
A molecular dynamics simulation study of thermal conductivity of plumbene
Rafat
Mohammadi
 *a,
Behrad
Karimi
a,
John
Kieffer
b and
Daniel
Hashemi
c
*a,
Behrad
Karimi
a,
John
Kieffer
b and
Daniel
Hashemi
c
aDepartment of Mechanical Engineering, Faculty of Engineering, Arak University, Arak 38156-88349, Iran. E-mail: r-mohammadi@araku.ac.ir
bDepartment of Materials Science and Engineering, University of Michigan, Ann Arbor, MI 48109, USA
cDepartment of Physics and Optical Engineering, Rose-Hulman Institute of Technology, IN 47803, USA
First published on 31st October 2024
Abstract
We investigate the lattice thermal conductivity of plumbene using molecular dynamics simulations, overcoming existing limitations by optimizing the parameters of Tersoff and Stillinger–Weber potentials via artificial neural networks. Our findings indicate that at room temperature, the thermal conductivity of a 1050 Å × 300 Å plumbene sheet is approximately 8 W m−1 K−1, significantly lower (23%) than that of bulk lead. Our analysis elucidates that thermal conductivity is enhanced by increased sample length, while it is reduced by temperature. Moreover, plumbene samples with zigzag edges display superior thermal conductivity compared to those with armchair edges. In addition, the thermal conductivity of plumbene exhibits an increase at low tensile strains, whereas it decreases as the strains become larger. This investigation provides crucial insights into the thermal conductivity behavior of plumbene under varying conditions.
1. Introduction
Materials scientists are tirelessly working towards the development of advanced materials to meet the demands of future technological applications. Among these materials, two-dimensional (2D) materials, also referred to as monolayers or single-layer materials, exhibit exceptional physical properties that set them apart from their bulk counterparts. Graphene, the pioneering 2D material, stands out for its remarkable stability, conductivity, and flexibility.1 Plumbene, a lead-based counterpart to graphene, has recently emerged as a subject of intense interest due to its unique structure and potential for electronic applications.2–9Both graphene and plumbene possess a hexagonally arranged structure, but plumbene has a slightly buckled configuration, leading to increased overlap between its Sigma (r) and pi (p) orbitals. Plumbene presents three stable structures: the planar form, as well as the high-buckled (HB) and low-buckled (LB) honeycomb structures. Notably, the buckled configurations exhibit greater stability compared to the planar structure. The low-buckled plumbene has a lattice constant of 4.934 Å and a buckling height of 0.99 Å, whereas the high-buckled plumbene measures 3.692 Å and 2.55 Å for its lattice constant and buckling height, respectively.3,6 In a noteworthy experiment conducted by Yuhara et al. in 2019, low-buckled honeycomb structures of plumbene were initially synthesized.7
Plumbene holds immense promise as a versatile platform for fabricating novel topological insulators and large-gap quantum spin Hall (QSH) insulators.2–6 Furthermore, it has emerged as a potential candidate for hydrogen storage.9 With its distinct properties and growing scientific interest, plumbene represents an exciting avenue for exploring cutting-edge technological advancements. However, despite its potential, little is known about the different properties of plumbene. Lu et al.3 used Ab initio density functional theory (DFT) calculations to investigate the material's electronic and structural properties of functionalized atomic lead films. They demonstrated that the 2D Pb structure can have applications in topological electronic devices. Li et al.6 investigated electronic and topological properties of plumbene by combining tight-binding models with first-principles calculations. Constructive coupling effects of topological states found in the low-buckled plumbene, causing the system to be a normal insulator, opposite to topologically nontrivial states formed in other 2D group IVA monolayers (from graphene to stanene). Bechstedt et al.10 used a four-band model to investigate the thermal and electrical conductance of 2D electron gases in doped Xenes, including graphene, silicene, germanene, stanene, and plumbene. They showed that thermal conductance increases linearly or quadratically with temperature. Mahdavifar et al.11 used first-principles calculations to examine the electronic and mechanical properties of single-layer plumbene in different armchairs, zigzag, and biaxial directions under applying external strain and electric field and in the presence of a Stone–Wales defect. They found that the plumbene monolayer exhibits the lowest Young's modulus and ideal strength among group IVA mono-elemental monolayers. This is attributed to its relatively weaker bond length compared to its counterparts. They also suggested that plumbene can be used in flexible electronics and is a promising candidate for the quantum spin hall effect when its electronic properties are tuned using external fields. Zhang et al.12 calculated the superconducting critical temperature of HB plumbene using first-principles calculations. They showed that the critical temperature of HB plumbene can be tuned by adjustment of Fermi level.
Complementing the aforementioned first-principle calculations and experimental/theoretical research on plumbene, two distinct studies have leveraged molecular dynamics (MD) simulations to elucidate the properties of plumbene. Das et al.13 investigated the mechanical properties of plumbene, including yield strength, ultimate tensile strength, and Young's modulus using MD simulations and studied the effects of sample size, temperature, and strain rate on these properties. Das et al.14 also carried out MD simulations to predict the thermal properties of plumbene and studied the effects of sample size on thermal conductivity. This group examined the thermal conductivity of plumbene for samples with widths of 300 Å and lengths ranging from 600 Å to 1050 Å. Their results showed that plumbene thermal conductivity fluctuates between 0.04 W m−1 K−1 and 0.24 W m−1 K−1 for different sample lengths. The embedded atom method (EAM) potential with the parameters of the bulk lead was used in these two MD simulations of plumbene.
Tersoff and Stillinger–Weber (SW) potentials are the most common potentials used in previous MD simulations on other 2D materials of group IVA.15–20 However, the parameters of these potentials have not yet been determined for lead or plumbene. Therefore, Das et al. used the available EAM potential of bulk lead for MD simulation of plumbene and no details about the structural parameters such as the lattice constant, bond length, and buckling height of the simulated plumbene have been mentioned in their research.13,14 In addition, previous experimental observations15 and MD simulations15–20 of the thermal conductivity of other 2D materials of group IVA have shown that the thermal conductivity of the 2D materials of group IVA increases with increased length. While the simulated thermal conductivity of plumbene fluctuates with increased length in Das et al. research.14
In this study, we tackle the challenges associated with plumbene MD simulations by optimizing the necessary parameters of Tersoff and SW potentials using artificial neural networks (ANNs). Neural networks have demonstrated efficacy in predicting empirical interatomic potential parameters. Bukkapatnam et al.21 developed a method that combines the universal function approximation capability of multilayer neural networks to accelerate a genetic algorithm for fitting atomic system potentials. They applied this method to determine the interatomic potential parameters for silicon using the Tersoff functional form. The concept of integrating traditional interatomic potentials with neural networks was explored by Malshe et al.,22 who proposed a method for fitting the parameters of any arbitrary empirical potential to a database and constructed an adjustable Tersoff potential for a Si5 cluster. Pun et al.23 demonstrated significant improvements in the transferability of machine-learning potentials by combining a general physics-based model, specifically the analytical bond-order potential, with neural network regression. This approach was exemplified by the development of a general-purpose physically informed neural network potential for aluminum. Nwachukwu and Winczewski24 applied neural networks to develop a new parametrization of the Tersoff potential for penta-graphene, achieving a parameterization with enhanced accuracy and reduced relative errors in the calculated structural and mechanical properties compared to the original parameterization.
Furthermore, we employed non-equilibrium molecular dynamics (NEMD) simulations with the optimized potentials to investigate the lattice thermal conductivity of plumbene sheets with both armchair and zigzag configurations. Our simulations explore the impact of sample length, temperature, and tensile strain on plumbene's thermal conductivity.
It is worth noting that previous theoretical predictions in ref. 3, 4, 6 and 11 have classified low-buckled plumbene as a topologically trivial insulator. Considering the energetic stability and electronic properties, our study primarily focuses on analyzing the thermal conductivity of the low-buckled plumbene.
Plumbene's distinctive structure and spin–orbit interaction position it as a promising candidate for various electronic applications. However, to fully comprehend its thermal and mechanical properties, further research is required. MD simulations prove to be a valuable tool in this endeavor. Hence, our study aims to contribute to the understanding of plumbene's thermal conductivity under different conditions.
2. Simulation methods
In this study, various techniques were employed to investigate the thermal conductivity of plumbene. First, Ab initio molecular dynamics simulations were carried out to reproduce the buckling structure of plumbene. The necessary parameters of the SW and Tersoff potentials for plumbene were then optimized using an ANN. Finally, NEMD simulations were conducted to calculate the thermal conductivity of plumbene using the optimized SW and Tersoff potentials. The details of each of these methods are discussed in further detail in the following sections.2.1. Potential optimization
The Tersoff and SW potentials are widely recognized empirical approaches extensively employed in the field of MD simulations. These potentials have been developed based on many-body interactions and have been carefully calibrated using a variety of data sources, including Ab initio calculations, DFT, artificial intelligence, and experimental results. In the realm of MD simulations involving group IVA 2D materials, these potentials are among the most frequently used potentials.15–20 However, these potentials have not been previously utilized or optimized specifically for lead or plumbene.In this study, we focus on optimizing the parameters of the Tersoff and SW potentials specifically for plumbene. To achieve this, we begin by conducting Ab initio molecular dynamics simulations to accurately reproduce the buckling structure of plumbene. Subsequently, we employ an in-house code to train a multi-layered perceptron neural network for optimizing the Tersoff and SW potentials parameters.
Here, our Ab initio MD calculation shows that at room temperature the bond length is 2.99 Å, the lattice constant is 4.98 Å, and the buckling height is 1.06 Å, which are comparable with previous research.3,6
| R (Å) | 3.70 |
| B (eV) | 4.30 × 102 |
| A (eV) | 1.8501 × 103 |
| d | 1.62137 × 101 |
| n | 7.2402 × 10−1 |
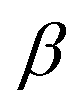
|
3.70 × 10−7 |
| h | −3.1998 × 10−1 |
| c | 3.016429 × 105 |
| D (Å) | 1.27016895 × 10−1 |
| λ 1 (1/Å) | 2.445200282 |
| λ 1 (1/Å) | 1.70 |
| ε (eV) | 1.95 |
| a | 1.80 |
| γ | 1.20 |
| p | 4.00 |
| λ | 3.10 × 101 |
| σ(Å) | 2.551 |
| A | 7.149556277 |
cos![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) θ0 θ0 |
−3.13992456 × 10−1 |
| B | 6.122245584 × 10−1 |
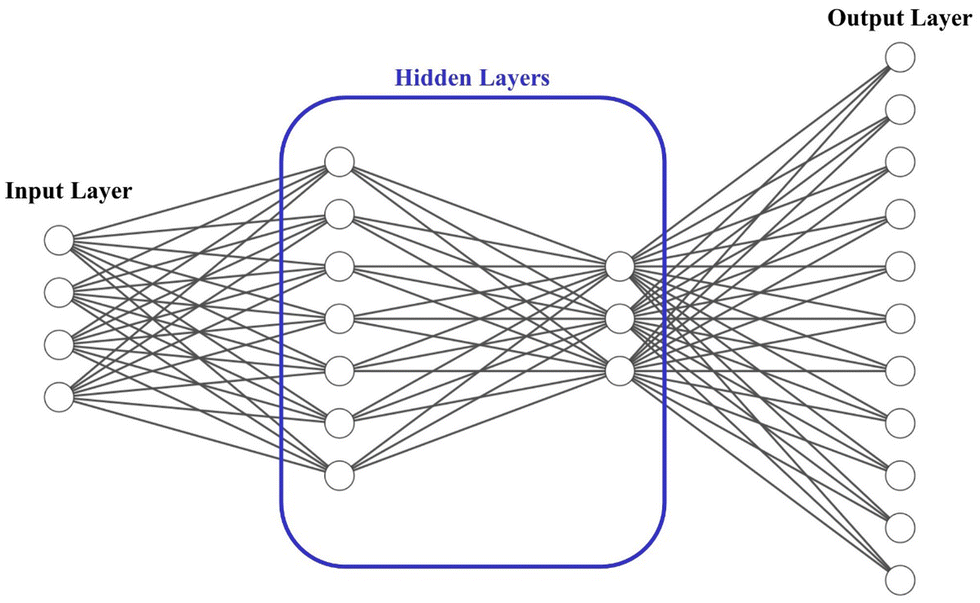 | ||
| Fig. 1 Designed neural network schematic. | ||
Initially, we generated parameters of Tersoff and SW potentials using random values. Subsequently, we performed MD simulations with these parameters to compute the structural properties of plumbene. These simulation results were then used as inputs for the ANN. This methodology is based on the approach outlined by Nwachukwu and Winczewski24 for parametrizing the Tersoff potential in penta-graphene using neural networks.
After evaluating various ANN configurations, we achieved the optimal balance between model complexity and accuracy by employing 2 hidden layers, with the first layer consisting of 7 nodes and the second layer comprising 3 nodes, utilizing the sigmoid tangent function. For the ANN training, the reference set was randomly divided into a training set (80% of the structures), an independent testing set (10%), and a validating set (10%) that was not used for the potential fitting. The training progress with optimization iterations is monitored through the evolution of the root mean squared error (RMSE), the mean absolute error (MAE), and the coefficient of determination (R2). The Levenberg–Marquardt algorithm was employed to design the network,31 with a specific number of iterations carried out to obtain optimal potential parameters. For both potentials, the MAE was low and R2 was close to 1 for all training, testing, and validating datasets. These results indicate the good generalization of the developed model and its capability to predict the potential parameters.
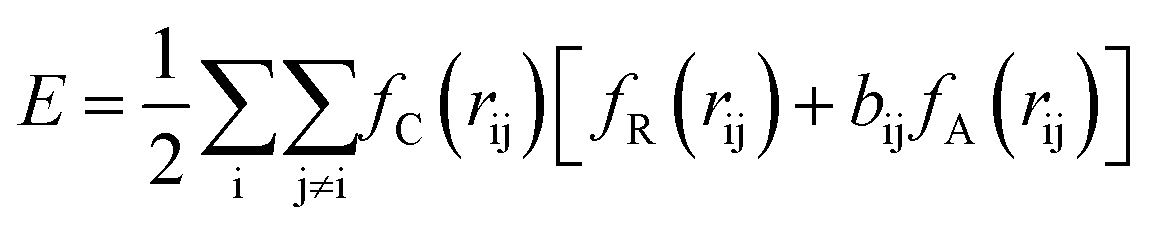 | (1) |
The function fC(rij) is the cut-off function that limits the range of interaction to the nearest neighbors. It is defined as:
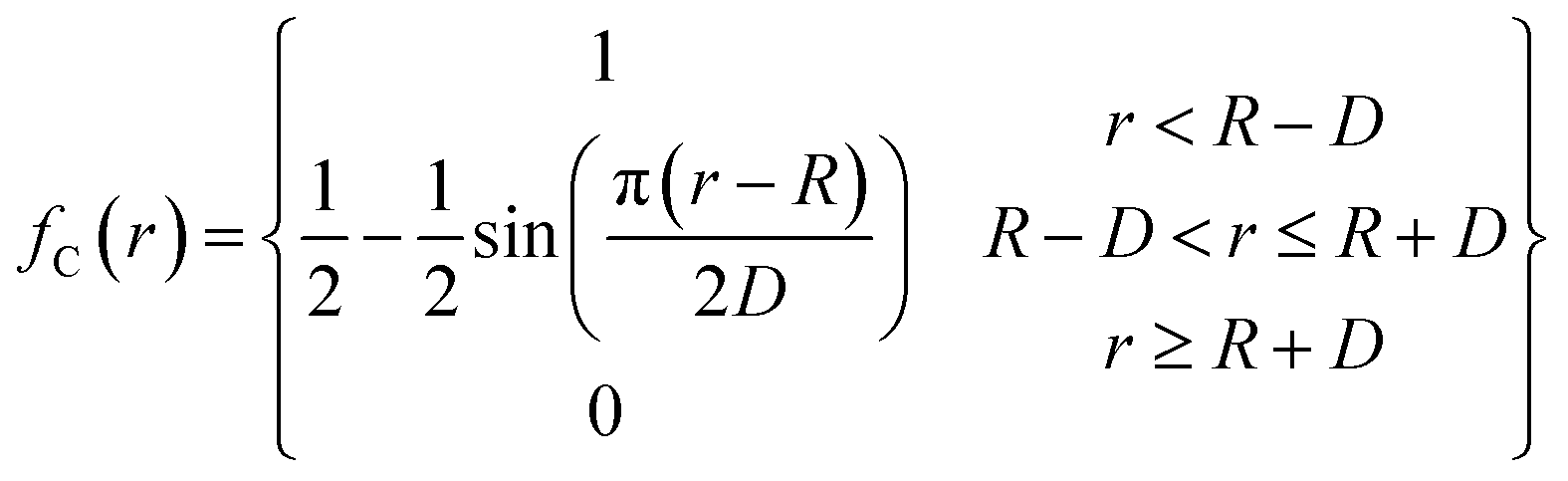 | (2) |
| fR(r) = Ae−λ1r | (3) |
| fA(r) = −Be−λ2r | (4) |
The term bij in eqn (1) describes the bond-order for a pair of atoms i–j, which describes the weakening of an i–j bond owing to the presence of the other bond i–k around atom i. This term explicitly accounts for angular interactions viaeqn (5–7):
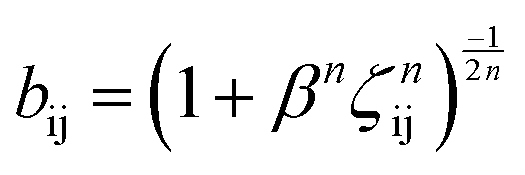 | (5) |
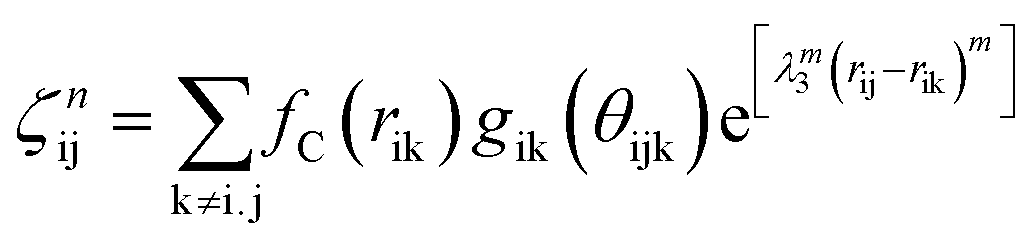 | (6) |
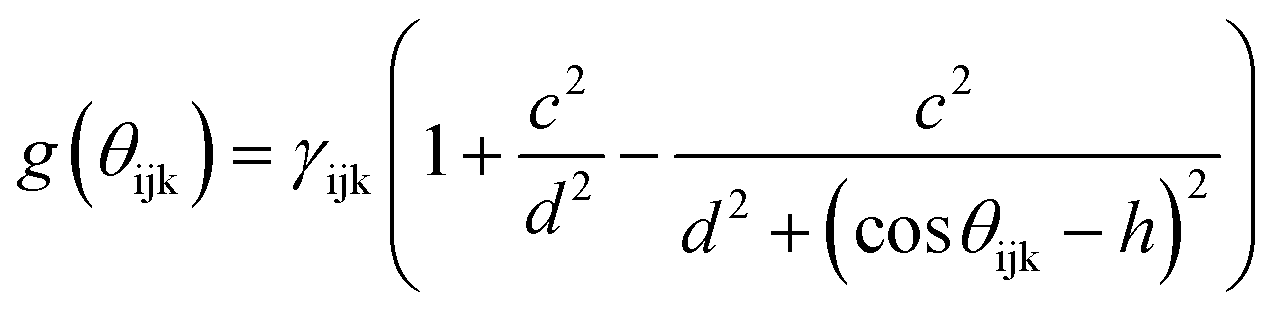 | (7) |
The parameters B, n, λ1, λ2, γ, c, d, and h are adjustable, while m = 3.0, λ2 = 0 and γ = 1.0 are kept constant. In all, the 11 independent parameters have been optimized. The calculated parameters by ANN are (h.R.D.A.B.λ1.λ2.β.n.c.d) and shown in Table 1.
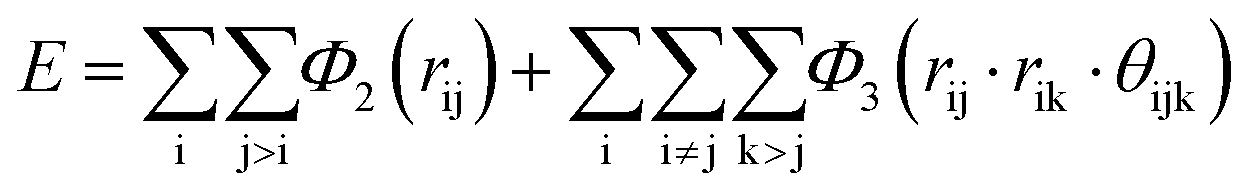 | (8) |
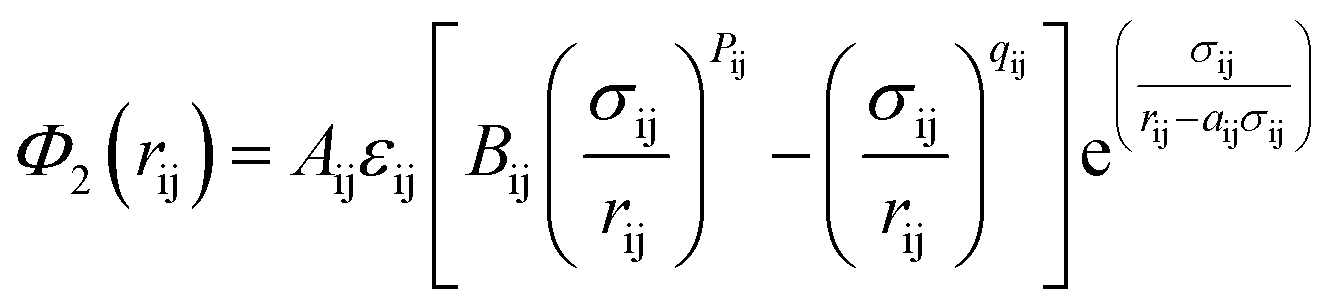 | (9) |
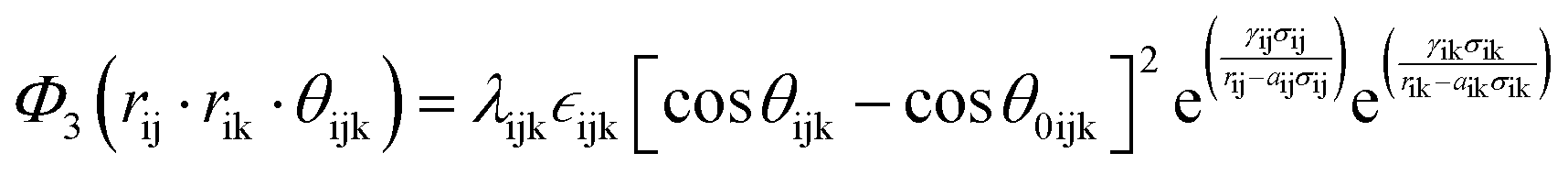 | (10) |
As indicated in eqn (9) and (10); to optimize the SW potential, the parameters ε, a, γ, A, σ, λ, cosθ0, B, and p could be calculated, while q = 0 is held constant. These parameters are optimized using the neural network and their values are shown in Table 2.
2.2. MD simulations
The large-scale atomic/molecular massively parallel simulator (LAMMPS)35 was used to perform molecular dynamics simulations, while Avogadro36 was used for atom visualization. NEMD simulation was employed to simulate thermal conductivity of plumbene using the optimized SW and Tersoff potentials. A view of the plumbene atomic structure is shown in Fig. 2(A). In this figure, the hexagonal structure of zigzag plumbene has been illustrated. Fig. 2(B) depicts the top and side views of LB plumbene. The buckling height, h, is also shown in Fig. 2(B). | ||
| Fig. 2 (A) Three-dimensional view of plumbene sheet. (B) The top and side views of buckled plumbene. | ||
Fig. 3 displays the simulation model for the thermal conductivity calculations. The simulation domain includes three regions, which are fixed walls, reservoir regions and the normal simulation region. At the two ends of the model, fixed atoms construct two fixed walls to prevent the atoms escaping from the system. Adjacent to the fixed walls, the hot and cold reservoirs (heat source and heat sink) are established to create the temperature gradient. When the simulation structure is in an equilibrium state, the velocities of the atoms in the reservoir regions are adjusted artificially in order to introduce a constant heat flux into the normal simulation region, which is located between the heat source and heat sink. At each time step, the velocities of the particles in the heat source and heat sink are scaled by the same factor so that the net kinetic energy is added or reduced with the same amount from the two reservoirs. This way, a constant heat flux is introduced into the simulation domain and a temperature gradient can be established along the heat flow direction for the simulation time. From the heat flow and the temperature gradient, the thermal conductivity of the plumbene can be obtained from the Fourier law. The model is divided into numerous slabs with a length of 20–25 Å along the X axis direction to evaluate the local temperatures.
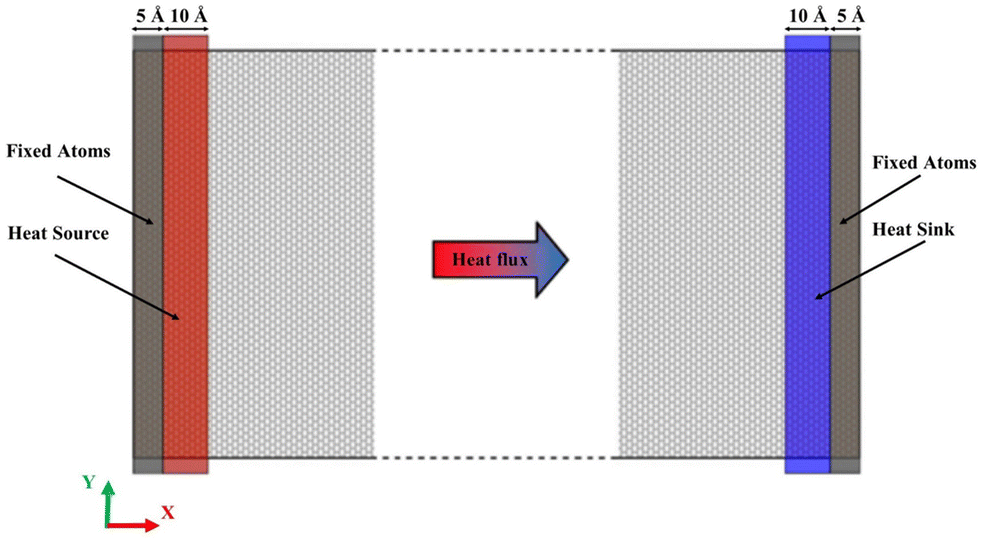 | ||
| Fig. 3 The simulation model for the NEMD calculations of the thermal conductivity. | ||
A time step of 1 fs was used in all simulations to simulate the thermal conductivity of plumbene. The simulation box was first relaxed in the NVT (number of particles, volume, and temperature) ensemble for 100 ps. The heat source and heat sink were then controlled by Langevin thermostats at constant temperatures, and finally by the NVE (Microcanonical (ensemble to calculate the heat flux along the plumbene until the temperature profile becomes linear over 1 ns. To calculate the in-plane thermal conductivity of plumbene, the Fourier law was applied using eqn (11).
 | (11) |
To comprehensively assess the interaction between lead atoms, we employ three different potentials. Firstly, we utilize the optimized SW and Tersoff potentials, which have been specifically optimized for this study. These potentials have been carefully calibrated to accurately capture the behavior of plumbene. Additionally, we incorporate the EAM potential used by Das et al.14 to explore its capability in reproducing the characteristic buckling hexagonal structure of plumbene. By employing these three distinct potentials, we aim to thoroughly analyze and understand the behavior of plumbene, allowing us to gain valuable insights into its structural properties.
In this study, we investigate the effects of temperature, strain, length, and width on the thermal conductivity of plumbene. To assess the impact of length on thermal conductivity, we vary the length from 300 Å to 1050 Å for a 300 Å wide sheet at 300 K. To study how width affects thermal conductivity, we use sheets with a fixed length of 300 Å and vary the width from 300 Å to 700 Å at room temperature. To examine the temperature dependence of thermal conductivity, we also computed the thermal conductivity with varying temperatures in the range of 300 K to 600 K while the size was kept fixed at 300 Å × 300 Å. Finally, to induce tensile strain, we move one fixed region at 0.5 Å ps−1 after equilibrium and expand the sample to the desired amount.
3. Evaluation of the potentials parameters optimization
To evaluate the accuracy of the optimized potentials parameters, we compare the structural properties of LB plumbene, which includes the average buckling height, average bond length, average lattice constant, and average atom bond angle obtained from Ab initio MD simulations, with the corresponding values calculated using the optimized potentials. The calculated structural parameters for LB plumbene at room temperature are tabulated in Table 3. Our computed structural parameters, obtained with the optimized SW and Tersoff potentials in MD simulations, demonstrate consistency with the results of Ab initio MD simulation. Furthermore, these parameters are compared with previously published results,2–4,6,9 showing good agreement.| Lattice constant (Å) | Bond length (Å) | Buckling height (Å) | Atom bond angle | |
|---|---|---|---|---|
| Current study, Ab initio MD simulation | 4.98 | 2.99 | 1.06 | 108.30° |
| Current study, MD simulation with optimized Tersoff potential | 4.81 | 2.96 | 1.07 | 109.73° |
| Current study, MD simulation with optimized SW potential | 4.93 | 2.99 | 0.99 | 108.85° |
| Zhao et al. (2016)2 | 4.93 | 3.00 | 0.93 | — |
| Lu et al. (2016)3 | 4.93 | — | — | — |
| Yu et al. (2017)4 | 4.93 | 3.00 | — | 108.34° |
| Li et al. (2019)6 | 4.93 | — | 0.99 | — |
| Vivek et al. (2021)9 | 5.08 | 3.09 | 0.95 | 108.06° |
The structural parameters of LB plumbene within the temperature range of 100 K to 500 K were also determined through Ab initio MD simulations. These calculated parameters were then compared with the results obtained from MD simulations, as depicted in Fig. 4. The graphs illustrate that all structural parameters exhibit an increase with rising temperature. Moreover, the NEMD simulations conducted using the optimized SW and Tersoff potentials yield consistent results with the Ab initio MD simulations. Notably, the SW potential demonstrates a closer correspondence to the Ab initio results, with maximum errors of only 0.4%, 1.08%, and 8% for the bond length, lattice constant, and buckling height, respectively. On the other hand, the Tersoff potential yields slightly larger discrepancies, with maximum errors of 1.5%, 3.8%, and 7% for the same parameters.
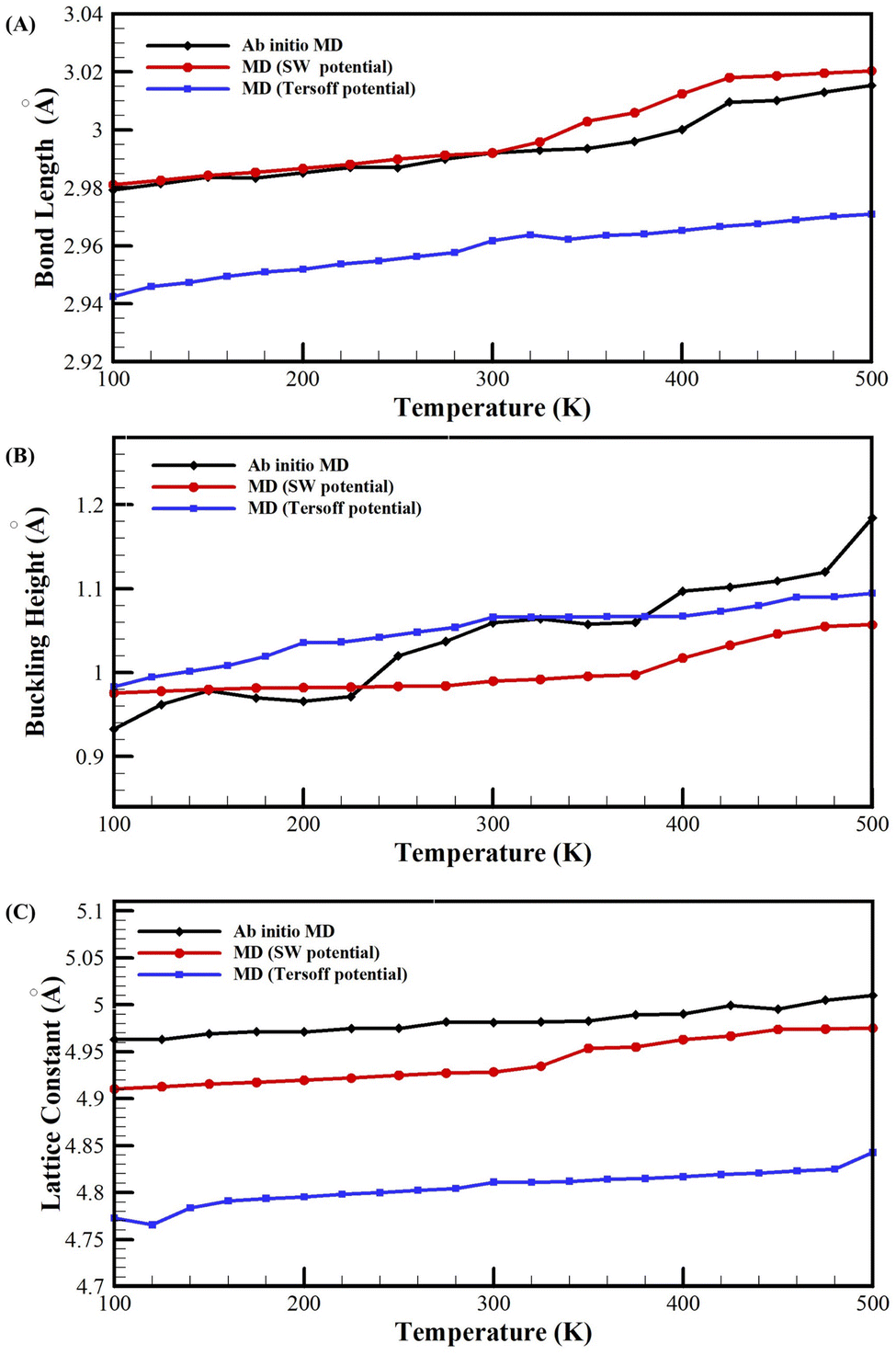 | ||
| Fig. 4 (A) The bond-length, (B) Buckling-height, and (C) Lattice-constant as a function of temperature from MD simulations with different potentials and Ab initio MD simulations. | ||
The findings presented in Table 3 and Fig. 4 affirm the consistency of the optimized SW and Tersoff potentials with the actual structure of LB plumbene. Furthermore, the optimized SW potential outperforms the optimized Tersoff potential in terms of accuracy.
The calculated phonon dispersion results of LB plumbene along the high-symmetry lines within the Brillouin zone obtained by both Tersoff and SW potentials are shown in Fig. 5, along with the phonon density of states (DOS). The primitive unit cell of plumbene consists of two atoms, resulting in three acoustic phonon modes (flexural out-of-plane acoustic (ZA), transverse acoustic (TA), longitudinal acoustic (LA)) and three optical phonon modes (flexural out-of-plane optical (ZO), transverse optical (TO), and longitudinal optical (LO)). These modes are marked near the Brillouin zone center (Γ point). It can be found that all branches have positive frequencies and no imaginary phonon modes, indicating that the structure of LB plumbene is thermodynamically stable.
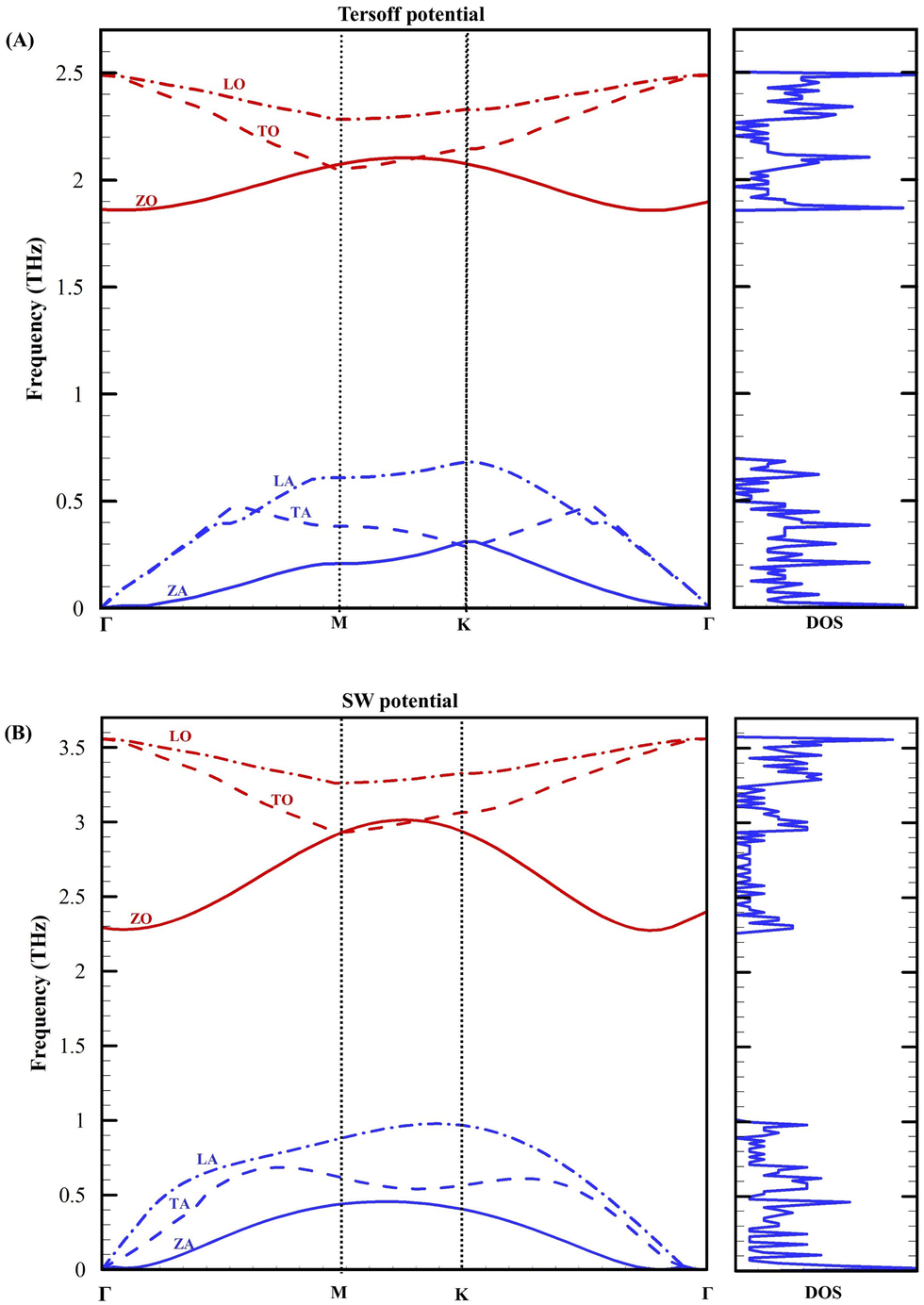 | ||
| Fig. 5 Phonon dispersion of LB plumbene along Γ–K–M–Γ direction and corresponding phonon DOS. (A) Tersoff potential, (B) SW potential. | ||
The calculated phonon dispersions of plumbene align with those of other 2D materials in group IVA37–41 due to their similar honeycombed lattice structures. In addition, they align with the reported phonon dispersions of plumbene obtained from first-principles calculations.42 These alignments confirm the accuracy of the optimized SW and Tersoff potentials. Specifically, acoustic frequencies are zero at the Γ point, and the ZA phonon mode displays quadratic dispersion, consistent with elastic theory,43 while the LA and TA branches exhibit linearity near the Γ point. For both potentials, the maximum frequency of acoustic modes is at K point and the minimum optical frequency is around the Γ point. Additionally, the acoustical and optical branches are well separated, similar to other structures of silicene, germanene, and stanene.40 In the gap frequencies between acoustic and optical modes, the phonon DOS is zero as shown in Fig. 5. The vibrational origin of the acoustic-optical gap, also known as the phonon bandgap, can be attributed to the buckled structure. As a result of the large acoustic-optical gap and the bunching of the acoustic phonon branches, phonon scattering significantly reduces.
Due to its heavier atomic mass and weaker bonding compared to other group IVA materials, plumbene exhibits lower phonon frequencies. In germanene, the highest phonon frequency is nearly equal to 300 cm−1 (9 THz), and in stanene, it is 180 cm−1 (5.4 THz).40 For plumbene, the highest phonon frequency is reported to be almost 15 meV (3.6 THz).42 Our simulation results for plumbene indicate that for the Tersoff potential, the highest phonon frequency is equal to 2.5 THz, and for the SW potential, it is equal to 3.5 THz, which is in good agreement with the reported results.42
The phonon bandgap for germanene and stanene is approximately between 50–70 cm−1 (1.5–2.1 THz),40 and for plumbene, it is 5.2 meV (1.3 THz).42 In agreement with these results, our simulation results for plumbene show that the phonon bandgap for both potentials is around 1.3 THz. In addition, increased ZA phonon scattering occurs for plumbene compared to germanene and stanene,40 due to the greater breaking of out-of-plane symmetry selection rules. These findings suggest that plumbene may exhibit the lowest lattice thermal conductivity among 2D materials in group IVA.
4. Results and discussion
The present study focuses on investigating the lattice thermal conductivity of plumbene using the Tersoff and SW potentials with the optimized parameters. Furthermore, we explore the influence of sample size, temperature, and strain on the thermal conductivity for different edge shapes of plumbene sheets.Fig. 6 illustrates the results for a plumbene sheet with a width of 300 Å and varying lengths at 300 K. Notably, the thermal conductivity values obtained from the SW potential tend to be higher than those from the Tersoff potential, which is consistent with its higher phonon frequencies observed in the phonon dispersions discussed in the previous section. However, since there are no experimental measurements available for plumbene's thermal conductivity, it is challenging to determine which set of outcomes is more accurate.
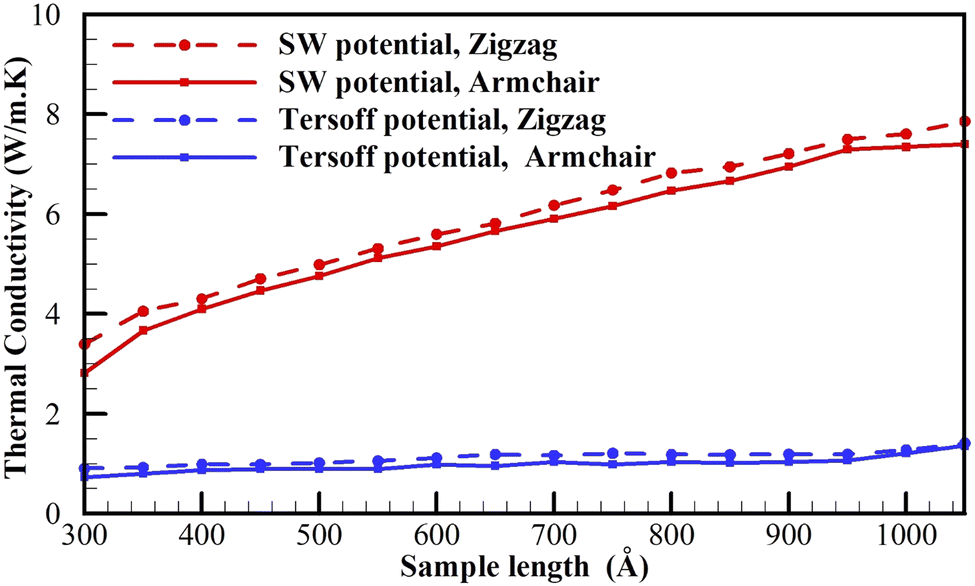 | ||
| Fig. 6 The thermal conductivities of plumbene sheets with a width of 300 Å as a function of length at 300 K. | ||
The observed thermal conductivity of plumbene falls within the range of 0.7–8 W m−1 K−1 at room temperature, which is considerably smaller than that of bulk lead (34.7 W m−1 K−1). Similar reductions in thermal conductivity have been observed in other 2D materials, such as silicene compared to bulk silicon44 and stanene compared to bulk tin.45 This substantial reduction in thermal conductivity can be attributed to increased phonon-surface scattering in low-dimensional semiconducting nanostructures.
Previous studies, utilizing density functional theory and many-body perturbation theory, have shown that electronic thermal conductivity in graphene at room temperature typically constitutes only around 10% of the total thermal conductivity of bulk graphene.46 While phonon thermal conductivity is dominant in graphene, constituting more than 90% of the total, the balance between electron and phonon mechanisms in other 2D materials remains uncertain and subject to debate. Some researchers have claimed that phonons are the primary heat carriers in 2D materials, both planer and buckled structures.19 However, other studies suggest that in some 2D materials like stanene, electronic thermal conductivity is significantly important, and it can even become dominant at room temperature.47 Consequently, it is plausible that plumbene may exhibit higher total thermal conductivity than our predictions for its lattice thermal conductivity.
Fig. 6 also highlights that, in general, the thermal conductivity of plumbene increases with sample length for both the Tersoff and SW potentials. Previous studies on other group IVA 2D materials have reported similar trends of thermal conductivity increasing with sample length.15–20,44,48 In addition, Fig. 6 shows that plumbene sheets with zigzag edges exhibit higher thermal conductivity than those with armchair edges implying a universal edge-shape dependence of thermal conductivity in the 2D-materials sheets.49–51
Fig. 7 depicts the variation of the thermal conductivity of plumbene with temperature for a 300 Å × 300 Å sample. This figure shows that increasing the temperature above 300 K, leads to a reduction in thermal conductivity for both armchair and zigzag configurations at almost similar rates. This is a common trend observed in other 2D materials.17,20,49 This phenomenon can be attributed to the Umklapp phonon–phonon scatterings. The obtained results show that the thermal conductivity of the zigzag configuration, obtained from SW potential, decreases from 3.4 W m−1 K−1 to 2 W m−1 K−1 when the temperature increases from 300 K to 600 K. Moreover, as shown in Fig. 7, zigzag configurations exhibit a higher thermal conductivity than the armchair configurations at all temperatures, which is in agreement with the behavior of other 2D nanostructures49,51
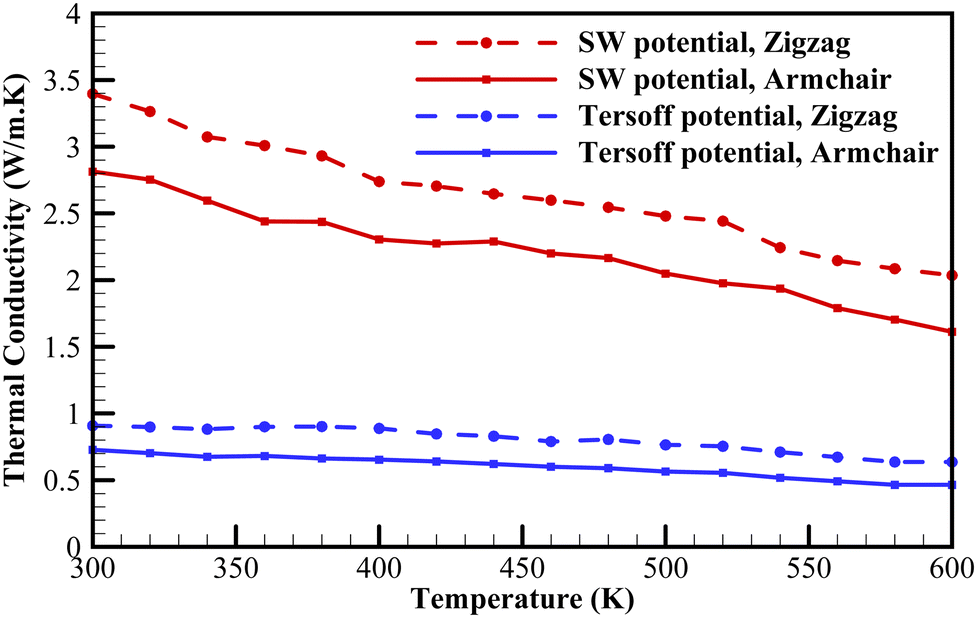 | ||
| Fig. 7 The thermal conductivities of a 300 Å × 300 Å plumbene sheet as a function of temperature. | ||
Previous studies have demonstrated that strain plays a significant role in shaping the thermal conductivity of various 2D materials. The application of stress or strain offers a mechanism to finely tune the thermal conductivity of materials, as evidenced by these investigations.44,52–56 Notably, the response of different 2D materials to tensile stress can exhibit distinct behaviors. For instance, in graphene, the thermal conductivity consistently decreases with increasing tensile strain.52–55 Conversely, in buckled silicene, an initial small increase in thermal conductivity is observed with the rise in tensile strain, followed by a subsequent decrease at larger strains.44,56 These findings emphasize the importance of strain in manipulating the thermal conductivity of 2D materials. By applying controlled tensile stress, it becomes possible to precisely modulate the thermal properties of these materials, allowing for tailored applications in various technological domains.
Here, we apply uniaxial tensile strain on the plumbene sheet of 300 Å × 300 Å at 300 K. Heat fluxes are applied in the strained direction. The calculated thermal conductivities in the strained direction obtained by both Tersoff and SW potentials are illustrated in Fig. 8. This figure shows that the Tersoff and SW potentials present different tensile failure limits of 3.4% and 20%, respectively. In addition, tensile strain decreases thermal conductivity monotonically when Tersoff potential is used, while the results of SW potential show that initially the thermal conductivity increases with increasing strain from 0 to 5% and with further increasing strain, the thermal conductivity starts to decrease. Since the optimized SW potential is able to predict the structural parameters better (as discussed in Section 3), and the buckled structure of plumbene is more similar to silicene than the planar structure of graphene, we believe the thermal behavior under strain which is obtained by SW potential would be more accurate. Earlier reported NEMD simulations of other nanostructures in literature also demonstrate that the thermal conductivity of 2D materials shows notably different temperature-dependence with various potentials.57,58
 | ||
| Fig. 8 Effect of strain on thermal conductivity of a 300 Å × 300 Å plumbene sheet at 300 K. | ||
By applying the optimized SW potential, the maximum thermal conductivity appears at 5% strain and its value is about 12% (2%) larger than that for the unstrained armchair (zigzag) configuration. At a tensile strain of 0.2, the thermal conductivity drops more than 64% (46%) compared to the strain-free value for the armchair (zigzag) configuration. The thermal behavior of plumbene under strain obtained by SW potential could be attributed to its initial buckled configuration. At small tensile strains, the buckled configuration becomes less buckled. As the buckled structure flattens, the scattering channels for phonons, especially out-of-plane modes are reduced, resulting in an enhanced thermal conductivity. At large tensile strains, the Pb–Pb bonds are severely stretched. Increasing the Pb–Pb bond length will weaken the interatomic interactions in the in-plane direction. As a consequence, in-plane stiffness decreases and so does the thermal conductivity.
5. Conclusion
In this study, we investigated the lattice thermal conductivity of low-buckled plumbene with different edge shapes by utilizing non-equilibrium molecular dynamics. The results revealed that no existing interatomic potential could precisely capture the unique features of plumbene. Consequently, we optimized the parameters of the Tersoff and Stillinger–Weber potentials using ANNs for a single-layer Pb sheet. This methodology enabled accurate reproduction of plumbene's low buckling structure as obtained from Ab initio MD calculations. Our calculated equilibrium buckling height of 0.99 (1.07) Å and lattice constant of 4.93 (4.81) Å, derived from the optimized SW (Tersoff) potential, match reasonably well with the values obtained from previous DFT simulations. However, the optimized SW potential demonstrated a superior capacity to predict accurate structural results. In addition, the phononic spectrum obtained from the optimized potentials was compared with first-principles results, showing that the optimized potentials are capable of predicting the correct phononic spectrum.We then applied the optimized SW and Tersoff potentials to calculate the lattice thermal conductivity of plumbene, exploring the relationship between plumbene's length, temperature, and strain with its thermal conductivity. The thermal conductivity of low-buckled plumbene calculated with the optimized SW potential was, on average, 4 times larger than the thermal conductivity obtained with the optimized Tersoff potential. The thermal conductivity of plumbene sheets (with widths of 300 Å and lengths ranging from 300 Å to 1050 Å) at room temperature ranged from 2.8–7.8 W m−1 K−1 with the optimized SW potential and from 0.7–1.4 W m−1 K−1 with the optimized Tersoff potential. Regardless of the results' magnitude, both the optimized SW and Tersoff potentials affirmed that an increase in the length of plumbene leads to an increase in its thermal conductivity.
Furthermore, we observed that an increase in temperature led to a decrease in plumbene's thermal conductivity. As the temperature increased from 300 K to 600 K, the thermal conductivity of a 300 Å × 300 Å sample with a zigzag configuration decreased from 3.4 W m−1 K−1 to 2 W m−1 K−1. Consistently, plumbene sheets with zigzag edges demonstrated higher thermal conductivity than those with armchair edges, aligning with the behavior of other 2D nanostructures.
We also examined the effect of applying uniaxial tensile strain on plumbene. When we employed the Tersoff potential, tensile strain resulted in a monotonous decrease in thermal conductivity, similar to the behavior of graphene under tensile strain. On the other hand, the results from the SW potential showed that thermal conductivity initially increased with strain from 0 to 5% but started to decrease with further strain, a behavior akin to silicene under tensile strain. Given the optimized SW potential's better prediction of plumbene's structural parameters and the closer resemblance of plumbene's buckled structure to silicene than to graphene's planar structure, we posit that the results obtained by the optimized SW potential in this research are more reliable.
Data availability
The Large-scale Atomic/Molecular Massively Parallel Simulator (LAMMPS) was used to perform molecular dynamics simulations, which can be found at https://www.sciencedirect.com/science/article/abs/pii/S002199918571039X with DOI: https://doi.org/10.1006/jcph.1995.1039.Conflicts of interest
There are no conflicts of interest to declare.References
- F. Zhang, K. Yang, G. Liu, Y. Chen, M. Wang, S. Li and R. Li, Recent advances on graphene: Synthesis, properties and applications, Composites, Part A, 2022, 160, 107051 CrossRef.
- H. Zhao, C. W. Zhang, W. X. Ji, R. W. Zhang, S. S. Li, S. S. Yan, B. M. Zhang, P. Li and P. J. Wang, Unexpected giant-gap quantum spin Hall insulator in chemically decorated plumbene monolayer, Sci. Rep., 2016, 6(1), 20152 CrossRef.
- Y. H. Lu, D. Zhou, T. Wang, S. A. Yang and J. Z. Jiang, Topological properties of atomic lead film with honeycomb structure, Sci. Rep., 2016, 6(1), 21723 CrossRef PubMed.
- X. L. Yu, L. Huang and J. Wu, From a normal insulator to a topological insulator in plumbene, Phys. Rev. B, 2017, 95(12), 125113 CrossRef.
- L. Zhang, H. Zhao, W. X. Ji, C. W. Zhang, P. Li and P. J. Wang, Discovery of a new quantum spin Hall phase in bilayer plumbene, Chem. Phys. Lett., 2018, 712, 78–82 CrossRef.
- Y. Li, J. Zhang, B. Zhao, Y. Xue and Z. Yang, Constructive coupling effect of topological states and topological phase transitions in plumbene, Phys. Rev. B, 2019, 99(19), 195402 CrossRef.
- J. Yuhara, B. He, N. Matsunami, M. Nakatake and G. Le Lay, Graphene's latest cousin: Plumbene epitaxial growth on a “nano WaterCube”, Adv. Mater., 2019, 31(27), 1901017 CrossRef.
- G. Bihlmayer, J. Sassmannshausen, A. Kubetzka, S. Blügel, K. von Bergmann and R. Wiesendanger, Plumbene on a magnetic substrate: a combined scanning tunneling microscopy and density functional theory study, Phys. Rev. Lett., 2020, 124(12), 126401 CrossRef.
- M. Sharma and R. Sharma, Plumbene: A next generation hydrogen storage medium, Int. J. Hydrogen Energy, 2021, 46(66), 33197–33205 CrossRef.
- F. Bechstedt, S. Grillo, O. Pulci and P. Gori, Thermal properties of Dirac fermions in Xenes: Model studies, Phys. Rev. B, 2021, 104(16), 165420 CrossRef.
- S. Mahdavifar and M. B. Tagani, Electronic and mechanical properties of Plumbene monolayer: A first-principle study, Phys. E, 2021, 134, 114837 CrossRef.
- B. Zhang, F. Guo, M. Zhu, L. Feng and Y. Zheng, The sensitive tunability of superconducting critical temperature in high-buckled plumbene by shifting Fermi level, Phys. E, 2021, 130, 114688 CrossRef.
- D. K. Das, J. Sarkar and S. K. Singh, Effect of sample size, temperature and strain velocity on mechanical properties of plumbene by tensile loading along longitudinal direction: A molecular dynamics study, Comput. Mater. Sci., 2018, 151, 196–203 CrossRef.
- D. K. Das, A. Mallick and S. K. Singh, Estimating thermal properties of plumbene by multiscale modeling using molecular dynamics simulation technique, Mech. Adv. Mater. Struct., 2022, 29(15), 2119–2125 CrossRef.
- X. Xu, L. F. Pereira, Y. Wang, J. Wu, K. Zhang, X. Zhao, S. Bae, C. Tinh Bui, R. Xie, J. T. Thong and B. H. Hong, Length-dependent thermal conductivity in suspended single-layer graphene, Nat. Commun., 2014, 5(1), 3689 CrossRef PubMed.
- T. Y. Ng, J. Yeo and Z. Liu, Molecular dynamics simulation of the thermal conductivity of shorts strips of graphene and silicene: a comparative study, Int. J. Mech. Mater. Des., 2013, 9, 105–114 CrossRef.
- J. M. Tagalog, C. G. Alipala, G. J. Paylaga, N. T. Paylaga and R. V. Bantaculo, Molecular dynamics simulation of the thermal conductivity of silicon-germanene nanoribbon (SiGeNR): a comparison with silicene nanoribbon (SiNR) and germanene nanoribbon (GeNR), Adv. Mater. Res., 2015, 1105, 285–289 Search PubMed.
- D. K. Das and J. Sarkar, Multiscale modeling of thermal properties of silicene using molecular dynamics, Mod. Phys. Lett. B, 2018, 32(27), 1850331 CrossRef.
- M. H. Rahman, E. H. Chowdhury, D. A. Redwan and S. Hong, Computational characterization of thermal and mechanical properties of single and bilayer germanene nanoribbon, Comput. Mater. Sci., 2021, 190, 110272 CrossRef.
- M. H. Rahman, M. S. Islam, M. S. Islam, E. H. Chowdhury, P. Bose, R. Jayan and M. M. Islam, Phonon thermal conductivity of the stanene/hBN van der Waals heterostructure, Phys. Chem. Chem. Phys., 2021, 23(18), 11028–11038 RSC.
- S. Bukkapatnam, M. Malshe, P. M. Agrawal, L. M. Raff and R. Komanduri, Parametrization of interatomic potential functions using a genetic algorithm accelerated with a neural network, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74(22), 224102 CrossRef.
- M. Malshe, R. Narulkar, L. M. Raff, M. Hagan, S. Bukkapatnam and R. Komanduri, Parametrization of analytic interatomic potential functions using neural networks, J. Chem. Phys., 2008, 129(4), 044111 CrossRef CAS.
- G. P. Pun, R. Batra, R. Ramprasad and Y. Mishin, Physically informed artificial neural networks for atomistic modeling of materials, Nat. Commun., 2019, 10(1), 2339 CrossRef.
- A. C. Nwachukwu and S. Winczewski, Application of the neural networks for developing new parametrization of the Tersoff potential for carbon, TASK Quarterly: scientific bulletin of Academic Computer Centre in Gdansk, 2020, 24, 299–333 Search PubMed.
- G. Kresse and J. J. Hafner, Ab initio molecular dynamics for open-shell transition metals, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48(17), 13115 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54(16), 11169 CrossRef CAS.
- P. E. Blöchl, Projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50(24), 17953 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77(18), 3865 CrossRef CAS.
- D. Hashemi and H. Iizuka, Magnetic properties of 3d transition metal (Sc–Ni) doped plumbene, RSC Adv., 2020, 10(12), 6884–6892 RSC.
- D. Hashemi and H. Iizuka, Substitutional 4d transition metal doping in atomically thin lead, RSC Adv., 2021, 11(11), 6182–6187 RSC.
- L. Prechelt, G. Montavon, G. B. Orr and K. R. Müller, Neural Networks: Tricks of the Trade. Lecture Notes in Computer Science, 2nd ed, Springer, 2012 Search PubMed.
- J. J. Tersoff, Modeling solid-state chemistry: Interatomic potentials for multicomponent systems, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 39(8), 5566 CrossRef.
- S. Plimpton, A. Thompson, S. Moore and A. Kohlmeyer, LAMMPS Documentation, Sandia National Laboratories. 2006 Search PubMed.
- F. H. Stillinger and T. A. Weber, Computer simulation of local order in condensed phases of silicon, Phys. Rev. B: Condens. Matter Mater. Phys., 1985, 31(8), 5262 CrossRef CAS PubMed.
- S. Plimpton, Fast parallel algorithms for short-range molecular dynamics, J. Comput. Phys., 1995, 117(1), 1–9 CrossRef.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, Avogadro: an advanced semantic chemical editor, visualization, and analysis platform, J. Cheminf., 2012, 4, 1–7 Search PubMed.
- E. N. Koukaras, G. Kalosakas, C. Galiotis and K. Papagelis, Phonon properties of graphene derived from molecular dynamics simulations, Sci. Rep., 2015, 5(1), 12923 CrossRef PubMed.
- X. J. Ge, K. L. Yao and J. T. Lü, Comparative study of phonon spectrum and thermal expansion of graphene, silicene, germanene, and blue phosphorene, Phys. Rev. B, 2016, 94(16), 165433 CrossRef.
- Y. D. Kuang, L. Lindsay, S. Q. Shi and G. P. Zheng, Tensile strains give rise to strong size effects for thermal conductivities of silicene, germanene and stanene, Nanoscale, 2016, 8(6), 3760–3767 RSC.
- B. Peng, H. Zhang, H. Shao, Y. Xu, G. Ni, R. Zhang and H. Zhu, Phonon transport properties of two-dimensional group-IV materials from ab initio calculations, Phys. Rev. B, 2016, 94(24), 245420 CrossRef.
- X. Yang, D. Han, H. Fan, M. Wang, M. Du and X. Wang, First-principles calculations of phonon behaviors in graphether: A comparative study with graphene, Phys. Chem. Chem. Phys., 2021, 23(1), 123–130 RSC.
- V. K. Dien, N. Thi Han, W. Bang-Li, K. I. Lin and M. F. Lin, Correlation between orbital hybridizations, phonon spectra, and thermal properties of graphene, germanene, and plumbene, Phys. Status Solidi RRL, 2023, 17(5), 2200469 CrossRef.
- J. Carrete, W. Li, L. Lindsay, D. A. Broido, L. J. Gallego and N. Mingo, Physically founded phonon dispersions of few-layer materials and the case of borophene, Mater. Res. Lett., 2016, 4(4), 204–211 CrossRef.
- Q. X. Pei, Y. W. Zhang, Z. D. Sha and V. B. Shenoy, Tuning the thermal conductivity of silicene with tensile strain and isotopic doping: A molecular dynamics study, J. Appl. Phys., 2013, 114, 3 CrossRef.
- B. Peng, H. Zhang, H. Shao, Y. Xu, X. Zhang and H. Zhu, Low lattice thermal conductivity of stanene, Sci. Rep., 2016, 6(1), 20225 CrossRef.
- T. Y. Kim, C. H. Park and N. Marzari, The electronic thermal conductivity of graphene, Nano Lett., 2016, 16(4), 2439–2443 CrossRef PubMed.
- H. Zhou, Y. Cai, G. Zhang and Y. W. Zhang, Quantum thermal transport in stanene, Phys. Rev. B, 2016, 94(4), 045423 CrossRef.
- S. Ghosh, D. L. Nika, E. P. Pokatilov and A. A. Balandin, Heat conduction in graphene: experimental study and theoretical interpretation, New J. Phys., 2009, 11(9), 095012 CrossRef.
- A. I. Khan, R. Paul and S. Subrina, Characterization of thermal and mechanical properties of stanene nanoribbons: a molecular dynamics study, RSC Adv., 2017, 7(80), 50485–50495 RSC.
- M. H. Rahman, E. H. Chowdhury, M. R. Shahadat and M. M. Islam, Engineered defects to modulate the phonon thermal conductivity of Silicene: A nonequilibrium molecular dynamics study, Comput. Mater. Sci., 2021, 191, 110338 CrossRef CAS.
- H. Y. Cao, Z. X. Guo, H. Xiang and X. G. Gong, Layer and size dependence of thermal conductivity in multilayer graphene nanoribbons, Phys. Lett. A, 2012, 376(4), 525–528 CrossRef CAS.
- Z. Guo, D. Zhang and X. G. Gong, Thermal conductivity of graphene nanoribbons, Appl. Phys. Lett., 2009, 95, 16 Search PubMed.
- X. Li, K. Maute, M. L. Dunn and R. Yang, Strain effects on the thermal conductivity of nanostructures, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81(24), 245318 CrossRef.
- N. Wei, L. Xu, H. Q. Wang and J. C. Zheng, Strain engineering of thermal conductivity in graphene sheets and nanoribbons: a demonstration of magic flexibility, Nanotechnology, 2011, 22(10), 105705 CrossRef.
- M. Guo, Y. Qian, H. Qi, K. Bi and Y. Chen, Experimental measurements on the thermal conductivity of strained monolayer graphene, Carbon, 2020, 157, 185–190 CrossRef CAS.
- M. Barati, T. Vazifehshenas, T. Salavati-fard and M. Farmanbar, Competing effects of strain and vacancy defect on thermal conductivity of silicene: A computational study, Comput. Mater. Sci., 2020, 173, 109407 CrossRef CAS.
- C. Si, X. D. Wang, Z. Fan, Z. H. Feng and B. Y. Cao, Impacts of potential models on calculating the thermal conductivity of graphene using non-equilibrium molecular dynamics simulations, Int. J. Heat Mass Transfer, 2017, 107, 450–460 CrossRef.
- X. Zhang, Z. Chen, H. Chen and L. Xu, Comparative studies of thermal conductivity for bilayer graphene with different potential functions in molecular dynamic simulations, Results Phys., 2021, 22, 103894 CrossRef.
| This journal is © the Owner Societies 2024 |