Strongly facet-dependent activity of iron-doped β-nickel oxyhydroxide for the oxygen evolution reaction†
Received
23rd January 2024
, Accepted 11th April 2024
First published on 12th April 2024
Abstract
Iron (Fe)-doped β-nickel oxyhydroxide (β-NiOOH) is a highly active, noble-metal-free electrocatalyst for the oxygen evolution reaction (OER), with the latter being the bottleneck in electrochemical water splitting for sustainable hydrogen production. The mechanisms underlying how the Fe dopant modulates this host material's water electro-oxidation activity are still not entirely clear. Here, we combine hybrid density functional theory (DFT) and Hubbard-corrected DFT to investigate the OER activity of the most thermodynamically favorable (and therefore, expected to be the majority) crystallographic facets of β-NiOOH, namely (0001) and (10![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0). By considering active sites involving both oxidation and reduction of the transition-metal active center during the redox cycle on these two different facets, we show that six-fold-lattice-coordinated Fe in β-NiOOH is redox inactive towards both oxidation and reduction while five-fold-lattice-coordinated Fe in β-NiOOH does exhibit redox activity. However, the determined redox activity of Fe (or lack of it) is not indicative of good (or bad) performance as a dopant on these two facets. Three of the four active sites investigated (oxo and hydroxo sites on (0001) and a hydrated site on (100)) exhibit only a marginal (<0.1 V) decrease or increase in the thermodynamic overpotential upon doping with Fe. Only one of the redox-active sites investigated, the hydroxo site on (100), exhibits a large attenuation in the thermodynamic overpotential upon doping (to ∼0.52 V from 0.86 V), although the doped overpotential is larger than that observed experimentally for Fe-doped NiOOH. Thus, although pure β-NiOOH facets containing four-, five-, or six-fold lattice-coordinated Ni sites have roughly equal OER activities, yielding similar OER onset potentials (shown in A. Govind Rajan, J. M. P. Martirez and E. A. Carter, J. Am. Chem. Soc., 2020, 142, 3600–3612), only those facets containing four-fold lattice-coordinated Fe (e.g., as shown in J. M. P. Martirez and E. A. Carter, J. Am. Chem. Soc., 2019, 141, 693–705) would be active under analogous conditions for the Fe-doped material. It follows that, while undoped β-NiOOH demonstrates a roughly facet-independent oxygen evolution activity, the activity of Fe-doped β-NiOOH strongly depends on the crystallographic facet. Our study further motivates the investigation of strategies for the selective growth of facets with low iron coordination number to enhance the water splitting activity of Fe-doped β-NiOOH.
0). By considering active sites involving both oxidation and reduction of the transition-metal active center during the redox cycle on these two different facets, we show that six-fold-lattice-coordinated Fe in β-NiOOH is redox inactive towards both oxidation and reduction while five-fold-lattice-coordinated Fe in β-NiOOH does exhibit redox activity. However, the determined redox activity of Fe (or lack of it) is not indicative of good (or bad) performance as a dopant on these two facets. Three of the four active sites investigated (oxo and hydroxo sites on (0001) and a hydrated site on (100)) exhibit only a marginal (<0.1 V) decrease or increase in the thermodynamic overpotential upon doping with Fe. Only one of the redox-active sites investigated, the hydroxo site on (100), exhibits a large attenuation in the thermodynamic overpotential upon doping (to ∼0.52 V from 0.86 V), although the doped overpotential is larger than that observed experimentally for Fe-doped NiOOH. Thus, although pure β-NiOOH facets containing four-, five-, or six-fold lattice-coordinated Ni sites have roughly equal OER activities, yielding similar OER onset potentials (shown in A. Govind Rajan, J. M. P. Martirez and E. A. Carter, J. Am. Chem. Soc., 2020, 142, 3600–3612), only those facets containing four-fold lattice-coordinated Fe (e.g., as shown in J. M. P. Martirez and E. A. Carter, J. Am. Chem. Soc., 2019, 141, 693–705) would be active under analogous conditions for the Fe-doped material. It follows that, while undoped β-NiOOH demonstrates a roughly facet-independent oxygen evolution activity, the activity of Fe-doped β-NiOOH strongly depends on the crystallographic facet. Our study further motivates the investigation of strategies for the selective growth of facets with low iron coordination number to enhance the water splitting activity of Fe-doped β-NiOOH.
Introduction
The electrochemical oxidation of water to produce oxygen, also known as the oxygen evolution reaction (OER), is a challenging reaction studied by many chemists, materials scientists, and engineers.1 As the anodic half-reaction in water splitting, the OER is vital to the carbon-dioxide-free synthesis of hydrogen gas via renewable electricity.2 Layered transition-metal oxides, hydroxides, and oxyhydroxides, in both their undoped and doped forms, can be used for electrocatalyzing the OER under alkaline conditions.2,3 Layered materials offer distinctive advantages, such as the presence of highly active sites and large surface-to-volume ratios, in electrochemical applications.4–6 Nickel oxyhydroxide (NiOOH), particularly in its β and γ phases, is a material that has been investigated extensively, both theoretically7–14 and experimentally,15–24 for OER electrocatalysis. Experiments have shown that doping NiOOH with iron (Fe) substantially reduces the OER overpotential of this material,25–27 with theory offering explanations of these observations.28–30 Theory also explained the superiority of doping with Fe, as compared to other transition-metal dopants, for enhancing the OER activity of NiOOH.31,32 The mechanisms by which Fe modulates the host material's water oxidation activity on various crystallographic facets were the subject of several previous studies and remain under investigation by various research groups.12,15,25,28,33–37
Recently, Martirez and Carter presented a real-space partitioning scheme28 to compute OER free energetics on an electrocatalyst surface by combining more accurate, but computationally demanding, hybrid density functional theory (DFT)38,39 calculations with less expensive Hubbard-U-corrected DFT (DFT+U)40 calculations. Whereas the latter technique only statically corrects for electron self-interaction errors within the transition-metal ions, the former adds a fraction of nonlocal electron exchange interactions in evaluating the self-consistent wavefunctions and energies, thereby correcting for spurious electron self-interaction in a global and systematic manner. Accordingly, hybrid DFT more accurately models the free-energy changes associated with oxidation-state changes in transition-metal compounds during redox reactions than DFT+U does. Using this scheme, Martirez and Carter found that a four-fold lattice-oxygen-coordinated Fe reactive site manifested a record-low thermodynamic OER overpotential of 0.14 V on the (21) facet of Fe-doped β-NiOOH,28 a minority but not insignificant facet41 (vide infra) of the hexagonal platelets observed to form.42,43 Subsequently, Govind Rajan et al. used the same approach to reveal facet-independent OER activity for undoped β-NiOOH. Specifically, they found that the hydroxo and hydrated active sites on the undoped (0001) and (100) facets, respectively, exhibit thermodynamic OER overpotentials of 0.44 and 0.43 V, in close agreement with the 0.48 V value on the hydroxo active site on the undoped (21) facet.12 Examining whether such facet-independence of the OER activity also holds for Fe-doped β-NiOOH is important, given the much greater activity of the latter. Efforts to understand facet and active-site dependence of the OER activity and mechanism will yield better microkinetic models44–46 and mechanism-enumeration strategies.47
Previously, Martirez and Carter considered the surface free energies of various crystallographic facets of β-NiOOH in the presence of explicit and implicit solvation, and showed that the solvated (0001) and (100) facets together constitute ∼70% of its surface, whereas the (21) facet contributes only ∼1.5% of the surface as per the thermodynamic Wulff construction.41 Later, Tkalych et al. investigated the OER activity of Fe-, Co-, and Mn-doped β-NiOOH(0001) using DFT+U calculations and showed that the presence of these dopants only marginally reduced the thermodynamic OER overpotential as compared to undoped β-NiOOH.48 However, so far, the OER activities of the various active sites that can exist on the two most-abundant facets of Fe-doped β-NiOOH—(0001) and (100)—have not been studied at the same hybrid DFT level of theory used in predicting the record-low thermodynamic OER overpotential of 0.14 V on the (21) facet.28 In this work, we address this knowledge gap by examining the thermodynamics of the various elementary steps involved in the OER mechanism on Fe-doped β-NiOOH(0001)/(100) using hybrid DFT. We investigate various OER mechanisms to systematically determine pathways with the lowest-possible thermodynamic overpotential on each active site considered. We find that the redox activity of Fe on the surface of β-NiOOH depends on its extent of coordination with lattice oxygen atoms. While six-fold lattice-coordinated Fe is found to be redox inactive, five-fold lattice-coordinated Fe changes oxidation states during the OER mechanism, thereby being the apparent primary active site during the OER. Combining our findings with those of Martirez and Carter, who showed four-fold lattice-coordinated Fe to be redox active (in their study of the (21) facet of Fe-doped β-NiOOH),28 we conclude that Fe needs to have a coordination number less than six to demonstrate redox activity that enables the OER on β-NiOOH surfaces. Overall, we rule out any major role for the (0001) and (100) facets in the OER activity of Fe-doped β-NiOOH, although previous theoretical work predicted these facets to be active during the OER on undoped β-NiOOH.10,12 Our conclusion that sites with low lattice coordination number are required for OER activity on Fe-doped β-NiOOH is consistent with prior electrochemical analysis based on various surface probe techniques (vide infra).49–52
Computational methods
Calculating the free energies of the species involved in the OER
In this work, we utilized the computational hydrogen electrode (CHE) model for electrochemical thermodynamics.53–55 Accordingly, the standard Gibbs free energy of a proton–electron couple is defined as 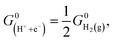 where
where 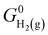 denotes the standard Gibbs free energy of gaseous hydrogen. One obtains the standard Gibbs free energy of H2(g),
denotes the standard Gibbs free energy of gaseous hydrogen. One obtains the standard Gibbs free energy of H2(g), 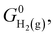 from:
from:
| G0i(T) = EDFTi + ZPEDFTi + (H0(T) − H0(0 K) − TS0(T))expi, |
where i = H2(g), EDFTi denotes the DFT energy of species i (here, an isolated H2 molecule in vacuum), ZPEDFTi the zero-point energy of species i, H0(T) the enthalpy at temperature T, and S0(T) the entropy at temperature T. Note that 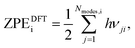 where Nmodes,i denotes the number of vibrational modes of species i, h is Planck's constant, and νji denotes the normal mode frequencies of species i. We assumed room temperature (T = 298.15 K) in all our calculations and obtained experimental values of H0(T) and S0(T) from the NIST-JANAF thermochemical database.56 Subsequently, we determined the free energy of H2O(l),
where Nmodes,i denotes the number of vibrational modes of species i, h is Planck's constant, and νji denotes the normal mode frequencies of species i. We assumed room temperature (T = 298.15 K) in all our calculations and obtained experimental values of H0(T) and S0(T) from the NIST-JANAF thermochemical database.56 Subsequently, we determined the free energy of H2O(l), 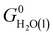 , using the following equation:
, using the following equation:
| G0H2O(l) = EDFTi + ZPEDFTi + (H0(T) − H0(0 K) − TS0(T))expi + ΔG0g→l(T), |
where i = H2O(g) and ΔG0g→l(T) denotes the experimental condensation free energy of water.56 We obtained the normal modes required for calculating the ZPE by evaluating the Hessian matrix from a central-difference scheme and a displacement of ±0.02 Å along all three directions relative to the optimized atomic positions. Next, we calculated the Gibbs free energy of O2(g) using the equation below, derived by enforcing the free energy change of 1.23 eV per electron needed for the OER at room temperature:
Finally, we determined the standard Gibbs free energies of the adsorbed species, which include the DFT energies (EDFTi) as well as vibrational contributions (Evib,i − TSvib,i), as follows:
| G0i = EDFTi + Evib,i − TSvib,i |
We calculated these quantities using the harmonic lattice model, as described below:57
where Ni denotes the number of surface atoms in the ith intermediate, νji denotes the jth frequency of the ith intermediate, 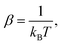 and kB is the Boltzmann constant. Subsequently, we calculated the standard Gibbs free energy change of reaction, ΔG0r,k, of each elementary step k of a mechanism using the species free energies. Following that, we determined the thermodynamic OER overpotential from the difference between the largest electroactive ΔG0r,k and 1.23 eV, per electron transferred, where 1.23 eV is the standard Gibbs free energy change per electron incurred during the OER at room temperature.47 Note that an electroactive step is one in which an electron is transferred so that the free energy of the reaction can be modulated by the application of electric potential. We did not consider the non-electroactive steps (i.e., those that do not involve the explicit transfer of an electron) in the evaluation of the thermodynamic overpotential. The electroactive step with the largest ΔG0r,k requirement is referred to as the potential-determining step (PDS).
and kB is the Boltzmann constant. Subsequently, we calculated the standard Gibbs free energy change of reaction, ΔG0r,k, of each elementary step k of a mechanism using the species free energies. Following that, we determined the thermodynamic OER overpotential from the difference between the largest electroactive ΔG0r,k and 1.23 eV, per electron transferred, where 1.23 eV is the standard Gibbs free energy change per electron incurred during the OER at room temperature.47 Note that an electroactive step is one in which an electron is transferred so that the free energy of the reaction can be modulated by the application of electric potential. We did not consider the non-electroactive steps (i.e., those that do not involve the explicit transfer of an electron) in the evaluation of the thermodynamic overpotential. The electroactive step with the largest ΔG0r,k requirement is referred to as the potential-determining step (PDS).
Details regarding the DFT calculations
We performed DFT calculations using the all-electron, frozen-core projector augmented-wave (PAW) formalism58,59 in the Vienna ab initio simulation package (VASP)60 to solve self-consistently for the valence electrons (3d and 4s for Ni/Fe; 2s and 2p for O; and 1s for H) and employed the frozen-core approximation via VASP's standard PAW potentials for the core electrons. We chose the Perdew–Burke–Ernzerhof (PBE) generalized gradient approximation functional61 for evaluating the exchange–correlation (XC) energy, along with a Hubbard correction (+U) based on Dudarev et al.'s approach,40 applied to account more properly for the localized nature of the 3d electrons and their attendant exchange interactions. Specifically, we used an effective U value of 5.5 eV for Ni as determined from linear response theory7 and a value of 4.3 eV for Fe calculated from electrostatically embedded unrestricted Hartree–Fock theory.62 For the plane wave basis set, we used a kinetic energy cutoff of 750 eV for the larger slabs and a smaller cutoff of 650 eV for the smaller slabs to manage the computational cost of the latter, for which we utilize hybrid DFT. The DFT-PBE+U simulations used a 3 × 3 × 1 k-point mesh for reciprocal space sampling, selected via the Monkhorst–Pack method.63,64 More details regarding the DFT calculations are provided in the ESI,† Section S1.
We evaluated the atom-projected magnetic moments using the LORBIT = 11 tag in VASP, which we used to assign oxidation states to the transition-metal cations in the system (see the ESI,† Tables S1 and S2 for surface atom oxidation state analysis for each facet) and to the oxygen species in some cases. We set the initial guesses for the magnetic moments for each system using the MAGMOM tag based on the expected oxidation state and number of unpaired electrons for each ion. We then allowed the system to self-consistently find the most stable spin state. We used the “our own n-layered integrated molecular orbital and molecular mechanics” formalism, also known as the ONIOM method pioneered by the K. Morokuma group,65 to incorporate a higher level of theory, namely hybrid DFT using the Heyd–Scuseria–Ernzerhof (HSE06) XC functional38,39,66 (with a lower fraction of exact exchange, α = 15%, than the standard form, which was shown to be more suitable for bulk late transition-metal oxides,67,68 and previously validated and applied to pure and Fe-doped NiOOH12,28,31,32), as a correction to the DFT-PBE+U energies. The equations underlying the ONIOM approach and details regarding the vibrational frequency and free-energy calculations are provided in the ESI,† Section S2.
Results and discussion
Reaction intermediates and plausible mechanisms involved in the OER
We considered the single-site, associative OER mechanism8,12,28,54,55,69 on each active site investigated in this work. In this mechanism, two water molecules adsorb and deprotonate four times through a series of proton-coupled electron transfer (PCET) steps at a single active site, leading to the coupling of two oxygen atoms and the evolution of oxygen gas.47 The intermediates involved in this mechanism are typically adsorbed water (*OH2), hydroxo (*OH), oxo (*O), hydrogen peroxide (*O(OH)H), hydroperoxo (*OOH), and superoxo/peroxo/molecular oxygen (*O2), as considered in our previous work.12 We denote a vacancy or a free adsorption site at the catalyst's active site simply as *. Note that these are the adsorbates considered as initial guess structures prior to geometry optimization. Upon relaxation within DFT-PBE+U and DFT-HSE06, in some cases, these intermediates may rearrange, e.g., dissociate or form a bond with a surface lattice atom, to form different species. Whenever drastic deviations from the above generic structures occur, we explain them within the associated discussion of the reaction mechanism.
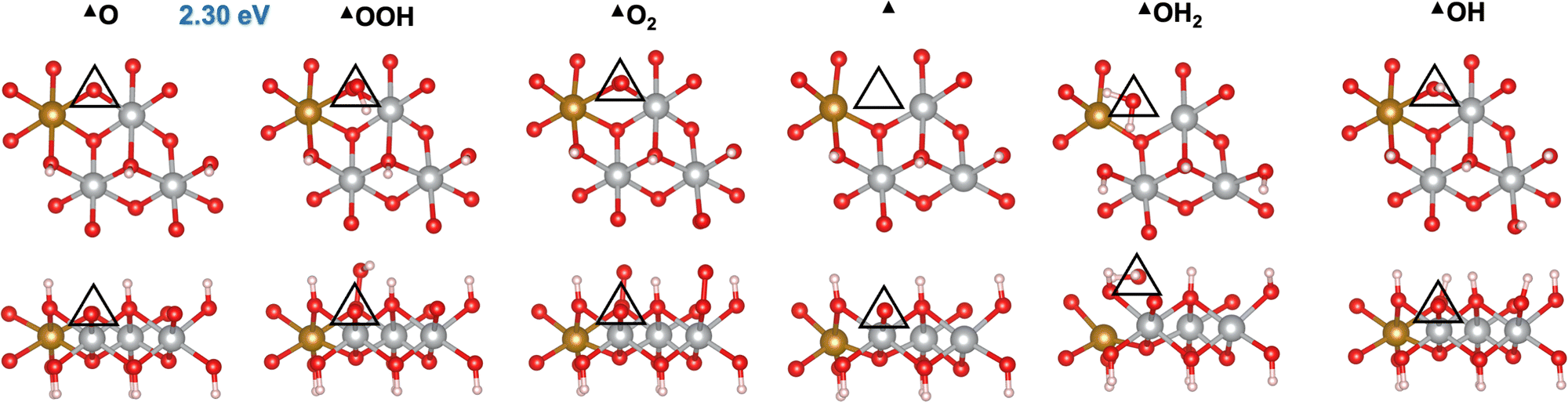
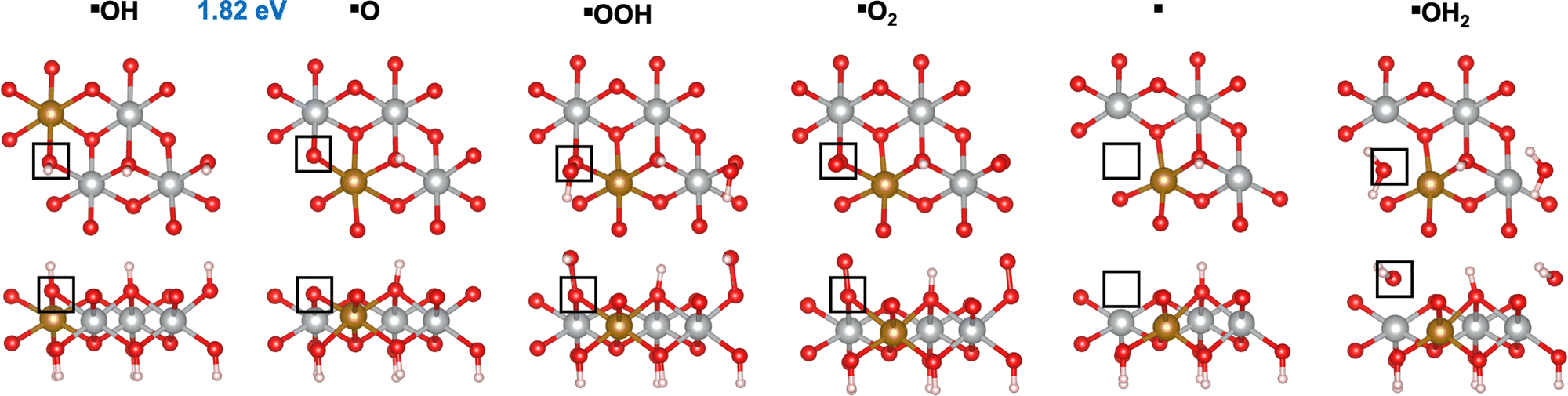
Fig. 1A shows a typical catalytic reaction cycle, labeled as mechanism M1, in black arrows. The M1 cycle starts from a lattice O (*O) on the surface and proceeds as follows:12,47
| | | *O + H2O → *OOH + (H+ + e−) | (R1) |
| | | *OOH → *O2 + (H+ + e−) | (R2) |
| | | *OH2 → *OH + (H+ + e−) | (R5) |
Here
(R1), (R2), (R5), and (R6) each involve a PCET step whereas
(R3) and (R4) denote oxygen desorption and water adsorption, respectively.
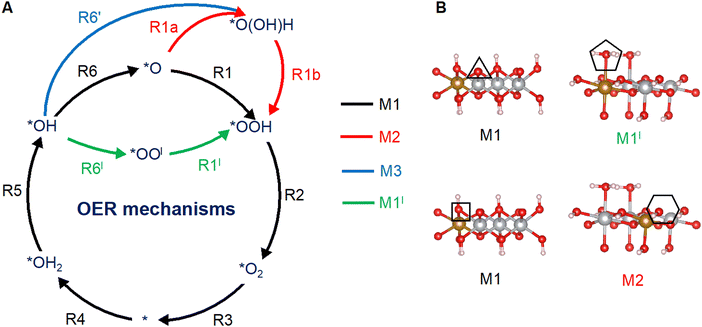 |
| | Fig. 1 OER mechanisms (A) and active sites (B) investigated in this work on the (0001) and (100) facets of Fe-doped β-NiOOH. (A) Mechanism M1 (black arrows) is the prototypical OER pathway involving steps (R1)–(R6). In mechanism M2, step (R1) is broken up into two steps: (R1a) and (R1b) (red arrows). In mechanism M3, steps (R6) and (R1) are replaced by (R6′) (blue arrow) and (R1b) (red arrow), respectively. Mechanism M1l denotes a lattice-oxygen-assisted mechanism, wherein steps (R6) and (R1) are replaced by (R6l) and (R1l), respectively (green arrows). (B) Left panels: *O (top) and *OH (bottom) sites on (0001), marked by a triangle and a square, respectively. Right panels: *OH2 (top) and *OH (bottom) sites on (100), marked by a pentagon and a hexagon, respectively. Only the side views of the top atomic layers are shown. Brown sphere: Fe, grey: Ni, red: O, and pink: H. The most-favored mechanism, i.e., the one with the lowest thermodynamic overpotential is shown below each active site. | |
Depending on the structure of the resting state of the surface (the initial state of the active site), i.e., whether the surface exhibits (1) *O, (2) *OH2, or (3) *OH species, the OER mechanism can begin with, respectively, either (1) the dissociative addition of a water molecule onto *O to produce *OOH via PCET (R1), (2) the deprotonation of *OH2 to produce *OH via PCET (R5), or (3) the deprotonation of *OH to produce *O via PCET (R6). All three scenarios are considered in the present work. Indeed, while the OER on the (0001) facet of β-NiOOH starts with (R1) or (R6) [at the *O (▲) and *OH (■) sites, respectively; see Fig. 1B], the OER on the (100) facet of β-NiOOH begins with (R5) or (R6) [at the *OH2 (![[pentagon filled]](https://www.rsc.org/images/entities/char_e13d.gif) ) and *OH (
) and *OH (![[hexagon filled, flat-side down]](https://www.rsc.org/images/entities/char_e039.gif) ) sites, respectively; see Fig. 1B].
) sites, respectively; see Fig. 1B].
In addition to the conventional mechanism M1 (steps (R1)–(R6)), we also considered an alternative mechanism (M2) in which (R1) is split into two steps, a non-electrochemical step followed by an electrochemical step:
| | | *O(OH)H → *OOH + (H+ + e−) | (R1b) |
Fig. 1A shows these steps marked with red arrows. Mechanism M2 therefore would involve seven elementary steps ((R1a), (R1b), and (R2)–(R6)). Another alternative mechanism (M3) bypasses the *O intermediate altogether, whereby (R6) is replaced with the following:
| | | *OH + H2O → *O(OH)H + (H+ + e−) | (R6′) |
(
Fig. 1A, blue arrow) and
(R1) is replaced by
(R1b) as above. Naturally, mechanism M3 occurs only on an active site at which *O is not the resting state. Finally, a lattice-oxygen-assisted mechanism M1
l involves the replacement of steps
(R6) and (R1) by the following steps:
| | | *OH + Ol → *OOl + (H+ + e−) | (R6l) |
| | | *OOl + H2O → *OOH + Ol + (H+ + e−) | (R1l) |
(
Fig. 1A, green arrows). Although there may be more than one plausible mechanism (M1/M1
l–M3) for each active site, we only report the one with the lowest thermodynamic overpotential in the main text for each site. Tables S3–S6 of the ESI
† provide the free energies corresponding to the steps involved in the (alternative) mechanism(s) with higher thermodynamic overpotentials or higher endergonic non-electroactive steps for each of the four active sites considered herein. As seen in
Fig. 1B, we find different OER mechanisms to be operative at the various active sites we considered, supporting the need to systematically examine a variety of reaction pathways.
47
OER mechanisms on Fe-doped β-NiOOH(0001)
We first investigated the OER mechanisms on the *O and *OH sites (labeled ▲ and ■ in Fig. 2 and 3, respectively) on the (0001) facet of β-NiOOH. Moving forward, ▲ and ■ replace the generic designation * to differentiate sites on (0001) when needed. Govind Rajan et al.12 previously studied the OER activity of these sites on undoped β-NiOOH by using the same level of theory employed here and showed that these are the only plausible active sites for the OER on the (0001) facet of β-NiOOH.12 As for the pure surface, the lowest-overpotential OER mechanism starts either from *O (step (R1), Table 1) or *OH (step (R6), Table 2), followed by the formation of *OOH, *O2, lattice O vacancy (*), and *OH2 species, as also considered in previous studies.8,12 Note that, on both the ▲ and ■ sites of the Fe-doped (0001) facet, the formation of O2 first goes through a superoxo (O2−) intermediate, whereas on both active sites of the Fe-doped (100) facet, molecular oxygen forms directly without going through superoxo (vide infra). As described in the Methods section, we calculated the free energies using the ONIOM system-partitioning scheme65 where we combine DFT-HSE06 (α = 15%)38,39 with DFT-PBE+U calculations.40,61Fig. 2 and 3 display the top and side views of the DFT-HSE06 (α = 15%)-relaxed structures on the three-layer slab model of the intermediates involved in the OER mechanism at these two active sites (starting from *O and *OH, respectively).
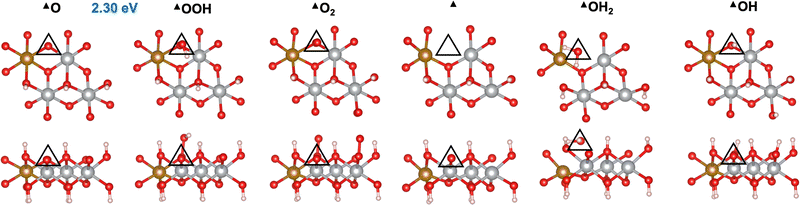 |
| | Fig. 2 Atomic structures of the intermediates involved in the OER mechanism with the lowest overpotential on the Fe-doped *O site (▲) on β-NiOOH(0001) relaxed using DFT-HSE06 (α = 15%). The ONIOM Gibbs free energy change corresponding to the PDS is shown in blue. Top panels: Top-down view. Bottom panels: Side view. The empty triangles mark the location of oxygen-containing species at the active site. We show only the top layer for clarity. Brown sphere: Fe, grey: Ni, red: O, and pink: H. | |
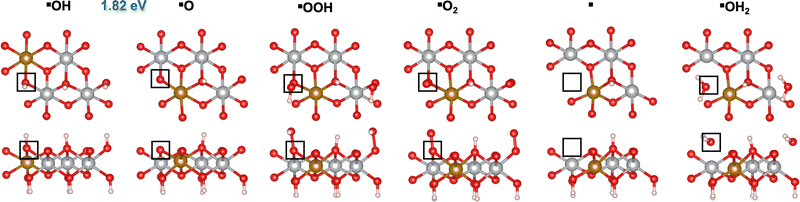 |
| | Fig. 3 Atomic structures of the intermediates involved in the OER mechanism with the lowest overpotential on the Fe-doped *OH site (■) on β-NiOOH(0001) relaxed using DFT-HSE06 (α = 15%). The ONIOM Gibbs free energy change corresponding to the PDS is shown in blue. Top panels: Top-down view. Bottom panels: Side view. The empty squares mark the location of oxygen-containing species at the active site. We show only the top layer for clarity. Brown sphere: Fe, grey: Ni, red: O, and pink: H. | |
Table 1 Reaction Gibbs free energies for the steps comprising the OER mechanism with the lowest overpotential (M1) on the pure and Fe-doped *O site (▲) on β-NiOOH(0001), calculated using ONIOM-DFT-HSE06 (α = 15%). Corresponding atomic structures for the Fe-doped case are in Fig. 2. The PDS is shown in italics. The steps are labeled as discussed above
| Step |
Reaction |
Pure β-NiOOH ΔG0r (eV)a |
Fe-doped β-NiOOH |
| ΔG0r (eV) |
Product o.s.b (Fe,Ni,Ni,Ni) |
|
Re-calculated from the paths explored in ref. 12. The new predicted overpotential on pure β-NiOOH is 0.15 V higher than in the previous work, due to a denser k-mesh used in the present work (see the ESI, Section S1).12
o.s. refers to oxidation state of the Fe cation dopant and three nearby Ni cations, given in the columns (first column being Fe). Up (↑) and down (↓) arrows indicate, respectively, an increase or decrease in the cation (Fe or Ni) oxidation state after the reaction. The charges are balanced in the reactions by the changes in the oxidation state of the intermediates and the cations.
η is the overpotential defined as (max PCET ΔG0r − 1.23 eV)/e.
Note that ▲O2 in (R2) and (R3) is a superoxo species.
|
|
R1
|
▲
O + H
2
O →
▲
OOH + (H
+
+ e
−
)
|
2.37 (η = 1.14 V)
|
2.30 (η = 1.07 V)
|
3
|
2↓
|
3
|
3
|
| R2 |
▲OOH → ▲O2 + (H+ + e−)d |
1.00 |
0.97 |
3 |
2 |
3 |
3 |
| R3 |
▲O2 → O2 + ▲d |
0.16 |
0.21 |
3 |
2 |
2↓ |
3 |
| R4 |
H2O + ▲ → ▲OH2 |
0.18 |
0.00 |
3 |
2 |
2 |
3 |
| R5 |
▲OH2 → ▲OH + (H+ + e−) |
0.33 |
0.23 |
3 |
3↑ |
2 |
3 |
| R6 |
▲OH → ▲O + (H+ + e−) |
0.89 |
1.21 |
3 |
3 |
3↑ |
3 |
Table 2 Reaction Gibbs free energies for the steps comprising the OER mechanism with the lowest overpotential (M1) on the pure and Fe-doped *OH site (■) on β-NiOOH(0001), calculated using ONIOM-DFT-HSE06 (α = 15%). Corresponding atomic structures for the Fe-doped case are in Fig. 3. PDSs are shown in italics. The steps are labeled as discussed above
| Step |
Reaction |
Pure β-NiOOH ΔG0r![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a (eV) a (eV) |
Fe-doped β-NiOOH |
| ΔG0r (eV) |
Product o.s.b (Fe,Ni,Ni,Ni) |
|
Re-calculated from the paths explored in ref. 12. The new predicted overpotential on pure β-NiOOH is 0.07 V higher than in the previous work, due to a denser k-mesh used in the present work (see the ESI, Section S1).12
o.s. refers to oxidation state of the Fe cation dopant and three nearby Ni cations, given in the columns (first column being Fe). Up (↑) and down (↓) arrows indicate, respectively, an increase or decrease in the cation (Fe or Ni) oxidation state after the reaction. The charges are balanced in the reactions by the changes in the oxidation state of the intermediates and the cations.
η is the overpotential defined as (max PCET ΔG0r − 1.23 eV)/e.
Note that ■O2 in (R2) and (R3) is a superoxo species.
|
|
R6
|
■
OH →
■
O + (H
+
+ e
−
)
|
1.74 (η = 0.51 V)
|
1.62 |
3 |
4↑ |
3 |
3 |
|
R1
|
■
O + H
2
O →
■
OOH + (H
+
+ e
−
)
|
1.61 |
1.82 (η = 0.59 V)
|
3
|
3↓
|
3
|
3
|
| R2 |
■OOH → ■O2+ (H+ + e−)d |
1.16 |
1.23 |
3 |
3 |
3 |
3 |
| R3 |
■O2 → O2 + ■d |
−0.03 |
−0.06 |
3 |
2↓ |
3 |
3 |
| R4 |
H2O + ■ → ■OH2 |
0.05 |
−0.08 |
3 |
2 |
3 |
3 |
| R5 |
■OH2 → ■OH + (H+ + e−) |
0.38 |
0.39 |
3 |
3↑ |
3 |
3 |
Further, the standard Gibbs free energies for the various mechanistic steps comprising the OER on the two Fe-doped active sites are compared to the undoped cases in Tables 1 and 2, respectively, at the ONIOM-DFT-HSE06 (α = 15%) level of theory. Therein, we also list the oxidation states of the surface Ni and Fe ions for each intermediate, deduced by comparing to the range of possible net atom-projected electronic magnetic moments of the cations in the various intermediates considered (vide supra), as obtained from DFT-HSE06 (α = 15%) (Table S1, ESI†).28 The corresponding DFT-PBE+U values of the reaction free energies are reported in the ESI,† Tables S7 and S8.
From Table 1, we see that the predicted thermodynamic OER overpotential on the Fe-doped *O site (▲) is 1.07 V, lower than the 1.14 V of the pure compound, although the PDS remains the same in the two cases. The value of 1.14 V differs from the 0.99 V reported in our previous publication12 due to the use of a denser k-point mesh for Fock sampling in the current work (see the ESI,† Section S1). In contrast, we predict the thermodynamic OER overpotential on the Fe-doped *OH (■) site (Table 2) to be 0.59 V, which is higher than the value of 0.51 V on undoped β-NiOOH(0001). In this case, the PDS changed from oxidative deprotonation of *OH (step (R6)) on the undoped material to the conversion of *O to *OOH (step (R1)) on the doped material. Interestingly, Fe doping collapses the PDS to be the same elementary reaction on both sites. Examining the oxidation states of the Ni and the Fe ions (Tables 1 and 2), we see that the Fe ion is redox inactive during the entire OER cycle and maintains an oxidation state of +3 throughout. In contrast, the Ni ion undergoes reduction from Ni3+ to Ni2+ during the PDS on the ▲ site (▲O + H2O → ▲OOH + (H+ + e−)) and from Ni4+ to Ni3+ during the same PDS on the ■ site (■O + H2O → ■OOH + (H+ + e−)). These differences in terms of which oxidation states are changing are likely responsible for the large difference in overpotential despite the PDS being the same. The energy required to reduce Ni3+ to Ni2+ is larger than that required to reduce Ni4+ to Ni3+; therefore, the former reduction increases the cost to oxidize *O to *OOH (a net two-electron oxidation step), as also seen in our previous work on the undoped surface.12 Notably, the PDS on undoped β-NiOOH is the same for the ▲ site as in both Fe-doped cases whereas it is different (■OH → ■O + (H+ + e−)) at the ■ site. In the latter case, the presence of Fe on the surface modulates the energy of *O, stabilizing it, such that although it raises energy requirement for *OOH formation (increasing the free energy requirement to 1.82 eV from only 1.61 eV in the absence of Fe), it lowers the potential requirement for *OH oxidation. The increase in ΔG0r for *OOH formation translates into the increase in the OER overpotential at the ■ site of the (0001) facet of Fe-doped β-NiOOH. Overall, we find that Fe-doped β-NiOOH(0001) exhibits relatively high overpotentials for the OER and therefore is unlikely to be the main active site during water splitting. This is in contrast to our previous work in which we showed that the (0001) facet on undoped β-NiOOH is as active for the OER as other crystallographic facets due to their similar overpotentials.12
OER mechanisms on Fe-doped β-NiOOH(10![[1 with combining macron]](https://www.rsc.org/images/entities/h3_char_0031_0304.gif) 0)
0)
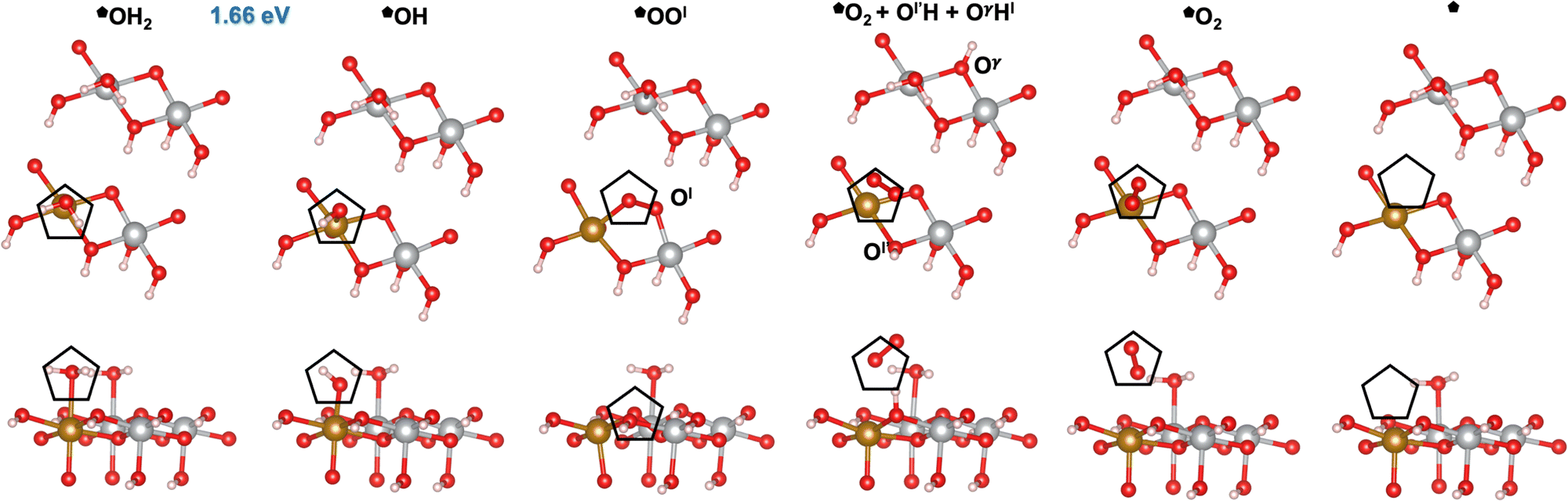
Next, we investigated the OER mechanisms on the *OH2 and the *OH sites (labeled and in Fig. 4 and 5, respectively) on the (100) facet of β-NiOOH. On this facet, *OOH and *O(OH)H intermediates underwent significant structural changes upon geometry optimization. Fig. 4 and 5 display the top and side views of the intermediates involved at these two active sites, relaxed using DFT-HSE06 (α = 15%). Note that, as shown previously by Martirez and Carter, β-NiOOH(100) is covered by half a monolayer of water molecules at equilibrium,41 thus explaining the two water molecules seen on the undoped surface with four cation sites exposed in the slab model. This is unlike the (0001) facet, which is free of any strongly adsorbed water molecules at equilibrium (water binding on (0001) is limited to only hydrogen-bonding with either an O or H due to the absence of undercoordinated cations).41
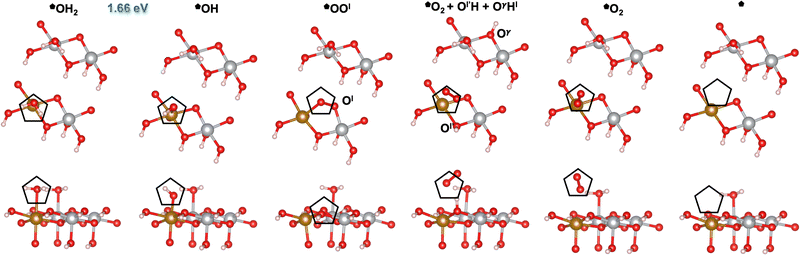 |
| | Fig. 4 Atomic structures of the intermediates involved in the OER mechanism on the Fe-doped hydrated site () with the lowest overpotential on β-NiOOH(100), relaxed using DFT-HSE06 (α = 15%). The intermediate labels are abbreviated from the labels provided in Table 3. The ONIOM Gibbs free energy change corresponding to the PDS is shown in blue. Top panels: Top-down view. Bottom panels: Side view. The empty pentagons mark the location of oxygen-containing species at the active site. Ol and Ol′ denote lattice oxygen atoms in the same layer, whereas Oγ denotes a lattice O in the adjacent layer of β-NiOOH(100). Both Ol′ and Oγ participate in the proton transfer events. We show only the top layer for clarity. Brown sphere: Fe, grey: Ni, red: O, and pink: H. | |
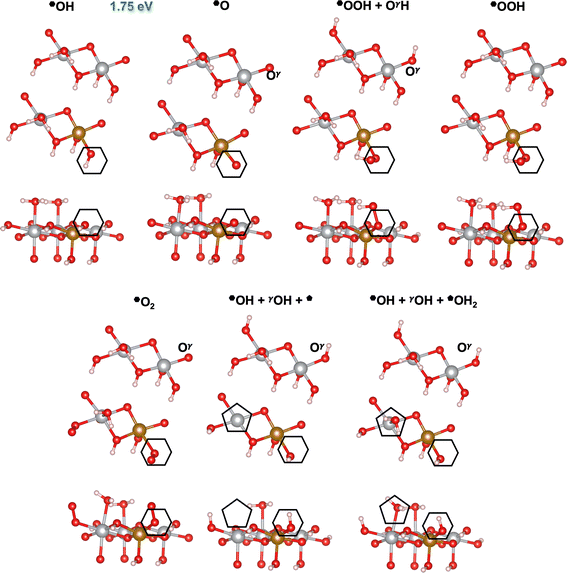 |
| | Fig. 5 Atomic structures of the intermediates involved in the OER mechanism with the lowest overpotential on the Fe-doped hydroxo site () on β-NiOOH(100) relaxed using DFT-HSE06 (α = 15%). The intermediate labels are abbreviated from the labels in Table 4. The ONIOM Gibbs free energy change corresponding to the PDS is shown in blue. Top panels: Top-down view. Bottom panels: Side view. The empty hexagons mark the location of oxygen-containing species at the active site. We show only the top layer for clarity. Brown sphere: Fe, grey: Ni, red: O, and pink: H. | |
Similar to our findings for undoped β-NiOOH(100),12 we find a lattice-oxygen(Ol)-assisted mechanism at the *OH2 site () to have the lowest overpotential on Fe-doped β-NiOOH(100). Therefore, as for the pure surface, here we start from *OH2 to assess the formation of *OH (step (R5), Table 3), which is followed by the formation of *OOl (denoted this way to emphasize that the *O binds to an O from the lattice (l)) instead of a surface *O. In forming OOl, two oxygen-containing species are oxidized to −1 (as in peroxides) with one electron going into the electrode and the other to the adjacent Fe4+ site (step (R6l), Table 3; the l in the superscript of (R6) indicates the involvement of a lattice oxygen in the conversion of *OH to *OOl). Following the formation of *OOl (wherein the O from *OH is bonded to Fe and the Ol from the lattice is bonded to Ni), a series of proton transfers facilitates the further oxidation of *OOl to a molecular *O2 physisorbed side-on to Fe (step (R1l), Table 3). Specifically, step (R1l) involves water attacking *OOl to form *O2, wherein one water proton is lost to solution and the other is captured by a lattice oxygen Ol′ in the same catalyst layer to form Ol′H. Concurrently, the lattice proton previously attached to Ol′, labeled Hl, protonates a lattice oxygen in an adjacent catalyst layer (Oγ) forming OγHl (Fig. 4). Ni3+ is also reduced to Ni2+ during this PCET process. In another PCET, where Ol′H deprotonates but receives back Hl from OγHl, the Ni2+ formed prior oxidizes back to Ni3+ and the very weakly interacting O2 rotates prior to subsequent desorption (step (R2), Table 3). This is followed by two non-electroactive steps, namely, O2 release (step (R3), Table 3) and H2O adsorption (step (R4), Table 3). Note again that a lattice O atom (Ol′ or Oγ) also protonates or deprotonates in some steps.
Table 3 Reaction Gibbs free energies for the steps comprising the OER mechanism with the lowest overpotential (M1l) on the pure and Fe-doped *OH2 site () on β-NiOOH(100), calculated using ONIOM-DFT-HSE06 (α = 15%). The superscript l denotes the involvement of a lattice oxygen in mechanism M1. Corresponding atomic structures for the Fe-doped case are in Fig. 4, shown there with abbreviated labels. PDSs are shown in italics. The steps are labeled as discussed above
| Step |
Reactiona |
Pure β-NiOOH ΔG0rb (eV) |
Fe-doped β-NiOOH |
| ΔG0r (eV) |
Product o.s.c (Fe,Ni,Ni,Ni) |
|
Ol and Ol′ denote lattice oxygen atoms in the same layer of β-NiOOH(100), whereas Oγ denotes a lattice O in the adjacent layer. Hl denotes a lattice proton, whether in the same or adjacent catalyst layer. Both Ol′ and Oγ participate in the proton transfer events.
Re-calculated from the paths explored in ref. 12. The new predicted overpotential on pure β-NiOOH is 0.01 V lower than in the previous work, due to a denser k-mesh used in the present work (see the ESI, Section S1).12
o.s. refers to oxidation state of the Fe cation dopant and three nearby Ni cations, given in the columns (first column being Fe). Up (↑) and down (↓) arrows indicate, respectively, an increase or decrease in the ion oxidation state as compared to the previous step. The charges are balanced in the reactions by the changes in the oxidation state of the intermediates and the cations.
η is the overpotential defined as (max PCET ΔG0r − 1.23 eV)/e.
Note that O2 in (R1l), (R2), and (R3) is a molecular oxygen species.
|
|
R5
|
OH
2
→
OH + (H
+
+ e
−
)
|
1.65 (η = 0.42 V)
|
1.60 |
4↑ |
3 |
3 |
3 |
|
R6
l
|
OH + O
l
→
OO
l
+ (H
+
+ e
−
)
|
1.36 |
1.66 (η = 0.43 V)
|
3↓
|
3
|
3
|
3
|
| R1l |
H2O + OOl + Ol′Hl + Oγ → O2 + Ol + Ol′H + OγHl + (H+ + e−) |
0.57 |
0.21 |
3 |
2↓ |
3 |
3 |
| R2 |
O2 + Ol′H + OγHl → O2 + Ol′Hl + Oγ + (H+ + e−)e |
1.32 |
1.44 |
3 |
3↑ |
3 |
3 |
| R3 |
O2 → O2 + e |
−0.14 |
−0.10 |
3 |
3 |
3 |
3 |
| R4 |
H2O + → OH2 |
0.17 |
0.11 |
3 |
3 |
3 |
3 |
As for the *OH site () on Fe-doped on β-NiOOH(100), starting from *OH (R6), the intermediates involved in the most favorable (lowest overpotential) mechanism are: *O; *OH; *OOH plus a protonated lattice oxygen in an adjacent layer (*OOH + OγH), which arises spontaneously from geometry optimization of *H2O2 (note this structural change leads to differences in steps (R1a), (R1b), and (R6′), see Table 4); *OOH + Oγ (formed when OγH loses its proton to solution), *O2, and a free (five-fold-coordinated) cation site (*), which can adsorb H2O (*H2O). The mechanisms are different at undoped versus Fe-doped sites (Table 4), unlike for the site where only the PDS changes but the mechanisms are identical (vide supra). On the undoped active site, the OER proceeds via M3 whereas at the Fe-doped active site, M2 is the most preferred mechanism. However, as for the site, lattice O atoms also participate in proton transfer at the site.
Table 4 Reaction Gibbs free energies for the steps comprising the OER mechanisms with the lowest overpotentials on the pure (M3) and Fe-doped (M2) *OH site () on β-NiOOH(100), calculated using ONIOM-DFT-HSE06 (α = 15%). Corresponding atomic structures for the Fe-doped case are in Fig. 5, shown there with abbreviated labels. The PDS is shown in italics. The steps are labeled as discussed above
| Step |
Pure β-NiOOH |
Step |
Fe-doped β-NiOOH |
| Reactiona |
ΔG0rb (eV) |
Reactiona |
ΔG0r (eV) |
Product o.s.c (Fe,Ni,Ni,Ni) |
|
Oγ denotes a lattice O in the adjacent layer, which participates in the proton transfer events.
Re-calculated from the paths explored in ref. 12. The new predicted overpotential on pure β-NiOOH is 0.13 V higher than in the previous work, due to a denser k-mesh used in the current work (see the ESI, Section S1).12
o.s. refers to oxidation state of the Fe cation dopant and three nearby Ni cations, given in the columns (first column being Fe). Up (↑) and down (↓) arrows indicate, respectively, an increase or decrease in the ion oxidation state as compared to the previous step. The charges are balanced in the reactions by the changes in the oxidation state of the intermediates and the cations.
Note that steps (R6′), (R1a), and (R1b) shown here differ from what is mentioned in the section “Reaction intermediates and plausible mechanisms involved in the OER”. This is because O(OH)H relaxes upon geometry optimization to OOH plus a protonated lattice oxygen in the adjacent catalyst layer, i.e., OOH + OγH.
η is the overpotential defined as (max PCET ΔG0r − 1.23 eV)/e.
Note that O2 in (R2) and (R3) is a molecular oxygen species on the Fe-doped active site but is a superoxo species at the undoped active site.
|
|
R6′d |
OH + H
2
O + O
γ
→
OOH + O
γ
H + (H
+
+ e
−
)
|
2.09 (η = 0.86 V)
|
R6
|
OH →
O + (H
+
+ e
−
)
|
1.75 (η = 0.52 V)
|
4
|
3↑
|
3
|
3
|
| R1b |
OOH + OγH → OOH + Oγ + (H+ + e−) |
1.81 |
R1ad |
O + H2O + Oγ → OOH + OγH |
0.30 |
4 |
2↓ |
2↓ |
3 |
| R2 |
OOH → O2 + (H+ + e−)f |
0.75 |
R1bd |
OOH + OγH → OOH + Oγ + (H+ + e−) |
1.67 |
4 |
2 |
3↑ |
3 |
| R3 |
O2 → O2 + f |
−0.11 |
R2 |
OOH → O2 + (H+ + e−)f |
0.86 |
3↓ |
2 |
3 |
3 |
| R4 |
H2O + + Oγ → OH + OγH |
−0.75 |
R3 |
O2 + OH2 + Oγ → O2 + OH + OγH + f |
−1.21 |
3 |
2 |
3 |
3 |
| R5 |
OH + OγH → OH + Oγ + (H+ + e−) |
1.14 |
R4 |
H2O + → OH2 |
0.24 |
4↑ |
2 |
2↓ |
3 |
|
|
|
|
R5 |
OH + OγH → OH + Oγ + (H+ + e−) |
1.32 |
4 |
2 |
3↑ |
3 |
On the site of β-NiOOH(100), the undoped catalyst forms *OOH directly from *OH and its reaction with water via PCET (where the former deprotonates via PCET while the latter binds to the resulting *O and transfers a proton to a lattice Oγ; step (R6′), Table 4).12 In contrast, on the Fe-doped catalyst, the lattice *OH simply undergoes deprotonation via PCET to produce *O in its first step (step (R6), Table 4). The tendency of Fe4+ to stably support *O and the inability of Ni4+ to do so (preferring to directly form a peroxo-like species) was found earlier on the (21) facet.28 Indeed, for the doped case, *OOH forms instead after a non-electroactive step in which the resulting *O reacts with water, where the latter loses a proton to a lattice Oγ and concurrently two Ni3+ ions are reduced to Ni2+ (step (R1a), Table 4). This step is followed by the deprotonation of OγH and the oxidation of one of the Ni2+ sites back to Ni3+ (step (R1b), Table 4). Subsequently, adsorbed molecular oxygen forms (step (R2), Table 4), which then desorbs in a highly exergonic step to produce an empty site (step (R3), Table 4). Along with O2 desorption, OH2 heterolytically dissociates with the OH migrating to the site while the proton transfers to Oγ (also part of step (R3), Table 4). The final steps involve the adsorption of another water molecule at the site and another deprotonation of OγH (steps (R4) and (R5), Table 4). Of note, step (R4), although non-electroactive overall, involves charge transfer between cations, wherein Fe3+ oxidizes to Fe4+, as in the initial state of the electrocatalyst, while one of the Ni3+ ions reduces to Ni2+.
We predict the PDSs on the Fe-doped β-NiOOH(100) surface to be OH + Ol → OOl + (H+ + e−) on the site, with an overpotential of 0.43 V (Fig. 4 and Table 3), and OH → O + (H+ + e−) on the site, with an overpotential of 0.52 V (Fig. 5 and Table 4). Both PDSs involve deprotonation of *OH, however a cation is reduced (Fe4+ to Fe3+) at the former site whereas a cation is oxidized (Ni2+ to Ni3+) in the latter. The standard Gibbs free energies for the various mechanistic steps comprising the OER on the and active sites are listed in Tables 3 and 4, respectively, calculated at the ONIOM-DFT-HSE06 (α = 15%) level of theory. As above, we assigned the ionic oxidation states for each intermediate by comparing to the atom-projected magnetic moments of the cations in the various intermediates considered (vide supra) from DFT-HSE06 (α = 15%) (Table S2, ESI†). The corresponding DFT-PBE+U values for the Gibbs free energies are reported in Tables S9 and S10 in the ESI.†
Interestingly, the OER overpotential on the Fe-doped site of 0.43 V is nearly identical to the value (0.42 V) on the same facet and site on undoped β-NiOOH, despite the change in PDS (Table 3). Thus, the activity of the site on β-NiOOH(100) is quantitatively unaffected by Fe doping. This overpotential insensitivity to Fe doping for this site exists even though the Fe ion is redox active during the OER cycle on the (100) facet, undergoing a reduction from Fe4+ to Fe3+ during the PDS at the site, unlike on the (0001) facet where it does not undergo any oxidation-state changes during the OER mechanism (vide supra). Thus, the presence of redox activity in the dopant ion does not necessarily lead to a reduction in the overall OER overpotential of the catalyst, although it can alter the nature of the PDS. In contrast, the OER overpotential on the site drops significantly from 0.86 V to 0.52 V upon Fe doping, along with a change in the nature of the PDS (Table 4). Here as well, the Fe ion is redox active during the OER cycle and undergoes reduction from Fe4+ to Fe3+ and back. However, it is the Ni ion that changes oxidation state during the PDS, oxidizing from Ni2+ to Ni3+. Considering our prior predictions for OER on the (21) facet,28 it is evident that the effectiveness of the Fe dopant in modulating the OER activity depends on multiple factors, including the active site, the crystallographic facet, and on the mechanism, i.e., the chemistry involved in each elementary step, which in turn determines the exact metal cation oxidation-state changes.
Inactivity of the (0001) and (100) facets for the OER on Fe-doped β-NiOOH
Based on the above findings, we infer that the lowest thermodynamic OER overpotentials on the (0001) and the (100) facet of Fe-doped β-NiOOH are 0.59 V and 0.43 V, respectively. These values are significantly higher than the 0.14 V predicted for the (21) facet of Fe-doped β-NiOOH.28 We thus conclude that the OER activity of Fe-doped β-NiOOH arises primarily from the (21) facet, despite its smaller area fraction in a polycrystalline catalyst, with the more area-fraction-dominant (0001) and the (100) facets less active during the OER (although they may contribute to the activity at higher applied overpotentials). This facet sensitivity is unlike what happens on undoped β-NiOOH, wherein these three facets have similar thermodynamic overpotentials, leading to a roughly facet-independent activity for the material (with η(0001) = 0.51 V and η(100) = 0.42 V from this work, and η(21) = 0.48 V from ref. 28), as concluded in ref. 12. Our results also indicate that the six-fold-lattice-coordinated Fe in β-NiOOH (on the (0001) facet) is redox inactive during the OER cycle, whereas the five-fold-lattice-coordinated Fe (on the (100) facet) and four-fold-lattice-coordinated Fe (on the (21) facet) are redox active during water splitting. Nevertheless, as evidenced by the hydrated site on the (100) facet, the redox activity of Fe is a necessary but not sufficient condition for a reduction in the overall OER overpotential on Fe-doped β-NiOOH, although it may influence the nature of the PDS. Overall, these findings further motivate28 the development of strategies that can selectively produce four-fold-lattice-coordinated Fe ions on the β-(Fe,Ni)OOH surface, i.e., synthesis of highly faceted crystals. For example, recent experimental work has explored vertically grown70 and reconstructed71 β-(Fe,Ni)OOH to enhance the OER activity on the material.
Discussion of our simulation results in the context of experiments
Our conclusion that sites with low lattice coordination number are required for OER activity is consistent with prior electrochemical OER analysis with various surface probe techniques by Hunter et al. (non-aqueous anodic polarization with infrared spectroscopy),49 Hao et al. (Raman spectroscopy with molecular probes),50 Jia et al. (extended X-ray absorption fine structure, X-ray photoelectron spectroscopy, and X-ray absorption spectroscopy),51 and Farhat et al. (controlled Fe deposition with scanning electron microscopy imaging and energy-dispersive X-ray spectroscopy)52 involving either the β or γ phase of NiOOH, to name a few. Specifically, in the study by Hunter et al., the authors experimentally confirmed the presence of a cis-dioxo-Fe(VI), i.e., Fe(VI)(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)2, intermediate (presumed via further oxidation of Fe(IV)O) by applying oxidizing potentials in the absence of water (using a nonaqueous solvent to suppress O2 evolution and stabilize oxidized intermediates). The same authors suggested that Fe(IV)O can only form at edge and corner sites (lattice coordination number less than 6) and cis-dioxo-Fe(VI) can only form at corner sites where Fe lattice O coordination is much lower.49 Although it is not possible to form cis-dioxo-Fe(VI) on the (100) facet where the Ni/Fe is five-fold-lattice-coordinated, it has been shown to form on the Fe-doped (21) facet in previous work from our group, albeit at much higher potentials and therefore not competitive against the O2 evolution pathway via Fe(IV)O (consistent with experiments showing that the cis-dioxo species forms when O2 release is eliminated and the electrode is subjected to high overpotentials).28 Moreover, the lowest overpotential OER mechanism on the Fe-doped (21) facet28 and the OER mechanism on the *OH () site of the Fe-doped (100) facet involve the Fe(IV)O intermediate, also seen experimentally by Hunter et al.49 at edge/corner sites, thus indicating the crucial role played by coordinatively unsaturated Fe in the OER activity of Fe-doped β-NiOOH. Likewise, Hao et al. identified an FeO intermediate as well as the participation of a bridging lattice O in the formation of the O2 species during the OER mechanism,50 which are involved in the most favorable OER mechanism on Fe-doped β-NiOOH(21) predicted by Martirez and Carter.28 Finally, both Jia et al. and Farhat et al. demonstrated that abundant undercoordinated Fe sites lead to higher OER current densities, with the latter concluding that Fe in solution is essential to replenish undercoordinated Fe and thus maintain activity, once again supporting the role of undercoordinated metal sites in the OER mechanism.51,52
O)2, intermediate (presumed via further oxidation of Fe(IV)O) by applying oxidizing potentials in the absence of water (using a nonaqueous solvent to suppress O2 evolution and stabilize oxidized intermediates). The same authors suggested that Fe(IV)O can only form at edge and corner sites (lattice coordination number less than 6) and cis-dioxo-Fe(VI) can only form at corner sites where Fe lattice O coordination is much lower.49 Although it is not possible to form cis-dioxo-Fe(VI) on the (100) facet where the Ni/Fe is five-fold-lattice-coordinated, it has been shown to form on the Fe-doped (21) facet in previous work from our group, albeit at much higher potentials and therefore not competitive against the O2 evolution pathway via Fe(IV)O (consistent with experiments showing that the cis-dioxo species forms when O2 release is eliminated and the electrode is subjected to high overpotentials).28 Moreover, the lowest overpotential OER mechanism on the Fe-doped (21) facet28 and the OER mechanism on the *OH () site of the Fe-doped (100) facet involve the Fe(IV)O intermediate, also seen experimentally by Hunter et al.49 at edge/corner sites, thus indicating the crucial role played by coordinatively unsaturated Fe in the OER activity of Fe-doped β-NiOOH. Likewise, Hao et al. identified an FeO intermediate as well as the participation of a bridging lattice O in the formation of the O2 species during the OER mechanism,50 which are involved in the most favorable OER mechanism on Fe-doped β-NiOOH(21) predicted by Martirez and Carter.28 Finally, both Jia et al. and Farhat et al. demonstrated that abundant undercoordinated Fe sites lead to higher OER current densities, with the latter concluding that Fe in solution is essential to replenish undercoordinated Fe and thus maintain activity, once again supporting the role of undercoordinated metal sites in the OER mechanism.51,52
Conclusions
In this work, we assessed the thermodynamic feasibility of multiple OER mechanisms on the Fe-doped (0001) and (100) facets of β-NiOOH, a promising alkaline-medium water-oxidation electrocatalyst. The assessment utilized a high level of electronic-structure theory, namely, hybrid DFT based on the HSE06 XC functional, as a correction to Hubbard-corrected DFT calculations, via the ONIOM system-partitioning scheme. Unlike undoped β-NiOOH for which multiple facets exhibit similar overpotentials, we established that Fe-doped β-NiOOH demonstrates facet-dependent OER activity, with much higher overpotentials on the majority facets than on a previously proposed minority facet possessing highly undercoordinated metal cations that exhibits a record-low overpotential. We systematically examined four different associative OER mechanisms, including the conventional mechanism via the *O, *OOH, *O2, *, *OH2, and *OH intermediates (M1), two alternative mechanisms involving adsorbed hydrogen peroxide (*O(OH)H) as an intermediate (M2 and M3), and one assisted by a lattice oxygen (M1l). We found that different mechanisms operate at the various active sites considered, indicating the need for a careful examination of potentially competing OER pathways on candidate catalyst materials, as Martirez and Carter did for the (21) facet of pure and Fe-doped β-NiOOH,50 we did for the (0001) and (100) facets of pure β-NiOOH,12 and here for both the (0001) and (100) facets of Fe-doped β-NiOOH.
Through our analysis, we found that Fe dopant cations exhibit three distinct behaviors in the host β-NiOOH lattice: (i) remaining redox inactive and simply modifying the OER free energetics as a spectator (at the *O (▲) and *OH (■) active sites on the (0001) facet); (ii) exhibiting redox activity during the OER mechanism and the active site for the PDS (at the *OH2 () site on the (100) facet); or (iii) bringing about changes in the oxidation states of Ni ions in the host β-NiOOH lattice during the PDS (at the *OH () site on the (100) facet). Fe remains redox inactive when it has a coordination number of six in the β-NiOOH lattice, e.g., on the (0001) facet, but demonstrates redox activity when it is five-fold lattice-coordinated (e.g., on the (100) facet), as observed in this work, or when it is four-fold lattice-coordinated (e.g., on the (21) facet), as shown by Martirez and Carter.28
Although it appears that no link exists between redox participation of Fe and the OER overpotential on Fe-doped β-NiOOH on the two facets studied here, we found that Fe can change the PDS on both the (0001) and (100) facets even though the preferred pathway initiated by deprotonation remains the same for the doped and pure case, except for the *OH (■) site on the (100) facet. This finding contrasts with the (21) facet, wherein both the preferred pathway and OER overpotential significantly change. Note that on β-NiOOH(0001) the thermodynamic overpotential decreases (by 0.07 V) on the *O site (▲), whereas it increases (by 0.08 V) on the *OH site (■) with Fe doping. On the other hand, on β-NiOOH(100), Fe doping only marginally impacts the overpotential on the *OH2 site () (by 0.01 V), whereas it reduces significantly (by 0.34 V) on the *OH site (). These findings indicate that the effect of Fe doping on the PDS and the overpotential on various active sites of β-NiOOH is a complex function of the site chemistry and the local coordination environment. Overall, we predict relatively high overpotentials of 0.59 V and 0.43 V for the OER on the (0001) and (100) facets, respectively, as compared to the predicted value of 0.14 V on the (21) facet of Fe-doped β-NiOOH. This observation reveals a strongly facet-dependent OER activity on Fe-doped β-NiOOH in contrast to the facet-independent OER activity seen on undoped β-NiOOH. In summary, using hybrid DFT calculations of higher accuracy, our work conclusively rules out the possibility of the OER primarily occurring on the majority facets of Fe-doped β-NiOOH and motivates the discovery of synthesis strategies for ensuring crystallographic facets where Fe ions can possess lower coordination numbers on β-NiOOH.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
EAC acknowledges financial support from the Air Force Office of Scientific Research under AFOSR Award No. FA9550-14-1-0254. AGR acknowledges financial support from the National Supercomputing Mission (DST/NSM/R&D_HPC_Applications/2021/07), which is coordinated by the Department of Science and Technology (DST) and the Ministry of Electronics and Information Technology (MeitY). Additionally, AGR thanks the Infosys Foundation, Bengaluru, for an Infosys Young Investigator grant. The research described in this paper was also supported in part under the Laboratory Directed Research and Development (LDRD) Program at Princeton Plasma Physics Laboratory, a national laboratory operated by Princeton University for the U.S. Department of Energy under Prime Contract No. DE-AC02-09CH11466. The authors acknowledge supercomputing support from Princeton University's Terascale Infrastructure for Groundbreaking Research in Engineering and Science (TIGRESS) and the Supercomputer Education and Research Centre (SERC) at the Indian Institute of Science.
References
- N.-T. Suen, S.-F. Hung, Q. Quan, N. Zhang, Y.-J. Xu and H. M. Chen, Electrocatalysis for the Oxygen Evolution Reaction: Recent Development and Future Perspectives, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- A. Govind Rajan, J. M. P. Martirez and E. A. Carter, Why Do We Use the Materials and Operating Conditions We Use for Heterogeneous (Photo)Electrochemical Water Splitting?, ACS Catal., 2020, 10, 11177–11234 CrossRef CAS.
- M. Gong and H. Dai, A Mini Review of NiFe-Based Materials as Highly Active Oxygen Evolution Reaction Electrocatalysts, Nano Res., 2015, 8, 23–39 CrossRef CAS.
- A. K. Verma, A. M. Verma and A. Govind Rajan, Theoretical Understanding of Electrochemical Phenomena in 2D Electrode Materials, Curr. Opin. Electrochem., 2022, 36, 101116 CrossRef CAS.
- A. Jain, Z. Wang and J. K. Nørskov, Stable Two-Dimensional Materials for Oxygen Reduction and Oxygen Evolution Reactions, ACS Energy Lett., 2019, 4, 1410–1411 CrossRef CAS.
- C. Luan, G. Liu, Y. Liu, L. Yu, Y. Wang, Y. Xiao, H. Qiao, X. Dai and X. Zhang, Structure Effects of 2D Materials on α-Nickel Hydroxide for Oxygen Evolution Reaction, ACS Nano, 2018, 12, 3875–3885 CrossRef CAS PubMed.
- Y.-F. Li and A. Selloni, Mechanism and Activity of Water Oxidation on Selected Surfaces of Pure and Fe-Doped NiOX, ACS Catal., 2014, 4, 1148–1153 CrossRef CAS.
- A. J. Tkalych, H. L. Zhuang and E. A. Carter, A Density Functional +U Assessment of Oxygen Evolution Reaction Mechanisms on β-NiOOH, ACS Catal., 2017, 7, 5329–5339 CrossRef CAS.
- A. J. Tkalych, H. L. Zhuang and E. A. Carter, Correction to “A Density Functional +U Assessment of Oxygen Evolution Reaction Mechanisms on β-NiOOH”, ACS Catal., 2018, 8, 6070 CrossRef CAS.
- A. D. Doyle, M. Bajdich and A. Vojvodic, Theoretical Insights to Bulk Activity Towards Oxygen Evolution in Oxyhydroxides, Catal. Lett., 2017, 147, 1533–1539 CrossRef.
- H. Xiao, H. Shin and W. A. Goddard, Synergy between Fe and Ni in the Optimal Performance of (Ni,Fe)OOH Catalysts for the Oxygen Evolution Reaction, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 5872–5877 CrossRef CAS PubMed.
- A. Govind Rajan, J. M. P. Martirez and E. A. Carter, Facet-Independent Oxygen Evolution Activity of Pure β-NiOOH: Different Chemistries Leading to Similar Overpotentials, J. Am. Chem. Soc., 2020, 142, 3600–3612 CrossRef CAS PubMed.
- A. Govind Rajan, J. M. P. Martirez and E. A. Carter, Coupled Effects of Temperature, Pressure, and pH on Water Oxidation Thermodynamics and Kinetics, ACS Catal., 2021, 11, 11305–11319 CrossRef CAS.
- Y.-F. Li, J.-L. Li and Z.-P. Liu, Structure and Catalysis of NiOOH: Recent Advances on Atomic Simulation, J. Phys. Chem. C, 2021, 125, 27033–27045 CrossRef CAS.
- L. Trotochaud, J. K. Ranney, K. N. Williams and S. W. Boettcher, Solution-Cast Metal Oxide Thin Film Electrocatalysts for Oxygen Evolution, J. Am. Chem. Soc., 2012, 134, 17253–17261 CrossRef CAS PubMed.
- I. J. Godwin and M. E. G. Lyons, Enhanced Oxygen Evolution at Hydrous Nickel Oxide Electrodes via Electrochemical Ageing in Alkaline Solution, Electrochem. Commun., 2013, 32, 39–42 CrossRef CAS.
- R. L. Doyle, I. J. Godwin, M. P. Brandon and M. E. G. Lyons, Redox and Electrochemical Water Splitting Catalytic Properties of Hydrated Metal Oxide Modified Electrodes, Phys. Chem. Chem. Phys., 2013, 15, 13737 RSC.
- D. Kang, T. W. Kim, S. R. Kubota, A. C. Cardiel, H. G. Cha and K.-S. Choi, Electrochemical Synthesis of Photoelectrodes and Catalysts for Use in Solar Water Splitting, Chem. Rev., 2015, 115, 12839–12887 CrossRef CAS PubMed.
- M. S. Burke, L. J. Enman, A. S. Batchellor, S. Zou and S. W. Boettcher, Oxygen Evolution Reaction Electrocatalysis on Transition Metal Oxides and (Oxy)Hydroxides: Activity Trends and Design Principles, Chem. Mater., 2015, 27, 7549–7558 CrossRef CAS.
- O. Diaz-Morales, D. Ferrus-Suspedra and M. T. M. Koper, The Importance of Nickel Oxyhydroxide Deprotonation on Its Activity towards Electrochemical Water Oxidation, Chem. Sci., 2016, 7, 2639–2645 RSC.
- A. C. Garcia, T. Touzalin, C. Nieuwland, N. Perini and M. T. M. Koper, Enhancement of Oxygen Evolution Activity of Nickel Oxyhydroxide by Electrolyte Alkali Cations, Angew. Chem., Int. Ed., 2019, 58, 12999–13003 CrossRef CAS PubMed.
- L. Francàs, S. Corby, S. Selim, D. Lee, C. A. Mesa, R. Godin, E. Pastor, I. E. L. Stephens, K.-S. Choi and J. R. Durrant, Spectroelectrochemical Study of Water Oxidation on Nickel and Iron Oxyhydroxide Electrocatalysts, Nat. Commun., 2019, 10, 5208 CrossRef PubMed.
- M. Han, C. Wang, J. Zhong, J. Han, N. Wang, A. Seifitokaldani, Y. Yu, Y. Liu, X. Sun and A. Vomiero,
et al., Promoted Self-Construction of β-NiOOH in Amorphous High Entropy Electrocatalysts for the Oxygen Evolution Reaction, Appl. Catal., B, 2022, 301, 120764 CrossRef CAS.
- Y. Ou, L. P. Twight, B. Samanta, L. Liu, S. Biswas, J. L. Fehrs, N. A. Sagui, J. Villalobos, J. Morales-Santelices and D. Antipin,
et al., Cooperative Fe Sites on Transition Metal (Oxy)Hydroxides Drive High Oxygen Evolution Activity in Base, Nat. Commun., 2023, 14, 7688 CrossRef CAS PubMed.
- M. W. Louie and A. T. Bell, An Investigation of Thin-Film Ni–Fe Oxide Catalysts for the Electrochemical Evolution of Oxygen, J. Am. Chem. Soc., 2013, 135, 12329–12337 CrossRef CAS PubMed.
- L. Trotochaud, S. L. Young, J. K. Ranney and S. W. Boettcher, Nickel–Iron Oxyhydroxide Oxygen-Evolution Electrocatalysts: The Role of Intentional and Incidental Iron Incorporation, J. Am. Chem. Soc., 2014, 136, 6744–6753 CrossRef CAS PubMed.
- S. Klaus, Y. Cai, M. W. Louie, L. Trotochaud and A. T. Bell, Effects of Fe Electrolyte Impurities on Ni(OH)2/NiOOH Structure and Oxygen Evolution Activity, J. Phys. Chem. C, 2015, 119, 7243–7254 CrossRef CAS.
- J. M. P. Martirez and E. A. Carter, Unraveling Oxygen Evolution on Iron-Doped β-Nickel Oxyhydroxide: The Key Role of Highly Active Molecular-like Sites, J. Am. Chem. Soc., 2019, 141, 693–705 CrossRef CAS PubMed.
- V. Fidelsky and M. C. Toroker, The Secret behind the Success of Doping Nickel Oxyhydroxide with Iron, Phys. Chem. Chem. Phys., 2017, 19, 7491–7497 RSC.
- M. Kumar, S. Piccinin and V. Srinivasan, Direct and Indirect Role of Fe Doping in NiOOH Monolayer for Water Oxidation Catalysis, Chem. Phys. Chem., 2022, 23 Search PubMed.
- J. M. P. Martirez and E. A. Carter, Noninnocent Influence of Host β-NiOOH Redox Activity on Transition-Metal Dopants’ Efficacy as Active Sites in Electrocatalytic Water Oxidation, ACS Catal., 2020, 10, 2720–2734 CrossRef CAS.
- J. M. P. Martirez and E. A. Carter, Secondary Transition-Metal Dopants for Enhanced Electrochemical O2 Formation and Desorption on Fe-Doped β-NiOOH, ACS Energy Lett., 2020, 5, 962–967 CrossRef CAS.
- W. Wang, B. Jiang, W. Xiong, H. Sun, Z. Lin, L. Hu, J. Tu, J. Hou, H. Zhu and S. Jiao, A New Cathode Material for Super-Valent Battery Based on Aluminium Ion Intercalation and Deintercalation, Sci. Rep., 2013, 3, 3383 CrossRef PubMed.
- H. Shin, H. Xiao and W. A. Goddard, In Silico Discovery of New Dopants for Fe-Doped Ni Oxyhydroxide (Ni1−xFexOOH) Catalysts for Oxygen Evolution Reaction, J. Am. Chem. Soc., 2018, 140, 6745–6748 CrossRef CAS PubMed.
- V. Fidelsky, V. Butera, J. Zaffran and M. C. Toroker, Three Fundamental Questions on One of Our Best Water Oxidation Catalysts: A Critical Perspective, Theor. Chem. Acc., 2016, 135, 162 Search PubMed.
- L.-F. Li, Y.-F. Li and Z.-P. Liu, Oxygen Evolution Activity on NiOOH Catalysts: Four-Coordinated
Ni Cation as the Active Site and the Hydroperoxide Mechanism, ACS Catal., 2020, 10, 2581–2590 Search PubMed.
- Y. Zhou and N. López, The Role of Fe Species on NiOOH in Oxygen Evolution Reactions, ACS Catal., 2020, 10, 6254–6261 Search PubMed.
- A. V. Krukau, O. A. Vydrov, A. F. Izmaylov and G. E. Scuseria, Influence of the Exchange Screening Parameter on the Performance of Screened Hybrid Functionals, J. Chem. Phys., 2006, 125, 224106 CrossRef PubMed.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, Hybrid Functionals Based on a Screened Coulomb Potential, J. Chem. Phys., 2003, 118, 8207–8215 CrossRef CAS.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton, Electron-Energy-Loss Spectra and the Structural Stability of Nickel Oxide: An LSDA+U Study, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505–1509 CrossRef CAS.
- J. M. P. Martirez and E. A. Carter, Effects of the Aqueous Environment on the Stability and Chemistry of β-NiOOH Surfaces, Chem. Mater., 2018, 30, 5205–5219 CrossRef CAS.
- F. Flevet and M. Flglarz, Preparation and Study by Electron Microscopy of the Development of Texture with Temperature of a Porous Exhydroxide Nickel Oxide, J. Catal., 1975, 39, 350–356 CrossRef.
- L. Gourrier, S. Deabate, T. Michel, M. Paillet, P. Hermet, J.-L. Bantignies and F. Henn, Characterization of Unusually Large “Pseudo-Single Crystal” of β-Nickel Hydroxide, J. Phys. Chem. C, 2011, 115, 15067–15074 CrossRef CAS.
- A. Govind Rajan and E. A. Carter, Microkinetic Model for pH- and Potential-Dependent Oxygen Evolution during Water Splitting on Fe-Doped β-NiOOH, Energy Environ. Sci., 2020, 13, 4962–4976 RSC.
- C. Hareli and M. Caspary Toroker, Water Oxidation Catalysis for NiOOH by a Metropolis Monte Carlo Algorithm, J. Chem. Theory Comput., 2018, 14, 2380–2385 CrossRef PubMed.
- J. Huang, M. Li, M. J. Eslamibidgoli, M. Eikerling and A. Groß, Cation Overcrowding Effect on the Oxygen Evolution Reaction, JACS Au, 2021, 1, 1752–1765 CrossRef CAS PubMed.
- A. Govind Rajan and E. A. Carter, Discovering Competing Electrocatalytic Mechanisms and Their Overpotentials: Automated Enumeration of Oxygen Evolution Pathways, J. Phys. Chem. C, 2020, 124, 24883–24898 CrossRef.
- A. J. Tkalych, J. M. P. Martirez and E. A. Carter, Effect of Transition-Metal-Ion Dopants on the Oxygen Evolution Reaction on NiOOH(0001), Phys. Chem. Chem. Phys., 2018, 20, 19525–19531 RSC.
- B. M. Hunter, N. B. Thompson, A. M. Müller, G. R. Rossman, M. G. Hill, J. R. Winkler and H. B. Gray, Trapping an Iron(VI) Water-Splitting Intermediate in Nonaqueous Media, Joule, 2018, 2, 747–763 CrossRef CAS.
- Y. Hao, Y. Li, J. Wu, L. Meng, J. Wang, C. Jia, T. Liu, X. Yang, Z.-P. Liu and M. Gong, Recognition of Surface Oxygen Intermediates on NiFe Oxyhydroxide Oxygen-Evolving Catalysts by Homogeneous Oxidation Reactivity, J. Am. Chem. Soc., 2021, 143, 1493–1502 CrossRef CAS PubMed.
- C. Jia, C. Zhen, L. Yin, H. Zhu, P. Du, A. Han, G. Liu and H.-M. Cheng, Topologic Transition-Induced Abundant Undercoordinated Fe Active Sites in NiFeOOH for Superior Oxygen Evolution, Nano Energy, 2023, 106, 108044 CrossRef CAS.
- R. Farhat, J. Dhainy and L. I. Halaoui, OER Catalysis at Activated and Codeposited NiFe-Oxo/Hydroxide Thin Films Is Due to Postdeposition Surface-Fe and Is Not Sustainable without Fe in Solution, ACS Catal., 2020, 10, 20–35 CrossRef CAS.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- J. Rossmeisl, A. Logadottir and J. K. Nørskov, Electrolysis of Water on (Oxidized) Metal Surfaces, Chem. Phys., 2005, 319, 178–184 CrossRef CAS.
- J. Rossmeisl, Z.-W. Qu, H. Zhu, G.-J. Kroes and J. K. Nørskov, Electrolysis of Water on Oxide Surfaces, J. Electroanal. Chem., 2007, 607, 83–89 CrossRef CAS.
- NIST-JANAF Thermochemical Tables https://janaf.nist.gov/.
-
T. L. Hill, An Introduction to Statistical Thermodynamics, Dover Publications Inc., 1986 Search PubMed.
- P. E. Blöchl, Projector Augmented-Wave Method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- G. Kresse and D. Joubert, From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- G. Kresse and J. Furthmüller, Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- N. J. Mosey, P. Liao and E. A. Carter, Rotationally Invariant Ab Initio Evaluation of Coulomb and Exchange Parameters for DFT+U Calculations, J. Chem. Phys., 2008, 129, 014103 CrossRef PubMed.
- H. J. Monkhorst and J. D. Pack, Special Points for Brillouin-Zone Integrations, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 5188–5192 CrossRef.
- J. D. Pack and H. J. Monkhorst, “Special Points for Brillouin-Zone Integrations”—a Reply, Phys. Rev. B: Condens. Matter Mater. Phys., 1977, 16, 1748–1749 CrossRef.
- L. W. Chung, W. M. C. Sameera, R. Ramozzi, A. J. Page, M. Hatanaka, G. P. Petrova, T. V. Harris, X. Li, Z. Ke and F. Liu,
et al., The ONIOM Method and Its Applications, Chem. Rev., 2015, 115, 5678–5796 CrossRef CAS PubMed.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, Erratum: “Hybrid Functionals Based on a Screened Coulomb Potential” [J. Chem. Phys. 118, 8207 (2003)], J. Chem. Phys., 2006, 124, 219906 CrossRef.
- J. He and C. Franchini, Screened Hybrid Functional Applied to 3d0 → 3d8 Transition-Metal Perovskites LaMO3 (M = Sc-Cu): Influence of the Exchange Mixing Parameter on the Structural, Electronic, and Magnetic Properties, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 235117 CrossRef.
- Y. Meng, X.-W. Liu, C.-F. Huo, W.-P. Guo, D.-B. Cao, Q. Peng, A. Dearden, X. Gonze, Y. Yang and J. Wang,
et al., When Density Functional Approximations Meet Iron Oxides, J. Chem. Theory Comput., 2016, 12, 5132–5144 CrossRef CAS PubMed.
- D. Friebel, M. W. Louie, M. Bajdich, K. E. Sanwald, Y. Cai, A. M. Wise, M.-J. Cheng, D. Sokaras, T.-C. Weng and R. Alonso-Mori,
et al., Identification of Highly Active Fe Sites in (Ni,Fe)OOH for Electrocatalytic Water Splitting, J. Am. Chem. Soc., 2015, 137, 1305–1313 CrossRef CAS PubMed.
- B. Kim, M. K. Kabiraz, J. Lee, C. Choi, H. Baik, Y. Jung, H.-S. Oh, S.-I. Choi and K. Lee, Vertical-Crystalline Fe-Doped β-Ni Oxyhydroxides for Highly Active and Stable Oxygen Evolution Reaction, Matter, 2021, 4, 3585–3604 CrossRef CAS.
- X. Liu, K. Ni, B. Wen, R. Guo, C. Niu, J. Meng, Q. Li, P. Wu, Y. Zhu and X. Wu,
et al., Deep Reconstruction of Nickel-Based Precatalysts for Water Oxidation Catalysis, ACS Energy Lett., 2019, 4, 2585–2592 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional details regarding the DFT calculations and the ONIOM-like free energy framework, atom-projected magnetic moments on Ni and Fe using the DFT-HSE06 (α = 15%) level of theory; Gibbs free energies of the elementary steps involved in alternative OER mechanisms with higher overpotentials or free energies of nonelectroactive steps at various active sites on the (0001) and the (100) facets of Fe-doped β-NiOOH at the ONIOM-DFT-HSE (α = 15%) level of theory; and Gibbs free energies of the elementary steps involved in the OER at various active sites on the (0001) and the (100) facets of Fe-doped β-NiOOH at the DFT-PBE+U level of theory on the N-layer slab models. See DOI: https://doi.org/10.1039/d4cp00315b |
|
| This journal is © the Owner Societies 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
John Mark P.
Martirez
*ab,
John Mark P.
Martirez
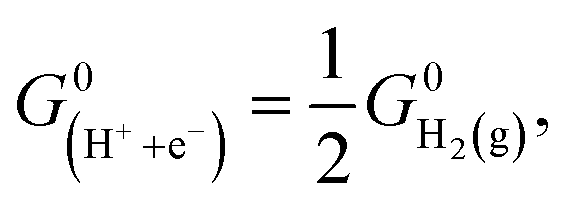 where
where  denotes the standard Gibbs free energy of gaseous hydrogen. One obtains the standard Gibbs free energy of H2(g),
denotes the standard Gibbs free energy of gaseous hydrogen. One obtains the standard Gibbs free energy of H2(g), 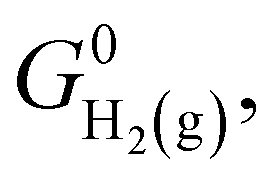 from:
from: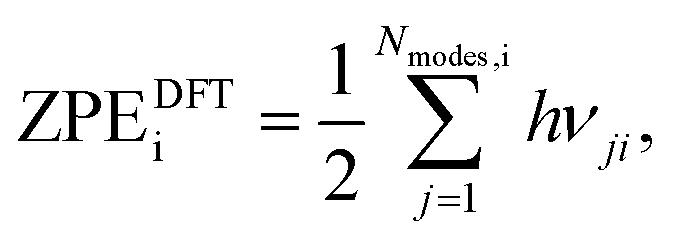 where Nmodes,i denotes the number of vibrational modes of species i, h is Planck's constant, and νji denotes the normal mode frequencies of species i. We assumed room temperature (T = 298.15 K) in all our calculations and obtained experimental values of H0(T) and S0(T) from the NIST-JANAF thermochemical database.56 Subsequently, we determined the free energy of H2O(l),
where Nmodes,i denotes the number of vibrational modes of species i, h is Planck's constant, and νji denotes the normal mode frequencies of species i. We assumed room temperature (T = 298.15 K) in all our calculations and obtained experimental values of H0(T) and S0(T) from the NIST-JANAF thermochemical database.56 Subsequently, we determined the free energy of H2O(l), 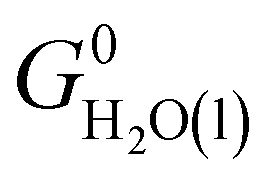 , using the following equation:
, using the following equation:
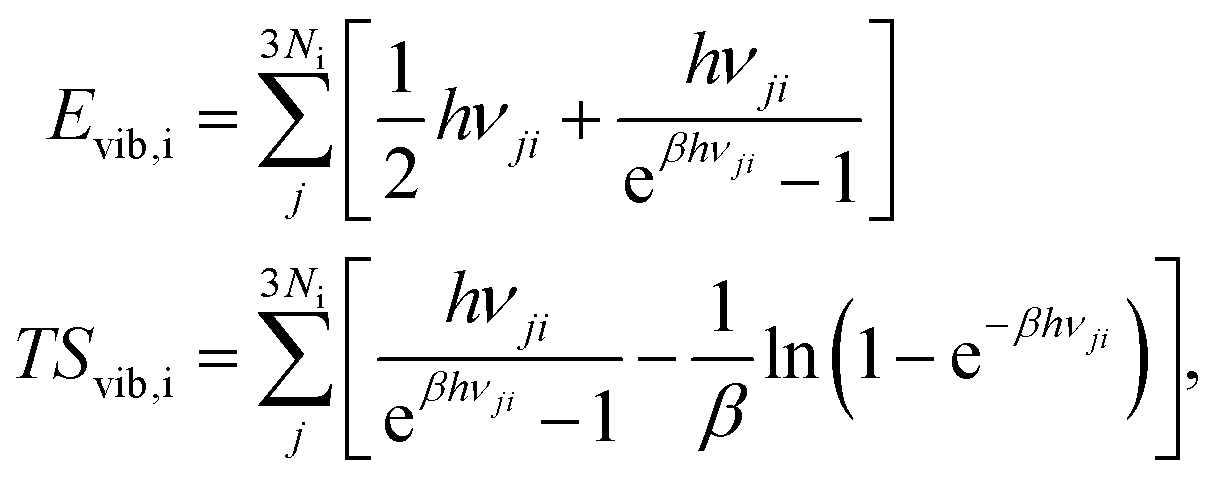
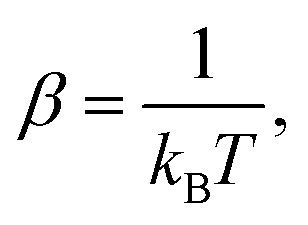 and kB is the Boltzmann constant. Subsequently, we calculated the standard Gibbs free energy change of reaction, ΔG0r,k, of each elementary step k of a mechanism using the species free energies. Following that, we determined the thermodynamic OER overpotential from the difference between the largest electroactive ΔG0r,k and 1.23 eV, per electron transferred, where 1.23 eV is the standard Gibbs free energy change per electron incurred during the OER at room temperature.47 Note that an electroactive step is one in which an electron is transferred so that the free energy of the reaction can be modulated by the application of electric potential. We did not consider the non-electroactive steps (i.e., those that do not involve the explicit transfer of an electron) in the evaluation of the thermodynamic overpotential. The electroactive step with the largest ΔG0r,k requirement is referred to as the potential-determining step (PDS).
and kB is the Boltzmann constant. Subsequently, we calculated the standard Gibbs free energy change of reaction, ΔG0r,k, of each elementary step k of a mechanism using the species free energies. Following that, we determined the thermodynamic OER overpotential from the difference between the largest electroactive ΔG0r,k and 1.23 eV, per electron transferred, where 1.23 eV is the standard Gibbs free energy change per electron incurred during the OER at room temperature.47 Note that an electroactive step is one in which an electron is transferred so that the free energy of the reaction can be modulated by the application of electric potential. We did not consider the non-electroactive steps (i.e., those that do not involve the explicit transfer of an electron) in the evaluation of the thermodynamic overpotential. The electroactive step with the largest ΔG0r,k requirement is referred to as the potential-determining step (PDS).