig. 1c and d) suggest that some Ru is still present on the topmost layer for both the CVD-Ru3/HDS-RF-TiO2 and CS-Ru3/HDS-RF-TiO2 samples, even after heating. However, the CO-TPD results, as well as the TD-LEIS and STEM results for CVD-Ru3(CO)12–HDS-RF-TiO2, both suggest there is no Ru on the surface layer. This discrepancy is simply attributed to the limitation of the depth analysis by ARXPS. Analysis of the differences in the At% determined experimentally and in the ARXPS model supports this (see ESI,† Fig. S12), indicating that there may have been effects related to the roughness or non-monotonic nature of the sputtered RF-TiO2 substrates on the ARXPS results. Based on this, the ARXPS depth profiles are treated as less quantitative regarding the composition of the outermost layer than LEIS.
The encapsulation of unheated CS-Ru3/HDS-RF-TiO2 is not expected, because conditions typically reported to induce encapsulation involve high temperature heating of the oxide substrate under UHV19,22,24–32,110 or exposure to H2.20,21,114–116 For cluster-specific examples, Ovari et al.72 used LEIS to show that Rh clusters are present on a TiO2(110) surface layer until 700 K, however after heating to 900 K the clusters are encapsulated. Similarly, in another example for Pd clusters on TiO2(110) the encapsulation started at ∼553 K, where the authors suggested that the activation of defect mobility due to heating is required for encapsulation to occur, due to movement in the surface layer promoting encapsulation compared to a static layer.19 Kaden et al. also found encapsulation of Pd clusters at elevated temperatures112 and Ir clusters upon deposition at higher deposition energies.117 Because no heating is required for the encapsulation of CS-Ru3 by sputtered RF-TiO2, this implies a different encapsulation mechanism which is not based on defect mobility typically only occurring at elevated temperatures.19,66,118 It is possible that the increased reactivity of the titania surface due to sputter-induced defects promoted the encapsulation reaction with the Ru. However, the exact mechanism for encapsulation cannot be determined from the TD-LEIS and ARXPS results.
To our knowledge, this type of titania encapsulation of small Ru clusters has not been previously reported in the literature, although some encapsulation studies using larger Ru materials have been conducted.119,120 In fact, there is a previous measurement by Zhao et al. of Ru3(CO)12 supported on TiO2(110) where the surface is heat treated under UHV and encapsulation did not occur, as evidenced by the availability of Ru–CO binding sites.39 The differences between this study and other literature can be attributed to the differences between the titania substrates; TiO2(110) was used in the referenced cluster encapsulation studies,19,72 which does not have the same surface properties as the HDS-RF-TiO2 used in this study. Variance in encapsulation behaviour with different cluster/substrate system is well known in the literature.19,26,28,121,122
For CVD-Ru3(CO)12/HDS-RF-TiO2, both the de-ligation of CO and encapsulation of the clusters occurred when the samples are heated. This naturally raises a question about how the ligand removal is related to cluster encapsulation. In the TD-XPS results, CO/Ru atomic ratios indicated most CO ligands are removed by heating to 423 K (Fig. S8, ESI†), but there may have still been one or a few ligands attached until higher temperatures. In the synchrotron ARXPS depth profile (Fig. 1c), the clusters are all on the surface layer at 423 K but encapsulation began upon heating to 523 K and reached the full depth at 648 K. TD-LEIS similarly showed encapsulation is completed (i.e. no Ru in the surface layer) at 660 K ± 120 K. Because ARXPS showed encapsulation did not start until 523 K, it is concluded that most of the ligands need to be removed for encapsulation, but complete ligand removal is not required. Therefore, there is no existing temperature range where completely bare Ru clusters can exist on the surface layer without encapsulation. This outcome must be kept in mind when considering catalytic applications, because it is often desired to remove the ligands and expose bare clusters for catalysis.39,40,54,55
Implication for catalysis
Given the catalytic potential for supported Ru clusters,8–17 these encapsulated Ru clusters are a strong candidate for future studies on encapsulated catalysts for reactions such as catalytic CO hydrogenation and photocatalytic water splitting. The effect of the closed titania overlayer on the catalytic and photocatalytic activities of the Ru clusters would need to be tested explicitly. The encapsulation of the Ru3 clusters can have benefits or disadvantages for catalytic processes which have to be weighed against each other. A potential disadvantage of the encapsulation is that the reactants cannot get into contact with the Ru clusters if the overlayer is too thick. However, if the overlayer is thin enough the presence of the Ru clusters can still influence the local electronic and geometric structure including that of the overlayer on top of the clusters and result in a catalytic effect.A potential further benefit of the titania overlayer could be increasing the stability of the clusters without suppressing the catalytic reaction, which has been shown in a study by Negishi et al.35 where improved activity was found for Au clusters with overlayers ∼3 times thicker than those in this study. Furthermore, in a future study the activity of supported Ru clusters can be compared between ligated clusters (below ∼423 K) and the de-ligated, encapsulated clusters (complete encapsulation occurs at ∼648–660 K). In both cases the clusters are covered with other materials; ligands in the first and a covering layer in the second. Testing the catalytic activity of covered clusters is worthwhile because catalytic effects have been shown in some cases for ligated clusters, even if ligands are still attached.123
Conclusions
In this study Ru3 clusters were deposited by CVD and CS onto sputter-treated TiO2 substrates, as well as TiO2(110) and SiO2. The cluster deposition method was found to influence the cluster surface properties before heat treatment; TD-XPS showed that the oxidation state of Ru on TiO2 varied for as-deposited clusters depending on the deposition method and type of TiO2 substrate (RF-TiO2 or TiO2(110)). After heat treating to 723–873 K, the oxidation states for Ru become identical, within experimental accuracy, for all the analysed TiO2 systems including RF-TiO2 and TiO2(110). The results suggest that for catalytic purposes, the specific deposition process likely will not result in significant differences if the clusters are to be heated.Depth profiling was performed using ARXPS, TD-LEIS, and STEM, and the results provided evidence for the encapsulation of CVD-deposited Ru3 by an overlayer of sputter-treated TiO2 substrate material, showing that no Ru was left on the topmost layer after heat treatment to 660 K ± 120 K. The minimum overlayer thickness was measured by TD-LEIS to be 0.35 nm ± 0.08 nm for CVD-Ru3(CO)12/HDS-RF-TiO2, equivalent to an average of 1.74 ML ± 0.41 ML of titania. ARXPS showed encapsulation starting between 423–573 K, reaching a maximum by 648 K. Combined with the TD-XPS results, this suggests that most of the CO ligands need to be removed for encapsulation to occur, but complete de-ligation is not required. ARXPS provided evidence that bare CS-Ru3 on HDS-RF-TiO2 was encapsulated as-deposited. Cluster encapsulation may provide an advantage for catalysis or photocatalysis applications by modifying the cluster properties, based on recent studies.35
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work is supported by the US Army project FA5209-16-R-0017. Part of the work is supported by the Australian Solar Thermal Research Institute (ASTRI), a project supported by the Australian Government, through the Australian Renewable Energy Agency (ARENA). The work at the University of Utah was partly supported by the Air Force Office of Scientific Research, under AFOSR grants FA9550-19-1-0261 and FA9550-22-1-0381. The authors acknowledge the facilities, and the scientific and technical assistance, of the Microscopy Australia (formerly known as AMMRF) nodes at Flinders University and the University of Adelaide, and the Australian National Fabrication Facility (ANFF) at Flinders University. The authors acknowledge Flinders Microscopy and Microanalysis and Adelaide Microscopy for their expertise. The authors acknowledge the support from the Australian Synchrotron (grant M15819) and the support of Dr Bruce Cowie from the Australian Synchrotron.References
- D. Lee, R. L. Donkers, G. Wang, A. S. Harper and R. W. Murray, J. Am. Chem. Soc., 2004, 126, 6193–6199 CrossRef CAS PubMed.
- J. Jung, H. Kim and Y.-K. Han, J. Am. Chem. Soc., 2011, 133, 6090–6095 CrossRef CAS PubMed.
- G. Ramakrishna, O. Varnavski, J. Kim, D. Lee and T. Goodson, J. Am. Chem. Soc., 2008, 130, 5032–5033 CrossRef CAS PubMed.
- M. Valden, X. Lai and D. W. Goodman, Science, 1998, 281, 1647–1650 CrossRef CAS PubMed.
- C. Xu, X. Lai, G. Zajac and D. Goodman, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 56, 13464 CrossRef CAS.
- C. P. Joshi, M. S. Bootharaju and O. M. Bakr, J. Phys. Chem. Lett., 2015, 6, 3023–3035 CrossRef CAS PubMed.
- A. Sanchez, S. Abbet, U. Heiz, W. D. Schneider, H. Hakkinen, R. N. Barnett and U. Landman, J. Phys. Chem. A, 1999, 103, 9573 CrossRef CAS.
- C. Moreno-Castilla, M. A. Salas-Peregrín and F. J. López-Garzón, J. Mol. Catal. A: Chem., 1995, 95, 223–233 CrossRef CAS.
- P. Panagiotopoulou, Appl. Catal., A, 2017, 542, 63–70 CrossRef CAS.
- R. Mutschler, E. Moioli and A. Züttel, J. Catal., 2019, 375, 193–201 CrossRef CAS.
- G. D. Weatherbee and C. H. Bartholomew, J. Catal., 1984, 87, 352–362 CrossRef CAS.
- K. Asakura and Y. Iwasawa, J. Chem. Soc., Faraday Trans., 1990, 86, 2657–2662 RSC.
- F. Solymosi, A. Erdöhelyi and M. Kocsis, J. Chem. Soc., Faraday Trans. 1, 1981, 77, 1003–1012 RSC.
- W. R. Hastings, C. J. Cameron, M. J. Thomas and M. C. Baird, Inorg. Chem., 1988, 27, 3024–3028 CrossRef CAS.
- C. S. Kellner and A. T. Bell, J. Catal., 1982, 75, 251–261 CrossRef CAS.
- R. D. Gonzalez and H. Miura, J. Catal., 1982, 77, 338–347 CrossRef CAS.
- E. Kikuchi, H. Nomura, M. Matsumoto and Y. Morita, Appl. Catal., 1983, 7, 1–9 CrossRef CAS.
- Q. Wang and K. Domen, Chem. Rev., 2020, 120, 919–985 CrossRef CAS PubMed.
- Q. Fu, T. Wagner, S. Olliges and H.-D. Carstanjen, J. Phys. Chem. B, 2005, 109, 944–951 CrossRef CAS PubMed.
- S. Tauster, S. Fung and R. L. Garten, J. Am. Chem. Soc., 1978, 100, 170–175 CrossRef CAS.
- G. L. Haller and D. E. Resasco, Advances in catalysis, Elsevier, 1989, vol. 36, pp. 173–235 Search PubMed.
- F. Pesty, H.-P. Steinrück and T. E. Madey, Surf. Sci., 1995, 339, 83–95 CrossRef CAS.
- A. S. Alotabi, C. T. Gibson, G. F. Metha and G. G. Andersson, ACS Appl. Energy Mater., 2021, 4, 322–330 CrossRef CAS.
- A. Berkó, I. Ulrych and K. Prince, J. Phys. Chem. B, 1998, 102, 3379–3386 CrossRef.
- O. Dulub, W. Hebenstreit and U. Diebold, Phys. Rev. Lett., 2000, 84, 3646 CrossRef CAS PubMed.
- R. Bennett, C. Pang, N. Perkins, R. Smith, P. Morrall, R. Kvon and M. Bowker, J. Phys. Chem. B, 2002, 106, 4688–4696 CrossRef CAS.
- R. Bennett, P. Stone and M. Bowker, Catal. Lett., 1999, 59, 99–105 CrossRef CAS.
- Y. Gao, Y. Liang and S. Chambers, Surf. Sci., 1996, 365, 638–648 CrossRef CAS.
- H. R. Sadeghi and V. E. Henrich, J. Catal., 1988, 109, 1–11 CrossRef CAS.
- H. R. Sadeghi and V. E. Henrich, Appl. Surf. Sci., 1984, 19, 330–340 CrossRef CAS.
- S. Labich, E. Taglauer and H. Knözinger, Top. Catal., 2000, 14, 153–161 CrossRef CAS.
- D. Mullins and K. Zhang, Surf. Sci., 2002, 513, 163–173 CrossRef CAS.
- Q. Fu and T. Wagner, J. Phys. Chem. B, 2005, 109, 11697–11705 CrossRef CAS PubMed.
- H. Feng, J. Lu, P. C. Stair and J. W. Elam, Catal. Lett., 2011, 141, 512–517 CrossRef CAS.
- W. Kurashige, R. Kumazawa, D. Ishii, R. Hayashi, Y. Niihori, S. Hossain, L. V. Nair, T. Takayama, A. Iwase, S. Yamazoe, T. Tsukuda, A. Kudo and Y. Negishi, J. Phys. Chem. C, 2018, 122, 13669–13681 CrossRef CAS.
- S. Vajda, M. J. Pellin, J. P. Greeley, C. L. Marshall, L. A. Curtiss, G. A. Ballentine, J. W. Elam, S. Catillon-Mucherie, P. C. Redfern and F. Mehmood, Nat. Mater., 2009, 8, 213–216 CrossRef CAS PubMed.
- Y. Negishi, M. Mizuno, M. Hirayama, M. Omatoi, T. Takayama, A. Iwase and A. Kudo, Nanoscale, 2013, 5, 7188–7192 RSC.
- G. A. Rizzi, A. Magrin and G. Granozzi, Phys. Chem. Chem. Phys., 1999, 1, 709–711 RSC.
- X. Zhao, J. Hrbek and J. A. Rodriguez, Surf. Sci., 2005, 575, 115–124 CrossRef CAS.
- D. Meier, G. Rizzi, G. Granozzi, X. Lai and D. Goodman, Langmuir, 2002, 18, 698–705 CrossRef CAS.
- T. Cai, Z. Song, Z. Chang, G. Liu, J. Rodriguez and J. Hrbek, Surf. Sci., 2003, 538, 76–88 CrossRef CAS.
- F. Yang, S. Kundu, A. B. Vidal, J. Graciani, P. J. Ramírez, S. D. Senanayake, D. Stacchiola, J. Evans, P. Liu and J. F. Sanz, Angew. Chem., Int. Ed., 2011, 50, 10198–10202 CrossRef CAS PubMed.
- P. Serp, P. Kalck and R. Feurer, Chem. Rev., 2002, 102, 3085–3128 CrossRef CAS PubMed.
- J. A. Rodriguez, J. Dvorak, T. Jirsak and J. Hrbek, Surf. Sci., 2001, 490, 315–326 CrossRef CAS.
- R. Psaro and S. Recchia, Catal. Today, 1998, 41, 139–147 CrossRef CAS.
- M. Xu and F. Zaera, J. Vac. Sci. Technol., A, 1996, 14, 415–424 CrossRef CAS.
- A. C. Papageorgiou, K. Diller, S. Fischer, F. Allegretti, F. Klappenberger, S. C. Oh, O. Z. Sağlam, J. Reichert, A. Wiengarten and K. Seufert, J. Phys. Chem. C, 2016, 120, 8751–8758 CrossRef CAS.
- E. Mohimi, Z. V. Zhang, S. Liu, J. L. Mallek, G. S. Girolami and J. R. Abelson, J. Vac. Sci. Technol., A, 2018, 36, 041507 CrossRef.
- G. E. Johnson, R. Colby and J. Laskin, Nanoscale, 2015, 7, 3491–3503 RSC.
- T. Choudhary, C. Sivadinarayana, C. C. Chusuei, A. Datye, J. Fackler Jr and D. Goodman, J. Catal., 2002, 207, 247–255 CrossRef CAS.
- H. S. Al Qahtani, R. Higuchi, T. Sasaki, J. F. Alvino, G. F. Metha, V. B. Golovko, R. Adnan, G. G. Andersson and T. Nakayama, RSC Adv., 2016, 6, 110765–110774 RSC.
- G. G. Andersson, V. B. Golovko, J. F. Alvino, T. Bennett, O. Wrede, S. M. Mejia, H. S. Al Qahtani, R. Adnan, N. Gunby and D. P. Anderson, J. Chem. Phys., 2014, 141, 014702 CrossRef PubMed.
- H. S. Al Qahtani, K. Kimoto, T. Bennett, J. F. Alvino, G. G. Andersson, G. F. Metha, V. B. Golovko, T. Sasaki and T. Nakayama, J. Chem. Phys., 2016, 144, 114703 CrossRef PubMed.
- G. Krishnan, N. Eom, R. M. Kirk, V. B. Golovko, G. F. Metha and G. G. Andersson, J. Phys. Chem. C, 2019, 123, 6642–6649 CrossRef CAS.
- G. Krishnan, H. S. Al Qahtani, J. Li, Y. Yin, N. Eom, V. B. Golovko, G. F. Metha and G. G. Andersson, J. Phys. Chem. C, 2017, 121, 28007–28016 CrossRef CAS.
- L. Howard-Fabretto, T. J. Gorey, G. Li, S. Tesana, G. F. Metha, S. L. Anderson and G. G. Andersson, Nanoscale Adv., 2021, 3, 3537–3553 RSC.
- F. X. Xiao, S. F. Hung, J. Miao, H. Y. Wang, H. Yang and B. Liu, Small, 2015, 11, 554–567 CrossRef CAS PubMed.
- I. X. Green, W. Tang, M. Neurock and J. T. Yates, Science, 2011, 333, 736–739 CrossRef CAS PubMed.
- A. C. Reber, S. N. Khanna, F. S. Roberts and S. L. Anderson, J. Phys. Chem. C, 2016, 120, 2126–2138 CrossRef CAS.
- H. S. Al Qahtani, G. F. Metha, R. B. Walsh, V. B. Golovko, G. G. Andersson and T. Nakayama, J. Phys. Chem. C, 2017, 121, 10781–10789 CrossRef CAS.
- F. S. Roberts, S. L. Anderson, A. C. Reber and S. N. Khanna, J. Phys. Chem. C, 2015, 119, 6033–6046 CrossRef CAS.
- J.-Y. Ruzicka, F. Abu Bakar, C. Hoeck, R. Adnan, C. McNicoll, T. Kemmitt, B. C. Cowie, G. F. Metha, G. G. Andersson and V. B. Golovko, J. Phys. Chem. C, 2015, 119, 24465–24474 CrossRef CAS.
- K. Nakata, S. Sugawara, W. Kurashige, Y. Negishi, M. Nagata, S. Uchida, C. Terashima, T. Kondo, M. Yuasa and A. Fujishima, Int. J. Photoenergy, 2013, 2013, 456583 CrossRef.
- K. Katsiev, G. Harrison, Y. Al-Salik, G. Thornton and H. Idriss, ACS Catal., 2019, 9, 8294–8305 CrossRef CAS.
- P. López-Caballero, A. W. Hauser and M. A. Pilar de Lara-Castells, J. Phys. Chem. C, 2019, 123, 23064–23074 CrossRef PubMed.
- U. Diebold, Surf. Sci. Rep., 2003, 48, 53–229 CrossRef CAS.
- H. Jensen, A. Soloviev, Z. Li and E. G. Søgaard, Appl. Surf. Sci., 2005, 246, 239–249 CrossRef CAS.
- E. Wahlström, N. Lopez, R. Schaub, P. Thostrup, A. Rønnau, C. Africh, E. Lægsgaard, J. Nørskov and F. Besenbacher, Phys. Rev. Lett., 2003, 90, 026101 CrossRef PubMed.
- I. Hadjoub, T. Touam, A. Chelouche, M. Atoui, J. Solard, M. Chakaroun, A. Fischer, A. Boudrioua and L.-H. Peng, Appl. Phys. A: Mater. Sci. Process., 2016, 122, 78 CrossRef.
- P. J. Cumpson, Appl. Surf. Sci., 1999, 144, 16–20 CrossRef.
- C. Wang and G. G. Andersson, Surf. Sci., 2011, 605, 889–897 CrossRef CAS.
- L. Óvári and J. Kiss, Appl. Surf. Sci., 2006, 252, 8624–8629 CrossRef.
- G. Li, B. Zandkarimi, A. C. Cass, T. J. Gorey, B. J. Allen, A. N. Alexandrova and S. L. Anderson, J. Chem. Phys., 2020, 152, 024702 CrossRef CAS PubMed.
- T. J. Gorey, Y. Dai, S. L. Anderson, S. Lee, S. Lee, S. Seifert and R. E. Winans, Surf. Sci., 2020, 691, 121485 CrossRef CAS.
- B. Zandkarimi, T. J. Gorey, G. Li, J. Munarriz, S. L. Anderson and A. N. Alexandrova, Chem. Mater., 2020, 32, 8595–8605 CrossRef CAS.
- T. J. Gorey, B. Zandkarimi, G. Li, E. T. Baxter, A. N. Alexandrova and S. L. Anderson, J. Phys. Chem. C, 2019, 123, 16194–16209 CrossRef CAS.
- E. T. Baxter, M.-A. Ha, A. C. Cass, H. Zhai, A. N. Alexandrova and S. L. Anderson, J. Phys. Chem. C, 2018, 122, 1631–1644 CrossRef CAS.
- E. T. Baxter, M.-A. Ha, A. C. Cass, A. N. Alexandrova and S. L. Anderson, ACS Catal., 2017, 7, 3322–3335 CrossRef CAS.
- Y. Dai, T. J. Gorey, S. L. Anderson, S. Lee, S. Lee, S. Seifert and R. E. Winans, J. Phys. Chem. C, 2017, 121, 361–374 CrossRef CAS.
- F. S. Roberts, M. D. Kane, E. T. Baxter and S. L. Anderson, Phys. Chem. Chem. Phys., 2014, 16, 26443–26457 RSC.
- M. D. Kane, F. S. Roberts and S. L. Anderson, Int. J. Mass Spectrom. Ion Processes, 2014, 370, 1–15 CrossRef CAS.
- L. Ovári, L. Bugyi, Z. Majzik, A. Berkó and J. Kiss, J. Phys. Chem. C, 2008, 112, 18011–18016 CrossRef.
- C. C. Chusuei, X. Lai, K. Luo and D. Goodman, Top. Catal., 2000, 14, 71–83 CrossRef CAS.
- S. L. Tait, L. T. Ngo, Q. Yu, S. C. Fain Jr and C. T. Campbell, J. Chem. Phys., 2005, 122, 064712 CrossRef PubMed.
- S. Lee, C. Fan, T. P. Wu and S. Anderson, J. Phys. Chem. B, 2005, 109, 11340–11347 CrossRef CAS PubMed.
- J. Kiss, L. Óvári, A. Oszkó, G. Pótári, M. Tóth, K. Baán and A. Erdóhelyi, Catal. Today, 2012, 181, 163–170 CrossRef CAS.
- S. A. Tenney, W. He, J. S. Ratliff, D. R. Mullins and D. A. Chen, Top. Catal., 2011, 54, 42–55 CrossRef CAS.
- S. A. Tenney, W. He, C. C. Roberts, J. S. Ratliff, S. I. Shah, G. S. Shafai, V. Turkowski, T. S. Rahman and D. A. Chen, J. Phys. Chem. C, 2011, 115, 11112–11123 CrossRef CAS.
- L. Óvári, A. Berkó, N. Balázs, Z. Majzik and J. Kiss, Langmuir, 2010, 26, 2167–2175 CrossRef PubMed.
- Z.-j Wang, F. Yang, S. Axnanda, C.-j Liu and D. W. Goodman, Appl. Catal., A, 2011, 391, 342–349 CrossRef CAS.
- Y. Niu, P. Schlexer, B. Sebok, I. Chorkendorff, G. Pacchioni and R. E. Palmer, Nanoscale, 2018, 10, 2363–2370 RSC.
- R. P. Galhenage, K. Xie, W. Diao, J. M. M. Tengco, G. S. Seuser, J. R. Monnier and D. A. Chen, Phys. Chem. Chem. Phys., 2015, 17, 28354–28363 RSC.
- H. H. Brongersma, T. Grehl, P. A. van Hal, N. C. Kuijpers, S. G. Mathijssen, E. R. Schofield, R. A. Smith and H. R. ter Veen, Vacuum, 2010, 84, 1005–1007 CrossRef CAS.
- A. Rafati, R. ter Veen and D. G. Castner, Surf. Interface Anal., 2013, 45, 1737–1741 CrossRef CAS PubMed.
- N. Belsey, D. Cant, C. Minelli, J. Araujo, B. Bock, P. Bruener, D. Castner, G. Ceccone, J. Counsell and P. Dietrich, J. Phys. Chem. C, 2016, 120, 24070–24079 CrossRef CAS PubMed.
- V. Jeyalakshmi, R. Mahalakshmy, K. Krishnamurthy and B. Viswanathan, Indian J. Chem., Sect. A: Inorg., Bio-inorg., Phys., Theor. Anal. Chem., 2012, 51A, 1263–1283 CAS.
- J. Daughtry, A. Alotabi, L. Howard-Fabretto and G. G. Andersson, Nanoscale Adv., 2020, 3, 1077–1086 RSC.
- A. Haseeb, M. Hasan and H. H. Masjuki, Surf. Coat. Technol., 2010, 205, 338–344 CrossRef CAS.
- Q. Ye, P. Liu, Z. Tang and L. Zhai, Vacuum, 2007, 81, 627–631 CrossRef CAS.
- V. N. Popok, I. Barke, E. E. B. Campbell and K.-H. Meiwes-Broer, Surf. Sci. Rep., 2011, 66, 347–377 CrossRef CAS.
- O. Baschenko and A. Nesmeev, J. Electron Spectrosc. Relat. Phenom., 1991, 57, 33–46 CrossRef.
- H. H. Brongersma, Charact. Mater., 2012, 1–23 Search PubMed.
- H. H. Brongersma, M. Draxler, M. De Ridder and P. Bauer, Surf. Sci. Rep., 2007, 62, 63–109 CrossRef CAS.
- G. Lu, A. Linsebigler and J. T. Yates Jr, J. Phys. Chem., 1994, 98, 11733–11738 CrossRef CAS.
- S. Abrahams and J. Bernstein, J. Chem. Phys., 1971, 55, 3206–3211 CrossRef CAS.
- W. E. Kaden, W. A. Kunkel and S. L. Anderson, J. Chem. Phys., 2009, 131, 114701 CrossRef CAS PubMed.
- L. W. Hoffman, G. G. Andersson, A. Sharma, S. R. Clarke and N. H. Voelcker, Langmuir, 2011, 27, 6759–6767 CrossRef CAS PubMed.
- L. Sutton, Tables of interatomic distances and configuration in molecules and ions, Chemical Society, 1965 Search PubMed.
- L. Mezey and J. Giber, Jpn. J. Appl. Phys., 1982, 21, 1569 CrossRef CAS.
- M. Aizawa, S. Lee and S. L. Anderson, Surf. Sci., 2003, 542, 253–275 CrossRef CAS.
- A. Howard, C. Mitchell, D. Morris, R. Egdell and S. Parker, Surf. Sci., 2000, 448, 131–141 CrossRef CAS.
- W. E. Kaden, W. A. Kunkel, F. S. Roberts, M. Kane and S. L. Anderson, Surf. Sci., 2014, 621, 40–50 CrossRef CAS.
- M. Aizawa, S. Lee and S. L. Anderson, J. Chem. Phys., 2002, 117, 5001–5011 CrossRef CAS.
- S. Tauster, S. Fung, R. Baker and J. Horsley, Science, 1981, 211, 1121–1125 CrossRef CAS PubMed.
- S. Bernal, F. Botana, J. Calvino, C. López, J. Pérez-Omil and J. Rodríguez-Izquierdo, J. Chem. Soc., Faraday Trans., 1996, 92, 2799–2809 RSC.
- S. Bernal, J. Calvino, M. Cauqui, J. Gatica, C. L. Cartes, J. P. Omil and J. Pintado, Catal. Today, 2003, 77, 385–406 CrossRef CAS.
- W. E. Kaden, W. A. Kunkel and S. L. Anderson, J. Chem. Phys., 2009, 131, 114701 CrossRef CAS PubMed.
- E. Hebenstreit, W. Hebenstreit, H. Geisler, C. Ventrice Jr, P. Sprunger and U. Diebold, Surf. Sci., 2001, 486, L467–L474 CrossRef CAS.
- T. Komaya, A. T. Bell, Z. Wengsieh, R. Gronsky, F. Engelke, T. S. King and M. Pruski, J. Catal., 1994, 149, 142–148 CrossRef CAS.
- T. Sham, T. Ohta, T. Yokoyama, Y. Kitajima, M. Funabashi, N. Kosugi and H. Kuroda, J. Chem. Phys., 1988, 88, 475–477 CrossRef CAS.
- K. Schierbaum, S. Fischer, M. Torquemada, J. De Segovia, E. Roman and J. Martin-Gago, Surf. Sci., 1996, 345, 261–273 CrossRef CAS.
- M. Della Negra, N. M. Nicolaisen, Z. Li and P. J. Møller, Surf. Sci., 2003, 540, 117–128 CrossRef CAS.
- T. Yoskamtorn, S. Yamazoe, R. Takahata, J.-I. Nishigaki, A. Thivasasith, J. Limtrakul and T. Tsukuda, ACS Catal., 2014, 4, 3696–3700 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The ESI provides detailed descriptions of the instruments and the experimental techniques applied. The raw data for the C 1s/Ru 3d and Ti 2p regions, and the TD-XPS and LEIS data are also presented, as well as additional STEM images. See DOI: https://doi.org/10.1039/d4cp00263f |
| This journal is © the Owner Societies 2024 |