
Quantum chemical modeling of enantioselective sulfoxidation and epoxidation reactions by indole monooxygenase VpIndA1†
Qinrou
Li
 ab,
Shiqing
Zhang
bc,
Fufeng
Liu
a,
Hao
Su
bc and
Xiang
Sheng
*bc
ab,
Shiqing
Zhang
bc,
Fufeng
Liu
a,
Hao
Su
bc and
Xiang
Sheng
*bc
aCollege of Biotechnology, Tianjin University of Science and Technology, Tianjin, 300457, P. R. China
bTianjin Institute of Industrial Biotechnology, Chinese Academy of Sciences, Tianjin 300308, P. R. China. E-mail: shengx@tib.cas.cn
cNational Center of Technology Innovation for Synthetic Biology, National Engineering Research Center of Industrial Enzymes and Key Laboratory of Engineering Biology for Low-Carbon Manufacturing, Tianjin 300308, P. R. China
First published on 2nd May 2024
Abstract
Indole monooxygenases (IMOs) are enzymes from the family of Group E monooxygenases, requiring flavin adenine dinucleotide (FAD) for their activities. IMOs play important roles in both sulfoxidation and epoxidation reactions. The broad substrate range and high selectivity of IMOs make them promising biocatalytic tools for synthesizing chiral compounds. In the present study, quantum chemical calculations using the cluster approach were performed to investigate the reaction mechanism and the enantioselectivity of the IMO from Variovorax paradoxus EPS (VpIndA1). The sulfoxidation of methyl phenyl sulfide (MPS) and the epoxidation of indene were chosen as the representative reactions. The calculations confirmed that the FADOOH intermediate is the catalytic species in the VpIndA1 reactions. The oxidation of MPS adopts a one-step mechanism involving the direct oxygen-transfer from FADOOH to the substrate and the proton transfer from the –OH group back to FAD, while the oxidation of indene follows a stepwise mechanism involving a carbocation intermediate. It was computationally predicted that VpIndA1 prefers the formation of (S)-product for the MPS sulfoxidation and (1S,2R)-product for the indene epoxidation, consistent with the experimental observations. Importantly, the factors controlling the stereo-preference of the two reactions are identified. The findings in the present study provide valuable insights into the VpIndA1-catalyzed reactions, which are essential for the rational design of this enzyme and other IMOs for industrial applications. It is also worth emphasizing that the quantum chemical cluster approach is again demonstrated to be powerful in studying the enantioselectivity of enzymatic reactions.
1. Introduction
The synthesis of chiral compounds is one of the most attractive fields within the scientific community. The generally low efficiency and low yield in obtaining enantiomerically enriched chemicals pose challenges to asymmetric synthesis. Employing enzymatic reactions emerges as an effective and environmentally benign strategy to overcome the limitations of traditional asymmetric synthesis methods.1–6 Indole monooxygenases (IMOs), a subclass of Group E monooxygenases, were identified in nature to catalyze the oxidation of indole and styrene in their degradation pathways.7–9 These enzymes attract much attention owing to their capacities in catalyzing the enantioselective sulfoxidation and epoxidation reactions.10 The broad substrate scope and high enantioselectivity of IMOs further increase their potential as biocatalytic tools for synthesizing chiral products.11IMOs are members of the flavoprotein monooxygenase (FPMO) family, relying on the cofactor flavin adenine dinucleotide (FAD) for their catalytic abilities.12 The typical catalytic cycle for FPMOs includes the activation of O2 forming a flavin-OO(H) intermediate and incorporating an oxygen atom into the substrate.10,13,14 In contrast to more studied members of the FPMO family, such as Baeyer–Villiger monooxygenases (BVMOs) and p-hydroxybenzoate hydroxylase (PHBH),15–17 IMOs have received comparatively less attention. To promote the application of IMOs, it is crucial to conduct further investigations into the specific reactions catalyzed by these enzymes and to explore the factors responsible for the stereoselectivity in detail.
Recently, an IMO from the bacterium Variovorax paradoxus EPS (VpIndA1) was identified12 and the crystal structures of VpIndA1 were subsequently reported in various forms.18 Structure analysis showed that the binding pocket comprises a number of nonpolar residues such as Phe50, Phe191, Phe201, Leu174, and Phe385.18 Additionally, two ionizable residues (Glu218 and Asp300) that directly interact with each other are also found in the vicinity of the active site (Fig. 1).
 | ||
| Fig. 1 Active site of the wide-type VpIndA1 in complex with the FAD cofactor (PDB: 7Z4X). | ||
The VpIndA1 was demonstrated to be capable of converting methyl phenyl sulfide (MPS) to the (S)-enantiomer of methyl phenyl sulfoxide (MPSO) in the sulfoxidation reaction and converting indene to the (1S,2R)-enantiomer of indene oxide (IO) in the epoxidation reaction.12,18 The reactions were proposed to follow the generally accepted catalytic cycle of FPMOs, wherein the dioxygen is activated to form a peroxide intermediate with flavin.14 Based on the molecular mechanics (MM) force field calculations, the stereo-preferences of VpIndA1 were rationalized by the geometric arrangements and the binding energies of different modes of the substrate.18 Advantageous variants were then designed for improved substrate acceptance and stereoselectivity.18
Although the FPMO-catalyzed reactions have been extensively studied by using different experimental techniques,10–23 there remains a debate regarding the protonation state of the peroxide in the intermediate after the activation of dioxygen by flavin. Namely, it is not clear that the catalytically relevant species is a flavin C4a-hydroperoxide (FADOOH) or a flavin C4a-peroxide (FADOO−) intermediate. By using the double-mixing stopped-flow technique, it was demonstrated that both species are involved in the reaction of the cyclohexanone oxidation catalyzed by cyclohexanone monooxygenase (CHMO), but only the FADOO− is capable of oxygenating cyclohexanone.19 In contrast, on the basis of the rapid acid quench in conjunction with the stopped-flow absorbance and fluorescence, it was suggested that the intermediate participating in the styrene epoxidation by styrene monooxygenase (SMO) is FADOOH.20 The involvement of this intermediate in the catalysis was also proposed for the hydroxylation reactions catalyzed by other FPMOs.21–23 For VpIndA1, the focused FPMO in the present study, the catalytically relevant species in the reaction, remains an open question. Furthermore, VpIndA1 exhibits high stereoselectivity toward various substrates.11 However, the origins of the enantioselectivity of VpIndA1 are obscure.
Various computational chemical methods, such as the hybrid quantum mechanics/molecular mechanics (QM/MM) method, the molecular dynamics (MD) simulation, and the quantum chemical cluster approach, have been used to model FPMO-catalyzed reactions.24–27 Herein, the cluster approach is employed to investigate the enantioselective sulfoxidation and epoxidation reactions catalyzed by VpIndA1. This method has been validated as a powerful tool in studying the enantioselectivity of widely distributed enzymes.28–31 As the addition of O2 to FAD has been established to be rapid both experimentally and computationally,18,32,33 the current study specifically delves into the oxidation processes of MPS and indene after the activation of O2 by FAD (Scheme 1). For the resulting peroxide intermediate, both protonated and deprotonated states are considered. Importantly, the enantioselectivities of VpIndA1 toward MPS and indene are perfectly reproduced by the calculations, and the factors favoring the formation of the (S)-enantiomer of MPSO in the sulfoxidation reaction and the (1S,2R)-enantiomer of IO in the epoxidation reaction are rationalized by detailed analysis on the optimized structures of the transition states and intermediates.
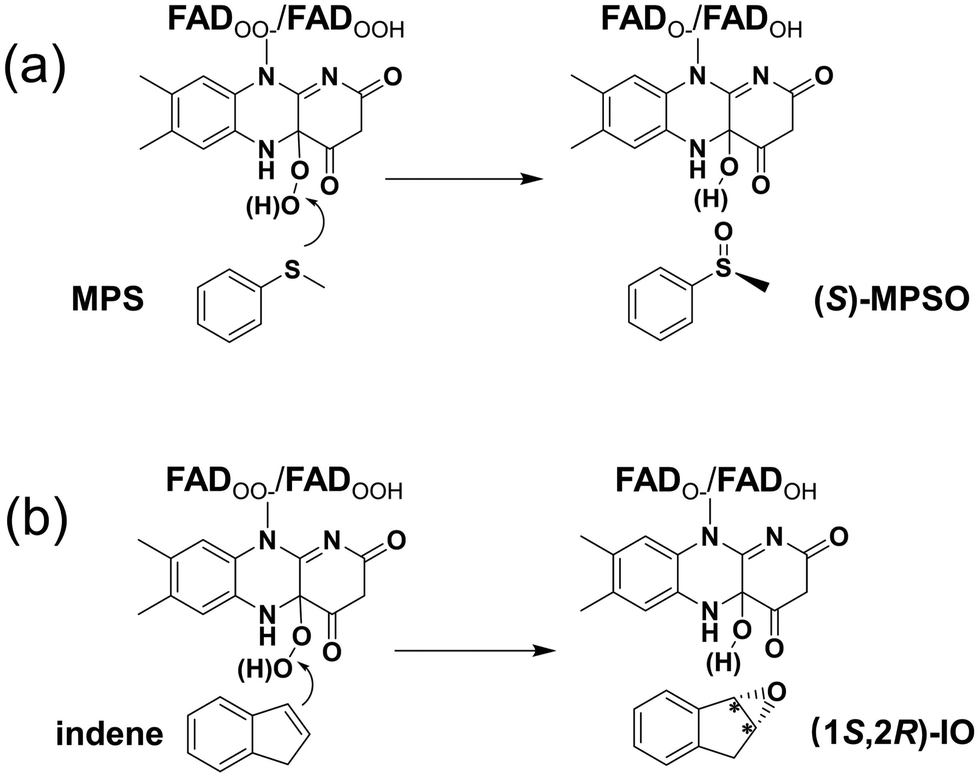 | ||
| Scheme 1 The VpIndA1-catalyzed reactions investigated in the present study: (a) the enantioselective sulfoxidation of methyl phenyl sulfide (MPS) and (b) the enantioselective epoxidation of indene. | ||
2. Computational methods
2.1 Technical details
All the calculations in the current study were performed by using the Gaussian16 C.01 program34 with the B3LYP-D3(BJ) density functional method.35–38 Geometry optimizations were carried out with the 6-31G(d,p) basis set. At the same level as geometry optimization, the single-point energies using the SMD solvation model39 with ε = 4 were calculated to estimate the effect of surroundings. Frequency calculations were performed to obtain zero-point energies (ZPEs) and to characterize the nature of the intermediates and transition states (TSs), ensuring the presence of only positive frequencies for intermediates and only one imaginary frequency for TSs. The animations illustrating the vibrations corresponding to the imaginary frequency are provided in the Supplementary files. Intrinsic reaction coordinate (IRC) analysis was conducted to validate that the TSs connect the right local minima (Fig. S17 and S18, ESI†). Single-point calculations with the large basis set 6-311+G(2d,2p) were carried out to obtain more accurate electronic energies, which were then corrected for SMD solvation and ZPE corrections. The obtained final energies are reported in the present study. All the figures of the optimized structures were prepared by using Open-Source Pymol (https://pymol.org/).2.2 Active site model
The active site model of the enzyme bound with the cofactor and substrate was designed based on the structure of wide-type VpIndA1 crystallized with FAD (PDB ID: 7Z4X).18 The substrate, MPS for the sulfoxidation reaction or indene for the epoxidation reaction, was manually incorporated into the respective model, and the FAD was modified to its FADOO− or FADOOH form. The residues making up the binding sites of FAD and substrate are included in the active site model (Ser45, Ser46, Cys48, Phe50, Ile75, Leu172, Leu174, Val189, Phe191, Phe201, Phe203, Val216, Glu218, Asp300, Pro301, Ile302, Thr303, Gly304, Gln305, Asn308 and Phe385), as schematically drawn in Fig. 2.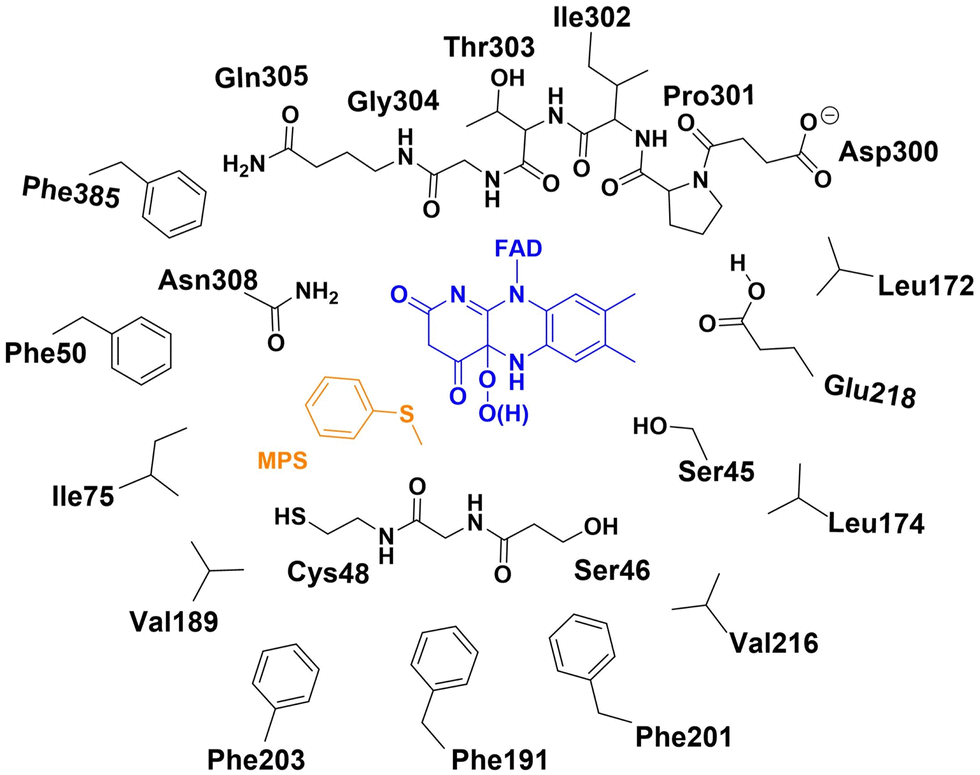 | ||
| Fig. 2 The schematic representation of the active site model for the enzyme bound with FADOO−/FADOOH and MPS. | ||
According to the results from the constant pH molecular dynamics (CpHMD) simulations and the PROPKA sever, the Glu218 residue was modeled in the protonated state and the Asp300 residue was modeled in the ionized form (see the details in the ESI†). In the present study, different protonation states were considered for the peroxide intermediate after O2 activation by FAD, which is the starting structure for the investigation of mechanism and enantioselectivity. Specifically, the two examined species are C(4a)-hydroperoxide (FADOOH) and C(4a)-peroxide (FADOO−). The chirality of C(4a) was determined by analyzing the crystal structure (PDB ID: 7Z4X),18 wherein the hydroperoxide/peroxide group is oriented toward the binding pocket of the substrate. For the model with FADOOH and FADOO−, the overall charge of the system is −1 and −2, respectively. The active site model comprises 351 atoms or 350 atoms for the system with MPS depending on the protonation state of the peroxide intermediate, and 345 atoms or 344 atoms for the system with indene.
The FADOO−/FADOOH and amino acids were truncated in the cluster model, and the hydrogen atoms saturating the truncated carbon were added manually. The truncated carbon and some associated hydrogen atoms were fixed during the geometry optimization processes to avoid unrealistic deviation from the crystal structure (see the fixed atoms in Fig. S1, ESI†). To ensure that the most favorable pathway is reported, geometries with different conformations of the substrate and active site residues were optimized for all the species along the reaction pathways and the lowest-energy one was reported for each species.
3. Results and discussion
As mentioned above, the protonation state of the peroxide in the intermediate after the activation of dioxygen by flavin remains uncertain. Considering this ambiguity, both the protonated (FADOOH) and deprotonated (FADOO−) forms of the peroxide intermediate were considered in the investigation of the mechanism and enantioselectivity of VpIndA1 toward MPS. Interestingly, calculations starting with FADOOH perfectly reproduced the experimental results, specifically favoring the (S)-MPSO enantiomer of the product (Fig. 3 and 4). Conversely, the computationally predicted outcome based on FADOO−, yielding (R)-MPSO, is inconsistent with the experimental observation (Fig. S2, ESI†). In the case of indene, the computational results starting from FADOOH and FADOO− both reproduced the trend of the experimental stereoselective outcome; however, the pathway with FADOOH exhibits a lower barrier compared to that with FADOO− (Fig. S3, ESI†). It can thus be concluded that it is more possible for the protonated FADOOH to be the catalytically involved species in the catalysis. In the following section, the calculation results of the reactions stemming from FADOOH are discussed in detail and the results starting with FADOO− are briefly presented. | ||
| Fig. 3 Optimized structures of the lowest-energy E:MPS complexes with the “Phenyl-left” mode (a) and with the “Phenyl-right” mode (b). The subscript S (in E:MPSS and E:MPSS') and the subscript R (in E:MPSR and E:MPSR') denote the configurations of the respective products originating from this ES complex. The energies, which are provided in parentheses in kcal mol−1, are relative to E:MPSS. For clarity, most of the hydrogen atoms are omitted in the figure. Selected distances are given in Å. | ||
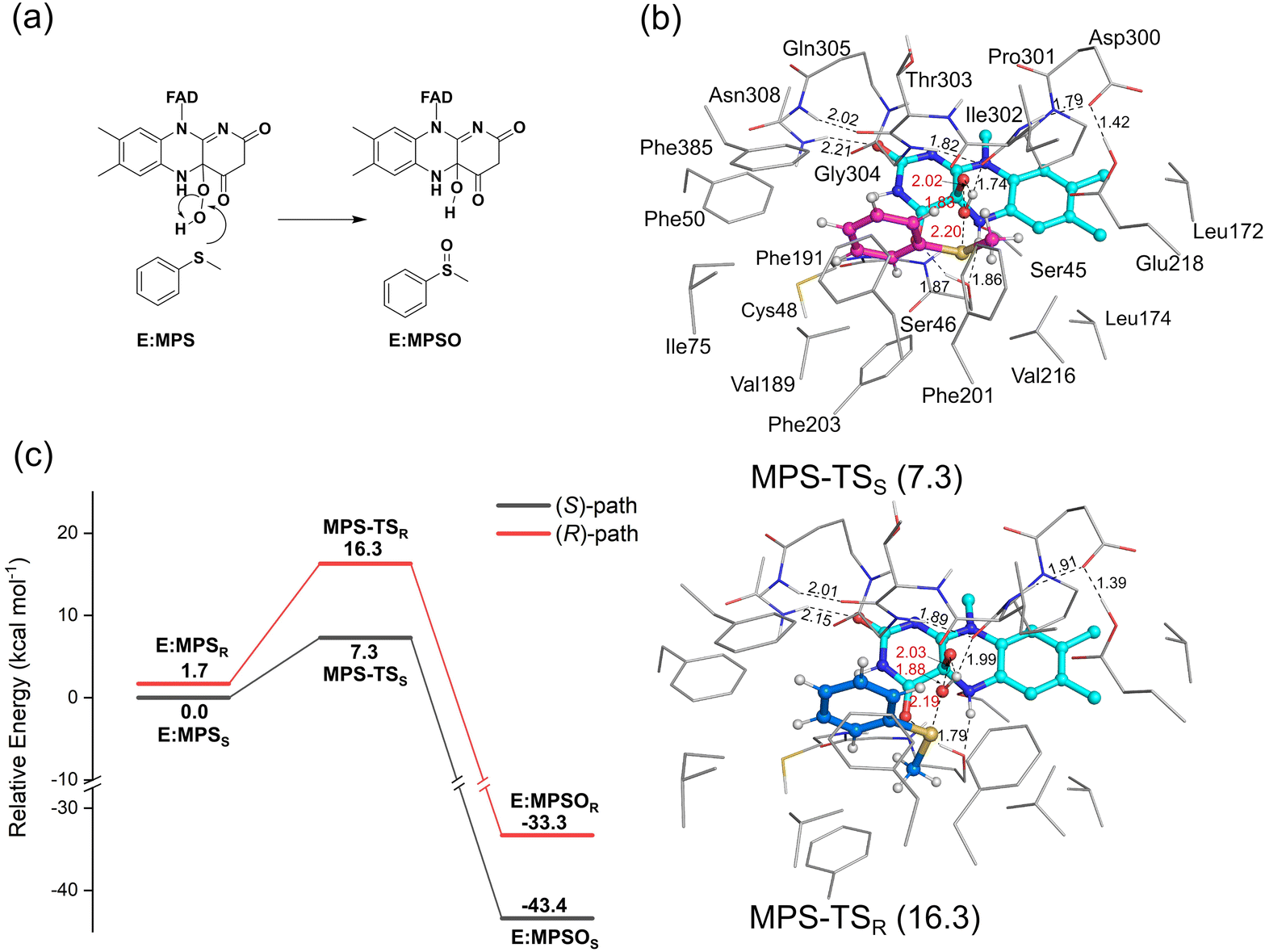 | ||
| Fig. 4 (a) Mechanism of the VpInA1-catalyzed sulfoxidation of MPS and (b) the optimized structures of MPS-TSS and MPS-TSR. (c) The calculated energy profiles for the VpInA1-catalyzed sulfoxidation of MPS, in which the pathways leading to the formation of (S)- and (R)-MPSO are shown in black and red, respectively. The energies relative to E:MPSS are provided in parentheses in kcal mol−1. For clarity, most of the hydrogen atoms are omitted in the figure. Selected distances are given in Å. | ||
3.1 Binding mode of MPS in the active site of VpInA1
To achieve the lowest-energy binding mode, a large number of structures with different conformations of the active site residues and substrates in the active site were optimized and their energies were compared (Fig. 3 for the lowest-energy structures and Fig. S4 for the others, ESI†). The obtained structures can be classified into two types depending on the direction of the MPS phenyl group pointing toward, named “Phenyl-left” mode and “Phenyl-right” mode. The phenyl ring faces toward Phe50 in the “Phenyl-left” mode (Fig. 3a) and toward Phe201 in the “Phenyl-right” mode (Fig. 3b). Depending on how the methyl group of MPS faces the peroxide in the FADOOH intermediate, either S- or R-products will be preferred. For each mode, it was found that the methyl group of MPS can be located at different positions, which decides whether the re or the si face to be attacked by the peroxide in the following oxidation process. For example, in the “Phenyl-left” mode, the binding of the methyl group in the vicinity of Ile302 leads to the peroxide group being located at the si face of the sulfur, and the binding in the vicinity of Phe203 results in the peroxide group being at the re face, for which the oxygen-transfer from the peroxide gives S- and R-products, respectively.A number of enzyme-substrate complexes with relatively low energies were considered for the studies of the sulfoxidation mechanism and enantioselectivity. It was shown that the pathways starting from all the considered structures with “Phenyl-right” mode are associated with prohibitively high barriers and this mode is thus not productive (Fig. S5, ESI†). In contrast, the pathways with the “Phenyl-left” mode were calculated to have reasonable barriers. The lowest-energy structures for the complexes leading to the formation of R- and S-products (called E:MPSR and E:MPSS, respectively) are shown in Fig. 3a. These two structures show high similarities in the hydrogen bond networks between FAD and the active site residues (Ser46 and Asn308). However, in E:MPSS, the methyl group of MPS is located close to Ile302, while in E:MPSR, it orients toward Phe203, leading to an unfavorable steric hindrance. This results in a higher energy of E:MPSR than E:MPSS by 1.7 kcal mol−1.
3.2 Mechanism and enantioselectivity of VpInA1 toward MPS
As mentioned above, a number of starting structures were considered for the following study on the oxidation mechanism and selectivity. The calculations showed that the VpInA1-catalyzed sulfoxidation is a concerted process that involves the attack of the –OH group of the FADOOH at the sulfur of MPS, leading to the S–O bond formation and the O–O bond cleavage. Simultaneously, the proton of –OH transfers back to FAD, giving FADOH in the enzyme-product complexes. This concerted mechanism is consistent with the previous proposal on the other enzymes from the same family.24,25,40 We also attempted to obtain an intermediate without involving the proton transfer, in which case the reaction would follow a stepwise mechanism. However, upon optimization, the proton spontaneously transfers from the –OH group back to FAD. The calculated energies of the lowest-energy transition states (TSs) corresponding to the formation of (S)-MPSO (MPS-TSS) and (R)-MPSO (MPS-TSR) are 7.3 kcal mol−1 and 16.3 kcal mol−1, respectively, relative to E:MPSS (Fig. 4a). Namely, the formation of the (S)-product is much more favored by the reaction compared to the (R)-enantiomer. This trend is indeed consistent with the experimentally observed stereopreference for S-enantiomer with a measured ee value of higher than 99%. Other optimized TS structures with higher energies are provided in Fig. S6, ESI.† The resulting enzyme-product complexes (E:MPSOS and E:MPSOR) have energies of −43.4 kcal mol−1 and −33.3 kcal mol−1 relative to E:MPSS for the S- and R-enantiomers, respectively (Fig. S7, ESI†).By scrutinizing the optimized structures of MPS-TSS and MPS-TSR, the key factors influencing the enantioselectivity of VpInA1 are unveiled. First, it should be emphasized that the phenyl group of the substrate is restricted in the binding pocket by the π–π interactions with surrounding aromatic residues (Phe50, Phe191, Phe201 and Phe385) and consequently occupies similar positions in MPS-TSR and MPS-TSS. However, the methyl group exhibits distinct orientations and can thus develop different interactions with nearby groups in two transition states. In MPS-TSR, unfavorable steric hindrances are identified between the methyl group of the substrate and the flavin group of the cofactor, as well as the Phe203 residue (Fig. 4b for the optimized structures and Fig. S8 (ESI†) for the schematic representation). Furthermore, the transferring –OH group experiences greater stabilization in MPS-TSS than in MPS-TSR, as evidenced by the shorter distance of the hydrogen bond with the backbone carbonyl group of Pro301 in the former (1.74 Å) than the latter (1.99 Å). It is interesting to note that in PpStyA, a styrene monooxygenase (SMO) from Pseudomonas putida that exhibits R-enantiopreference toward MPS, the substrate binding pocket is mainly composed of less bulky residues (see a structural comparison in Fig. S9, ESI†),41,42 allowing the substrate to dynamically adjust its conformation during the reaction. Specifically, the equivalent position of Phe203 in VpInA1 is found to be occupied by a small amino acid isoleucine. These differences provide additional support for the rationale behind the enantioselectivity of VpInA1 toward MPS.
3.3 Mechanism and enantioselectivity of VpInA1 toward indene
Following the strategy in the study of the sulfoxidation reaction, a number of structures with different orientations of indene were considered in the calculations to ensure that the lowest-energy binding mode was obtained. Similar to that for MPS, the binding modes of indene to VpInA1 can also be classified into two types: “Phenyl-left” mode and “Phenyl-right” mode, in which the phenyl ring of indene faces toward Phe50 and Phe201, respectively (Fig. S10 and S11 for optimized structures, ESI†). Depending on how the methylene group of indene orientates, the binding modes can lead to the formation of either (1S,2R)- or (1R,2S)-IO. Taking the “Phenyl-left” mode as an illustration, when the methylene group of indene is directed toward Phe203, it leads to the formation of (1S,2R)-IO, whereas an orientation toward Ile302 yields (1R,2S)-IO.Similar to the sulfoxidation reaction catalyzed by VpInA1, the lowest-energy E:indene1R,2S complex, which results in the formation of (1R,2S)-IO, falls into the “Phenyl-left” type (Fig. S10, ESI†). The calculated energy of E:indene1S,2R leading to the other enantiomer (1S,2R)-IO is 0.9 kcal mol−1 higher than that of E:indene1R,2S. Structure analysis showed that the two complexes have comparable hydrogen bond networks within the active site. However, the difference in the orientation of the methylene group of indene in the two structures leads to an undesirable steric hindrance between the methylene group and nearby residues in E:indene1S,2R, which is not present in E:indene1R,2S. Again, similar to the sulfoxidation reaction, the pathways of indene epoxidation with the “Phenyl-right” mode are associated with prohibitively high barriers (Fig. S12, ESI†) and this mode is not productive here either.
The calculations reveal that the indene epoxidation catalyzed by VpInA1 follows a stepwise mechanism involving a carbocation intermediate. Specifically, the reaction pathway initiates with the –OH group of FADOOH attacking the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of indene, forming a carbocation intermediate, succeeded by a proton transfer from the –OH group back to FAD. According to the calculations, the proton transfer is a barrier-less process and the final product has an energy of ca. 20 kcal mol−1 lower than that of the intermediate, indicating that the carbocation intermediate will rapidly convert to the final product once formed. The lowest-energy TSs resulting in the formation of (1R,2S)-product (indene-TS1R,2S) and (1S,2R)-product (indene-TS1S,2R) have energies of 16.6 kcal mol−1 and 13.4 kcal mol−1, respectively, relative to E:indene1R,2S (Fig. 5, see other optimized TS structures in Fig. S13, ESI†). Namely, the pathway leading to the (1S,2R)-product is 3.2 kcal mol−1 lower than that of the (1R,2S)-product. Experimentally, the (1S,2R)-enantiomer was indeed the preferred product with an ee value of 35%. The computational trend is thus in agreement with the experimental results.
C bond of indene, forming a carbocation intermediate, succeeded by a proton transfer from the –OH group back to FAD. According to the calculations, the proton transfer is a barrier-less process and the final product has an energy of ca. 20 kcal mol−1 lower than that of the intermediate, indicating that the carbocation intermediate will rapidly convert to the final product once formed. The lowest-energy TSs resulting in the formation of (1R,2S)-product (indene-TS1R,2S) and (1S,2R)-product (indene-TS1S,2R) have energies of 16.6 kcal mol−1 and 13.4 kcal mol−1, respectively, relative to E:indene1R,2S (Fig. 5, see other optimized TS structures in Fig. S13, ESI†). Namely, the pathway leading to the (1S,2R)-product is 3.2 kcal mol−1 lower than that of the (1R,2S)-product. Experimentally, the (1S,2R)-enantiomer was indeed the preferred product with an ee value of 35%. The computational trend is thus in agreement with the experimental results.
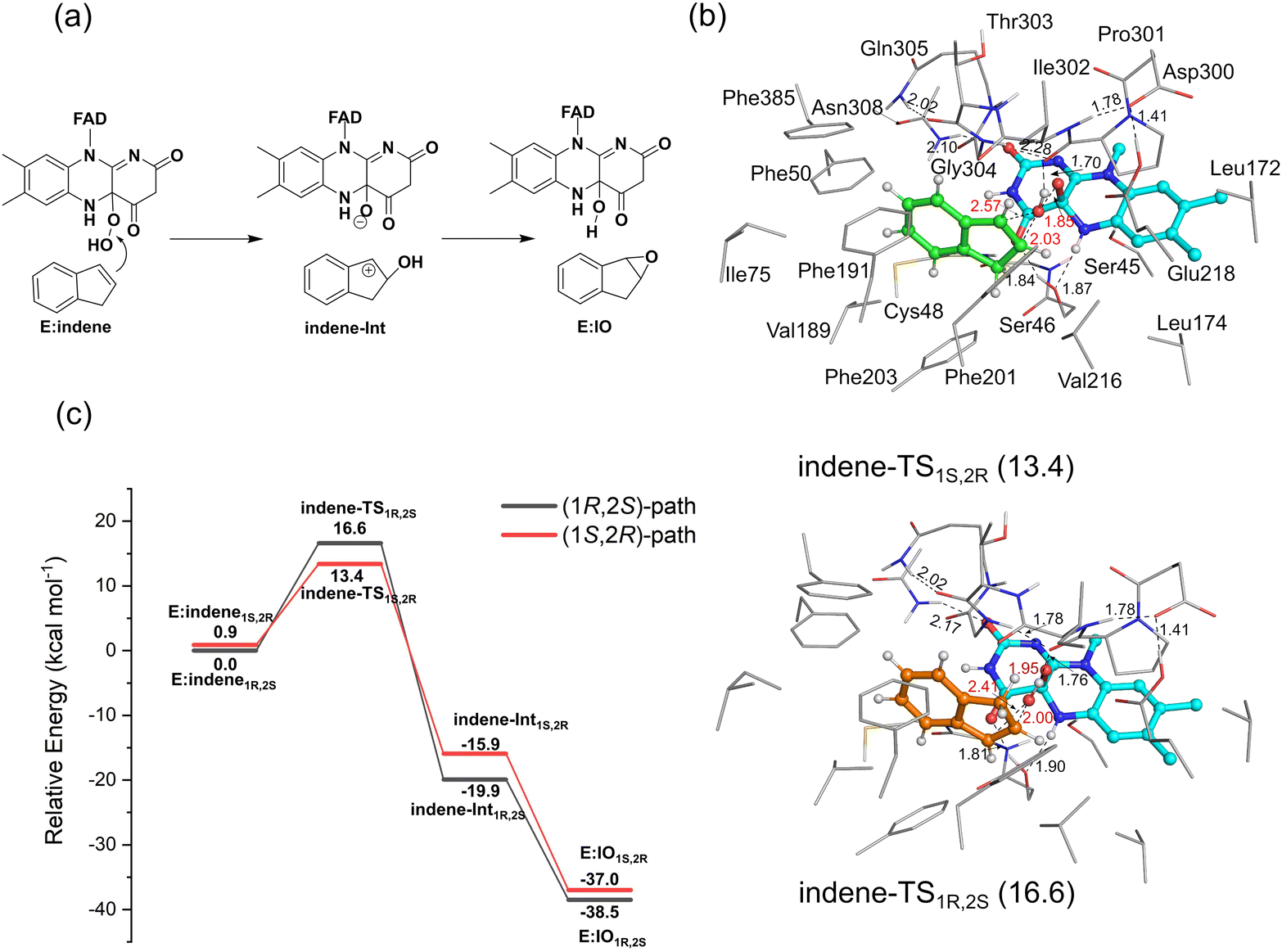 | ||
| Fig. 5 (a) Mechanism of the VpInA1-catalyzed indene epoxidation and (b) the optimized structures of indene-TS1R,2S and indene-TS1S,2R. The energies, provided in parentheses in kcal mol−1, are all referenced relative to E:indene1R,2S. (c) The calculated energy profiles for the VpInA1-catalyzed indene epoxidation, in which the pathways leading to the formation of (1R,2S)- and (1S,2R)-IO are shown in black and red, respectively. For clarity, most of the hydrogen atoms are omitted in the figure. Selected distances are given in Å. | ||
Experimentally, the Phe191Met/Phe201Leu/Ile302Val mutant exhibited an increased stereo-selectivity with an ee value of 99.80%.18 Using the same active site model and methodology mentioned above, the reaction pathways of this mutant were also studied (Fig. S16 for the optimized structures, ESI†). The calculated energy difference between the two TSs increases from 3.2 kcal mol−1 for the wide-type enzyme to 7.2 kcal mol−1 for the mutant, reproducing thus the experimental trend. These results emphasize the robust capabilities of the quantum chemical cluster approach in elucidating the enantioselectivity of enzymatic reactions.
It is worth noting that the trends observed in the energies of E:indene and E:IO in the pathways leading to two enantiomers are reversed compared to the TSs that dictate the enantiopreference of the reaction. Therefore, relying solely on the energies of E:indene and E:IO for rationalizing the enantioselectivity of VpInA1 is misleading. This highlights the importance of investigating the entire reaction pathway to accurately pinpoint the factors controlling selectivity. Similar conclusions have also been drawn from the quantum chemical studies on other enzymes.43,44
4. Conclusions
The broad substrate range and high selectivity of indole monooxygenases (IMOs) make them valuable candidates for asymmetric synthesis. Nevertheless, the unclear reaction mechanisms, along with the ambiguous origin of the enantioselectivity, have hindered their industrial applications. In the present work, the sulfoxidation and indene epoxidation catalyzed by the IMO from Variovorax paradoxus EPS (VpIndA1), an enzyme from the family of IMOs, are studied by using the quantum chemical cluster approach.The calculations show that the protonated FADOOH rather than the deprotonated FADOO− is the catalytically relevant species in the VpIndA1-catalyzed reactions. For both methyl phenyl sulfide (MPS) and indene substrates, the preferred binding mode is that the phenyl ring on the substrate is orientated toward Phe50. Mechanistic investigations reveal distinct pathways for the substrate oxidation by VpInA1. The oxidation of MPS follows a one-step mechanism, consisting of the direct oxygen transfer from FADOOH to the substrate, accompanied by proton transfer of the –OH group back to FAD. In contrast, the oxidation of indene proceeds via a stepwise mechanism involving a carbocation intermediate. Upon analyzing the optimized structures of the corresponding transition states, it can be concluded that the aromatic residues within the active site, especially Phe203, play significant roles in controlling the enantioselectivity of VpIndA1.
The details of the reaction mechanisms obtained in the present study provide valuable information on the IMO-catalyzed reactions. It holds considerable significance in facilitating the systematic design of enzyme variants with tailored properties. The current study also emphasizes the robust capabilities of the quantum chemical cluster approach in elucidating the reaction mechanism and selectivity of enzymes.
Author contributions
X. S. designed the project and supervised the project; Q. L. performed research and analyzed data; S. Z, F. L., and H. S. contributed also to the calculations and data analysis; Q. L. and X. S. wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the National Key R&D Program of China (2021YFA0911500), the Tianjin Synthetic Biotechnology Innovation Capacity Improvement Project (TSBICIP-CXRC-026) and the National Natural Science Foundation of China (22103095) for the financial support. A portion of the calculations in this study were performed on the ORISE Supercomputer.References
- C. M. Clouthier and J. N. Pelletier, Chem. Soc. Rev., 2012, 41, 1585–1605 RSC.
- C. K. Winkler, J. H. Schrittwieser and W. Kroutil, ACS Cent. Sci., 2021, 7, 55–71 CrossRef CAS PubMed.
- M. Hall, RSC Chem. Biol., 2021, 2, 958–989 RSC.
- S. Wu, R. Snajdrova, J. C. Moore, K. Baldenius and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2021, 60, 88–119 CrossRef CAS PubMed.
- D. Yi, T. Bayer, C. P. S. Badenhorst, S. Wu, M. Doerr, M. Höhne and U. T. Bornscheuer, Chem. Soc. Rev., 2021, 50, 8003–8049 RSC.
- A. R. Alcántara, P. Domínguez de María, J. A. Littlechild, M. Schürmann, R. A. Sheldon and R. Wohlgemuth, ChemSusChem, 2022, 15, e202102709 CrossRef PubMed.
- K. E. O'Connor, A. D. Dobson and S. Hartmans, Appl. Environ. Microbiol., 1997, 63, 4287–4291 CrossRef PubMed.
- G.-H. Lin, H.-P. Chen and H.-Y. Shu, PLoS One, 2015, 10, e0138798 CrossRef PubMed.
- M. Sadauskas, J. Vaitekūnas, R. Gasparavičiūtė and R. Meškys, Appl. Environ. Microbiol., 2017, 83, e01453–e01417 CrossRef CAS PubMed.
- C. E. Paul, D. Eggerichs, A. H. Westphal, D. Tischler and W. J. H. van Berkel, Biotechnol. Adv., 2021, 51, 107712 CrossRef CAS PubMed.
- T. Heine, A. Scholtissek, S. Hofmann, R. Koch and D. Tischler, ChemCatChem, 2020, 12, 199–209 CrossRef CAS.
- D. Tischler, R. Schwabe, L. Siegel, K. Joffroy, S. R. Kaschabek, A. Scholtissek and T. Heine, Molecules, 2018, 23, 809 CrossRef PubMed.
- A. Mattevi, Trends Biochem. Sci., 2006, 31, 276–283 CrossRef CAS PubMed.
- E. Romero, J. R. Gómez Castellanos, G. Gadda, M. W. Fraaije and A. Mattevi, Chem. Rev., 2018, 118, 1742–1769 CrossRef CAS PubMed.
- H. Leisch, K. Morley and P. C. K. Lau, Chem. Rev., 2011, 111, 4165–4222 CrossRef CAS PubMed.
- S. Montersino, D. Tischler, G. T. Gassner and W. J. H. van Berkel, Adv. Synth. Catal., 2011, 353, 2301–2319 CrossRef CAS.
- M. J. L. J. Fürst, A. Gran-Scheuch, F. S. Aalbers and M. W. Fraaije, ACS Catal., 2019, 9, 11207–11241 CrossRef.
- J. Kratky, D. Eggerichs, T. Heine, S. Hofmann, P. Sowa, R. H. Weiße, D. Tischler and N. Sträter, Angew. Chem., Int. Ed., 2023, 62, e202300657 CrossRef CAS PubMed.
- D. Sheng, D. P. Ballou and V. Massey, Biochemistry, 2001, 40, 11156–11167 CrossRef CAS PubMed.
- A. Kantz and G. T. Gassner, Biochemistry, 2011, 50, 523–532 CrossRef CAS PubMed.
- M. G. Taylor and V. Massey, J. Biol. Chem., 1990, 265, 13687–13694 CrossRef CAS PubMed.
- J. Sucharitakul, P. Chaiyen, B. Entsch and D. P. Ballou, J. Biol. Chem., 2006, 281, 17044–17053 CrossRef CAS PubMed.
- R. Baron, C. Riley, P. Chenprakhon, K. Thotsaporn, R. T. Winter, A. Alfieri, F. Forneris, W. J. H. van Berkel, P. Chaiyen, M. W. Fraaije, A. Mattevi and J. A. McCammon, Proc. Natl. Acad. Sci., 2009, 106, 10603–10608 CrossRef CAS PubMed.
- A. C. C. Barbosa, R. P. P. Neves, S. F. Sousa, M. J. Ramos and P. A. Fernandes, ACS Catal., 2018, 8, 9298–9311 CrossRef CAS.
- Y. Özkılıç and N. Ş. Tüzün, J. Phys. Chem. A, 2019, 123, 3149–3159 CrossRef PubMed.
- A. Rodríguez Benítez, S. E. Tweedy, S. A. Baker Dockrey, A. L. Lukowski, T. Wymore, D. Khare, C. L. Brooks, III, B. A. Palfey, J. L. Smith and A. R. H. Narayan, ACS Catal., 2019, 9, 3633–3640 CrossRef PubMed.
- Y. Dong, T. Li, S. Zhang, J. Sanchis, H. Yin, J. Ren, X. Sheng, G. Li and M. T. Reetz, ACS Catal., 2022, 12, 3669–3680 CrossRef CAS.
- F. Himo, J. Am. Chem. Soc., 2017, 139, 6780–6786 CrossRef CAS PubMed.
- X. Sheng and F. Himo, J. Am. Chem. Soc., 2019, 141, 11230–11238 CrossRef CAS PubMed.
- X. Sheng, M. Kazemi, F. Planas and F. Himo, ACS Catal., 2020, 10, 6430–6449 CrossRef CAS.
- X. Sheng and F. Himo, Acc. Chem. Res., 2023, 56, 938–947 CrossRef CAS PubMed.
- J. Sucharitakul, C. Tongsook, D. Pakotiprapha, W. J. H. van Berkel and P. Chaiyen, J. Biol. Chem., 2013, 288, 35210–35221 CrossRef CAS PubMed.
- S. Visitsatthawong, P. Chenprakhon, P. Chaiyen and P. Surawatanawong, J. Am. Chem. Soc., 2015, 137, 9363–9374 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- R. D. Bach and O. Dmitrenko, J. Phys. Chem. B, 2003, 107, 12851–12861 CrossRef CAS.
- U. E. Ukaegbu, A. Kantz, M. Beaton, G. T. Gassner and A. C. Rosenzweig, Biochemistry, 2010, 49, 1678–1688 CrossRef CAS PubMed.
- J. Nikodinovic-Runic, L. Coulombel, D. Francuski, N. D. Sharma, D. R. Boyd, R. M. O. Ferrall and K. E. O’Connor, Appl. Microbiol. Biotechnol., 2013, 97, 4849–4858 CrossRef CAS PubMed.
- S. Moa and F. Himo, J. Inorg. Biochem., 2017, 175, 259–266 CrossRef CAS PubMed.
- M. Prejanò, X. Sheng and F. Himo, ChemistryOpen, 2022, 11, e202100250 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp00552j |
| This journal is © the Owner Societies 2024 |