
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Molecular machines working at interfaces: physics, chemistry, evolution and nanoarchitectonics
Katsuhiko
Ariga
 *ab,
Jingwen
Song
c and
Kohsaku
Kawakami
cd
*ab,
Jingwen
Song
c and
Kohsaku
Kawakami
cd
aResearch Center for Materials Nanoarchitectonics, National Institute for Materials Science (NIMS), 1-1 Namiki, Tsukuba 305-0044, Japan. E-mail: ARIGA.Katsuhiko@nims.go.jp
bGraduate School of Frontier Sciences, The University of Tokyo, 5-1-5 Kashiwa-no-ha, Kashiwa 277-8561, Japan
cResearch Center for Functional Materials, National Institute for Materials Science (NIMS), 1-1 Namiki, Tsukuba 305-0044, Ibaraki, Japan
dGraduate School of Pure and Applied Sciences, University of Tsukuba, 1-1-1 Tennodai, Tsukuba 305-8577, Ibaraki, Japan
First published on 9th April 2024
Abstract
As a post-nanotechnology concept, nanoarchitectonics combines nanotechnology with advanced materials science. Molecular machines made by assembling molecular units and their organizational bodies are also products of nanoarchitectonics. They can be regarded as the smallest functional materials. Originally, studies on molecular machines analyzed the average properties of objects dispersed in solution by spectroscopic methods. Researchers' playgrounds partially shifted to solid interfaces, because high-resolution observation of molecular machines is usually done on solid interfaces under high vacuum and cryogenic conditions. Additionally, to ensure the practical applicability of molecular machines, operation under ambient conditions is necessary. The latter conditions are met in dynamic interfacial environments such as the surface of water at room temperature. According to these backgrounds, this review summarizes the trends of molecular machines that continue to evolve under the concept of nanoarchitectonics in interfacial environments. Some recent examples of molecular machines in solution are briefly introduced first, which is followed by an overview of studies of molecular machines and similar supramolecular structures in various interfacial environments. The interfacial environments are classified into (i) solid interfaces, (ii) liquid interfaces, and (iii) various material and biological interfaces. Molecular machines are expanding their activities from the static environment of a solid interface to the more dynamic environment of a liquid interface. Molecular machines change their field of activity while maintaining their basic functions and induce the accumulation of individual molecular machines into macroscopic physical properties molecular machines through macroscopic mechanical motions can be employed to control molecular machines. Moreover, research on molecular machines is not limited to solid and liquid interfaces; interfaces with living organisms are also crucial.
 Katsuhiko Ariga | Katsuhiko Ariga received his PhD degree from the Tokyo Institute of Technology in 1990. He joined the National Institute for Materials Science (NIMS) in 2004 and is currently the leader of the Supermolecules Group and a senior scientist with special missions of Research Centre for Materials Nanoarchitectonics (MANA), NIMS. He is also appointed as a professor in The University of Tokyo. |
 Jingwen Song | Jingwen Song received her PhD degree in 2021 from The University of Tokyo under the guidance of Professor Katsuhiko Ariga. She also studied in the Supermolecules Group at the World Premier International (WPI) Research Centre for Materials Nanoarchitectonics (MANA), National Institute for Materials Science (NIMS) from 2018 to 2021. She is currently a postdoctoral researcher in the Medical Soft Matter group, NIMS. |
 Kohsaku Kawakami | Kohsaku Kawakami received his PhD degree in Chemical Engineering in 2000 from Kyoto University. After working for pharmaceutical companies including Shionogi and Merck, he joined National Institute for Materials Science in 2006. He is currently a group leader of the Medical Soft Matter group and a director of the Material Open Platform for Pharmaceutical Science. He also serves as a professor at the University of Tsukuba. |
1. Introduction
Materials science has been developed to solve various social problems such as environmental,1 energy,2 and biomedical issues.3 It has become clear that the regulations of microstructures and nanostructures in materials have significant impacts on their functions. In materials science, precise structural control has become increasingly important,4 in addition to the development of traditional scientific disciplines such as organic chemistry,5 inorganic chemistry,6 polymer chemistry,7 coordination chemistry,8 supramolecular chemistry,9 biochemistry,10 and other materials sciences.11 In other words, the performance of the same material can be greatly enhanced by precisely controlling its nanostructure. Especially, the development of nanotechnology since the 20th century has encouraged this trend of materials research. Techniques that allow high-resolution observation of atoms and molecules,12 as well as have the ability to measure the properties of single molecules,13 reveal the special properties of nanostructures. At the same time, methodologies have also been developed in supramolecular chemistry and coordination chemistry to fabricate precise nanostructured materials from molecules and ions by skilfully utilizing molecular recognition phenomena14 and molecular assembly formation.15 Just as nanotechnology has changed the course of science as a breakthrough concept, some concepts should be proposed that integrate these trends. As a post-nanotechnology concept,16 nanoarchitectonics has been proposed as the integration of nanotechnology with organic chemistry, supramolecular chemistry, materials science, microfabrication engineering, and biotechnology to develop functional materials (Fig. 1).17 Nanoarchitectonics elevates nanotechnology to an advanced materials science. Further development by incorporating new technologies such as materials informatics is also anticipated.18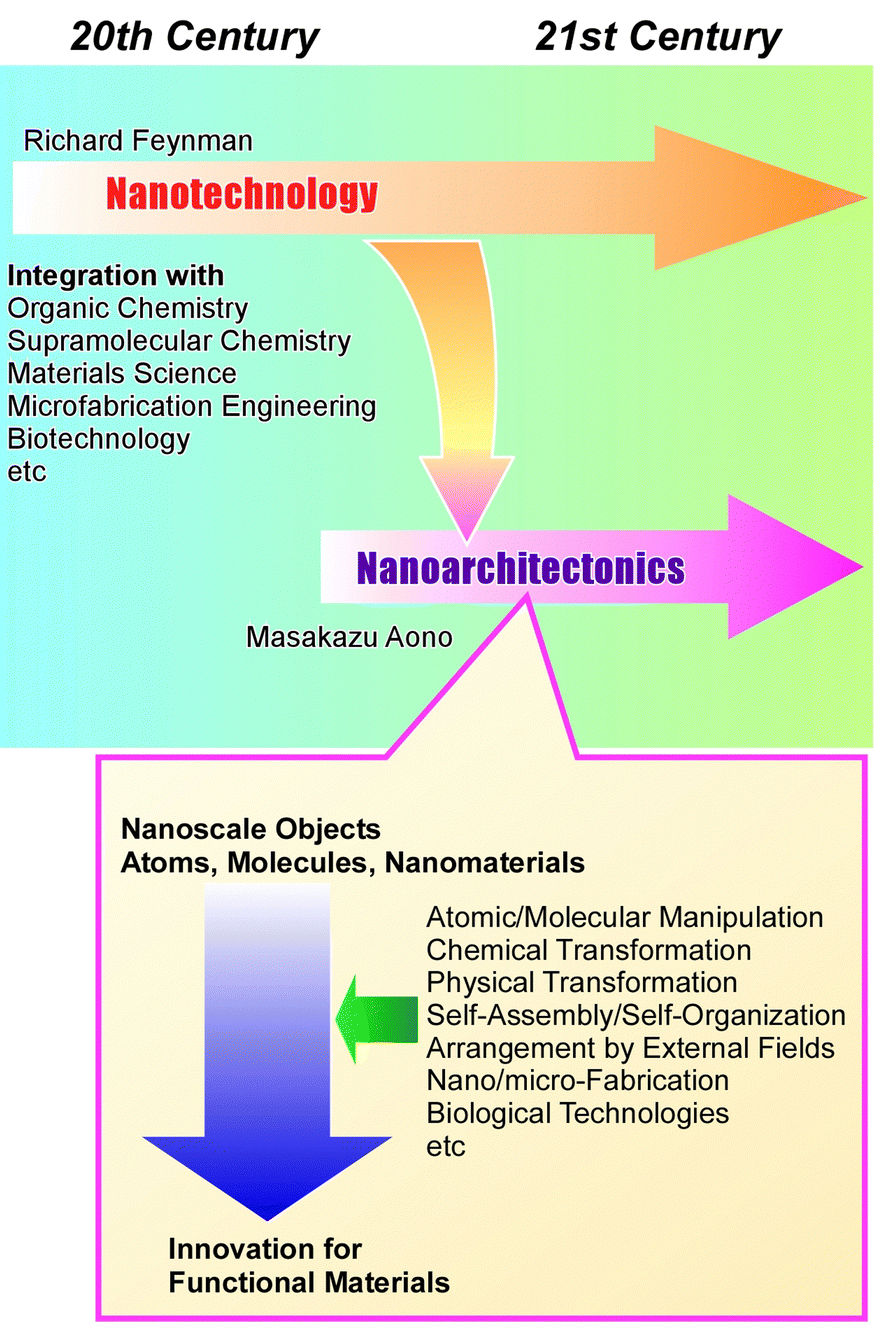 | ||
| Fig. 1 Outline of the nanoarchitectonics concept as the post-nanotechnology concept. | ||
As Richard Feynman proposed nanotechnology in the 20th century,19 nanoarchitectonics was proposed by Masakazu Aono in the 21st century (or the very end of the 20th century).20 Nanoarchitectonics is not based on a simple unitary assembly based on equilibrium as in self-assembly, but a combination of options from atomic/molecular manipulation, chemical transformation (organic synthesis), physical transformation, self-assembly/self-organization, arrangement by external fields, nano/micro-fabrication, biological technologies, etc. to architect functional materials.21 Compared to equilibrium-based aggregate formation, asymmetric and hierarchical structures can be easily created.22 Since all matter is originally made of atoms and molecules, this methodology is applicable regardless of materials or applications. It can be likened to the Theory of Everything in physics,23 and can be called the Method for Everything in materials science.24 The application of this methodology extends from the basics, such as material synthesis25 and structural control,26 to applied fields, such as sensors,27 devices,28 environment,29 energy,30 basic biochemistry,31 and biomedical applications.32
Here, molecular machines are considered as contents of nanoarchitectonics. Molecular machines are molecules or molecular assemblies that perform specific movements and have properties defined like general machines. Molecular machines made by assembling molecular units and their organizational bodies are also products of nanoarchitectonics.33 Looking at the progress of molecular machine research, it sometimes looks like the evolutionary process of living organisms.34 Organisms originated and evolved in the oceans and then exploded on land. Also, organisms that functioned in single cells developed their functions by becoming multicellular. These elements can be seen in the nanoarchitectonics of molecular machines. Interestingly, we see such medium-induced evolution in molecular machines (Fig. 2). Evolution of molecular machines is driven by technical advancements in analyses and fabrications. Originally, studies on molecular machines have analyzed the average properties of objects dispersed in solution by spectroscopic methods.35 This is exactly how unicellular organisms arose in the ocean. Functional molecular systems perform diverse functions by forming supramolecules and aggregates.36 This is like the emergence of multicellular organisms in the ocean. The development of analytical techniques through nanotechnology has made it possible to observe individual molecules at high resolution. Molecular machines, nanocars, and other structures have been observed on solid substrates.37 This is like tracing the history of organisms moving from the oceans to the land. Studies at the interface of molecular machines facilitated this evolution.
 | ||
| Fig. 2 Progress of molecular machine research, it sometimes looks like the evolutionary process based on medium-induced evolution. | ||
The evolution of molecular machines also requires further advances. High-resolution observation of molecular machines is usually done under high vacuum and cryogenic conditions. However, for their practical use, molecular machines need to be able to function under ambient conditions. It is also necessary to work with macroscopic stimuli and motions. These conditions are met in dynamic interfacial environments such as the surface of water at room temperature.38 At the air–water interface, amphiphilic molecules form monolayer structures according to molecular machine research under ambient conditions.39 In that environment, the molecular recognition ability through hydrogen bonding and other means has been demonstrated to be much higher than that in water.40 In addition, monolayers on the water surface can deform laterally on a macroscopic scale such as tens of centimeters, and this movement is coupled with nanometer-sized conformational changes of molecular machines in the monolayer.41 The force applied to the molecule at that time is only a few pN to several tens of pN per molecule,42 which is much smaller than the force required for photoisomerization as molecular actions are typically used in supramolecular control of conventional molecular machines. In other words, the nanoarchitectonics approach of assembling molecular machines as a monolayer at a liquid interface enables delicate machine manipulation similar to that of living organisms.43 From this perspective, liquid interfaces are becoming a new playground for molecular machine research.
Molecular machines evolve while changing the environment in which they operate. This review summarizes the trends of molecular machines that continue to evolve under the concept of nanoarchitectonics in interfacial environments. Some recent examples of molecular machines and supramolecular functional structures in solution are briefly introduced first. This is followed by an overview of studies of molecular machines and similar supramolecular structures in various interfacial environments. The interfacial environments are classified into (i) solid interfaces, (ii) liquid interfaces, and (iii) various material and biological interfaces. In addition to the importance of interfaces as a place to accurately evaluate one of the molecular machines, interfaces that form a group and perform a harmonized function will be discussed.
2. Molecular and supramolecular machines in solutions
The basis of molecular machine research is the observation and evaluation of the function of molecules with well-defined structures built based on organic and supramolecular chemistry, which were originally studied in solution systems. That research trend continues, and many molecular machines and supramolecular systems have been developed based on great molecular designs.44 However, it is not the purpose of this review to introduce them all exhaustively. Here, an overview is provided of the importance of architecting supramolecular systems that function like machines at the nano-level, supplemented with a few examples.Molecular assembly structures with machine-like or similar functions have been proposed one after another. For example, one-dimensional (1D) channel structures with a specific size of space, entrance and exit, are thought to be able to transport guest molecules from the entrance to the exit. A recent review by Ogoshi and co-workers describes the fabrication of molecular-scale continuous 1D channels from pillar[n]arenes.45 By covalently linking pillar[n]arene units, continuous and discrete 1D channels can be synthesized. Molecular cages are also being considered to demonstrate the specific properties of isolated molecules. When a substrate is confined in an isolated cavity, it is placed in a distinctly different situation than in a bulk solution. A recent review article by Takezawa and Fujita discusses trends in coordination-type molecular cages (Fig. 3).46 By confining molecules within a cage, the cavity can trap, arrange, fold, compress, and twist the guest molecules. Sawada and Fujita use metallo-peptide chains to architect such self-assembled nanospaces.47 Based on the cooperative process of peptide self-folding and metal coordination, they have succeeded in building unprecedented and geometrically well-defined nanostructures by passing multiple metal-peptide rings. Haino and co-workers have used calix[4]arene oligomers as a nanospace continuum and have performed conformational analysis of these oligomers.48 These examples illustrate recent developments in the art of architecting nanospace as a functional field through skilful consideration of molecular designs.
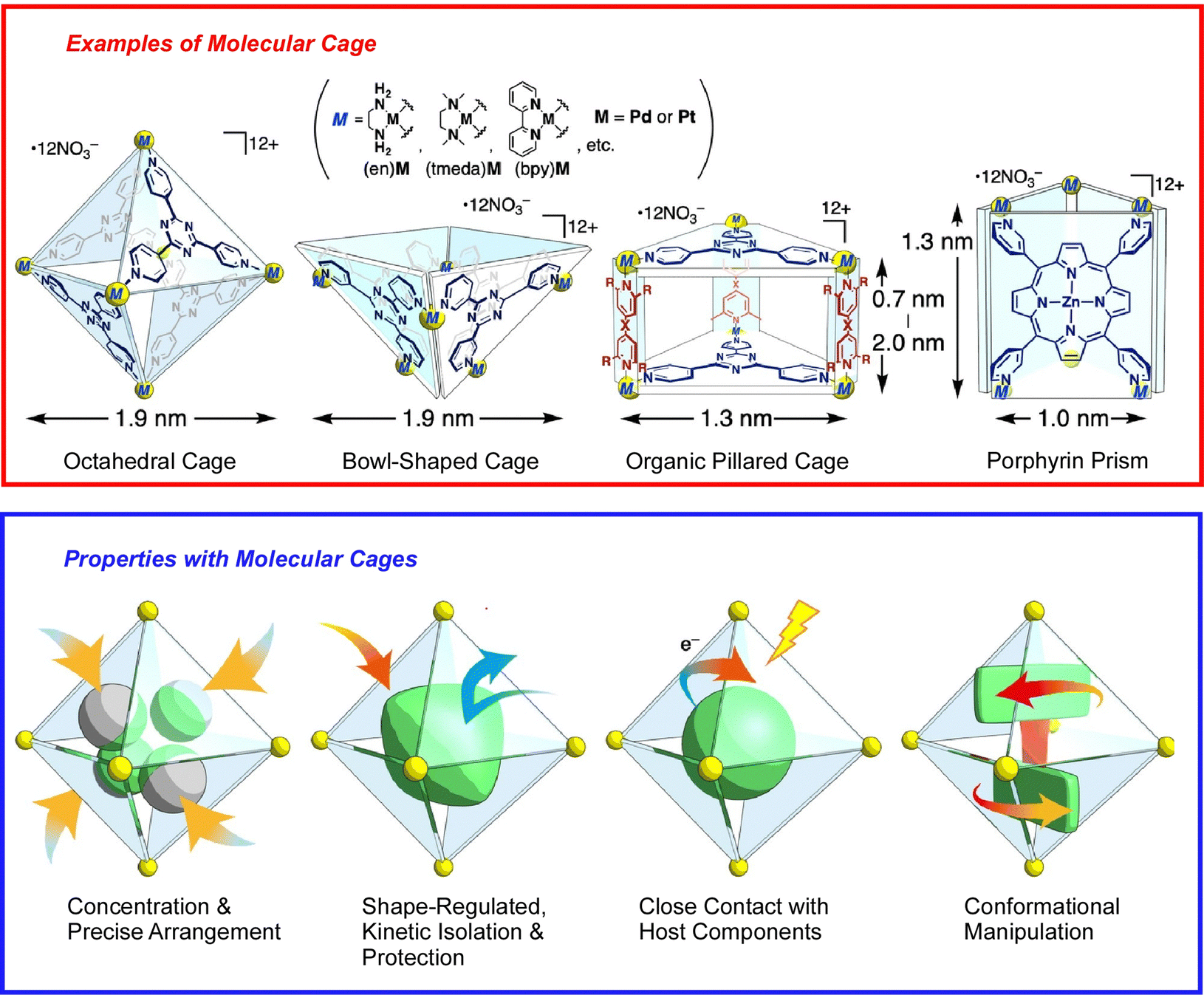 | ||
| Fig. 3 Coordination-type molecular cages with capability of confining molecules for trapping, arrangement, folding, compression, and twisting. Reprinted with permission from ref. 46. Copyright 2021 Oxford University Press. | ||
Unique examples of molecular memory machines utilizing the helical structure of polymers have also been reported. A recent review article by Yashima and Maeda describes synthetic helical polymers with static and dynamic memory functions of polymer helicity.49 Based on molecular interaction and external stimuli, polymer conformation is often trapped in the local energy minimum thermodynamically. This behavior works like memorization of particular conformation such as fixed helicity. The helix induction and subsequent dynamic and static memory strategies do not require specific chiral monomers or chiral catalysts and initiators for the synthesis of helical polymers. Under this condition, both right-handed and left-handed helical polymers can be synthesized. The two systems shown in Fig. 4 are the first to demonstrate helix induction and subsequent static memory, possessing complementary properties in terms of the time required for memory and the stability of the static helicity memory. The process shown on the right panels requires a longer time than the system in the left panel for helix induction and subsequent static helix memory at higher temperatures. The latter has a higher helix-induction barrier, which allows for further modification while maintaining static helical memory, further improving the stability of static helical memory. According to the merits of these complementary static memory properties, they have successfully developed a switchable chiral stationary phase for HPLC, a chiral catalyst, and a highly sensitive colorimetric/fluorescent chiral sensor.
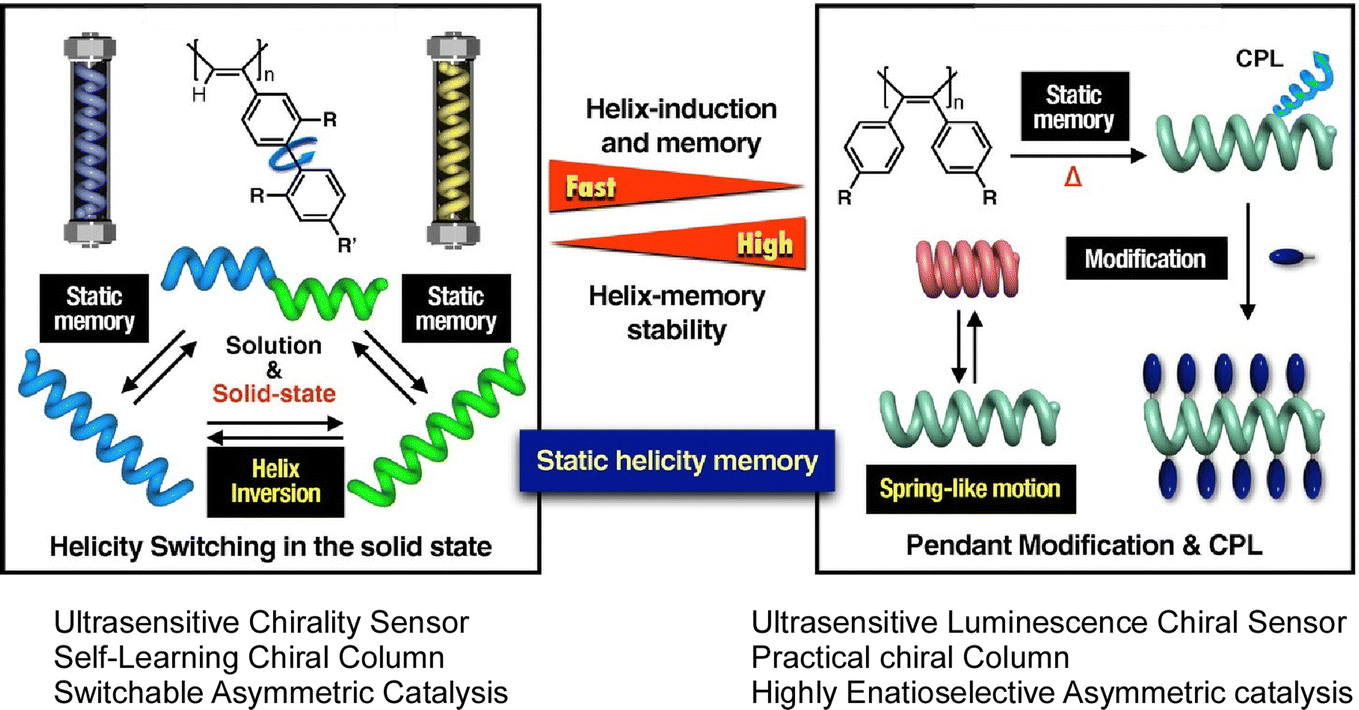 | ||
| Fig. 4 Synthetic helical polymers with unique dynamic and static helicity memory functions. The left system requires a longer time at higher temperatures than the right system with helix induction and subsequent static helix memory occurring more rapidly. The right system has a higher helix-induction barrier for further modification while maintaining static helical memory. Reprinted with permission from ref. 49. Copyright 2021 Oxford University Press. | ||
There have been a series of excellent molecular machine developments that have greatly advanced the working of conventional molecular machines. As a molecular machine for producing molecules, Leigh and co-workers reported a programmable stereo-crossing synthesis machine.50 It is a robot-like molecular machine that moves substrates between different activation sites to obtain different products. The molecular robot can be programmed to stereoselectively generate any of the four diastereoisomers from the addition of thiols and alkenes to α,β-unsaturated aldehydes in a one-pot, sequential manner. Such programmable molecular machines are expected to play an important role in chemical synthesis and molecular manufacturing. Controlled synthesis of rotaxanes by molecular pumps has also been reported by Astumian, Li, Stoddart, and co-workers who used artificial molecular pumps coupled with cyclic redox-driven processes to supply rings in pairs to produce higher energy poly[n]rotaxanes to appear as an assembled line.51 This programmable strategy allows the precise incorporation of 2, 4, 6, 8, and 10 rings with 8+, 16+, 24+, 32+, and 40+ charges, respectively, into polymer dumbbells with 6 cation charges.
Molecular systems that perform a wide variety of functions under rational control are being continuously investigated. Developing a rotary system that couples directional motions is an important challenge for driving several molecular machines in a coordinated manner. Feringa and co-workers have developed molecular rotary motors based on more detailed designs of rotor molecules.52 Well-regulated multiple kinetic barriers in isomerization and synchronous motion allowed the motor to slide and rotate during the entire motor rotary cycle. Wang, Kermagoret, Bardelang, and co-workers reported a system that can coordinate guest trapping into cyclic molecular space in water.53 They report a system in which guest trapping can be tuned (Fig. 5). In the synthetic macrocyclic compound cucurbit[7]uril, they showed that guest molecules can be exchanged via kinetic trapping and by pH, light, and redox stimuli to modulate the energy well and kinetic barrier. This macrocyclic host molecule may be an excellent cyclic molecule for building sophisticated molecular machines that work in water, since the rate constants and binding constants for the associated guest can be tuned.
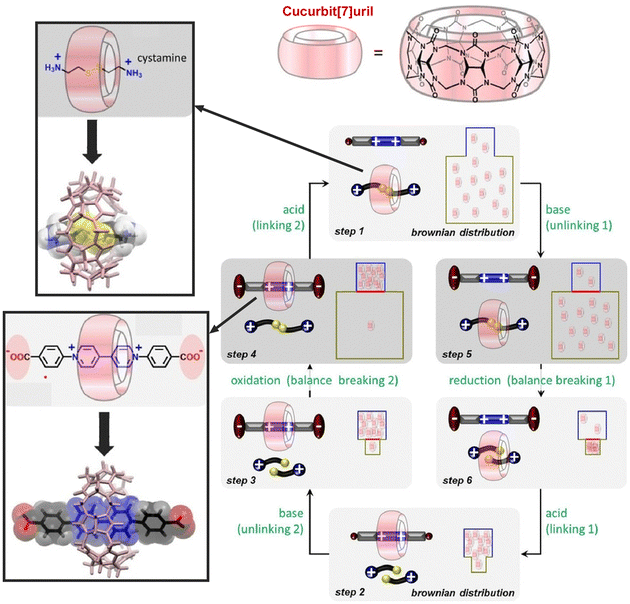 | ||
| Fig. 5 A system to coordinate guest trapping into cyclic molecular space of synthetic macrocyclic compound cucurbit[7]uril in which guest molecules can be exchanged via kinetic trapping and by pH, light, and redox stimuli to modulate the energy well and kinetic barrier. Reprinted with permission from ref. 53. Copyright 2021 Wiley-VCH. | ||
Stoddart and co-workers reported a molecular pump using a rotaxane structure.54 It uses redox energy and precise regulation of noncovalent interactions to pump a positively charged ring out of solution and wrap it around an oligomethylene chain to partially act as an entanglement with kinetic traps. The redox-active viologen unit at the center of the dumbbell molecular pump contributes to the ratcheting mechanism by performing the dual roles of attracting and then repelling the ring during the redox cycle. This artificial molecular pump performs the work repeatedly in a two-cycle operation and can drive the ring from equilibrium to a locally higher concentration. As a molecular pump, Leigh and co-workers reported a system using a catalytic reaction.55 In this system, crown ether macrocyclic compounds can be continuously pumped up the molecular axis from bulk solution in the presence of a fuel. The system uses an autonomous ratcheting mechanism by a chemical reaction, and works for directional preference in the motion of molecular rings on the axle molecule. This ratcheting action has been demonstrated to pump up to three macrocyclic compounds from bulk solution onto the axle under conditions with continuous barrier formation and removal. The use of catalysis to drive artificial molecular pumps may open the door to highly functional molecular machine systems by architecting functional systems of catalysis and molecular machines. The group also reported a catalyst-driven motor consisting of 1-phenylpyrrole 2,2′-dicarboxylic acid as a type of molecular machine that consumes chemical fuels.56 It has been shown that the energy of the chemical fuel is continuously transferred to cause repeated 360° rotations around the covalent N–C bond connecting the two aromatic rings.
As mentioned above, the development of molecular machines working in solution systems, the origin of molecular machines, continues without fading. This is because there remains ample potential for nanoarchitectonics at the molecular level, where functional molecular units are continuously wisely designed and assembled.
3. Molecular machines working at solid interfaces
A solid-state interface is an environment where nano-level analysis, from molecular growth observation to single-molecule functional measurement, is highly advanced.57 A solid interface is the place where molecular nanoarchitectonics such as local probe chemistry58 and on-surface synthesis59 are being developed as fusion research of nanotechnology and organic synthesis. The ability to analyze molecular motion and function at a high level makes it a powerful site for observation and analysis of molecular machines as molecular-level nanoarchitectonics.3.1. Various basic examples
Takami, Jiang, Weiss, and co-workers have successfully used scanning tunneling microscopy (STM) to manipulate the double-decker molecules at the liquid–solid interface of 1-phenyloctane solvent and graphite.60 A heteroleptic single double-decker molecule with lutetium in octaethylporphyrin–naphthalocyanine sandwich was decomposed by a voltage pulse from a probe, removing the upper octaethylporphyrin ligand and leaving the lower naphthalocyanine ligand at the surface. A decomposed molecular domain was formed within the double-decker molecular domain. A molecular “sliding block puzzle” was formed on a graphite surface by a cascade manipulation of double-decker molecules. Hipps and coworkers used STM to study the details of the self-assembly of a double-decker complex composed of yttrium and a phthalocyanine ring at the solution–solid interface.61 Their results show that molecule–substrate interactions, bias voltages, tunneling currents, and STM probes play important roles in the control and stabilization of molecular assembly. Selective rotation of molecular rotors was reported by Doltsinis, Glorius, Fuchs, and co-workers.62 The N-heterocyclic carbene rotor developed has a mesityl N-substituent on one side and a chiral naphthylethyl substituent on the other, and rotation in one direction is driven by inelastic tunneling of electrons to the STM chip (Fig. 6). Electrons preferentially tunnel through the mesityl N-substituent, while the chiral naphthyl substituent controls the directionality. Such N-heterocyclic carbene-based surface rotators offer new possibilities for the design and construction of functional molecular systems with excellent stability. | ||
| Fig. 6 Selective rotation of N-heterocyclic carbene-based surface molecular rotators driven by inelastic tunneling of electrons to the STM chip. Reprinted with permission from ref. 62. Copyright 2020 American Chemical Society. | ||
Single-molecule fluorescence microscopy is a promising technique for monitoring the action of single-molecule machines at room temperature. One strategy would be to attach multiple dyes to the same single-molecule machine. Tour, Wang, and co-workers examined the optical properties of single-molecule machines loaded with two different BODIPY dyes (Fig. 7).63 They found that at the air–glass interface, the single molecule with two dyes attached had a higher tendency to exhibit single-object-like photobleaching and appeared as if it were a single dye. Furthermore, the fluorescence intensity of the two-dye system did not scale with the number of attached dyes. This non-scaling nature of the fluorescence intensity was interpreted with a self-quenching model based on the intramolecular fluorescence resonance energy transfer (FRET) process or the singlet–triplet annihilation process. This study clarified the fluorescence properties of a single molecule of two dyes bound together, providing hints for design guidelines for fluorescence-labeled nanomachine molecules. The group also investigated the use of a triplet quencher, a cyclooctatetraene group, to improve the photostability of molecular machines attached to solid surfaces.64 The presence of cyclooctatetraene groups depletes the triplet state and prolongs the photobleaching time of the first dye. The cyclooctatetraene group on the second dye protects the breakdown product from the photodegradation of the first dye and can further improve the photostability of the second dye. This successful molecular architecture of functional groups can significantly extend the photodegradation lifetime of the molecular machine.
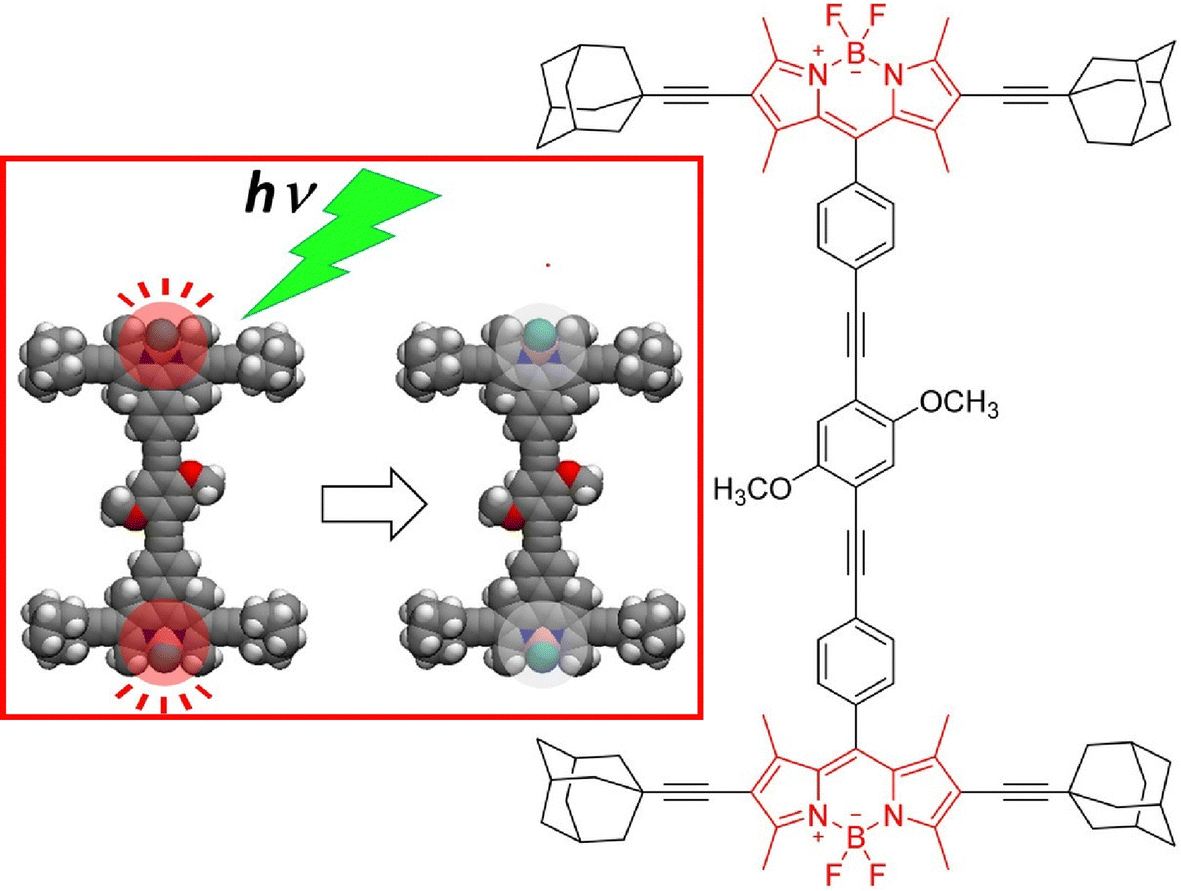 | ||
| Fig. 7 A single-molecule machine loaded with two different BODIPY dyes with a higher tendency to exhibit single-object-like photobleaching at the air–glass interface. Reprinted with permission from ref. 63. Copyright 2016 American Chemical Society. | ||
Nanoarchitecture control at the interface using molecular machine structures is another promising approach to enhance functionalization. Attempts have been made to build multilayered molecular machine structures using appropriate thin film techniques, such as the layer-by-layer (LbL) method.65 Unger, Schalley, and co-workers have succeeded in building chemically switchable single and multilayers of rotaxanes in a regular pattern on gold surfaces (Fig. 8).66 The ordered arrays of stimuli-responsive rotaxanes have well-controlled axial shuttling and are expected to be useful for coupled mechanical motion. Monolayers and multilayers of rotaxane were shown to switch reversibly between the two ordered states by linear dichroism effects in the angle-resolved near-edge X-ray absorption fine structure (NEXAFS) spectra. Such concerted switching processes were observed only when the surfaces were well-packed, and such effects were not seen on low-density surfaces with no lateral ordering. The system showed the importance of not only single molecular machines but also their successful nanoarchitectonics at the two-dimensional (2D) interface to achieve collective functionality.
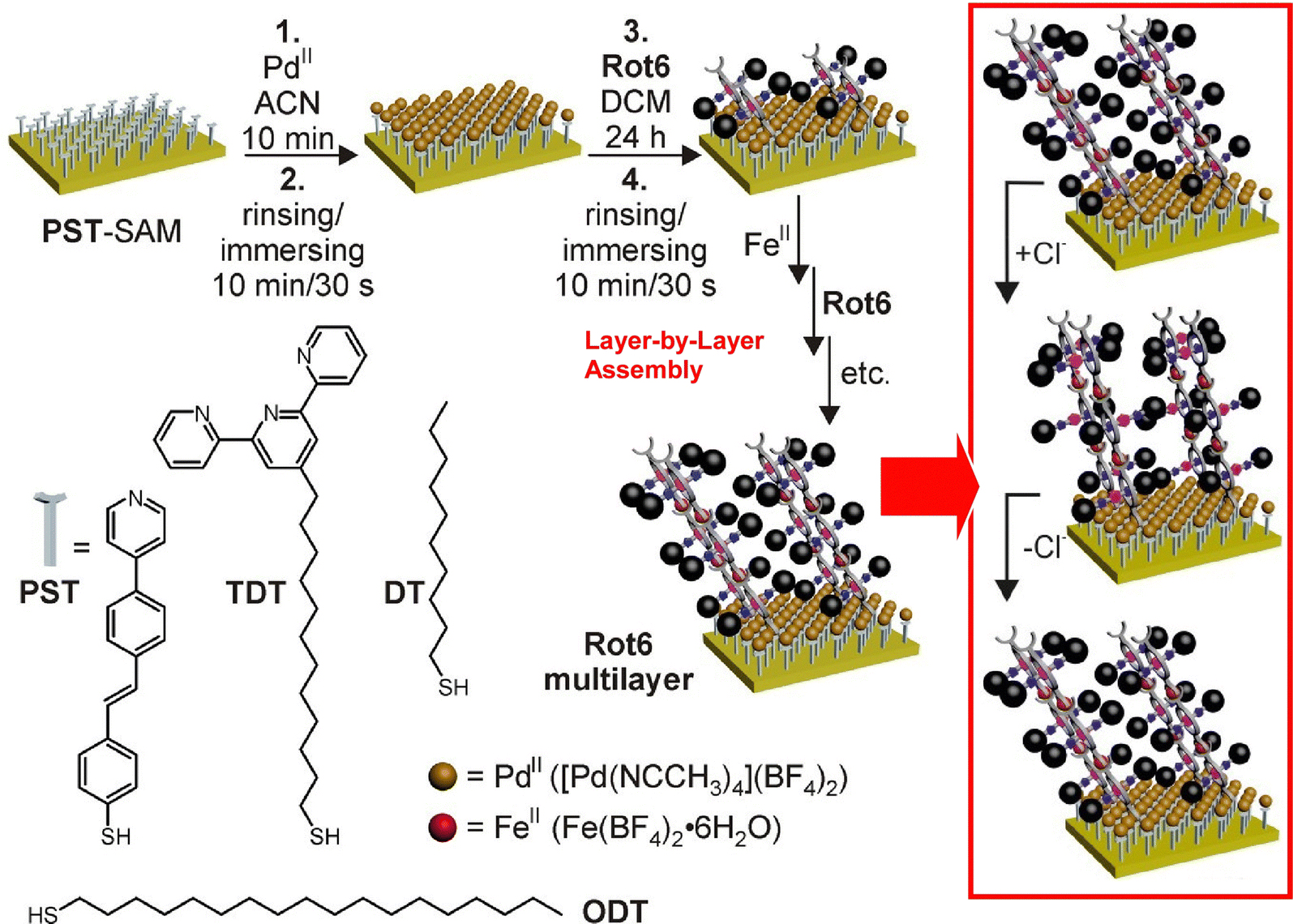 | ||
| Fig. 8 Layer-by-layer (LbL) nanoarchitectonics of chemically switchable single and multilayers of rotaxanes in a regular pattern on gold surfaces with well-controlled axial shuttling. Reproduced under terms of the CC-BY license from ref. 66, 2015 American Chemical Society. | ||
3.2. Endohedral machines
Single-molecule devices based on the electronic actuation of atomic/cluster motions confined in a fullerene-like cage have also been investigated. Carravetta and co-workers observed quantized translational and rotational motions of molecular hydrogen in a cage of C60.67 Theoretical models showed that the H2 inside C60 was a three-dimensional (3D) quantum rotor moving in a nearly spherical potential. Based on the discovery of the superatomic state of the C60 molecule, Petek and co-workers studied the factors affecting the hybridization of energy and wave functions into nearly free electronic bands in molecular solids.68 In particular, they examined how the energy of superatomic states depends on factors such as the degree of cohesion into 1D/3D solids, cage size, and exohedral and endohedral doping by metal atoms. Sufficient effects of the ionization potential of an inner-shell atom can induce the formation of a superatomic state with a conduction band in the middle of the gap between the LUMO and the HOMO of the C60 molecule. With plane-wave density functional theory studies, a new paradigm of intermolecular interactions beyond the conventional electronic interactions between spn hybridized orbitals in organic molecular solids is revealed, which may lead to the design of quantum structures and materials having extraordinary electronic and optical properties.Petek and co-workers used STM imaging and electronic structure theory to realize a single-molecule switch based on tunnel electron-driven rotation of triangular Sc3N clusters in an icosahedral C80 fullerene cage (Fig. 9).69 They demonstrated tunnel electron-induced multi-state hierarchical switching based on enantiomerization of a single endohedral fullerene molecule, suggesting that the antisymmetric stretching vibration of the Sc3N cluster is the gateway for energy transfer from tunnel electrons to cluster rotation. By selecting the conductive substrate and molecular buffer layer, the interface potential and the associated charge transfer can be tuned to control molecular switching. Furthermore, the potential applied from the STM probe or molecular leads could be used to control the charge, spin, and phonon degrees of freedom of the molecular switches. Hierarchical switching of conductivity between multiple steady states is advantageous in integrating single molecular switches with inner fullerenes into molecular devices with multiple logical states. Based on the atom-like building blocks and multi-level switching ability, endohedral fullerene-based devices become suitable for molecular-scale integration into parallel computing structures. Seideman and co-workers have also demonstrated atoms/clusters in fullerene cages.70 A novel methodology for single-molecule devices was proposed on the basis of electronic actuation of atom/cluster motion within the fullerene cage. Inelastic electron tunneling via resonance of lithium atoms in a gold–lithium@C60–gold junction is slightly perturbed by the motion of embedded atoms as the lithium atoms vibrate with large amplitude against the fullerene wall and the fullerene cage bounces between the gold electrodes.
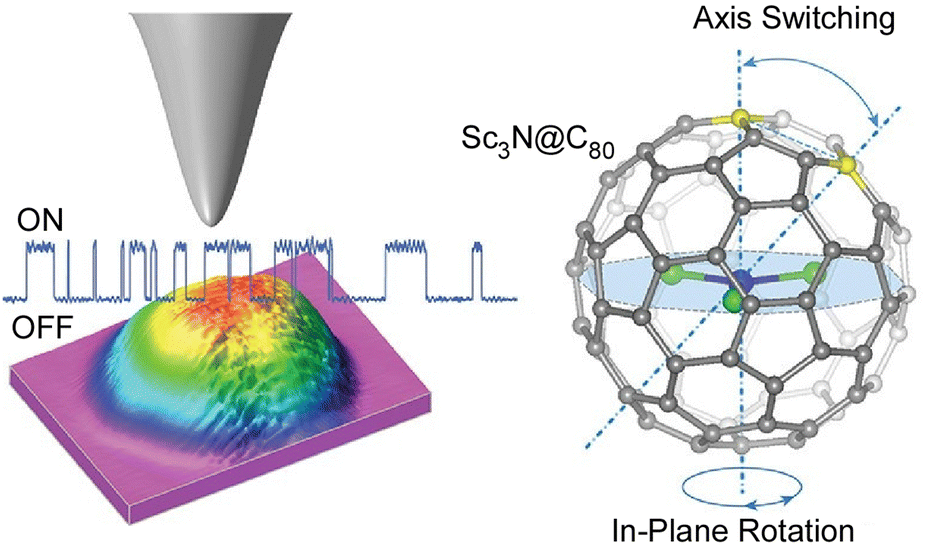 | ||
| Fig. 9 A single-molecule switch based on tunnel electron-driven rotation of triangular Sc3N clusters in an icosahedral C80 fullerene cage. Reprinted with permission from ref. 69a. Copyright 2011 American Chemical Society. | ||
As a non-fullerene endohedral cage molecular device, Clever and co-workers utilized a Pd-based coordination capsule as a nanospace: substituents attached via C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds behave as molecular rotors in the cage.71 The pronounced donor–acceptor nature of the substituents lowers the rotational barrier and allows electronic control of the rotational velocity within the cage. This is a new type of endohedral push–pull rotor built in self-assembly. When the guest is encapsulated, it is found to slow down the rotation. Such dynamic push–pull systems could be used to form stimulus-responsive supramolecular systems, catalytic cavities, functional materials, and so on.
C double bonds behave as molecular rotors in the cage.71 The pronounced donor–acceptor nature of the substituents lowers the rotational barrier and allows electronic control of the rotational velocity within the cage. This is a new type of endohedral push–pull rotor built in self-assembly. When the guest is encapsulated, it is found to slow down the rotation. Such dynamic push–pull systems could be used to form stimulus-responsive supramolecular systems, catalytic cavities, functional materials, and so on.
3.3. Single and supramolecular molecular rotors
As seen in some of the aforementioned examples, the behavior of rotation is single molecules and supermolecules is fundamental and can be easily subjected to evaluation in molecular machines. There have been various studies on the precise analysis of molecular rotors and molecular motors on solid interfaces. In this section, examples of studies on the rotational motion of basic molecular units such as single molecules and supramolecular complexes on solid substrates are discussed.Deshlahra, Sykes, and co-workers reported single-molecule observations of N-methylbutylamine molecular rotors supported on Cu(111) surfaces, using tunneling electrons from the STM tip to excite the vibrational modes of the molecules that drive their rotational motion.72Fig. 10 shows a schematic of the molecular adsorption site of N-methylbutylamine, showing ways of the molecular rotation around the N–Cu bond that serves as the axis of the molecular rotor. The figure also exemplifies a typical large STM image where both isolated single molecules and small clusters are present. It was found that the enantiomers of N-methylbutylamine interconvert at high speed in the gas phase, but on the Cu(111) surface, each enantiomer can retain its chirality because a single pair of nitrogen atoms is bound to Cu. By tuning the electron flux, the individual rotational motions between six stable molecular orientations on the Cu(111) surface could be monitored in real-time. In most of the STM tips used to electrically excite the rotators, one enantiomer rotated faster than the other. This supports the fact that the STM tip itself may be chiral, and that the diastereomers resulting from the interaction between the chiral STM tip and the chiral molecules may cause significant physical differences in the rotational speed of the R and S molecular rotors.
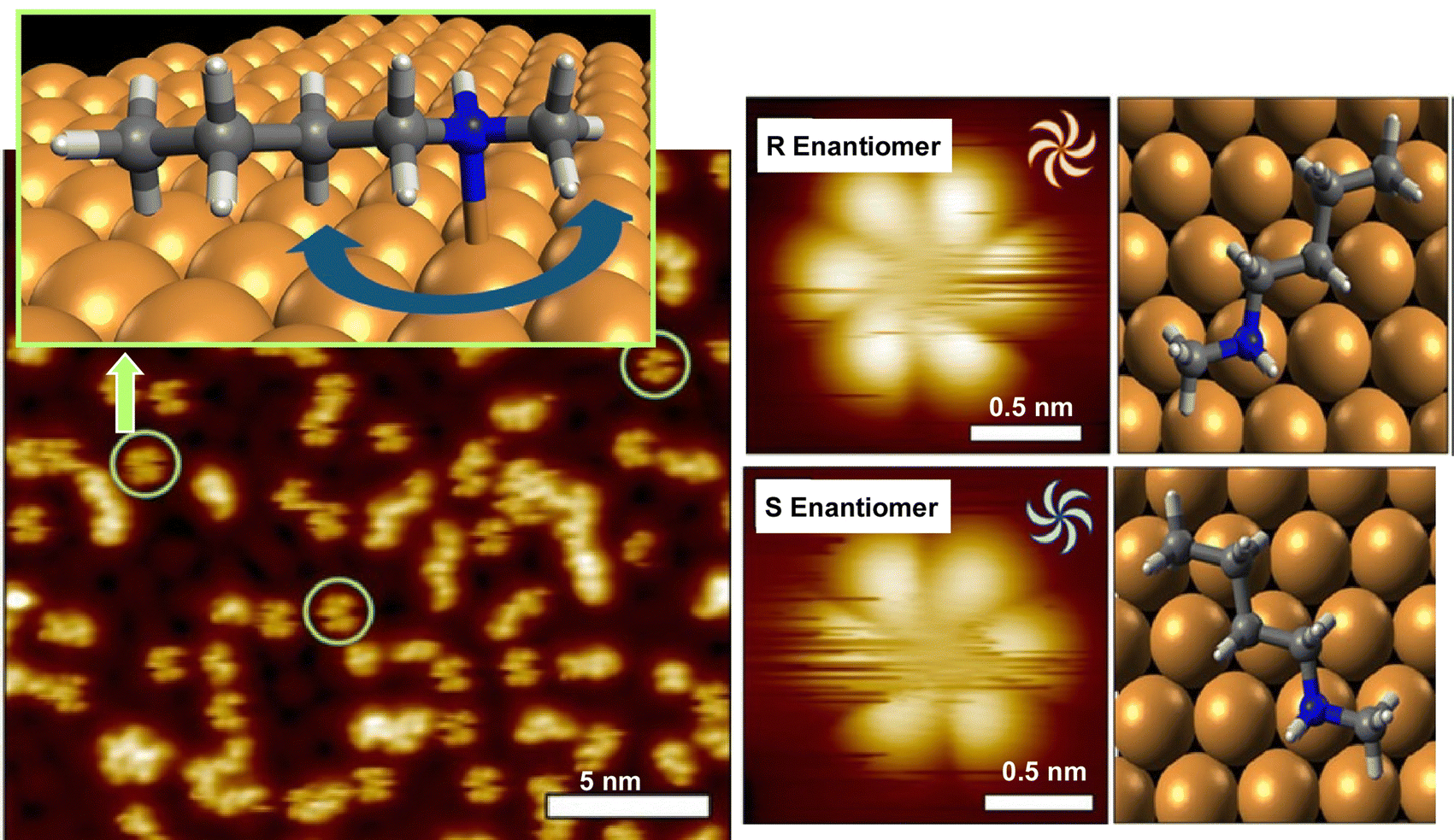 | ||
| Fig. 10 Single-molecule observations of N-methylbutylamine molecular rotors supported on Cu(111) surfaces, using tunneling electrons from the STM tip to excite the vibrational modes of the molecules that drive their rotational motion. Reprinted with permission from ref. 72a. Copyright 2021 American Chemical Society. | ||
It has been established that the permeability of electrons passing through a chiral molecule depends on the spin of the electrons. This phenomenon is called chiral-induced spin selectivity. Cohen, Feringa, Naaman, and co-workers have measured multi-state switching of spin selectivity in electron transfer through molecular motors based on four different helical arrangements using magnetoconductive atomic force microscopy.73 The results revealed that the helix state determines the molecular structure on the surface. The modulation of spin selectivity by controlling the helical state opens up the possibility of tuning spin selectivity on demand with high spatio-temporal precision. Regardless of whether the driving force for the rotation of the molecular rotor is activated by heat, an electric field, or light, the rotation is determined by the intrinsic rotational potential landscape. Thus, the essence of controlling the motion of molecular rotors is to tune the potential landscape. Du, Gao, and co-workers have used tetra-tert-butyl-nickel phthalocyanine on an Au(111) substrate as a molecular rotor.74 They demonstrated that both the sample bias and the STM tip–molecule distance can modulate the rotational potential of the molecular rotor. The mechanism for changing the rotational potential was found to be a combination of van der Waals interactions and the interaction between molecular dipoles and an electric field.
Surface-supported molecular motors are considered as nanomechanical devices in various nanoscale applications. However, molecular motors are usually prepared with covalent bonds and cannot be reconstituted without chemical reactions. Hill, Joachim, Uchihashi, and co-workers reported a supramolecularly recombinable molecular rotor (Fig. 11).75 This supramolecular rotor is a platinum–porphyrin supramolecular dimer assembly that can create a chiral image in the form of an association on a surface. Using the tunneling current of STM, it was demonstrated that a Pt–porphyrin supramolecular dimer assembly supported on an Au(111) surface can be rotated in a highly directional manner. The direction of rotation of this molecular rotor is decided only by the surface chirality of the dimer. More interestingly, the chirality can be inverted in situ by a process involving rearrangements within the dimer. This is possible because the rotor structure is made by supramolecular nanoarchitectonics. This result paves the way for building complex molecular machines on surfaces to mimic versatile biological supramolecular rotors on smaller scales.
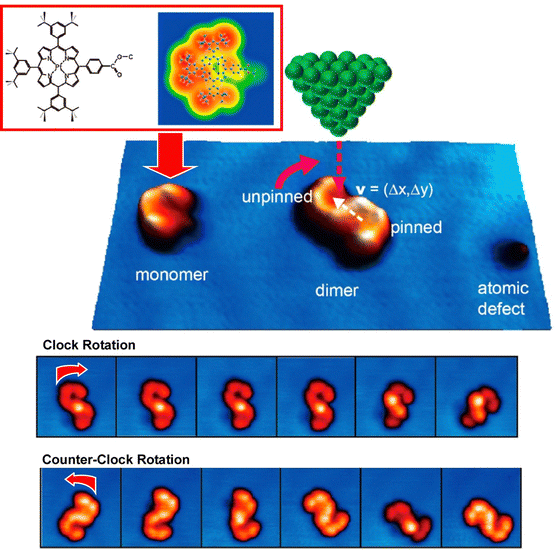 | ||
| Fig. 11 A supramolecularly recombinable molecular rotor of Pt–porphyrin supramolecular dimer supported on an Au(111) surface with rotation behaviors in a highly directional manner, where the direction of rotation of this molecular rotor is determined solely by the surface chirality of the dimer. Reprinted with permission from ref. 75. Copyright 2015 American Chemical Society. | ||
Single-molecule imaging and manipulation using optical microscopy are commonly used to study biomolecular machines, and an effort toward synthetic molecular machines has been reported by Noji and co-workers.76 In this study, a single-molecule optical microscopy technique was used for the observation of a synthetic molecular rotor, a double-decker porphyrin. Magnetic beads (approximately 200 nm) were attached to the double-decker porphyrin, and their rotational dynamics were captured with a temporal resolution of 0.5 ms. As shown in Fig. 12, the double-decker porphyrin was fixed to the amino-modified glass substrate surface and alkyne-modified magnetic beads. The rotational dynamics of the beads was visualized by optical microscopy under halogen lamp illumination. Double-decker porphyrins exhibit rotational diffusion of 90° steps, which is consistent with a four-fold symmetric structure. When the porphyrin was forced to rotate with magnetic tweezers, four stable stopping angles, also separated by 90°, were observed. These results indicate that single-molecule optical microscopy is useful for elucidating the elementary properties of synthetic molecular machines.
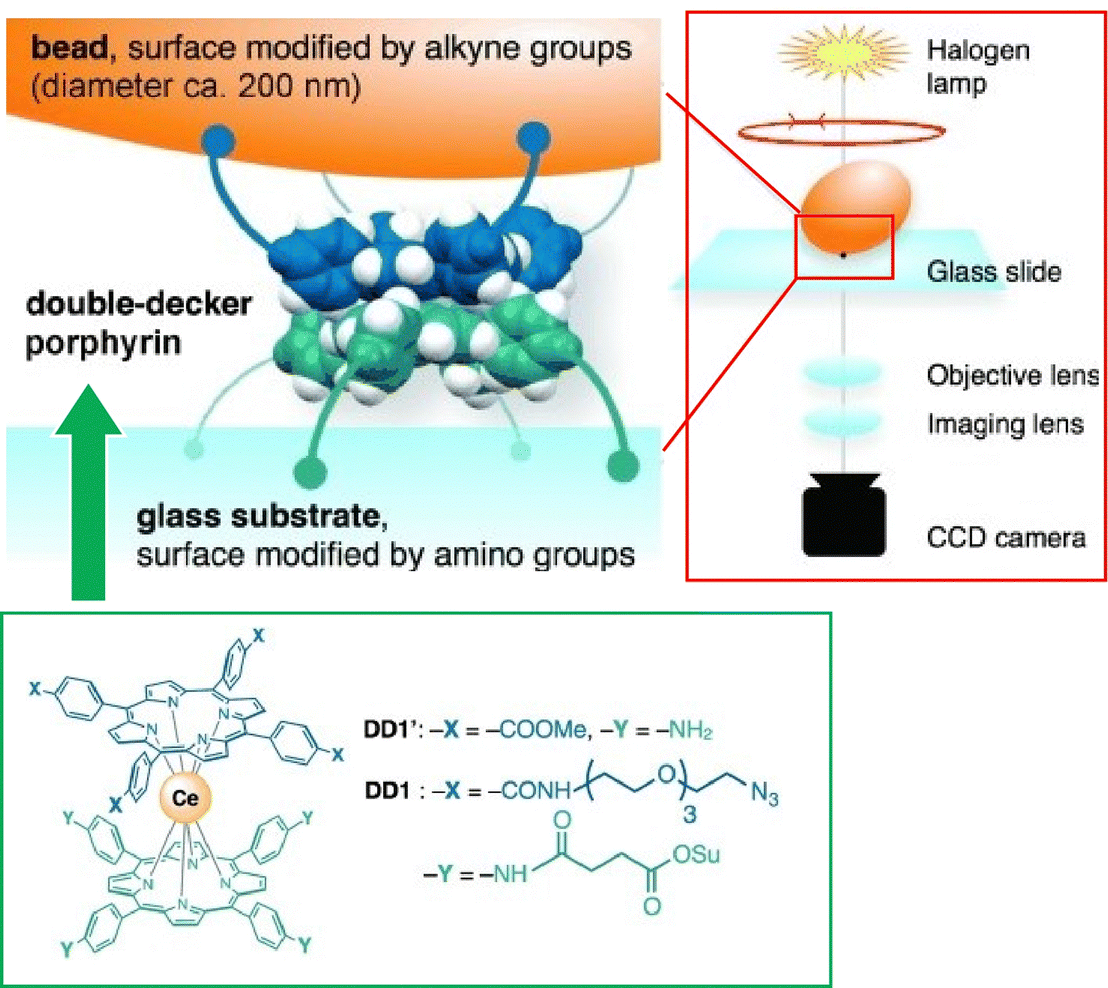 | ||
| Fig. 12 Single-molecule imaging and manipulation using optical microscopy for evaluation of a synthetic molecular rotor, a double-decker porphyrin with magnetic bead. Reprinted with permission from ref. 76. Copyright 2014 Wiley-VCH. | ||
3.4. Rotors to gears
Molecular machines and functional molecules do not work alone, but when they are assembled and function together, the functions they exhibit become dramatically more sophisticated. In biological systems, various advanced functions are expressed by skilfully coordinating biomolecular machines and biofunctional molecules.77 Even in the basic science of molecular observation on a solid substrate, the linkage of molecular machines can be analyzed. For example, it has been reported that molecular rotors move like gears in coordination.Soe et al. used low-temperature STM to observe two molecular rotors functioning as molecular gears in coordinated motion.78 Two molecular gears (Fig. 13(A)), each with a diameter of 1.2 nm and a tooth count of 6, were fixed on copper adatoms separated by only 1.9 nm on a lead surface. The stepwise rotation of the first gear in the train around the axis of the Cu adatom causes the second gear to rotate, as in a macroscopic gear train. The first gear, and then this second gear, until the axis of the two Cu adatoms is positioned exactly on the lead surface, making the molecular tooth-to-tooth dynamics completely reversible function. Once assembled, the first molecular gear of the train can be rotated stepwise by the STM operating the handle. In the example of the three gears working together in Fig. 13(B), the stepwise rotation of the first molecular gear of the train is transmitted first to the second molecular gear to move the others. To achieve this complex molecular mechanism, it is most important to place and center the two molecular gears on their respective Cu adatoms. This centering allows the two molecular gears to avoid lateral movement because they are self-stabilized by the intertwined teeth and handles. As a result, rotational motion is transmitted in steps of approximately 30°. Transmission of rotational motion from the first molecular gear to the next requires precise optimization of the atomic axis of each gear and the positioning of each molecular gear to conform to the deflection of the molecular teeth between the molecular gears with reliable reversibility and reproducibility. Such rotational transmission could be applied in the future to build molecular-scale Pascal mechanical calculators, to measure the power of single molecular rotors using different molecular gear train lengths, and to input data into atomic-scale quantum circuits by mechanically switching single molecule switches for each input digit.
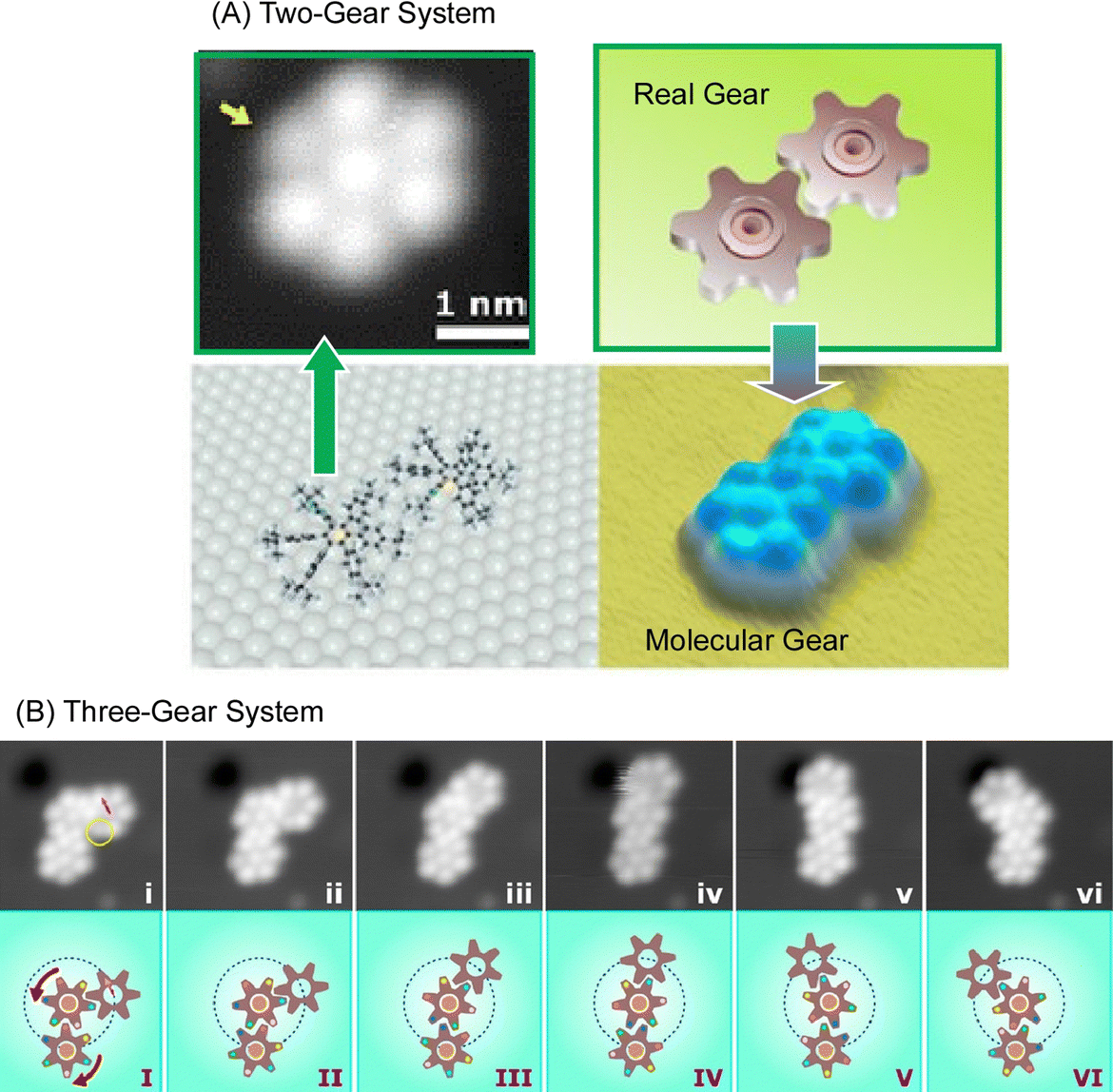 | ||
| Fig. 13 Multiple molecular rotors functioning as molecular gears in coordinated motion: (A) two-gear system; (B) three-gear system. Reprinted with permission from ref. 78. Copyright 2019 American Chemical Society. | ||
The development of molecular gear systems in which multiple rotors work in tandem is an important element in the construction of nanoscale machines. However, molecular gears have a high degree of freedom of motion, and it is important to evaluate whether the individual rotors in a molecular gear train are moving with proper repatriation. To evaluate the correlated motion of neat molecular gears, Rapenne and co-workers designed a molecular rotor with a hydrotris(indazolyl)borate anchor to prevent surface diffusion and a central ruthenium atom as a fixed axis of rotation.79 Then, a series of star-shaped organometallic molecular gears consisting of azimuthal pentaporphyrin cyclopentadienyl gears specifically labeled to monitor their motion by time-resolved STM were designed and assembled. Asymmetrization of the gears was first performed sterically. It was done by introducing one tooth longer than the other four teeth. The electronic properties of one paddle were tuned by changing the properties of the porphyrin scaffold and the central metal. To achieve this structural diversity, the modular synthetic method is used by successive cross-coupling reactions to the penta(p-halogenophenyl)cyclopentadienyl ruthenium(II) main constituent with a pre-activated p-iodophenyl group. This figure schematically illustrates the key parameters to be considered in the design of molecular gear trains that achieve correlated intermolecular rotation in response to external stimuli. A virtual star-shaped gear train (blue) incorporating labeled teeth green is mounted on a rotating axis (red) with fixed intermolecular distances and viewed from above before and after an external stimulus that induces correlated motion. Molecular-level nanoarchitectonics techniques based on organic synthesis are important to bring out the advanced functionality of such molecular machines.
For regulation of molecular rotor motions upon dipole moment interference in an array of molecular rotors, dipolar molecular rotor compounds were designed, synthesized, and adsorbed as a self-assembled 2D array on an Ag(111) surface by Herges and co-workers.80 In this molecular rotor, a 4,8,12-trioxatriangulene platform known to be physisorbed on metal surfaces, a phenyl group that bonds to the central carbon atom and acts as a pivot joint to reduce barriers to rotation, and a small dipole unit at the top, a pyridine and pyridazine units are arranged. Theoretical calculations and STM observation studies suggest that the dipoles of adjacent rotators interact spatially through a pair of energetically favorable head–tail nanoarchitectures.
3.5. Molecular cars (nanocars)
One of the molecular machines developed with the most intelligent of molecular nanoarchitectonics would be the molecular car (nanocar). By skilfully combining molecular shapes and properties, we can assemble molecules that look like cars or function like cars.81 This is a technology to move them like a car while observing them at high resolution on a solid interface. In fact, the Nanocar Race, a world-class event in which nanocars compete for speed on a solid interface, has been held twice so far (2017 and 2022).82 Normal car races have been held since the early 1900s. Since nanocars are two billionths of the size of a normal car, humans have reduced the size of race cars by 9 orders of magnitude in just over 100 years. Here some recent examples of research on nanocars and related molecular machines are discussed.Kudernac, Ruangsupapichat, and co-workers synthesized a nanocar molecule with four rotating motors as wheels.83 This nanocar molecule undergoes continuous and distinct conformational changes upon successive electronic and vibrational excitations. STM observations confirmed that the nanocar molecules travel in one direction on the Cu(111) surface by activating the conformational changes in the rotors by inelastic electron tunneling. The system can follow a linear or random surface trajectory or be stationary by tuning the chirality of the individual motor units. This nanocar architecture will contribute significantly to the study of more sophisticated molecular machine systems with directional motion. A nanocar with a fullerene wheel design is also being developed by Kelly, Tour, and coworkers.84 Nanocars and nanotrucks, novel single-molecule nanomachines with fullerenes as wheels, have been designed and synthesized (Fig. 14). These nanovehicles consist of three basic components, a molecular chassis, freely rotating alkynyl axles, and spherical fullerene wheels. The C60-based spherical wheels and alkyne-based freely rotating axles allow molecular structures to roll freely on gold surfaces at the nanoscale. The rolling motion observed by STM was confirmed to be the same as that of macroscopic entities rolling perpendicular to the axis. It was also proved that the wheel-like rolling motion by fullerenes is not bar-slip or sliding translation. This study emphasizes that precise molecular design and synthesis can control the directionality of the motion of molecular-sized nanoarchitectures.
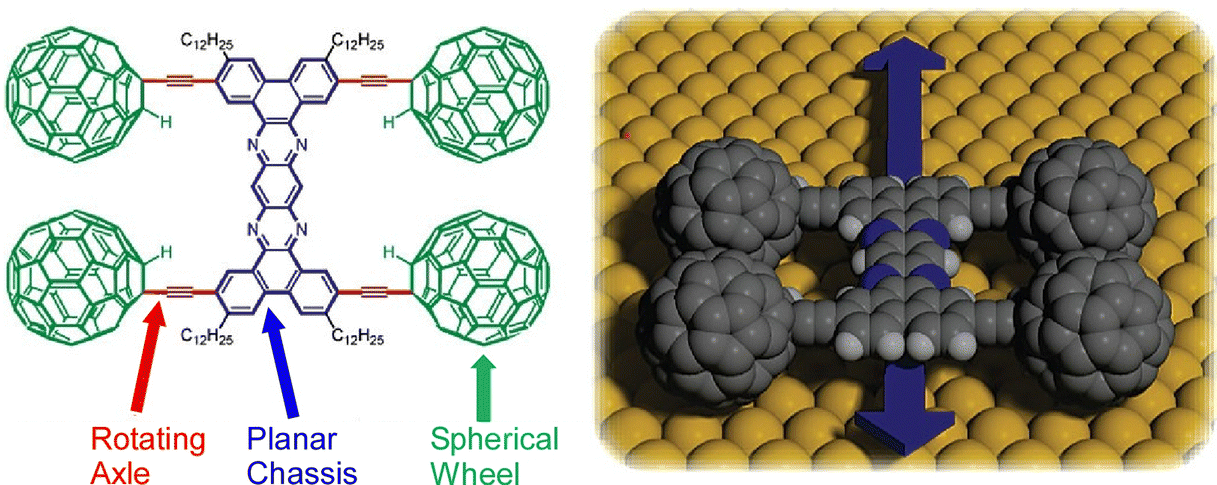 | ||
| Fig. 14 A nanocar with spherical fullerene wheels, freely rotating alkynyl axles, and a molecular chassis. Reprinted with permission from ref. 84. Copyright 2006 American Chemical Society. | ||
Following these pioneering studies of nanocars, consideration has recently been given to the mechanisms of diffusive motions of surface rolling molecules. Pishkenari and co-workers used classical all-atom molecular dynamics to investigate the effect of different-size vacancies on the motion of nanocars, nanotracks, and C60 on gold surfaces at different temperatures.85 At 200 K, holes or vacancies appear as obstacles in the path of C60, and at higher temperatures, C60 can enter these holes. However, larger pores become permanent traps for C60. On the other hand, molecules designed as nanotracks have relatively rigid bodies, which limits the movement of the wheels. Vacancies in the grain boundaries are likely to repel or trap nanocars, preventing their free movement. Therefore, a single-crystal substrate is essential for unimpeded movement of nanocars and surface-rolling molecules such as C60. Abbasi-Pérez, Kantorovich, and co-workers used molecular walkers (1,3-bis(imidazol-1-ylmethyl)-5(1-phenylethyl)benzene) along a Cu column on a Cu(110) surface with analysis by velocity-based Monte Carlo dynamics simulations calculated using ab initio density functional theory.86 The molecular walker moves to the nearest equivalent lattice site by a so-called inchworm mechanism in which it first walks on its hind legs and then on its front legs. The molecule diffuses by a two-step mechanism and has its inherent asymmetry.
Most studies of nanocar motion have been conducted in ultrahigh vacuum and cryogenic quiescent environments to precisely observe molecular images. On the other hand, some observations have been made under ambient conditions. Tour, Wang, and coworkers observed the thermal diffusion of four adamantane-wheeled nanocars on an air–glass interface at room temperature using line-scan imaging with improved temporal resolution (Fig. 15).87 This high-speed imaging technique revealed that the four-wheeled nanocar diffuses quasi-randomly in two dimensions on the glass surface. They tend to follow linear diffusion trajectories on short time scales, which is consistent with the diffusion of a wheel's rotational mode. The nanocar molecules lose directionality with time, suggesting that other diffusion modes, such as pivoting motion, may also contribute to thermal diffusion at room temperature.
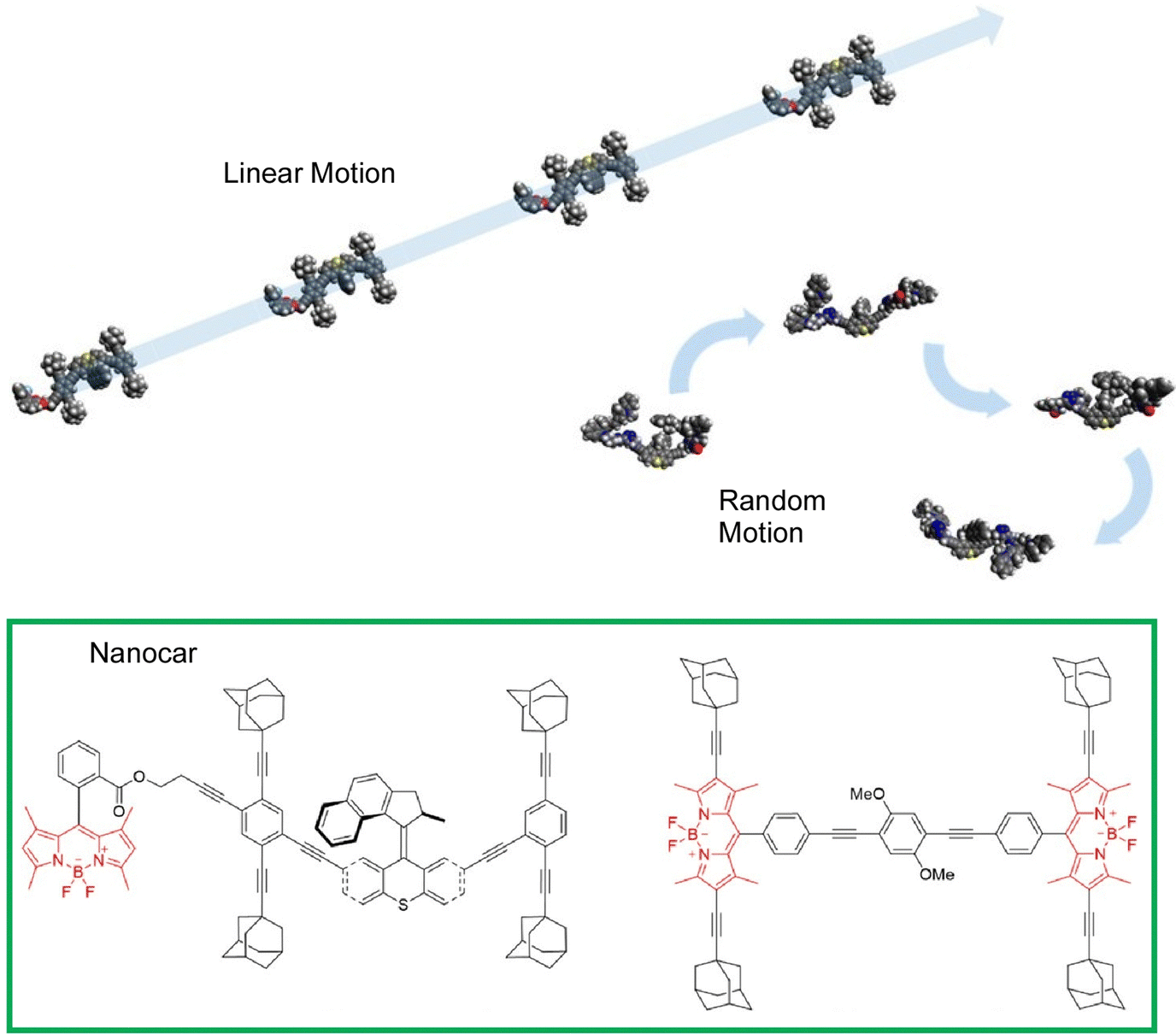 | ||
| Fig. 15 Thermal diffusion of four adamantane-wheeled nanocars on an air–glass interface at room temperature, where the nanocars tend to follow linear diffusion trajectories on short time scales, and lose directionality with time. Reprinted with permission from ref. 87. Copyright 2018 American Chemical Society. | ||
Further developments of nanocar structures have also been reported. Grill, Tour, and coworkers designed, synthesized, and observed the motion of a second-generation motorized nanocar (Fig. 16).88 In designing the nanocar, a light-driven unidirectional molecular motor was used instead of the oligo(phenyleneethynylene) chassis. Because C60 quenched the photoisomerization process of the molecular motor, a p-carborane wheel was chosen. The paddle-shaped rotor is intended to interact with the surface to generate a force that propels the nanocar during irradiation. The nanocar was deposited on a Cu(111) surface and single molecule imaging was performed at 5.7 K by STM observation. This motorized nanocar paves the way for the development of nanocars with various drive schemes. Xu, Li, Zeng, and co-workers have successfully fabricated the most simplified two-wheeled nanocar that is constructed by coordination interactions.89 The nanocar bridges two enantiomeric motors as wheels around a central pseudosquare plane Ag(I), which ensures that the cars move in the same direction when observed from the outside. The individual motors are assembled via metal coordination into a hierarchical motorized nanocar system, which may also perform robotic functions such as metal transport and release.
 | ||
| Fig. 16 A second-generation motorized nanocar using a light-driven unidirectional molecular motor. Reprinted with permission from ref. 88. Copyright 2012 American Chemical Society. | ||
4. Molecular machines working at liquid interfaces
While high resolution, such as molecular imaging by STM, is possible at solid interfaces, this is generally difficult to achieve at liquid interfaces. Due to such limitations, molecular machine studies at interfaces have been dominated by those at solid interfaces. However, it is easier to provide more dynamic perturbations at the liquid interface, which can also be a site of accumulation of molecular machines.90 These qualities make the liquid interface an increasingly attractive medium for molecular machine research.The various advantages of liquid interfaces are well recognized in supramolecular chemistry and materials science research. Liquid interfaces, such as gas–liquid and liquid–liquid interfaces, provide a meeting place for different media and their secondary sites for the synthesis of molecular thin films,91 organic semiconductor molecular films,92 2D nanocarbons,93 metal–organic frameworks (MOFs),94 covalent organic frameworks (COFs),95 and so on. For example, as summarized in a recent review by Wan and co-authors,96 insoluble COF structures, such as those made as powders by solvothermal synthesis, can be built as 2D films at a liquid interface, enabling applications in many fields such as photoelectric conversion, fluorescence, electronic devices, chemical sensing, electrocatalysis, and molecular separation. Alonso and Mikhailov theorize about active protein machines floating at liquid surfaces.97 Periodic mechanical motion within the machine, a microscopic swimmer, leads to a molecular propulsive force applied at the air–liquid interface. When the energy supplied to the machine exceeds a certain threshold, the flat interface line becomes unstable. Then, a region of interfacial turbulence through irregular traveling waves and propagating machine clusters results. An example of the use of the LB method at the air–water interface for integrated thin films of molecular shuttles has also been reported by Credi and co-workers.98 As described above, the liquid interface provides a unique venue for research ranging from materials science to the behavior of molecular machines.
It is necessary to consider molecular machine manipulation methods that make better use of the characteristics of liquid interfaces. The characteristics of liquid interfaces, such as the air–water interface, are that the size of the structure varies greatly depending on the directions (a vertical direction to the interface and a lateral direction along the interface) and that it can be changed dynamically. When considering a monolayer deployed at the air–water interface, the mechanical deformation of the monolayer, such as compression and expansion in the lateral direction (in-plane direction), is usually macro-sized, as large as several tens of centimeters. Since the molecules in the monolayer are confined in the nanometer-sized film thickness direction, structural changes such as conformational changes can be as large as nanometers or sub-nanometers. At the liquid interface, visible mechanical mutations can be coupled with molecular-level motion.99 If molecular machines are integrated at the liquid interface as monolayers, macroscopic manipulations, such as hand motions, can be used to control the conformations and consequent functions of the molecular machines.100 This is named hand-operating nanotechnology.101
One example is shown in Fig. 17.102 The molecular machine used in this example is the steroid cyclophane. Steroid cyclophane has a cyclic core composed of 1,6,20,25-tetraaza[6.1.6.1]paracyclophane with four steroidal moieties (cholic acid) connected via a flexible L-lysine spacer. Steroid cyclophanes in the monolayer change their conformation when deployed on the air–water interface to form a monolayer and subjected to macroscopic mechanical displacement from the lateral direction. At low pressure, it takes on an open conformation with the hydrophilic surface of the cholic acid attached to the water surface. Under high surface pressure, the spacer folds back to form a more compact cavity conformation in which the cholic acid moiety is collected. When the conformation of the steroid cyclophane changes from open to cavity, the steroid cyclophane can catch fluorescent guest molecules (6-(p-toluidino)naphthalene-2-sulfonate, TNS) that are dissolved in the aqueous phase. Surface reflectance fluorescence measurement of the entrapped guest molecules revealed that the guest molecules are repeatedly captured and released in synchronization with the repeated compression and expansion of the monolayer, which is several tens of centimeters in size. This indicates that the molecular machine can be driven by macroscopic mechanical motions that are equivalent to those of our hand motions. This is made possible by the highly anisotropic and dynamic nature of the liquid interface.
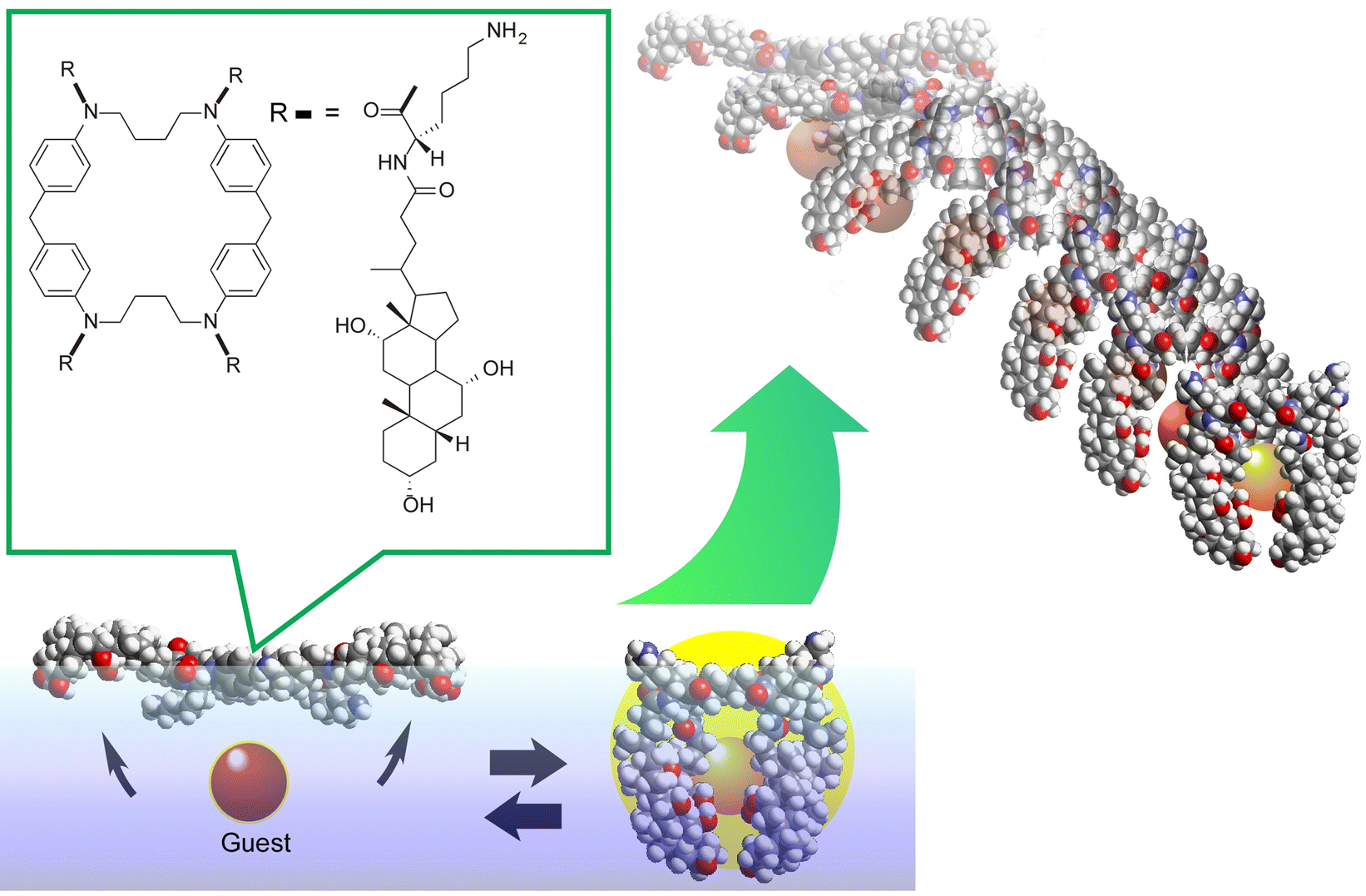 | ||
| Fig. 17 The molecular machine, steroid cyclophane, that changes from an open conformation to a cavity conformation and can catch fluorescent guest molecules upon lateral compression of its monolayer at the air–water interface. | ||
The force exerted on the molecules in the monolayer by compression and expansion at the air–water interface is very small. This makes it possible to delicately control molecular conformation for regulation of molecular recognition at the interface, because molecular recognition efficiency can be sensitively altered by host molecule conformations. This control allows enantioselective binding of amino acids103 and discrimination of nucleobases104 that differ by only one methyl group, with the simple and versatile stimulus of mechanical force. Sakakibara et al. have used this technique in indicator displacement assay (IDA).105 This method takes advantage of the phenomenon of competitive binding of the indicator and guest to complementary sites on the host. The IDA method has become widely used in the development of multicomponent optical sensors and enantioselective assays because of its ability to accurately measure the total concentration of the guest (or analyte).106Fig. 18 presents a method called mechanically controlled indicator displacement assay (MC-IDA) at the air–water interface. This method uses a deformable monolayer of fluorescently labeled receptor molecules. The conformation of the molecule can be changed by externally inducing lateral compression and expansion of the monolayer to control molecular recognition. Mechanical compression of the monolayer of the receptor molecule causes fluorescence resonance energy transfer (FRET) between the receptor and the indicator, which switches fluorescence emission on. The addition of a guest displaces the indicator, effectively quenching the FRET process (switch-off). These behaviors are dependent on the conformation of the receptor. It was demonstrated that controlling it mechanistically at the air–water interface allows for very sensitive molecular detection.
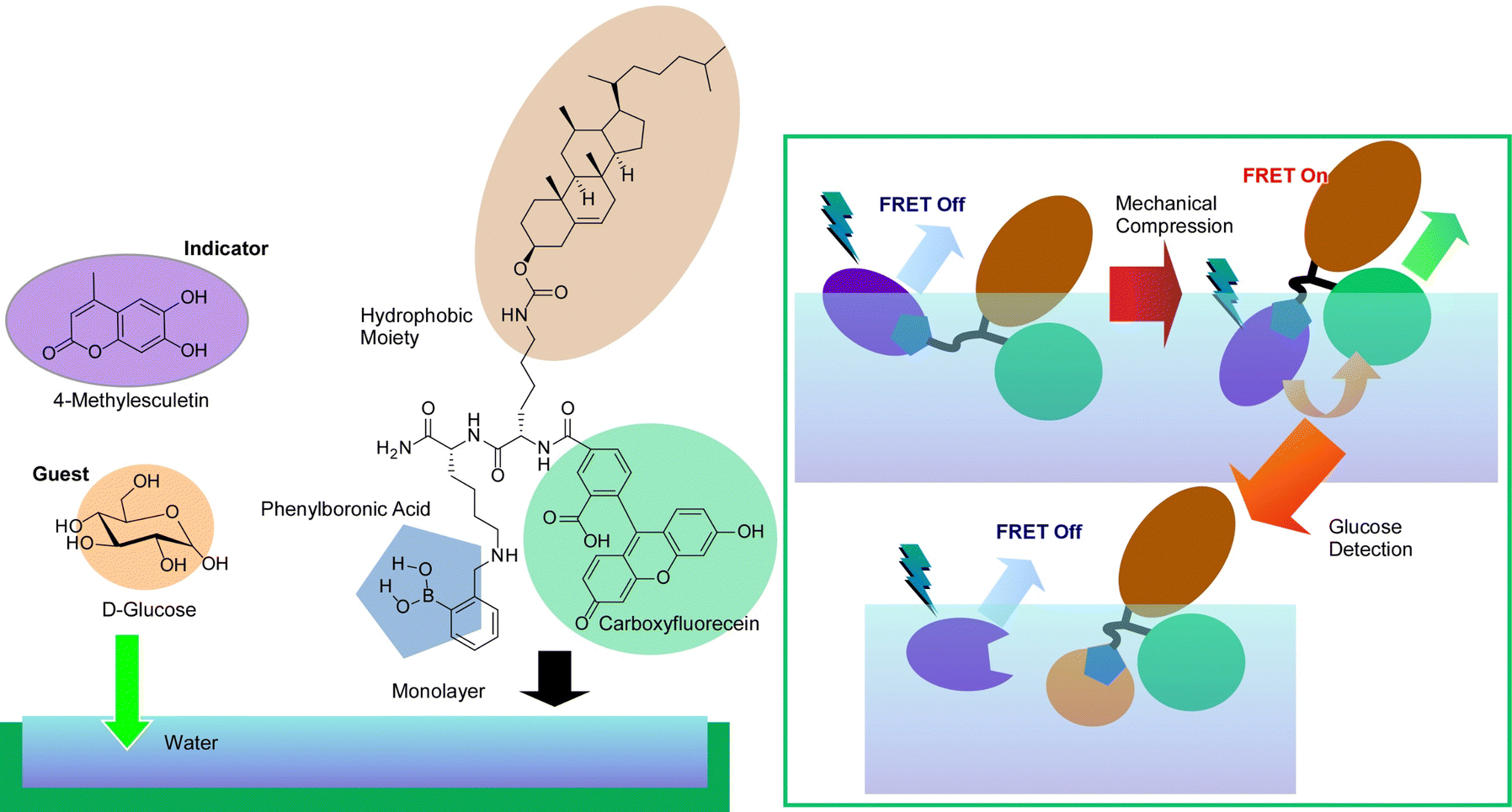 | ||
| Fig. 18 A method called mechanically controlled indicator displacement assay (MC-IDA) at the air–water interface with fluorescence resonance energy transfer (FRET) between the receptor and the indicator to switch on and switching off upon addition of a guest with quenching the FRET process. | ||
Liquid interfaces, such as the air–water interface, are a suitable place to accumulate molecular machine actions and output them as large effects. Gengler, Feringa, and co-workers have observed the photoswitching effect in monolayers of bis(thiaxanthylidene)-based photoswitching amphiphilic molecular machines, and similarly in a mixed monolayer with a phospholipid of dipalmitoylphosphatidylcholine (DPPC) at the air–water interface (Fig. 19).107 Efficient photoisomerization of the central core of the amphiphilic molecular machine from the antifolded to the congruent form was found to change the surface pressure in either direction depending on the initial molecular packing. Monolayers of photoswitchable amphiphilic molecular machines provide a powerful venue for the investigation of the fundamental science of cooperativity and amplification of motion of molecular machines at liquid interfaces. Cooperative switching systems that can convert light energy into mechanical motion will be of great interest for various applications including molecular sensing, nanoscale energy converters, optical devices, and data storage.
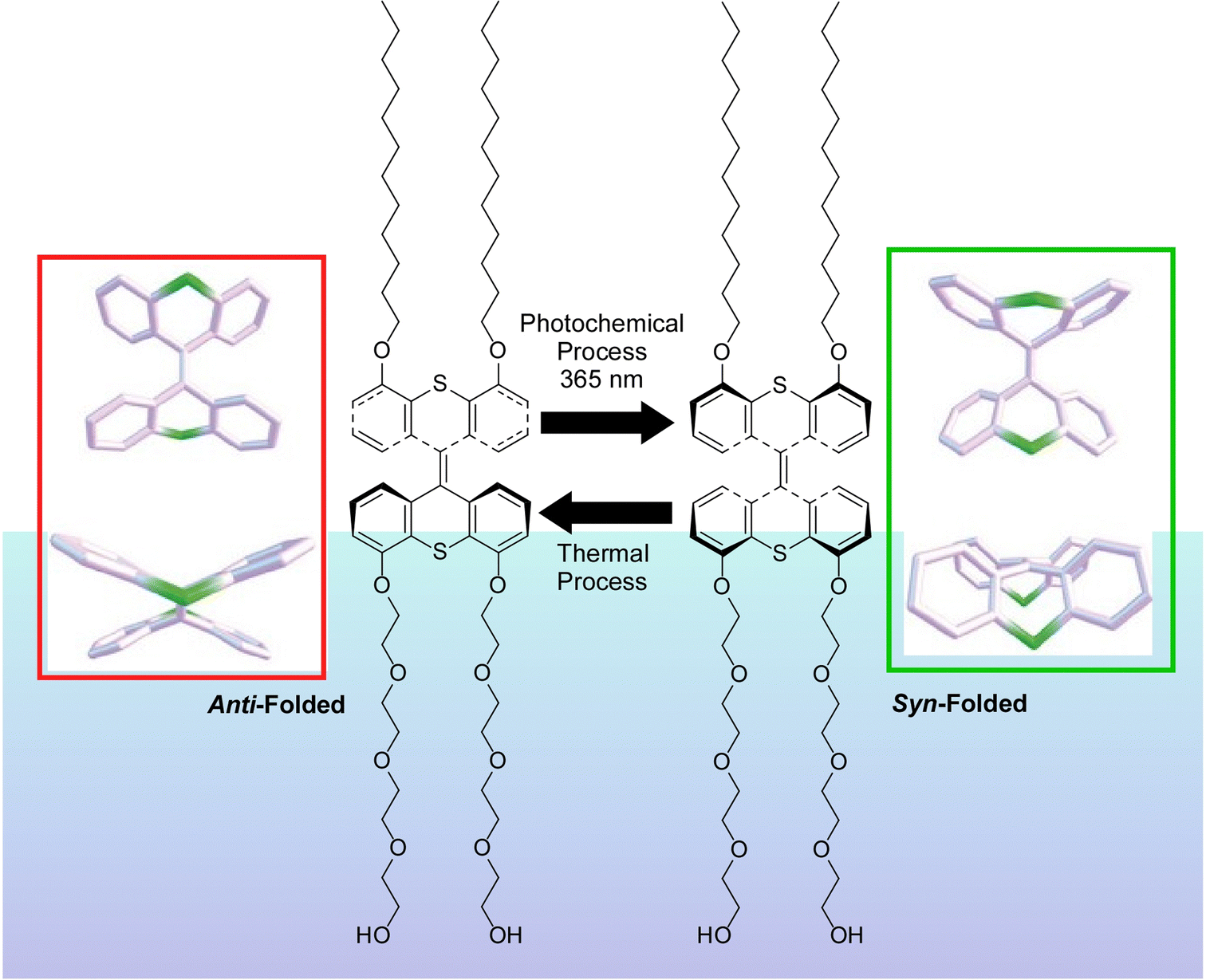 | ||
| Fig. 19 Photoswitching effect of bis(thiaxanthylidene)-based photoswitching amphiphilic molecular machines, in single-component monolayer or a mixed monolayer with dipalmitoylphosphatidylcholine (DPPC) at the air–water interface. Reprinted with permission from ref. 107. Copyright 2017 Wiley-VCH. | ||
Thus, at the air–water interface, macroscopic mechanical properties and conformational nanoscale phenomena of molecular machines can be linked. Contrary to the above example, at the air–water interface, macroscopic mechanical stimuli can subtly control the conformation of molecules, from −90° to −80° (Fig. 20).108 This 2D interface is an ideal site for mechanochemistry with molecular conformational tuning since it allows for increased molecular orientation and large changes in occupied surface areas per molecule. The proposed system provides a good opportunity to correlate input energy and the molecular output quantitatively. In this case, the corresponding driving force is supplied by 2D mechanical compression. The input energy in this process can be thermodynamically calculated from isotherms between surface pressures and molecular areas (a 2D version of the pressure–volume isotherm). Integration of the isotherm with molecular area changes up to a surface pressure of 10 mN m−1 results in an energy change of 0.179 ± 0.002 kcal![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) mol−1. Separately. energy resulting from molecular conformational changes can be calculated by density functional theory. Surprisingly, input energy thermodynamically provided by the mechanical process and output energy that is finally provided to molecules are very close to each other. Applying small mechanical energies (<1 kcal mol−1) to the monolayer, the area of the monolayer changes significantly (>50%) and the conformation of binaphthyl can be fine-tuned accordingly. As described above, molecular simulations show that the mechanical energy is converted in proportion to the torsional energy. Unexpectedly high conversion efficiency is experimentally and theoretically proved. Molecular dynamics simulations of monolayers also confirm a gradual shift in the global average torsion angle of the monolayer. At 2D liquid interfaces, macroscopic factors such as mechanical stimuli and thermodynamic parameters can be translated into precise conformational adjustments of molecules. The liquid interface is a favorable environment for tuning the structure and function of molecular machines.
mol−1. Separately. energy resulting from molecular conformational changes can be calculated by density functional theory. Surprisingly, input energy thermodynamically provided by the mechanical process and output energy that is finally provided to molecules are very close to each other. Applying small mechanical energies (<1 kcal mol−1) to the monolayer, the area of the monolayer changes significantly (>50%) and the conformation of binaphthyl can be fine-tuned accordingly. As described above, molecular simulations show that the mechanical energy is converted in proportion to the torsional energy. Unexpectedly high conversion efficiency is experimentally and theoretically proved. Molecular dynamics simulations of monolayers also confirm a gradual shift in the global average torsion angle of the monolayer. At 2D liquid interfaces, macroscopic factors such as mechanical stimuli and thermodynamic parameters can be translated into precise conformational adjustments of molecules. The liquid interface is a favorable environment for tuning the structure and function of molecular machines.
 | ||
| Fig. 20 Subtle control of the conformation of binaphthyl molecules, from −90° to −80°, upon macroscopic mechanical stimuli at the air–water interface where application of small mechanical energies (<1 kcal mol−1) results in fine tuning of the conformation of binaphthyl accordingly. Reprinted with permission from ref. 108. Copyright 2015 Wiley-VCH. | ||
The above example reported that a continuous (analogous) conformational change of an amphiphilic molecule, binaphthyl, can be mechanically induced at the air–water interface. Unlike that, Mori et al. have successfully induced a digital conformational change at the air–water interface by dissolving simple binaphthyl molecules in a matrix lipid monolayer and depositing binaphthyl pseudocrystals by mechanical compression and expansion of the monolayer (Fig. 21).109 In this example, the amphiphilic racemic dipalmitoylphosphatidylcholine (rac-DPPC) was used as a representative matrix lipid to form a stable mixed film at the water–air interface. By mechanically compressing and expanding the monolayer, the binaphthyl derivatives underwent a repeated phase transition between a homogeneous liquid state and a metastable nanocrystalline form. Accordingly, a reversible cisoid–transoid structural transition with left-handed and right-handed helicity inversion was achieved. The lipid matrix at the air–water interface promotes the formation of metastable nanocrystals with an open trans configuration of binaphthyl molecules. The crystallization and dissolution of metastable binaphthyl crystals and the associated structural changes are reversible and can be observed repeatedly. Since this phenomenon is based on a phase transition, the structural change of the binaphthyl moiety is not continuous, but discontinuous at the transition point. This can be described as a digital control of the structure of the molecular machine at liquid interfaces.
 | ||
| Fig. 21 Digital conformational changes of the binaphthyl derivatives at the air–water interface in the matrix lipid monolayer of the amphiphilic racemic dipalmitoylphosphatidylcholine (rac-DPPC), where a reversible cisoid–transoid structural transition with left-handed and right-handed helicity inversion can be achieved. Reprinted with permission from ref. 109. Copyright 2017 Wiley-VCH. | ||
Since the air–water interface is an ideal place to manipulate molecules, various optical information can also be modulated by using molecules whose mechanical deformation is coupled with optical properties. As one example, Nakanishi et al. studied the behavior of FLAP (flexible aromatic photofunctional systems) molecules at the air–water interface (Fig. 22).110 FLAP is a series of hybrid π-conjugated molecules that combine rigidity and flexibility. The FLAP molecules exhibit different dynamic behaviors depending on the surrounding environment, and thus emit a wide variety of colors in thin films, solutions, and crystals.111 Compared to chromogenic molecular rotors, the fluorescence has a non-negligible polarity dependence, but it shows a more sensitive fluorogenic response in the low viscosity range.112 New syntheses of nitrogen-containing FLAP molecules and the viscosity dependence of their photophysical properties have also been investigated.113 Polarity-independent flapping viscous probes with hydrophobic/hydrophilic substituents have been synthesized as FLAP molecules to be investigated at the air–water interface. Fluorescence related to internal motion and orientation was observed in situ at the air–water interface, and it was found that the internal motion of the molecule was suppressed at the interface. Compared to conventional molecular rotor-type viscous probes, FLAP exhibits diverse behaviors because it modulates not only molecular motion but also molecular orientation. Understanding molecular behavior at the interface is also of biological significance because such an interface provides a cell membrane-like field where various biomolecules work.
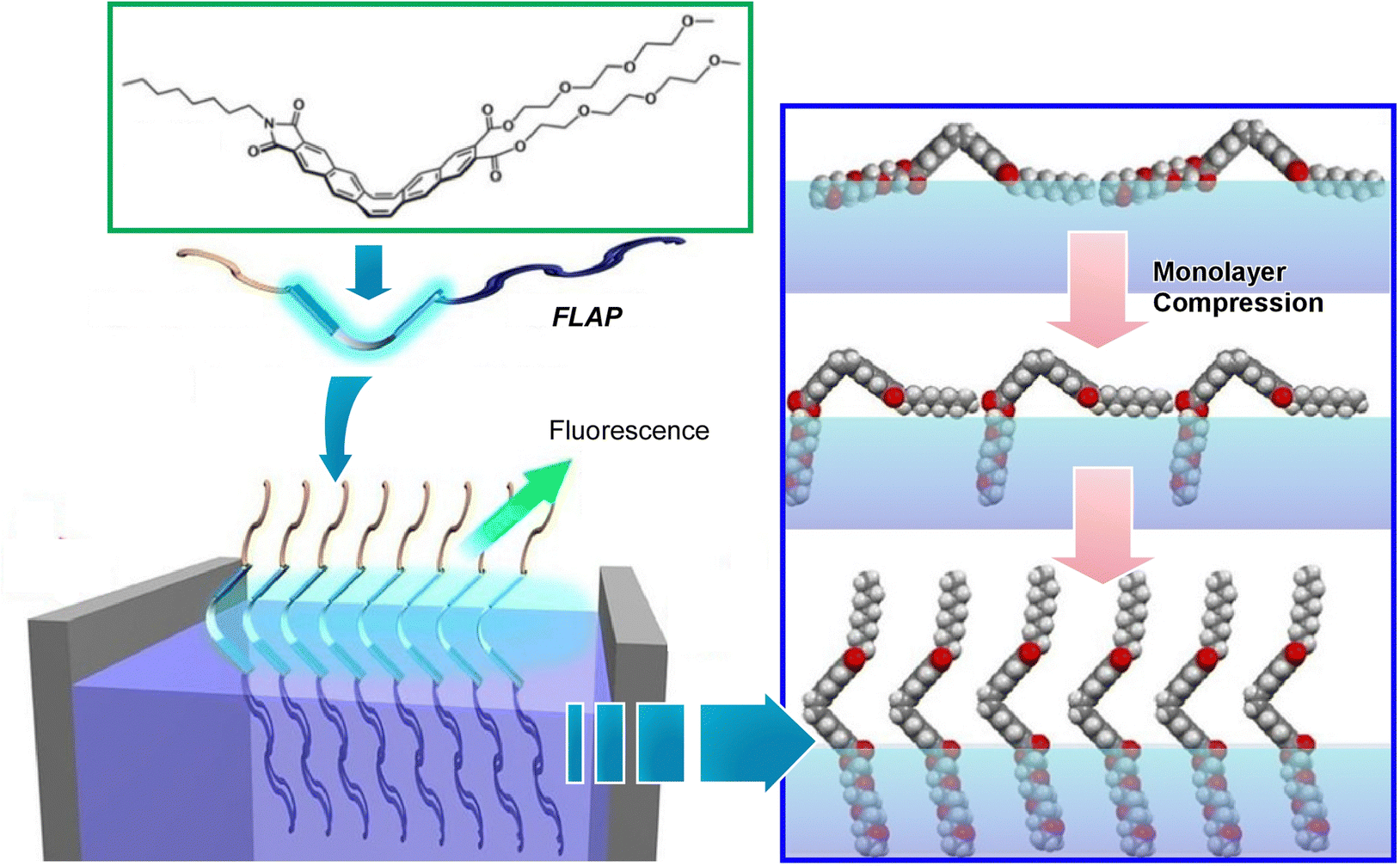 | ||
| Fig. 22 Behavior of FLAP molecules of hybrid π-conjugated molecules with rigidity and flexibility at the air–water interface, where fluorescence related to internal motion and orientation was observed in situ. Reprinted with permission from ref. 110. Copyright 2019 Wiley-VCH. | ||
Adachi et al. synthesized a double-paddle dinuclear platinum complex containing a pyrazole ring linked by an alkyl spacer and dynamically manipulated its orientation and luminescence properties at the water–air interface (Fig. 23).114 It was found that mechanical compression causes the platinum complex plane to change from perpendicular to parallel upon their monolayer compression and simultaneously emit light from the air–water interface. The control of the luminescence intensity by molecular floating was described by the term of submarine emission, because emission happens upon molecular appearance from water to air. The air–water interface is suitable as an asymmetric field where the conformation and motion of molecules can be controlled by applying only a small force. When one of the paddles is hydrogen bonded at the water surface and the other is in the gas phase, hydrogen bonding at the water surface can occur even though both paddles of molecules are identical. By using the air–water interface as an asymmetric field, a 2D chirality can be induced. Asymmetric reactions and motions may be controlled by applying mechanical forces at the air–water interface. In a similar molecular design, Adachi et al. recently proposed the concept of coordination amphiphilicity based on ligand properties and molecular topology.115 In a series of molecular designs, the importance of the molecular design of coordination compounds in the formation of monolayers at the air–water interface was investigated. Molecular modeling and the association constants of the compounds suggested that the appropriate hydrophilicity of the coordination surfaces and intermolecular interactions, including hydrogen bonding, are important factors in the formation of monolayers.
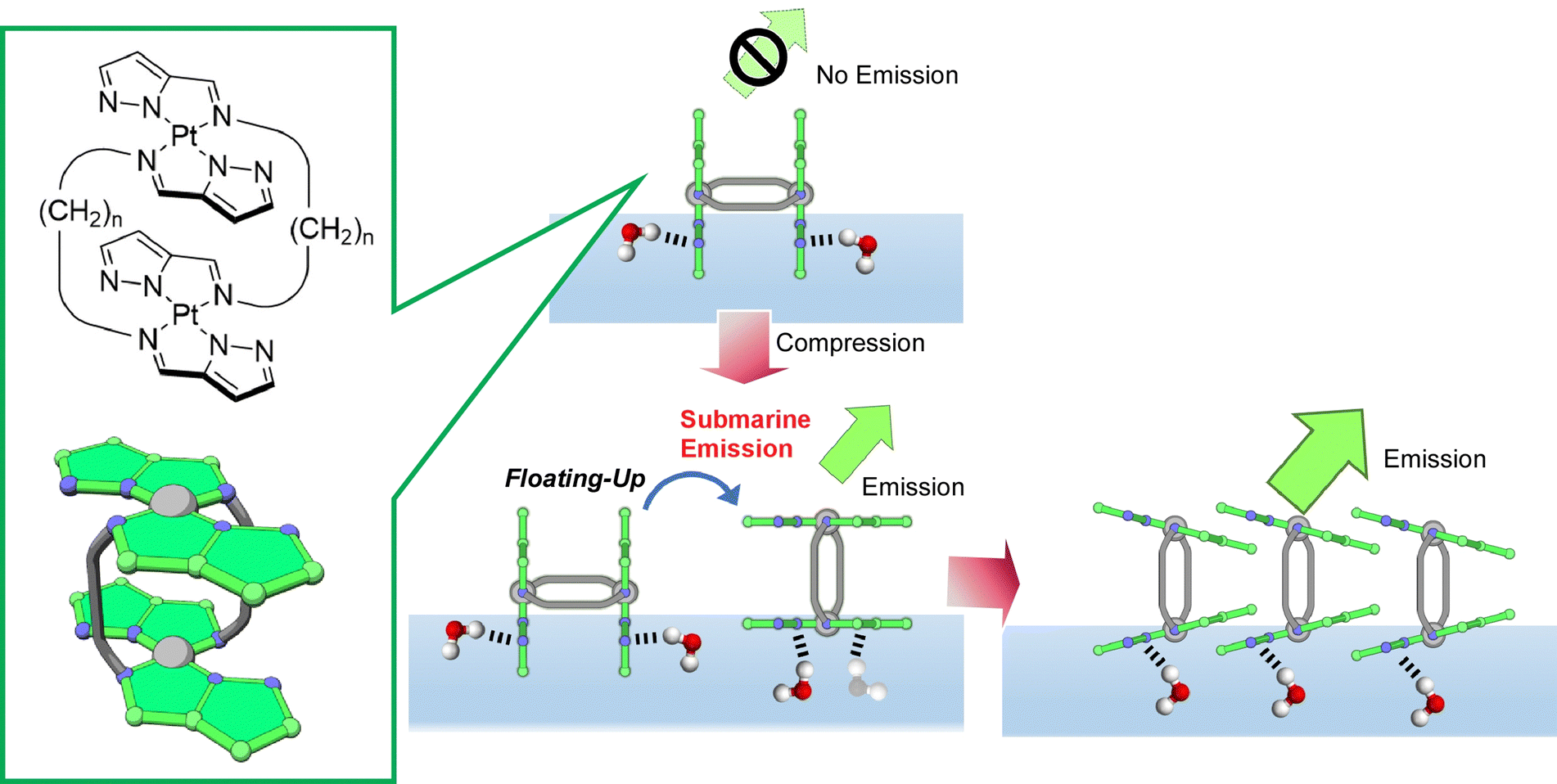 | ||
| Fig. 23 The submarine emission upon conformational and orientational changes of a double-paddle dinuclear platinum complex upon their monolayer compression accompanied by simultaneous light emission from the air–water interface. | ||
The behavior of various other molecular machines at the air–water interface has also been examined. Their behavior is influenced by mechanical compression of the monolayer from the lateral direction and coexisting lipid molecular films. For example, the molecular rotor, 4-farnesyloxyphenyl-4,4-difluoro-4-bora-3a,4a-diaza-s-indacene (BODIPY-ISO) rotation, was shown to be dynamically controlled, depending on the type of coexisting molecules.116 Molecules of bisbinaphthyl durene, in which two binaphthyl groups are joined via a central durene moiety, can form a wide variety of conformers.117 Preference of the formed conformer type was determined depending on the surrounding environment. This can be controlled in detail by controlling the environment, such as the state of miscibility with lipids in solution, on a solid substrate, or at the air–water interface. On the other hand, the molecular aggregation state of monobinaphthyl durene can be tuned by applying different mechanical stimuli to control its conformation at the air–water interface.118 For example, the molecular conformation and aggregation state of monobinaphthyl durene could be continuously controlled by applying mechanical stimuli such as vortex motion at the air–water interface. These results provide various insights into the regulation of the dynamic behavior of molecular machines driven by mechanical stimuli at the liquid interfaces.
5. Advanced interfaces
5.1. Material interfaces
Not only model-like interfaces such as clean solid interfaces that can be observed by STM and liquid interfaces such as undisturbed air–water interfaces, but also more advanced interfaces such as interfaces inside materials and biomembrane surfaces are being considered for the application of molecular machines. These studies will lead to the important step of the practical application of molecular machines. The last sections below summarize examples of research on analogous applications of molecular machines at interfaces in materials and biotechnology.Materials that are nanoarchitectonics with regular structures include MOFs,119 COFs,120 and other porous materials.121 Those materials form nanospaces controlled at the nanometer level. It is very interesting to confine the function of molecular machines inside such materials. By incorporating molecular machines into the backbone of porous framework structures, nano-actuators, highly controlled molecular transport, and host–guest phenomena in elaborately structured spaces are expected to be achieved. Krause and co-workers have developed a COF structure incorporating diamine-based light-driven molecular motors (Fig. 24).122 Indeed, they succeeded in synthesizing a crystalline 2D COF with stacked hexagonal layers containing 20 mol% molecular motors. Molecular motor dynamics were investigated by in situ spectroscopy and various features and issues were discussed.
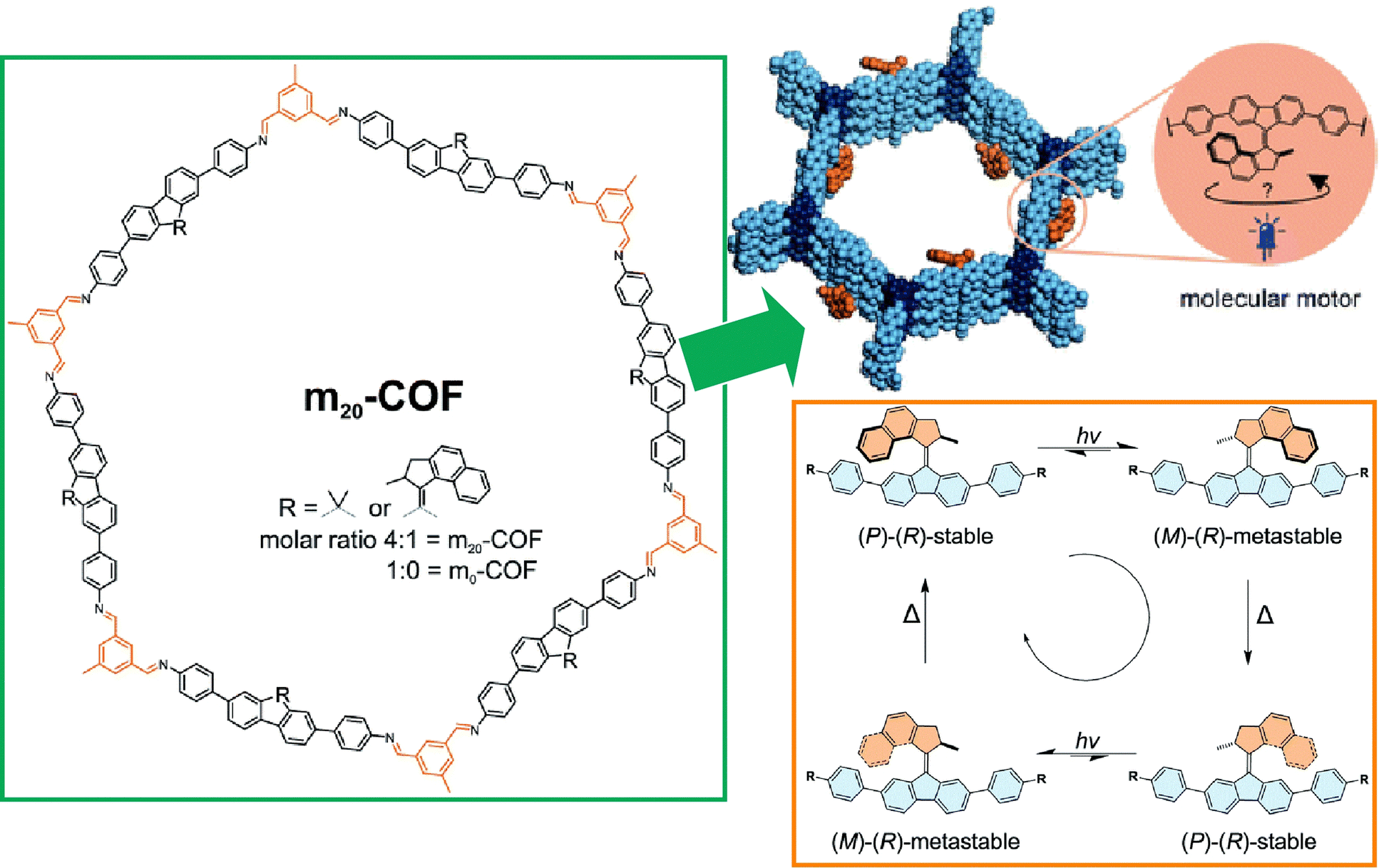 | ||
| Fig. 24 A COF structure incorporating diamine-based light-driven molecular motors. Reproduced under terms of the CC-BY license from ref. 122, 2022 Royal Society of Chemistry. | ||
Accumulation of molecular machine motions into macroscopic motions is one challenge for the practical application of molecular machines. It is an important next step toward future actuators and soft robotics. The key to solving this challenge is to effectively integrate synthetic molecular machines in hierarchically aligned structures and to collect many individual molecular motions in a coordinated manner. Preparing such hierarchical structures is a hallmark of nanoarchitectonics, and nanoarchitectonics concepts can be useful for this purpose. Feringa, Chen, and co-workers reported the incorporation of molecular motor molecules into liquid crystal network phases and evaluated their complex motions upon single wavelength illumination (Fig. 25).123 In this case, the molecular motors play multiple roles in the network as cross-linkers, actuators, and chiral dopants. The collective rotational motion of the motors demonstrated different types of motion of the polymer films, including bending, wavy motion, high-speed unidirectional motion on the surface, and synchronized helical motion. Polymeric liquid crystal films, including racemic ones, can move over surfaces in high-speed wave-like motions. On the other hand, films containing enantiomerically pure motors are capable of complex helical motions upon UV irradiation. A medium such as liquid crystals, which possess flexibility while having an organized structure, is useful in creating responsive materials that amplify the functionality of molecular machines. The same group has also developed a supramolecular system consisting of 95% water, formed by the hierarchical self-assembly of photo-responsive amphiphilic molecular motors.124 Molecular motors first assemble into nanofibers, which are then aligned into bundles that form a string several centimeters long. Light irradiation induces rotational motion of the molecular motors, which propagates and accumulates, causing the fibers to contract toward the light source.
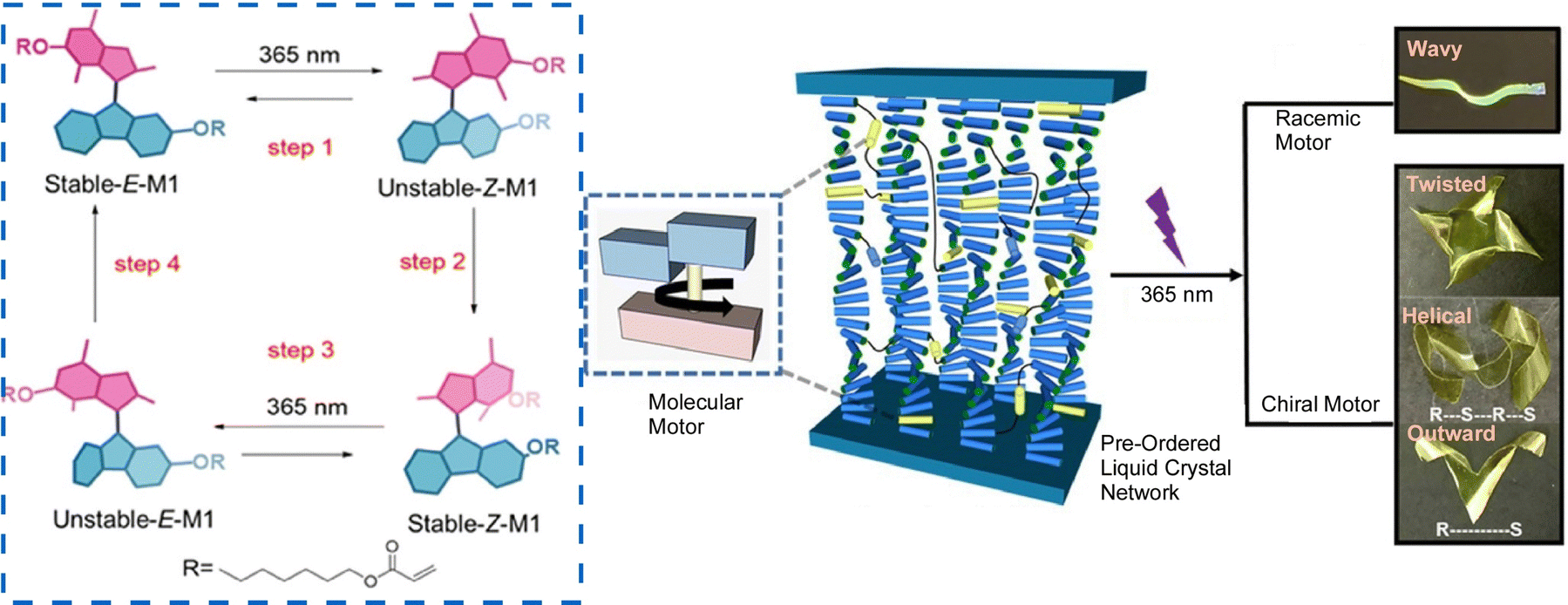 | ||
| Fig. 25 Liquid crystal network incorporating molecular motors with complex motions upon single wavelength illumination, including bending, wavy motion, high-speed unidirectional motion on the surface, and synchronized helical motion. Reproduced under terms of the CC-BY license from ref. 123, 2021 American Chemical Society. | ||
The incorporation of molecular machines in molecular assemblies, such as lipid bilayers, has also been investigated. Such systems can also be envisioned for biological applications. Barboiu, Giuseppone, and co-workers modified a hydrophobic light-driven rotational motor with two 18-crown-6 macrocyclic compounds and incorporated it into a phospholipid bilayer (Fig. 26).125 This ion channel exhibits selective transport of alkali cations across the phospholipid bilayer in the dark. Furthermore, UV irradiation strongly increases its transport activity by up to 400%. The actuation dynamics of the molecular motor provide energy to the ion channel. Increased motional dynamics help to overcome the activation barrier required for ion transfer between macrocyclic molecules along the artificial pore. The increased ion transport activity is due to the non-equilibrium actuation dynamics of the light-driven rotary motor. Molecular design may also allow for architecting dynamic superstructures that are ion-selective and permeable.
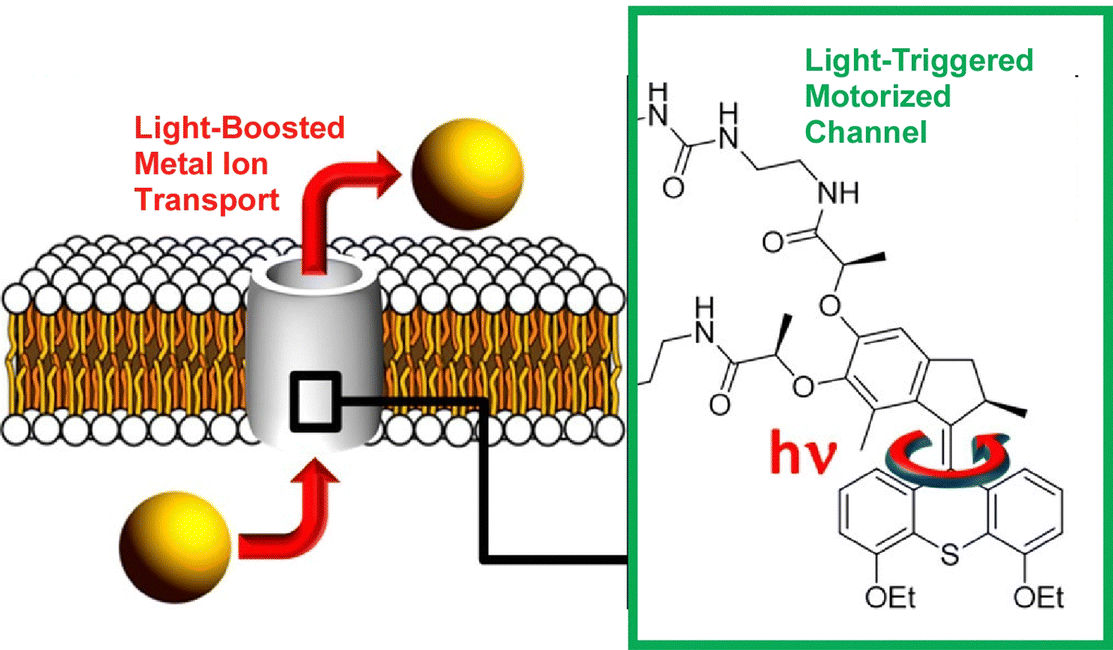 | ||
| Fig. 26 Hydrophobic light-driven rotational motor with two 18-crown-6 macrocyclic compounds incorporated into a phospholipid bilayer, where the ion channel exhibits selective transport of alkali cations across the phospholipid bilayer in the dark and UV irradiation strongly increases its transport activity by up to 400%. Reprinted with permission from ref. 125. Copyright 2021 American Chemical Society. | ||
5.2. Bio-interfaces
As mentioned above, molecular machines can be incorporated into lipid bilayers, which are also cell membrane models, to control their membrane permeability. Recent studies have also reported systems in which molecular motors are incorporated into the plasma membrane of real living cells. García-López et al. demonstrated that the nanomechanical action of molecular machines can be used to perforate the cell bilayer.126 By physically adsorbing a molecular motor onto the lipid bilayer and subsequently activating the motor with ultraviolet light, they were able to pierce the cell membrane. The nanomechanical action of molecular motors can selectively target specific cell surface recognition sites by using molecular machines with short peptides attached. They have also used two-photon excitation of near-infrared light for molecular motor rotation upon appropriate motor design (Fig. 27).127 The used wavelengths of light only cause minimal damage to normal cells. The molecular motors were able to combine cell-specific targeting by peptides with biologically safe activation by near-infrared light. It would be possible to rapidly and specifically kill undesirable cell types such as cancer cells using biologically safe wavelengths of light. Using advanced molecular machines, Gram-positive and Gram-negative bacteria, including methicillin-resistant Staphylococcus aureus, could be killed in minutes using an artificial molecular machine that could be activated by visible light (405 nm).128 This could be a distinctive antimicrobial therapy that greatly surpasses conventional antibiotics. Visible light-activated antimicrobial agents have an unconventional mode of antimicrobial action and are unlikely to cause bacterial resistance.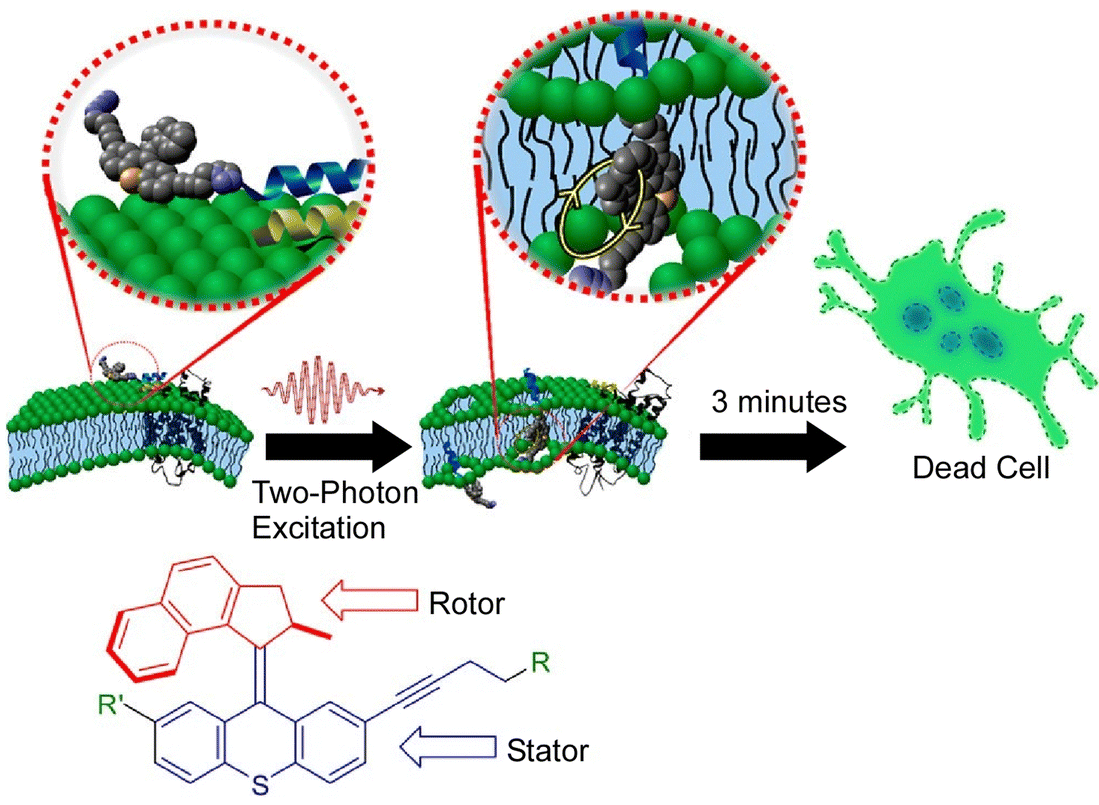 | ||
| Fig. 27 Two-photon excitation of near-infrared light for molecular motor rotation to rapidly kill specific cell types using biologically safe wavelengths of light. Reprinted with permission from ref. 127. Copyright 2019 American Chemical Society. | ||
The development of artificial ion-channel molecular machines with high activity, selectivity, and gating capabilities is of medical interest. Qu, Bao, and co-workers have developed an ion-channel molecular machine that penetrates the cell membrane (Fig. 28).129 A high-speed motor with a crown ether group was introduced into the stator of the two arms. This bola-amphiphile type molecular design allows this molecular machine to penetrate lipid bilayers. The rotational motion of the motor provided additional working energy to accelerate ion transport in the channel, thus realizing the optical gating function. The light-driven rotational motion promoted K+ efflux, generated reactive oxygen species, and ultimately activated caspase-3-dependent apoptosis. Excellent cytotoxicity and cancer cell selectivity were imparted. The design of this dynamic molecular machine is expected to demonstrate the ability to regulate intracellular K+ levels and indicate a novel strategy for drug discovery in the treatment of cancer and other diseases.
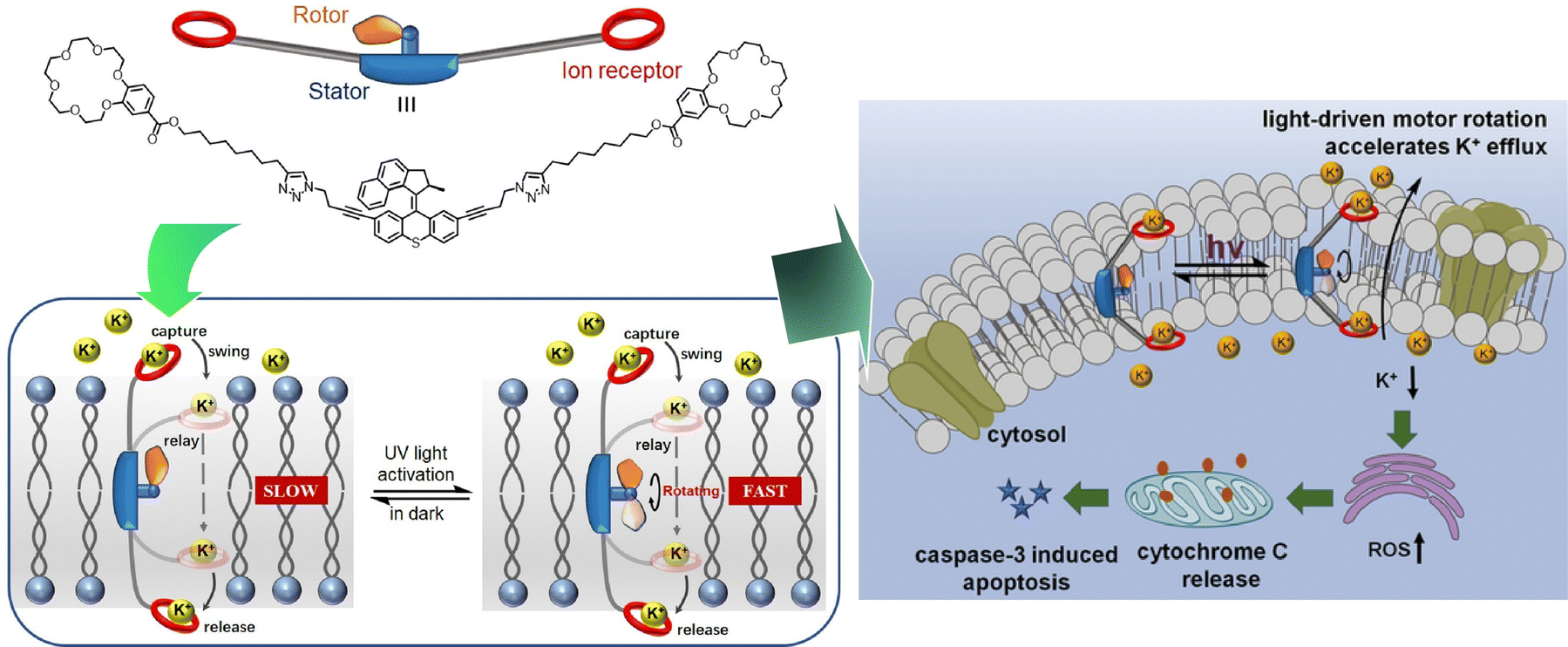 | ||
| Fig. 28 An ion-channel molecular machine at the cell membrane with light-driven rotational motion promoting K+ efflux, generated reactive oxygen species, and ultimately activated caspase-3-dependent apoptosis. Reprinted with permission from ref. 129. Copyright 2022 Wiley-VCH. | ||
6. Perspectives: meaning of the study of molecular machines at interfaces and future evolution
Molecular machines were developed through their clever molecular design, and in the early studies, their motion and function were determined by spectral analysis of molecules dissolved in solution. Subsequent significant developments in nanotechnology have made it possible to observe high-resolution molecular imaging on solid substrates, and molecular manipulation on solid interfaces has been actively studied.130 In addition, basic considerations on machine operations and functions have been made thermodynamically and kinetically.131 The detailed analysis has been fed back to molecular design and the development of advanced molecular machines such as molecular gears and nanocars.In this paper, we have seen several examples of the movement and function of molecular machines in interfacial environments. With the development of probe microscopy, we are now able to capture the movement of molecular machines in realistic images as if we were looking at them with our own eyes. Finally, the authors would like to emphasize that this is extremely significant progress in the development of molecular machine science. Molecular machines were originally initiated as chemistry in solution, and the example shown in Fig. 29(A) is a typical early example of molecular machines.132 In response to reactions with external substances or external stimuli, molecular rotors exhibit rotation in a constant direction. Such a motion has been confirmed by spectroscopic techniques such as NMR in solution. Then, why can molecular rotors move in a certain direction? To answer such a question, we have to examine the problem from an energetic point of view. Fig. 29(B) plots the calculated energy profile of the rotation of a triptycene rotor against a helicene stator in a model molecule of the molecular rotor, plotted against the rotation angle.133 The asymmetry of the energy profile with energy dissipation would result in a preferential rotation in one direction. This ratcheting mechanism occurs in coexistence with thermal fluctuations in solution under ambient conditions. This situation is problematic for further understanding of the phenomena. Although the motions of molecular machines can be evaluated by calculations, experimental confirmation of such a detailed motion in solution is usually very tough. In contrast, conditions such as on the surface, at cryogenic temperatures, and in an ultrahigh vacuum, where the fluctuation factor is eliminated, are extremely favorable for understanding the nature of such molecular machines. Investigation the motions at a surface/interface is very important to elucidate the mechanism of action of molecular machines. Clean solid surfaces provide an ideal field. Liquid and membrane interfaces provide a field for studies that bridge the gap between solution and biological systems. These are the most meaningful points in studies of molecular machines at interfaces.
 | ||
| Fig. 29 (A) Typical example of a molecular rotor with unidirectional rotation upon external stimuli; (B) a model of rotor unit and calculated energy as a function of dihedral angle between the front part and the back part. Reprinted with permission from ref. 133. Copyright 2001 American Chemical Society. | ||
Switching working environments is similar to the evolutionary process of organisms that evolved from water to land. Furthermore, molecular machines are expanding their activities from the static environment of a solid interface to the more dynamic environment of a liquid interface and the interface between materials and living organisms.134 In addition to the aspect that molecular machines change their field of activity while maintaining their basic functions, as in the workings of molecular machines in cell membranes, there is also the characteristic that molecular machines function collectively in dynamic environments. This makes it possible to reflect the accumulation of individual molecular machines into macroscopic physical properties and, conversely, to control molecular machines through macroscopic mechanical motions. According to these backgrounds, this review overviews and discusses the trends in molecular machine research by classifying them into (i) solid interfaces, (ii) liquid interfaces, and (iii) various material and biological interfaces.
Of course, all the features cannot be explained in this short review article. Molecular machines and related phenomena including extremely long distance motion,135 1D motion along a specific motion,136 pulling of flexible chains,137 and translation upon the effect of Brownian motions138 have been reported. However, another important direction on molecular machines can be suggested below. Molecular machine research is not only changing the interfacial environment, but also evolving from individual organisms to collective systems that work in tandem. This is similar to the process of evolution of living organisms from unicellular to multicellular organisms. The collective function of molecular machines is currently achieved by accumulating a single type of molecular machine in an environment such as a liquid interface, and then harmonizing them to make them function together. For more advanced functions such as those seen in biological systems,139 it will be important to form a system in which multiple types of functional molecular machines work in tandem. It is important not only to develop individual molecular machines but also to rationally organize them into an appropriate environment such as an interface. In other words, the organization of molecular machines into interfaces through nanoarchitectonics will be the key to future development. The organization of molecular machines of multiple species is expected to make the system more complex. Although it has been difficult to build such complex systems using conventional methods, the introduction of artificial intelligence technologies140 may provide a solution to this problem. We can expect molecular machines to evolve further with the addition of new essences such as the setting of an interface, organization by nanoarchitectonics, and analysis by artificial intelligence.
Author contributions
Katsuhiko Ariga: conceptualization, writing – original draft, writing – review and editing, and funding acquisition. Jingwen Song: conceptualization and review and editing. Kohsaku Kawakami: project administration and review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was partially supported by the Japan Society for the Promotion of Science KAKENHI (grant numbers JP20H00392 and JP23H05459).Notes and references
- (a) R. Kaur and S. Liu, Prog. Surf. Sci., 2016, 91, 136–153 CrossRef CAS; (b) N. Kasmuri, N. A. A. Tarmizi and A. Mojiri, Environ. Sci. Pollut. Res., 2022, 29, 30820–30836 CrossRef CAS PubMed; (c) A. Chapman, E. Ertekin, M. Kubota, A. Nagao, K. Bertsch, A. Macadre, T. Tsuchiyama, T. Masamura, S. Takaki, R. Komoda, M. Dadfarnia, B. Somerday, A. T. Staykov, J. Sugimura, Y. Sawae, T. Morita, H. Tanaka, K. Yagi, V. Niste, P. Saravanan, S. Onitsuka, K.-S. Yoon, S. Ogo, T. Matsushima, G. Tumen-Ulzii, D. Klotz, D. H. Nguyen, G. Harrington, C. Adachi, H. Matsumoto, L. Kwati, Y. Takahashi, N. Kosem, T. Ishihara, M. Yamauchi, B. B. Saha, M. A. Islam, J. Miyawaki, H. Sivasankaran, M. Kohno, S. Fujikawa, R. Selyanchyn, T. Tsuji, Y. Higashi, R. Kirchheim and P. Sofronis, Bull. Chem. Soc. Jpn., 2022, 95, 73–103 CrossRef CAS; (d) J. Wang, F. Matsuzawa, N. Sato, Y. Amano and M. Machida, Bull. Chem. Soc. Jpn., 2023, 96, 1088–1098 CrossRef CAS.
- (a) D. Guo, R. Shibuya, C. Akiba, S. Saji, T. Kondo and J. Nakamura, Science, 2016, 351, 361–365 CrossRef CAS PubMed; (b) T. Minato and T. Abe, Prog. Surf. Sci., 2017, 92, 240–280 CrossRef CAS; (c) K. Maeda, F. Takeiri, G. Kobayashi, S. Matsuishi, H. Ogino, S. Ida, T. Mori, Y. Uchimoto, S. Tanabe, T. Hasegawa, N. Imanaka and H. Kageyama, Bull. Chem. Soc. Jpn., 2022, 95, 26–37 CrossRef CAS; (d) A. Yoshino, Bull. Chem. Soc. Jpn., 2022, 95, 195–197 CrossRef CAS; (e) P. A. Shinde, Q. Abbas, N. R. Chodankar, K. Ariga, M. A. Abdelkareem and A. G. Olabi, J. Energy Chem., 2023, 79, 611–638 CrossRef CAS.
- (a) T.-Y. Liao, A. Biesiekierski, C. C. Berndt, P. C. King, E. P. Ivanova, H. Thissen and P. Kingshott, Prog. Surf. Sci., 2022, 97, 100654 CrossRef CAS; (b) S. Quader, K. Kataoka and H. Cabral, Adv. Drug Delivery Rev., 2022, 182, 114115 CrossRef CAS PubMed; (c) T. Niwa, T. Tahara, C. E. Chase, F. G. Fang, T. Nakaoka, S. Irie, E. Hayashinaka, Y. Wada, H. Mukai, K. Masutomi, Y. Watanabe, Y. Cui and T. Hosoya, Bull. Chem. Soc. Jpn., 2023, 96, 283–290 CrossRef CAS; (d) S. Mohanan, C. I. Sathish, T. J. Adams, S. Kan, M. Liang and A. Vinu, Bull. Chem. Soc. Jpn., 2023, 96, 1188–1195 CrossRef CAS.
- (a) T. Kawawaki, Y. Kataoka, M. Hirata, Y. Iwamatsu, S. Hossain and Y. Negishi, Nanoscale Horiz., 2021, 6, 409–448 RSC; (b) T. Itagaki, Y. Ito and M. Ueda, J. Colloid Interface Sci., 2022, 617, 129–135 CrossRef CAS PubMed; (c) H. Imahori, Bull. Chem. Soc. Jpn., 2023, 96, 339–352 CrossRef CAS; (d) S. d Kalyana Sundaram, M. M. Hossain, M. Rezki, K. Ariga and S. Tsujimura, Biosensors, 2023, 13, 1018 CrossRef CAS PubMed; (e) H. Masai, Bull. Chem. Soc. Jpn., 2023, 96, 1196–1205 CrossRef CAS.
- (a) K. Y. Cheung, K. Watanabe, Y. Segawa and K. Itami, Nat. Chem., 2021, 13, 255–259 CrossRef CAS PubMed; (b) H. Fujimoto, K. Yasui and M. Tobisu, Bull. Chem. Soc. Jpn., 2023, 96, 872–886 CrossRef CAS; (c) T. Ema, Bull. Chem. Soc. Jpn., 2023, 96, 693–701 CrossRef CAS; (d) A. Minami, Bull. Chem. Soc. Jpn., 2023, 96, 1216–1223 CrossRef CAS.
- (a) H. Hosono and M. Kitano, Advances in materials and applications of inorganic electrides, Chem. Rev., 2021, 121, 3121–3185 CrossRef CAS PubMed; (b) H. Tokoro, K. Nakabayashi, S. Nagashima, Q. Song, M. Yoshikiyo and S. Ohkoshi, Bull. Chem. Soc. Jpn., 2022, 95, 538–555 CrossRef CAS; (c) D. Wu, K. Kusada, Y. Nanba, M. Koyama, T. Yamamoto, T. Toriyama, S. Matsumura, O. Seo, I. Gueye, J. Kim, L. S. R. Kumara, O. Sakata, S. Kawaguchi, Y. Kubota and H. Kitagawa, J. Am. Chem. Soc., 2022, 144, 3365–3369 CrossRef CAS PubMed; (d) M. S. Islam, Y. Shudo and S. Hayami, Bull. Chem. Soc. Jpn., 2022, 95, 1–25 CrossRef CAS.
- (a) J. Zha, N. Batisse, D. Claves, M. Dubois, L. Frezet, A. P. Kharitonov and L. N. Alekseiko, Prog. Surf. Sci., 2016, 91, 57–71 CrossRef CAS; (b) K. Oka, B. Winther-Jensen and H. Nishide, Adv. Energy Mater., 2021, 11, 2003724 CrossRef CAS; (c) S. Watanabe and K. Oyaizu, Bull. Chem. Soc. Jpn., 2023, 96, 1108–1128 CrossRef CAS; (d) Y. Furukawa and D. Shimokawa, Bull. Chem. Soc. Jpn., 2023, 96, 1243–1251 CrossRef CAS.
- (a) L. Dong, Z. A. Gao and N. Lin, Prog. Surf. Sci., 2016, 91, 101–135 CrossRef CAS; (b) Y. Gu, J.-J. Zheng, K. Otake, M. Shivanna, S. Sakaki, H. Yoshino, M. Ohba, S. Kawaguchi, Y. Wang, F. Li and S. Kitagawa, Angew. Chem., Int. Ed., 2021, 60, 11688–11694 CrossRef CAS PubMed; (c) Y. Shan, G. Zhang, W. Yin, H. Pang and Q. Xu, Bull. Chem. Soc. Jpn., 2022, 95, 230–260 CrossRef CAS; (d) S. Horike, Bull. Chem. Soc. Jpn., 2023, 96, 887–898 CrossRef CAS.
- (a) V. Humblot, S. M. Barlow and R. Raval, Prog. Surf. Sci., 2004, 76, 1–19 CrossRef CAS; (b) S. Datta, Y. Kato, S. Higashiharaguchi, K. Aratsu, A. Isobe, T. Saito, D. D. Prabhu, Y. Kitamoto, M. J. Hollamby, A. J. Smith, R. Dalgliesh, N. Mahmoudi, L. Pesce, C. Perego, G. M. Pavan and S. Yagai, Nature, 2020, 583, 400–405 CrossRef CAS PubMed; (c) E. Mieda, Y. Morishima, T. Watanabe, H. Miyake and S. Shinoda, Bull. Chem. Soc. Jpn., 2023, 96, 538–544 CrossRef CAS; (d) S. Shoji, V. Stepanenko, F. Würthner and H. Tamiaki, Bull. Chem. Soc. Jpn., 2022, 95, 1083–1085 CrossRef CAS.
- (a) E. Moore, H. Thissen and N. H. Voelcker, Prog. Surf. Sci., 2013, 88, 213–236 CrossRef CAS; (b) S. Asamitsu, S. Obata, Z. Yu, T. Bando and H. Sugiyama, Molecules, 2019, 24, 429 CrossRef PubMed; (c) K. Murayama, H. Okita and H. Asanuma, Bull. Chem. Soc. Jpn., 2023, 96, 1179–1187 CrossRef CAS; (d) M. Fukuyama, Bull. Chem. Soc. Jpn., 2023, 96, 1252–1257 CrossRef CAS.
- (a) S. Dong, H. Jiao, Z. Wang, J. Zhang and X. Cheng, Prog. Surf. Sci., 2022, 97, 100663 CrossRef CAS; (b) Z.-Z. Pan, W. Lv, Q.-H. Yang and H. Nishihara, Bull. Chem. Soc. Jpn., 2022, 95, 611–620 CrossRef CAS; (c) T. Murata, K. Minami, T. Yamazaki, T. Sato, H. Koinuma, K. Ariga and N. Matsuki, Bull. Chem. Soc. Jpn., 2023, 96, 29–34 CrossRef CAS; (d) M. Ishii, Y. Yamashita, S. Watanabe, K. Ariga and J. Takeya, Nature, 2023, 622, 285–291 CrossRef CAS PubMed; (e) T. Adschiri, S. Takami, M. Umetsu, S. Ohara, T. Naka, K. Minami, D. Hojo, T. Togashi, T. Arita, M. Taguchi, M. Itoh, N. Aoki, G. Seong, T. Tomai and A. Yoko, Bull. Chem. Soc. Jpn., 2023, 96, 133–147 CrossRef CAS.
- (a) Y. Sugimoto, P. Pou, M. Abe, P. Jelinek, R. Pérez, S. Morita and Ó. Custance, Nature, 2007, 446, 64–67 CrossRef CAS PubMed; (b) B.-C. Huang, C.-C. Hsu, Y.-H. Chu and Y.-P. Chiu, Prog. Surf. Sci., 2022, 97, 100662 CrossRef CAS; (c) K. Tada, Y. Hinuma, S. Ichikawa and S. Tanaka, Bull. Chem. Soc. Jpn., 2023, 96, 373–380 CrossRef CAS; (d) Y. Hashikawa and Y. Murata, Bull. Chem. Soc. Jpn., 2023, 96, 943–967 CrossRef CAS.
- (a) K. Kimura, K. Miwa, H. Imada, M. Imai-Imada, S. Kawahara, J. Takeya, M. Kawai, M. Galperin and Y. Kim, Nature, 2019, 570, 210–213 CrossRef CAS PubMed; (b) Y. Kim, K. Motobayashi, T. Frederiksen, H. Ueba and M. Kawai, Prog. Surf. Sci., 2015, 90, 85–143 CrossRef CAS; (c) K. Kawai, M. Fujitsuka and A. Maruyama, Acc. Chem. Res., 2021, 54, 1001–1010 CrossRef CAS PubMed; (d) S. Fan, T. Takada, A. Maruyama, M. Fujitsuka and K. Kawai, Bull. Chem. Soc. Jpn., 2022, 95, 1697–1702 CrossRef CAS.
- (a) Y. Tamura, H. Takezawa and M. Fujita, J. Am. Chem. Soc., 2020, 142, 5504–5508 CrossRef CAS PubMed; (b) S. Negi, M. Hamori, Y. Kubo, H. Kitagishi and K. Kano, Bull. Chem. Soc. Jpn., 2023, 96, 48–56 CrossRef CAS; (c) T. Matsuno and H. Isobe, Bull. Chem. Soc. Jpn., 2023, 96, 406–419 CrossRef CAS.
- (a) K. Ariga, M. Nishikawa, T. Mori, J. Takeya, L. K. Shrestha and J. P. Hill, Adv. Mater., 2019, 20, 51–95 CAS; (b) R. Kubota, Bull. Chem. Soc. Jpn., 2023, 96, 802–812 CrossRef CAS; (c) Y. Yamamoto, S. Kushida, D. Okada, O. Oki, H. Yamagishi and Hendra, Bull. Chem. Soc. Jpn., 2023, 96, 702–710 CrossRef CAS.
- K. Ariga, Nanoscale Horiz., 2021, 6, 364–378 RSC.
- (a) K. Ariga, Q. Ji, J. P. Hill, Y. Bando and M. Aono, NPG Asia Mater., 2012, 4, e17 CrossRef; (b) M. Aono and K. Ariga, Adv. Mater., 2016, 28, 989–992 CrossRef CAS PubMed.
- (a) W. Chaikittisilp, Y. Yamauchi and K. Ariga, Adv. Mater., 2022, 34, 2107212 CrossRef CAS PubMed; (b) L. R. Oviedo, V. R. Oviedo, M. O. Martins, S. B. Fagan and W. L. da Silva, J. Nanopart. Res., 2022, 24, 157 CrossRef.
- (a) R. P. Feynman, California Inst. Technol. J. Eng. Sci., 1960, 4, 23–36 Search PubMed; (b) M. Roukes, Sci. Am., 2001, 285, 48–57 CrossRef CAS PubMed.
- (a) K. Ariga, Q. Ji, W. Nakanishi, J. P. Hill and M. Aono, Mater. Horiz., 2015, 2, 406–413 RSC; (b) K. Ariga and M. Aono, Jpn. J. Appl. Phys., 2016, 55, 1102A6 CrossRef.
- K. Ariga, J. Li, J. Fei, Q. Ji and J. P. Hill, Adv. Mater., 2016, 28, 1251–1286 CrossRef CAS PubMed.
- K. Ariga, X. Jia, J. Song, J. P. Hill, D. T. Leong, Y. Jia and J. Li, Angew. Chem., Int. Ed., 2020, 59, 15424–15446 CrossRef CAS PubMed.
- R. B. Laughlin and D. Pines, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 28–31 CrossRef CAS PubMed.
- (a) K. Ariga and R. Fakhrullin, Bull. Chem. Soc. Jpn., 2022, 95, 774–795 CrossRef CAS; (b) K. Ariga, Bull. Chem. Soc. Jpn., 2024, 97, uoad001 CrossRef.
- (a) W. Nakanishi, K. Minami, L. K. Shrestha, Q. Ji, J. P. Hill and K. Ariga, Nano Today, 2014, 9, 378–394 CrossRef CAS; (b) M. Eguchi, A. S. Nugraha, A. E. Rowan, J. Shapter and Y. Yamauchi, Adv. Sci., 2021, 8, 2100539 CrossRef CAS PubMed; (c) Y. Oaki and K. Sato, Nanoscale Adv., 2022, 4, 2773–2781 RSC.
- (a) K. Ariga, T. Mori, T. Kitao and T. Uemura, Adv. Mater., 2020, 32, 1905657 CrossRef CAS PubMed; (b) K. Ariga and M. Shionoya, Bull. Chem. Soc. Jpn., 2021, 94, 839–859 CrossRef CAS; (c) Y. Wang, D. Niu, G. Ouyang and M. Liu, Nat. Commun., 2022, 13, 1710 CrossRef CAS PubMed; (d) T. Yoshida and M. Ogawa, Nanoscale, 2022, 14, 7480–7483 RSC.
- (a) S. Ishihara, J. Labuta, W. Van Rossom, D. Ishikawa, K. Minami, J. P. Hill and K. Ariga, Phys. Chem. Chem. Phys., 2014, 16, 9713–9746 RSC; (b) M. Pandeeswar, S. P. Senanayak and T. Govindaraju, ACS Appl. Mater. Interfaces, 2016, 8, 30362–30371 CrossRef CAS PubMed; (c) M. Komiyama, T. Mori and K. Ariga, Bull. Chem. Soc. Jpn., 2018, 91, 1075–1111 CrossRef CAS; (d) R. Moradi, N. P. Khalili, N. L. W. Septiani, C.-H. Liu, E. Doustkhah, Y. Yamauchi and S. V. Rotkin, Small, 2022, 18, 2104847 CrossRef CAS PubMed.
- (a) J. M. Giussi, M. L. Cortez, W. A. Marmisollé and O. Azzaroni, Chem. Soc. Rev., 2019, 48, 814–849 RSC; (b) K. Ariga, M. Ito, T. Mori, S. Watanabe and J. Takeya, Nano Today, 2019, 28, 100762 CrossRef; (c) K. Ariga, T. Makita, M. Ito, T. Mori, S. Watanabe and J. Takeya, Beilstein J. Nanotechnol., 2019, 10, 2014–2030 CrossRef CAS PubMed; (d) K. Terabe, T. Tsuchiya and T. Tsuruoka, Adv. Electron. Mater., 2022, 8, 2100645 CrossRef CAS; (e) T. Tsuchiya, T. Nakayama and K. Ariga, Appl. Phys. Express, 2022, 15, 100101 CrossRef CAS.
- (a) T.-A. Pham, A. Qamar, T. Dinh, M. K. Masud, M. Rais-Zadeh, D. G. Senesky, Y. Yamauchi, M.-T. Nguyen and H.-P. Phan, Adv. Sci., 2020, 7, 2001294 CrossRef CAS PubMed; (b) X. Tang, L. Zhou, H. Yu, Y. Dai, J. Ouyang, Z. Liu, Y. Wang, Z. Le and A. A. Adesina, Sep. Purif. Technol., 2021, 278, 119604 CrossRef; (c) A. M. Abd-Elnaiem, R. F. A. El-Baki, F. Alsaaq, S. Orzechowska and D. Hamad, J. Inorg, Organomet. Polym. Mater., 2022, 32, 1191–1205 CrossRef CAS.
- (a) J. Kim, J. H. Kim and K. Ariga, Joule, 2017, 1, 739–768 CrossRef CAS; (b) J. Na, D. Zheng, J. Kim, M. Gao, A. Azhar, J. Lin and Y. Yamauchi, Small, 2022, 18, 2102397 CrossRef CAS PubMed; (c) X. Liu, T. Chen, Y. Xue, J. Fan, S. Shen, M. S. A. Hossain, M. A. Amin, L. Pan, X. Xu and Y. Yamauchi, Coord. Chem. Rev., 2022, 459, 214440 CrossRef CAS; (d) W. Meng, H. He, L. Yang, Q. Jiang, B. Yuliarto, Y. Yamauchi, X. Xu and H. Huang, Chem. Eng. J., 2022, 450, 137932 CrossRef CAS; (e) G. Chen, S. K. Singh, K. Takeyasu, J. P. Hill, J. Nakamura and K. Ariga, Sci. Technol. Adv. Mater., 2022, 23, 413–423 CrossRef CAS PubMed; (f) P. A. Shinde, N. R. Chodankar, H.-J. Kim, M. A. Abdelkareem, A. A. Ghaferi, Y.-K. Han, A. G. Olabi and K. Ariga, ACS Energy Lett., 2023, 8, 4474–4487 CrossRef CAS.
- (a) Q. Zou, K. Liu, M. Abbas and X. Yan, Adv. Mater., 2016, 28, 1031–1043 CrossRef CAS PubMed; (b) K. Ariga and R. Fakhrullin, RSC Adv., 2021, 11, 18898–18914 RSC; (c) X. Shen, J. Song, C. Sevencan, D. T. Leong and K. Ariga, Sci. Technol. Adv. Mater., 2022, 23, 199–224 CrossRef PubMed; (d) V. Karthick, L. K. Shrestha, V. G. Kumar, P. Pranjali, D. Kumar, A. Palf and K. Ariga, Nanoscale, 2022, 14, 10630–10647 RSC; (e) S. Banerjee and J. Pillai, Expert Opin. Drug Metab. Toxicol., 2019, 15, 499–515 CrossRef PubMed; (f) X. Jia, J. Chen, W. Lv, H. Li and K. Ariga, Cell Rep. Phys. Sci., 2023, 4, 101251 CrossRef CAS.
- (a) S. Sharma, M. K. Masud, Y. V. Kaneti, P. Rewatkar, A. Koradia, M. S. A. Hossain, Y. Yamauchi, A. Popat and C. Salomon, Small, 2021, 17, 2102220 CrossRef CAS PubMed; (b) D. B. Momekova, V. E. Gugleva and P. D. Petrov, ACS Omega, 2021, 6, 33265–33273 CrossRef CAS PubMed; (c) W. Hu, J. Shi, W. Lv, X. Jia and K. Ariga, Sci. Technol. Adv. Mater., 2022, 23, 393–412 CrossRef CAS PubMed; (d) J. Song, K. Kawakami and K. Ariga, Curr. Opin. Colloid Interface Sci., 2023, 65, 101702 CrossRef CAS; (e) L. Sutrisno and K. Ariga, NPG Asia Mater., 2023, 15, 21 CrossRef CAS.
- (a) X. Liang, L. Li, J. Tang, M. Komiyama and K. Ariga, Bull. Chem. Soc. Jpn., 2020, 93, 581–603 CrossRef CAS; (b) P. Qiao, Q. Shi, S. Zhang, X. Zhang, Y. Yang, B. Liu, X. Wang, Q. Luo and L. Wang, Mater. Today Chem., 2022, 24, 100893 CrossRef CAS.
- K. Ariga, T. Mori and J. P. Hill, Soft Matter, 2017, 8, 15–20 RSC.
- (a) J.-P. Sauvage, Angew. Chem., Int. Ed., 2017, 56, 11080–11093 CrossRef CAS PubMed; (b) J. F. Stoddart, Angew. Chem., Int. Ed., 2017, 56, 11094–11125 CrossRef CAS PubMed; (c) B. L. Feringa, Angew. Chem., Int. Ed., 2017, 56, 11060–11078 CrossRef CAS PubMed.
- (a) S. Saha, E. Johansson, A. H. Flood, H.-R. Tseng, J. I. Zink and J. F. Stoddart, Chem. – Eur. J., 2005, 11, 6846–6858 CrossRef CAS PubMed; (b) S. Corra, M. Curcio, M. Baroncini, S. Silvi and A. Credi, Adv. Mater., 2022, 32, 1906064 CrossRef PubMed.
- (a) C. Manzano, W.-H. Soe, H. S. Wong, F. Ample, A. Gourdon, N. Chandrasekhar and C. Joachim, Nat. Mater., 2009, 8, 576–579 CrossRef CAS PubMed; (b) K. Qu, P. Duan, J.-Y. Wang, B. Zhang, Q.-C. Zhang, W. Hong and Z.-N. Chen, Nano Lett., 2021, 21, 9729–9735 CrossRef CAS PubMed.
- K. Ariga, T. Mori, S. Ishihara, K. Kawakami and J. P. Hill, Chem. Mater., 2014, 26, 519–532 CrossRef CAS.
- K. Ariga, M. Ishii and T. Mori, Chem. – Eur. J., 2020, 26, 6461–6472 CrossRef CAS PubMed.
- (a) K. Ariga and T. Kunitake, Acc. Chem. Res., 1998, 31, 371–378 CrossRef CAS; (b) K. Ariga, Phys. Chem. Chem. Phys., 2020, 22, 24856–24869 RSC.
- K. Ariga, T. Mori and J. P. Hill, Adv. Mater., 2012, 24, 158–176 CrossRef CAS PubMed.
- (a) K. Ariga, Small Sci., 2021, 1, 2000032 CrossRef CAS; (b) K. Ariga, Small Methods, 2022, 6, 2101577 CrossRef PubMed.
- (a) K. Ariga, Chem. Sci., 2020, 11, 10594–10604 RSC; (b) K. Ariga, Nanoscale, 2022, 14, 10610–10629 RSC.
- (a) V. Balzani, A. Credi, F. Raymo and J. Stoddart, Angew. Chem., Int. Ed., 2000, 39, 3348–3391 CrossRef CAS; (b) T. Nagasaki and S. Shinkai, J. Inclusion Phenom. Macrocyclic Chem., 2007, 58, 205–219 CrossRef CAS; (c) F. Nicoli, E. Paltrinieri, M. T. Bakić, M. Baroncini, S. Silvi and A. Credi, Coord. Chem. Rev., 2021, 428, 213589 CrossRef CAS.
- K. Kato, S. Ohtani, S. Fa and T. Ogoshi, Bull. Chem. Soc. Jpn., 2021, 94, 2319–2328 CrossRef CAS.
- H. Takezawa and M. Fujita, Bull. Chem. Soc. Jpn., 2021, 94, 2351–2369 CrossRef CAS.
- T. Sawada and M. Fujita, Bull. Chem. Soc. Jpn., 2021, 94, 2342–2350 CrossRef CAS.
- M. Morie, R. Sekiya and T. Haino, Bull. Chem. Soc. Jpn., 2021, 94, 2792–2799 CrossRef CAS.
- E. Yashima and K. Maeda, Bull. Chem. Soc. Jpn., 2021, 94, 2637–2661 CrossRef CAS.
- S. Kassem, A. T. L. Lee, D. A. Leigh, V. Marcos, L. I. Palmer and S. Pisano, Nature, 2017, 549, 374–378 CrossRef CAS PubMed.
- Y. Qiu, B. Song, C. Pezzato, D. Shen, W. Liu, L. Zhang, Y. Feng, Q.-H. Guo, K. Cai, W. Li, H. Chen, M. T. Nguyen, Y. Shi, C. Cheng, R. D. Astumian, X. Li and J. F. Stoddart, Science, 2020, 368, 1247–1253 CrossRef CAS PubMed.
- P. Štacko, J. C. M. Kistemaker, T. van Leeuwen, M.-C. Chang, E. Otten and B. L. Feringa, Science, 2017, 356, 964–968 CrossRef PubMed.
- X. Yang, Q. Cheng, V. Monnier, L. Charles, H. Karoui, O. Ouari, D. Gigmes, R. Wang, A. Kermagoret and D. Bardelang, Angew. Chem., Int. Ed., 2021, 60, 6617–6623 CrossRef CAS PubMed.
- C. Cheng, P. R. McGonigal, S. T. Schneebeli, H. Li, N. A. Vermeulen, C. Ke and J. F. Stoddart, Nat. Nanotechnol., 2015, 10, 547–553 CrossRef CAS PubMed.
- S. Amano, S. D. P. Fielden and D. A. Leigh, Nature, 2021, 594, 529–534 CrossRef CAS PubMed.
- S. Borsley, E. Kreidt, D. A. Leigh and B. M. W. Roberts, Nature, 2022, 604, 80–85 CrossRef CAS PubMed.
- (a) E. Kazuma, J. Jung, H. Ueba, M. Trenary and Y. Kim, Science, 2018, 360, 521–526 CrossRef CAS PubMed; (b) B. Doppagne, T. Neuman, R. Soria-Martinez, L. E. P. López, H. Bulou, M. Romeo, S. Berciaud, F. Scheurer, J. Aizpurua and G. Schull, Nat. Nanotechnol., 2020, 15, 207–211 CrossRef CAS PubMed.
- (a) S. Kawai, O. Krejčí, T. Nishiuchi, K. Sahara, T. Kodama, R. Pawlak, E. Meyer, T. Kubo and A. S. Foster, Sci. Adv., 2006, 6, eaay8913 CrossRef PubMed; (b) S. Kawai, H. Sang, L. Kantorovich, K. Takahashi, K. Nozaki and S. Ito, Angew. Chem., Int. Ed., 2020, 59, 10842–10847 CrossRef CAS PubMed.
- (a) P. Ruffieux, S. Wang, B. Yang, C. Sánchez-Sánchez, J. Liu, T. Dienel, L. Talirz, P. Shinde, C. A. Pignedoli, D. Passerone, T. Dumslaff, X. Feng, K. Müllen and R. Fasel, Nature, 2016, 531, 489–492 CrossRef CAS PubMed; (b) S. Clair and D. G. de Oteyza, Chem. Rev., 2019, 119, 4717–4776 CrossRef CAS PubMed; (c) K. Sun, K. Sugawara, A. Lyalin, Y. Ishigaki, K. Uosaki, O. Custance, T. Taketsugu, T. Suzuki and S. Kawai, ACS Nano, 2023, 17, 24355–24362 CrossRef CAS PubMed.
- T. Takami, T. Ye, B. K. Pathem, D. P. Arnold, K. Sugiura, Y. Bian, J. Jiang and P. S. Weiss, J. Am. Chem. Soc., 2020, 132, 16460–16466 CrossRef PubMed.
- S. Rana, J. Jiang, K. V. Korpany, U. Mazur and K. W. Hipps, J. Phys. Chem. C, 2021, 125, 1421–1431 CrossRef CAS.
- J. Ren, M. Freitag, C. Schwermann, A. Bakker, S. Amirjalayer, A. Rühling, H.-Y. Gao, N. L. Doltsinis, F. Glorius and H. Fuchs, Nano Lett., 2020, 20, 5922–5928 CrossRef CAS PubMed.
- T. Jin, V. García-López, F. Chen, J. Tour and G. Wang, J. Phys. Chem. C, 2016, 120, 26522–26531 CrossRef CAS.
- T. Jin, V. García-López, P.-T. Chiang, S. Kuwahara, J. M. Tour and G. Wang, J. Phys. Chem. C, 2019, 123, 3011–3018 CrossRef CAS.
- (a) G. Rydzek, Q. Ji, M. Li, P. Schaaf, J. P. Hill, F. Boulmedais and K. Ariga, Nano Today, 2015, 10, 138–167 CrossRef CAS; (b) Z. Zhang, J. Zeng, J. Groll and M. Matsusaki, Biomater. Sci., 2022, 10, 4077–4094 RSC; (c) K. Ariga, Y. Lvov and G. Decher, Phys. Chem. Chem. Phys., 2022, 24, 4097–4115 RSC; (d) K. Ariga, Chem. Mater., 2023, 35, 5233–5254 CrossRef CAS; (e) K. Ariga, J. Song and K. Kawakami, Chem. Commun., 2024, 60, 2152–2167 RSC.
- T. Heinrich, C. H.-H. Traulsen, M. Holzweber, S. Richter, V. Kunz, S. K. Kastner, S. O. Krabbenborg, J. Huskens, W. E. S. Unger and C. A. Schalley, J. Am. Chem. Soc., 2015, 137, 4382–4390 CrossRef CAS PubMed.
- S. Mamone, M. Ge, D. Hüvonen, U. Nagel, A. Danquigny, F. Cuda, M. C. Grosse, Y. Murata, K. Komatsu, M. H. Levitt, T. Rõõm and M. Carravetta, J. Chem. Phys., 2009, 130, 081103 CrossRef CAS PubMed.
- J. Zhao, M. Feng, J. Yang and H. Petek, ACS Nano, 2009, 3, 853–864 CrossRef CAS PubMed.
- (a) T. Huang, J. Zhao, M. Feng, A. A. Popov, S. Yang, L. Dunsch and H. Petek, Nano Lett., 2011, 11, 5327–5332 CrossRef CAS PubMed; (b) T. Huang, J. Zhao, M. Feng, A. A. Popov, S. Yang, L. Dunsch and H. Petek, Chem. Phys. Lett., 2012, 552, 1–12 CrossRef CAS.
- R. Jorn, J. Zhao, H. Petek and T. Seideman, ACS Nano, 2011, 5, 7858–7865 CrossRef CAS PubMed.
- M. Krick, J. Holstein, C. Würteleb and G. H. Clever, Chem. Commun., 2016, 52, 10411–10414 RSC.
- (a) T. A. Balema, Y. Liu, N. A. Wasio, A. M. Larson, D. A. Patel, P. Deshlahra and E. C. H. Sykes, J. Phys. Chem. C, 2021, 125, 3584–3589 CrossRef CAS; (b) H. L. Tierney, A. E. Baber, E. C. H. Sykes, A. Akimov and A. B. Kolomeisky, J. Phys. Chem. C, 2009, 113, 10913–10920 CrossRef CAS.
- Q. Zhu, W. Danowski, A. K. Mondal, F. Tassinari, C. L. F. van Beek, G. H. Heideman, K. Santra, S. R. Cohen, B. L. Feringa and R. Naaman, Adv. Sci., 2021, 8, 2101773 CrossRef CAS PubMed.
- H.-L. Lu, Y. Cao, J. Qi, A. Bakker, C. A. Strassert, X. Lin, K.-H. Ernst, S. Du, H. Fuchs and H.-J. Gao, Nano Lett., 2018, 18, 4704–4709 CrossRef CAS PubMed.
- P. Mishra, J. P. Hill, S. Vijayaraghavan, W. Van Rossom, S. Yoshizawa, M. Grisolia, J. Echeverria, T. Ono, K. Ariga, T. Nakayama, C. Joachim and T. Uchihashi, Nano Lett., 2015, 15, 4793–4798 CrossRef CAS PubMed.
- T. Ikeda, T. Tsukahara, R. Iino, M. Takeuchi and H. Noji, Angew. Chem., Int. Ed., 2014, 53, 10082–10085 CrossRef CAS PubMed.
- (a) R. L. Jack, G. Buchanan, A. Dubini, K. Hatzixanthis, T. Palmer and F. Sargent, EMBO J., 2004, 23, 3962–3972 CrossRef CAS PubMed; (b) S. Eberhard, G. Finazzi and F.-A. Wollman, Annu. Rev. Genet., 2008, 42, 463–515 CrossRef CAS PubMed.
- W.-H. Soe, S. Srivastava and C. Joachim, J. Phys. Chem. Lett., 2019, 10, 6462–6467 CrossRef CAS PubMed.
- S. Abid, Y. Gisbert, M. Kojima, N. Saffon-Merceron, J. Cuny, C. Kammerer and G. Rapenne, Chem. Sci., 2021, 12, 4709–4721 RSC.
- S. Hamer, J.-S. von Glasenapp, F. Röhricht, C. Li, R. Berndt and R. Herges, Chem. – Eur. J., 2021, 27, 17452–17458 CrossRef CAS PubMed.
- (a) J.-F. Morin, Y. Shirai and J. M. Tour, Org. Lett., 2006, 8, 1713–1716 CrossRef CAS PubMed; (b) G. Vives and J. M. Tour, Acc. Chem. Res., 2009, 42, 473–487 CrossRef CAS PubMed; (c) R. Pawlak, T. Meier, N. Renaud, M. Kisiel, A. Hinaut, T. Glatzel, D. Sordes, C. Durand, W.-H. Soe, A. Baratoff, C. Joachim, C. E. Housecroft, E. C. Constable and E. Meyer, ACS Nano, 2017, 11, 9930–9940 CrossRef CAS PubMed.
- (a) Y. Shirai, K. Minami, W. Nakanishi, Y. Yonamine, C. Joachim and K. Ariga, Jpn. J. Appl. Phys., 2016, 55, 1102A2 CrossRef; (b) G. Rapenne and C. Joachim, Nat. Rev. Mater., 2017, 2, 17040 CrossRef.
- T. Kudernac, N. Ruangsupapichat, M. Parschau, B. Maciá, N. Katsonis, S. R. Harutyunyan, K.-H. Ernst and B. L. Feringa, Nature, 2011, 479, 208–211 CrossRef CAS PubMed.
- Y. Shirai, A. J. Osgood, Y. Zhao, Y. Yao, L. Saudan, H. Yang, C. Yu-Hung, L. B. Alemany, T. Sasaki, J.-F. Morin, J. M. Guerrero, K. F. Kelly and J. M. Tour, J. Am. Chem. Soc., 2006, 128, 4854–4864 CrossRef CAS PubMed.
- A. Nemati, H. N. Pishkenari, A. Meghdari and S. S. Ge, J. Phys. Chem. C, 2020, 124, 16629–16643 CrossRef CAS.
- D. Abbasi-Pérez, H. Sang, L. Pérez-Garcí, A. Floris, D. B. Amabilino, R. Raval, J. M. Recio and L. Kantorovich, Chem. Sci., 2019, 10, 5864–5874 RSC.
- T. Jin, V. García-López, S. Kuwahara, P.-T. Chiang, J. M. Tour and G. Wang, J. Phys. Chem. C, 2018, 122, 19025–19036 CrossRef CAS.
- P.-T. Chiang, J. Mielke, J. Godoy, J. M. Guerrero, L. B. Alemany, C. J. Villagomez, A. Saywell, L. Grill and J. M. Tour, ACS Nano, 2012, 6, 592–597 CrossRef CAS PubMed.
- M. Li, S. Li, K. Zhang, X. Chi, H. Zhou, H.-B. Xu, Y. Zhang, Q. Li, D. Wang and M.-H. Zeng, Nanoscale, 2021, 13, 16748–16754 RSC.
- (a) K. Ariga, T. Mori and J. Li, Langmuir, 2019, 35, 3585–3599 CrossRef CAS PubMed; (b) K. Ariga, Langmuir, 2020, 36, 7158–7180 CrossRef CAS PubMed.
- (a) O. N. Oliveira Jr., L. Caseli and K. Ariga, Chem. Rev., 2022, 122, 6459–6513 CrossRef CAS PubMed; (b) K. Ariga, Acc. Mater. Res., 2022, 3, 404–410 CrossRef CAS; (c) S. Negi, M. Hamori, H. Kitagishi and K. Kano, Bull. Chem. Soc. Jpn., 2022, 95, 1537–1545 CrossRef CAS.
- (a) M. Ito, Y. Yamashita, Y. Tsuneda, T. Mori, J. Takeya, S. Watanabe and K. Ariga, ACS Appl. Mater. Interfaces, 2020, 12, 56522–56529 CrossRef PubMed; (b) M. Ito, Y. Yamashita, T. Mori, M. Chiba, T. Futae, J. Takeya, S. Watanabe and K. Ariga, Langmuir, 2022, 38, 5237–5247 CrossRef CAS PubMed.
- (a) T. Mori, H. Tanaka, A. Dalui, N. Mitoma, K. Suzuki, M. Matsumoto, N. Aggarwal, A. Patnaik, S. Acharya, L. K. Shrestha, H. Sakamoto, K. Itami and K. Ariga, Angew. Chem., Int. Ed., 2018, 57, 9679–9683 CrossRef CAS PubMed; (b) J. Song, T. Murata, K.-C. Tsai, X. Jia, F. Sciortino, R. Ma, Y. Yamauchi, J. P. Hill, L. K. Shrestha and K. Ariga, Adv. Mater. Interfaces, 2022, 9, 2102241 CrossRef CAS.
- (a) R. Makiura, S. Motoyama, Y. Umemura, H. Yamanaka, O. Sakata and H. Kitagawa, Nat. Mater., 2010, 9, 565–571 CrossRef CAS PubMed; (b) R. Sakamoto, T. Yagi, K. Hoshiko, S. Kusaka, R. Matsuoka, H. Maeda, Z. Liu, Q. Liu, W.-Y. Wong and H. Nishihara, Angew. Chem., Int. Ed., 2017, 56, 3526–3530 CrossRef CAS PubMed; (c) T. Ohata, K. Tachimoto, K. J. Takeno, A. Nomoto, T. Watanabe, I. Hirosawa and R. Makiura, Bull. Chem. Soc. Jpn., 2023, 96, 274–282 CrossRef CAS.
- (a) K. Dey, M. Pal, K. C. Rout, S. Kunjattu H, A. Das, R. Mukherjee, U. K. Kharul and R. Banerjee, J. Am. Chem. Soc., 2017, 139, 13083–13091 CrossRef CAS PubMed; (b) M. Matsumoto, L. Valentino, G. M. Stiehl, H. B. Balch, A. R. Corcos, F. Wang, D. C. Ralph, B. J. Mariñas and W. R. Dichtel, Chem, 2018, 4, 308–317 CrossRef CAS.
- B. Bai, D. Wang and L.-J. Wan, Bull. Chem. Soc. Jpn., 2021, 94, 1090–1098 CrossRef CAS.
- S. Alonso and A. S. Mikhailov, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2009, 79, 061906 CrossRef PubMed.
- M. Clemente-León, A. Credi, M.-V. Martínez-Díaz, C. Mingotaud and J. F. Stoddart, Adv. Mater., 2006, 18, 1291–1296 CrossRef.
- K. Ariga, Y. Yamauchi, T. Mori and J. P. Hill, Adv. Mater., 2013, 25, 6477–6512 CrossRef CAS PubMed.
- (a) K. Ariga, T. Mori, W. Nakanishi and J. P. Hill, Phys. Chem. Chem. Phys., 2017, 19, 23658–23676 RSC; (b) K. Ariga, ChemNanoMat, 2020, 6, 870–880 CrossRef CAS.
- (a) K. Ariga, K. Minami, M. Ebara and J. Nakanishi, Polym. J., 2016, 48, 371–389 CrossRef CAS; (b) L. K. Shrestha, T. Mori and K. Ariga, Curr. Opin. Colloid Interface Sci., 2018, 35, 68–80 CrossRef CAS.
- (a) K. Ariga, Y. Terasaka, D. Sakai, H. Tsuji and J. Kikuchi, J. Am. Chem. Soc., 2000, 122, 7835–7836 CrossRef CAS; (b) K. Ariga, T. Nakanishi, Y. Terasaka, H. Tsuji, D. Sakai and J. Kikuchi, Langmuir, 2005, 21, 976–981 CrossRef CAS PubMed.
- T. Michinobu, S. Shinoda, T. Nakanishi, J. P. Hill, K. Fujii, T. N. Player, H. Tsukube and K. Ariga, J. Am. Chem. Soc., 2006, 128, 14478–14479 CrossRef CAS PubMed.
- T. Mori, K. Okamoto, H. Endo, J. P. Hill, S. Shinoda, M. Matsukura, H. Tsukube, Y. Suzuki, Y. Kanekiyo and K. Ariga, J. Am. Chem. Soc., 2010, 132, 12868–12870 CrossRef CAS PubMed.
- K. Sakakibara, L. A. Joyce, T. Mori, T. Fujisawa, S. H. Shabbir, J. P. Hill, E. V. Anslyn and K. Ariga, Angew. Chem., Int. Ed., 2012, 51, 9643–9646 CrossRef CAS PubMed.
- (a) S. L. Wiskur, H. Ait-Haddou, J. J. Lavigne and E. V. Anslyn, Acc. Chem. Res., 2001, 34, 963–972 CrossRef CAS PubMed; (b) A. C. Sedgwick, J. T. Brewster II, T. Wu, X. Feng, S. D. Bull, X. Qian, J. L. Sessler, T. D. James, E. V. Anslyn and X. Sun, Chem. Soc. Rev., 2021, 50, 9–38 RSC.
- J. Cheng, P. Štacko, P. Rudolf, R. Y. N. Gengler and B. L. Feringa, Angew. Chem., Int. Ed., 2017, 56, 291–296 CrossRef CAS PubMed.
- D. Ishikawa, T. Mori, Y. Yonamine, W. Nakanishi, D. L. Cheung, J. P. Hill and K. Ariga, Angew. Chem., Int. Ed., 2015, 54, 8988–8991 CrossRef CAS PubMed.
- T. Mori, D. Ishikawa, Y. Yonamine, Y. Fujii, J. P. Hill, I. Ichinose, K. Ariga and W. Nakanishi, ChemPhysChem, 2017, 18, 1470–1474 CrossRef CAS PubMed.
- W. Nakanishi, S. Saito, N. Sakamoto, A. Kashiwagi, S. Yamaguchi, H. Sakai and K. Ariga, Chem. – Asian J., 2019, 14, 2869–2876 CrossRef CAS PubMed.
- (a) Y. Goto, S. Omagari, R. Sato, T. Yamakado, R. Achiwa, N. Dey, K. Suga, M. Vacha and S. Saito, J. Am. Chem. Soc., 2021, 143, 14306–14313 CrossRef CAS PubMed; (b) T. Yamakado and S. Saito, J. Am. Chem. Soc., 2022, 144, 2804–2815 CrossRef CAS PubMed.
- R. Kimura, H. Kitakado, A. Osuka and S. Saito, Bull. Chem. Soc. Jpn., 2020, 93, 1102–1106 CrossRef CAS.
- K. Suga, T. Yamakado and S. Saito, Bull. Chem. Soc. Jpn., 2021, 94, 1999–2002 CrossRef CAS.
- J. Adachi, T. Mori, R. Inoue, M. Naito, N. H.-T. Le, S. Kawamorita, J. P. Hill, T. Naota and K. Ariga, Chem. – Asian J., 2020, 15, 406–414 CrossRef CAS PubMed.
- J. Adachi, M. Naito, S. Sugiura, N. H.-T. Le, S. Nishimura, S. Huang, S. Suzuki, S. Kawamorita, N. Komiya, J. P. Hill, K. Ariga, T. Naota and T. Mori, Bull. Chem. Soc. Jpn., 2022, 95, 889–897 CrossRef CAS.
- T. Mori, H. Chin, K. Kawashima, H. T. Ngo, N.-J. Cho, W. Nakanishi, J. P. Hill and K. Ariga, ACS Nano, 2019, 13, 2410–2419 CAS.
- M. Ishii, T. Mori, W. Nakanishi, J. P. Hill, H. Sakai and K. Ariga, Helicity manipulation of a double-paddled binaphthyl in a two-dimensional matrix field at the air–water interface, ACS Nano, 2020, 14, 13294–13303 CrossRef CAS PubMed.
- M. Ishii, T. Mori, W. Nakanishi, J. P. Hill, H. Sakai and K. Ariga, Langmuir, 2022, 38, 6481–6490 CrossRef CAS PubMed.
- (a) S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS PubMed; (b) H. Furukawa, K. E. Cordova, M. O’Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed; (c) S. Moribe, Y. Takeda, M. Umehara, H. Kikuta, J. Ito, J. Ma, Y. Yamada and M. Hirano, Bull. Chem. Soc. Jpn., 2023, 96, 321–327 CrossRef CAS; (d) B. Ay, R. Takano and T. Ishida, Bull. Chem. Soc. Jpn., 2023, 96, 1129–1138 CrossRef CAS.
- (a) R. Liu, K. T. Tan, Y. Gong, Y. Chen, Z. Li, S. Xie, T. He, Z. Lu, H. Yang and D. Jiang, Chem. Soc. Rev., 2021, 50, 120–242 RSC; (b) S. Li, J. Zou, L. Tan, Z. Huang, P. Liang and X. Meng, Chem. Eng. J., 2022, 446, 137148 CrossRef CAS; (c) Y. Charles-Blin, T. Kondo, Y. Wu, S. Bandow and K. Awaga, Bull. Chem. Soc. Jpn., 2022, 95, 972–977 CrossRef CAS; (d) C. Wang, Z. Zhang, Y. Zhu, C. Yang, J. Wu and W. Hu, Adv. Mater., 2022, 34, 2102290 CrossRef CAS PubMed.
- (a) B. Karimi, N. Ganji, O. Pourshiani and W. R. Thiel, Prog. Mater. Sci., 2022, 125, 100896 CrossRef CAS; (b) R. G. Shrestha, S. Maji, A. K. Mallick, A. Jha, R. M. Shrestha, R. Rajbhandari, J. P. Hill, K. Ariga and L. K. Shrestha, Bull. Chem. Soc. Jpn., 2022, 95, 1060–1067 CrossRef CAS; (c) A. Perez-Calm, L. K. Shrestha, J. R. Magana, J. Esquena, L. M. Salonen, R. G. Shrestha, R. Ma, K. Ariga and C. Rodriguez-Abreu, Bull. Chem. Soc. Jpn., 2022, 95, 1687–1696 CrossRef CAS; (d) C. Xu, C. Lei, Y. Wang and C. Yu, Angew. Chem., Int. Ed., 2022, 61, e202112752 CrossRef CAS PubMed.
- C. Stähler, L. Grunenberg, M. W. Terban, W. R. Browne, D. Doellerer, M. Kathan, M. Etter, B. V. Lotsch, B. L. Feringa and S. Krause, Chem. Sci., 2022, 13, 8253–8264 RSC.
- J. Hou, G. Long, W. Zhao, G. Zhou, D. Liu, D. J. Broer, B. L. Feringa and J. Chen, J. Am. Chem. Soc., 2022, 144, 6851–6860 CrossRef CAS PubMed.
- J. Chen, F. K.-C. Leung, M. C. A. Stuart, T. Kajitani, T. Fukushima, E. van der Giessen and B. L. Feringa, Nat. Chem., 2018, 10, 132–138 CrossRef CAS PubMed.
- W.-Z. Wang, L.-B. Huang, S.-P. Zheng, E. Moulin, O. Gavat, M. Barboiu and N. Giuseppone, J. Am. Chem. Soc., 2021, 143, 15653–15660 CrossRef CAS PubMed.
- V. García-López, F. Chen, L. G. Nilewski, G. Duret, A. Aliyan, A. B. Kolomeisky, J. T. Robinson, G. Wang, R. Pal and J. M. Tour, Nature, 2017, 548, 567–572 CrossRef PubMed.
- D. Liu, V. García-López, R. S. Gunasekera, L. G. Nilewski, L. B. Alemany, A. Aliyan, T. Jin, G. Wang, J. M. Tour and R. Pal, ACS Nano, 2019, 13, 6813–6823 CrossRef CAS PubMed.
- A. L. Santos, D. Liu, A. K. Reed, A. M. Wyderka, A. van Venrooy, J. T. Li, V. D. Li, M. Misiura, O. Samoylova, J. L. Beckham, C. Ayala-Orozco, A. B. Kolomeisky, L. B. Alemany, A. Oliver, G. P. Tegos and J. M. Tour, Sci. Adv., 2022, 8, eabm2055 CrossRef CAS PubMed.
- H. Yang, J. Yi, S. Pang, K. Ye, Z. Ye, Q. Duan, Z. Yan, C. Lian, Y. Yang, L. Zhu, D.-H. Qu and C. Bao, Angew. Chem., Int. Ed., 2022, 61, e202204605 CrossRef CAS PubMed.
- (a) A. Barragán, T. Nicolás-García, K. Lauwaet, A. Sánchez-Grande, J. L. Urgel, J. Björk, E. M. Pérez and D. Écija, Angew. Chem., Int. Ed., 2023, 62, e202212395 CrossRef PubMed; (b) K. H. Au-Yeung, S. Sarkar, T. Kühne, O. Aiboudi, D. A. Ryndyk, R. Robles, N. Lorente, F. Lissel, C. Joachim and F. Moresco, ACS Nano, 2023, 17, 3128–3134 CrossRef CAS PubMed.
- (a) C. Pezzato, C. Cheng, J. F. Stoddart and R. D. Astumian, Chem. Soc. Rev., 2017, 46, 5491–5507 RSC; (b) R. D. Astumian, Acc. Chem. Res., 2018, 51, 2653–2661 CrossRef CAS PubMed.
- T. R. Kelly, H. De Silva and R. A. Silva, Nature, 1999, 401, 150–152 CrossRef CAS PubMed.
- T. R. Kelly, Acc. Chem. Res., 2001, 34, 514–522 CrossRef CAS PubMed.
- (a) J. Shen, C. Ren and H. Zeng, Acc. Chem. Res., 2022, 55, 1148–1159 CrossRef CAS PubMed; (b) K. Ariga, Micromachines, 2021, 14, 25 CrossRef PubMed.
- D. Civita, M. Kolmer, G. J. Simpson, A.-P. Li, S. Hecht and L. Grill, Science, 2020, 370, 957–960 CrossRef CAS PubMed.
- S. Kawai, A. Benassi, E. Gnecco, H. Söde, R. Pawlak, X. Feng, K. Müllen, D. Passerone, C. A. Pignedoli, P. Ruffieux, R. Fasel and E. Meyer, Science, 2010, 351, 957–961 CrossRef PubMed.
- (a) R. Pawlak, J. G. Vilhena, A. Hinaut, T. Meier, T. Glatzel, A. Baratoff, E. Gnecco, R. Pérez and E. Meyer, Nat. Commun., 2019, 10, 685 CrossRef CAS PubMed; (b) J. G. Vilhena, R. Pawlak, P. D’Astolfo, X. Liu, E. Gnecco, M. Kisiel, T. Glatzel, R. Pérez, R. Häner, S. Decurtins, A. Baratoff, G. Prampolini, S.-X. Liu and E. Meyer, Phys. Rev. Lett., 2020, 128, 216102 CrossRef PubMed.
- (a) S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Chem. Rev., 2015, 115, 10081–10206 CrossRef CAS PubMed; (b) M. J. Skaug, C. Schwemmer, S. Fringes, C. D. Rawlings and A. W. Knoll, Science, 2018, 359, 1505–1508 CrossRef CAS PubMed.
- (a) J. P. Dekker and E. J. Boekema, Biochim. Biophys. Acta, Bioenerg., 2005, 1706, 12–39 CrossRef CAS PubMed; (b) D. A. Bryant and D. P. Canniffe, J. Phys. B: At., Mol. Opt. Phys., 2018, 51, 033001 CrossRef.
- (a) G. Nakada, Y. Igarashi, H. Imai and Y. Oaki, Adv. Theory Simul., 2019, 2, 1800180 CrossRef CAS; (b) P. M. Pflüger and F. Glorius, Angew. Chem., Int. Ed., 2020, 59, 18860–18865 CrossRef PubMed; (c) K. Nakaguro, Y. Mitsuta, S. Koseki, T. Oshiyama and T. Asada, Bull. Chem. Soc. Jpn., 2023, 96, 1099–1107 CrossRef CAS.
| This journal is © the Owner Societies 2024 |