Modulation of luminescence properties of circularly polarized thermally activated delayed fluorescence molecules with axial chirality by donor engineering†
Received
25th January 2024
, Accepted 4th March 2024
First published on 6th March 2024
Abstract
Multifunctional thermally activated delayed fluorescence (TADF) materials are currently a trending research subject for luminescence layer materials of organic light-emitting diodes (OLEDs). Among these, circularly polarized thermally activated delayed fluorescence (CP-TADF) materials have the advantage of being able to directly achieve highly efficient circularly polarized luminescence (CPL). The simultaneous integration of outstanding luminescence efficiency and excellent luminescence asymmetry factor (glum) is a major constraint for the development of CP-TADF materials. Therefore, on the basis of first-principles calculations in conjunction with the thermal vibration correlation function (TVCF) method, we study CP-TADF molecules with different donors to explore the feasibility of using the donor substitution strategy for optimizing the CPL and TADF properties. The results indicate that molecules with the phenothiazine (PTZ) unit as the donor possess small energy difference, a great spin–orbit coupling constant and a rapid reverse intersystem crossing rate, which endow them with remarkable TADF features. Meanwhile, compared with the reported molecules, the three designed molecules exhibit better CPL properties with higher glum values. Effective molecular design strategies by donor engineering to modulate the CPL and TADF properties are theoretically proposed. Our findings reveal the relationship between molecular structures and luminescence properties of CP-TADF molecules and further provide theoretical design strategies for optimizing the CPL and TADF properties.
1. Introduction
Organic light-emitting diodes (OLEDs) with light weight and bendable properties have been extensively used in optoelectronic lighting and display fields widely.1–3 As one of the most popular choices for light-emitting layer materials in OLEDs, thermally activated delayed fluorescence (TADF) materials can utilize the reverse intersystem crossing (RISC) process to achieve efficient use of excitons, which is due to the small energy difference between their singlet excited states and the triplet excited states.4–8 Recently, multifunctional TADF materials have emerged to meet the needs of society. These include multi-stimulus-responsive TADF materials,9 circularly polarized thermally activated delayed fluorescence (CP-TADF) materials,10–12 and aggregation-induced TADF materials.13 Among these, CP-TADF materials show promising development prospects due to their circularly polarized luminescence (CPL), and are widely used in the fields of three-dimensional displays and information encryption.14,15 Moreover, CPL materials are independent of the polarizing film and 1/4 wave plate, avoiding complex device structures and the loss of luminance intensity.16 In recent years, more and more attention has been paid to CPL research. In 1997, Meijer et al. discovered the circularly polarized electroluminescence (CPEL) effect in chiral polymers.17 Since then, various CPL materials have been found. However, all of these materials inevitably exhibit efficiency roll-off, which limits the further development of circularly polarized organic light-emitting diodes (CP-OLEDs).
To address this issue, Imagawa et al. combined a chiral skeleton with a TADF molecule for the first time, which had a sufficiently small energy difference (ΔEST) between the first singlet excited state (S1) and the first triplet excited state (T1). Moreover, fast RISC could be achieved efficiently, which guaranteed high exciton utilization.18 They synthesized CP-TADF molecules with central chirality, namely 12-(2-(diphenylamino)phenyl)-12-hydroxynaphthacen-5(12H)-one (DPHN), with which CPL can be achieved with large luminescence asymmetry factor (glum) and considerable exciton utilization. Recently, Chen et al. realized that the large distance between the chiral skeleton and the emitting unit makes the perturbation of the chiral unit small, which limits the enhancement of the CPL properties. Therefore, they designed axially chiral CP-TADF molecules, (−)-(S)-Cz-Ax-CN and (+)-(R)-Cz-Ax-CN, synthesized by coupling two fluorophores 3-(9H-carbazol-9-yl)benzonitrile (Cz-CN). The CP-TADF molecules achieved a glum of 10−2 orders of magnitude and a photoluminescence quantum yield (PLQY) of 68.2%.19 In 2020, Zheng et al. achieved better CPL and TADF properties by replacing one of the carbazole (Cz) units with phenoxazine (PXZ) based on the same chiral skeleton.20 Therefore, achieving CPL while maintaining high exciton utilization has become a popular research topic in CP-TADF materials.
By changing the donor unit, it is possible to balance the properties of both CPL and TADF. This motivated us to explore the strategy of developing efficient molecules by designing and testing the luminescence properties of CP-TADF molecules with different donor units. The molecular structures in the ground and excited states are optimized using the BMK functional coupled with the 6-31G(d) basis set based on the density functional theory (DFT) and time-dependent density functional theory (TD-DFT) methods, respectively. The photophysical properties including molecular transition properties, intersystem crossing (ISC) and RISC processes, and CPL properties have been investigated in detail theoretically. The effects of donor substitutions on these properties have also been studied. The aim of our study is to propose design strategies for modulating CPL and TADF properties and offer rational suggestion for designing efficient CP-TADF molecules.
2. Theoretical methods and computational details
First, we construct all structures of molecules and use the polarizable continuum method by the Integral Equation Formalism model (IEFPCM) to simulate the liquid phase environment in toluene.21,22 In order to make our theoretical calculations more reliable, we use different functionals (B3LYP, BMK, M062X, PBE0, and WB97XD)23–25 combined with the 6-31G(d) basis set26 for the optimization and frequency analysis of the S1 state of Cz-Ax-CN in toluene. Fluorescence emission wavelengths are obtained and the corresponding results are listed in Table 1. It is found that the computational data (449 nm) by BMK can well match the experiment result (458 nm). In the DFT and TD-DFT calculations, some approximate methods are adopted to simplify the calculations, such as the case of two-electron integrals and Tamm–Dancoff approximation. Thus, some acceptable differences between theoretical calculations and experimental measurements are generated, such as the difference between 449 nm (computational data) and 458 nm (experiment result). This makes us to think that using the 6-31G(d) basis set to calculate the emission wavelength and other photophysical properties of molecules is reliable, and this method has been recognized in related research studies.27,28 Therefore, for the subsequent discussion, we employ the BMK functional with the 6-31G(d) basis set. We optimize the structure of S0 based on the structure we constructed. Then, based on the optimized S0 structure, the S1 and T1 states of the four chiral TADF molecules are optimized by using DFT and TD-DFT to obtain the corresponding molecular structures. All the molecular structures are with no imaginary frequency, which determines the stability of the structures, so we can use the obtained structures to analyze the photophysical properties of the molecules. All the calculations are carried out using the Gaussian 16 program suite.29
Table 1 The fluorescence emission wavelengths (nm) of Cz-Ax-CN in toluene calculated using different functionals and experimentally measured wavelengths
|
|
B3LYP |
BMK |
M062X |
PBE0 |
WB97XD |
Exp. |
| Cz-Ax-CN |
572 |
449 |
408 |
534 |
379 |
458 |
Decay rates should also be obtained. The main formulas used in this study are summarized below.
The fluorescence radiative rate is obtained using the Einstein spontaneous emission equation:30
| | 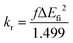 | (1) |
Here,
f represents the oscillator strength of S
1, Δ
Efi represents the vertical emission energy (cm
−1) between the S
1 and the ground states (S
0) and
kr is defined as the radiative decay rate (s
−1).
31
For the non-radiative process between S1 and S0, the non-radiative decay rate (knr) is obtained from the Fermi's golden rule and Condon approximation:
| | 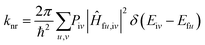 | (2) |
Here,
Piv represents the Boltzmann distribution function of the initial state and
Ĥfu,iv represents the interaction between two Born–Oppenheimer states. The
Ĥ can be expressed as:
| | | ĤΨiν = ĤBOΦi(r, Q)Θiν(Q) + ĤSOΦi(r, Q)Θiν(Q) | (3) |
ĤBO represents the non-adiabatic coupling.
ĤSO represents the spin–orbit coupling (SOC) obtained by the quadratic response function method in the Dalton program.
32,33r and
Q stand for the normal coordinates of electrons and nuclei, respectively.
Φ and
Θ stand for the electron wave function and nuclear vibration wave function, respectively. Then, according to the Fourier transform of the delta function, we calculate
knr from S1 to S0 as:
| | 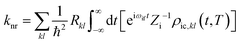 | (4) |
Here,
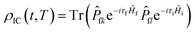
represents the thermal vibration correlation function (TVCF) in the non-radiative process. All calculation methods and application of these formulas are also used in Shuai, Peng, Cui and our previous works.
34–43 The MOMAP program is used to perform these calculations.
44 The calculations of the SOC constant and frequency analyses are performed based on the optimized molecular structures of T
1.
Then, in order to characterize the geometric structures of different states, we calculate the root mean squared displacement (RMSD). The calculation formula is as follows:
| | 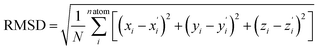 | (5) |
Here i is the atomic number, and xi and 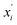 are the x-coordinates of the ith atom in the first and the second structures, respectively. y and z have a similar meaning to x. Through this analysis, we can preliminarily predict and compare the photophysical properties of molecules such as the knr.
are the x-coordinates of the ith atom in the first and the second structures, respectively. y and z have a similar meaning to x. Through this analysis, we can preliminarily predict and compare the photophysical properties of molecules such as the knr.
Furthermore, to quantitatively characterize the CPL properties, the glum values can be calculated with the equation:
| | 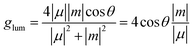 | (6) |
Here,
μ and
m are the electronic transition dipole moment and magnetic transition dipole moment, respectively, and
θ is the angle between them.
Then, the frontier molecular orbital (FMO) analyses and natural transition orbital (NTO) analyses are both achieved using Multiwfn.45
3. Results and discussion
Based on the proposed molecule Cz-Ax-CN synthesized by Chen's group,20 we chose three different donor moieties, 9,10-dihydro-9,9-dimethylacridine (DMAC), PTZ and PXZ, to design new axial chiral TADF molecules named DMAC-Ax-CN, PTZ-Ax-CN and PXZ-Ax-CN by donor substitution engineering, and the corresponding structures are shown in Fig. 1. Then, molecular structures and transition properties as well as radiative and non-radiative decay processes are studied, and the CPL and TADF properties are revealed.
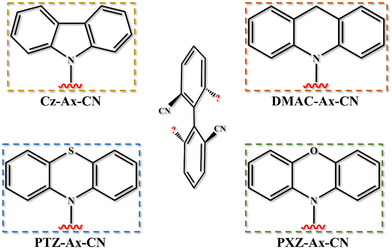 |
| | Fig. 1 The structures of the skeleton and donors for all molecules. | |
3.1. Molecular structures and RMSD values
Identifying the geometry of the CP-TADF molecule is the first step in studying its photophysical properties. Therefore, the structures of the ground and excited states of the four molecules are optimized by applying the BMK functional and the 6-31G(d) basis set and the stability of the structures is ensured by frequency analysis. To compare the structural differences of the molecules in different states, we calculate the RMSD values between the S0, S1 and T1 states of the four molecules. The corresponding results are displayed in Fig. 2. The results show that the RMSD value between S0 and S1 is smaller for the Cz-Ax-CN (0.163 Å) and PTZ-Ax-CN (0.193 Å) molecules than the other two molecules (0.853 Å and 0.387 Å). This suggests that these two molecules may have smaller non-radiative quenching rates, which is consistent with our subsequent calculations of knr. It is found that the RMSD value between S1 and T1 is smaller for all molecules. Additionally, the RMSD value of the PTZ-Ax-CN (0.022 Å) molecules is significantly smaller than that of the other three molecules (0.064 Å, 0.086 Å and 0.026 Å), indicating a smaller recombination energy, which may result in a more desirable RISC process.
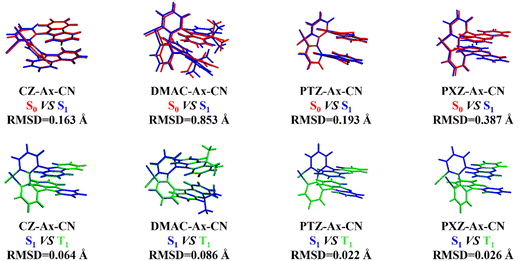 |
| | Fig. 2 Geometry comparisons and RMSD values of all molecules between S0 (red), S1 (blue), and T1 (green) in toluene, respectively. | |
3.2. Thermally activated delayed fluorescence properties
The performance of TADF materials can be evaluated using the kRISC and kr values. The variations in the molecular geometry result in differences in the electronic structure, such as energy levels and transition properties, which ultimately affect the TADF process. Thus, we examine these characteristics by investigating the excited state and luminescence properties of the four chiral TADF molecules.
3.2.1. Energy gaps and the frontier molecular orbitals.
ΔEST has a significant impact on the TADF properties of a molecule.46,47 A small energy difference promotes the RISC process, resulting in a more efficient TADF. To obtain the adiabatic energy of the molecules at S1 and T1 states, we optimize the molecular structures to calculate the energy difference. The corresponding data are presented in Table 2. To provide a more intuitive comparison of the energy differences between the four molecules, the ΔEST diagram is shown in Fig. S1 (ESI†). The results indicate that the energy difference of the three designed molecules (0.06 eV, 0.04 eV and 0.05 eV) is significantly smaller than that of the original molecule (0.12 eV), making it more likely for exciton transitions from T1 to S1. Therefore, it is conducive to achieving efficient TADF and luminescence efficiency. To investigate the reason for the change in the energy difference, we calculate the FMOs and the corresponding results are shown in Fig. 3. According to the equation:| | 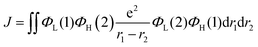 | (8) |
where J is the exchange energy of the two unpaired electrons at the excited states, and ES and ET are adiabatic energies of S1 and T1, respectively. It can be observed that the HOMO of all molecules is primarily located on the donor unit, whereas the LUMO is predominantly situated on the acceptor group and chiral skeleton. Moreover, the overlap of molecular frontier orbitals is quantitatively compared using the Multiwfn program, and the corresponding results are listed in Table S1 (ESI†). Therefore, there is more distinct separation of the HOMO and LUMO for the three designed molecules (26.43%, 25.58% and 25.60%) compared to the original molecule (28.31%). This distribution of molecular orbitals is one of the reasons for the decrease in the energy difference for TADF molecules.48
Table 2 Detailed data for Cz-Ax-CN, DMAC-Ax-CN, PTZ-Ax-CN and PXZ-Ax-CN. λem (nm) is the emission wavelength of the molecule, ES is the adiabatic energy of the S1 state, ET is the adiabatic energy of the T1 state, ΔEST is the adiabatic energy difference between the S1 state and the T1 state of the molecules, and f is the oscillator strength of the molecules in toluene
|
|
λ
em (nm) |
E
S (eV) |
E
T (eV) |
ΔEST (eV) |
f
|
| Cz-Ax-CN |
449.20 |
3.08 |
2.96 |
0.12 |
0.0347 |
| DMAC-Ax-CN |
537.27 |
2.87 |
2.81 |
0.06 |
0.0094 |
| PTZ-Ax-CN |
600.68 |
2.48 |
2.44 |
0.04 |
0.0031 |
| PXZ-Ax-CN |
603.16 |
2.46 |
2.41 |
0.05 |
0.0090 |
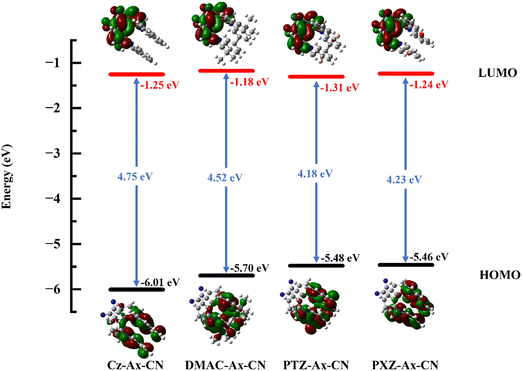 |
| | Fig. 3 Energies and distributions of the HOMO and LUMO for all molecules in toluene. | |
To further determine the transition properties of the molecule,49 we calculate the NTO based on its structures for S1 and T1 and the results are presented in Fig. 4, and the enlarged images are shown in Fig. S2 and S3 (ESI†). Chen's group concluded that according to the local-excited (LE) proportion, excited states can be categorized into three types of excitations: the charge-transfer (CT) state (0–40%), the hybrid local-excited and charge-transfer (HLCT) state (40–75%), and the LE state (75–100%).50–52 According the result, the LE components of the S1 state for the four molecules are 32.11%, 27.19%, 25.79% and 27.07%, indicating that the S1 state of all four molecules is the CT state. However, it is worth noting that the T1 state of all three designed molecules is the CT state and the T1 state of the CZ molecule is the HLCT state. According to previous investigation of Penfold,53–56 when the T1 state of the molecule is the CT state, such a molecule usually has a smaller ΔEST. Combining our previous calculations of ΔEST and current orbital information, our calculations fit well with that conclusion. For the TADF molecule, the electronic properties are more favorable for smaller ΔEST and facilitate a more desirable RISC process. Moreover, by regulating the geometric structures by donor engineering, which affects the electronic structure and the ΔEST, it is expected to regulate the TADF properties.
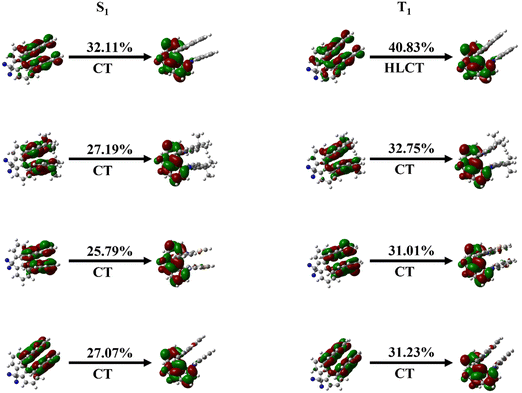 |
| | Fig. 4 Natural transition orbitals (NTOs) of the S1 and T1 states for Cz-Ax-CN, DMAC-Ax-CN, PTZ-Ax-CN and PXZ-Ax-CN in toluene, respectively. | |
3.2.2. Spin–orbit coupling and decay rates.
In addition to the ΔEST, the TADF properties are also related to the SOC between S1 and T1.57,58 The SOC values of the molecule are calculated and the results are presented in Table 3. Among the four molecules, PTZ-Ax-CN (0.134 cm−1) has a larger SOC value than the other three (0.055 cm−1, 0.066 cm−1 and 0.050 cm−1), suggesting a more efficient RISC process. To enhance the intuitive comparison of the conversion processes of the molecules, we calculate the kISC and kRISC values between S1 and T1 using data of SOC and ΔEST. The corresponding results are listed in Table 4 and are plotted as shown in Fig. S4 (ESI†). It is found that the three designed molecules (3.76 × 106 s−1, 7.67 × 106 s−1 and 1.73 × 105 s−1) exhibit a significantly higher kRISC compared to the original molecule (5.62 × 104 s−1). This is closely related to their smaller ΔEST and larger SOC, which ensures ideal TADF properties. To investigate the energy consumption process, we calculate the knr of the four molecules. According to the data in Table 4, we discover that the knr of the three designed molecules (6.82 × 107 s−1, 3.31 × 107 s−1 and 5.62 × 107 s−1) is slightly increased compared to that of the Cz-Ax-CN (2.45 × 107 s−1), which is caused by the stronger vibrational coupling effect with smaller energy difference between S1 and S0 (shown in Fig. S1, ESI†). In addition, the values of kr are calculated by combining the oscillator strength (f) obtained from the optimized S1, and the corresponding data are shown in Table 4. The Cz-Ax-CN has a higher kr (1.15 × 107 s−1) due to its stronger f (0.0347). Moreover, the DMAC-Ax-CN (0.0094), PTZ-Ax-CN (0.0031), and PXZ-Ax-CN (0.0090) have smaller f, and they also demonstrate considerable kr (2.17 × 106 s−1, 5.73 × 105 s−1 and 1.65 × 106 s−1). Thus, it can be seen that all four molecules have large kr and efficient emission can be achieved. In summary, based on the donor engineering strategy, three new efficient TADF molecules are proposed.
Table 3 Calculated SOC constants between S1 and T1 states for Cz-Ax-CN, DMAC-Ax-CN, PTZ-Ax-CN and PXZ-Ax-CN in toluene based on optimized T1 structures
| Unit: cm−1 |
〈T1|Ĥsoc|S1〉 |
| Cz-Ax-CN |
0.055 |
| DMAC-Ax-CN |
0.066 |
| PTZ-Ax-CN |
0.134 |
| PXZ-Ax-CN |
0.050 |
Table 4 Rate constants of radiative and non-radiative processes from S1 to S0 states as well as the ISC and RISC rates between S1 and T1 states for all studied molecules in toluene
|
|
k
ISC (s−1) |
k
RISC (s−1) |
k
r (s−1) |
k
nr (s−1) |
| Cz-Ax-CN |
4.94 × 105 |
5.62 × 104 |
1.15 × 107 |
2.45 × 107 |
| DMAC-Ax-CN |
6.47 × 106 |
3.76 × 106 |
2.17 × 106 |
6.82 × 107 |
| PTZ-Ax-CN |
1.90 × 107 |
7.67 × 106 |
5.73 × 105 |
3.31 × 107 |
| PXZ-Ax-CN |
1.17 × 106 |
1.73 × 105 |
1.65 × 106 |
5.62 × 107 |
3.3. Circularly polarized luminescence properties
In order to evaluate the CPL properties of the molecules, the electron circular dichroism spectra (ECD) are calculated using the Multiwfn program based on the optimized ground state and the corresponding results are shown in Fig. 5.59,60 The plotted lines in all molecules are symmetric about the X-axis, indicating the presence of the cotton effect and verifying the success of the chiral perturbation strategy. This suggests that the molecules are chiral and can achieve circularly polarized luminescence. To compare the CPL properties of the four molecules, we also conduct vertical emission calculations based on the S1 structure. Combining the result of vertical excitation and vertical emission, we calculate the circularly polarized absorption asymmetry factor (gabs) and the circularly polarized luminescence asymmetry factor (glum), respectively, and the data are listed in Table S2 (ESI†) and Table 5. Based on these data, it can be observed that the angles between the electronic transition dipole moment (μ) and magnetic transition dipole moment (m) of the four molecules are approximately 180°, with cosine values of −1. This indicates that the two dipole moments are oriented in opposite directions. Research has shown that the CPL properties of chiral molecules can be measured using the glum. The larger the absolute value of this factor, the better the circularly polarized properties of the molecule. Combined with the theoretical formulas, it is evident that the value of the glum is primarily influenced by three factors: the μ, m and θ. Our previous study has indicated that the angle between the two dipole moments has the greatest impact on the luminescence asymmetry factor,61 and we aim to obtain a dipole moment angle with a larger cosine value. From this perspective, for these four axially chiral molecules with some symmetry, their dipole moment angles are almost always optimal, demonstrating the advantage of this design strategy. Simplifying the calculation formula, we can get 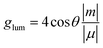 , i.e., the glum is proportional to the absolute value of the μ and inversely proportional to the absolute value of the m. As the μ is two orders of magnitude larger than that of the m, decreasing the former may be an effective strategy for obtaining better circular polarization properties. To verify this, we plotted a line graph of the glum and the μ, as shown in Fig. 6. This is consistent with our conjecture and confirms the effect of μ on the nature of CPL. We find that the three designed molecules have a more desirable glum than the original molecules due to their smaller μ. PTZ-Ax-CN has the smallest μ and the largest glum. Thus, using the donor engineering strategy, the CPL properties are modulated. Compared with the reported molecule (Cz-Ax-CN), three new efficient CP-TADF molecules (DMAC-Ax-CN, PTZ-Ax-CN and PXZ-Ax-CN) with large RISC rates and large glum values are theoretically proposed.
, i.e., the glum is proportional to the absolute value of the μ and inversely proportional to the absolute value of the m. As the μ is two orders of magnitude larger than that of the m, decreasing the former may be an effective strategy for obtaining better circular polarization properties. To verify this, we plotted a line graph of the glum and the μ, as shown in Fig. 6. This is consistent with our conjecture and confirms the effect of μ on the nature of CPL. We find that the three designed molecules have a more desirable glum than the original molecules due to their smaller μ. PTZ-Ax-CN has the smallest μ and the largest glum. Thus, using the donor engineering strategy, the CPL properties are modulated. Compared with the reported molecule (Cz-Ax-CN), three new efficient CP-TADF molecules (DMAC-Ax-CN, PTZ-Ax-CN and PXZ-Ax-CN) with large RISC rates and large glum values are theoretically proposed.
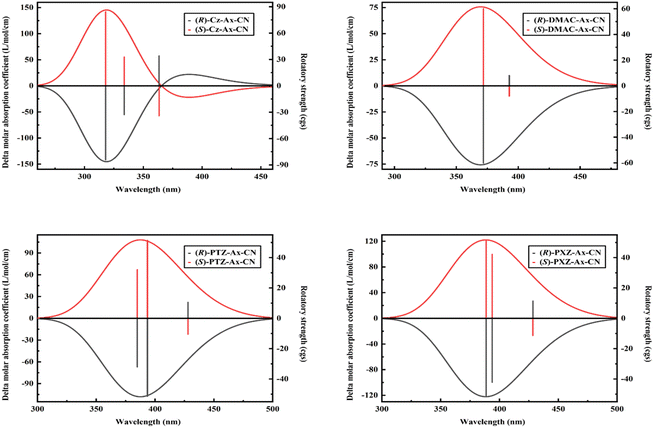 |
| | Fig. 5 ECD spectra of Cz-Ax-CN, DMAC-Ax-CN, PTZ-Ax-CN and PXZ-Ax-CN in toluene. The rotatory strength is also illustrated. | |
Table 5 The data of luminescence asymmetry factor (glum) for Cz-Ax-CN, DMAC-Ax-CN, PTZ-Ax-CN and PXZ-Ax-CN in toluene
|
|
|μ| (×10−18 esu cm) |
|m| (×10−20 erg G−1) |
cos![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) θ θ |
g
lum (×10−2) |
| Cz-Ax-CN |
1.806 |
0.471 |
−1 |
−1.04 |
| DMAC-Ax-CN |
1.028 |
0.379 |
−1 |
−1.47 |
| PTZ-Ax-CN |
0.623 |
0.348 |
−1 |
−2.23 |
| PXZ-Ax-CN |
1.058 |
0.382 |
−1 |
−1.45 |
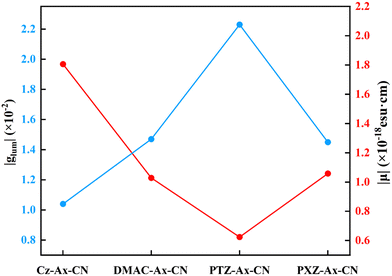 |
| | Fig. 6 Calculated luminescence asymmetry factor (glum) and transition electric dipole moments (μ) of the molecules. | |
4. Conclusion
In summary, through first-principles calculations with the TVCF method, this work validates the design strategy for efficient CP-TADF molecules by donor engineering. The results show that modulation of the donor unit can achieve a smaller ΔEST and a smaller μ for the axial chiral TADF molecule. The former facilitates the RISC process and thus promotes the efficient utilization of the triple excitons, which is essential for effective TADF emission. The latter ensures the desired glum, which indicates that the molecule can achieve better CPL properties. For three designed molecules, we also observed that the S1 and T1 all possess CT features, and this results in smaller ΔEST required for an efficient RISC process. Additionally, we have computationally verified the relationship between the μ and the glum. Therefore, it is possible to modulate the μ by regulating the donor unit of CP-TADF materials to achieve a large glum. Thus, three new efficient molecules with dominant CPL and TADF properties are theoretically proposed, and wise molecular design strategies by donor engineering to modulate the CPL and TADF properties are illustrated. This work reveals the inner relationship between molecular structures and luminescence properties, and could provide theoretical guidance for designing new efficient CP-TADF molecules.
Author contributions
S. L.: writing-original draft; S. L.: visualization; Y. G.: methodology; L. L.: conceptualization and formal analysis; C.-K. W.: supervision; J. F.: writing-review & editing; Y. S.: writing-review & editing.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant no. 11874241, 12274266, 11874242, 21933002 and 12374269). Thanks to Professor Yingli Niu for his great help in the usage of MOMAP.
References
- T. M. Figueira-Duarte and K. Mullen, Pyrene-Based Materials for Organic Electronics, Chem. Rev., 2011, 111, 7260–7314 CrossRef CAS PubMed.
- M. A. McCarthy, B. Liu, E. P. Donoghue, I. Kravchenko, D. Y. Kim, F. So and A. G. Rinzler, Low-Voltage, Low-Power, Organic Light-Emitting Transistors for Active Matrix Displays, Science, 2011, 332, 570–573 CrossRef CAS PubMed.
- L. J. Rothberg and A. J. Lovinger, Status of and Prospects for Organic Electroluminescence, J. Mater. Res., 1996, 11, 3174–3187 CrossRef CAS.
- C. W. Tang and S. A. VanSlyke, Organic Electroluminescent Diodes, Appl. Phys. Lett., 1987, 51, 913–915 CrossRef CAS.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Highly Efficient Organic Light-Emitting Diodes from Delayed Fluorescence, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- S. Hirata, Y. Sakai, K. Masui, H. Tanaka, S. Y. Lee, H. Nomura, N. Nakamura, M. Yasumatsu, H. Nakanotani, Q. Zhang, K. Shizu, H. Miyazaki and C. Adachi, Highly Efficient Blue Electroluminescence Based on Thermally Activated Delayed Fluorescence, Nat. Mater., 2015, 14, 330–336 CrossRef CAS PubMed.
- D. D. Zhang, X. Z. Song, M. H. Cai, H. Kaji and L. Duan, Versatile Indolocarbazole-Isomer Derivatives as Highly Emissive Emitters and Ideal Hosts for Thermally Activated Delayed Fluorescent OLEDs with Alleviated Efficiency Roll-Off, Adv. Mater., 2018, 30, 1705406 CrossRef PubMed.
- S. Y. Shao and L. X. Wang, Through-Space Charge Transfer Polymers for Solution-Processed Organic Light-Emitting Diodes, Aggregate, 2020, 1, 45–56 CrossRef.
- M. L. Yang, I. S. Park, Y. Miyashita, K. Tanaka and T. Yasuda, Mechanochromic Delayed Fluorescence Switching in Propeller-Shaped Carbazole-Isophthalonitrile Luminogens with Stimuli-Responsive Intramolecular Charge-Transfer Excited States, Angew. Chem., Int. Ed., 2020, 59, 13955–13961 CrossRef CAS PubMed.
- P. Xue, X. Wang, W. J. Wang, J. Y. Zhang, Z. J. Wang, J. B. Jin, C. Zheng, P. Li, G. H. Xie and R. F. Chen, Solution-Processable Chiral Boron Complexes for Circularly Polarized Red Thermally Activated Delayed Fluorescent Devices, ACS Appl. Mater. Interfaces, 2021, 13, 47826–47834 CrossRef CAS PubMed.
- Y. C. Xu, Q. Y. Wang, X. X. Song, Y. Wang and C. L. Li, New Fields, New Opportunities and New Challenges: Circularly Polarized Multiple Resonance Thermally Activated Delayed Fluorescence Materials, Chem. – Eur. J., 2022, 29, e202203414 CrossRef PubMed.
- G. J. Li, F. Zhan, W. W. Lou, D. Wang, C. Deng, L. N. Cao, Y. N. Yang, Q. S. Zhang and Y. B. She, Efficient Deep-Blue Organic Light-Emitting Diodes Employing Difluoroboron-Enabled Thermally Activated Delayed Fluorescence Emitters, J. Mater. Chem. C, 2020, 8, 17464–17473 RSC.
- Y. Fu, Z. C. Ye, J. P. Xiao, L. Y. Liao, L. F. Chen, Y. X. Mu, S. M. Ji, Z. J. Zhao, H. L. Zhang and Y. P. Huo, Large Effects of Tiny Structural Changes on the AIE-TADF Type Xanthone Derivatives in Mechano-Responsive Luminescence and Electroluminescence, Dyes Pigm., 2022, 205, 110550 CrossRef CAS.
- Y. J. Kim, B. Y. Ahn, K. B. Kim and Y. G. Kim, Potential Application of Spintronic Light-Emitting Diode to Binocular Vision for Three-Dimensional Display Technology, J. Korean Phys. Soc., 2006, 49, 505–508 Search PubMed.
- Y. T. Sang, J. L. Han, T. H. Zhao, P. F. Duan and M. H. Liu, Circularly Polarized Luminescence in Nanoassemblies: Generation, Amplification, and Application, Adv. Mater., 2020, 32, 1900110 CrossRef CAS PubMed.
- D. W. Zhang, M. Li and C. F. Chen, Recent Advances in Circularly Polarized Electroluminescence Based on Organic Light-Emitting Diodes, Chem. Soc. Rev., 2020, 49, 1331–1343 RSC.
- E. Peeters, M. P. T. Christiaans, R. A. J. Janssen, H. F. M. Schoo, H. P. J. Dekkers and E. W. Meijer, Circularly Polarized Electroluminescence from a Polymer Light-Emitting Diode, J. Am. Chem. Soc., 1997, 119, 9909–9910 CrossRef CAS.
- T. Imagawa, S. Hirata, K. Totani, T. Watanabe and M. Vacha, Thermally Activated Delayed Fluorescence with Circularly Polarized Luminescence Characteristics, Chem. Commun., 2015, 51, 13268–13271 RSC.
- M. Li, Y. F. Wang, D. D. Zhang, L. Duan and C. F. Chen, Axially Chiral TADF-Active Enantiomers Designed for Efficient Blue Circularly Polarized Electroluminescence, Angew. Chem., Int. Ed., 2020, 59, 3500–3504 CrossRef CAS PubMed.
- Z. L. Tu, Z. P. Yan, X. Liang, L. Chen, Z. G. Wu, Y. Wang, Y. X. Zheng, J. L. Zuo and Y. Pan, Axially Chiral Biphenyl Compound-Based Thermally Activated Delayed Fluorescent Materials for High-Performance Circularly Polarized Organic Light-Emitting Diodes, Adv. Sci., 2020, 7, 2000804 CrossRef CAS PubMed.
- J. Z. Fan, S. Qiu, L. L. Lin and C. K. Wang, First-Principles Investigation on Triazine Based Thermally Activated Delayed Fluorescence Emitters, Chin. J. Chem. Phys., 2016, 29, 291–296 CrossRef CAS.
- J. Tomasi, B. Mennucci and R. Cammi, Quantum Mechanical Continuum Solvation Models, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS PubMed.
- P. J. Stephens, C. F. Chabalowski, F. J. Devlin and K. J. Jalkanen, Ab Initio Calculation of Vibrational Circular Dichroism Spectra Using Large Basis Set MP2 Force Fields, Chem. Phys. Lett., 1994, 225, 247–257 CrossRef CAS.
- C. Adamo and V. Barone, Toward Reliable Density Functional Methods without Adjustable Parameters: The PBE0 Model, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- A. D. Boese and J. M. Martin, Development of Density Functionals for Thermochemical Kinetics, J. Chem. Phys., 2004, 121, 3405–3416 CrossRef CAS PubMed.
- S. Hirata and M. Head-Gordon, Time-Dependent Density Functional Theory within the Tamm-Dancoff Approximation, Chem. Phys. Lett., 1999, 314, 291–299 CrossRef CAS.
- S. Y. Lin, Q. Ou and Z. G. Shuai, Computational Selection of Thermally Activated Delayed Fluorescence (TADF) Molecules with Promising Electrically Pumped Lasing Property, ACS Mater. Lett., 2022, 4, 487–496 CrossRef CAS.
- S. Y. Lin, Q. Ou, Q. Peng and Z. G. Shuai, Aggregation-Enhanced Thermally Activated Delayed Fluorescence Efficiency for Two-Coordinate Carbene–Metal–Amide Complexes: A QM/MM Study, J. Phys. Chem. Lett., 2021, 12, 2944–2953 CrossRef CAS PubMed.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Rev. A.03, 2016 Search PubMed.
- Z. Shuai, Thermal Vibration Correlation Function Formalism for Molecular Excited State Decay Rates, Chin. J. Chem., 2020, 38, 1223–1232 CrossRef CAS.
- D. E. Mccumber, Einstein Relations Connecting Broadband Emission and Absorption Spectra, Phys. Rev., 1964, 136, A954 CrossRef.
- K. Aidas, C. Angeli, K. L. Bak, V. Bakken, R. Bast, L. Boman, O. Christiansen, R. Cimiraglia, S. Coriani and P. Dahle, The Dalton Quantum Chemistry Program System, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 269–284 CAS.
- Dalton. A Molecular Electronic Structure Program. Release Dalton2013, 2014, available from: https://daltonprogram.org.
- Z. G. Shuai and Q. Peng, Excited States Structure and Processes: Understanding Organic Light-Emitting Diodes at the Molecular Level, Phys. Rep., 2014, 537, 123–156 CrossRef CAS.
- Y. J. Sun, H. Geng, Q. Peng and Z. G. Shuai, Computational Study on the Charge Transport and Optical Spectra of Anthracene Derivatives in Aggregates, Chem. Phys. Chem., 2020, 21, 952–957 CrossRef CAS PubMed.
- P. A. Yin, Q. Wan, Y. L. Niu, Q. Peng, Z. M. Wang, Y. X. Li, A. J. Qin, Z. G. Shuai and B. Z. Tang, Theoretical and Experimental Investigations on the Aggregation-Enhanced Emission from Dark State: Vibronic Coupling Effect, Adv. Electron. Mater., 2020, 6, 2000255 CrossRef CAS.
- Y. J. Gao, W. K. Chen, T. T. Zhang, W. H. Fang and G. L. Cui, Theoretical Studies on Excited-State Properties of Au(III) Emitters with Thermally Activated Delayed Fluorescence, J. Phys. Chem. C, 2018, 122, 27608–27619 CrossRef CAS.
- J. Y. Chen, X. X. Xiao, S. Li, Y. Duan, G. Wang, Y. Liao, Q. Peng, H. B. Fu, H. Geng and Z. G. Shuai, A Novel Strategy toward Thermally Activated Delayed Fluorescence from A Locally Excited State, J. Phys. Chem. Lett., 2022, 13, 2653–2660 CrossRef CAS PubMed.
- M. Z. Li, F. Y. Li, Q. Zhang, K. Zhang, Y. Z. Song, J. Z. Fan, C. K. Wang and L. L. Lin, Theoretical Verification of Intermolecular Hydrogen Bond Induced Thermally Activated Delayed Fluorescence in SOBF-OMe*, Chinese Phys. B, 2021, 30, 123302 CrossRef CAS.
- L. L. Lv, K. Yuan, C. D. Si, G. F. Zuo and Y. C. Wang, Mechanism Study of TADF and Phosphorescence in Dinuclear Copper (I) Molecular Crystal Using QM/MM Combined with An Optimally Tuned Range-Separated Hybrid Functional, Org. Electron., 2020, 81, 105667 CrossRef CAS.
- L. Y. Peng, W. K. Chen, Z. W. Li, Y. J. Gao and G. L. Cui, QM/MM Studies on Thermally Activated Delayed Fluorescence of A Dicopper Complex in the Solid State, J. Phys. Chem. C, 2021, 125, 27372–27380 CrossRef CAS.
- K. Zhang, J. Z. Fan, C. K. Wang and L. L. Lin, Highly Efficient T-Shaped Deep-Red Thermally Activated Delayed Fluorescence Emitters: Substitution Position Effect, Phys. Chem. Chem. Phys., 2021, 23, 21883–21892 RSC.
- H. P. Zou, Y. Y. Ma, H. L. Liu, Q. F. Mu, K. Zhang, Y. Z. Song, L. L. Lin, C. K. Wang and J. Z. Fan, A QM/MM Study on Through Space Charge Transfer-Based Thermally Activated Delayed Fluorescence Molecules in the Solid State, J. Mater. Chem. C, 2022, 10, 517–531 RSC.
- Y. L. Niu, W. Q. Li, Q. Peng, H. Geng, Y. P. Yi, L. J. Wang, G. J. Nan, D. Wang and Z. G. Shuai, MOlecular MAterials Property Prediction Package (MOMAP) 1.0: A Software Package for Predicting the Luminescent Properties and Mobility of Organic Functional Materials, Mol. Phys., 2018, 116, 1078–1090 CrossRef CAS.
- T. L. Wu, S. Y. Liao, P. Y. Huang, Z. S. Hong, M. P. Huang, C. C. Lin, M. J. Cheng and C. H. Cheng, Exciplex Organic Light-Emitting Diodes with Nearly 20% External Quantum Efficiency: Effect of Intermolecular Steric Hindrance between the Donor and Acceptor Pair, ACS Appl. Mater. Interfaces, 2019, 11, 19294–19300 CrossRef CAS PubMed.
- T. Chen, L. Zheng, J. Yuan, Z. F. An, R. F. Chen, Y. Tao, H. H. Li, X. J. Xie and W. Huang, Understanding the Control of Singlet-Triplet Splitting for Organic Exciton Manipulating: A Combined Theoretical and Experimental Approach, Sci. Rep., 2015, 5, 1–11 Search PubMed.
- S. P. Huang, Q. S. Zhang, Y. Shiota, T. Nakagawa, K. Kuwabara, K. Yoshizawa and C. Adachi, Computational Prediction for Singlet-and Triplet-Transition Energies of Charge-Transfer Compounds, J. Chem. Theory Comput., 2013, 9, 3872–3877 CrossRef CAS PubMed.
- P. Samanta, D. Kim, V. Coropceanu and J. Brédas, Up-Conversion Intersystem Crossing Rates in Organic Emitters for Thermally Activated Delayed Fluorescence: Impact of the Nature of Singlet vs Triplet Excited States, J. Am. Chem. Soc., 2017, 139, 4042–4051 CrossRef CAS PubMed.
- S. Xu, Q. Q. Yang, Y. F. Wan, R. F. Chen, S. Wang, S. Y. Si, B. C. Yang, D. Liu, C. Zheng and W. Huang, Predicting Intersystem Crossing Efficiencies of Organic Molecules for Efficient Thermally Activated Delayed Fluorescence, J. Mater. Chem. C, 2019, 7, 9523–9530 RSC.
- R. F. Chen, Y. T. Tang, Y. F. Wan, T. Chen, C. Zheng, Y. Y. Qi, Y. F. Cheng and W. Huang, Promoting Singlet/triplet Exciton Transformation in Organic Optoelectronic Molecules: Role of Excited State Transition Configuration, Sci. Rep., 2017, 7, 6225 CrossRef PubMed.
- L. F. Chen, S. T. Zhang, H. Li, R. F. Chen, L. Jin, K. Yuan, H. H. Li, P. Lu, B. Yang and W. Huang, Breaking the Efficiency Limit of Fluorescent OLEDs by Hybridized Local and Charge-Transfer Host Materials, J. Phys. Chem. Lett., 2018, 9, 5240–5245 CrossRef CAS PubMed.
- T. T. Liu, X. J. Chen, J. Zhao, W. C. Wei, Z. Mao, W. Wu, S. B. Jiao, Y. Liu, Z. Y. Yang and Z. G. Chi, Hybridized Local and Charge-Transfer Excited State Fluorophores Enabling Organic Light-Emitting Diodes with Record High Efficiencies Close to 20%, Chem. Sci., 2021, 12, 5171–5176 RSC.
- M. K. Etherington, J. Gibson, H. F. Higginbotham, T. J. Penfold and A. P. Monkman, Revealing the Spin-Vibronic Coupling Mechanism of Thermally Activated Delayed Fluorescence, Nat. Commun., 2016, 7, 1–7 RSC.
- J. Gibson, A. P. Monkman and T. J. Penfold, The Importance of Vibronic Coupling for Efficient Reverse Intersystem Crossing in Thermally Activated Delayed Fluorescence Molecules, ChemPhysChem, 2016, 17, 2956–2961 CrossRef CAS PubMed.
- Y. Y. Ma, K. Zhang, Y. C. Zhang, Y. Z. Song, L. L. Lin, C. K. Wang and J. Z. Fan, Effects of Secondary Acceptors on Excited-State Properties of Sky-Blue Thermally Activated Delayed Fluorescence Molecules: Luminescence Mechanism and Molecular Design, J. Phys. Chem. A, 2020, 125, 175–186 CrossRef PubMed.
- R. S. Nobuyasu, J. S. Ward, J. Gibson, B. A. Laidlaw, Z. Ren, P. Data, A. S. Batsanov, T. J. Penfold, M. R. Bryce and F. B. Dias, The Influence of Molecular Geometry on the Efficiency of Thermally Activated Delayed Fluorescence, J. Mater. Chem. C, 2019, 7, 6672–6684 RSC.
- J. Gibson and T. Penfold, Nonadiabatic Coupling Reduces the Activation Energy in Thermally Activated Delayed Fluorescence, Phys. Chem. Chem. Phys., 2017, 19, 8428–8434 RSC.
- M. Y. Wong and E. Zysman-Colman, Purely Organic Thermally Activated Delayed Fluorescence Materials for Organic Light-Emitting Diodes, Adv. Mater., 2017, 29, 1605444 CrossRef PubMed.
- T. Lu and F. Chen, Calculation of Molecular Orbital Composition, Acta Chim. Sin., 2011, 69, 2393–2406 CAS.
- K. Shizu, H. Noda, H. Tanaka, M. Taneda, M. Uejima, T. Sato, K. Tanaka, H. Kaji and C. Adachi, Highly Efficient Blue Electroluminescence Using Delayed-Fluorescence Emitters with Large Overlap Density between Luminescent and Ground States, J. Phys. Chem. C, 2015, 119, 26283–26289 CrossRef CAS.
- S. L. Liu, S. S. Liu, Y. Gao, H. Lan, L. L. Lin, C. K. Wang, J. Z. Fan and Y. Z. Song, Acceptor engineering for modulating circularly polarized luminescence and thermally activated delayed fluorescence properties, Mater. Today Chem., 2023, 33, 101700 CrossRef CAS.
|
| This journal is © the Owner Societies 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ,
Chuan-Kui
Wang
,
Jianzhong
Fan
,
Chuan-Kui
Wang
,
Jianzhong
Fan



 represents the thermal vibration correlation function (TVCF) in the non-radiative process. All calculation methods and application of these formulas are also used in Shuai, Peng, Cui and our previous works.34–43 The MOMAP program is used to perform these calculations.44 The calculations of the SOC constant and frequency analyses are performed based on the optimized molecular structures of T1.
represents the thermal vibration correlation function (TVCF) in the non-radiative process. All calculation methods and application of these formulas are also used in Shuai, Peng, Cui and our previous works.34–43 The MOMAP program is used to perform these calculations.44 The calculations of the SOC constant and frequency analyses are performed based on the optimized molecular structures of T1.

 are the x-coordinates of the ith atom in the first and the second structures, respectively. y and z have a similar meaning to x. Through this analysis, we can preliminarily predict and compare the photophysical properties of molecules such as the knr.
are the x-coordinates of the ith atom in the first and the second structures, respectively. y and z have a similar meaning to x. Through this analysis, we can preliminarily predict and compare the photophysical properties of molecules such as the knr.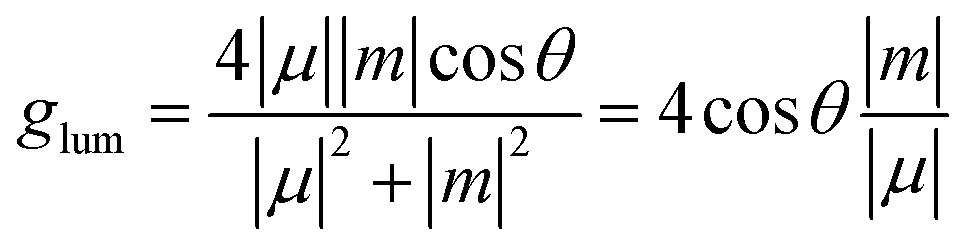





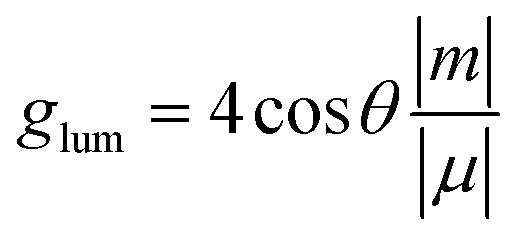 , i.e., the glum is proportional to the absolute value of the μ and inversely proportional to the absolute value of the m. As the μ is two orders of magnitude larger than that of the m, decreasing the former may be an effective strategy for obtaining better circular polarization properties. To verify this, we plotted a line graph of the glum and the μ, as shown in Fig. 6. This is consistent with our conjecture and confirms the effect of μ on the nature of CPL. We find that the three designed molecules have a more desirable glum than the original molecules due to their smaller μ. PTZ-Ax-CN has the smallest μ and the largest glum. Thus, using the donor engineering strategy, the CPL properties are modulated. Compared with the reported molecule (Cz-Ax-CN), three new efficient CP-TADF molecules (DMAC-Ax-CN, PTZ-Ax-CN and PXZ-Ax-CN) with large RISC rates and large glum values are theoretically proposed.
, i.e., the glum is proportional to the absolute value of the μ and inversely proportional to the absolute value of the m. As the μ is two orders of magnitude larger than that of the m, decreasing the former may be an effective strategy for obtaining better circular polarization properties. To verify this, we plotted a line graph of the glum and the μ, as shown in Fig. 6. This is consistent with our conjecture and confirms the effect of μ on the nature of CPL. We find that the three designed molecules have a more desirable glum than the original molecules due to their smaller μ. PTZ-Ax-CN has the smallest μ and the largest glum. Thus, using the donor engineering strategy, the CPL properties are modulated. Compared with the reported molecule (Cz-Ax-CN), three new efficient CP-TADF molecules (DMAC-Ax-CN, PTZ-Ax-CN and PXZ-Ax-CN) with large RISC rates and large glum values are theoretically proposed.

