
Direct reduction of NO into N2 catalyzed by fullerene-supported rhodium clusters†
Ruomeng
Li
,
Ya-Ke
Li
*,
Jianzhi
Xu
 and
Gao-Lei
Hou
*
and
Gao-Lei
Hou
*
MOE Key Laboratory for Non-Equilibrium Synthesis and Modulation of Condensed Matter, School of Physics, Xi’an Jiaotong University, Xi’an, 710049 Shaanxi, China. E-mail: yake.li@xjtu.edu.cn; gaolei.hou@xjtu.edu.cn
First published on 3rd May 2024
Abstract
Catalytic conversion of NO has long been a focus of atmospheric pollution control and diesel vehicle exhaust treatment. Rhodium is one of the most effective metals for catalyzing NO reduction, and understanding the nature of the active sites and underlying mechanisms can help improve the design of Rh-based catalysts towards NO reduction. In this work, we investigated the detailed catalytic mechanisms for the direct reduction of NO to N2 by fullerene-supported rhodium clusters, C60Rh4+, with density functional theory calculations. We found that the presence of C60 facilitates the smooth reduction of NO into N2 and O2, as well as their subsequent desorption, recovering the catalyst C60Rh4+. Such a process fails to be completed by free Rh4+, emphasizing the critical importance of C60 support. We attribute the novel performance of C60Rh4+ to the electron sponge effect of C60, providing useful guidance for designing efficient catalysts for the direct reduction of NO.
Introduction
Nitric oxide (NO) is an air pollutant associated with severe environmental issues, posing great health risks to human beings. Its treatment has received extensive efforts,1,2 and current methods for NO control, such as absorption, decomposition, and reduction, always encounter shortcomings such as process complexity and by-product generation.3 The three-way catalyst (TWC), which employs noble metals such as Pt, Pd, and Rh, stands out as an effective catalyst for reducing NO emissions in automobile exhaust. In particular, rhodium excels in NO chemical adsorption and dissociation, showing great activity and selectivity in NO reduction into N2.4,5Previous studies investigated the dissociation and adsorption of NO on the surfaces of Rh(100), Rh(111), and Rh(110) surfaces and found that N atoms bind and desorb at 450–650 K, and O atoms desorb in O2 form at 1000–1400 K.6–18 It is shown that Rh-doped metal catalysts exhibit more superior catalytic performance than pure metal catalysts for converting NO, emphasizing the critical importance of rhodium.19,20 However, rhodium is scarce and expensive, limiting its widespread utilization.1,3,4 In this regard, recently emerged single-atom catalysts (SACs) and single-cluster catalysts (SCCs) that offer high atomic efficiency21–23 may provide an interesting approach for employing Rh-based catalysts in NO conversion.
Mass spectrometric experiments and theoretical calculations on the reactions of free rhodium clusters with NO have elucidated the important role of Rh in N–O bond activation.24–33 For instance, Ghosh et al. found that Rh clusters containing one to five atoms exhibit stronger bonding with NO compared to Rh(100) and Rh(111), rendering Rh clusters to be good catalysts for NO reduction.25 Xie et al. studied the NO adsorption and reduction on the Rh7+ cluster and found that the reaction follows a three-step process: initial adsorption of NO, subsequent NO decomposition into N and O atoms, and then, the reaction of the N atom with a second adsorbed NO, leading to the formation of N2 molecules and rhodium oxides.27 Romo-Ávila et al. investigated the NO dissociation on Rhn± clusters (n = 3, 4, 6, and 13) and found that the dissociation of N–O is more feasible on square facets than on triangular atomic environments.28 They also found the energy barrier for breaking the N–O bond depends on the charge states of the rhodium clusters, with cationic clusters being more reactive than neutrals and anions, in agreement with an earlier experimental study.24
Previously, Mackenzie and co-workers found that once two NO molecules co-adsorb on Rhx± clusters, NO first dissociates to generate mobile N atoms that can then combine to form N2 molecules; resultant rhodium dioxide clusters RhxO2± further adsorb NO, and the process of NO reduction to N2 replicates, yielding higher rhodium oxides.24 In these cases, catalytic reactions stopped with the formation of rhodium oxides and no O2 release, that will, in the end, lead to the deactivation of the catalysts.24,26,27,31,33 Then, a question arises: if a catalytic cycle cannot be completed on free rhodium clusters, how about when supported on certain substrates, considering that in practice most metal catalysts are dispersed on supports like metal oxides and porous carbon materials? In this regard, Chen et al. recently showed that Rh-doped anionic clusters, RhCe2O3–5−, with Rh acting as the “active center” and Ce2O3–5 as the “support”, can effectively prompt NO reduction into N2 by introducing CO molecules.34
In this work, we report the study on the reduction of NO to N2 catalyzed by C60Rh4+ clusters, with those by free Rh4+ and C60Rh+ clusters as comparison. These clusters were chosen because (i) German et al. recently investigated the structures of C60Rhn+ (n = 1–6) clusters using infrared multiple photon dissociation spectroscopy and found that C60Rh4+ has a square Rh4+ moiety, different from the pyramidal structure for free Rh4+,35 while square Rh4+ has been shown to be more reactive for N–O dissociation than a triangular one28 and (ii) C60 has been shown to have a novel geometric and electronic effect in promoting catalytic reactions.36–39 We find that it is more energetically favorable for C60Rh4+ than C60Rh+ in catalyzing NO conversion, and the presence of C60 promotes O2 desorption, allowing the completion of the reaction cycle. We aim to gain insights into the nature of catalytic active sites and reaction mechanisms to guide the improvement of Rh-based catalysts for NO reduction.
Theoretical methods
All DFT calculations were carried out using the Gaussian 09 program40 package to investigate the structures of Rh4+, C60Rh+, and C60Rh4+ and their reaction mechanisms with NO. Three functionals, BPW91,41,42 PBE,43 and B3LYP,44,45 were tested for optimizing the structures of Rh4+, C60Rh+, and C60Rh4+. The 6-31G(d) basis set was used for N, O, and C, and the SDD46 basis set for Rh. It was found that PBE and BPW91 could well reproduce the structures of C60Rh+ and C60Rh4+ determined previously (see Fig. S1–S4 in ESI†).35 Since BPW91 has been found to be superior in simulating the infrared spectra of fullerene-metal clusters in terms of efficiency and accuracy,36,37,47,48 it was chosen to optimize all the geometries of transition states and intermediates during the catalytic NO conversion process. Harmonic frequency analysis was performed to make sure all transition states have only one imaginary frequency and the intermediates have none. Intrinsic reaction coordinate (IRC) calculations were conducted to validate the calculated transition state structures and to confirm the nature and stability of reaction pathways.49 The natural bond orbital (NBO) analysis was performed with NBO 3.1 as implemented in Gaussian 09.50Results and discussion
We first confirm that BPW91 can provide reliable structures for Rh4+, C60Rh+, and C60Rh4+ as determined from previous experiments (see Fig. S1–S4 in ESI†).35 The most stable structure of C60Rh4+ has a quartet sate with the four Rh atoms adsorbed on one hexagonal surface of fullerene, forming a square shape parallel to the hexagon, as shown in Fig. 1. The adsorbed NO molecule binds with C60Rh4+ through a Rh–N bond due to the high affinity of small rhodium clusters towards N atoms.20,25 This step forms the first intermediate IN1 with an exothermic energy of 55.7 kcal mol−1, smaller than the calculated binding energy of 88.8 kcal mol−1 between C60 and Rh4+ in C60Rh4+. Subsequently, the N–O bond undergoes dissociation, which is facilitated by the two adjacent Rh atoms. The energy barrier associated with this process is 49.6 kcal mol−1, resulting in the formation of a Rh–N bond and a Rh–O bond in IN2 (IN1 → TS1 → IN2). Then, a second NO molecule adsorbs on IN2, resulting in the formation of the intermediate IN3 via forming another Rh–N bond. This process exothermically releases an energy of 55.8 kcal mol−1, similar to the energy released by the first NO adsorption. Then, the N–O bond is cleaved by the other two adjacent Rh atoms. Thereafter, all four Rh sites are fully occupied by two N atoms and two O atoms (IN3 → TS2 → IN4). The energy barrier for this process is 56.8 kcal mol−1, about 7 kcal mol−1 higher than the first N–O bond dissociation, presumably due to the steric effect of the pre-adsorbed N and O atoms from the first NO molecule. This process behaves as the rate-determining step in the catalytic NO reduction by the C60Rh4+ cluster. | ||
| Fig. 1 Reaction pathway of NO reduction on C60Rh4+ calculated at the BPW91/6-31G(d)&SDD level of theory. R stands for reactants, IN for intermediates, TS for transition states, and P for products. All energies are provided relative to the energies of the reactants, i.e., C60Rh4+ and two NO molecules. | ||
Upon combination of the two dissociated nitrogen atoms on the “surface” of the rhodium cluster by overcoming a barrier of 14.3 kcal mol−1, the N2 moiety forms in a very stable intermediate IN5 that is 114.9 kcal mol−1 below the energy of the reactants. Subsequently, N2 desorbs from the cluster with an endothermic energy of 18.7 kcal mol−1, forming the intermediate IN6 (IN4 → TS3 → IN5 → IN6). Then, the two O atoms on the “surface” of the rhodium cluster interact with each other to form an O2 moiety in IN7 through a barrier of 11.9 kcal mol−1 (IN6 → TS4 → IN7). Afterwards, the O2 molecule desorbs by adsorbing an energy of 51.1 kcal mol−1, completing a reaction cycle for the direct reduction of NO to N2 catalyzed by C60Rh4+, i.e.,  . Overall, the calculation shows that this reaction has an exothermic energy of 42.2 kcal mol−1, in excellent agreement with the experimental value of 41.4 kcal mol−1.3 During the reaction, the four Rh atoms serve as the active sites for the activation and conversion of two NO molecules.
. Overall, the calculation shows that this reaction has an exothermic energy of 42.2 kcal mol−1, in excellent agreement with the experimental value of 41.4 kcal mol−1.3 During the reaction, the four Rh atoms serve as the active sites for the activation and conversion of two NO molecules.
To elucidate the important role of multiple Rh sites for NO reduction, we calculate the reaction pathway of two NO molecules reacting with a C60-fullerene-supported single rhodium atom, i.e., C60Rh+ (see Fig. S5, ESI†). It seems that although the reaction cycle can be completed, first and second N–O bond dissociations encounter significant energy barriers, amounting to 103.2 and 89.7 kcal mol−1, respectively. Such barriers are even larger than the calculated binding energy of 82.3 kcal mol−1 between C60 and Rh+ in C60Rh+,35 which may result in the destruction of the C60Rh+ cluster first before the reaction can proceed, precluding its usefulness in practice.
The above discussion raises an interesting question: What is the role of C60 in the reaction? Can the same reaction cycle be completed without the presence of C60? To answer these questions, we investigated the adsorption and dissociation pathway of two NO molecules on a free Rh4+ cluster and presented it in Fig. 2. The most stable structure of Rh4+ is a pyramid with a doublet state. The first NO molecule is adsorbed on Rh4+ through a Rh–N bond, forming the intermediate IN1 with an energy release of 62.1 kcal mol−1. Subsequently, the N–O bond undergoes dissociation facilitated by two adjacent Rh sites, resulting in the formation of a Rh–O bond and a Rh–N–Rh ring in IN2 (IN1 → TS1 → IN2). The energy barrier for this process is calculated to be 29.2 kcal mol−1, consistent with the calculated value of 27.7 kcal mol−1 for N–O bond dissociation of NO on Rh4+ reported by Romo-Ávila et al.28 Then, the cluster adsorbs a second NO molecule, forming a very stable intermediate, IN3, which is about 130 kcal mol−1 below the total energy of reactants and releases an energy of 68.3 kcal mol−1. Similar to the first NO dissociation, a second Rh–N–Rh ring and a Rh–O–Rh ring form in IN4 via TS2. The barrier for this step is 55.4 kcal mol−1, in agreement with the value of 57.7 kcal mol−1 reported for the second N–O bond dissociation.27 This step is the rate-determining step in the reaction of NO molecules with the Rh4+ cluster.
 | ||
| Fig. 2 Reaction pathway of NO reduction on Rh4+ calculated at the BPW91/6-31G(d)&SDD level of theory. R stands for reactants, IN for intermediates, TS for transition states, and P for products. All energies are provided relative to the energies of the reactants, i.e., Rh4+ and two NO molecules. | ||
In the following process, the two N atoms on the cluster surface migrate and encounter each other to combine to form a N2 moiety by overcoming an energy barrier of 39.8 kcal mol−1 (IN4 → TS3 → IN5). Subsequently, N2 desorbs from the cluster surface by absorbing 15.6 kcal mol−1 energy, resulting in the formation of the product Rh4O2+ (IN5 → TS4 → IN6 → P). Our calculated results on the reaction of Rh4+ with NO match the previous theoretical results on the reaction between NO and Rhn+ (n = 4, 6, or 7) clusters.26–28 However, due to the geometric constraint, as the two O atoms in Rh4O2+ are spatially distant, the combination of the two O atoms to form O2 is hindered at normal temperatures. For example, Torres et al. calculated the desorption energy of O2 from Rh6O2+, i.e., ΔE = E(Rh6+) + E(O2) − E(Rh6+O2), to be approximately 3.07 eV (70.8 kcal mol−1), almost 20 kcal mol−1 higher than that in the presence of C60.26 Such a process is expected to occur only at high temperatures, for instance, in the range of 1000–1400 K.18 These analyses highlight the unique role of C60 in facilitating the completion of the reaction cycle of NO + NO → N2 + O2.
To more properly uncover the geometric effect and electronic effect when comparing the catalytic performance of C60Rh4+ and Rh4+, we also calculated the reaction pathway of NO reduction on square Rh4+, a less stable isomer for the Rh4+ cluster but possessing a similar geometry of the Rh4+ moiety in C60Rh4+. Fig. S6 in the ESI† presents the result, and it can be seen that although it seems the NO reduction into N2 and O2 on square Rh4+ can be fulfilled, the energy barriers for the two-NO-bond cleavage are 54.5 and 61.6 kcal mol−1, both about 5 kcal mol−1 higher than the barriers on C60Rh4+. This clearly shows that the electronic effect is in play. Overall, both geometric and electronic effects play roles in the catalytic NO reduction by C60Rh4+, as the most stable Rh4+ becomes square from pyramidal when it is supported on C60, demonstrating the important support effect.
To uncover why C60 can have such a role in the catalytic reduction of NO to N2, we conducted natural bond orbital (NBO) analysis. Fig. 3 shows the calculated NBO partial charges of the Rh4+ moiety and C60 of NO reduction catalyzed by C60Rh4+, and the charge changes for the catalytic reactions of NO by pyramid Rh4+, square Rh4+ and C60Rh+ are shown in Fig. S7–S9 (ESI†), respectively. It can be seen that the metal moiety shows alternating charges increasing and decreasing due to the cooperative effect of the electrophilic reactant NO, its dissociated N and O atoms, and the C60 support. Before the formation of IN4, the C60 support progressively donates electrons to metal and the reactant and reaches a maximum positive charge of 0.33 e; afterwards, before releasing the N2 molecule (IN6), the charges on Rh4 and C60 show exactly reverse trends; then, the positive charge on C60 keeps decreasing, meaning that C60 has been accepting electrons from metal and/or the reactant. Thus, during the whole process, C60 alternatively donates and accepts electrons, behaving as an “electron sponge”, as we previously proposed.37,38 Specifically, for the O2 desorption process, the charge changes seem to reduce the interaction between Rh4+ and O atoms and simultaneously increase the binding energy between Rh4+ and C60. Hence, the presence of C60 enables the O atoms to become “surface” O rather than “lattice” O, making the desorption of the O2 molecule easier. In the case of free Rh4+, the reactant seems to extract electrons from metal due to their higher electron negativity compared to metal, thus increasing the binding strength between metal and reactant. Without the electronic buffering effect of C60, such an enhanced binding strength would make desorption of the formed molecules difficult.
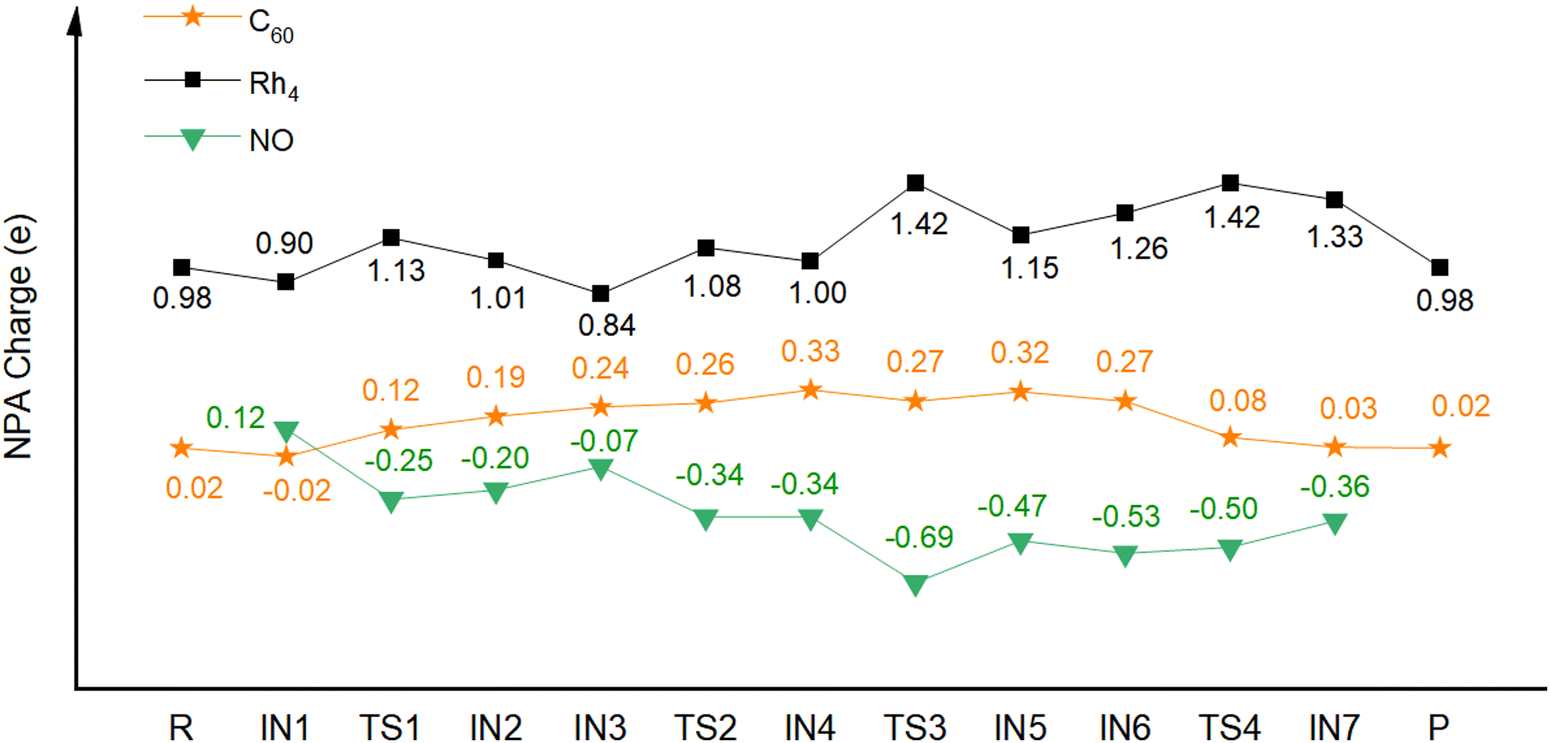 | ||
| Fig. 3 Calculated NBO partial charges (e) of the Rh4+ cluster (black square) and C60 (orange star) of NO reduction catalyzed by C60Rh4+. | ||
Conclusions
In summary, we studied the catalytic NO reduction into N2 by rhodium clusters, in particular Rh4+, with and without the presence of C60-fullerene support. The Rh atoms serve as the active sites for the reduction of two NO molecules, and the C60 support plays an important role in regenerating the Rh4+ catalyst with relatively smooth O2 desorption, which is difficult for free Rh4+ without the presence of C60 as it forms a stable rhodium oxide cluster, Rh4O2+. The introduction of C60 support not only induces a geometric transformation for the ground state of free Rh4+ from a trigonal pyramid to a square form but also can donate to or accept from the metal electrons during the NO catalytic reduction. This work demonstrates the electron sponge behavior of C60, and the ability of C60 to donate and accept charges at distinct steps within the reaction between NO and Rh clusters sheds light on the intricate electronic interactions involved, offering valuable insights for designing efficient catalysts in the realm of direct reduction of NO.Author contributions
G.-L. H. conceived and designed the research. R. L., Y.-K. L., and J. X. performed the research. R. L., Y.-L., and G.-L. H. wrote the manuscript. All authors approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (92261101), Innovation Capability Support Program of Shaanxi Province (2023-CX-TD-49), and Research Foundation–Flanders FWO (G033724N). G.-L. H. acknowledges the support from the Fundamental Research Funds for Central Universities, China. Part of the computational resources and services used in this work were provided by the VSC (Flemish Supercomputer Center), funded by the Research Foundation–Flanders (FWO) and Flemish Government–Department EWI.Notes and references
- H. Gandhi, G. Graham and R. W. McCabe, J. Catal., 2003, 216, 433–442 CrossRef CAS.
- J. M. Bakker and F. Mafuné, Phys. Chem. Chem. Phys., 2022, 24, 7595–7610 RSC.
- S. Roy, M. Hegde and G. Madras, Appl. Energy, 2009, 86, 2283–2297 CrossRef CAS.
- K. C. Taylor, Catal. Rev., 1993, 35, 457–481 CrossRef CAS.
- M. Shelef and G. Graham, Catal. Rev., 1994, 36, 433–457 CrossRef CAS.
- R. Baird, R. Ku and P. Wynblatt, Surf. Sci., 1980, 97, 346–362 CrossRef CAS.
- T. Root, L. Schmidt and G. B. Fisher, Surf. Sci., 1983, 134, 30–45 CrossRef CAS.
- P. Ho and J. White, Surf. Sci., 1984, 137, 103–116 CrossRef CAS.
- L. A. DeLouise and N. Winograd, Surf. Sci., 1985, 159, 199–213 CrossRef CAS.
- T. Root, G. B. Fisher and L. Schmidt, J. Chem. Phys., 1986, 85, 4679–4686 CrossRef CAS.
- T. Root, G. B. Fisher and L. Schmidt, J. Chem. Phys., 1986, 85, 4687–4695 CrossRef CAS.
- M. Bowker, Q. Guo and R. Joyner, Surf. Sci., 1991, 257, 33–40 CrossRef CAS.
- M. Van Tol and B. Nieuwenhuys, Appl. Surf. Sci., 1993, 67, 188–197 CrossRef CAS.
- H. Borg, J. J. Reijerse, R. Van Santen and J. Niemantsverdriet, J. Chem. Phys., 1994, 101, 10052–10063 CrossRef CAS.
- Y. J. Kim, S. Thevuthasan, G. S. Herman, C. H. Peden, S. A. Chambers, D. N. Belton and H. Permana, Surf. Sci., 1996, 359, 269–279 CrossRef CAS.
- D. Loffreda, D. Simon and P. Sautet, J. Chem. Phys., 1998, 108, 6447–6457 CrossRef CAS.
- A. Beniya, T. Koitaya, H. Kondoh, K. Mukai, S. Yoshimoto and J. Yoshinobu, J. Chem. Phys., 2009, 131, 084704 CrossRef PubMed.
- M. Hopstaken and J. Niemantsverdriet, J. Phys. Chem. B, 2000, 104, 3058–3066 CrossRef CAS.
- K. Sato, T. Yoshinari, Y. Kintaichi, M. Haneda and H. Hamada, Appl. Catal., B, 2003, 44, 67–78 CrossRef CAS.
- A. Arab, M. Nahali and F. Gobal, J. Alloys Compd., 2017, 695, 1924–1929 CrossRef CAS.
- A. Wang, J. Li and T. Zhang, Nat. Rev. Chem., 2018, 2, 65–81 CrossRef CAS.
- J.-C. Liu, Y. Tang, Y.-G. Wang, T. Zhang and J. Li, Nat. Sci. Rev., 2018, 5, 638–641 CrossRef CAS.
- X. Li, S. Mitchell, Y. Fang, J. Li, J. Perez-Ramirez and J. Lu, Nat. Rev. Chem., 2023, 7, 754–767 CrossRef CAS PubMed.
- M. L. Anderson, M. S. Ford, P. J. Derrick, T. Drewello, D. P. Woodruff and S. R. Mackenzie, J. Phys. Chem. A, 2006, 110, 10992–11000 CrossRef CAS PubMed.
- P. Ghosh, R. Pushpa, S. de Gironcoli and S. Narasimhan, J. Chem. Phys., 2008, 128, 194708 CrossRef PubMed.
- M. Torres, F. Aguilera-Granja, L. Balbás and A. Vega, J. Phys. Chem. A, 2011, 115, 8350–8360 CrossRef CAS PubMed.
- H. Xie, M. Ren, Q. Lei and W. Fang, J. Phys. Chem. A, 2011, 115, 14203–14208 CrossRef CAS PubMed.
- S. Romo-Ávila and R. Guirado-López, J. Phys. Chem. A, 2012, 116, 1059–1068 CrossRef PubMed.
- Y. Tawaraya, S. Kudoh, K. Miyajima and F. Mafune, J. Phys. Chem. A, 2015, 119, 8461–8468 CrossRef CAS PubMed.
- H.-Q. Yang, H.-Q. Fu, B.-F. Su, B. Xiang, Q.-Q. Xu and C.-W. Hu, J. Phys. Chem. A, 2015, 119, 11548–11564 CrossRef CAS PubMed.
- F. Mafuné, M. Takenouchi, K. Miyajima and S. Kudoh, J. Phys. Chem. A, 2016, 120, 356–363 CrossRef PubMed.
- T. Nagata, K. Kawada, X. Chen, M. Yamaguchi, K. Miyajima and F. Mafuné, Phys. Chem. Chem. Phys., 2021, 23, 26721–26728 RSC.
- Y.-X. Zhao, X.-G. Zhao, Y. Yang, M. Ruan and S.-G. He, J. Chem. Phys., 2021, 154, 180901 CrossRef CAS PubMed.
- J.-J. Chen, S.-D. Wang, Z.-Y. Li, X.-N. Li and S.-G. He, J. Am. Chem. Soc., 2023, 145, 18658–18667 CrossRef CAS PubMed.
- E. German, G.-L. Hou, J. Vanbuel, J. M. Bakker, J. A. Alonso, E. Janssens and M. J. López, Carbon, 2022, 197, 535–543 CrossRef CAS.
- G.-L. Hou, T. Yang, M. Li, J. Vanbuel, O. V. Lushchikova, P. Ferrari, J. M. Bakker and E. Janssens, Angew. Chem., Int. Ed., 2021, 60, 27095–27101 CrossRef CAS PubMed.
- M. Li, T. Yang, J. M. Bakker, E. Janssens and G.-L. Hou, Cell Rep. Phys. Sci., 2022, 3, 100910 CrossRef CAS.
- G.-L. Hou and E. Janssens, Fundam. Res., 2023 DOI:10.1016/j.fmre.2023.10.011.
- J. Zheng, L. Huang, C. H. Cui, Z. C. Chen, X. F. Liu, X. Duan, X. Y. Cao, T. Z. Yang, H. Zhu, K. Shi, P. Du, S. W. Ying, C. F. Zhu, Y. G. Yao, G. C. Guo, Y. Yuan, S. Y. Xie and L. S. Zheng, Science, 2022, 376, 288–292 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision E.01, Gaussian, Inc, Wallingford CT, 2013 Search PubMed.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS PubMed.
- J. P. Perdew, in Electronic Structure of Solids '91, ed. P. Ziesche and H. Eschrig, Akademie Verlag, Berlin, 1991, p. 11 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- D. Andrae, U. Haeussermann, M. Dolg, H. Stoll and H. Preuss, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS.
- J. Xu, J. M. Bakker, O. V. Lushchikova, P. Lievens, E. Janssens and G.-L. Hou, J. Am. Chem. Soc., 2023, 145, 22243–22251 CrossRef CAS PubMed.
- J. Xu, A. Li, X. Li and G.-L. Hou, Mon. Not. R. Astron. Soc., 2023, 525, 3061–3074 CrossRef CAS.
- K. Fukui, Acc. Chem. Res., 1981, 14, 363–368 CrossRef CAS.
- A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735–746 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: calculated isomers of Rh4+, C60Rh4+, and C60Rh+; additional reaction pathways; calculated NBO partial charges; cartesian coordinates. See DOI: https://doi.org/10.1039/d4cp01398k |
| This journal is © the Owner Societies 2024 |