
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The windmill, the dragon, and the frog: geometry control over the spectral, magnetic, and electrochemical properties of cobalt phthalocyanine regioisomers†
Nikolai
Tkachenko
 a,
Viacheslav
Golovanov
ad,
Aleksandr
Penni
a,
Sami
Vesamäki
a,
M. R.
Ajayakumar
a,
Atsuya
Muranaka
b,
Nagao
Kobayashi
c and
Alexander
Efimov
*a
a,
Viacheslav
Golovanov
ad,
Aleksandr
Penni
a,
Sami
Vesamäki
a,
M. R.
Ajayakumar
a,
Atsuya
Muranaka
b,
Nagao
Kobayashi
c and
Alexander
Efimov
*a
aTampere University, Korkeakoulunkatu 10, 33720 Tampere, Finland. E-mail: alexandre.efimov@tuni.fi
bMolecular Structure Characterization Unit, RIKEN Center for Sustainable Resource Science, Wako, Saitama 351-0198, Japan
cFaculty of Textile Science and Technology, Shinshu University, Ueda 386-8567, Japan
dSouth-Ukrainian National University, Staroportofrankovskaya str. 26, 65020, Odessa, Ukraine
First published on 10th June 2024
Abstract
For the first time, we have prepared non-aggregating phthalocyanine cobalt complexes as a set of resolved positional isomers. These compounds comprise a unique test bed for the structure–properties studies, as their optical and electrochemical properties are influenced by the planarity of the phthalocyanine macrocycle, which can be controlled by the positional isomerism of the bulky aromatic substituents at the α-phthalo sites. We support our conclusions with molecular modelling studies, which show a perfect match between the calculated and experimentally determined spectral/electrochemical values. We challenge a common perception that the NMR spectra of cobalt phthalocyanines cannot be measured due to the paramagnetic nature of Co(II). We suggest instead that the key factors affecting the NMR spectral resolution are molecular aggregation and π–π stacking. These interactions are suppressed by the bulky peripheral substituents on the cobalt phthalocyanines prepared, making these isomeric compounds an excellent tool for paramagnetic NMR studies.
Introduction
Phthalocyanines (Pcs) are widely utilised in areas as diverse as catalysis, the dye industry, medicinal chemistry, sensing, and materials science.1 Cobalt phthalocyanine complexes (CoPcs) are uniquely suited for electrocatalytic CO2 reduction.2,3 CoPcs generally suffer from severe molecular aggregation which dramatically reduces their catalytic activity; however, in cases when the aggregation is suppressed, their efficiency becomes remarkably high.4–6 It has been recently emphasised that a non-aggregating CoPc acting as a single-atom catalyst would be of ultimate value for numerous chemical transformations and CO2 conversion in particular.7,8Solubility and aggregation properties of Pcs can be controlled by peripheral substitution. Alkoxy, aryloxy, or tert-butyl substituents are typically used to improve the solubility of these compounds. There are a number of strategies for the synthesis of non-aggregating Pcs with defined substitution patterns based on thermally and/or sterically controlled reaction methodologies.9–13 Common examples of such substituted Pcs include tetrakis[tert-butyl]phthalocyanine, octakis[butoxy]phthalocyanine, and various tetrakis and octakis phenoxy- and thiophenyl-bearing derivatives. There are grounds to expect that in some cases the properties of the positional isomers may differ significantly. Whereas the normal conformation of a Pc's aromatic core is approximately flat, the presence of substituents may induce in-plane and out-of-plane distortions.14,15 Appropriate peripheral substitution can therefore be employed to achieve desired opto-electronic, magnetic, and catalytic properties.16 Peripherally tetrasubstituted Pcs present an additional challenge in that they are often produced as mixtures of regioisomers, which are rarely separated to isolate individual compounds, and hence the materials obtained remain poorly characterised. Furthermore, the separation of isomeric Pcs may be difficult, if possible at all.17 It is thus not surprising that most studies have been performed with structurally the simplest and most readily available Pcs, for which positional isomerism is either non-existent or unimportant.
Reliable prediction of the geometries, spectral properties, and HOMO/LUMO energies for these species via molecular modelling would be immensely valuable. Ab initio density functional theory (DFT) calculations18 can address many important aspects, such as structurally induced effects on the magnetic, optical, and subsequently catalytic characteristics of CoPcs.7,19 In the context of aggregation-induced deactivation of CoPc catalysts, computational modelling20,21 can provide valuable insights into the mutual arrangement of the metal phthalocyanine molecules, opening a pathway for improving the catalytic performance.
Investigation of Pcs by NMR spectroscopy is in general problematic due to the possible presence of regioisomers, molecular aggregation via strong π–π stacking, and low solubility. NMR characterisation of CoPcs is even more challenging due to the paramagnetic nature of the Co(II) ion, a reason to which the poor resolution and broadness of the peaks is usually attributed.22–24 On the other hand, paramagnetic NMR studies are becoming more common, and some excellent publications have recently appeared, in which Co(II) complexes containing aromatic ligands are investigated.25–29 However, when the system under study contained a porphyrin core, the NMR spectrum was unresolved.26 When a paramagnetic atom is introduced into a structure, crucial spatial information can be gained via hyperfine chemical shifts, pseudocontact shifts, and changes in relaxation time constants.
The synthesis and investigation of non-aggregating and isomerically pure CoPcs have hitherto remained underexplored. In this work, we present a sterically controlled approach to the preparation of a series of isomerically pure non-aggregating CoPcs and perform a detailed paramagnetic NMR and opto-electronic analysis of these compounds.
Results and discussion
The structures of the three phthalocyanines prepared and studied in this work are shown in Fig. 1. The three isomeric molecules have the same general structure but differ in the position of the four bulky di[tert-butyl]phenyl (t-Bu2Ph) substituents at the α-phthalo sites. The compounds are denoted as “windmill” (C4h symmetry), “dragon” (Cs symmetry) and “frog” (C2v symmetry). Usually, when bulky α-phthalo substituents are employed, formation of only the most symmetrical windmill isomer is expected.10,30,31 Herein, we demonstrate that the frog and dragon isomers can also be obtained, although energetically unfavourable intramolecular steric interactions present in their structures can be envisioned to lower the yields.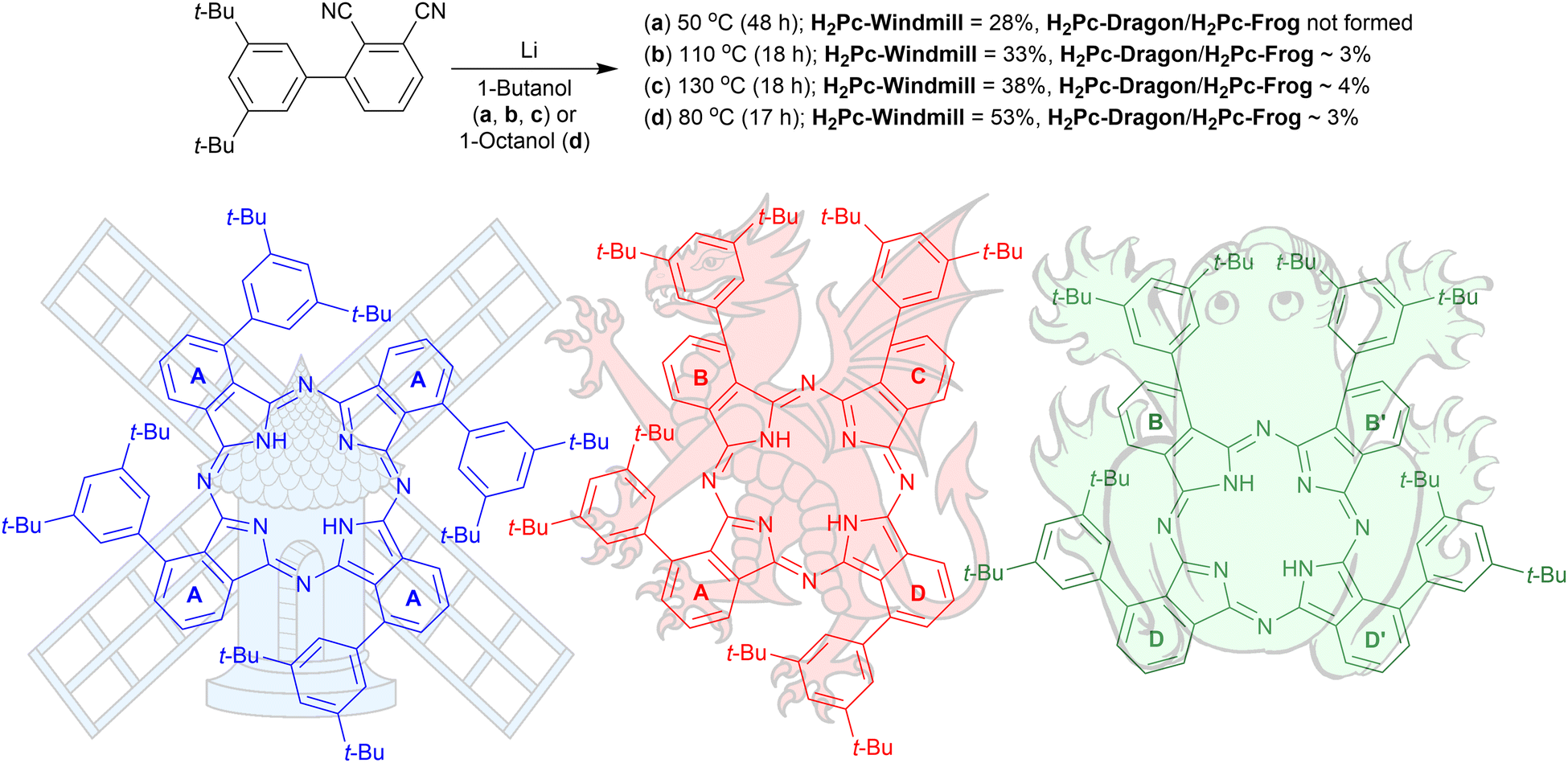 | ||
| Fig. 1 Synthesis of the three isomeric phthalocyanines. The isoindole rings are denoted as (a)–(d) for interpreting the NMR spectra. | ||
Synthesis
3′,5′-Di-tert-butyl-[1,1′-biphenyl]-2,3-dicarbonitrile was prepared as described earlier10 and used for the condensation reaction (Fig. 1) in the presence of lithium butoxide in butanol at 55 °C for 48 h under an argon atmosphere. The reaction provided a 28% isolated yield for the windmill isomer; however, the reaction did not yield any noticeable amounts of the other regioisomers. When the reaction temperature was increased to 110 °C or 135 °C, the formation of the other two Pcs was observed. These were later identified as the frog and dragon isomers, and the three compounds can be resolved by TLC on silica, with their mobility decreasing in the order windmill > dragon > frog. Performing the reaction in octanol at 80 °C proved to maximize the yields of all the three isomeric Pcs. Preparative separation of the three isomers was astonishingly simple. After condensation, the reaction mixture was washed with water to remove the lithium ions, and the organic solvent was removed under reduced pressure. The residual green solid was suspended in hexane, and the resulting green solution was pipetted off. What remained in the solid residue was, to our great surprise, a single isomer fraction, attributed to the windmill Pc by NMR analysis. The final purification of the isomers required a short column using dichloromethane/hexane as eluent or preparative TLC. Metal insertions were done by heating the cobalt salts with phthalocyanine free base at 110 °C in dimethylformamide for 19 h under argon.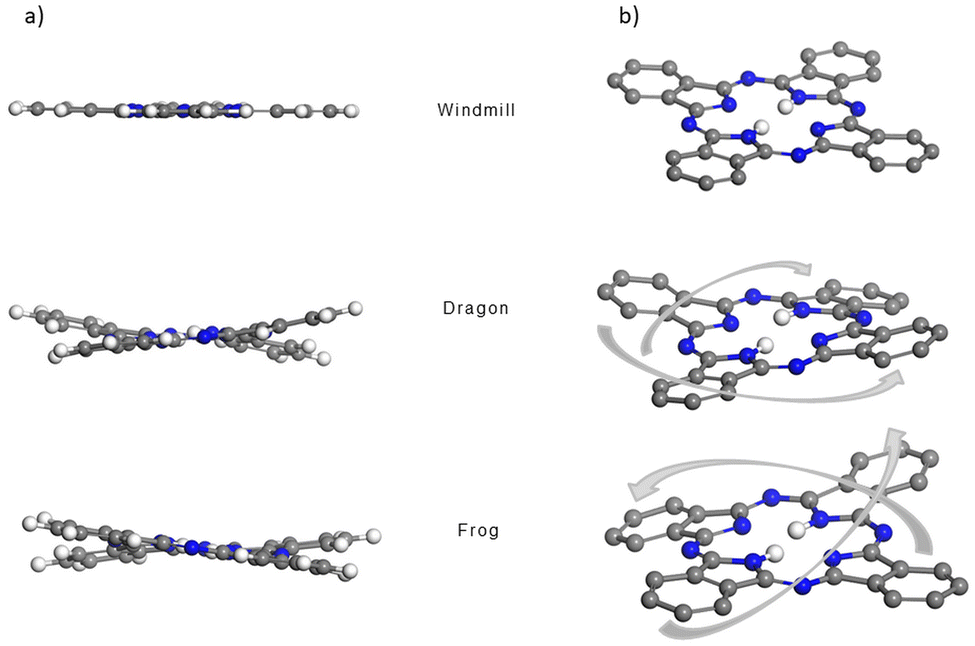 | ||
| Fig. 2 Side (a) and isometric (b) view of the molecular core of the windmill (top), dragon (middle) and frog (bottom) isomers, visualizing the saddle shape distortions of the macrocycle for the dragon and frog molecules. The t-Bu2Ph substituents and outer hydrogen atoms are removed for clarity. C, H and N atoms are represented as grey, white and blue spheres, respectively. | ||
NMR studies
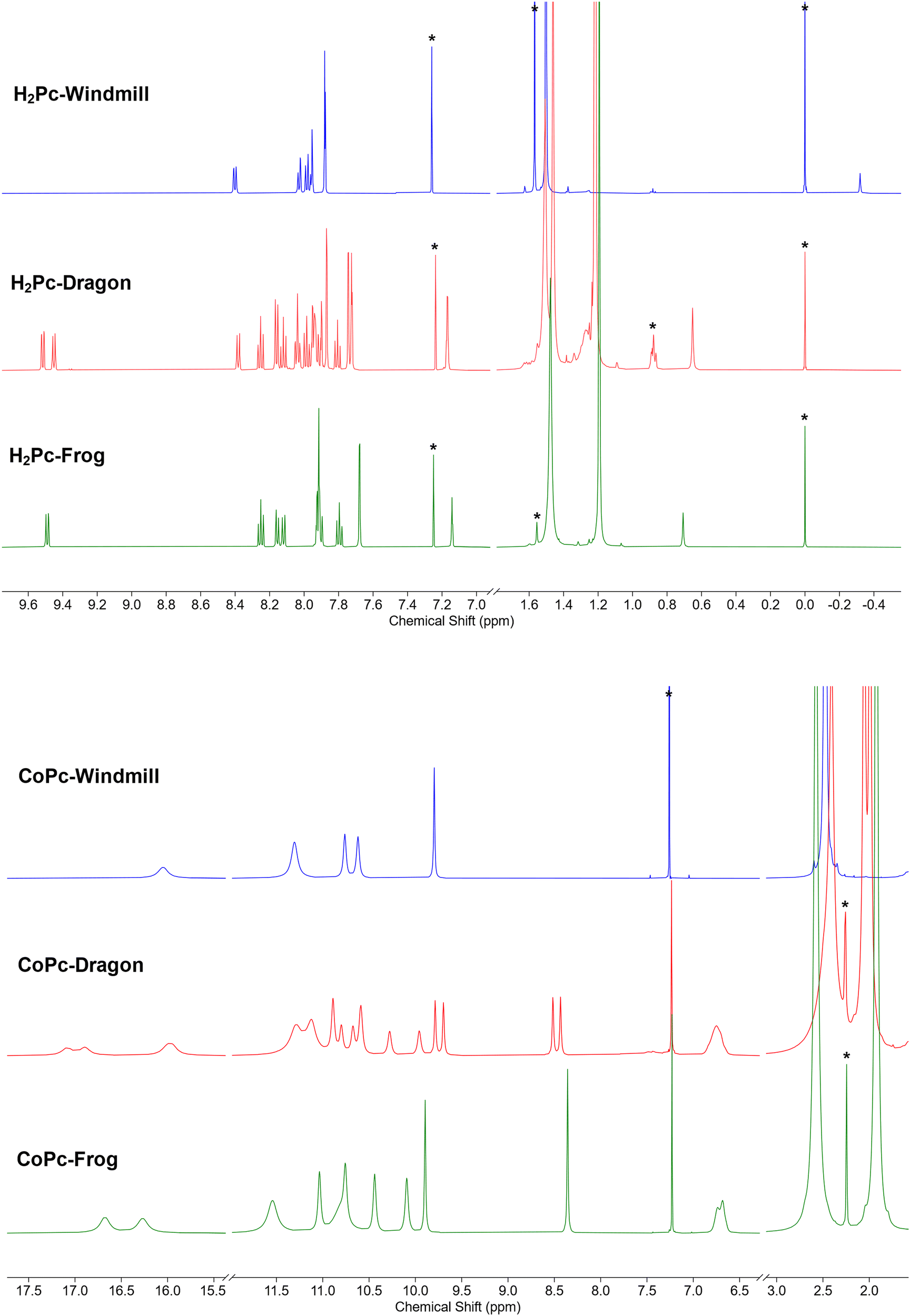 | ||
| Fig. 3 (a) NMR spectra of the free base phthalocyanines. (b) NMR spectra of the Co complexes. | ||
A more complex, although still rather clear, spectrum was observed for the third isomer (Fig. 3a, green trace). The two coupled systems at 9.49–8.25–8.16 and 8.12–7.80–7.90 ppm were assigned to the two groups of the phthalo protons. The phenyl rings produced coupled signals at 7.68–7.14 and 7.93–7.91 ppm. Tert-Butyl groups gave two signals of 36 protons each at 1.48 and 1.19 ppm. The NH protons signal appeared at 0.71 ppm. Such a symmetry was attributed to the frog isomer, although the exact assignment of the signals remained to be done. The 2D NOESY spectrum (Fig. S2, ESI†) showed correlations at 7.91–1.48, 8.12–1.48 ppm, and 7.68–1.19 and 7.14–1.19 ppm, and no correlation between peaks at 9.49 and the alkyl groups. Therefore, the peak at 9.49 ppm was assigned to the α-phthalo protons of the rings D and D′ (see Fig. 1), and peaks at 8.25 and 8.16 ppm belonged to the same rings D/D′, as well as phenyls 7.93–7.91 ppm and tert-butyls at 1.48 ppm. The peak at 8.12 was hence attributed to the alfa-phthalo protons of the rings B/B′, 7.80 and 7.90 ppm were related to the corresponding β and β′ protons, and the phenyls and tert-butyls were found at 7.68, 7.14 and 1.19 ppm, respectively. This upfield shift of the alkyl and p-phenyl signals is supported by the DFT model, in which the B and B′ phenyl rings are stretched away from the macrocycle, tilted and overlap each other, thus putting the protons into a shielding region above the phenyl planes.
The dragon phthalocyanine, obtained as a middle fraction on normal phase chromatography, had the most complex NMR pattern among the three isomers (Fig. 3a, red trace). The assignment was based on interpretations described above. In the 2D COSY spectrum (see Fig. S3, ESI†) the phthalo protons formed four separate coupled systems, namely 9.52–8.25–8.16, 9.45–8.12–8.03, 8.38–7.98–8.04, and 8.16–7.81–7.93 ppm. For these groups, only β-phthalo hydrogens can be identified from the COSY spectrum, while the alfa and β′-hydrogens are difficult to differentiate. Based on signal assignment for the other two isomers, the highest chemical shift in each group was attributed to the alfa-phthalo protons. In the 2D NOESY spectrum (Fig. S4, ESI†), correlations can be seen for the pairs of signals at 8.38–1.51, 8.16–1.46, 7.87–1.46, 7.95–1.51, 1.22–7.17 and 1.22–7.74 ppm. Hence, it was concluded that di(tert-butyl)phenyl group with the signals at 7.95 and 1.51 ppm belongs to the ring D and correlates with the alfa-phthalo proton of the ring A at 8.38, while the ring A has the di(tert-butyl)phenyl signals at 1.46–7.87 ppm which correlate with the alfa-phthalo proton of the ring B at 8.16 ppm. Signals at 9.52 and 9.45 ppm do not give correlations to the aliphatic groups at all, and hence were assigned to the alfa-phthalo protons of the rings D and C, respectively. Additional proof for the correctness of this assignment was obtained from the HMBC spectrum (Fig. S5, ESI†), where the β-phthalo proton at 8.12 ppm and the o-phenyl proton at 7.74 ppm of the ring C give correlations to the same q-carbon on the isoindole ring at 140.2 ppm. Similarly, the β-phthalo proton at 8.25 ppm and the o-phenyl proton at 7.94 ppm of the ring D show correlations to the same q-carbon on the isoindole ring at 141.7 ppm. For the rings A and B, the situation is identical as the β-phthalo and o-phenyl protons reveal correlations to the q-carbons on the respective isoindole rings. Insufficient resolution along the 13C axis in the HSQC and HMBC spectra precluded complete assignment of the 13C resonances. The tert-butyl hydrogens are split into broadened peaks at 1.51 (ring D), 1.46 (ring A), and 1.22 ppm (rings B and C). It is notable that, similarly to the situation for the frog isomer, the NH protons give a singlet at 0.65 ppm, representing unusually strong deshielding.
Simulations of the 1H NMR chemical shift for the NH protons are in qualitative agreement with the experimental observations. For the hydrogen atoms inside of the fully symmetrical windmill core, a signal at −9.95 ppm is predicted, whereas in the real spectrum this resonance is located at −0.32 ppm. For the distorted dragon and frog isomers, the calculated chemical shifts for the NH protons move downfield to 0.9 ppm, whereas in the real spectra these signals are found at 0.65 and 0.71 ppm, respectively. Deshielding of the NH protons for the isomers with a distorted macrocyclic aromatic ring is in line with the Hirshfeld population analysis, which shows an approx. 8% diminution of the electron density on the hydrogens inside the macrocycle for the frog and dragon phthalocyanines compared to the windmill isomer.
A quick proof of the structure was achieved by high-resolution ESI-TOF mass spectrometry. Being measured in positive polarity, all three complexes gave the same results. The three isotopic multiplets could be observed (Fig. S6b, ESI†), corresponding to the molecular ions M+/[M + H]+, [M + Na]+ and [M + K]+ within the 3 mDa accuracy (<5 ppm). The lightest m/z signal came from an overlap of M+ and [M + H]+ molecular ions, which is quite typical for metal porphyrinoids. The m/z data testify for Co(II)Pc neutral complex formation rather than Co(III)Pc monocation, which would not produce single-charged adducts with proton, sodium or potassium.
Encouraged by the unusually high solubility of the complexes obtained, we carried out NMR measurements. The samples were prepared as solutions in deuterochloroform with concentrations of ca. 30 mg ml−1. The spectral window was initially set from +50 to −50 ppm, and eventually decreased to 20 to −2 ppm. To our surprise, all the three isomeric complexes produced broadened, yet sufficiently well-resolved, signals which could be integrated (Fig. 3b). The spectrum of the windmill CoPc showed a peak of four protons at 16.05 ppm, a peak of eight protons at 11.30 ppm and a pair of sharper singlets at 10.76 and 10.62 ppm with the integral of eight, and a sharp peak at 9.80 ppm. The tert-butyls’ signal was clearly visible though shifted downfield to 2.47 ppm. Even though the sample was made obviously at the solubility limit, we did not notice any difference with the spectrum of a more dilute sample, except for the signal intensity. The 13C NMR spectrum of cobalt windmill (Fig. S7, ESI†) suffered from poor sensitivity, however produced sharp peaks both in the aromatic and aliphatic regions. The same trends can be observed, as the peaks were shifted by a few ppm to the lower field. In the HSQC and HMBC spectra of Co-windmill (Fig. S8 and S9, ESI†) correlations for the protons at 11–9 ppm and 2.47 ppm can be traced, however the signal at 16.05 ppm gave no cross-peaks.
1H NMR spectra of the dragon and frog were also well resolved and showed patterns similar to their free base counterparts, though with the signals shifted downfield.
Graphical signal assignments for the free base phthalocyanines and their Co complexes are given in Fig. 4.
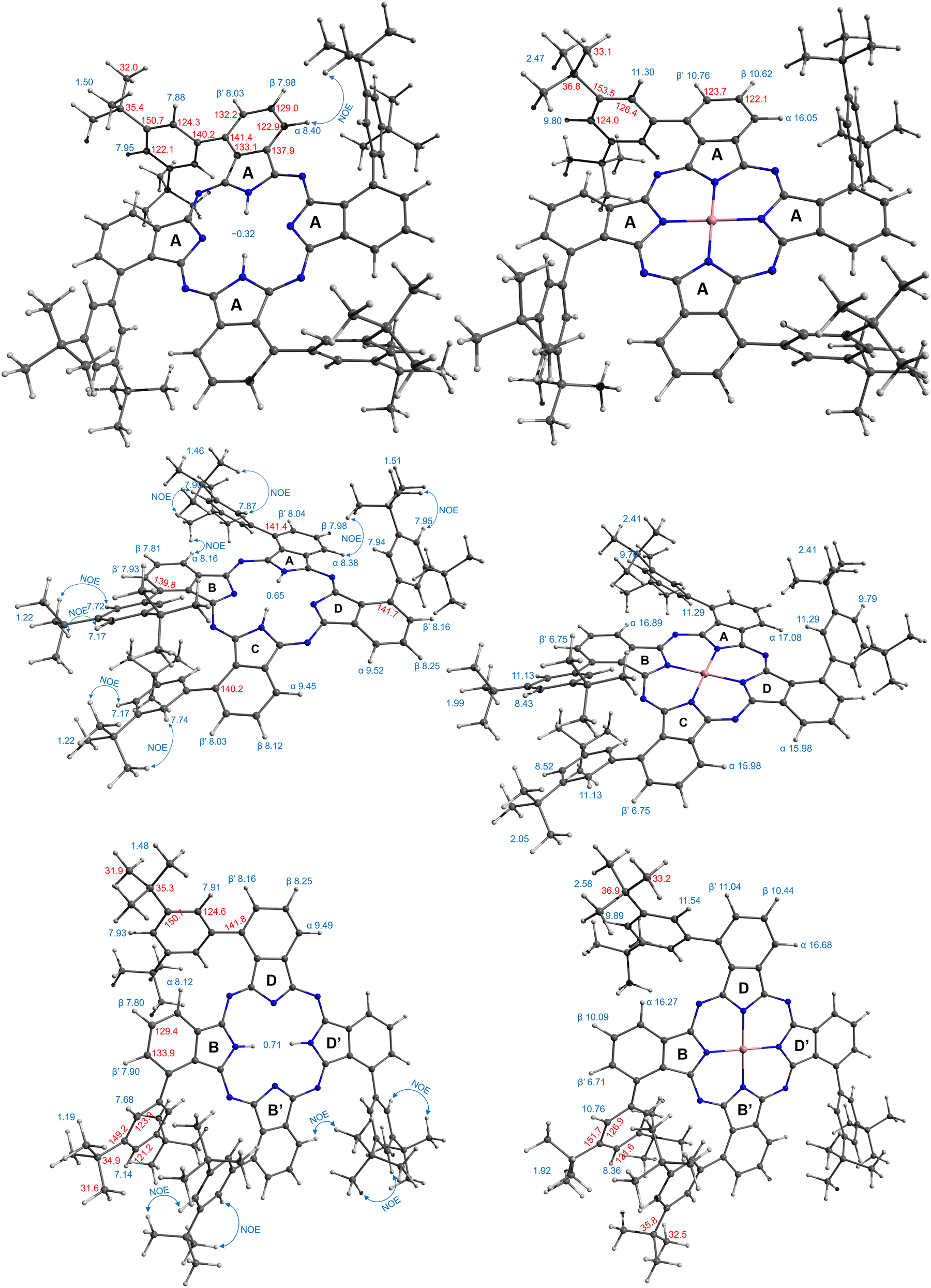 | ||
| Fig. 4 Assignment of NMR signals for the compounds under study. | ||
The T1 relaxation times were measured for the protons of free bases and cobalt complexes using the inversion recovery pulse sequence with 10–14 points and processing the data with JEOL Delta 5.2 software. For the free base compounds the relaxation constants ranged from 0.5 s for NH protons to almost 2 s for the β-phthalo H with no major deviation among the isomers. For the cobalt complexes the T1 values ranged from milliseconds for the broadest to tens of milliseconds for the sharpest peaks. The obtained data for the three isomers are summarized in Table 1.
| CoPc-windmill | CoPc-dragon | ||||||
| δ, ppm | T 1, ms | Distance, Å | Assignment | δ, ppm | T 1, ms | Distance, Å | Assignment |
| 16.05 | 5 | 5.9 | α | 17.08 | 5.9 | 5.5 | α-A |
| 11.30 | 9 | 6.6 | o-Ph | 16.89 | 6.1 | 5.7 | α-B |
| 10.76 | 27 | 7.6 | β′ | 15.98 | 6 | 6.1 | α-C,D |
| 10.62 | 26 | 7.6 | β | 11.29 | 6.9 | 6.6 | o-Ph-A,D |
| 9.80 | 69 | 8.9 | p-Ph | 11.13 | 6.9 | 6.6 | o-Ph-B,C |
| 2.47 | 57 | 8.9 | t-Bu | 10.89 | 24 | 7.5 | β/β′ |
| CoPc-frog | 10.80 | 25 | 7.5 | β/β′ | |||
| δ, ppm | T 1, ms | Distance, Å | Assignment | 10.67 | 25 | 7.3 | β/β′ |
| 16.68 | 6.3 | 6.0 | α-D | 10.59 | 26 | 7.9 | β/β′ |
| 16.27 | 5.9 | 5.7 | α-B | 10.28 | 25 | 7.6 | β/β′ |
| 11.54 | 8.3 | 6.6 | o-Ph-D | 9.96 | 26 | 7.9 | β/β′ |
| 11.04 | 26 | 7.6 | β'-D | 9.79 | 65 | 9.6 | p-Ph-D |
| 10.76 | 6.4 | 6.6 | o-Ph-B | 9.70 | 66 | 9.4 | p-Ph-A |
| 10.44 | 27 | 7.6 | β-D | 8.52 | 81 | 10.1 | p-Ph-C |
| 10.09 | 26 | 7.5 | β-B | 8.43 | 82 | 9.8 | p-Ph-B |
| 9.89 | 64 | 8.6 | p-Ph-D | 6.75 | 45 | 7.7 | β′-B,C |
| 8.36 | 83 | 9.5 | p-Ph-B | 2.41 | 48 | 9.2 | t-Bu-A,D |
| 6.71 | 45 | 7.6 | β'-B | 2.05 | 35 | 10.4 | t-Bu-C |
| 2.58 | 47 | 8.9 | t-Bu-D | 1.99 | 36 | 8.7 | t-Bu-B |
| 1.92 | 37 | 9.6 | t-Bu-B | ||||
The obtained values clearly felt into three categories: 5–10 ms, 20–30 ms, and 30–70 ms, and somewhat correlate to the chemical shifts. Detailed analysis however required information regarding the distance from a proton to the cobalt ion, which was obtained from the geometry modelling by DFT calculations. The results are summarized in Table 1.
Simplest interpretation could be made for the windmill isomers. All four of its rings are equivalent, and only six signals are observed in the spectrum. The shortest relaxation 5 ms fits well with the shortest distance 5.86 Å and highest chemical shift 16.1 ppm of the alfa-phthalo proton. The two β-phthalo hydrogens (10.6 and 10.8 ppm) are almost at the same distance (7.6 Å) and their relaxation constants 26 and 27 ms are also very similar. Rather unexpectedly, the o-phenyl protons appear at 11.3 ppm and have a relaxation constant of 9 ms, but it makes sense as they are located 6.6 Å from cobalt. The p-phenyl protons are at a longer distance 8.9 Å, which makes them less shifted 9.8 ppm and relaxing longer 69 ms. The tert-butyl protons are at the same distance in average as the p-phenyl, and their relaxation time is similar. When plotting the dependence of T1vs. distance in a log–log scale (Fig. 5), one obtains a straight line showing the dependence of r−6, as expected from theory.27 The frog isomer had two types of the rings. The rings B had the phenyl groups pointing towards each other. This made the p-phenyl protons stretched away from the phthalocyanine (9.5 Å to cobalt), with longer relaxations (83 ms) and higher chemical shifts (8.5 ppm). The p-phenyl hydrogen of the ring D is closer to cobalt at 8.6 Å, relaxing faster at 64 ms and shifted downfield to 9.9 ppm. The other sets of protons demonstrated the same trend – less deformed ring D gave higher chemical shifts, shorter relaxations, and shorter distances to cobalt especially so for the tert-butyl groups. Same large difference was observed for the signals of the β′-phthalo protons of the rings B, which underwent an upfield shift to 6.7 ppm. A plausible explanation would be again the deformation of the phthalocyanine ring, making the proton going outside of the plane.
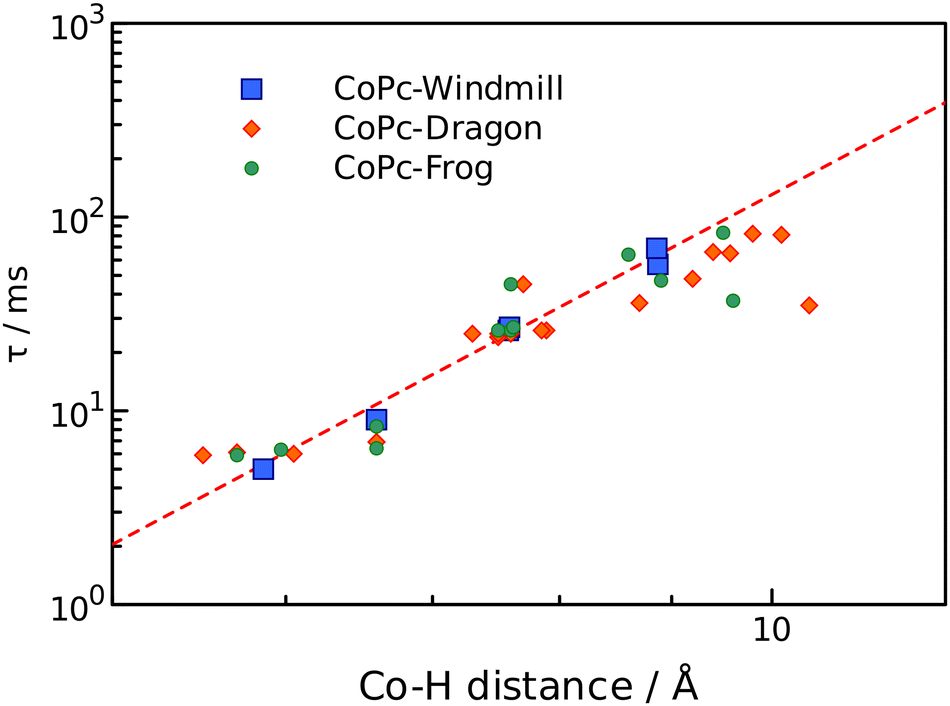 | ||
| Fig. 5 Relaxation times as a function of r−6 distance to cobalt ion. Windmill, squares; dragon, diamonds; frog, circles. | ||
For the dragon isomers, mapping the signals was the most complex. The proton peaks were easily separable into groups identified by relaxation times, but their attribution to the phthalo rings required employing the data for windmill and frog as a calibration graph (Fig. 5) and arranging the signals according to the proximity to the cobalt ion. The alfa-phthalo protons with relaxation time 5–6 ms resided at 5–6 Å, the o-phenyl rings were next in distance with similar relaxations.
Assigning the β protons would not be reliable with the available data, except for the “bent” β′-phthalo atoms of the rings B and C, which were shifted downfield similarly to the signals of the frog isomer. Also, the signals of tert-butyl groups followed nicely the trend mentioned above – stretched away, longer relaxing, upfield shifted signals for the rings B and C, and nearly identical to the windmill signals of the rings A and D.
Fig. 5 shows that the most significant deviation from the theoretical prediction is for the protons which are lying off the phthalocyanine plane, especially for the deformed tert-butyl groups. On the other hand, these distances were determined as an average value and are variable in fact. Also, this might indicate that the effect of the dz2 orbital of cobalt ion might be anisotropic. Either way, the proposed synthesized set of isomers proved to be an excellent testbed for probing the paramagnetic effect on a variety of distances and orientational angles.
Optical properties
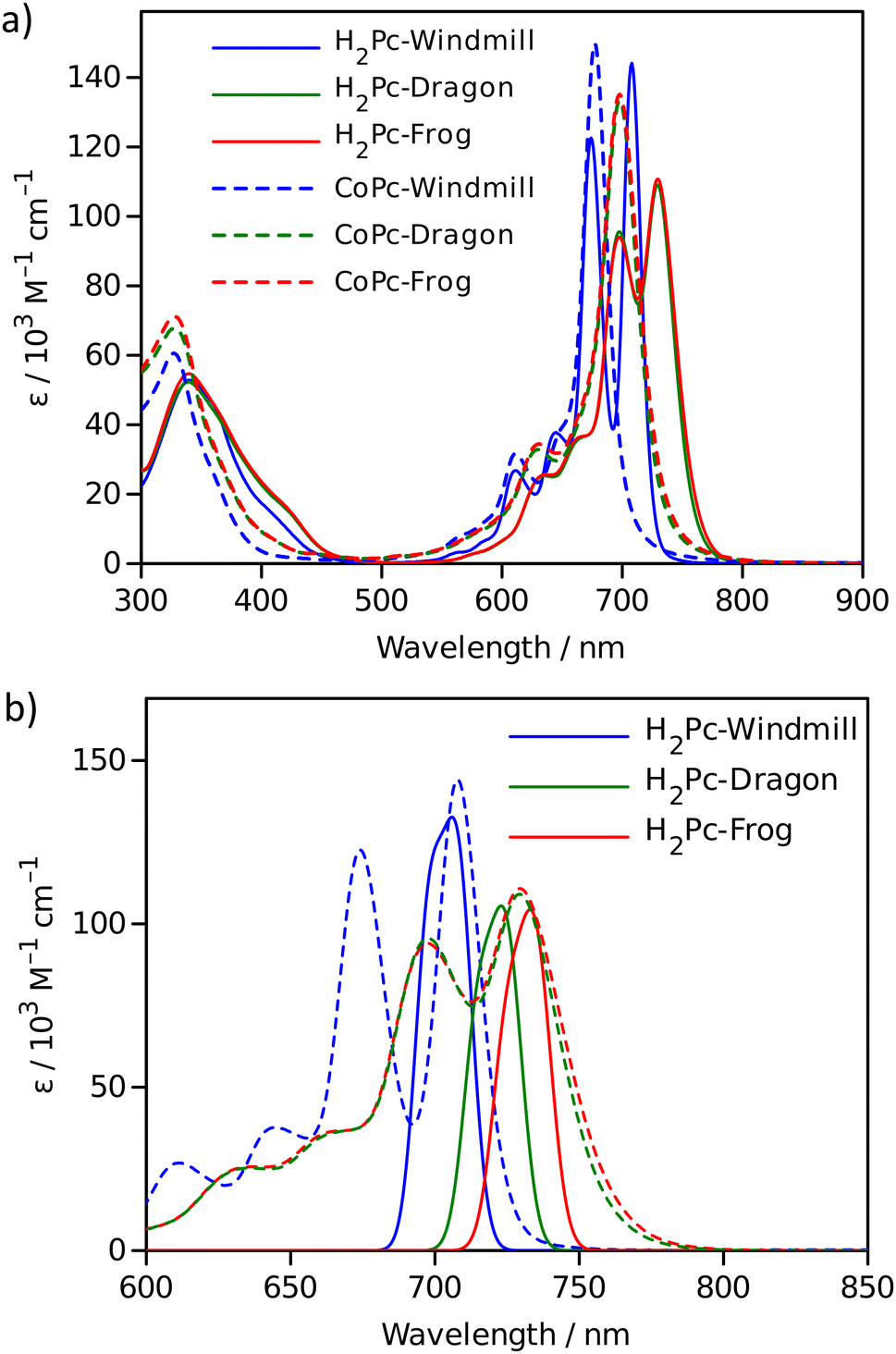 | ||
| Fig. 6 Absorption spectra: (a) measured in toluene for free bases (solid lines) and cobalt complexes (dotted lines); (b) free base spectra (dotted lines) overlapped with the calculated absorption bands (solid lines). | ||
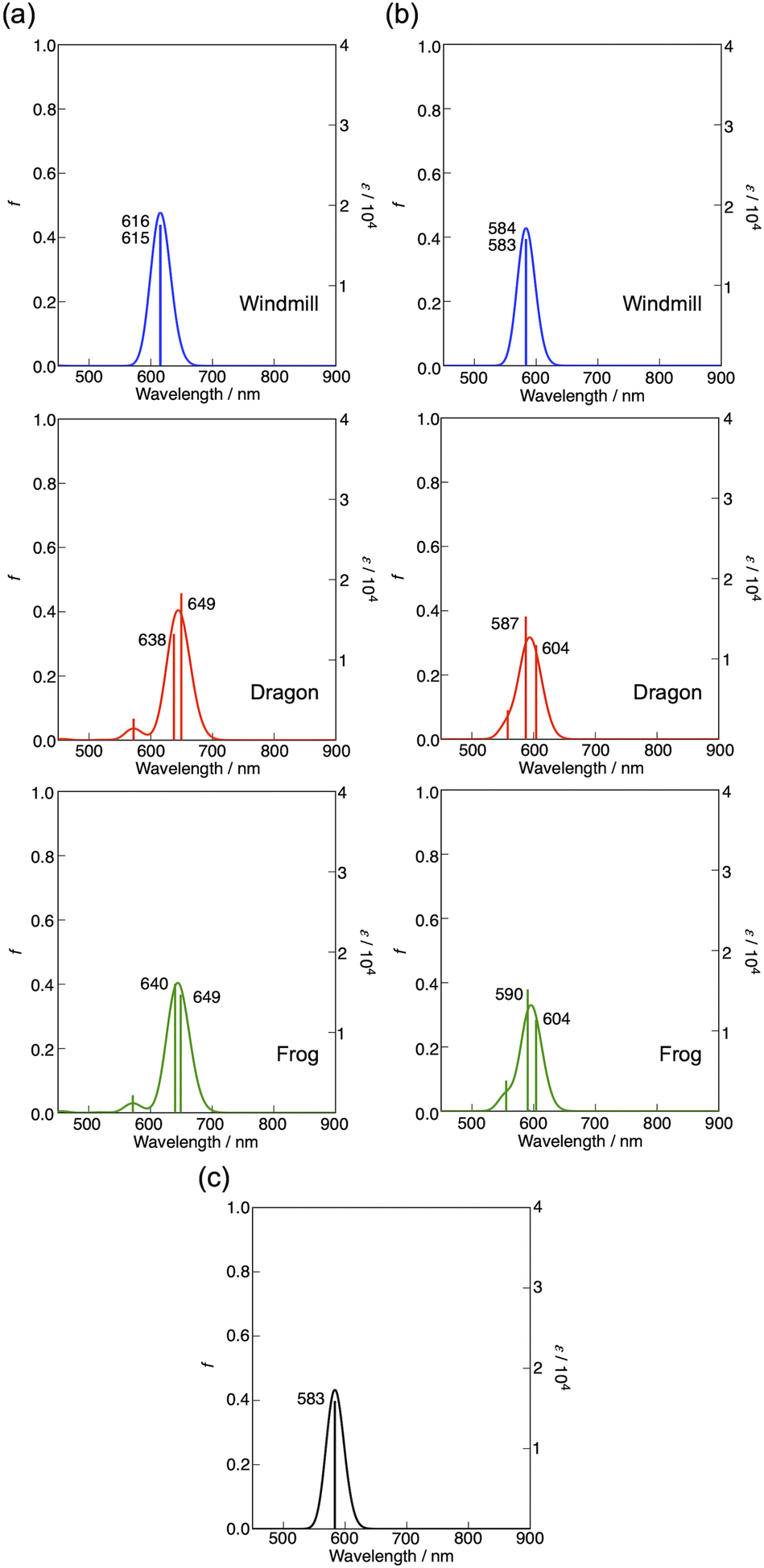
The effect of macrocycle distortion on absorption spectra can be clearly seen in a gradual shift of the Q bands. Also, it can be noted that there is virtually no difference in absorption spectra of frog and dragon compounds. The Q band red shift for frog and dragon isomers is roughly 22 nm for both the free base and Co phthalocyanine (Table S1, ESI†). This agrees well with theoretical modelling prediction (Fig. 6). The computational studies demonstrate conclusively that the large Q band red shifts (22 nm) seen experimentally for very nonplanar (dragon, frog) molecules compared to symmetrical (windmill) moiety are primarily the result of the phthalocyanine core structural deformations, which is in line with previous observations.32
The shift is accompanied by a gradual band broadening from 21 nm for windmill isomer to 28–30 nm for dragon and frog isomers of Co compounds, and even larger broadening from 20 nm to 33–36 nm for the free base compounds. The broadening observed for the Co complexes can be mainly attributed to the departure of symmetry from C4h since the excited state of the frog and dragon is not degenerate.
The apparent molar absorption coefficient (ε) at the Q band maximum of the Co complexes is clearly larger for high symmetry windmill isomer when compared to those of dragon and frog isomers. This is ascribable to the splitting of the Qx and Qy bands in the latter two isomers. Accordingly, the oscillator strength (proportional to the product ε × Δλ) is roughly the same for different isomers, being in the range f = 0.2–0.25, which corresponds to “allowed” transitions. This is reasonable as optical properties of the phthalocyanines are determined by the conjugated π-systems of the macrocycles and should not differ much for compounds with the same chromophore.
It was shown that in some Co porphyrin derivatives a so-called charge transfer (CT) transition corresponding to a shift of electron density from the porphyrin macrocycle to the central metal, Co, can be observed, though the oscillator strength of the transition (and thus the molar absorption coefficient) is weak.33 The NIR absorption spectra of the Co phthalocyanines were measured at sample concentration of 1 mM in 1 cm cuvettes and are presented in Fig. S11 (ESI†), which shows distinct and relatively narrow absorption bands just above 1000 nm wavelength. The molar absorption coefficient for this band is roughly 300 M−1 cm−1, and the oscillator strength of this transition can be estimated to be close to f ≈ 4 × 10−4, which qualifies formally this transition as forbidden one. It can be noted that the reported molar absorption coefficient for similar transition measured for CoTPP is 75 M−1 cm−1 (at 856 nm) but the band widths was much larger,33 so the oscillator strength must be roughly the same. Similarly to the Q-band positions of different isomers, the NIR band position are also red-shifted for distorted isomers. The NIR band of windmill is at 1008 nm (1.23 eV) and at 1028 (1.21 eV) and 1034 nm (1.20 eV) for dragon and frog isomers, respectively. The bandwidth (FWHM) for windmill is 38 nm but broadened to 65 and 59 nm for dragon and frog, respectively.
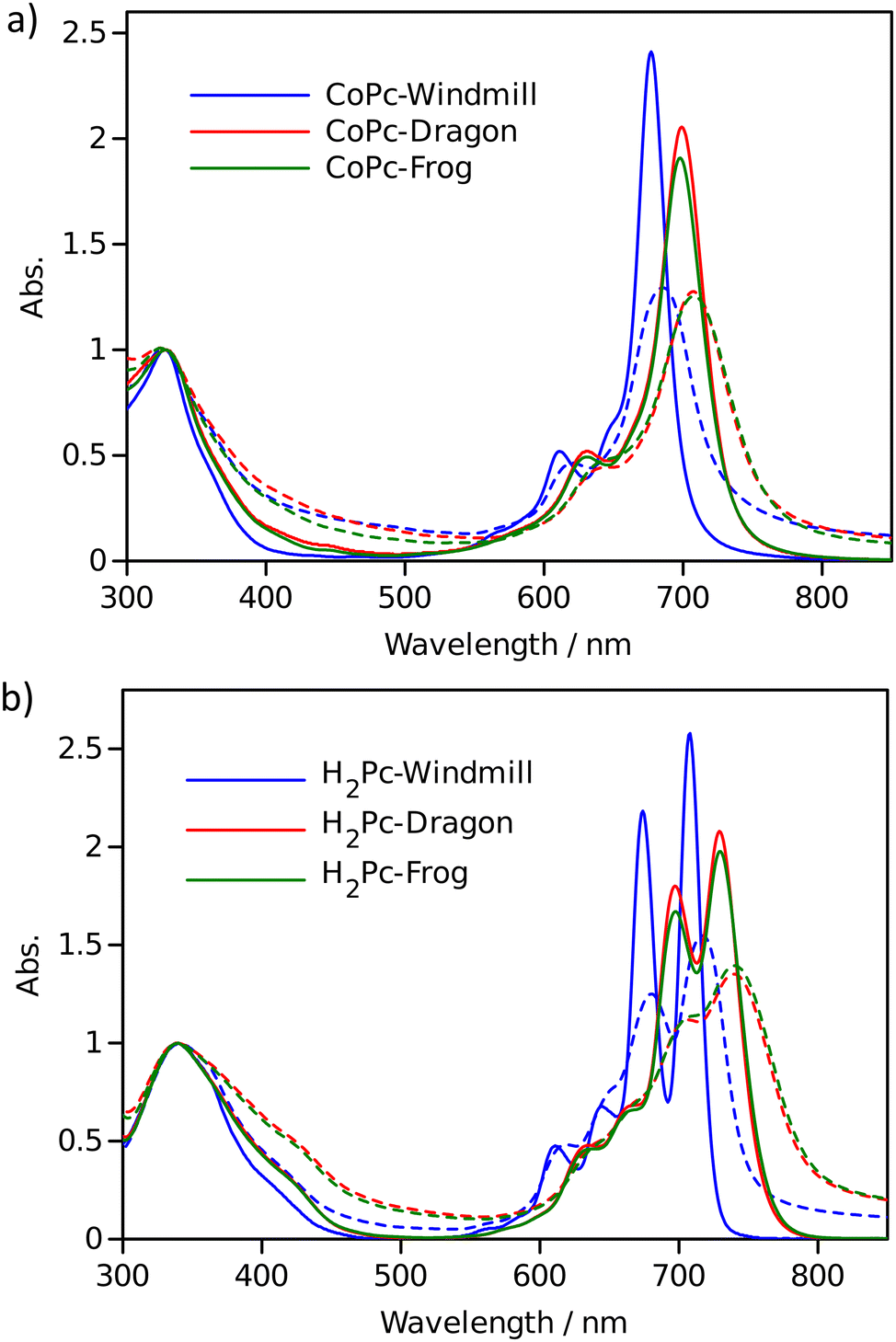 | ||
| Fig. 7 Comparison of the absorption spectra in solution (solid lines) and films (dotted lines) for Co complexes (a) and free bases (b). The absorbance at the Soret band was set as unity. | ||
Magnetic circular dichroism spectroscopy and spectroelectrochemistry
The magnetic circular dichroism (MCD) spectrum of Co windmill isomer was measured to understand its electronic structure. As seen in Fig. 8a, the complex exhibited an intense negative-to-positive MCD sign sequence corresponding to the Q band. The observed MCD signal, which can be assigned to the Faraday A term, is characteristic of phthalocyanine metal complexes with high symmetry.34,35 We studied the electrochemistry of Co windmill molecule to gain an insight into the valency of the cobalt ion. Fig. 8b shows the changes of the electronic absorption spectra during the electrochemical reduction of Co windmill at a potential negative of the first reduction. A new absorption band appeared at 479 nm is attributable to the metal-to-ligand charge transfer (MLCT) transition(s) of Co(I) phthalocyanine.36 We therefore confirmed that the cobalt ion in its initial neutral form was divalent.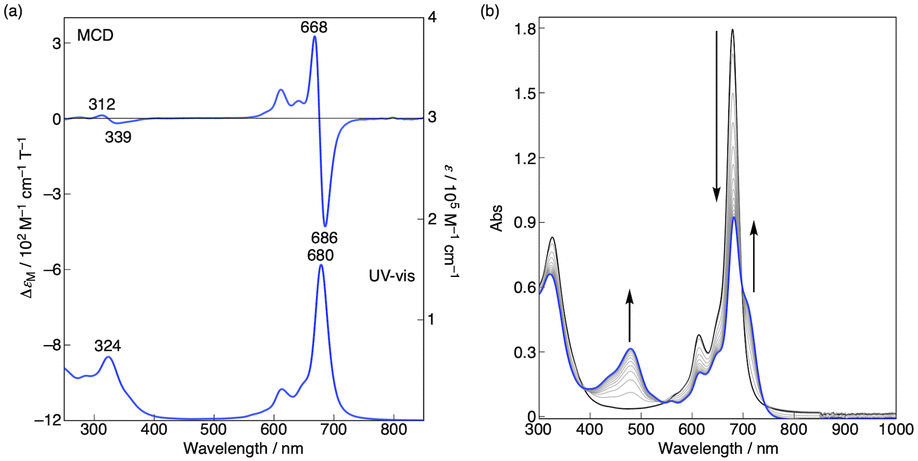 | ||
| Fig. 8 (a) MCD (top) and electronic absorption (bottom) spectra of Co windmill complex in CHCl3 at room temperature. (b) Electronic absorption spectra during the electrochemical reduction of windmill at –0.8 V (vs. Ag/AgCl) in benzonitrile with 0.1 M n-Bu4NPF6 under a nitrogen atmosphere. | ||
 | ||
| Fig. 9 (a) Calculated absorption spectra for the optimized structures of Co complex (a), three isomers in which peripheral groups are replaced by hydrogen atoms (b), and non-substituted CoPc (c). | ||
As seen in this figure, compared with unsubstituted flat CoPc (583 nm), the Q bands of windmill, dragon, and frog were estimated at 616, ca. 644 (average of two calculated Q transitions), and ca. 645 nm, respectively. Accordingly, the effect of four phenyl groups at α-positions plus deformation effect was calculated to be about 33, 61, and 62 nm, respectively. Fig. 9b shows the results on molecules without phenyl-substituents but the extent of deformation of the Pc plane is the same as that in Fig. 9a. In this case, the Q band position was calculated at ca. 584, 596, and 597 nm, respectively in the above order. Thus, the effect of deformation is amounted to be ca. 1, 13, and 13 nm while that by four phenyl-substituents at α positions were 32, 48, and 48 nm for the windmill, dragon, and frog isomers, respectively. Thus, the effect of phenyl substituents at the α positions for the dragon and frog isomers is similar and much larger than that for the windmill isomer. Since, the effect of substituents and deformation of the Pc plane on the absorption spectra are known to work synergistically for ZnPcs whose deformation is well quantified,32 we may be able to conclude that the extent of deformation for the dragon and frog-isomers is by chance similar. As calculated here, spectral shift due to four phenyl substituents is larger when they are linked to the well deformed Pc plane, and this is exactly what was observed for deformed ZnPcs.32
| Pc2− | Pc1− | Co(I) | Pc1+ | Pc2+ | |
|---|---|---|---|---|---|
| H2Pc-windmill | −1.72 | −1.38 | 0.33 | 0.70 | |
| H2Pc-dragon | −1.71 | −1.34 | 0.27 | 0.65 | |
| H2Pc-frog | −1.72 | −1.33 | 0.25 | 0.63 | |
| CoPc-windmill | −1.63 | −0.78 | 0.26 | 0.82 | |
| CoPc-dragon | −1.65 | −0.86 | 0.19 | 0.69 | |
| CoPc-frog | −1.44 | −0.85 | 0.19 | 0.68 |
The energy positions of the HOMO states obtained from electrochemical measurements are presented in Fig. 10. The most prominent feature that can be deduced from these results is destabilization of the HOMO orbitals for nonplanar dragon and frog isomers as compared with windmill molecule. The similar upward shift in energy has been observed both for free base and cobalt-substituted complexes.
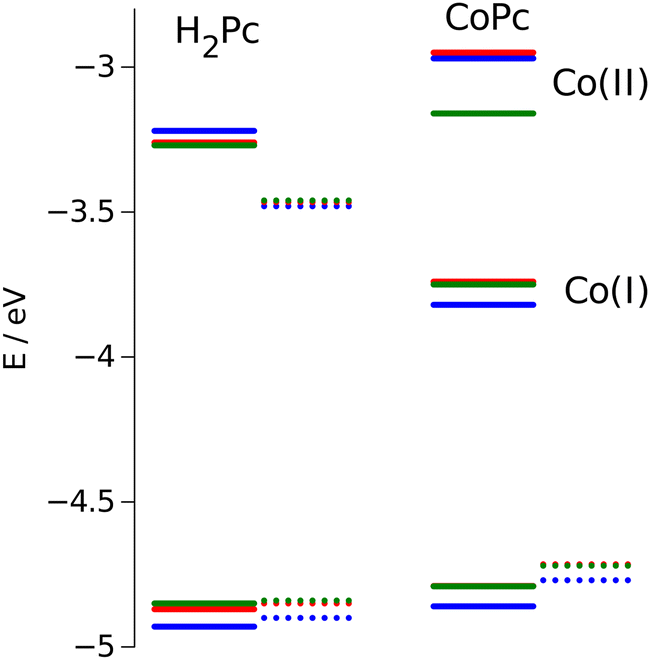 | ||
| Fig. 10 Calculated HOMO/LUMO energies (dotted lines) vs. measured values (solid lines). Values for windmill are in blue, for dragon in green, and for frog isomer are in red. | ||
The calculation of the HOMO eigenvalues depicted in the same figure are sustainable with experimental observations. We notice that upward shifts of the HOMO orbitals on ca. 0.05 eV for the complexes with distorted core provided by PBE functional is even quantitatively corroborating the experimental value.
Obviously, the twisting of the macrocycle should lead to the changes in its HOMO position. Indeed, the flat aromatic core possesses π-bonding orbitals of the same energy, while the broken symmetry induces perturbation in the π-orbitales that violates their equivalence. This conclusion is in agreement with results reported by other authors.16,41–43 In these reports, the red shift of the Q and Soret bands is associated with destabilization of the HOMO a1u and a2u orbitals, especially for ruffled and saddled porphyrins.
Electron spin resonance (ESR) spectroscopy
Low-spin Co(II) phthalocyanines and porphyrins are known to be ESR active at low temperature and their ESR spectra are sensitive to the environment.43 We therefore measured the low temperature ESR spectrum of Co windmill isomer. As shown in Fig. 11a, the complex exhibited ESR signals at g⊥ = 2.276 and g‖ = 2.001. These g values are comparable to those of the Co(II) complex of 1,4,8,11,15,18,22,25-octaphenylphthalocyanine (g⊥ = 2.2896 and g‖ = 1.9603).44 Although the 59Co (I = 7/2) hyperfine splitting is often observed for the ESR spectra of low-spin Co(II) phthalocyanines and porphyrins, our complex did not exhibit hyperfine structure.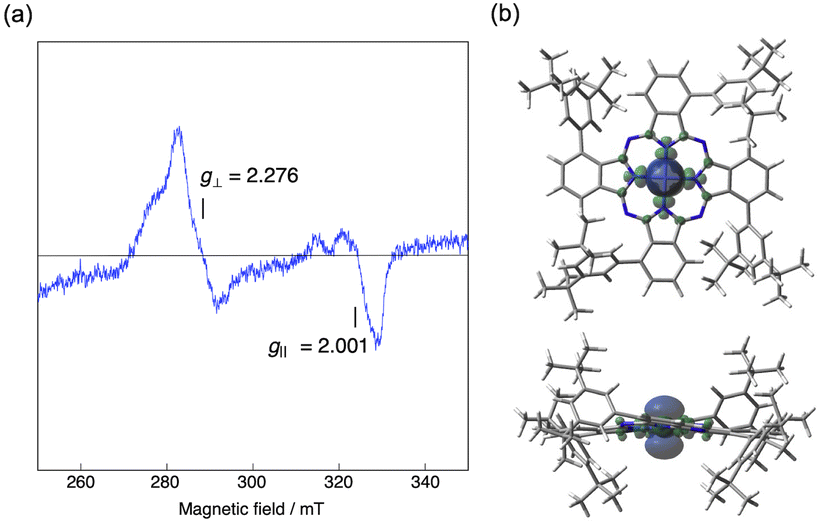 | ||
| Fig. 11 (a) ESR spectrum of Co windmill in benzonitrile at 77 K under Ar. The sample concentration was 0.5 mM. (b) Spin density map (isoval = 0.0004, blue: positive spin, green: negative spin) of the low-spin doublet state of Co windmill calculated at the UB3LYP/6-31G(d,p) level. | ||
To understand better this ESR/NMR activity, we have calculated the spin density of the low-spin doublet state of Co(II) in windmill molecule. As is clearly seen in Fig. 11b, the spin density is high only in the close vicinity of Co(II) ion, and does not spread into the outer benzene moiety, suggesting that there is almost no interaction between the Co(II) ion and benzene protons of the Pc periphery. This appears to be the reason that we observed its 1H NMR signals just like those of diamagnetic compounds. Thus, though the ESR data and spectroelectrochemical experiment revealed that the oxidation state of Co is +2, we recorded 1H NMR spectrum which is similar to those of diamagnetic compounds.
Photophysics
The excitation wavelength was set to 610 nm, close to the second minor Q band. This allows monitoring spectral perturbations of the main Q band and keeping excitation photon access energy at a reasonable minimum (610 nm over 680 nm of the Q band maximum). As anticipated, the transient absorption (TA) response of Co compounds was short-lived. There were fast perturbations within roughly 200 fs which were attributed to instrument artifacts (e.g. pump–probe interference22,23,46–53) and possibly excited state thermalization, however the rest of response can be fitted globally to bi-exponential decay model. The first time constant was around 1 ps and the second around 10 ps for all three compounds in toluene. An example of the decay associated spectra (DAS) for CoPc-windmill in toluene is shown in Fig. S15 (ESI†).
The doublet excited state may decay yielding the quartet state via inter-system crossing process or resulting in the “charge transfer” state when the electron density from the phthalocyanine macrocycle is shifted to the central metal, Co. The latter was reported for a number of Co porphyrins, namely when the initially excited doublet 2Q(π,π*) decays to 2CT(π,d) with the electron localized on dz2 orbital of Co, which is conversion of Co(II) to Co(I).54 We expect similar process to take place in our Co phthalocyanine compounds. The existence of the CT state is confirmed by the NIR band as discussed above (see Fig. S11, ESI†). The charge transfer, or the electron density shift to Co must be sensitive to the medium polarity, whereas the inter-system crossing is virtually insensitive to the solvent polarity. Therefore, the TA measurements were repeated in benzonitrile (PhCN), and the obtained time constants are summarized in Table 3. Apparently, the relaxation is slower in PhCN, which agrees with the relaxation through the charge transfer state.54 Thus, the proposed reaction scheme is
| Co(II)Pc + hv → Co(II)PcD1 → Co(I)Pc+ → Co(II)Pc |
| Compound | Solvent | τ 1, ps | τ 2, ps |
|---|---|---|---|
| CoPc-windmill | Toluene | 1.5 ± 0.1 | 9.2 ± 0.2 |
| PhCN | 2.26 ± 0.05 | 24.2 ± 0.6 | |
| CoPc-dragon | Toluene | 0.83 ± 0.01 | 7.7 ± 1 |
| PhCN | 1.40 ± 0.03 | 24.7 ± 0.5 | |
| CoPc-frog | Toluene | 1.75 ± 0.03 | 12.3 ± 0.3 |
| PhCN | 0.83 ± 0.02 | 19.3 ± 0.5 |
Experimental/methods and materials
General notes
Solvents and reagents were obtained commercially from TCI Europe, Merck-Sigma-Aldrich-VWR and used as received unless otherwise noticed. NMR spectra were recorded on a JEOL JNM-ECZ500R 500 MHz instrument and processed with JEOL Delta 5.2 software. Chemical shifts are reported in parts per million (ppm) and referenced to TMS (0.00 ppm) for 1H NMR spectra and CDCl3 (77.16 ppm) for 13C NMR spectra. T1 measurements were done via an inversion recovery method with the arrayed delay between 2π and π pulses, followed by fitting with a nonlinear inversion recovery algorithm built in the JEOL Delta 5.2 software. High-resolution ESI-TOF mass spectra were measured on a JEOL ESI-TOF JMS-T100LP instrument using the reserpine as a mass correction reference. The UV/vis absorption spectra were recorded with a Shimadzu UV-1800 spectrophotometer. The fluorescence emission spectra were recorded with FLS-1000 spectrofluorimeter (Edinburg Instruments UK). The raw signals were corrected using an instrument response function provided by the manufacturer.The magnetic circular dichroism (MCD) spectra were recorded with a JASCO J-1500 spectropolarimeter equipped with a JASCO PMCD-593 permanent magnet that produces parallel and antiparallel magnetic fields of up to 1.2 Tesla. The magnitude of the MCD signal is expressed in terms of magneto-molar circular dichroic absorption ΔεM/M−1 cm−1 T−1.
Spectroelectrochemistry experiments were performed with a Hokuto Denko HZ-3000 system and JASCO V-670 spectrometer. A 1 mm quartz cell with a platinum-mesh working electrode, a platinum-wire counter electrode, and a Ag/AgCl reference electrode were used to obtain the absorption spectra of the reduced species.
The electron spin resonance (ESR) spectra were recorded with a JEOL JES-RE3X spectrometer. Mn2+ was used as an internal standard.
The transient absorption spectroscopy measurements were done using a femtosecond pump–probe system. All samples were dissolved in tolunene and placed in 2 mm cuvette. A Libra F laser system (Coherent Inc.) was employed to create fundamental light pulses at 800 nm at a repetition rate of 1 kHz. The pulse energy was 1 mJ, and the pulse duration was approximately 100 fs. The fundamental beam was split in two, and the majority of the beam energy was directed to a Topas C optical parametric amplifier (Light Conversion Ltd) to produce excitation pulses at the desired wavelength. Ca. 10% of the fundamental beam was delivered to a white continuum generator (sapphire crystal) for sample probing. The probe beam was split in two to record reference and signal responses. The measurement system (ExciPro, CDP Inc.) was equipped with a silicon CCD array for measurements in the visible part of the spectrum, and an InGa diode array for the near-infrared (NIR) wavelengths. The measurements were carried out by comparing responses with and without excitation using a chopper synchronized with the fundamental laser pulses. The spectra were typically acquired by recording 5000 shots, averaging over 5 s. The recorded wavelength ranges were: visible (460–740 nm) and NIR (880–1100 nm).
Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). The green fraction was evaporated to dryness under reduced pressure to give a mixture of the three isomeric phthalocyanines. The solid residue was suspended in hexane, filtered off, rinsed with hexane, and dried under vacuum to afford the title compound as a turquoise powder. Yield 337 mg, 53%. The hexane washings were evaporated to dryness and retained for subsequent isolation of H2Pc-dragon and H2Pc-frog (vide infra). 1H NMR (500 MHz, CDCl3): δ = 8.40 (dd, J = 7.3, 1.0 Hz, 4H), 8.03 (dd, J = 7.3, 1.0 Hz, 4H), 7.98 (t, J = 7.3 Hz, 4H), 7.95 (t, J = 1.8 Hz, 4H), 7.88 (d, J = 1.8 Hz, 8H), 1.50 (s, 72H), −0.32 (s, 2H) ppm; 13C NMR (126 MHz, CDCl3): δ = 150.7, 141.4, 140.2, 133.1, 132.2, 129.0, 124.3, 122.9, 122.1, 35.4, 32.0 ppm; HRMS (ESI): m/z calcd for C88H98N8Na+: 1289.78067 [M + Na]+, found: 1289.78027.
:1 to 2:1). After evaporation of appropriately pooled fractions, the residues were washed with ethanol, acetonitrile and methanol and dried under vacuum.
:1 to 1:1). After evaporation of appropriately pooled fractions, CoPc-windmill and CoPc-frog were washed with acetonitrile and methanol and dried under vacuum, and CoPc-dragon was additionally purified by preparative TLC (hexane/CH2Cl2 1:1).
:1). The green fraction was evaporated to dryness under reduced pressure to give a mixture of the three isomeric phthalocyanines. The solid residue was suspended in hexane, filtered off, rinsed with hexane, and dried under vacuum to afford the title compound as a turquoise powder. Yield 337 mg, 53%. The hexane washings were evaporated to dryness and retained for subsequent isolation of H2Pc-dragon and H2Pc-frog (vide infra). 1H NMR (500 MHz, CDCl3): δ = 8.40 (dd, J = 7.3, 1.0 Hz, 4H), 8.03 (dd, J = 7.3, 1.0 Hz, 4H), 7.98 (t, J = 7.3 Hz, 4H), 7.95 (t, J = 1.8 Hz, 4H), 7.88 (d, J = 1.8 Hz, 8H), 1.50 (s, 72H), −0.32 (s, 2H) ppm; 13C NMR (126 MHz, CDCl3): δ = 150.7, 141.4, 140.2, 133.1, 132.2, 129.0, 124.3, 122.9, 122.1, 35.4, 32.0 ppm; HRMS (ESI): m/z calcd for C88H98N8Na+: 1289.78067 [M + Na]+, found: 1289.78027.
:1 to 2:1). After evaporation of appropriately pooled fractions, the residues were washed with ethanol, acetonitrile and methanol and dried under vacuum.
:1 to 1:1). After evaporation of appropriately pooled fractions, CoPc-windmill and CoPc-frog were washed with acetonitrile and methanol and dried under vacuum, and CoPc-dragon was additionally purified by preparative TLC (hexane/CH2Cl2 1:1).
DFT calculations
Most DFT calculations were performed with the DMol3 package.55,56 The Perdew–Burke–Ernzerhof exchange–correlation functional (PBE)57 was used together with Grimme's dispersion correction.58 For all atoms, a DNP type basis set including a polarization function was used.59 Geometries were fully optimized with the conductor-like screening model (COSMO)60,61 to account for solvation effects. Frequency calculations were carried out to check the Gibbs free energies and to obtain the optical absorption spectra for the molecular structures under study using time-dependent density functional theory (TDDFT). Calculation of NMR chemical shifts and nuclear spin–spin coupling constants was done with the Amsterdam density functional program (ADF 2022.103).62,63 The TZ2P-J basis set was used for the NMR calculations. The electronic absorption spectra and spin density map for the metalated molecules were calculated for the optimized geometries of the low-spin doublet CoPcs at the UB3LYP/6-31G(d,p) level using the Gaussian 16 package.39 Gaussian bands with a half width at half maximum of 500 cm−1 were used to produce the calculated absorption spectra.Conclusions
In porphyrinoid chemistry it has been commonly accepted that the NMR spectra of cobalt complexes are ill-resolved. To the best of our knowledge, 1262 articles dealing with cobalt phthalocyanines have been published since the year 1934. Most of these reports provide no NMR data for the CoPcs. Typically, in a work studying a Pc core with a range of metal ions, e.g. Zn, Ni, Co, NMR measurements for the Co complex are not performed.22,23,46–53 Still, there are 32 articles in which NMR measurements for the CoPcs were carried out. On examination of the published data, one finds that in some of these publications only lists of chemical shifts are given; in cases when the experimental spectra are shown, the resolution is poor.11,64–73 In other publications however, the data are presented both as lists of chemical shifts and experimental traces, and the spectral resolution is mostly good enough for a proper integration.44,74–92 The NMR spectra display signals of the peripheral groups distant to the cobalt centre, and, in some cases, broadened peaks of the phthalo protons. Upon closer inspection of the structures, one observes that the first category of CoPcs with poorly resolved NMR spectra encompasses either unsubstituted Pcs or Pcs with linear or planar substituents. To the second group of CoPcs with sufficiently resolved NMR spectra belong molecules with ample peripheral substitution, large aryl side groups, or other sterically imposing fragments that restrict co-planar intermolecular interactions between the Pc cores.In our case, the CoPcs investigated show normal 1H and 13C NMR spectra similar to those of diamagnetic Pc compounds. The spectroelectrochemical and low-temperature ESR measurements proved that the oxidation state of the cobalt ion in CoPc-windmill is +2, hence the complex is paramagnetic. Considering the high-quality NMR spectra, this may indicate that the spin density could be localised in close vicinity of the metal centre, thus not significantly affecting the rest of the molecule. To confirm this hypothesis, we performed spin–density calculations, which showed that the cobalt's unpaired electron is confined to the metal centre so as to not interact appreciably with the protons on the Pc's periphery. This observation would imply that, considering the examples described above,44,74–78,80–92 the main factor affecting the spectral resolution must be the phthalocyanines’ aggregation.
The isomeric compounds synthesised represent an ideal model to study the effects of peripheral substitution and macrocycle bending on the physical properties of Pcs. DFT modelling of the three isomers predicted a flat macrocyclic core for the windmill isomer and a distorted saddle-shaped macrocycle for the dragon and frog isomers, in line with the UV-vis and NMR spectra and the paramagnetic relaxation enhancement measurements. The Q band in the absorption spectra undergoes a pronounced red shift and broadening on macrocycle bending and/or symmetry lowering. Since the three isomers bear the same weakly electron donating peripheral groups (t-Bu2Ph) at the α-phthalo positions, these spectral perturbations cannot be attributed to the electron donating/accepting properties of the substituents.
We envisage that the discovered lack of aggregation might be highly beneficial for possible catalytic applications.5,6,8,24,44,64,73,90,91,93–97 It can be seen that the bulky t-Bu2Ph groups do not prevent access to the Co atom in the centre of the macrocycle. Small molecules like carbon dioxide can freely approach the metal centre, and we foresee that well-known catalytic properties of Co porphyrins and phthalocyanines can be fully exploited with all the three isomers studied herein. This, however, goes beyond the scope of the present work. It should nonetheless be noted that all these catalytic reactions rely on converting Co(II) to Co(I) by an external source of electrons. Our electrochemical measurements clearly demonstrate that this property is retained for all the three isomeric CoPcs studied. A transition to the Co(I)Pc excited state was detected by measuring absorption spectra in the near-IR range, although its oscillator strength was found to be very weak, prohibiting direct access to this state, e.g. for photocatalytic applications. The Co(I)Pc state was observed in the relaxation pathway of the photoexcited CoPc as revealed by fsTA experiments. The lifetime of this state is short, roughly 10–20 ps, in agreement with the previously investigated Co phthalocyanines and porphyrins, and can potentially be increased by coupling the CoPc with a light-activated electron donor or immobilising it on an electrode and applying a required potential.
In summary, we have demonstrated a dramatic difference in the behaviour of phthalocyanine's positional isomers bearing bulky weakly electron donating substituents. The sterically hindering side groups determine the bending of the macrocycle thus affecting the spectral and redox properties and providing a unique insight into the structure–properties relationship. The suppressed molecular aggregation makes the prepared compounds an excellent tool for paramagnetic NMR studies, which may be useful for investigating catalytic reactions, e.g. on Co(II). The windmill phthalocyanine is a unique framework for the coordinated metal ion, which prevents aggregation while allowing small molecules to approach the central atom. Such isolation of the coordinated metal centre makes the windmill Pc a remarkable structure for prospective use in single-atom catalysis—an area of active investigation by many research groups worldwide.
Author contributions
A. P., S. V., A. E. – synthesis. A. P., S. V., M. R. A., A. E – NMR studies. M. R. A – electrochemical measurements. N. T. – optical measurements. V. G., A. M. – molecular modelling and computations. N. K., A. M. – MCD and ESR studies. N. T., V. G., A. M. – funding acquisitions. All authors contributed to writing and editing of the manuscript. A. E. – project administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding from Jane & Aatos Erkko Foundation (JAES/LACOR-31228007701); Research Council of Finland, Flagship on Photonics Research and Innovation (PREIN, 320164) is gratefully acknowledged. Vyacheslav Golovanov is grateful to the SAR Europe, program MSCA4Ukraine (grant No. 1233501), and the Research Council of Finland (grant No. 353861) for financial support. This work was partly supported by JSPS (Japan) KAKENHI(B) (No. 20H02728) (to A. M.). We thank Dr Yugo Oshima (RIKEN) for measuring the ESR spectra. Computational resources were provided by the CSC-IT Centre for Science, Espoo, Finland and HOKUSAI (RIKEN), Japan.Notes and references
- A. R. Battersby, Nat. Prod. Rep., 2000, 17, 507–526 RSC.
- M. Usman, M. Humayun, M. D. Garba, L. Ullah, Z. Zeb, A. Helal, M. H. Suliman, B. Y. Alfaifi, N. Iqbal, M. Abdinejad, A. A. Tahir and H. Ullah, Nanomaterials, 2021, 11, 2029 CrossRef CAS.
- C. J. Kaminsky, S. Weng, J. Wright and Y. Surendranath, Nat. Catal., 2022, 5, 430–442 CrossRef CAS.
- P. Cao, X. Quan, X. Nie, K. Zhao, Y. Liu, S. Chen, H. Yu and J. G. Chen, Nat. Commun., 2023, 14, 172 CrossRef CAS.
- Q. Chang, Y. Liu, J.-H. Lee, D. Ologunagba, S. Hwang, Z. Xie, S. Kattel, J. H. Lee and J. G. Chen, J. Am. Chem. Soc., 2022, 144, 16131–16138 CrossRef CAS.
- J. Choi, P. Wagner, S. Gambhir, R. Jalili, D. R. MacFarlane, G. G. Wallace and D. L. Officer, ACS Energy Lett., 2019, 4, 666–672 CrossRef CAS.
- S. Ren, E. W. Lees, C. Hunt, A. Jewlal, Y. Kim, Z. Zhang, B. A. W. Mowbray, A. G. Fink, L. Melo, E. R. Grant and C. P. Berlinguette, J. Am. Chem. Soc., 2023, 145, 4414–4420 CrossRef CAS.
- Y. Fu, D. Xu, Y. Wang, X. Li, Z. Chen, K. Li, Z. Li, L. Zheng and X. Zuo, ACS Sustainable Chem. Eng., 2020, 8, 8338–8347 CrossRef CAS.
- N. Kobayashi, H. Ogata, N. Nonaka and E. A. Luk’yanets, Chem. – Eur. J., 2003, 9, 5123–5134 CrossRef CAS.
- J. Ranta, T. Kumpulainen, H. Lemmetyinen and A. Efimov, J. Org. Chem., 2010, 75, 5178–5194 CrossRef CAS.
- S. Mori, M. Nagata, Y. Nakahata, K. Yasuta, R. Goto, M. Kimura and M. Taya, J. Am. Chem. Soc., 2010, 132, 4054–4055 CrossRef CAS.
- L. Tejerina, M. V. Martínez-Díaz and T. Torres, Org. Lett., 2015, 17, 552–555 CrossRef CAS.
- K. D. Mulholland, S. Yoon, C. C. Rennie, E. K. Sitch, A. I. McKay, K. Edkins and R. M. Edkins, Chem. Commun., 2020, 56, 8452–8455 RSC.
- C. J. Kingsbury and M. O. Senge, Coord. Chem. Rev., 2021, 431, 213760 CrossRef CAS.
- G. F. Manbeck and E. Fujita, J. Porphyrins phthalocyanines, 2015, 19, 45–64 CrossRef CAS.
- R. E. Haddad, S. Gazeau, J. Pécaut, J.-C. Marchon, C. J. Medforth and J. A. Shelnutt, J. Am. Chem. Soc., 2003, 125, 1253–1268 CrossRef CAS.
- B. Görlach, M. Dachtler, T. Glaser, K. Albert and M. Hanack, Chem. – Eur. J., 2001, 7, 2459–2465 CrossRef.
- V. Golovanov, V. Golovanova and T. T. Rantala, J. Phys. Chem. Solids, 2016, 89, 15–22 CrossRef CAS.
- Nat. Commun., 2021, 12, 5884 DOI:10.1038/s41467-021-26130-0.
- K. Virkki, H. Hakola, M. Urbani, L. Tejerina, M. Ince, M. V. Martínez-Díaz, T. Torres, V. Golovanova, V. Golovanov and N. V. Tkachenko, J. Phys. Chem. C, 2017, 121, 9594–9605 CrossRef CAS.
- V. Golovanov, V. Golovanova, B. Nazarchuk and T. T. Rantala, Appl. Surf. Sci., 2023, 616, 156514 CrossRef CAS.
- M. Canlıca, J. Photochem. Photobiol., A, 2019, 384, 112043 CrossRef.
- A. Thimiopoulos, A. Vogiatzi, E. D. Simandiras, G. A. Mousdis and N. Psaroudakis, Inorg. Chim. Acta, 2019, 488, 170–181 CrossRef CAS.
- S. Roy, M. Miller, J. Warnan, J. J. Leung, C. D. Sahm and E. Reisner, ACS Catal., 2021, 11, 1868–1876 CrossRef CAS.
- M. Lehr, T. Paschelke, E. Trumpf, A.-M. Vogt, C. Näther, F. D. Sönnichsen and A. J. McConnell, Angew. Chem., Int. Ed., 2020, 59, 19344–19351 CrossRef CAS.
- A. Swartjes, P. B. White, J. P. J. Bruekers, J. A. A. W. Elemans and R. J. M. Nolte, Nat. Commun., 2022, 13, 1846 CrossRef CAS.
- H. Amouri, L. Mimassi, M. N. Rager, B. E. Mann, C. Guyard-Duhayon and L. Raehm, Angew. Chem., Int. Ed., 2005, 44, 4543–4546 CrossRef CAS.
- I. S. Tidmarsh, B. F. Taylor, M. J. Hardie, L. Russo, W. Clegg and M. D. Ward, New J. Chem., 2009, 33, 366–375 RSC.
- A. J. McConnell, C. M. Aitchison, A. B. Grommet and J. R. Nitschke, J. Am. Chem. Soc., 2017, 139, 6294–6297 CrossRef CAS.
- N. Kobayashi, R. Higashi and T. Tomura, BCSJ, 1997, 70, 2693–2698 CrossRef CAS.
- T. Sugimori, S. Okamoto, N. Kotoh, M. Handa and K. Kasuga, Chem. Lett., 2000, 1200–1201 CrossRef CAS.
- T. Fukuda, S. Homma and N. Kobayashi, Chem. – Eur. J., 2005, 11, 5205–5216 CrossRef CAS.
- A. Antipas and M. Gouterman, J. Am. Chem. Soc., 1983, 105, 4896–4901 CrossRef CAS.
- N. Kobayashi, A. Muranaka and J. Mack, Circular Dichroism and Magnetic Circular Dichroism Spectroscopy for Organic Chemists, The Royal Society of Chemistry, 2011 Search PubMed.
- J. Mack, M. J. Stillman and N. Kobayashi, Coord. Chem. Rev., 2007, 251, 429–453 CrossRef CAS.
- W. A. Nevin, M. R. Hempstead, W. Liu, C. C. Leznoff and A. B. P. Lever, Inorg. Chem., 1987, 26, 570–577 CrossRef CAS.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- N. Kobayashi and T. Fukuda, J. Am. Chem. Soc., 2002, 124, 8021–8034 CrossRef CAS.
- C. J. Kingsbury and M. O. Senge, Coord. Chem. Rev., 2021, 431, 213760 CrossRef CAS.
- S. Gentemann, C. J. Medforth, T. P. Forsyth, D. J. Nurco, K. M. Smith, J. Fajer and D. Holten, J. Am. Chem. Soc., 1994, 116, 7363–7368 CrossRef CAS.
- L. Ukrainczyk, M. Chibwe, T. J. Pinnavaia and S. A. Boyd, J. Phys. Chem., 1994, 98, 2668–2676 CrossRef CAS.
- T. Honda, T. Kojima and S. Fukuzumi, J. Am. Chem. Soc., 2012, 134, 4196–4206 CrossRef CAS.
- K. Ishii and N. Kobayashi, in The porphyrin handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, New York, 2003, vol. 16, ch. 102, pp. 1–42 Search PubMed.
- A. Günsel, A. Kobyaoğlu, A. T. Bilgiçli, B. Tüzün, B. Tosun, G. Arabaci and M. N. Yarasir, J. Mol. Struct., 2020, 1200, 127127 CrossRef.
- H. Y. Y. Akkurt, A. ihsan Okur and A. Gül, J. Porphyrins phthalocyanines, 2012, 16, 192–199 CrossRef CAS.
- S. Arucu, M. B. Sağlam and A. R. Özkaya, J. Mol. Struct., 2019, 1198, 126883 CrossRef CAS.
- M. Canlıca, J. Mol. Struct., 2020, 1214, 128160 CrossRef.
- Y. Gök, H. Z. Gök, M. K. Yılmaz, M. Farsak and İ. Ü. Karayiğit, Polyhedron, 2018, 153, 128–138 CrossRef.
- M. Kandaz, M. N. U. Yaraşir, A. Koca and Ö. Bekaroğlu, Polyhedron, 2002, 21, 255–263 CrossRef CAS.
- R. Medyouni, L. Mansour, F. Zaghroub, L. Baklouti and N. Hamdi, J. Chem. Res., 2016, 40, 216–223 CrossRef CAS.
- A. Y. Tolbin, M. S. Savelyev, A. Y. Gerasimenko, L. G. Tomilova and N. S. Zefirov, Phys. Chem. Chem. Phys., 2016, 18, 15964–15971 RSC.
- H. Z. Yu, J. S. Baskin, B. Steiger, C. Z. Wan, F. C. Anson and A. H. Zewail, Chem. Phys. Lett., 1998, 293, 1–8 CrossRef CAS.
- B. Delley, J. Chem. Phys., 1990, 92, 508–517 CrossRef CAS.
- B. Delley, J. Chem. Phys., 2000, 113, 7756–7764 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS.
- Y. Inada and H. Orita, J. Comput. Chem., 2008, 29, 225–232 CrossRef CAS.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
- A. Klamt, V. Jonas, T. Bürger and J. C. W. Lohrenz, J. Phys. Chem. A, 1998, 102, 5074–5085 CrossRef CAS.
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- G. Schreckenbach and T. Ziegler, Int. J. Quantum Chem., 1997, 61, 899–918 CrossRef CAS.
- A. Aykanat, Z. Meng, R. M. Stolz, C. T. Morrell and K. A. Mirica, Angew. Chem., Int. Ed., 2022, 61, e202113665 CrossRef CAS.
- M. D. Hartle, S. K. Sommer, S. R. Dietrich and M. D. Pluth, Inorg. Chem., 2014, 53, 7800–7802 CrossRef CAS.
- J. Jeong, R. S. Kumar, N. Mergu and Y.-A. Son, J. Mol. Struct., 2017, 1147, 469–479 CrossRef CAS.
- G. Lu, Y. Zhou, Y. Xiang and D. Xia, J. Porphyrins phthalocyanines, 2010, 14, 904–910 CrossRef CAS.
- K. A. Mirica, Z. Meng and R. M. Stolz, US Pat., US2020/0361976A1, 2020.
- F. Molla, A. Yazıcı, N. A. Selçuki, A. Altındal, B. Salih and Ö. Bekaroğlu, J. Porphyrins Phthalocyanines, 2020, 24, 662–674 CrossRef CAS.
- M. Peterson, C. Hunt, Z. Wang, S. E. Heinrich, G. Wu and G. Ménard, Dalton Trans., 2020, 49, 16268–16277 RSC.
- V. G. P. Ribeiro, J. P. F. Mota, A. E. Costa Júnior, N. M. A. Lima, P. B. A. Fechine, J. C. Denardin, L. Carbone, E. Bloise, G. Mele and S. E. Mazzetto, Molecules, 2019, 24, 3284 CrossRef CAS.
- S. Şener, Heterocycl. Commun., 2020, 26, 37–45 CrossRef.
- S. Ren, E. W. Lees, C. Hunt, A. Jewlal, Y. Kim, Z. Zhang, B. A. W. Mowbray, A. G. Fink, L. Melo, E. R. Grant and C. P. Berlinguette, J. Am. Chem. Soc., 2023, 145, 4414–4420 CrossRef CAS.
- N. G. Bichan, E. N. Ovchenkova and T. N. Lomova, Russ. J. Inorg. Chem., 2018, 63, 1453–1460 CrossRef CAS.
- N. G. Bichan, E. N. Ovchenkova, V. A. Mozgova, N. O. Kudryakova and T. N. Lomova, Polyhedron, 2022, 222, 115908 CrossRef CAS.
- R. Decréau, M. Chanon and M. Julliard, Inorg. Chim. Acta, 1999, 293, 80–87 CrossRef.
- B. Ertem, A. Bilgin, H. Kantekin and Y. Gök, Polyhedron, 2008, 27, 2186–2192 CrossRef CAS.
- A. A. Filippova, A. A. Voronina and A. S. Vashurin, Russ. J. Inorg. Chem., 2017, 62, 777–782 CrossRef CAS.
- S. Gorun, A. I. Loas, K. Griswold, L. Lapok, H. H. Patel and R. Gerdes, WO2012061344A1, 2012.
- C. Huang, K. Wang, J. Sun and J. Jiang, Dyes Pigm., 2014, 109, 163–168 CrossRef CAS.
- M. Kimura, M. Sugawara, S. Sato, T. Fukawa and T. Mihara, Chem. – Asian J., 2010, 5, 869–876 CrossRef CAS.
- S. Lin, L. Ji, L. Jing, J. Lu, H. Wang, S. Sun, B. Kong and X. Zhang, Inorg. Chim. Acta, 2015, 434, 24–30 CrossRef CAS.
- S. Makhseed, M. Al-Sawah, J. Samuel and H. Manaa, Tetrahedron Lett., 2009, 50, 165–168 CrossRef CAS.
- I. Nar, A. Atsay, H. P. Karaoğlu, A. Altındal and E. Hamuryudan, Dalton Trans., 2018, 47, 15017–15023 RSC.
- M. Nemakal, S. Aralekallu, I. Mohammed, M. Pari, K. R. Venugopala Reddy and L. K. Sannegowda, Electrochim. Acta, 2019, 318, 342–353 CrossRef CAS.
- E. N. Ovchenkova, N. G. Bichan and T. N. Lomova, Dyes Pigm., 2016, 128, 263–270 CrossRef CAS.
- E. N. Ovchenkova, N. G. Bichan, A. A. Tsaturyan, N. O. Kudryakova, M. S. Gruzdev, F. E. Gostev, I. V. Shelaev, V. A. Nadtochenko and T. N. Lomova, J. Phys. Chem. C, 2020, 124, 4010–4023 CrossRef CAS.
- E. A. Safonova, J. A. Wytko, J. Weiss, E. A. Ugolkova, N. N. Efimov, V. V. Minin, Y. G. Gorbunova and A. Yu Tsivadze, J. Porphyrins Phthalocyanines, 2020, 24, 1083–1092 CrossRef CAS.
- B. R. Schrage, W. Zhou, L. A. Harrison, D. E. Nevonen, J. R. Thompson, K. E. Prosser, C. J. Walsby, C. J. Ziegler, D. B. Leznoff and V. N. Nemykin, Inorg. Chem., 2022, 61, 20177–20199 CrossRef CAS.
- A. S. Vashurin, Russ. Chem. Bull., 2016, 65, 2220–2228 CrossRef CAS.
- G. Zhang, L. Zhang, D. Zhang, Q. Wu, Y. Sasaki, Y. Hisaeda, K. Yasuhara, J. Kikuchi and X.-M. Song, Inorg. Chem. Commun., 2020, 115, 107866 CrossRef CAS.
- S. A. Znoiko, M. A. Serova, A. A. Uspenskaya, A. V. Zav’yalov, V. E. Maizlish and G. P. Shaposhnikov, Russ. J. Gen. Chem., 2016, 86, 2501–2507 CrossRef CAS.
- P. Cao, X. Quan, X. Nie, K. Zhao, Y. Liu, S. Chen, H. Yu and J. G. Chen, Nat. Commun., 2023, 14, 172 CrossRef CAS.
- W. Fan, Z. Duan, W. Liu, R. Mehmood, J. Qu, Y. Cao, X. Guo, J. Zhong and F. Zhang, Nat. Commun., 2023, 14, 1426 CrossRef CAS.
- C. J. Kaminsky, S. Weng, J. Wright and Y. Surendranath, Nat. Catal., 2022, 5, 430–442 CrossRef CAS.
- M. Usman, M. Humayun, M. D. Garba, L. Ullah, Z. Zeb, A. Helal, M. H. Suliman, B. Y. Alfaifi, N. Iqbal, M. Abdinejad, A. A. Tahir and H. Ullah, Nanomaterials, 2021, 11, 2029 CrossRef CAS.
- T. Wang, L. Guo, H. Pei, S. Chen, R. Li, J. Zhang and T. Peng, Small, 2021, 17, 2102957 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp01564a |
| This journal is © the Owner Societies 2024 |