
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpecific interaction between the DSPHTELP peptide and various functional groups†
Haeun
Kwon‡
a,
Seongeon
Jin‡
b,
Jina
Ko
a,
Jungki
Ryu
 acde,
Ja-Hyoung
Ryu
*b and
Dong Woog
Lee
*a
acde,
Ja-Hyoung
Ryu
*b and
Dong Woog
Lee
*a
aSchool of Energy and Chemical Engineering, Ulsan National Institute of Science and Technology (UNIST), 50 UNIST-gil, Ulsan 44919, Republic of Korea. E-mail: dongwoog.lee@unist.ac.kr
bDepartment of Chemistry, School of Natural Sciences, Ulsan National Institute of Science and Technology (UNIST), 50 UNIST-gil, Ulsan 44919, Republic of Korea. E-mail: jhryu@unist.ac.kr
cEmergent Hydrogen Technology R&D Center, Ulsan National Institute of Science and Technology (UNIST), Ulsan 44919, Republic of Korea
dGraduate School of Carbon Neutrality, Ulsan National Institute of Science and Technology (UNIST), Ulsan 44919, Republic of Korea
eCenter for Renewable Carbon, Ulsan National Institute of Science and Technology (UNIST), Ulsan 44919, Republic of Korea
First published on 15th July 2024
Abstract
M13 bacteriophages serve as a versatile foundation for nanobiotechnology due to their unique biological and chemical properties. The polypeptides that comprise their coat proteins, specifically pVIII, can be precisely tailored through genetic engineering. This enables the customized integration of various functional elements through specific interactions, leading to the development of innovative hybrid materials for applications such as energy storage, biosensing, and catalysis. Notably, a certain genetically engineered M13 bacteriophage variant, referred to as DSPH, features a pVIII with a repeating DSPHTELP peptide sequence. This sequence facilitates specific adhesion to single-walled carbon nanotubes (SWCNTs), primarily through π–π and hydrophobic interactions, though the exact mechanism remains unconfirmed. In this study, we synthesized the DSPHTELP peptide (an 8-mer peptide) and analyzed its interaction forces with different functional groups across various pH levels using surface forces apparatus (SFA). Our findings indicate that the 8-mer peptide binds most strongly to CH3 groups (Wad = 13.74 ± 1.04 mJ m−2 at pH 3.0), suggesting that hydrophobic interactions are indeed the predominant mechanism. These insights offer both quantitative and qualitative understanding of the molecular interaction mechanisms of the 8-mer peptide and clarify the basis of its specific interaction with SWCNTs through the DSPHTELP M13 bacteriophage.
Introduction
The M13 bacteriophage, belonging to the filamentous bacteriophage family, is characterized by approximately 2700 copies of the major coat protein (pVIII) and is terminated at each end with about five pairs of four distinct minor proteins (pIII, pVI, pVII, and pIX).1,2 Owing to its biocompatibility, efficient production, target-specific reactivity, high surface-to-volume ratio, and flexibility, the M13 bacteriophage stands out as an ideal candidate for assembling and fabricating a variety of functional nanomaterials.3–5 Notably, phage display technology enables the facile and versatile modification of the M13 phage to bind selectively to target substances, facilitating the creation of tailor-made functional nanoparticles for epitope mapping, naive binding peptide selection, and drug discovery.6–11Due to these advantages, researchers have employed the M13 bacteriophage in a wide array of applications, including rechargeable batteries that integrate peptides for binding active materials,12–15 extracellular matrix-based cell platforms,16–19 piezoelectric devices,20 sensors,21–27etc. Specifically, DSPH phages within the M13 bacteriophage, characterized by a repeating DSPHTELP sequence in the pVIII coat proteins, have shown high specificity in adhering to single-walled carbon nanotubes (SWCNTs). This specificity has been harnessed for highly efficient applications in dye-sensitized solar cells (DSSCs),28 sodium29 and lithium-ion batteries,30 and second near-infrared (NIR) window fluorescence imaging.31 Despite the broad utility, the precise adhesion mechanisms, which are critical to the success of these applications, largely remain speculative, with a clear understanding of the biomolecular interaction mechanisms posing a challenge for the systematic design of efficient target material binding. Therefore, we investigated the interaction between the DSPHTELP peptide and various functional groups to understand the adhesion mechanism of this specific sequence and analyze the similarities and differences to the adhesion mechanism of the actual DSPH phage.
The poor understanding is attributed to the intricate nature of biomolecule interaction mechanisms, which may involve a complex interplay of hydrogen bonding, van der Waals forces, electrostatic interactions, steric repulsion, hydration forces, cation–π interactions, and hydrophobic interactions.32 In recent studies utilizing surface forces apparatus (SFA), we explored the interactions between the M13 bacteriophage (specifically the DSPHTELP-peptide) and various functionalized self-assembled monolayers (SAMs) to shed light on the interaction mechanisms, identifying π and hydrophobic interactions as predominant.33 However, definitive conclusions were elusive due to the potential involvement of other coat proteins in the adhesion process and significant phage swelling at higher pH levels (∼8.5), which emphasized steric over other interactions.
To mitigate these uncertainties observed in our previous work, this study synthesizes and examines the interactions of only the DSPHTELP-peptide (an 8-mer peptide mirroring the sequence of the pVIII protein, but devoid of other bacteriophage biomolecules such as other coat proteins and nucleotides). We analyzed the interactions between this peptide and four differently functionalized self-assembled monolayers (SAMs)—carboxylic (–COOH), amine (–NH2), methyl (–CH3), and phenyl (–C6H5)—using SFA. Our findings reveal the strongest adhesion of the peptide to CH3-SAMs, underscoring the pivotal role of hydrophobic interactions in molecular binding processes.
Experimental methods
Materials
All the amino acids (aspartic acid; D, serine; S, proline; P, histidine; H, threonine; T, glutamic acid; E, leucine; L) required for the peptide synthesis and O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HBTU) were purchased from Apex Bio, Houston and Chem-Impex, Chicago. Piperidine and diisopropylethylamine were purchased from Alfa Aesar. Dimethylformamide was purchased from Thermo Fisher Scientific. 3-(Triethoxysilyl)propyl isocyanate, 10-carboxy-1-decanethiol (95%) for COOH-SAM, 11-amino-1-undecanethiol hydrochloride (99%) for NH2-SAM, 1-undecanethiol (98%) for CH3-SAM and 2-phenylethanethiol (98%) for C6H5-SAM were purchased from Sigma-Aldrich.Synthesis of the DSPHTELP-peptide
The peptide was synthesized using the standard Fmoc-(9-fluorenylmethoxycarbonyl) solid-phase peptide synthesis (SPPS) method, scaled to 0.106 mmol. Rink amide MBHA resin was used. The Fmoc on the resin and each amino acid were removed using a DMF/piperidine mixture. In each step of synthesis, Fmoc-protected amino acid was added using O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HBTU) and diisopropylethylamine (DIPEA) as a coupling agent. Finally, the peptide was cleaved from the resin using cleavage cocktail (TFA/triisopropylsilane/deionized water mixture at a 9.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.5:0.5 ratio) and the products will be precipitated in cold ether (Fig. 1). Precipitates were purified using high-performance liquid chromatography (HPLC, Agilent Technologies, USA) with a C18 reverse column in an acetonitrile/water mixture (Fig. S1, ESI†).
:0.5:0.5 ratio) and the products will be precipitated in cold ether (Fig. 1). Precipitates were purified using high-performance liquid chromatography (HPLC, Agilent Technologies, USA) with a C18 reverse column in an acetonitrile/water mixture (Fig. S1, ESI†).
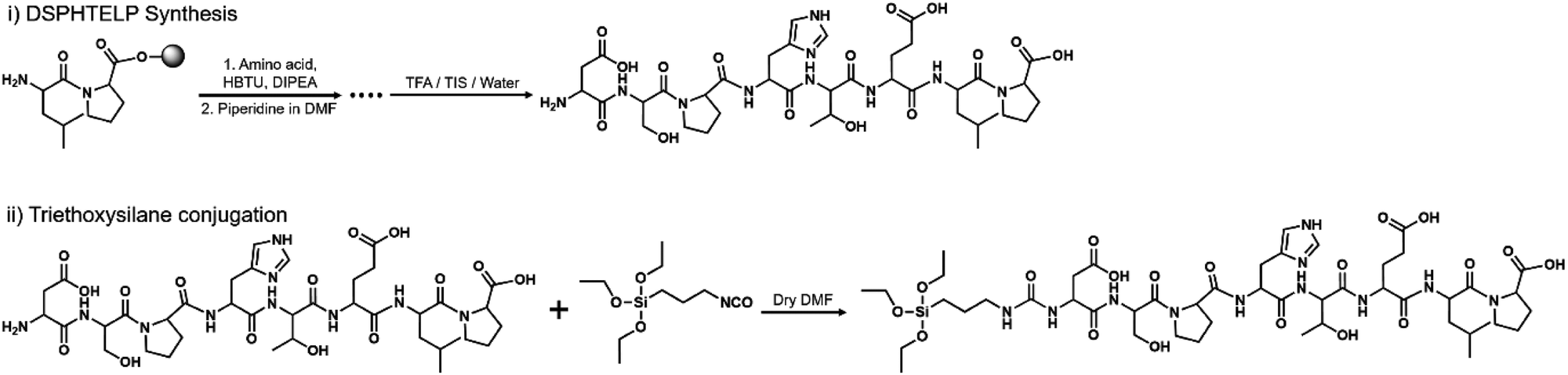 | ||
| Fig. 1 Schematic representation of the synthesis of 8-mer peptides. 8-Mer peptides were synthesized by solid-phase peptide synthesis at a 0.106 mmol scale. The N-terminal of the synthesized amino acid backbones was conjugated with triethoxysilane in anhydrous DMF solvent. | ||
DSPHTELP-peptide conjugation with triethoxysilane
The purified DSPHTELP peptide (20 mg, 0.0224 mmol) was dissolved in anhydrous DMF (1 mg mL−1). Then, 3-(triethoxysilyl)propyl isocyanate (6 mg, 0.0224 mmol) was added into the solution. After stirring at room temperature overnight, crude products were purified by size exclusion chromatography. The synthesis of peptides will be confirmed with matrix-assisted laser desorption/ionization (MALDI-TOF/TOF, Ultraflex III) as depicted in Fig. S2 (ESI†).Preparation of the DSPHTELP-peptide surface on mica
A cleaved clean mica (Grade #1, S&J Trading, Floral Park, NY, USA) with a back-silver coating of approximately 50 nm thickness was adhered to a cylindrical disk using UV glue for 50 minutes. The back-silvered mica attached to the disk was treated with O2 plasma at 100 W for 3 minutes.34The synthesized triethoxysilane–DSPHTELP peptide solution (100 μL) in DMF solvent (1 mg mL−1) was diluted in 20 mL of DMF. 50 μL of the peptide solution was dropped around the treated mica immersed in 20 mL of DMF solvent at 700 rpm. After 1 hour, weakly bound peptides were removed using DMF solvent and dried with nitrogen gas. The evenly coated peptide surface was confirmed using a Veeco Multimode V AFM in the standard tapping mode (Fig. S3, ESI†).
Preparation of self-assembled monolayer (SAM) films with different end-functional groups
Surfaces with four different functional groups were prepared by using alkanethiol monolayers deposited on atomically smooth gold surfaces. Initially, the smooth gold surface (∼45nm in thickness) was prepared through electron-beam evaporation on cleaved clean mica (Grade #1, S&J Trading, Floral Park, NY, USA). Subsequently, the gold-coated surface was affixed to a cylindrical glass disk (radius, R ∼ 2cm) using UV glue (NOA 81, Norland Products, NJ, USA), gold surface down, and allowed to cure for 50 minutes.35,36 The mica was gently peeled off from the glued surface in ethanol solution, leaving behind an atomically smooth gold surface. The gold-coated disk was then immersed in a 1mM alkanethiol–ethanol solution for 18 hours. The strong gold–sulfur interaction facilitated the effective binding of each self-assembled monolayer (SAM) to the gold surface.33,37,38 To eliminate excess SAM molecules, a low-intensity sonication was applied for 30 seconds, followed by washing with ethanol and nitrogen gas drying.39
Measurement of interaction forces using SFA
The interaction between the 8-mer peptide and each functionalized SAM was measured using surface forces apparatus (SFA2000, SurForce LLC, Santa Barbara, USA). The SFA is an instrument that directly measures the absolute distance and interaction forces between two surfaces with resolutions of 0.1 nm and 10 nN, respectively.34 The SFA offers the advantage of measuring a wide range of forces, and through a careful analysis of the force–distance curves, it can provide valuable insights on the interaction mechanism. Consequently, the SFA has been effectively utilized to quantify the interaction forces between various surfaces coated with proteins,40 lipid bilayers,41 biopolymers,42–45 and supramolecules.46The disk coated with SAM was mounted at the top of the chamber, while the disk coated with the peptide was mounted at the bottom in a cross-cylindrical geometry. 60 μL of filtered buffer solution (pH 3.0 or pH 8.5 KNO3) was injected between the surfaces and equilibrated for 30 minutes (Fig. 2). Two surfaces were brought into contact by using a fine-control motor at an approach velocity of ∼5 nm s−1. Then a short (5 s) or long (1 h) contact time (tc) was allowed to check the molecular rearrangement during the contact, followed by separation of two surfaces at a separation velocity of ∼5 nm s−1.
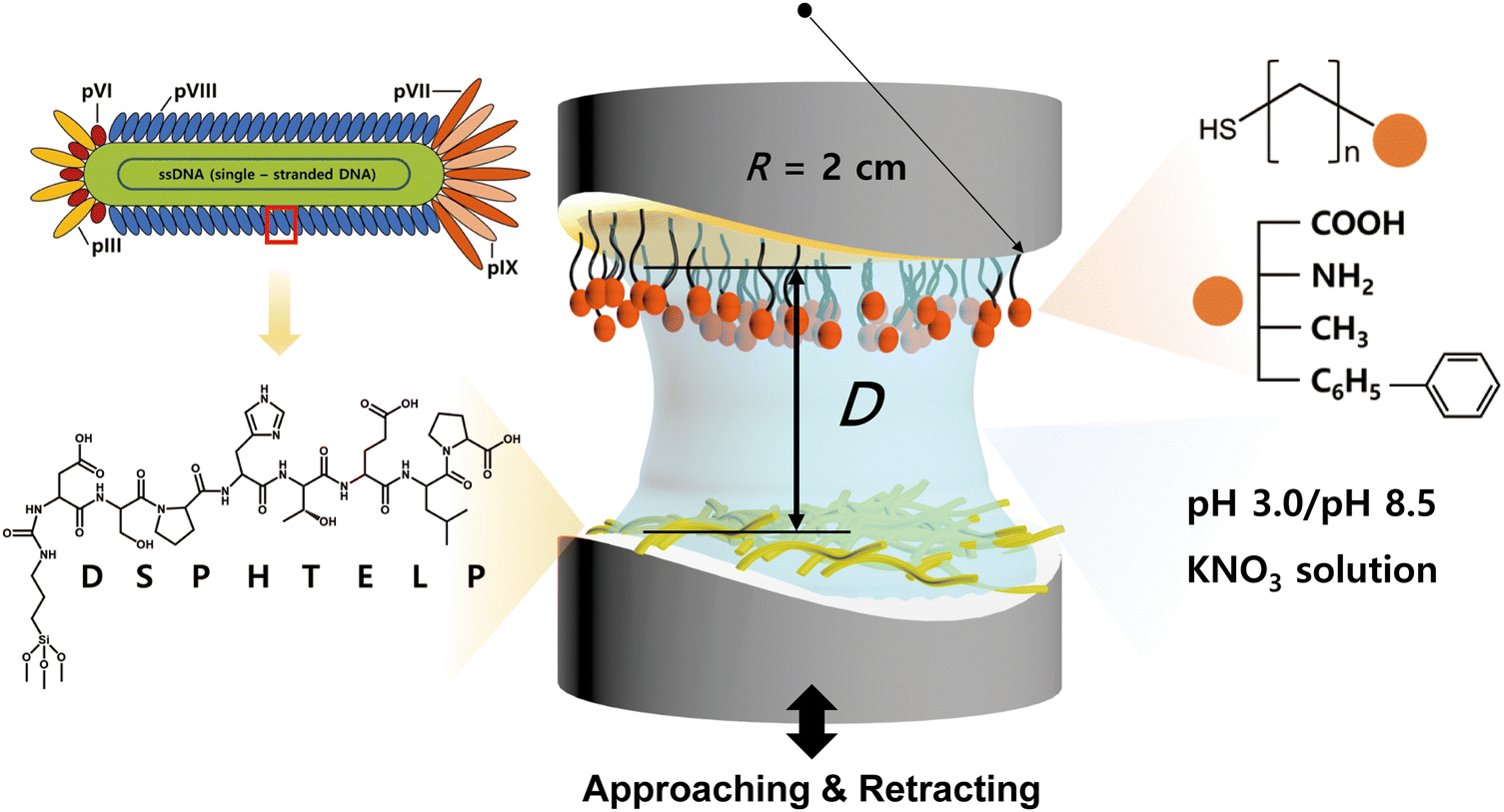 | ||
| Fig. 2 Schematic of surface forces apparatus (SFA) for measuring the interaction forces between the 8-mer peptide layer (bottom surface) and four different alkanethiols’ SAM layers (top surface) under various conditions. | ||
The absolute distance (D) between two surfaces was determined using multiple beam interferometry (MBI) by observing the fringes of equal chromatic order (FECO).47 The interaction forces (F) were measured by observing the deflection of the lower surface's double cantilever spring with a spring constant of 2451.7 N m−1. The adhesion force, Fad, was defined as the absolute value of the lowest F, before jump-out, where R is the radius of the cylindrical disk, typically around 2 cm.42,48 The Fad was converted into an interaction energy per unit of contact area (Wad = 2Fad/3πR) according to the Johnson–Kendall–Roberts model49 which is typically applied to soft materials with significant deformations. All experiments were conducted at room temperature (T ∼ 23 °C) and repeated at least three times.
Results and discussion
Interaction force measurements between the DSPHTELP-peptide and SAMs
The binding constant, Ka, is generally used to quantitatively compare the binding between two molecules50 and can be measured using surface plasmon resonance (SPR) or isothermal titration calorimetry (ITC). However, the goal of this study is to evaluate the “adhesion energy per unit area” of the 8-mer peptide to surfaces with various functional groups and compare it with our previous work,33 which was conducted using SFA.We utilized the SFA to measure the force–distance profiles between the synthesized 8-mer peptide and four types of functionalized SAMs at two pH levels: low (pH 3.0) and high (pH 8.5). The selection of SAMs with distinct terminal functional groups allows for the exploration of various interaction forces (e.g., hydrogen bonding, π–π, cation–π, hydrophobic, and electrostatic interactions) which are pivotal in understanding the material interaction mechanisms.33,45,51,52
The charge states of both the SAMs and the peptide (pI value: ∼ 4.1) are known to vary with pH, necessitating the analysis at both low and high pH for a holistic view of the interaction mechanisms. Notably, the charge variations of amino acids within the peptide, influenced by pH changes, are critical to this analysis.53–55 Among the seven amino acids present in the 8-mer peptide, aspartic acid (D), glutamic acid (E), and histidine (H) exhibit ionizable characteristics, which lead to fluctuations in charge dependent on the pH environment. As depicted in Fig. 3f and 4f, H (pKa ∼ 6.00) carries a positive charge at pH 3.0, while it becomes neutral at pH 8.5. On the other hand, D (pKa ∼ 3.65) and E (pKa ∼ 4.25) are neutral at pH 3.0 but carry a negative charge at pH 8.5. Proline (P, pKa ∼ 1.99) retains a negative charge on its carboxyl group at both pH 3.0 and pH 8.5.33
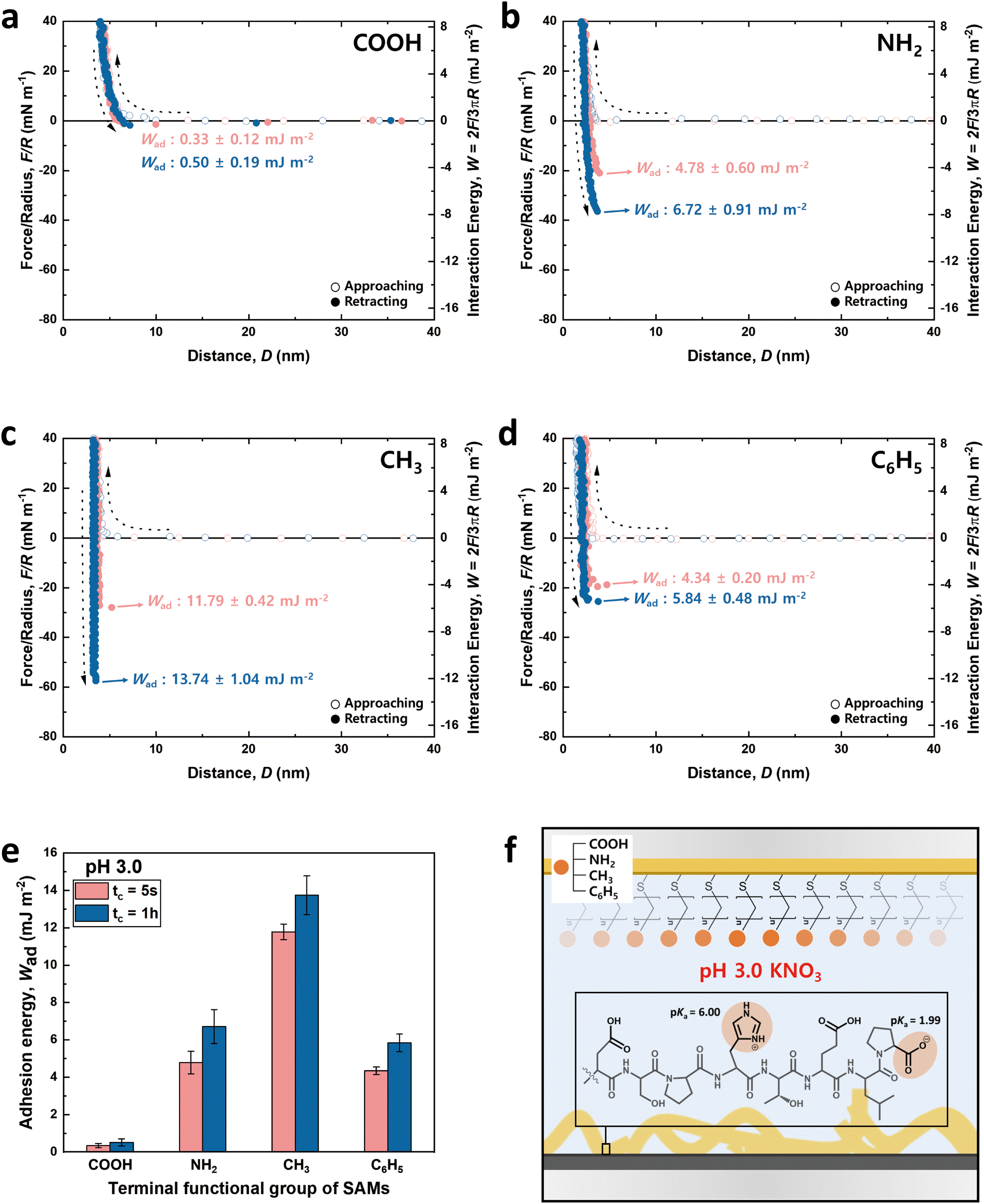 | ||
| Fig. 3 Approaching and retracting force–distance profiles between the 8-mer peptide and functionalized SAM surfaces at pH 3.0. Four different functionalized SAMs are utilized: (a) –COOH, (b) –NH2, (c) –CH3, and (d) –C6H5. (e) Bar graphs show the adhesion energy of the 8-mer peptide against four different SAMs as a function of contact time; average ± standard error bars (n > 3). (f) A peptide schematic showing the charged state of amino acids at pH 3.0. | ||
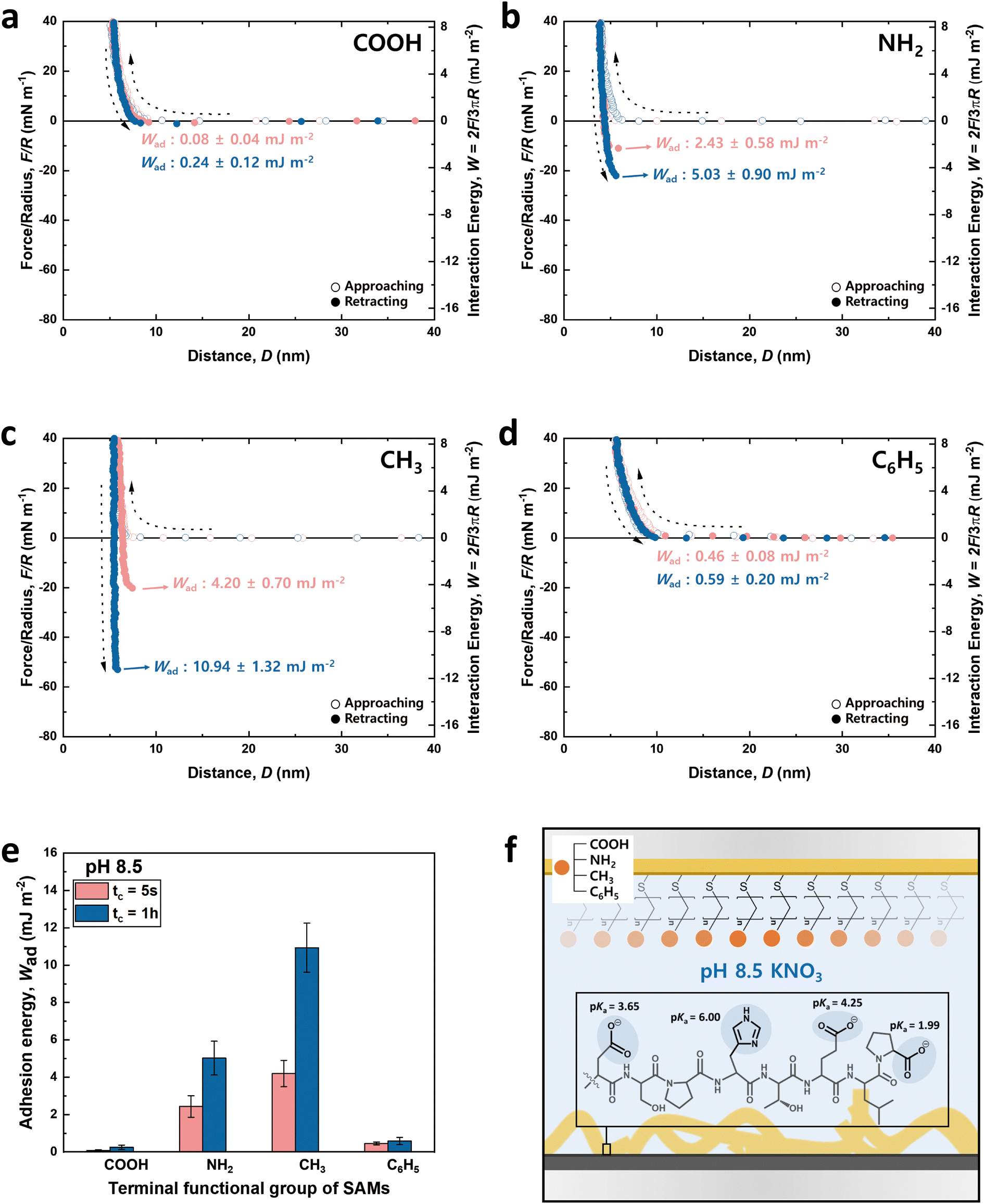 | ||
| Fig. 4 Approaching and retracting force–distance profiles between the 8-mer peptide and functionalized SAM surfaces at pH 8.5. Four different functionalized SAMs are utilized: (a) –COOH, (b) –NH2, (c) –CH3, and (d) –C6H5. (e) Bar graphs show the adhesion energy of the 8-mer peptide against four different SAMs as a function of contact time; average ± standard error bars (n > 3). (f) A peptide schematic showing the charged state of amino acids at pH 8.5. | ||
Furthermore, to consider the potential molecular rearrangement of peptides upon contact with SAM surfaces, we introduced both short and long contact times (tc). This approach is based on the hypothesis that molecules at the interface may reorganize into more energetically favorable configurations, a phenomenon observed as an increase in adhesion energy with prolonged tc.33,56
Fig. 3 and 4 illustrate the approaching and retracting force–distance profiles of the peptide and SAMs at pH 3.0 and 8.5, respectively. At pH 3.0, the peptide is overall neutral carrying one positive and one negative charge (Fig. 3f). The measured adhesion energies are reported as follows (Table 1): COOH-SAM (Wad = 0.33 ± 0.12 mJ m−2 at tc = 5 s, Wad = 0.50 ± 0.19 mJ m−2 at tc = 1 h); NH2-SAM (Wad = 4.78 ± 0.60 mJ m−2 at tc = 5 s, Wad = 6.72 ± 0.91 mJ m−2 at tc = 1 h); CH3-SAM (Wad = 11.79 ± 0.42 mJ m−2 at tc = 5 s, Wad = 13.74 ± 1.04 mJ m−2 at tc = 1 h); C6H5-SAM (Wad = 4.34 ± 0.20 mJ m−2 at tc = 5 s, Wad = 5.84 ± 0.48 mJ m−2 at tc = 1 h). Based on these results, the adhesion energy at pH 3.0 was the highest against CH3-SAM, followed by NH2–, C6H5–, and COOH– SAMs. Conversely, at pH 8.5 (Fig. 4f), where the peptide was negatively charged, the adhesion energy decreased across all SAMs compared to pH 3.0, with a similar preference order, showing the strongest adhesion with CH3-SAM and the weakest with COOH-SAMs. The adhesion energies at pH 8.5 are detailed as follows (Table 1): COOH-SAM (Wad = 0.08 ± 0.04 mJ m−2 at tc = 5 s, Wad = 0.24 ± 0.12 mJ m−2 at tc = 1 h); NH2-SAM (Wad = 2.43 ± 0.58 mJ m−2 at tc = 5 s, Wad = 5.03 ± 0.90 mJ m−2 at tc = 1 h); CH3-SAM (Wad = 4.20 ± 0.70 mJ m−2 at tc = 5 s, Wad = 10.94 ± 1.32 mJ m−2 at tc = 1 h); C6H5-SAM (Wad = 0.46 ± 0.08 mJ m−2 at tc = 5 s, Wad = 0.59 ± 0.20 mJ m−2 at tc = 1 h). These results highlight the significant influence of pH on the adhesion energy of the peptide with different SAMs, underscoring the role of charge in mediating these interactions.
| Adhesion energy, Wad (mJ m−2) | ||||
|---|---|---|---|---|
| pH 3.0 | pH 8.5 | |||
| 5 s | 1 h | 5 s | 1 h | |
| COOH | 0.33 ± 0.12 | 0.50 ± 0.19 | 0.08 ± 0.04 | 0.24 ± 0.12 |
| NH2 | 4.78 ± 0.60 | 6.72 ± 0.91 | 2.43 ± 0.58 | 5.03 ± 0.90 |
| CH3 | 11.79 ± 0.42 | 13.74 ± 1.04 | 4.20 ± 0.70 | 10.94 ± 1.32 |
| C6H5 | 4.34 ± 0.20 | 5.84 ± 0.48 | 0.46 ± 0.08 | 0.59 ± 0.20 |
Interaction force between the DSPHTELP-peptide and COOH-SAM
At pH 3.0, COOH-SAM (pKa value: ∼5.5) exposes COOH without deprotonation, allowing it to function as both a hydrogen bond donor and acceptor. This characteristic facilitates hydrogen bonding with the peptide, especially due to the presence of hydrogen bond-capable functional groups in aspartic acid (D), serine (S), histidine (H), threonine (T), and glutamic acid (E). However, at pH 8.5, COOH-SAM undergoes deprotonation to form COO−, functioning only as a hydrogen bond acceptor. Concurrently, the peptide acquires a negative charge, predominantly due to the D, E, and proline (P), diminishing the probability of hydrogen bonding and amplifying electrostatic repulsion with the COO− group. Consequently, adhesion to COOH-SAM is reduced at pH 8.5 (Wad = 0.24 ± 0.12 mJ m−2), compared to that at pH 3.0 (Wad = 0.50 ± 0.19 mJ m−2). Nevertheless, as illustrated in Fig. 3e and 4e, COOH-SAM demonstrates the weakest interaction energy among the four SAM types, suggesting that hydrogen bonding plays a minimal role in peptide adhesion.Interaction force between the DSPHTELP-peptide and NH2-SAM
At pH 3.0, the terminal NH2 group of NH2-SAM, with a pKa value around 7.5, predominantly exists in its protonated form (NH3+, ∼100%), serving as a cation source and hydrogen bond donor. Therefore, the protonated amine can engage in cation–π interactions with H, which acts as a π-source, and hydrogen bonding with D, S, H, T, and E. Moreover, the negatively charged carboxyl group of P (pKa = 1.99) can participate in electrostatic attraction with NH3+. As the pH increases to 8.5, the Henderson–Hasselbalch equation57,58 predicts a sharp decline in the proportion of protonated amine (to about 9%), markedly reducing cation–π interactions. Nonetheless, the rise in the concentration of unprotonated NH2 groups, capable of acting as both hydrogen bond donors and acceptors, possibly fosters enhanced hydrogen bonding with the amino acids of the peptide. Additionally, the presence of negatively charged D, E, and P in the peptide imparts an overall negative charge to it, facilitating electrostatic interactions. As a result, the adhesion energy was measured as Wad = 6.72 ± 0.91 mJ m−2 at pH 3.0 and Wad = 5.03 ± 0.90 mJ m−2 at pH 8.5.Interaction force between the DSPHTELP-peptide and CH3-SAM
CH3-SAM (water contact angle ∼ 101.0 ± 0.8°) displays the highest hydrophobicity among the four SAMs tested, making it an optimal candidate for assessing hydrophobic interactions. In our experiments, CH3-SAM showed the most substantial adhesion energy at both pH levels studied (Fig. 3e and 4e), underscoring the primacy of hydrophobic interactions in the binding of the DSPHTELP peptide. This high level of interaction is attributed mainly to the hydrophobic nature of L with its methyl group and P with its ring structure. Moreover, the adhesion energy was notably higher at pH 3.0 (Wad = 13.74 ± 1.04 mJ m−2) compared to that at pH 8.5 (Wad = 10.94 ± 1.32 mJ m−2). This variance can be attributed to the charge states of the peptide at different pH levels; it is neutral at pH 3.0, enhancing hydrophobic interactions, whereas at pH 8.5, the peptide carries a negative charge, which slightly diminishes these hydrophobic interactions. These findings suggest that the synthesized 8-mer peptide, despite containing only three non-polar amino acids (one L and two Ps), exhibits strong hydrophobic interactions due to the presence of hydrophobic moieties. Previous research has also confirmed that even net-hydrophilic substances can participate in strong hydrophobic interactions if hydrophobic moieties are present.45,59Interaction force between the DSPHTELP-peptide and C6H5-SAM
At low pH, the peptide carries a positive charge due to H, thus serving as a source for cation–π interactions. This interaction mechanism contributes to a robust adhesion with C6H5-SAM at pH 3.0 (Wad = 5.84 ± 0.48 mJ m−2). However, at the higher pH of 8.5, while H loses its positive charge and becomes neutral, it retains its π-structure, allowing for the possibility of π–π interactions. As a result, the adhesion energy at pH 8.5 is markedly lower (Wad = 0.59 ± 0.20 mJ m−2) than that at pH 3.0. This difference emphasizes the significance of cation–π interactions (with the peptide acting as a cation source) and suggests that π–π interactions contribute minimally to the overall adhesion energy.Effect of contact time on the adhesion energy
Previous studies have commonly observed an increase in adhesion energy with prolonged contact time among biomolecules and polymers.42,60–62 Consistent with these findings, our system displayed higher adhesion energies at longer contact times (tc = 1 h) compared to shorter ones (tc = 5 s) at both pH levels, 3.0 and 8.5. This trend suggests that the peptide undergoes a gradual reorientation or structural rearrangement to optimize its binding affinity towards the SAM surfaces.Moreover, the rise in adhesion energy attributable to extended contact was more pronounced at pH 8.5 than at pH 3.0. This phenomenon can be attributed to several factors: (1) at the elevated pH, aspartic acid (D) and glutamic acid (E) acquire a negative charge, inducing electrostatic repulsion between these amino acids. This repulsion may expand the structure of the peptide, increasing the mobility of the peptide. (2) A more substantial hydration layer may form around the peptide at higher pH levels, as the ionized segments of the peptide engage more intensely with water molecules. The substantial hydration layer formed at higher pH levels can initially impede adhesion by preventing direct contact between the peptide and the SAM. Over time, however, this layer may undergo changes or compression during prolonged contact, contributing to an increase in adhesion energy.
Comparing the interaction mechanism of the DSPHTELP-peptide and M13 bacteriophage
The adhesion observed between the 8-mer peptide and M13 bacteriophage, although similar, exhibited noticeable differences despite sharing the identical DSPHTELP sequence, as illustrated in Fig. 5. The differences in adhesion with NH2- and CH3-SAMs under both pH 3.0 and pH 8.5 conditions can be explained by the following factors: | ||
| Fig. 5 Bar graph showing the adhesion energy on the (a) 8-mer peptide and (b) M13 bacteriophage layers with four different SAMs,33 depending on contact time and pH; average ± standard error bars (n = 3 in each group). Schematic representations of the interactions between (c) CH3-SAM and the 8-mer peptide and (d) C6H5-SAM and the M13 bacteriophage. | ||
(1) The differences in adhesion compared to the M13 bacteriophage at both NH2- and CH3-SAMs, under pH 3.0 and pH 8.5, can be attributed to differences in size and density. The M13 bacteriophage, when coated as a monolayer on mica, showed a density of about 30 units per μm2, with coverage reaching approximately 82%. This indicates a less dense coating of the M13 bacteriophage (Fig. S4, ESI†),33 compared to the 8-mer peptide, which, due to its smaller size, forms a dense layer without significant void spaces (Fig. S3, ESI†). The denser arrangement of the peptide enhances its interaction with the SAM through increased hydrophobic and cation–π interactions, attributed mainly to the presence of L, P, and H. Particularly at pH 8.5, the peptide maintains adhesion to both NH2- and CH3-SAMs, unlike the M13 bacteriophage, which exhibits limited adhesion likely due to swelling.
(2) The reduced adhesion of the 8-mer peptide compared to the M13 bacteriophage on C6H5-SAM is attributed to the difference in amino acid mobility. Unlike the M13 bacteriophage, which is oriented along the pVIII and connected to ssDNA, the 8-mer peptide directly attaches to the mica surface. At pH 3.0, H in the peptide carries a positive charge, facilitating its binding to the negatively charged mica. However, upon binding to the mica surface, H can no longer act as a cation source for cation–π interactions, resulting in the lower adhesion of the peptide to C6H5-SAM. In contrast, for the M13 bacteriophage, histidine remains relatively free, as it is situated away from the mica surface. This greater freedom of histidine appears to enhance the effectiveness of the cation–π interaction between the M13 bacteriophage and C6H5-SAM.
Conclusion
In this study, we successfully synthesized an 8-mer peptide (DSPHTELP), designed to mimic the coat protein (pVIII) of DSPH M13 bacteriophages. Our investigation into the interaction mechanisms, facilitated by measuring the forces between the synthesized peptide and four different functionalized self-assembled monolayers (SAMs: COOH–, NH2–, CH3–, and C6H5–) using surface forces apparatus (SFA), led to several notable conclusions:(1) The adhesion order of the peptide with SAMs (CH3– > NH2– > C6H5– > COOH–) highlights hydrophobic interactions as the primary force, especially influenced by leucine (L) and proline (P), with histidine (H) playing a crucial role in cation–π interactions.
(2) The adhesion strengths were higher at pH 3.0 compared to pH 8.5 across all SAMs. This difference is attributed to the neutral charge of the peptide at pH 3.0, which enhances its hydrophobic properties. Additionally, H becomes positively charged at lower pH, facilitating cation–π interactions. The observation of increased adhesion over longer contact times suggests a beneficial structural rearrangement and reorientation of the peptide for optimal binding.
These findings not only provide detailed quantitative insights into the molecular interaction mechanisms but also highlight the utility of the DSPHTELP sequence at higher pH levels—a scenario where M13 bacteriophages previously faced limitations. The implications of this research extend to the design and development of innovative materials in fields such as nano-biomaterials and biomedicine, offering foundational knowledge for future advancements. In upcoming future works, we plan to investigate the effect of individual amino acids by inserting a point mutation (e.g., DSSHTELS) and also study other sequences, such as VSGSSPDS which are known to specifically adhere to gold.63,64
Author contributions
The research was conceived by all authors. Experiments were performed by H. K. and S. J. with the aid of J. K. The research was supervised by J. R., J.-H. R., and D. W. L. All authors contributed to the writing of the manuscript and the ESI.†Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Basic Science Research Program (NRF-2023R1A2C2004762 and RS-2023-00208386) and 2022 PRIMA QUEBEC-NRF Joint Research Program (NRF-2022K1A3A1A74099490) funded by the National Research Foundation (NRF) of Korea, and the 2024 Research Fund (1.240005.01) of Ulsan National Institute of Science and Technology (UNIST).References
- S. S. Sidhu, Biomol. Eng., 2001, 18, 57–63 CrossRef CAS PubMed.
- G. T. Hess, J. J. Cragnolini, M. W. Popp, M. A. Allen, S. K. Dougan, E. Spooner, H. L. Ploegh, A. M. Belcher and C. P. Guimaraes, Bioconjugate Chem., 2012, 23, 1478–1487 CrossRef CAS PubMed.
- C. Y. Chiang, C. M. Mello, J. Gu, E. C. Silva, K. J. Van Vliet and A. M. Belcher, Adv. Mater., 2007, 19, 826–832 CrossRef CAS.
- J.-M. Lee, Y. Lee, V. Devaraj, T. M. Nguyen, Y.-J. Kim, Y. H. Kim, C. Kim, E. J. Choi, D.-W. Han and J.-W. Oh, Biosens. Bioelectron., 2021, 188, 113339 CrossRef CAS PubMed.
- D. Marvin, L. Welsh, M. Symmons, W. Scott and S. Straus, J. Mol. Biol., 2006, 355, 294–309 CrossRef CAS PubMed.
- G. P. Smith and V. A. Petrenko, Chem. Rev., 1997, 97, 391–410 CrossRef CAS PubMed.
- S. S. Sidhu, Curr. Opin. Biotechnol, 2000, 11, 610–616 CrossRef CAS PubMed.
- S. S. Sidhu, G. A. Weiss and J. A. Wells, J. Mol. Biol., 2000, 296, 487–495 CrossRef CAS PubMed.
- G. A. Weiss, J. A. Wells and S. S. Sidhu, Protein Sci., 2000, 9, 647–654 CrossRef CAS PubMed.
- G. A. Weiss and S. S. Sidhu, J. Mol. Biol., 2000, 300, 213–219 CrossRef CAS PubMed.
- C. Kim, W. G. Kim and J. W. Oh, Polym. Sci. Technol., 2014, 25, 307–314 Search PubMed.
- B. Lee, Y. Ko, G. Kwon, S. Lee, K. Ku, J. Kim and K. Kang, Joule, 2018, 2, 61–75 CrossRef CAS.
- K. T. Nam, D.-W. Kim, P. J. Yoo, C.-Y. Chiang, N. Meethong, P. T. Hammond, Y.-M. Chiang and A. M. Belcher, Science, 2006, 312, 885–888 CrossRef CAS PubMed.
- K. T. Nam, R. Wartena, P. J. Yoo, F. W. Liau, Y. J. Lee, Y.-M. Chiang, P. T. Hammond and A. M. Belcher, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17227–17231 CrossRef CAS PubMed.
- Y. J. Lee, H. Yi, W.-J. Kim, K. Kang, D. S. Yun, M. S. Strano, G. Ceder and A. M. Belcher, Science, 2009, 324, 1051–1055 CrossRef CAS PubMed.
- L. Wu, L. A. Lee, Z. Niu, S. Ghoshroy and Q. Wang, Langmuir, 2011, 27, 9490–9496 CrossRef CAS PubMed.
- L. AndrewáLee, Chem. Commun., 2008, 5185–5187 Search PubMed.
- N. Uchida and T. Muraoka, Chem. Commun., 2023, 59, 9687–9697 RSC.
- Y. C. Shin, J. H. Lee, L. Jin, M. J. Kim, J.-W. Oh, T. W. Kim and D.-W. Han, Biomater. Res., 2014, 18, 1–7 CrossRef.
- B. Y. Lee, J. Zhang, C. Zueger, W.-J. Chung, S. Y. Yoo, E. Wang, J. Meyer, R. Ramesh and S.-W. Lee, Nat. Nanotechnol., 2012, 7, 351–356 CrossRef CAS PubMed.
- C. Mao, A. Liu and B. Cao, Angew. Chem., Int. Ed., 2009, 48, 6790–6810 CrossRef CAS PubMed.
- H. Jin, N. Won, B. Ahn, J. Kwag, K. Heo, J.-W. Oh, Y. Sun, S. G. Cho, S.-W. Lee and S. Kim, Chem. Commun., 2013, 49, 6045–6047 RSC.
- J.-W. Oh, W.-J. Chung, K. Heo, H.-E. Jin, B. Y. Lee, E. Wang, C. Zueger, W. Wong, J. Meyer and C. Kim, Nat. Commun., 2014, 5, 3043 CrossRef PubMed.
- S.-J. Kim, Y. Lee, E. J. Choi, J.-M. Lee, K. H. Kim and J.-W. Oh, Nano Convergence, 2023, 10, 1 CrossRef CAS PubMed.
- D. Seol, J.-S. Moon, Y. Lee, J. Han, D. Jang, D.-J. Kang, J. Moon, E. Jang, J.-W. Oh and H. Chung, Spectrochim. Acta, Part A, 2018, 197, 159–165 CrossRef CAS PubMed.
- J. Park, J.-M. Lee, H. Chun, Y. Lee, S. J. Hong, H. Jung, Y.-J. Kim, W.-G. Kim, V. Devaraj and E. J. Choi, Biosens. Bioelectron., 2021, 177, 112979 CrossRef CAS PubMed.
- I. Kim, J.-S. Moon and J.-W. Oh, Nano Convergence, 2016, 3, 1–17 CrossRef PubMed.
- X. Dang, H. Yi, M.-H. Ham, J. Qi, D. S. Yun, R. Ladewski, M. S. Strano, P. T. Hammond and A. M. Belcher, Nat. Nanotechnol., 2011, 6, 377–384 CrossRef CAS PubMed.
- M. Moradi, Z. Li, J. Qi, W. Xing, K. Xiang, Y.-M. Chiang and A. M. Belcher, Nano Lett., 2015, 15, 2917–2921 CrossRef CAS PubMed.
- M. Moradi, J. C. Kim, J. Qi, K. Xu, X. Li, G. Ceder and A. M. Belcher, Green Chem., 2016, 18, 2619–2624 RSC.
- H. Yi, D. Ghosh, M.-H. Ham, J. Qi, P. W. Barone, M. S. Strano and A. M. Belcher, Nano Lett., 2012, 12, 1176–1183 CrossRef CAS PubMed.
- D. Leckband and J. Israelachvili, Q. Rev. Biophys., 2001, 34, 105–267 CrossRef CAS PubMed.
- C. Lim, J. Ko, D. Jeon, Y. Song, J. Park, J. Ryu and D. W. Lee, Commun. Chem., 2019, 2, 96 CrossRef.
- J. Israelachvili, Y. Min, M. Akbulut, A. Alig, G. Carver, W. Greene, K. Kristiansen, E. Meyer, N. Pesika and K. Rosenberg, Rep. Prog. Phys., 2010, 73, 036601 CrossRef.
- L. Chai and J. Klein, Langmuir, 2007, 23, 7777–7783 CrossRef CAS PubMed.
- M. Valtiner, S. H. Donaldson Jr, M. A. Gebbie and J. N. Israelachvili, J. Am. Chem. Soc., 2012, 134, 1746–1753 CrossRef CAS PubMed.
- Q. Guo and F. Li, Phys. Chem. Chem. Phys., 2014, 16, 19074–19090 RSC.
- L. Newton, T. Slater, N. Clark and A. Vijayaraghavan, J. Mater. Chem. C, 2013, 1, 376–393 RSC.
- J. P. Folkers, P. E. Laibinis and G. M. Whitesides, Langmuir, 1992, 8, 1330–1341 CrossRef CAS.
- D. Leckband, F.-J. Schmitt, J. Israelachvili and W. Knoll, Biochemistry, 1994, 33, 4611–4624 CrossRef CAS PubMed.
- X. Banquy, D. W. Lee, K. Kristiansen, M. A. Gebbie and J. N. Israelachvili, Biomacromolecules, 2016, 17, 88–97 CrossRef CAS PubMed.
- D. W. Lee, C. Lim, J. N. Israelachvili and D. S. Hwang, Langmuir, 2013, 29, 14222–14229 CrossRef CAS PubMed.
- D. W. Lee, X. Banquy, S. Das, N. Cadirov, G. Jay and J. Israelachvili, Acta Biomater., 2014, 10, 1817–1823 CrossRef CAS PubMed.
- P. Zheng, L. Xiang, J. Chang, Q. Lin, L. Xie, T. Lan, J. Liu, Z. Gong, T. Tang and L. Shuai, Biomacromolecules, 2021, 22, 2033–2042 CrossRef CAS PubMed.
- Y. Song, J. Park, C. Lim and D. W. Lee, ACS Sustainable Chem. Eng., 2019, 8, 362–371 CrossRef.
- J. Park, J. Park, J. Lee, C. Lim and D. W. Lee, Nat. Commun., 2022, 13, 112 CrossRef CAS PubMed.
- J. Israelachvili, J. Colloid Interface Sci., 1973, 44, 259–272 CrossRef CAS.
- K. Kendall, J. Phys. D: Appl. Phys., 1971, 4, 1186 CrossRef.
- K. L. Johnson, K. Kendall and A. Roberts, Proc. R. Soc. London, Ser. A, 1971, 324, 301–313 CAS.
- A. Sharifi-Rad, J. Mehrzad, M. Darroudi, M. R. Saberi and J. Chamani, J. Biomol. Struct. Dyn., 2021, 39, 1029–1043 CrossRef CAS PubMed.
- S. Y. Lee, J. Lee, Y. Song, M. Valtiner and D. W. Lee, Nanoscale, 2021, 13, 19568–19577 RSC.
- B. G. Keselowsky, D. M. Collard and A. J. García, J. Biomed. Mater. Res., Part A, 2003, 66, 247–259 CrossRef PubMed.
- N. Wakabayashi, Y. Yano, K. Kawano and K. Matsuzaki, Eur. Biophys. J., 2017, 46, 121–127 CrossRef CAS PubMed.
- L. Kacprzyk, V. Rydengård, M. Mörgelin, M. Davoudi, M. Pasupuleti, M. Malmsten and A. Schmidtchen, Biochim. Biophys. Acta, Biomembr., 2007, 1768, 2667–2680 CrossRef CAS PubMed.
- T. D. Do, N. E. LaPointe, N. J. Economou, S. K. Buratto, S. C. Feinstein, J.-E. Shea and M. T. Bowers, J. Phys. Chem. B, 2013, 117, 10759–10768 CrossRef CAS PubMed.
- C. C. Dupont-Gillain, C. Fauroux, D. Gardner and G. Leggett, J. Biomed. Mater. Res., Part A, 2003, 67, 548–558 CrossRef PubMed.
- Q. Z. Wang, X. G. Chen, N. Liu, S. X. Wang, C. S. Liu, X. H. Meng and C. G. Liu, Carbohydr. Polym., 2006, 65, 194–201 CrossRef CAS.
- R. J. Tallarida, R. B. Murray, R. J. Tallarida and R. B. Murray, Manual of Pharmacologic Calculations: with Computer Programs, 1987, pp. 74–75 Search PubMed.
- J. Choi, D. S. Hwang, C. Lim and D. W. Lee, Carbohydr. Polym., 2024, 324, 121504 CrossRef CAS PubMed.
- C. Lim, D. W. Lee, J. N. Israelachvili, Y. Jho and D. S. Hwang, Carbohydr. Polym., 2015, 117, 887–894 CrossRef CAS PubMed.
- T. H. Anderson, J. Yu, A. Estrada, M. U. Hammer, J. H. Waite and J. N. Israelachvili, Adv. Funct. Mater., 2010, 20, 4196–4205 CrossRef CAS PubMed.
- P. Steffen, C. Verdier and C. Wagner, Phys. Rev. Lett., 2013, 110, 018102 CrossRef CAS PubMed.
- A. S. Khalil, J. M. Ferrer, R. R. Brau, S. T. Kottmann, C. J. Noren, M. J. Lang and A. M. Belcher, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 4892–4897 CrossRef CAS PubMed.
- J. Hou, X. Qian, Y. Xu, Z. Guo, B. Thierry, C.-T. Yang, X. Zhou and C. Mao, Biosens. Bioelectron., 2023, 237, 115423 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp01739k |
| ‡ These authors contributed equally to this work. |
| This journal is © the Owner Societies 2024 |