
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExtension of nature's NIR-I chromophore into the NIR-II region†
Kittipan
Siwawannapong
 a,
James R.
Diers
b,
Nikki Cecil M.
Magdaong
c,
Phattananawee
Nalaoh
d,
Christine
Kirmaier
c,
Jonathan S.
Lindsey
*a,
Dewey
Holten
*c and
David F.
Bocian
*b
a,
James R.
Diers
b,
Nikki Cecil M.
Magdaong
c,
Phattananawee
Nalaoh
d,
Christine
Kirmaier
c,
Jonathan S.
Lindsey
*a,
Dewey
Holten
*c and
David F.
Bocian
*b
aDepartment of Chemistry, North Carolina State University, Raleigh, NC 27695-8204, USA. E-mail: jlindsey@ncsu.edu
bDepartment of Chemistry, University of California, Riverside, CA 92521-0403, USA. E-mail: david.bocian@ucr.edu
cDepartment of Chemistry, Washington University, St. Louis, MO 63130-4889, USA. E-mail: holten@wustl.edu
dDepartment of Chemistry, University of Tennessee, Knoxville, TN 37996, USA
First published on 1st May 2024
Abstract
The development of chromophores that absorb in the near-infrared (NIR) region beyond 1000 nm underpins numerous applications in medical and energy sciences, yet also presents substantial challenges to molecular design and chemical synthesis. Here, the core bacteriochlorin chromophore of nature's NIR absorbers, bacteriochlorophylls, has been adapted and tailored by annulation in an effort to achieve absorption in the NIR-II region. The resulting bacteriochlorin, Phen2,1-BC, contains two annulated naphthalene groups spanning meso,β-positions of the bacteriochlorin and the 1,2-positions of the naphthalene. Phen2,1-BC was prepared via a new synthetic route. Phen2,1-BC is an isomer of previously examined Phen-BC, which differs only in attachment via the 1,8-positions of the naphthalene. Despite identical π-systems, the two bacteriochlorins have distinct spectroscopic and photophysical features. Phen-BC has long-wavelength absorption maximum (912 nm), oscillator strength (1.0), and S1 excited-state lifetime (150 ps) much different than Phen2,1-BC (1292 nm, 0.23, and 0.4 ps, respectively). These two molecules and an analogue with intermediate characteristics bearing annulated phenyl rings have unexpected properties relative to those of non-annulated counterparts. Understanding the distinctions requires extending concepts beyond the four-orbital-model description of tetrapyrrole spectroscopic features. In particular, a reduction in symmetry resulting from annulation results in electronic mixing of x- and y-polarized transitions/states, as well as vibronic coupling that together reduce oscillator strength of the long-wavelength absorption manifold and shorten the S1 excited-state lifetime. Collectively, the results suggest a heuristic for the molecular design of tetrapyrrole chromophores for deep penetration into the relatively unutilized NIR-II region.
Introduction
The near-infrared (NIR) spectral region is of great interest owing to a confluence of research objectives ranging from medical to energy sciences. The NIR is of interest in medicine given the deep penetration of NIR light into soft tissue, which holds potential for imaging and therapeutic applications.1 Interest in the NIR for energy sciences arises due to the large number of solar photons in this region.2,3 The NIR is now divided into the NIR-I (700–1000 nm) and the NIR-II (1000–1700 nm) regions.4 Most work to date has focused on the development of chromophores that absorb in the NIR-I region although the NIR-II region has become a target for active research.5–9 The recent emphasis on the NIR-II region reflects the longstanding progression of the field of photochemistry – including fundamental studies and chromophore function – across the ultraviolet (UV) and visible (VIS), and into the NIR spectral regions.Nature's chosen molecules for solar light harvesting are hydroporphyrins, namely chlorophylls and bacteriochlorophylls. The long-wavelength absorption band of chlorophylls lies predominantly in the red region (∼640–710 nm), whereas that of bacteriochlorophylls is in the NIR-I region (∼750–800 nm).10 Such values of the long-wavelength band are for the isolated pigments in organic solvents. Longer wavelengths of absorption are obtained in native pigment–protein assemblies.11–13 The long-wavelength absorption band of the native hydroporphyrins tends to have a large molar absorption coefficient, namely ∼100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1,10 making them well suited for diverse applications.
000 M−1 cm−1,10 making them well suited for diverse applications.
Efforts to extend the position of the long-wavelength absorption have been carried out to understand the limits of spectroscopic malleability of the core hydroporphyrin chromophores. A general rule of thumb for the molecular designer is that the more extended the π-system, the longer the wavelength.14 The notion of extended π-systems dates to fundamental studies of the relationship between color and constitution of synthetic dyes.15–18 Critical exceptions to this heuristic are known, however, including the effects of macrocycle type (porphyrin vs. chlorin vs. bacteriochlorin) and specific peripheral-substituent patterns on the spectra of tetrapyrrole chromophores.14,19
Over the years, we have been working to understand the effects of conjugated groups on the spectroscopic and photophysical features of synthetic bacteriochlorin analogues of native bacteriochlorophylls.20 Representative synthetic bacteriochlorins are shown in Chart 1. Each contains a gem-dimethyl group (8- and 18-positions) in the reduced ring to secure the chromophore from adventitious dehydrogenation in an aerobic environment. The bacteriochlorin H-BC contains no substituents at the β-pyrrole (2,3,12,13) or meso (5,10,15,20) positions. Annulation of conjugated groups across adjacent β-pyrrole positions (Im-BC-1) or β,meso-positions (Im-BC-2) with imides20 causes a significant bathochromic shift of the long-wavelength (S0 → S1) band from that of H-BC (714 nm) to 800 or 875 nm, respectively. Even more pronounced spectral shifts are observed upon incorporation of annulated arenes to give Phen-BC and Benz-BC, which exhibit Qy maxima at 912 and 1033 nm, respectively.21 The excited-state lifetimes are also shortened with increasing wavelength along this series: 3800 ps (H-BC), 3200 ps (Im-BC-1),20 1000 ps (Im-BC-2), 150 ps (Phen-BC), and 7 ps (Benz-BC).21
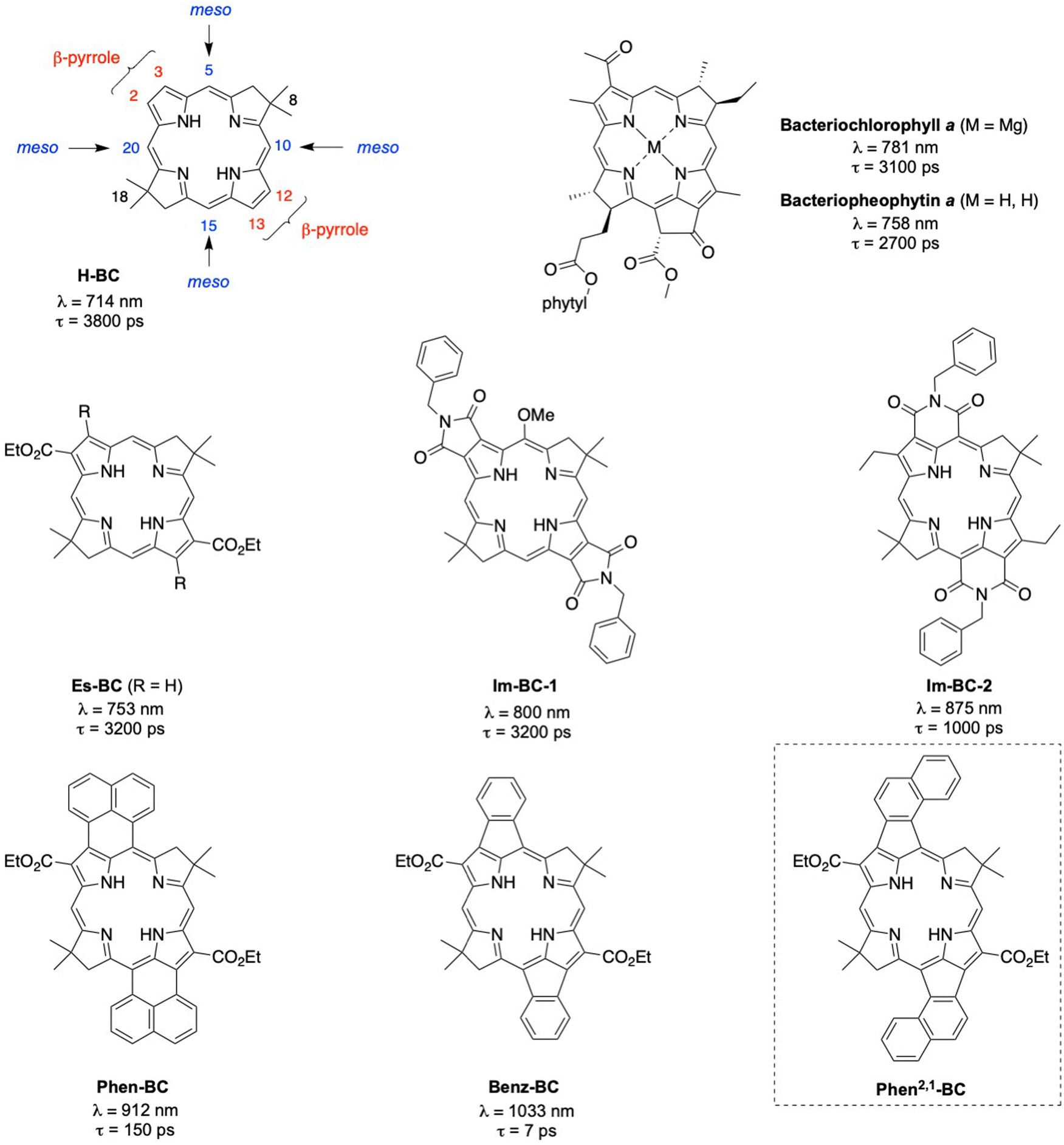 | ||
| Chart 1 Synthetic and natural bacteriochlorins. | ||
Recently, we performed a series of density functional theory (DFT) calculations that examined nearly 100 designs that might extend the S0 → S1 band deeper into the NIR-II region. One design that appeared synthetically tractable, Phen2,1-BC (Chart 1), was calculated to have a Qy band at 1277 nm. In this paper, the synthesis of Phen2,1-BC is described followed by photophysical characterization and theoretical analysis. Compound Phen2,1-BC exhibits a S0 → S1 absorption band at 1292 nm but with diminished intensity versus expectations. Bacteriochlorins Phen-BC and Phen2,1-BC are isomers that have an identical number of π-bonds, but differ in the sites of connection to the annulated naphthalene unit. Yet these two isomers exhibit profoundly different absorption spectra and excited-state lifetimes. Efforts to understand the photophysical features of Phen2,1-BC have led to new insights concerning both hydroporphyrin spectroscopy and molecular design of annulated hydroporphyrins with potentially deeper NIR absorption.
Results
Synthesis
The prior synthesis of annulated bacteriochlorins Phen-BC and Benz-BC relied on the approach shown in Scheme 1.21 A bromo-substituted dihydrodipyrrin (I)21 was converted via a Northern–Southern route22 to the corresponding 2,12-dibromobacteriochlorin (II). For annulation, the latter was treated with the arene, which contained one bromine atom and one boronic acid moiety that together define the spacing of adjacent annulation sites. The annulation entailed a two-step process: (1) Suzuki coupling23 of the 2,12-dibromobacteriochlorin (II) with the arene-boronic acid to attach the arene at the bacteriochlorin 2,12-positions, and (2) Heck coupling24 of the arene-bromide with the bacteriochlorin meso-carbons (5,15-positions). Thus, reaction with naphthalene III gave Phen-BC, whereas benzene IV gave Benz-BC. Attempts to use naphthalene V (prepared herein), however, gave a very poor yield in the Suzuki coupling process. A general challenge was to obtain sufficient 2,12-dibromobacteriochlorin (II) for expansive studies of reaction conditions that might ameliorate the coupling with V. An alternative route to 2,12-dibromobacteriochlorin (II) was explored beginning with pyrrole VI and synthon VII to obtain a bromo-substituted dihydrodipyrrin (VIII) for use via an Eastern–Western route,22 but this approach was not fruitful. The synthetic approaches viaII to obtain annulated bacteriochlorins were discontinued (see ESI,† Section S1).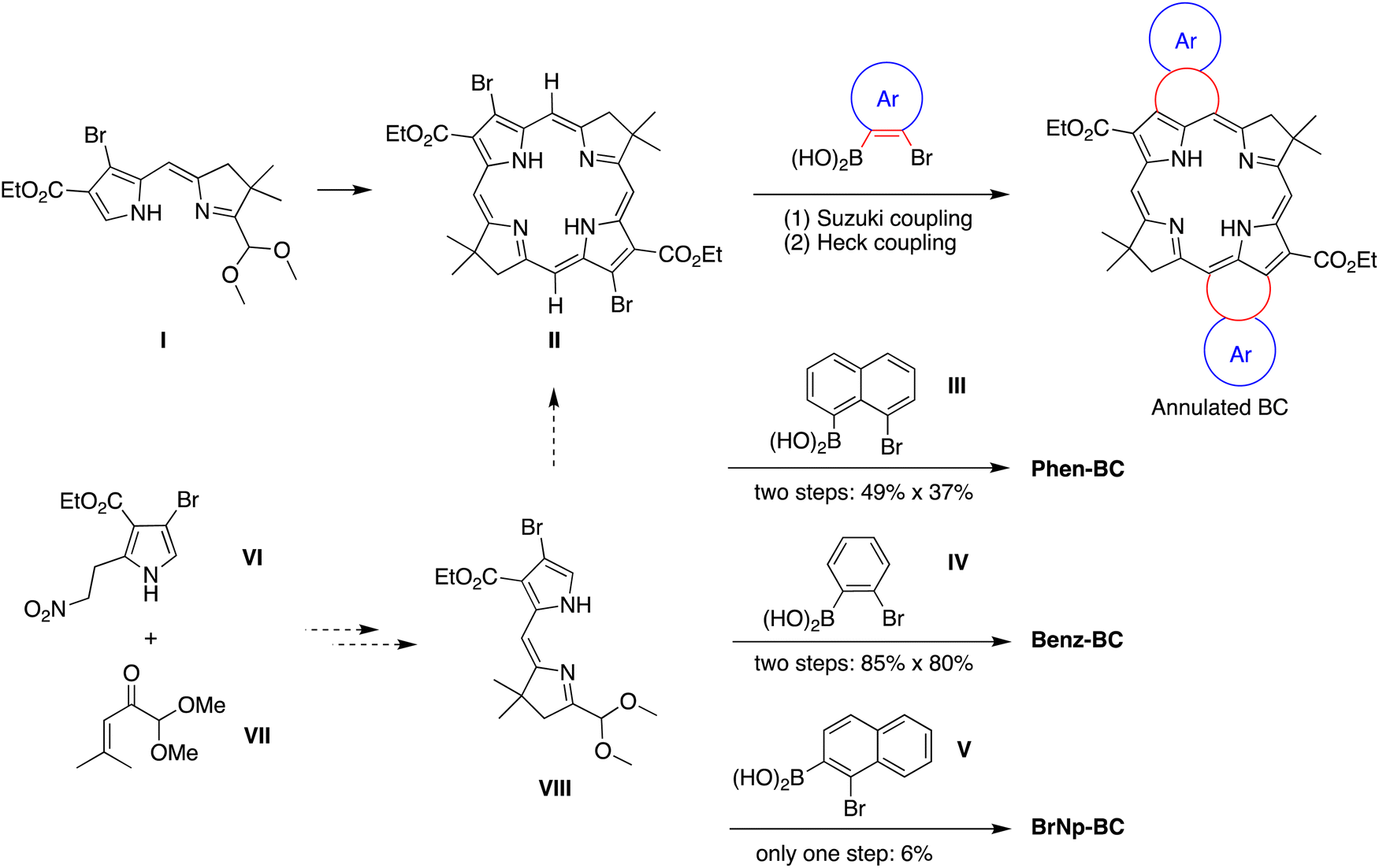 | ||
| Scheme 1 Synthesis routes to annulated bacteriochlorins. | ||
A new route was developed to gain access to the target bacteriochlorin Phen2,1-BC (Scheme 2). The route entails incorporation of a bromonaphthyl group at the beginning of the synthesis, and relies on the Eastern–Western approach to construct the bacteriochlorin macrocycle. After exploratory studies of a four-component approach to the requisite bromoarene-pyrrole,25 a two-component approach26 was pursued. Thus, reaction of 1-bromo-2-(2-nitrovinyl)naphthalene (1)27 and β-enamino ester 2 was carried out to afford pyrrole 3. Study of reaction conditions identified use of overnight reflux in toluene25 (containing 20 mol% FeCl3 relative to 1), which afforded 3 in 47% yield (see the ESI,† Section S1 for reaction surveys28). The structure of 3 was confirmed by X-ray crystallography (Fig. 1(A)).
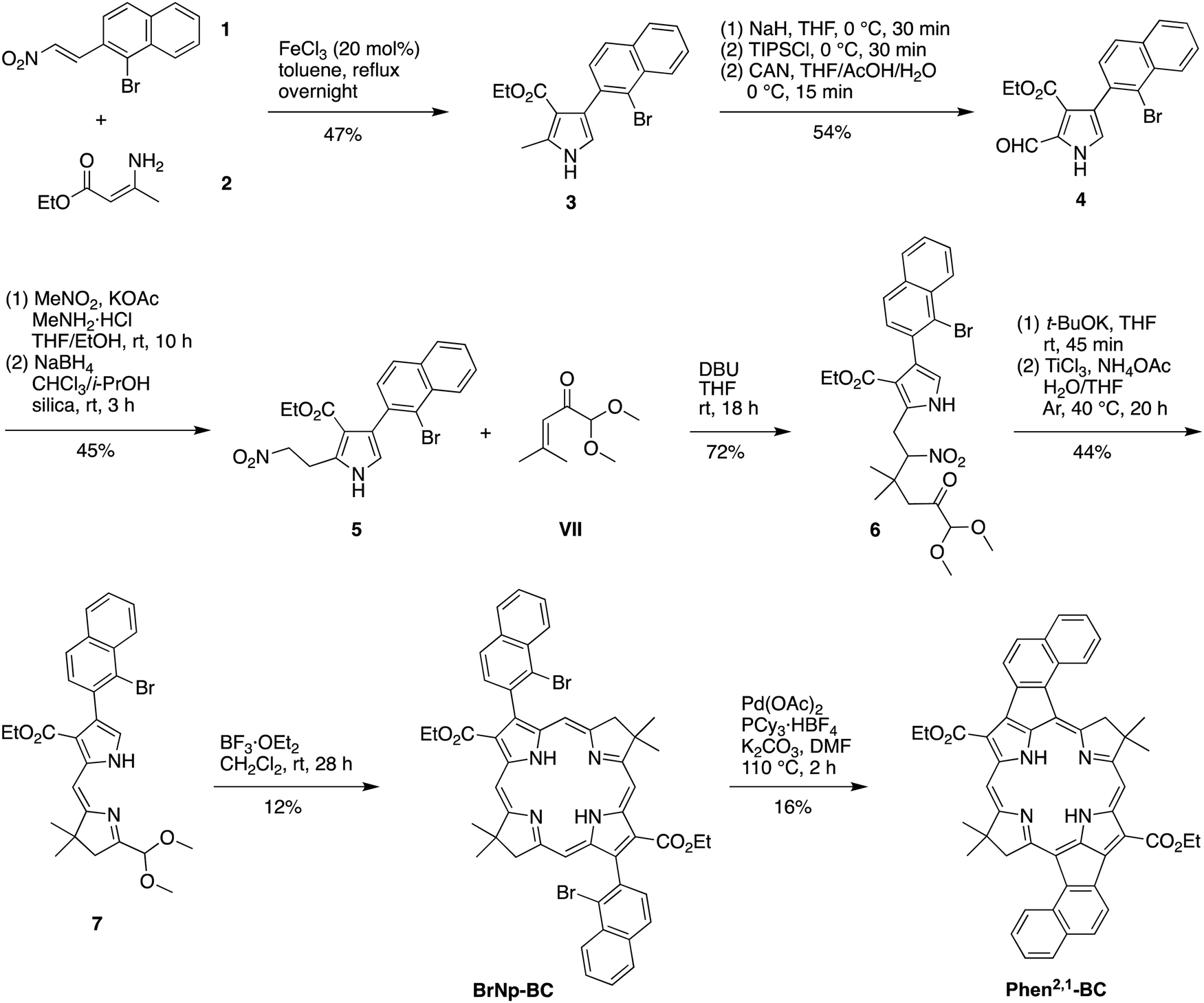 | ||
| Scheme 2 Synthesis of annulated bacteriochlorin Phen2,1-BC. | ||
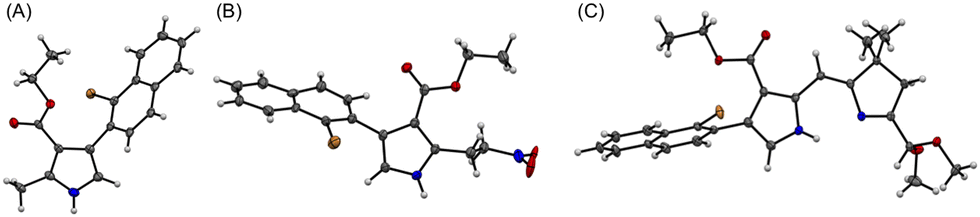 | ||
| Fig. 1 ORTEP drawing of the single crystal X-ray structures of (A) bromonaphthyl-bearing pyrrole 3, (B) the intermediate 5, and (C) dihydrodipyrrin 7. All ellipsoids are contoured at the 50% level. Disordered units are omitted for clarity (Label: C = grey, N = blue, O = red, Br = orange, H = white). | ||
The subsequent transformations to form the corresponding dihydrodipyrrin follow those for the Eastern–Western route. Thus, pyrrole 3 was converted29 to the N-TIPS derivative followed by oxidation of the 2-methyl group using ceric ammonium nitrate (CAN)30 to give the 2-carboxaldehyde 4 in 54% yield. Henry reaction31 with nitromethane and subsequent reduction29 with NaBH4 gave the 2-(2-nitroethyl)pyrrole 5 in 45% yield. The structure of 5 was confirmed by X-ray crystallography (Fig. 1(B)). Michael addition32 of 5 and α,β-unsaturated ketone VII33 under known conditions29,34 gave the nitrohexanone 6 in 72% yield. Cyclization29,34 of 6 using potassium tert-butoxide followed by addition of TiCl3 in the presence of ammonium acetate (McMurry–Melton reaction)35,36 gave dihydrodipyrrin 7 in 44% yield. The structure of 7 was confirmed by X-ray crystallography (Fig. 1(C)). Self-condensation29 of 7 was carried out with catalysis by BF3·OEt2 at room temperature, which gave (as expected)22 bacteriochlorin BrNp-BC as a single band (for evidence of atropisomers, see the Experimental section) in 12% yield.
The Heck reaction24 of BrNp-BC was carried out using Pd(OAc)2 and the ligand tricyclohexylphosphonium (PCy3) tetrafluoroborate in N,N-dimethylformamide (DMF) at 110 °C, the same conditions used previously21 for formation of Phen-BC and Benz-BC. The desired doubly annulated bacteriochlorin Phen2,1-BC was obtained in 16% yield. The composition of Phen2,1-BC was confirmed by accurate mass analysis, and the structure was analyzed by a battery of 2D NMR methods (ESI,† Section S11). Analysis by 1D 1H NMR spectroscopy (in CDCl3) showed a singlet at 3.21 ppm, attributed to the resonance of the N–H protons, which can be compared with those for annulated bacteriochlorins Phen-BC (1.97 ppm) and Benz-BC (2.89 ppm).21 In perspective, the resonance of the N–H protons in the ∼2–3 ppm region is unusual, given that most tetrapyrrole macrocycles (and bacteriochlorins) lacking annulation tend to resonate in the far upfield region (–1 to –3 ppm). Comparison of the 1H NMR chemical shifts of the three annulated bacteriochlorins with two benchmark bacteriochlorins is provided in Table 1. The change in chemical shifts suggests progressively weaker ring currents among the annulated bacteriochlorins in the series Phen-BC > Benz-BC > Phen2,1-BC.
| Hydrogens | Chemical shifts (ppm) | ||||
|---|---|---|---|---|---|
| H-BC 37 |
EtEs-BC
38
|
Benz-BC 21 | Phen-BC 21 | Phen2,1-BC | |
| a 2,12-Bis(carboethoxy)-3,13-diethyl-8,8,18,18-tetramethylbacteriochlorin (R = Et in Chart 1). | |||||
| meso-H | 8.73 | 8.64 | 8.71 | 8.35 | 7.77 |
| 8.83 | 9.66 | ||||
| gem-Dimethyl | 1.97 | 1.94 | 1.78 | 1.57 | 1.20 |
| N–H | −2.38 | −1.43 | 2.89 | 1.97 | 3.21 |
| pyrroline CH2 | 4.47 | 4.78 | 4.00 | 4.31 | 3.49 |
In summary, the synthesis of Phen2,1-BC described herein entailed development of a new route (Scheme 2) with the following advantages versus the prior route (Scheme 1): (1) use of one Pd-coupling reaction (Heck) rather than a sequence of two Pd coupling reactions (Suzuki and Heck), which required a large excess of the bromo-arene-boronic acid to avoid competitive coupling processes; (2) no requirement to synthesize a bromo-arene-boronic acid (which is facile for III and IV, but not for V or perhaps other annulation partners); and (3) avoidance of bromination of the pyrrole moiety leading to the dihydrodipyrrin. On the other hand, one disadvantage is the requirement to install the annulation motif (i.e., the bromonaphthalene unit) at the first step of the synthesis.
Absorption spectra
Fig. 2(A) shows electronic absorption spectra of six bacteriochlorins (Chart 1 and Scheme 2) in toluene at room temperature. The S0 → S1 vibronic manifold for each molecule is shaded. The spectra are labeled with the S0 → S1 oscillator strengths obtained from time-dependent DFT (TDDFT) calculations (vide infra).39 This method affords a S1(0,0) peak intensity ratio of 22 for BrNp-BCvs.Phen2,1-BC, in good agreement with the ratio of 21 for the measured molar absorption coefficients (see Experimental section). The S0 → S1 manifolds are normalized to the peak intensity in Fig. 2(B) (solid). The wavelengths and energy spacing of the spectral features are summarized in Table 2. The S1(0,0) band of non-annulated bacteriochlorins H-BC (714 nm), Es-BC (753 nm), and BrNp-BC (771 nm) is characteristically sharp and intensifies and shifts bathochromically along this series. At progressively higher energy are the weak S2(0,0) band (490–530 nm) and strong S3(0,0) (365–380 nm) and S4(0,0) (340–360 nm) bands. The characteristics of these features can be understood within Gouterman's four-orbital model (vide infra).19,40,41 The standard assignments for the non-annulated bacteriochlorins are S0 → S1 (Qy), S0 → S2 (Qx), S0 → S3 (Bx), and S0 → S4 (By). | ||
| Fig. 2 (A) Absorption spectra of H-BC (grey), Es-BC (black), BrNp-BC (violet), Phen-BC (blue), Benz-BC (gold), and Phen2,1-BC (red). The S0 → S1 vibronic manifold is shaded. The oscillator strength of the S0 → S1 manifold obtained from TDDFT calculations is given. (B) Absorption spectra (solid) and fluorescence spectra (dashed) of the bacteriochlorins normalized at the largest peak. The value in parenthesis is the absorption–fluorescence (Stokes) shift. | ||
| Property | Units | H-BC | Es-BC | BrNp-BC | Phen-BC | Benz-BC | Phen2,1-BC |
|---|---|---|---|---|---|---|---|
| a See Chart 1 and Scheme 2 for chemical structures. All measurements were made in toluene at room temperature. b No emission observed. c Excited-state properties are from the literature for Phen-BC,21Es-BC,42 and H-BC.43 d From TDDFT calculations. The S0 → S1 and S0 → S2 oscillator strengths are f1 and f2, respectively. | |||||||
| S1 absorption | |||||||
| S1(0,0) | nm | 714 | 753 | 771 | 912 | 1033 | 1292 |
| S1(1,0) | nm | 678 | 714 | 730 | 823 | 914 | 1110 |
| S1(2,0) | nm | 649 | 678 | 695 | 818 | 975 | |
| S1(1,0)–S1(0,0) | cm−1 | 744 | 725 | 728 | 1142 | 1260 | 1269 |
| S1(2,0)–S1(1,0) | cm−1 | 659 | 744 | 690 | 1239 | 1247 | |
| S2 absorption | |||||||
| S2(0,0) | nm | 489 | 523 | 529 | 675 | 718 | 812 |
| S2(1,0) | nm | 462 | 490 | 496 | 619 | 654 | 750 |
| S2(2,0) | nm | 433 | 460 | 463 | 571 | 600 | 678 |
| S2(1,0)–S2(0,0) | cm−1 | 1195 | 1288 | 1258 | 1340 | 1363 | 1018 |
| S2(2,0)–S2(1,0) | cm−1 | 1450 | 1331 | 1437 | 1358 | 1376 | 1223 |
| S1 emission | |||||||
| S1(0,0) | nm | 716 | 756 | 777 | 937 | 1042 | |
| S1(0,1) | nm | 757 | 801 | 824 | 1072 | 1221 | |
| S1(1,0)–S1(0,0) | cm−1 | 756 | 743 | 734 | 1344 | 1407 | |
| Differences | |||||||
| S1(0,0) Ab-Em | cm−1 | 39 | 53 | 100 | 293 | 84 | |
| S2(0,0)–S1 (0,0) Ab | cm−1 | 6444 | 5840 | 5933 | 3850 | 4247 | 4575 |
| Excited statec | |||||||
| Φ f | 0.14 | 0.19 | 0.029 | 0.004 | 2.3 × 10−4 | <3 × 10−5 | |
| τ S | ps | 3800 | 3200 | 500 | 150 | 7 | 0.4 |
| k IC | ps−1 | 17000 |
11000 |
4200 | 152 | 7 | 0.4 |
| Electronic structured | |||||||
| S1–S2 TDM θ12 | degrees | 90 | 73 | 89 | 55 | 46 | 39 |
| f 1 | 0.37 | 0.52 | 0.75 | 1.0 | 0.39 | 0.23 | |
| f 2 | 0.11 | 0.19 | 0.18 | 0.31 | 0.41 | 0.53 | |
| f 1/f2 | 3.4 | 2.7 | 4.2 | 3.2 | 0.95 | 0.43 | |
In contrast, TDDFT calculations show that the four-orbital model is not applicable for the annulated bacteriochlorins such as Benz-BC and Phen2,1-BC because the S0 → S1 and S0 → S2 transitions have mixed x–y polarization (vide infra). One indicator that the four-orbital model is not operative is the dramatically diminished intensity of the S0 → S1 manifold along the series Phen-BC > Benz-BC > Phen2,1-BC as the features move to longer wavelengths (Fig. 2 and Table 2). Moreover, the S1(1,0) band (1110 nm) and S2(1,0) band (750 nm) of Phen2,1-BC and the S1(1,0) band (914 nm) of Benz-BC are more intense than expected for a Franck–Condon progression. These observations indicate vibronic coupling significantly impacts the S0 → S1 and S0 → S2 manifolds of Phen2,1-BC and Benz-BC. The nature of the vibronic progressions are also altered by annulation. A ∼1300 cm−1 vibronic spacing (stretching vibrations19) dominates the S0 → S1 manifolds of Phen-BC, Benz-BC, Phen2,1-BC compared to the dominant spacing of ∼730 cm−1 (an accordion-like mode44) in the S0 → S1 manifold of H-BC, Es-BC, and BrNp-BC (Table 2). Spectra of Benz-BC and Phen2,1-BC at 77 K show, in addition to the primary ∼1300 cm−1 spacing, a weaker progression built on an ∼500 cm−1 mode (ESI,† Fig. S2 and Table S6). The latter could be an accordion-like mode down-shifted in energy due to the added mass of the annulated rings.
Fluorescence spectra and quantum yields
Fig. 2(B) zooms in on the S0 → S1 absorption manifolds (solid) of the six bacteriochlorins and shows the corresponding S1 → S0 fluorescence manifolds (dashed). Peak positions and energy spacings are summarized in Table 2. The typical small Stokes shifts between the S1(0,0) absorption and fluorescence maxima are found for H-BC (39 cm−1), Es-BC (53 cm−1), and BrNp-BC (100 cm−1). In each case, the fluorescence spectrum is mirror image to the absorption spectrum in terms of both relative intensities and dominant spacing (∼730 cm−1) of the vibronic bands.Annulated Phen-BC shows a moderate (293 cm−1) Stokes shift with mirror symmetry of the fluorescence and absorption manifolds. On the other hand, the intensity profile in the fluorescence manifold of Benz-BC is not mirror symmetric to that in the absorption manifold (Fig. 2(B), gold). This reinforces the view that the (1,0) band in the S0 → S1 absorption manifold of Benz-BC gains considerable strength via vibronic coupling. Turning to Phen2,1-BC, it is apparent from Fig. 2(B), and measurements extending to 1700 nm presented in ESI,† Section S4, that fluorescence from the new annulated bacteriochlorin is too weak to be observed. Thus, the fluorescence quantum yield (Φf) of Phen2,1-BC is <3 × 10−5, using meso-tetraphenylporphyrin in non-degassed toluene (Φf = 0.070)45 or Phen-BC in Ar-purged toluene (Φf = 0.004)21 as standards. Here, Φf = 2.3 × 10−4 for Benz-BC was determined, updating a value found previously where emission was barely observed and the vibronic manifold not resolved.21 The Φf values of two other bacteriochlorins determined previously are 0.14 for H-BC,43 and 0.13 for Es-BC.42 The low Φf value found here of 0.029 for BrNp-BC is attributed to a heavy atom effect of the bromines (Scheme 2) on S1 → T1 intersystem crossing, consistent with transient absorption studies (ESI,† Section S6).
Excited-state decay properties
 | ||
| Fig. 3 (A) Representative TA spectra for Phen2,1-BC at select delay times after excitation at 500 nm; (B) ground-state absorption spectrum and kinetic profile and dual-exponential fit at 875 nm. The vertical dashed lines in panel A mark the cutoff between the visible and NIR data, the latter using a 4× smaller ΔA scale (see left and right vertical axes). | ||
The TA data sets were subjected to global analysis presented in ESI,† Section S5 in which several kinetic models were tested. The analysis is most consistent with Model D (Fig. S7, ESI†), in which the lowest excited singlet state (S1) of Phen2,1-BC decays by internal conversion to S0 with a time constant of ∼0.4 ps. This rapid downhill (∼1 eV) decay produces a vibrationally “hot” S0 that “cools” with τ ∼12 ps by flow of the excess energy into the solvent. A time scale of ∼10 ps or longer has been found for vibrational cooling of large aromatic systems46–53 including tetrapyrroles.54,55 Also, the S1 excited state itself likely has not vibrationally relaxed prior to internal conversion because internal vibrational redistribution of such systems often takes >1 ps.46,47,50–52 The results for Phen2,1-BC can be compared with those for H-BC,43Es-BC,42BrNp-BC (ESI,† Section S6), and Phen-BC.21 The S1 excited-state lifetimes are 3.8 ns, 3.3 ns, 500 ps and 150 ps, and the rate constants for S1 → S0 internal conversion are (17 ns)−1, (11 ns)−1, (4.2 ns)−1 and (152 ps)−1, respectively (Table 2).
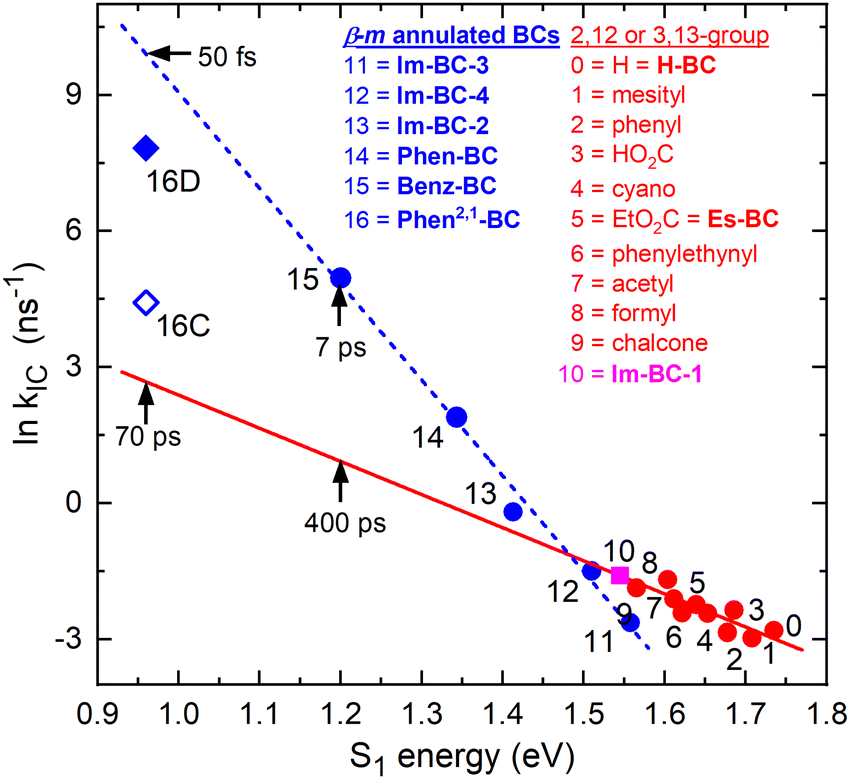 | ||
| Fig. 4 Energy-gap law plot showing the natural logarithm of the rate constant for S1 → S0 internal conversion versus the S1 energy for various bacteriochlorins. The blue line is a fit for bacteriochlorins bearing one or two imide moieties or annulated rings spanning adjacent meso and β-pyrrole positions, including Phen-BC and Benz-BC (Chart 1). The blue line projects a time constant of ∼50 fs for Phen2,1-BC. The red line is a fit for ten benchmark bacteriochlorins (ESI,† Chart S1) and one β,β-annulated imide-bacteriochlorin (point 10). | ||
In ESI,† Section S5, a less-favored interpretation of the TA data for Phen2,1-BC is considered in which the S1 → S0 internal conversion time is ∼12 ps (and prior relaxation within S1 taking ∼0.4 ps). This Model C gives the blue open diamond (denoted 16C) in Fig. 4. This time is shorter than the 70 ps extrapolated (red line) for “normal” bacteriochlorins. Similarly, the measured 7 ps for Benz-BC (point 15)21 is shorter than the extrapolated 400 ps. A smaller difference exists for Phen-BC (point 14). Collectively, the results obtained herein for Phen2,1-BC along with those for Phen-BC and Benz-BC show that β-meso annulation results in S1 → S0 internal conversion rates enhanced from those expected on the basis of energy-gap-law considerations for most bacteriochlorins.
Electronic-structure calculations
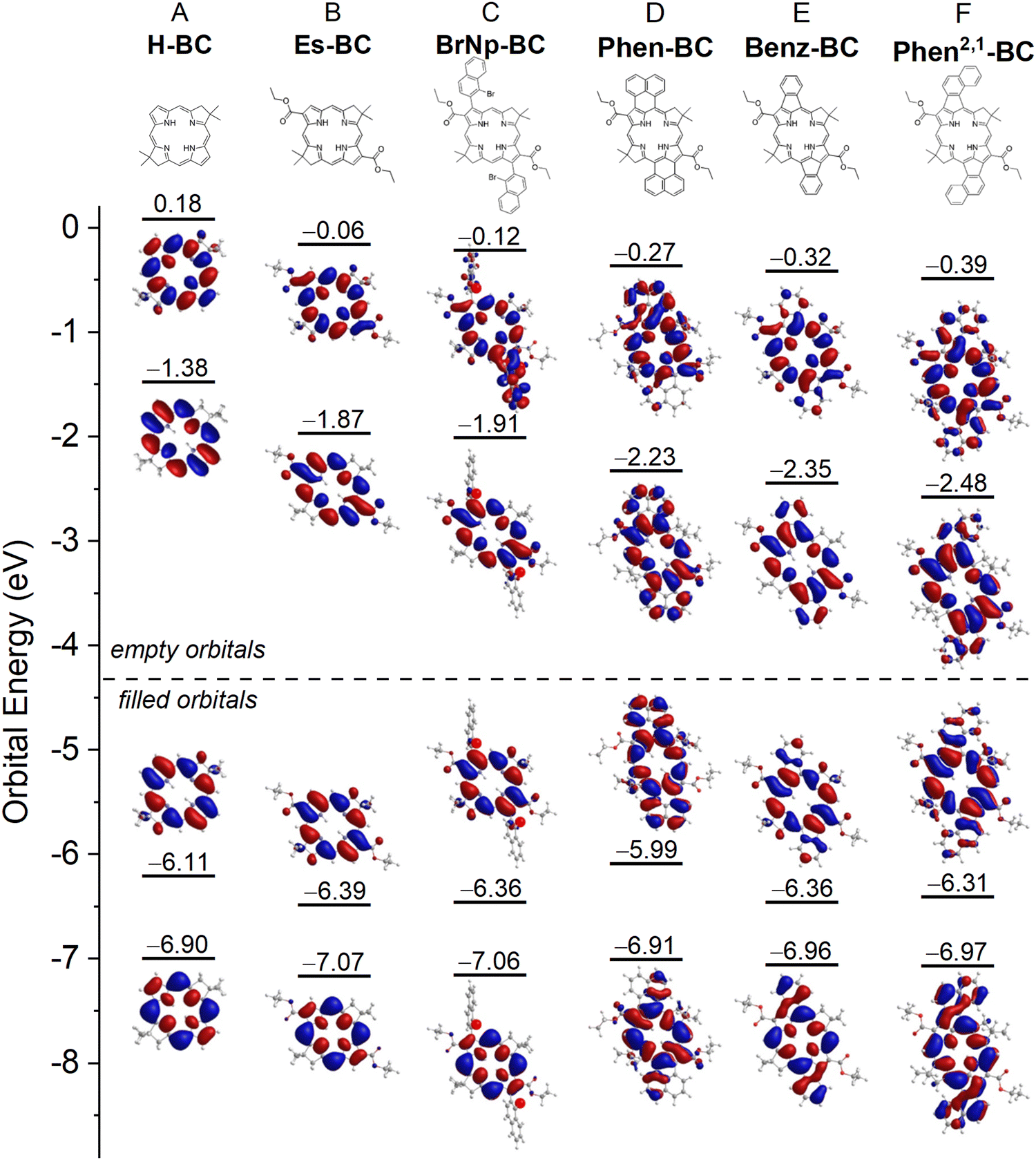 | ||
| Fig. 5 Frontier MO correlation diagram for six bacteriochlorins. | ||
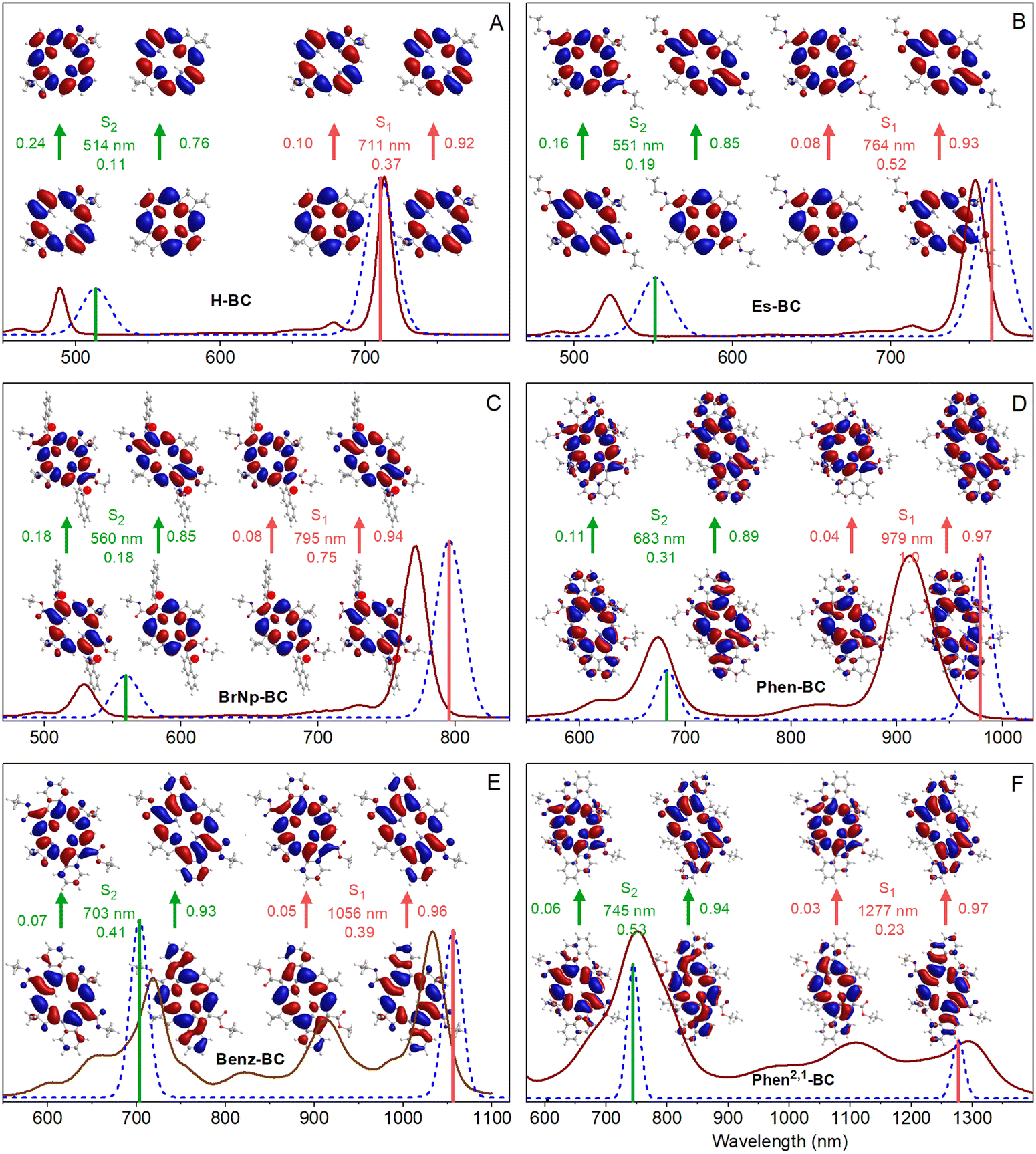 | ||
| Fig. 6 Comparison for (A) H-BC, (B) Es-BC, (C) BrNp-BC, (D) Phen-BC, (E) Benz-BC, and (F) Phen2,1-BC of the absorption spectra in toluene that are measured (brown line) or calculated via TDDFT (dashed blue lines and colored sticks) normalized at the S0 → S1(0,0) peak. Calculated spectral features were given 10-nm Gaussian skirts. Shown for the S0 → S1 and S0 → S2 transitions are the natural transition occupied and virtual orbitals along with the wavelength and oscillator strength of the transition obtained from TDDFT. Two NTO promotions contribute to each transition, with weights indicated by the values that flank the arrows. | ||
The TDDFT calculations reproduce well the measured spectra, with the caveat that these calculations do not account for vibronic satellite bands. In particular, the calculations capture the bathochromic shift of the S0 → S1 manifold along the series H-BC < Es-BC < BrNp-BC < Phen-BC < Benz-BC < Phen2,1-BC. The calculations also reproduce the increase in S1/S2 intensity ratio along the first three members and the reverse for the last three members: H-BC < Es-BC < BrNp-BC > Phen-BC > Benz-BC > Phen2,1-BC (Fig. 6 and last row of Table 2).
Fig. 6 also gives the natural transition orbitals (NTOs) for the S0 → S1 and S0 → S2 transitions. ESI,† Section S8 gives the NTOs for higher energy transitions. A pair of occupied and virtual (unoccupied) NTOs reveals the change in electron density involved in a transition.56 All six bacteriochlorins require two NTO pairs (contribution of two electron promotions), for which the relative weights are given next to the arrows in Fig. 6. The occupied NTOs for the S0 → S1 and S0 → S2 transitions of H-BC (Fig. 6(A)) are the same as the filled frontier MOs (HOMO and HOMO−1) (Fig. 5(A)). The same is true of the virtual NTOs compared to the empty MOs (LUMO and LUMO+1). This close matching of NTOs and MOs also holds for Es-BC (Fig. 6(B)vs.Fig. 5(B)) and BrNp-BC (Fig. 6(C)vs.Fig. 5(C)). This reflects the fact that for a given transition there is no common orbital involved in multiple electron promotions.
Setting aside electron delocalization onto the fused ring systems, the electron densities at the macrocycle positions in the MOs (Fig. 5) and NTOs (Fig. 6) for Phen-BC, Benz-BC, and Phen2,1-BC (panels D–F) are generally similar (but not identical) to those for the non-annulated analogues (panels A–C). This seems surprising given (1) the variation in S1/S2 intensity ratio described above (Fig. 6) and (2) substantial changes in the transition dipole moment (TDM) directions (Fig. 7). For H-BC, the S0 → S1 (Qy) transition lies along the NH–HN axis, which is the traditional y-axis for tetrapyrroles, and the S0 → S2 (Qx) transition is perpendicular to that (θ12 = 90° in Fig. 7(A)).19,57 For Es-BC, the S0 → S2 transition is rotated somewhat off perpendicular (θ12 = 73°) due to the 2,12-ester groups. The flanking 3,13-bis(bromonaphthalene) groups of BrNp-BC reintroduce near orthogonality (θ12 = 89°). The S1–S2 angle difference (θ12) decreases dramatically among the annulated bacteriochlorins: Phen-BC (55°) > Benz-BC (46°) > Phen2,1-BC (39°). The shift of S1 and S2 TDM directions from perpendicularity (and off the traditional x- and y-axes) reflects mixed x–y compositions of the S0 → S1 and S0 → S2 transitions (vide infra).
 | ||
| Fig. 7 Calculated TDM directions for the S0 → S1 and S0 → S2 transitions, the angle between them (θ12), and the ratio of oscillator strengths (f1/f2) for (A) H-BC, (B) Es-BC, (C) BrNp-BC, (D) Phen-BC, (E) Benz-BC, and (F) Phen2,1-BC. See Chart 1. The calculated ratio of oscillator strengths (f1/f2) for the two transitions is also given. | ||
In spite of the mixed x–y character implied by the TDM directions, the macrocycle electron distributions of the MOs and NTOs involved in the S0 → S1 transition for the three annulated bacteriochlorins (Fig. 5(D)–(F) and 6(D)–(F)) primarily have the characteristics of those for the non-annulated analogues (Fig. 5(A)–(C) and 6(A)–(C)), which are y-polarized (hence the traditional Qy designation for the non-annulated bacteriochlorins). Similarly, the MOs and NTOs involved in S0 → S2 for the three annulated bacteriochlorins are similar to those for the non-annulated analogs, which are x-polarized (normally designated Qx). Such findings are outside expectations and require in-depth analysis described in the following discussion.
Discussion
The creation of new NIR-II absorbers presents a host of challenges encompassing molecular design, synthesis, photophysics, and theory. The design of Phen2,1-BC emerged from prior knowledge of the properties of the annulated bacteriochlorins Phen-BC and Benz-BC along with subsequent extensive calculations aimed at attaining longer Qy maxima. Bacteriochlorins Phen-BC and Phen2,1-BC have identical numbers of π-bonds but are isomeric due to the different sites of connection on the annulated naphthalene rings. It warrants emphasis that the term annulation here refers to an appended group that is conjugated with the macrocycle at two points of attachment. Isomers Phen-BC and Phen2,1-BC have quite distinct photophysical features (Fig. 2 and Table 2) despite generally similar electron distributions in their frontier orbitals (Fig. 5 and 6). The electron-density distributions in the annulated bacteriochlorins are also generally similar to those of the non-annulated benchmarks. These observations prompt several fundamental questions: (1) how can one explain these seemingly incongruous findings (Fig. 2, 5 and 6)? (2) Is there a connection to the rapid internal conversion rates, and thus short S1 lifetimes, for the annulated bacteriochlorins (Fig. 4)? The resolution of these issues requires a reexamination of the applicability of the four-orbital model19,40,41 to tetrapyrrole architectures that include annulations. Accordingly, the subsequent sections are organized as follows: first, the four-orbital model applied to bacteriochlorins is presented; second, the electronic and spectral effects of x–y mixing are described; and third, the effects of vibronic coupling are considered. Taken together, the results provide new insights concerning hydroporphyrin designs that can afford (strong) absorption in the NIR-II region.The four-orbital model applied to bacteriochlorins
In Gouterman's four-orbital model,19,40,41 the S0 → S1 (Qy) transition derives from the asymmetric linear combination of the HOMO → LUMO and HOMO−1 → LUMO+1 excited-state configurations, both being y-polarized. Similarly, the S0 → S2 (Qx) transition reflects the asymmetric combination of HOMO−1 → LUMO and HOMO → LUMO+1, both being x-polarized. The configurational mixing decreases as the energy difference (ΔE) between excited-state configurations increases. For bacteriochlorins such as H-BC, ΔE is large, the mixing is small, and S0 → S1 transition has 92% HOMO → LUMO character (Table S7, ESI†). In progressing from H-BC to Es-BC and BrNp-BC (Chart 1), the substituents cause the HOMO → LUMO energy to drop (Fig. 5). The changes in spectral properties along this trio of compounds (Fig. 2) are consistent with the four-orbital-model (ESI,† Section S10). However, the model cannot explain the dramatic decrease in S0 → S1 intensity along the series Phen-BC > Benz-BC > Phen2,1-BC (Fig. 2 and Table 2).Electronic and spectral effects of X–Y mixing
The fundamental underpinning of Gouterman's four-orbital model19,40,41 is that the tetrapyrroles have electron promotions, optical transitions, and excited states that are classified by one of two distinct irreducible representations in the appropriate symmetry group: metalloporphyrins (D4h); free base porphyrins, free base bacteriochlorins and metallobacteriochlorins (D2h); and free base chlorins and metallochlorins (C2v). For all these cases, the promotions, transitions, and excited states relevant to the UV-Vis-NIR spectral properties have one of two symmetry designations, which transform as either x or y but not a mixture thereof.The actual symmetry (considering macrocycle and peripheral substituents) of the MOs (and NTOs) for H-BC, Es-BC, BrNp-BC, Phen-BC, Benz-BC, and Phen2,1-BC is not D4h, D2h, or C2v, but rather is C2h. The filled orbitals transform as au and the empty orbitals as bg. The promotions have bu symmetry and thus could lie anywhere in the x–y plane. Consequently, the excited-state configurations could have mixed x–y polarization. Such x–y mixing is reflected in a diminution in the S2–S1 angle θ12 from 90° and rotation of one or both of the TDM vectors off the traditional x and y axes (Fig. 7 and Table 2).
The S1 and S2 TDMs are orthogonal for H-BC and BrNp-BC (Fig. 7). The difference angle θ12 drops to 73° for Es-BC due to substitution at only the 2,12 and not the 3,13 positions that flank the NH–HN (y) axis. This reduction in θ12 by 17° for Es-BC seems to imply significant x–y mixing. The extent of mixing is then progressively more substantial as θ12 decreases along the series Phen-BC (55°) > Benz-BC (46°) > Phen2,1-BC (39°) (Fig. 7 and Table 2). Along the series of annulated bacteriochlorins, the S0 → S1 intensity also drops dramatically (Fig. 2 and Table 2). In contrast, a relatively small amount of x–y mixing is implied by the electron distributions at the macrocycle positions for the MOs and NTOs of Phen-BC, Benz-BC, and Phen2,1-BC, which are similar (but not identical) to those of H-BC, Es-BC, and BrNp-BC (Fig. 5 and 6).
In the simple empirical model for understanding the observations, the transitions/states are mixed by a coefficient α, whose square is given in eqn (1) and plotted in Fig. 8. The function α2 gives the amount of pure S1 that remains in the new S1 state upon mixing with pure S2. Similarly, α2 gives the amount of pure S2 that remains in the new S2 state upon mixing with pure S1. Consider two limiting cases. When θ12 = 90°, α2 = 1 and the new S1 retains 100% of the original pure y-polarized S1 character and the new S2 retains 100% of the original pure x-polarized S2 character, and both retain their original transition-dipole strengths. When θ12 = 0°, α2 = 0.5, mixing is maximal, and the new S1 and S2 transitions/states both are 50/50 mixtures of the original pure x- and y-polarized transitions/states. In principle, all the transition-dipole strength could reside in one new transition and the other would be silent. However, the actual ratio would depend on the specific case and the intensity ratio with no x–y mixing (e.g., that for H-BC).
| α2 = [cos(45° − |θ12|/2)]2 − 90° ≤ θ12 ≤ 90° | (1) |
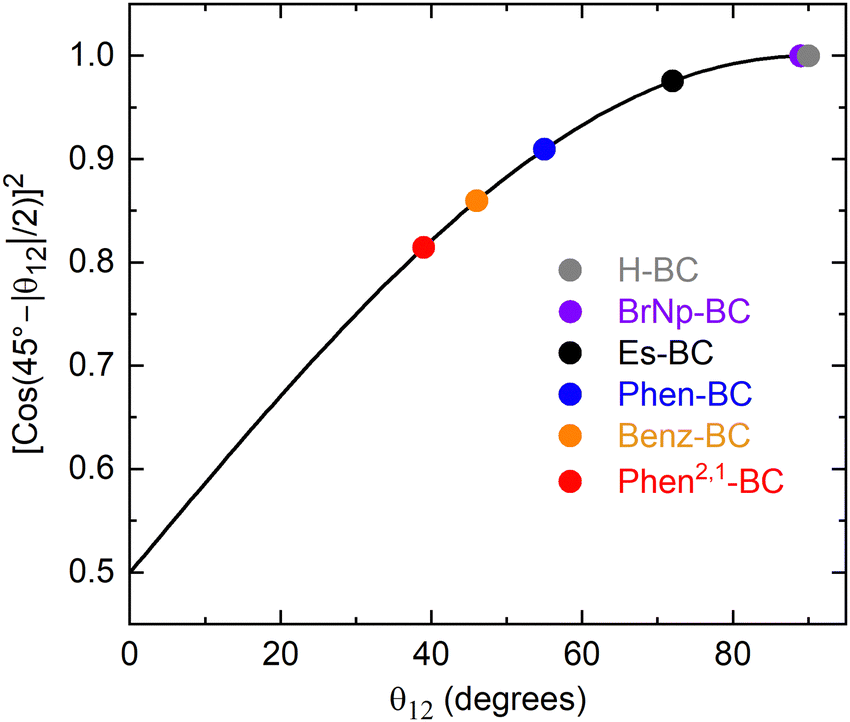 | ||
| Fig. 8 Plot of the square of the mixing coefficient α as a function of the absolute value of the angle θ12 between the S1 and S2 TDM vectors. | ||
The above-noted quandary is solved by inspection of Fig. 8 for the specific cases (colored circles) for the various bacteriochlorins (Chart 1). For Es-BC, the TDM difference angle θ12 is 17° off orthogonal (Fig. 7(B)), yet α2 = 0.98 and the S1 state remains 98% the pure y-polarized S1. For annulated Phen2,1-BC, θ12 drops to 39° and the S1 oscillator strength to f = 0.23 (Fig. 7 and Table 2), yet α2 = 0.82 and the S1 state remains 82% the pure y-polarized S1. Thus, the electron distributions at the macrocycle positions for the MOs and NTOs of Phen2,1-BC are quite similar to those of BrNp-BC, Es-BC, and H-BC (Fig. 5 and 6). In short, it takes only a small amount of x–y mixing to substantially impact spectral and electronic properties such as the relative intensities of the S0 → S1 and S0 → S2 transitions and their directions in the molecular framework.
Vibronic mixing involving two transitions/states results in dipole strength shifting from one to the other. The extent of mixing between S0 → S1 and S0 → S2 transitions will increase the closer the two excited states are in energy. Thus, the mixing will be greater for annulated bacteriochlorins Phen2,1-BC (∼4580 cm−1) and Benz-BC (∼4250 cm−1) than non-annulated analogues H-BC (∼6440 cm−1), BrNp-BC (∼5930 cm−1) and Es-BC (∼5840 cm−1) (Table 2). Key vibrations must also have the appropriate symmetry to mix the two transitions/states. The extent of electronic x–y mixing [indicated by θ12 between TDMs (Fig. 7 and Table 2)] and the extent of vibronic mixing [indicated intensity of the (1,0) absorption bands (Fig. 2)] increases along the series Phen-BC < Benz-BC < Phen2,1-BC.
There is a parallel decrease in the S1 excited-state lifetime along the same series (Table 2): Phen-BC (150 ps) > Benz-BC (7 ps) > Phen2,1-BC (0.4 ps). In all three cases, the S1 decay is dominated by S1 → S0 internal conversion, which is much more facile than expected based on the energy-gap law for non-radiative decay of normal bacteriochlorins (Fig. 4). The S1 → S0 internal conversion process depends on the coupling of electronic and vibrational degrees of freedom, namely the breakdown of the Born–Oppenheimer approximation.60–63 Thus, a reduction in symmetry links (1) the extent of electronic x–y mixing reflected in the angle between the S0 → S1 and S0 → S2 TDMs (Fig. 7), (2) the reduction in S0 → S1 intensity (Fig. 2), (3) vibronic activity in the absorption spectra (Fig. 2), (4) an increased rate of internal conversion (Fig. 4), and (5) a diminished ring current measured by NMR spectroscopy (Table 1).
Outlook
The synthesis of Phen2,1-BC described herein entailed development of a new route (Scheme 2) that is distinct from the prior route to annulated bacteriochlorins including Benz-BC and Phen-BC (Scheme 1), and may prove useful for construction of additional annulated bacteriochlorins. Annulation to form Phen2,1-BC, Benz-BC and Phen-BC impacts electronic structure, optical properties, and excited-state dynamics in unexpected ways. The desired bathochromic shift of the S0 → S1 transition into the NIR-II region is accompanied by diminished absorption strength and a shortened excited-state lifetime. These observables are connected to one another and to electronic x–y mixing and vibronic coupling involving the S0 → S1 and S0 → S2 transitions. The electronic and vibronic effects in turn arise from a reduction in symmetry entailed by the β-meso annulation motifs employed. The collective optical, excited-state, electronic and vibronic impacts of annulation were unexpected and have required analysis beyond the four-orbital model, which relies on symmetry partitioning of the relevant (orthogonal) x- and y-polarized electronic configurations.The observations and calculations described herein reveal that the key heuristic in achieving strong S0 → S1 absorption in the NIR-II region is to maintain x–y orthogonality. To this end, preliminary calculations on annulated bacteriochlorin (Phen2,1)2-BC shown in Chart 2 give an angle between the S0 → S1 and S0 → S2 transition dipole moments of 82°, and an S0 → S1 band at 1436 nm with a substantial oscillator strength of 0.33. It is anticipated that this architecture will also have an S1 lifetime consistent with that predicted from bacteriochlorins that possess a high degree of x–y orthogonality. The synthesis and photophysical characterization of (Phen2,1)2-BC and additional chromophores are required to assess the broad applicability of the picture that has emerged upon comparison of Phen2,1-BC and other annulated bacteriochlorins described herein.
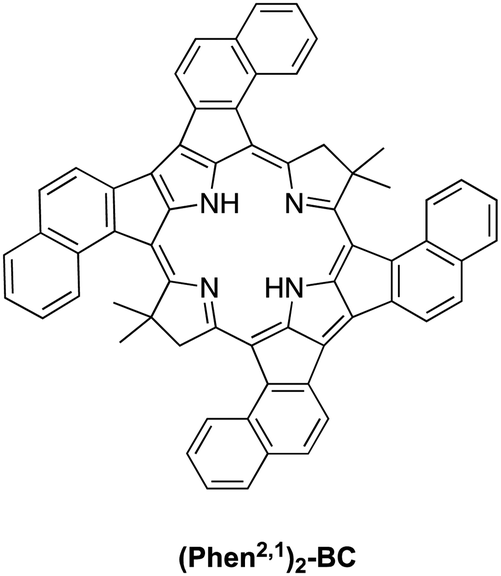 | ||
| Chart 2 Fictive bacteriochlorin for deep NIR-II penetration. | ||
Experimental section
Photophysical properties
Static absorption (Shimadzu UV-1800), static emission (Horiba Nanolog; PTI Quantamaster), and other measurements were performed at room temperature on dilute (μM), Ar-purged solutions of compounds in toluene. Some absorption spectra were acquired at 295 or 77 K on samples in 2-methyltetrahydrofuran. Determination of Φf values utilized samples with A ≤ 0.1 at the excitation wavelength and used meso-tetraphenylporphyrin in non-degassed toluene (Φf = 0.070)45 or Phen-BC (Φf = 0.004)21 as a standard. Transient absorption (TA) studies utilized an ultrafast laser system with (0.5–1 μJ) ∼100 fs excitation flashes from a 1 kHz Ti:sapphire laser system (Spectra Physics) and Helios spectrometer (Ultrafast Systems). TA data were analyzed using Surface Xplorer (Ultrafast Systems), CarpetView (Light Conversion), and custom routines in OriginPro (OriginLab).Density functional theory calculations
DFT calculations were performed with Gaussian 16 version C.01.39 Calculations used the PCM model for the arrays in toluene. Molecular geometries were fully optimized using the hybrid B3LYP functional and the basis set 6-31G*. These calculations used Gaussian 16 defaults. Electron-density distributions and energies of the molecular orbitals (MOs) were obtained using the long-range corrected ωB97XD functional and the basis set 6-31++G**. TDDFT calculations were performed using the long-range corrected ωB97XD functional and the basis set 6-31++G** with the Gaussian 16 defaults and the additional keyword n states = 16. The MO, NTO, and TDM images were created in Chemcraft.General synthetic methods
Tetrahydrofuran (THF) was freshly distilled from sodium/benzophenone ketyl. Silica (40 μm average particle size) was used for column chromatography. Other solvents (anhydrous or reagent-grade) were employed as received from commercial suppliers. 1H NMR, 10B NMR, and 13C{1H} NMR spectra were recorded at room temperature in CDCl3 unless noted otherwise. Deactivated silica for chromatographic purification of acid-labile compounds was prepared by treatment with 3% triethylamine in hexanes followed by washing with hexanes. Deactivated silica for TLC analysis of acid-labile compounds was prepared by running with 3% triethylamine in hexanes followed by drying of the plate prior to the analysis. Electrospray ionization mass spectrometry (ESI-MS) data were obtained in the positive-ion mode (unless noted otherwise) and are reported for the molecular ion or protonated molecular ion. Commercial compounds were used as received. The known compounds 1-bromo-2-(2-nitrovinyl)naphthalene (1),27 Michael acceptor VII,33 and dihydrodipyrrin I21 were prepared as described in the literature.Synthetic procedures
:1 volume HCl/volume THF) and stirred at room temperature for 2 h. The reaction mixture was diluted with water (10.0 mL) and extracted with ethyl acetate (3 × 15.0 mL). The combined organic extract was dried over anhydrous Na2SO4 and concentrated under reduced pressure. Column chromatography [silica, hexanes/ethyl acetate (5:1 to 7:3)] afforded a white solid (134.6 mg, 64%). m.p. 234–238 °C: 1H NMR (600 MHz, CD3OD) δ 8.22 (d, J = 8.46 Hz, 1H), 7.85–7.88 (m, 2H), 7.61 (t, 1H), 7.55 (t, 1H), 7.31 (d, J = 7.68 Hz, 1H); 13C{1H} NMR (150 MHz, CD3OD) δ 136.2, 132.8, 129.4, 129.3, 128.8, 128.6, 128.1, 127.7, 126.3. The aromatic quaternary C–B(OH)2 was not observed; 10B NMR (54 MHz, CDCl3) δ 29.6.
:1 to 4:1)] afforded a light-yellow solid (2.86 g, 47%). m.p. 175–176 °C; 1H NMR (500 MHz, CDCl3) δ 8.42 (br, 1H), 8.36 (d, J = 8.6 Hz, 1H), 7.83 (d, J = 8.2 Hz, 1H), 7.76 (d, J = 8.3 Hz, 1H), 7.55–7.60 (m, 1H), 7.47–7.51 (m, 1H), 7.40 (d, J = 8.4 Hz, 1H), 6.58 (d, J = 2.4 Hz, 1H), 4.00 (q, J = 7.2 Hz, 2H), 2.58 (s, 3H), 0.83 (t, J = 7.1 Hz, 3H); 13C{1H} NMR (125 MHz, CDCl3) δ 165.5, 135.9, 135.4, 133.4, 132.3, 129.6, 128.0, 127.7, 127.2, 126.6, 126.4, 126.0, 124.6, 115.9, 111.3, 59.2, 13.8, 13.7; ESI-MS obsd 358.0434, calcd 358.0437 [(M + H)+, M = C18H16BrNO2].
:1 to 5:1)] afforded an orange solid (1.88 g, 54%). m.p. 196–197 °C; 1H NMR (600 MHz, CDCl3) δ 10.29 (d, J = 0.7 Hz, 1H), 10.20 (br, 1H), 8.35 (d, J = 8.5 Hz, 1H), 7.86 (d, J = 8.0 Hz, 1H), 7.81 (d, J = 8.3 Hz, 1H), 7.59–7.63 (m, 1H), 7.52–7.56 (m, 1H), 7.40 (d, J = 8.3 Hz, 1H), 7.10 (dd, J = 2.9, 0.6 Hz, 1H), 4.13 (q, J = 7.2 Hz, 2H), 0.94 (t, J = 7.1 Hz, 3H); 13C{1H} NMR (150 MHz, CDCl3) δ 182.3, 163.4, 133.8, 133.4, 133.1, 132.3, 129.2, 128.8, 128.1, 127.8, 127.6, 126.9, 126.6, 124.7, 124.4, 121.2, 60.6, 13.7; ESI-MS obsd 372.0229, calcd 372.0230 [(M + H)+, M = C18H14BrNO3].
:4 to 1:9)] afforded a light-yellow solid (1.20 g, 45%). m.p. 129–130 °C; 1H-NMR (500 MHz, CDCl3) δ 8.62 (br, 1H), 8.35 (d, J = 8.4 Hz, 1H), 7.85 (d, J = 8.1 Hz, 1H), 7.77 (d, J = 8.3 Hz, 1H), 7.57–7.62 (m, 1H), 7.49–7.54 (m, 1H), 7.38 (d, J = 8.4 Hz, 1H), 6.69 (d, J = 2.6 Hz, 1H), 4.78–4.83 (m, 2H), 3.97 (q, J = 7.0 Hz, 2H), 3.63–3.69 (m, 2H), 0.75 (t, J = 7.2 Hz, 3H); 13C{1H} NMR (125 MHz, CDCl3) δ 165.1, 135.2, 133.4, 133.2, 132.2, 129.3, 128.0, 127.7, 127.3, 126.7, 126.5, 126.2, 124.8, 117.3, 112.1, 74.6, 59.6, 25.4, 13.6; ESI-MS obsd 417.0441, calcd 417.0444 [(M + H)+, M = C19H17BrN2O4].
:1)] afforded a light-yellow solid (224 mg, 72%). m.p. 48–51 °C; 1H NMR (500 MHz, CDCl3) δ 8.45 (br, 1H), 8.33 (d, J = 8.6 Hz, 1H), 7.83 (d, J = 8.2 Hz, 1H), 7.75 (d, J = 8.4 Hz, 1H), 7.55–7.60 (m, 1H), 7.47–7.52 (m, 1H), 7.36 (d, J = 8.2 Hz, 1H), 6.61 (d, J = 2.6 Hz, 1H), 5.28 (ABX, 3JAX = 11.8 Hz, 3JBX = 2.3 Hz, 1H), 4.43 (s, 1H), 3.91–4.04 (m, 2H), 3.85 (ABX, 2JAB = 14.6 Hz, 3JBX = 2.4 Hz, 1H), 3.44 (s, 3H), 3.43 (s, 3H), 3.38 (ABX, 2JAB = 14.4 Hz, 3JAX = 11.8 Hz, 1H), 2.76, 2.69 (AB, 2J = 18.6 Hz, 2H), 1.34 (s, 3H), 1.22 (s, 3H), 0.74 (t, J = 7.1 Hz, 3H); 13C{1H} NMR (125 MHz, CDCl3) δ 203.2, 164.8, 135.5, 133.4, 133.0, 132.2, 129.4, 128.0, 127.7, 127.2, 126.7, 126.4, 126.1, 124.8, 117.3, 112.2, 104.5, 94.8, 59.4, 54.99, 54.97, 44.7, 36.7, 26.6, 24.1, 23.7, 13.6; ESI-MS obsd 575.1381, calcd 575.1387 [(M + H)+, M = C27H31BrN2O7].
:1 to 9:1)] afforded a light-yellow solid (228 mg, 44%). m.p. 161–162 °C; 1H NMR (600 MHz, CDCl3) δ 11.35 (br, 1H), 8.36 (d, J = 8.5 Hz, 1H), 7.84 (d, J = 8.2 Hz, 1H), 7.76 (d, J = 8.3 Hz, 1H), 7.56–7.60 (m, 1H), 7.48–7.52 (m, 1H), 7.42 (d, J = 8.2 Hz, 1H), 6.97 (s, 1H), 6.81 (d, J = 2.5 Hz, 1H), 5.06 (s, 1H), 4.00 (q, J = 7.1 Hz, 2H), 3.48 (s, 6H), 2.68 (s, 2H), 1.29 (s, 6H), 0.78 (t, J = 1.1 Hz, 3H); 13C{1H} (125 MHz, CDCl3) δ 177.1, 165.3, 164.1, 136.1, 135.3, 133.4, 132.3, 129.6, 128.0, 127.7, 127.1, 126.5, 126.4, 126.0, 124.7, 118.9, 111.6, 105.9, 102.5, 59.2, 54.6, 48.3, 40.6, 29.0; ESI-MS obsd 525.1382, calcd 525.1384 [(M + H)+, M = C27H29BrN2O4].
:1)] afforded a dark-purple solid (34.4 mg, 12%). 1H NMR (600 MHz, CDCl3) δ 9.75 (br, 2H), 8.55 (d, J = 8.2 Hz, 2H), 8.29 (s, 2H), 8.07–8.13 (m, 4H), 7.86 (t, J = 8.4 Hz, 2H), 7.77 (t, J = 7.9 Hz, 2H), 7.72 (t, J = 7.5 Hz, 2H), 4.41–4.48 (m, 2H), 4.32–4.39 (m, 2H), 4.18–4.23 (m, 4H), 1.89–1.90 (m, 12H), 1.02 (t, J = 7.1 Hz, 6H), –1.05 (s, 2H); 13C{1H} NMR (150 MHz, CDCl3) δ 173.6, 165.5, 160.5, 138.51, 138.47, 135.8, 135.6, 134.1, 132.8, 132.40, 132.38, 129.9, 129.8, 128.41, 128.39, 127.90, 127.88, 127.84, 126.9, 126.72, 126.70, 125.68, 125.65, 120.41, 120.3, 99.4, 97.6, 60.6, 51.2, 46.2, 31.2, 31.0, 30.9, 30.8, 13.8; ESI-MS obsd 925.1763, calcd 925.1782 [(M + H)+, M = C50H44Br2N4O4]. λabs (toluene) 384, 359, 529, 771 nm. No evidence (by TLC analysis or 1H NMR spectroscopy) was observed for the presence of atropisomers. On the other hand, 13C{1H} NMR analysis showed 35 signals whereas 25 were expected, which is attributed to the presence of atropisomers. Similar observations were made for the analogous precursors to Phen-BC and Benz-BC.21
Phen2,1-BC
Following a procedure,21 a mixture of BrNp-BC (34.3 mg, 37.2 μmol), PCy3·HBF4 (41.0 mg, 111 μmol), and K2CO3 (25.6 mg, 185 μmol) in anhydrous DMF (3.7 mL) in a Schlenk flask was freeze-pumped-thawed for 3 cycles. Pd(OAc)2 (8.33 mg, 37.1 μmol) was added into the mixture, which was then heated in an oil bath at 110 °C for 2 h. The reaction mixture was allowed to cool to room temperature and then was diluted with dichloromethane (10.0 mL). The resulting mixture was washed with water (15.0 mL) and brine (15.0 mL). Each aqueous solution was extracted with ethyl acetate (2 × 15.0 mL). The combined organic extract was dried over anhydrous Na2SO4 and then concentrated under reduced pressure. The crude mixture was passed through a deactivated silica pad, eluted with ethyl acetate, and concentrated under reduced pressure. Preparative thin-layer chromatography [deactivated silica, hexanes/ethyl acetate (4:1)] afforded a dark-brown solid (4.5 mg, 16%). 1H NMR (600 MHz, CDCl3) δ 8.03 (d, J = 8.0 Hz, 2H), 7.95 (d, J = 8.5 Hz, 2H), 7.77 (s, 2H), 7.62 (d, J = 8.1 Hz, 2H), 7.43 (t, J = 7.6 Hz, 2H), 7.37 (d, J = 8.2 Hz, 2H), 7.43 (t, J = 7.3 Hz, 2H), 4.43 (q, J = 7.1 Hz, 4H), 3.49 (s, 4H), 3.21 (s, 2H), 1.49 (t, J = 7.0, 6H), 1.20 (s, 12H); 13C{1H} NMR (150 MHz, CDCl3) δ 174.2, 164.5, 159.2, 146.0, 143.7, 143.6, 142.2, 135.9, 134.7, 129.7, 129.2, 127.7, 125.6, 125.45, 125.40, 123.7, 117.9, 112.2, 101.1, 60.7, 52.3, 46.2, 28.3, 14.5; ESI-MS obsd 762.3184, calcd 762.3200 [(M + H)+, M = C50H42N4O4]. λabs (toluene) 374, 401, 455, 750, 1110, 1292 nm.
Molar absorption coefficients
The molar absorption coefficients were determined by preparing 0.20 mM Phen2,1-BC (0.97 mg) in toluene (6.6 mL) and BrNp-BC (0.98 mg) in toluene (5.3 mL). Known volumes of the mother solution (120, 180, 240, 300, and 360 μL for Phen2,1-BC and 20, 30, 40, 50, and 60 μL for BrNp-BC) were diluted with toluene to 3.0 mL for instrumental measurement. The absorption spectra were recorded at room temperature. The averages of three runs were calculated: BrNp-BC (ε384nm = 64500 M−1 cm−1; ε359nm = 100600 M−1 cm−1; ε529nm = 25100 M−1 cm−1; and ε771nm = 125900 M−1 cm−1); Phen2,1-BC (ε347nm = 32400 M−1 cm−1; ε401nm = 33200 M−1 cm−1; ε455nm = 39500 M−1 cm−1; ε750nm = 19000 M−1 cm−1; ε1110nm = 6000 M−1 cm−1; and ε1292nm = 6100 M−1 cm−1).
Single-crystal X-ray diffraction analyses
SCXRD data were obtained using a Bruker D8 Venture diffractometer at 100 K (MoKα = 0.71073 Å). The structural refinement and graphics were calculated and generated using APEX4, SHELXL, OLEX2, and MERCURY3 software.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by a grant from the Chemical Sciences, Geosciences and Biosciences Division, Office of Basic Energy Sciences, of the U.S. Department of Energy (DE-FG02-05ER15661), and by NC State University. Compound characterization was performed in part by the Molecular Education, Technology and Research Innovation Center (METRIC) at NC State University, which is supported by the State of North Carolina. XRD analyses were performed at University of Tennessee at Knoxville.References
- A. M. Smith, M. C. Mancini and S. Nie, Nat. Nanotechnol., 2009, 4, 710–711 CrossRef CAS PubMed.
- D. Meng, R. Zheng, Y. Zhao, E. Zhang, L. Dou and Y. Yang, Adv. Mater., 2022, 34, 2107330 CrossRef CAS PubMed.
- C. Han, B. K. Kundu, Y. Liang and Y. Sun, Adv. Mater., 2024, 36, 2307759 CrossRef CAS PubMed.
- S. Zhu, R. Tian, A. L. Antaris, X. Chen and H. Dai, Adv. Mater., 2019, 31, 1900321 CrossRef PubMed.
- Z. Guo, S. Park, J. Yoon and I. Shin, Chem. Soc. Rev., 2014, 43, 16–29 RSC.
- N.-N. Zhang, C.-Y. Lu, M.-J. Chen, X.-L. Xu, G.-F. Shu, Y.-Z. Du and J.-S. Ji, J. Nanobiotechnol., 2021, 19, 132 CrossRef PubMed.
- H. Li, Y. Kim, H. Jung, J. Y. Hyun and I. Shin, Chem. Soc. Rev., 2022, 51, 8957–9008 RSC.
- X. Liu, B. Yu, Y. Shen and H. Cong, Coord. Chem. Rev., 2022, 468, 214609 CrossRef CAS.
- B. Dunn, M. Hanafi, J. Hummel, J. R. Cressman, R. Veneziano and P. V. Chitnis, Bioengineering, 2023, 10, 954 CrossRef CAS PubMed.
- M. Taniguchi and J. S. Lindsey, Photochem. Photobiol., 2021, 97, 136–165 CrossRef CAS PubMed.
- M. Kobayashi, M. Akiyama, H. Kise and T. Watanabe, in Chlorophylls and Bacteriochlorophylls: Biochemistry, Biophysics, Functions and Applications, ed. B. Grimm, R. J. Porra, W. Rüdiger and H. Scheer, Springer, Dordrecht, The Netherlands, 2006, pp. 55–66 Search PubMed.
- H. Scheer, in Chlorophylls and Bacteriochlorophylls. Biochemistry, Biophysics, Functions and Application, ed. B. Grimm, R. J. Porra, W. Rüdiger and H. Scheer, Springer, Dordrecht, The Netherlands, 2006, pp. 1–26 Search PubMed.
- M. Kobayashi, S. Akutsu, D. Fujinuma, H. Furukawa, H. Komatsu, Y. Hotota, Y. Kato, Y. Kuroiwa, T. Watanabe, M. Ohnishi-Kameyama, H. Ono, S. Ohkubo and H. Miyashita, in Photosynthesis, ed. Z. Dubinsky, 2013, pp. 47–90 Search PubMed.
- J. W. Springer, K. M. Faries, J. R. Diers, C. Muthiah, O. Mass, H. L. Kee, C. Kirmaier, J. S. Lindsey, D. F. Bocian and D. Holten, Photochem. Photobiol., 2012, 88, 651–674 CrossRef CAS PubMed.
- A. Maccoll, Q. Rev., 1947, 1, 16–58 RSC.
- L. N. Ferguson, Chem. Rev., 1948, 43, 385–446 CrossRef CAS PubMed.
- K. Venkataraman, The Chemistry of Synthetic Dyes, Academic Press, New York, 1952, vol. I, pp. 323–400 Search PubMed.
- S. Dähne, Science, 1978, 199, 1163–1167 CrossRef PubMed.
- M. Gouterman, The Porphyrins, ed. D. Dolphin, Academic Press, New York, 1978, vol. 3, pp. 1–165 Search PubMed.
- P. Vairaprakash, E. Yang, T. Sahin, M. Taniguchi, M. Krayer, J. R. Diers, A. Wang, D. M. Niedzwiedzki, C. Kirmaier, J. S. Lindsey, D. F. Bocian and D. Holten, J. Phys. Chem. B, 2015, 119, 4382–4395 CrossRef CAS PubMed.
- H. Fujita, H. Jing, M. Krayer, S. Allu, G. Veeraraghavaiah, Z. Wu, J. Jiang, J. R. Diers, N. C. M. Magdaong, A. K. Mandal, A. Roy, D. M. Niedzwiedzki, C. Kirmaier, D. F. Bocian, D. Holten and J. S. Lindsey, New J. Chem., 2019, 43, 7209–7232 RSC.
- V.-P. Tran, P. Wang, N. Matsumoto, S. Liu, H. Jing, P. Nalaoh, K. Chau Nguyen, M. Taniguchi and J. S. Lindsey, J. Porphyrins Phthalocyanines, 2023, 27, 1502–1551 CrossRef CAS.
- R. Martin and S. L. Buchwald, Acc. Chem. Res., 2008, 41, 1461–1473 CrossRef CAS PubMed.
- I. P. Beletskaya and A. V. Cheprakov, Chem. Rev., 2000, 100, 3009–3066 CrossRef CAS PubMed.
- S. Maiti, S. Biswas and U. Jana, J. Org. Chem., 2010, 75, 1674–1683 CrossRef CAS PubMed.
- L. Li, M.-N. Zhao, Z.-H. Ren, J. Li and Z.-H. Guan, Synthesis, 2012, 532–549 CAS.
- A. Kolarovic, A. Käslin and H. Wennemers, Org. Lett., 2014, 16, 4236–4239 CrossRef CAS PubMed.
- D. Akbaslar, E. S. Giray and O. Algul, Mol. Diversity, 2021, 25, 2321–2338 CrossRef CAS PubMed.
- H. Jing, P. Wang, B. Chen, J. Jiang, P. Vairaprakash, S. Liu, J. Rong, C.-Y. Chen, P. Nalaoh and J. S. Lindsey, New J. Chem., 2022, 46, 5534–5555 RSC.
- M. Gałęzowski and D. T. Gryko, J. Org. Chem., 2006, 71, 5942–5950 CrossRef PubMed.
- F. A. Luzzio, Tetrahedron, 2001, 57, 915–945 CrossRef CAS.
- T. Tokoroyama, Eur. J. Org. Chem., 2010, 2009–2016 CrossRef CAS.
- O. Mass and J. S. Lindsey, J. Org. Chem., 2011, 76, 9478–9487 CrossRef CAS PubMed.
- D. T. M. Chung, P. V. Tran, K. Chau Nguyen, P. Wang and J. S. Lindsey, New J. Chem., 2021, 45, 13302–13316 RSC.
- J. E. McMurry and J. Melton, J. Org. Chem., 1973, 38, 4367–4373 CrossRef CAS.
- V.-P. Tran, N. Matsumoto, P. Nalaoh, H. Jing, C.-Y. Chen and J. S. Lindsey, Organics, 2022, 3, 262–274 CrossRef CAS.
- C.-Y. Chen, M. Taniguchi and J. S. Lindsey, J. Porphyrins Phthalocyanines, 2014, 18, 433–456 CrossRef CAS.
- M. Krayer, M. Ptaszek, H.-J. Kim, K. R. Meneely, D. Fan, K. Secor and J. S. Lindsey, J. Org. Chem., 2010, 75, 1016–1039 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian, Inc., Wallingford CT, 2019.
- M. Gouterman, J. Chem. Phys., 1959, 30, 1139–1161 CrossRef CAS.
- M. Gouterman, J. Mol. Spectrosc., 1961, 6, 138–163 CrossRef CAS.
- Y. Liu, S. Allu, M. N. Reddy, D. Hood, J. R. Diers, D. F. Bocian, D. Holten and J. S. Lindsey, New J. Chem., 2017, 41, 4360–4376 RSC.
- E. Yang, C. Kirmaier, M. Krayer, M. Taniguchi, H.-J. Kim, J. R. Diers, D. F. Bocian, J. S. Lindsey and D. Holten, J. Phys. Chem. B, 2011, 115, 10801–10816 CrossRef CAS PubMed.
- J. R. Diers, Q. Tang, C. J. Hondros, C.-Y. Chen, D. Holten, J. S. Lindsey and D. F. Bocian, J. Phys. Chem. B, 2014, 118, 7520–7532 CrossRef CAS PubMed.
- M. Taniguchi, J. S. Lindsey, D. F. Bocian and D. Holten, J. Photochem. Photobiol., C, 2021, 46, 100401 CrossRef CAS.
- W. Wild, A. Seilmeier, N. H. Gottfried and W. Kaiser, Chem. Phys. Lett., 1985, 119, 259–263 CrossRef CAS.
- E. R. Henry, W. A. Eaton and R. M. Hochstrasser, Proc. Natl. Acad. Sci. U. S. A., 1986, 83, 8982–8986 CrossRef CAS PubMed.
- J. R. Hill and D. D. Dlott, J. Chem. Phys., 1988, 89, 842–858 CrossRef CAS.
- T.-C. Chang and D. D. Dlott, J. Chem. Phys., 1989, 90, 3590–3602 CrossRef CAS.
- F. Laermer, T. Elsaesser and W. Kaiser, Chem. Phys. Lett., 1989, 156, 381–386 CrossRef CAS.
- A. Mokhtari, J. Chesnoy and A. Laubereau, Chem. Phys. Lett., 1989, 155, 593–598 CrossRef CAS.
- G. Angel, R. Gagel and A. Laubereau, Chem. Phys., 1989, 131, 129–134 CrossRef CAS.
- U. Sukowski, A. Seilmeier, T. Elsaesser and S. F. Fischer, J. Chem. Phys., 1990, 93, 4094–4101 CrossRef CAS.
- J. Rodriguez, C. Kirmaier and D. Holten, J. Chem. Phys., 1991, 94, 6020–6029 CrossRef CAS.
- O. Bilsel, S. N. Milam, G. S. Girolami, K. S. Suslick and D. Holten, J. Phys. Chem., 1993, 97, 7216–7221 CrossRef CAS.
- R. L. Martin, J. Chem. Phys., 2003, 118, 4775–4777 CrossRef CAS.
- A. K. Mandal, M. Taniguchi, J. R. Diers, D. M. Niedzwiedzki, C. Kirmaier, J. S. Lindsey, D. F. Bocian and D. Holten, J. Phys. Chem. A, 2016, 120, 9719–9731 CrossRef CAS PubMed.
- G. Herzberg and E. Teller, Z. Physikal. Chem. B, 1933, 21, 410–446 CrossRef.
- M. H. Perrin, M. Gouterman and C. L. Perrin, J. Chem. Phys., 1969, 50, 4137–4150 CrossRef CAS.
- J. B. Birks, Photophysics of Aromatic Molecules, Wiley-Interscience, London, 1970, pp. 142–192 Search PubMed.
- G. W. Robinson and R. P. Frosch, J. Chem. Phys., 1962, 37, 1962–1973 CrossRef CAS.
- W. Siebrand, J. Chem. Phys., 1967, 46, 440–447 CrossRef CAS.
- R. Englman and J. Jortner, Mol. Phys., 1970, 18, 145–164 CrossRef CAS.
- H. Cai and C. Han, CN107698486, 2018.
- L. Ling, Z. Song, H. Shan, C. Wang, S. Li, Y. Wang, J. Hu, Q. Chen, H. Zhang and Y. Yang, Chem. Commun., 2023, 59, 2739–2742 RSC.
- M. Lanzi, Q. Dherbassy and J. Wencel-Delord, Angew. Chem., Int. Ed., 2021, 60, 14852–14857 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Exploratory syntheses; photophysical data for bacteriochlorins; results of TDDFT calculations; 1H and 13C{1H} NMR spectra of new compounds; and single-crystal X-ray data. CCDC 2327783 (3), 2327784 (5), and 2327785 (7). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cp00779d |
| This journal is © the Owner Societies 2024 |