
Small but bright: origin of the enhanced luminescence of ultrasmall ZnGa2O4:Cr3+ in mesoporous silica nanoparticles
Wai-Tung
Shiu
 a,
Vania
Yoo
a,
Yihong
Liu
a,
Lo-Yueh
Chang
b,
Tahereh
Azizivahed
a,
Yining
Huang
a,
Paul J.
Ragogna
a and
Lijia
Liu
*a
a,
Vania
Yoo
a,
Yihong
Liu
a,
Lo-Yueh
Chang
b,
Tahereh
Azizivahed
a,
Yining
Huang
a,
Paul J.
Ragogna
a and
Lijia
Liu
*a
aDepartment of Chemistry, The University of Western Ontario, 1151 Richmond Street, London, Ontario N6A 5B7, Canada. E-mail: Lijia.liu@uwo.ca
bNational Synchrotron Radiation Research Center, 101 Hsin-Ann Road, Hsinchu, 30076, Taiwan
First published on 10th June 2024
Abstract
Chromium(III)-doped zinc gallate (CZGO) is one of the representative persistent luminescent phosphors emitting in the near-infrared (NIR) region. The emission wavelength it covers falls in the tissue-transparent window, making CZGO a promising optical probe for various biomedical applications. The PersL mechanism dictates that such a phenomenon is only profound in large crystals, so the preparation of CZGO with sizes small enough for biological applications while maintaining its luminescence remains a challenging task. Recent attempts to use mesoporous silica nanoparticles (MSN) as a template for growing nanosized CZGO have been successful. MSN is also a well-studied drug carrier, and incorporating CZGO in MSN further expands its potential in imaging-guided therapeutics. Despite the interest, it is unclear of how the addition of MSN would affect the luminescence properties of CZGO. In this work, we observed that forming a CZGO@MSN nanocomposite could enhance the luminescence intensity and extend the PersL lifetime of CZGO. X-ray absorption fine structure (XAFS) analysis was conducted to investigate the local structure of Zn2+, and an interaction between Zn2+ in CZGO and the MSN matrix was identified.
Introduction
Persistent luminescence (PersL) is a unique optical phenomenon present in some inorganic phosphors, where prolonged luminescence can be observed after the removal of the excitation source. The mechanism of PersL materials was first reviewed by H. F. Brito.1,2 The defects in various Eu2+-doped inorganic matrixes were investigated using density functional theory and a PersL mechanism was proposed. In brief, excited electrons were first trapped by defects in the inorganic phosphor and slowly tunneled to the excited states of the emission center. These excited electrons were then relaxed to the ground state, releasing energy in the form of light. To date, the PersL mechanism is still not fully understood. However, it is widely accepted that the afterglow is correlated to deep electron traps as a result of defects in the inorganic phosphor.3–6 The types of defects include structural vacancies, antisites, and substitutional impurities.1,3 A representative commercialized material with PersL property is Eu2+,Dy3+-doped SrAl2O4, which was first used in signage and coating applications for the long-lasting green emission properties.7 Recently, PersL phosphors with emission within the near-infrared (NIR) regions were actively explored for their potential applications in optical-based bioimaging. The NIR region is also known as the tissue-transparent window as biological tissues and fluids are the most transparent in this region. Additionally, continuous excitation is not required due to the long-lasting afterglow properties, which could eliminate interference from tissue autofluorescence. These properties make NIR-emitting PersL phosphors superior to other conventional bioimaging probes such as gold nanoparticles and organic dyes.8,9 Ever since the first demonstration of using MgSiO3:Eu2+,Dy3+,Mn2+ in bioimaging, many host-activator combinations were discovered to produce NIR PersL.10–13 In particular, chromium(III)-doped zinc gallate, abbreviated as CZGO, is one of the most investigated materials of this class. The characteristic emission wavelength for CZGO is ∼700 nm, which is related to the d–d forbidden transition of octahedrally situated Cr3+ dopant ion.14 The biocompatibility and toxicity of CZGO have also been evaluated, and the results support the safe use as in vivo optical probe for bioimaging.15,16In addition to the optical properties, particle size also plays an important role in nanoparticles used in biological applications. To ensure good clearance by the liver, the particle size should be constrained to less than 150 nm.17 CZGO can be synthesized with various methods, such as solid-state, hydrothermal, and sol–gel synthesis.18 The solid-state synthesis is a well-established and most effective method to synthesize PersL phosphors. However, the method requires the use of high temperature (>1000 °C), which often leads to crystal growth in the micron level. Nanosized PersL particles are often synthesized under milder conditions. For example, hydrothermal synthesis conducted at temperatures between 120 °C and 220 °C have shown promising results in synthesizing nanosized CZGO.16,19–21 It was also demonstrated that the particle size can be easily reduced by modifying the amount of metal ion precursors used in the synthesis. To date, the lowest reported particle size synthesizing using this method is less than 10 nm. One drawback of hydrothermally synthesized CZGO is the weak luminescence without any post-treatment and this is often overcome with high-temperature annealing treatment.5,14 However, the application of heat will also increase the particle size to ∼90 nm. Thus, it is urgent to find a solution to synthesize smaller, but brighter CZGO nanoparticles.
Although CZGO possess ideal optical properties for optical-based bioimaging, bare CZGO still lacks bioactivity without any functionalization. Surface modification can be used to improve the solubility and targeting ability of CZGO. Nonetheless, the attachment of molecules usually leads to a drastic decrease in luminescence. The limited surface area per particle also constrains the number of available attachments. As an alternative approach, incorporating CZGO into a bioactive matrix is a promising strategy to preserve the pristine optical properties while expanding its application in imaging-guided theragnosis.22,23 Our group previously investigated the use of amorphous calcium phosphate (ACP), an established drug carrier, as a matrix for CZGO loading.24 CZGO has high affinity to the ACP, and a composite can be successfully formed, but the morphology and particle size of the nanocomposite was not ideal. Mesoporous silica nanoparticles (MSN) are another class of drug carrier material with high surface area and pore volume.25 Studies have also proven it to have low toxicity, good biocompatibility, and high stability.26–28 When MSN is incorporated into nanoparticle synthesis, it can act as a morphology controlling template for the growth of uniform, small particles. To synthesize the nanoparticle@MSN composite, the MSN is first synthesized using the Stöber method. Metal ion precursors were then introduced to allow diffusion and crystallization within the mesopores of the MSN. The resulting composite combines the advantages of having superior optical properties from the nanoparticles and versatile functionality from the MSN.29–31
The CZGO@MSN nanocomposite was first synthesized in 2015 by the Han group and the rechargeability of the nanocomposite was demonstrated.32 Since then, multiple studies have also synthesized the nanocomposite with other dopants (Sn4+, and Yb3+) to enhance the optical properties.33–35 In these studies, CZGO were confined within the MSN template, and the overall particle sizes were between 50 nm to 100 nm. Microscopy images have also revealed the sizes of the CZGO nanoparticles were all below 10 nm, which is smaller than any other synthesis approaches reported so far. The toxicity of CZGO and MSN are both low, and it has been shown that the combination of the two components will also yield a product with low toxicity. In the form of CZGO-MSN core–shell nanostructures and composites, multiple cell viability tests have confirmed high cell survival rate even when a concentrated dose of sample was used.34,36,37 These properties make CZGO@MSN a promising material for biological applications. Regardless, to the best of our knowledge, there has been no discussion on why these ultrasmall CZGO nanoparticles exhibit luminescence and afterglow duration comparable or better than the ones of larger sizes. In this work, we investigated the origin of the strong luminescence from the ultrasmall CZGO in MSN. CZGO@MSN were synthesized following the method commonly used by the community, and for comparison, free-standing CZGO were synthesized without introducing the MSN template. The latter have a crystalline size much larger than the CZGO within the MSN. Interestingly, these CZGO@MSN exhibit much brighter luminescence and longer PersL duration compared to the free-standing CZGO. We identified the presence of an interaction between the MSN template and CZGO that facilitated the luminescence enhancement.
Experimental
Materials
Gallium nitrate hydrate [Ga(NO3)3·xH2O, 99.9%], chromium(III) acetate monohydrate [Cr(CH3COO)3,], hydrochloric acid [HCl, 37%, wt], and (3-aminopropyl)triethocysilane [APTES, 99 + %] were purchased from Sigma Aldrich. Zinc acetate dihydrate [Zn(CH3COO)2, 97 + %] was obtained from Alfa Aesar. Hexadecyltrimethylammonium bromide [CTAB, 99 + %] and ammonium hydroxide [NH4OH, 28–32%, wt] were bought from Thermo Fischer Scientific. Tetraethoxysilane [TEOS, 99 + %] were purchased from Acros organics.Synthesis of mesoporous silica nanoparticles (MSN)
MSN were prepared according to previous literature with modifications.33 In a 100 mL round bottom flask, 1.15 g of CTAB was dissolved in 56 mL of deionized water, 16 mL of ethanol, and 0.1 mL of (NH4)OH. The above solution was stirred for 30 minutes at 60 °C using an oil bath. 2.92 mL of TEOS was then added to the above mixture and stirred for 2 hours at 60 °C. After the solution was cooled to room temperature, a two-step centrifugation was performed. The mixture was first centrifuged at 9000 rpm for 15 minutes. The supernatant was collected and centrifuged at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 minutes. The product was washed with ethanol three times and dried at 60 °C for 12 hours. Calcination was then performed for 5 hours at 550 °C, with a heating rate of 2 °C min.−1.
000 rpm for 10 minutes. The product was washed with ethanol three times and dried at 60 °C for 12 hours. Calcination was then performed for 5 hours at 550 °C, with a heating rate of 2 °C min.−1.
Synthesis of the mesoporous silica-ZnGa2O4:Cr3+ (CZGO@MSN) nanocomposite
The CZGO@MSN composite and the CZGO nanoparticles were synthesized following an established protocol by Lin et al.33 In two separate vials, two stock solutions of 4 M Ga(NO3)3·xH2O solution and 0.5 M Cr(CH3COO)3 were made. A precursor solution was then obtained by combining 0.5 mL of the Ga(NO3)3·xH2O stock solution, 2 μL of the Cr(CH3COO)3 stock solution, and 0.2195 g of Zn(CH3COO)2. After the precursor powder was dissolved, 0.1 g of MSN was added to the precursor solution and stirred until all MSN particles were dispersed evenly in the precursor solution. The mixture was then dried in a vacuum oven at 60 °C for 16 hours. After, the product was ground and annealed at 600 °C for 2 hours with a heating rate of 5 °C minute−1. When the product was cooled to room temperature, it was ground again and annealed at 900 °C for 4 hours with a heating rate of 2 °C minute−1 to obtain the CZGO@MSN composite. The obtained product was then washed three times using 0.01 M HCl by centrifugation, which was conducted at 2000 rpm for 5 minutes. For comparison, bare CZGO nanoparticles were synthesized following the identical procedure without the addition of MSN.Synthesis of Zn-doped MSN
In a 100 mL round bottom flask, 1.15 g of CTAB, 56 mL of deionized water, 16 mL of ethanol, and 0.1 mL of (NH4)OH was added and stirred for 30 minutes at 60 °C. Then, 2.92 mL of TEOS and 0.238 g of Zn(CH3COO)2 was added to the flask. The above mixture was stirred for 2 hours at 60 °C, and cool down to room temperature. After, centrifugation was performed for 15 minutes at 9000 rpm. The supernatant was collected and underwent centrifugation at 10000 rpm for 10 minutes. This is followed by three ethanol washing steps. (10000 rpm, 5 minutes). The collected product was dried at 60 °C for 10 hours. Finally, calcination was then performed for 5 hours at 550 °C, with a heating rate of 2 °C minute−1 to obtain the final product.
Characterization
X-ray diffraction (XRD) was performed on a Inel CPS Powder diffractometer, equipped with a Cu Kα tube source. Fourier Transform Infrared Spectroscopy (FT-IR) was measured on a Perkin Elmer Spectrum Teo FT-IR Spectrometer. Transmission electron microscopy (TEM) was obtained using a Thermo Scientific Talos L120C spectrometer, with an attachment to perform energy dispersive X-ray spectroscopy (EDS). The obtained data was analyzed with ImageJ. The Brunauer–Emmett–Teller (BET) N2 adsorption–desorption measurements were conducted on a Micromeritics ASAP 2020 porosity analyzer at 77 K and the specific surface area was measured by the BET method. The samples were activated at 200 °C for 5 hours under vacuum prior to the measurement. Photoluminescence (PL) spectroscopy was performed using a StellaNet Inc UN318-1215 spectrometer and an AvaSpec-ULS2048XL-EVO detector. The samples were excited with 254 nm and 395 nm UV lights. The PersL images were taken using an in vivo imaging system (IVIS spectrum, PerkinElmer). The samples were first excited using a 254 nm UV light for 5 minutes, and then placed in the in vivo imaging system. The synchrotron X-ray absorption near-edge structure (XANES) data was obtained at the National Synchrotron Radiation Research Center (NSRRC) using beamline TLS 17C1 for the Zn K-edge and at the Canadian Light Source using the soft X-ray microcharacterization beamline (SXRMB) for the Si K-edge spectra. The Zn K-edge was collected using the fluorescence yield (FY) detection mode. The Si K-edge was collected using the total electron yield (TEY) and FY detection modes. The obtained data was analyzed using Athena from the Demeter package.38Results and discussion
The crystal structure of the synthesized samples was examined using XRD, listed as Fig. 1a. The diffraction pattern of CZGO@MSN was compared to bare CZGO. Well-defined peaks can be identified, and the diffraction patterns of both samples matched the standard diffraction pattern of ZnGa2O4 (JCPDS 00-038-1240). No impurity peaks were observed in the diffraction pattern of pure CZGO, indicating the synthesis method adapted in this study yielded the CZGO without any impurity phase. A broad peak was observed at 24°, showing the presence of amorphous MSN in the nanocomposite. Additionally, narrow diffraction peaks were observed in pure CZGO, suggesting high crystallinity in the sample. This is different for CZGO@MSN. The diffraction peaks were broader than the CZGO, showing less-ideal CZGO crystallinity in the nanocomposite. Fig. 1b displayed the FT-IR spectrum of CZGO@MSN compared to pure CZGO and MSN. CZGO exhibited an intense peak at 570 cm−1 and a weaker peak at 410 cm−1. The peaks were attributed to the Zn–O and Ga–O metal–oxygen bonding vibrations, respectively.39 In the FT-IR spectrum of pure MSN, the absorption bands were assigned to Si–O–Si stretching (1059 cm−1), Si–O stretching (808 cm−1) and Si–O bending (445 cm−1) vibrations.40 All the above bands were observed in the spectrum of CZGO@MSN, verifying the presence of both the MSN and CZGO components in the synthesized nanocomposite. The morphology of the synthesized samples was then studied using TEM, shown as Fig. 1c. Vacant pores of MSN appeared as a light grey color in the TEM image, while loaded pores were black, suggesting the CZGO particles were synthesized within the pores of the MSN template. The average particles size of pure CZGO is 75.6 nm, which is relatively large compared to the CZGO synthesized in the CZGO@MSN nanocomposite. It is also significantly larger than bare CZGO nanoparticles synthesized using the hydrothermal method. The size of hydrothermally synthesized CZGO can be tuned by the amount of metal ion precursor used and it was demonstrated to synthesize nanoparticles of 8 nm. Additionally, from our BET results, the pore size of the CZGO@MSN nanocomposite was averaged to be 3.2 nm. This is significantly smaller than bare CZGO, effectively demonstrating the role of MSN as a size regulator in the system. It is also noted that the overall particle size of CZGO@MSN decreased compared to the MSN alone. The particle size of MSN and CZGO@MSN is 70 nm and 47.5 nm, respectively.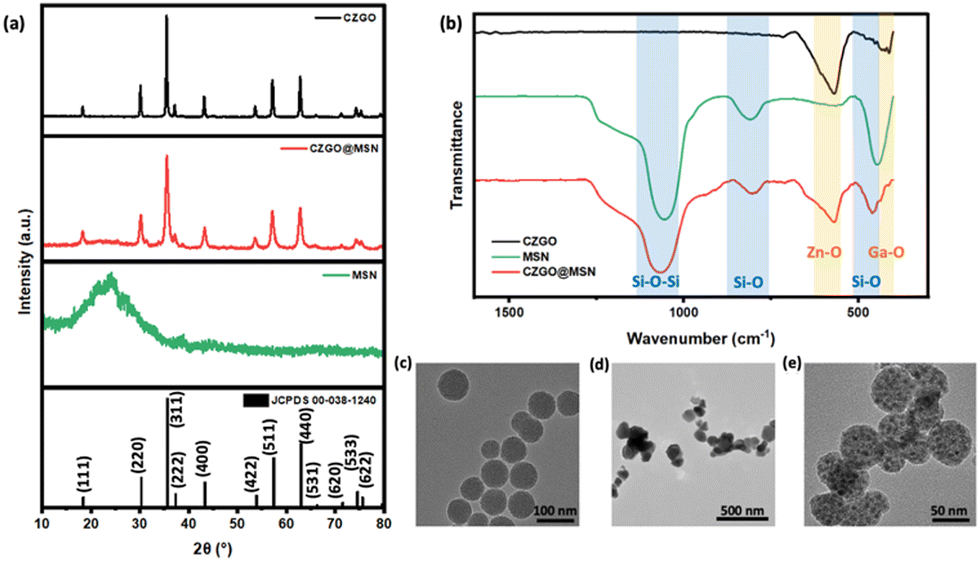 | ||
| Fig. 1 (a) The X-ray diffraction pattern of CZGO and CZGO@MSN compared to pure MSN and the standard diffraction of ZnGa2O4 (JCPDS 00-038-1240). (b) The FT-IR spectra of the synthesized products. (c)–(e) From left to right: the TEM images of MSN, bare CZGO, and CZGO@MSN. | ||
The elemental compositions of the synthesized samples were examined by EDS. The Ga/Zn ratios were determined by dividing the atomic weight percentage of the two elements. The detailed parameters can be found in Table 1. Interestingly, although the same amount of Zn and Ga precursors were used during the synthesis, the samples do not exhibit the same stoichiometric ratios. The Ga/Zn ratio of CZGO was 1.9, which is close to the ideal stoichiometry of ZnGa2O4. However, the Ga:Zn ratio of CZGO@MSN was 1.3, indicating an excess of Zn in the nanocomposite. We will get back to this point later.
| Sample | Atomic weight % | Ga/Zn | ||
|---|---|---|---|---|
| Zn | Ga | Si | ||
| CZGO | 34.0 ± 4.2 | 64.4 ± 4.2 | — | 1.9 |
| CZGO@MSN | 17.7 ± 2.1 | 23.7 ± 2.4 | 58.2 ± 2.4 | 1.3 |
To confirm the successful integration of CZGO in MSN, the pore size of the MSN before and after adding CZGO were investigated using the BET isotherm and the results were summarized in Table 2. A decrease in pore volume and surface area was observed as the nanocomposite was formed, which verified the formation of particles in the mesopores. But, as CZGO was formed, the pore size increased. Similar observations were reported by Li, where the pore size increased from 2.450 nm to 2.912 nm. The authors explained this phenomenon by CZGO forming in the nanopores of the mesopores as tiny, isolated particles.32 The N2 adsorption desorption isotherms were also shown as Fig. 2. Overall, the isotherm curve of the MSN was highly comparable to previously published literature.41 The isotherm curve has the shape of a typical Type IV isotherm and verified the mesoporous nature of our synthesized MSN. We then turned our attention to the isotherm of CZGO@MSN, where a two-step N2 desorption was recognized. The presence of this feature implied that the MSN was partially plugged by the CZGO.42
| Pore size (nm) | Pore volume (cm3 g−1) | Surface area (m2 g−1) | |
|---|---|---|---|
| MSN | 2.7 | 0.67 | 992.3 |
| CZGO@MSN | 3.2 | 0.12 | 147.9 |
 | ||
| Fig. 2 The N2 adsorption desorption plot of (a) MSN and (b) CZGO@MSN. | ||
The luminescence property of CZGO and CZGO@MSN were then examined. Fig. 3a show both samples emitted red luminescence under 254 nm UV excitation. While the red luminescence emission of CZGO@MSN is stronger. It is clearer to see from the PL spectrum (Fig. 3b). To ensure the PL results were comparable, 0.0300 g of sample was used for the PL measurement. The distance between the fibre optics light probe and the sample were kept constant during the data acquisition. Although both samples exhibited similar PL profile that are characteristic of Cr3+ emission in CZGO, the intensity of CZGO@MSN was almost 2.5 times higher than the one of bare CZGO. The strong peak at 697 nm was attributed to the characteristic red luminescence from the Cr3+ d–d forbidden transition. This peak, also referred to as the N2 line, is originated from Cr3+ adjacent to an antisite defect within the host lattice (e.g. Zn2+ replacing Ga3+, and vice versa). The shoulder at ∼688 nm is commonly known as the R line and is related to Cr3+ ions in an unperturbed octahedral site. The remaining features are phonon side bands.16,43 Recall the TEM images presented in Fig. 1, the particle size of the CZGO in CZGO@MSN is much smaller than the bare CZGO. Our observation here suggested the PL intensity is not always proportional to the particle size, and there are additional factors affecting the luminescence. It was previously reported that the luminescence intensity of CZGO is related to the presence of oxygen vacancies in the zinc gallate lattice, and the increased amount of oxygen vacancy would result in an intensity drop in the N2 line feature along with a stronger R line.44 A change in the R/N2 intensity also implies a modified crystal field strength of Cr3+, which can be achieved by adjusting precursor concentration or introducing co-dopants.14,45,46 In our case, the calculated intensity ratio of the R/N2 is very similar between the CZGO@MSN composite (0.76) and free-standing CZGO (0.72). A comparison can also be seen from the normalized PL spectra (Fig. 3c). Therefore the formation of CZGO@MSN doesn’t alter the structure surrounding the Cr3+ color center. On the other hand, it was reported that luminescence enhancement of CZGO can be achieved through post annealing,6 demonstrated by an overall increase of PL profile without change in the R/N2 intensity ratio. It was attributed to the improved crystallinity through heat treatment, which is different from our case. Therefore, the luminescence enhancement is unlikely related to oxygen vacancies present in CZGO or the change in the Cr3+ crystal field, and there were external factors affecting the luminescence intensity, which will be discussed below.
 | ||
| Fig. 3 (a) The optical photograph of CZGO and CZGO@MSN acquired by a cell phone camera (iPhone 13 Pro) under 254 nm excitation. (b) The PL spectra of CZGO and CZGO@MSN at the NIR region (254 nm excitation). (c) The PL spectra in (b) normalized to the intensity maximum. (d) The PLE spectra of CZGO and CZGO@MSN by monitoring the emission peak at 697 nm. (e) The PL spectra of CZGO@MSN with references within the visible region (254 nm excitation). (f) The PL spectra of CZGO and CZGO@MSN under 395 nm excitation. | ||
The PL excitation (PLE) spectra of CZGO and CZGO@MSN were obtained by monitoring the emission peak at 697 nm (Fig. 3d). The technique was used to understand the light absorbing property of the nanocomposite. The PL spectrum exhibited three peaks located at ∼270 nm, ∼420 nm, and ∼570 nm. The band below 300 nm is related to the band gap absorption of the ZnGa2O4 host lattice. The remaining features are associated with Cr3+ in an octahedral field (4A2 → 4T1g and 4A2 → 4T2g).19,47 Compared to bare CZGO, the peak position of CZGO@MSN shifted to a higher wavelength for the peak located ∼270 nm, which suggests the ZnGa2O4 host lattice exhibited a narrower band gap, compared to bare CZGO. Additionally, a 10 nm blueshift is present for the CZGO@MSN nanocomposite in the ∼420 nm peak, which is attributed to an increased local crystal field strength around the Cr3+ dopant as the MSN template was applied.
Previously, various reports have reported the formation of Zn-doped MSN using the sol–gel approach.48,49 In our case, we hypothesize that during the synthesis of CZGO@MSN, pure MSN was exposed to Zn2+ ions, and could form Zn-doped MSN as the nanocomposite was synthesized. Vacancies are often observed when a dopant is introduced to a system. When Zn2+ is doped into MSN, it could act as a trap center for nearby oxygen vacancies. This process will create defects on the surface and interface of the MSN, resulting to luminescence emission in the visible range.50Fig. 3e displays the PL emission spectrum of MSN, Zn-doped MSN, and CZGO@MSN, excited using a 254 nm UV light. Two peaks were located at ∼332 nm and ∼384 nm. The shorter wavelength (higher energy) peak has been observed in porous and mesoporous silica, and have been attributed to a silanol species (Si–OH) on the silica surface.51,52 The peak at longer wavelength (lower energy) is associated with oxygen vacancies in the silica network.53 In a recent study by Huang et al., the authors observed an enhancement of the 376 nm PL emission in amorphous silica nanoparticles and attributed to the modulation of surface groups (i.e. dihydroxylation of Si–OH groups to Si–O–Si groups).54 In our case, the Zn doping and the formation of CZGO@MSN has similar influence on the PL of MSN by enhancing both the 332 nm and the 384 nm emission intensity, which supported our hypothesis of forming Zn-doped MSN during the synthesis procedure. MSN in CZGO@MSN composite has more Si–OH and oxygen vancies due to Zn doping. To investigate the role of Zn-doped MSN in the nanocomposite, a PL spectrum was taken under the excitation of a 395 nm UV light, which is shown in Fig. 3f. The luminescence of CZGO@MSN was significantly higher, which confirms the presence of efficient energy transfer from the MSN to CZGO.
The PersL properties of CZGO and CZGO@MSN were examined using an in vivo imaging system. The samples were first excited using a 254 nm UV light for 5 minutes. The excitation source was then removed, and the luminescence images were captured using a charge-coupled device camera. Fig. 4a showed the images taken at selected time frames. The luminescence lifetime of both samples lasted for at least 180 minutes. The luminescence decay was also much slower when the CZGO particles were synthesized under the MSN template. A PersL decay curve was obtained by measuring the luminescence counts during the data acquisition period and is presented as Fig. 4b. The luminescence counts were significantly higher for the CZGO@MSN nanocomposite at 6 minutes, which is consistent with our PL data. Similar to other PersL phosphor materials, the luminescence started with a quick drop in intensity, followed by a slower decay, suggesting there are two components contributing to the afterglow.55–58 A bi-exponential fitting was also conducted on the PersL decay curve following the equation,

 | ||
| Fig. 4 (a) The PersL images of CZGO and CZGO@MSN, (b) PersL decay curve. | ||
XANES is an element-specific technique that is capable of probing the local environment of an element of interest in a compound. By scanning the X-ray energy across an absorption threshold of an element, a drastic increase in the absorption coefficient marks the energy required to promote the core electron transition to the previously unoccupied electronic state following the dipole transition rule. The spectral onset marks the transition energy, which is related to the oxidation state of the element. Fig. 5a displays the Zn K-edge XANES of CZGO and CZGO@MSN compared to Zn-doped MSN. The main resonance corresponds to the Zn 1s → 4p transition. Oscillations that immediately follow the main resonance is caused by the multiple scattering of the electrons, so the local geometry surrounding the target element can modulate these oscillations. In the case of Zn, the number of O that bond to the Zn, and Zn–O distance determines the shape of the XANES profile.59 Overall, the spectral features of CZGO and CZGO@MSN were highly similar, suggesting the local structure of Zn in CZGO and CZGO@MSN were nearly identical. A small shift in the main resonance could be observed and this was more apparent when the first derivative was applied (Fig. 5b). The peak maximum of Zn-doped MSN is located at the highest photon energy, this is followed by the CZGO@MSN nanocomposite, then CZGO. This confirms our hypothesis that Zn in CZGO@MSN contains two chemical environments, CZGO and Zn-doped MSN, respectively. This further supported by the Si K-edge XANES in Fig. 5c. The absorption peak of the Si K-edge probes the Si 1s → 3p transition, and the position is highly dependent on the oxidation states of the Si atom. The peak maxima of the synthesized was located at ∼1848.5 eV in our obtained data, suggesting is the characteristic feature of SiO2.60 A difference in the absorption peak intensity was also observed among these samples. The formation of oxygen vacancies will increase the Si 3p orbital occupancy, resulting in a lower peak intensity. Similar to our observation at the Zn K-edge XANES, the peak intensity of the CZGO@MSN nanocomposite in situated between Zn-doped MSN and pure MSN, which is further confirms the formation of Zn-doped MSN in the nanocomposite.
 | ||
| Fig. 5 The XANES spectra of the synthesized samples. (a) The Zn K-edge XANES. The inset shows the first derivative of Zn K-edge XANES at the main resonance region. (b) The Si K-edge XANES. | ||
With the formation of Zn-doped MSN using various techniques, we propose an interaction between the synthesized nanocomposite and the Zn-doped MSNs. The luminescence mechanism is illustrated in Fig. 6. The process begins with valance band electrons in Zn-doped MSN being promoted to an excited state with the simulation of UV light. These electrons will then migrate to the oxygen related defects in the MSN system. The oxygen defects in the system are capable of stabilizing the triplet state excitons and result in blue luminescence emission. Then, the light emitted by the Zn-doped MSN is re-absorbed by the Cr3+ dopant in CZGO to facilitate the d–d transition. The electrons are then shifted to the trapped states within the ZnGa2O4. Finally, the electrons tunnel back to the excited state of Cr3+ and relax back to the ground state, producing the red luminescence.
 | ||
| Fig. 6 The proposed interaction between Zn-doped MSN and CZGO@MSN. (1) excitation, (2) electron migrates to the defect, (3) radiative decay within Zn-doped MSN, (4) excitation of the Cr3+ center, (5) electron trapping, (6) electron tunneling, (7) NIR emission from the Cr3+ center. | ||
Conclusions
In summary, the optical properties of a CZGO@MSN nanocomposite were compared to bare CZGO synthesized under the same conditions. In contrast to the common conception that better luminescence properties are often associated with large crystals, we found brighter and stronger luminescence was observed for the CZGO@MSN nanocomposite in which CZGO is only ∼5 nm on average. XAFS analysis was conducted to study the environments of different ions. We discovered during the incorporation of CZGO in MSN through ion impingement, Zn2+ enters the MSN lattice, forming Zn-doped MSN. Compared to pristine non-luminescent MSN, Zn-doped MSN emits luminescence in the visible range, which falls within the light absorption window of CZGO. Upon excitation, Zn-doped MSN enables additional energy transfers to the CZGO emission channel, and it also acts as an additional trap site to extend the PersL lifetime of CZGO.Author contributions
Wai-Tung Shiu: formal analysis, investigation, writing – original draft. Vania Yoo: investigation. Yihong Liu: investigation, formal analysis. Lo-Yueh Chang: investigation. Tahereh Azizvahed: investigation. Yining Huang: writing – review and editing, supervision. Paul Ragogna: supervision, funding acquisition. Lijia Liu: conceptualization, supervision, funding acquisition, formal analysis, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is funded by the Natural Sciences and Engineering Research Council Canada (NSERC), Discovery Program (DG RGPIN-2020-06675) and Western Strategic Support for NSERC Success fund. Electron microscopy was performed at the Canadian Centre for Electron Microscopy, a Canada Foundation for Innovation Major Science Initiatives funded facility (also supported by NSERC and other government agencies). Part of the research described in this paper was performed at the Canadian Light Source, a national research facility of the University of Saskatchewan, which is supported by the Canada Foundation for Innovation (CFI), NSERC, the Canadian Institutes of Health Research (CIHR), the Government of Saskatchewan, and the University of Saskatchewan.References
- H. F. Brito, J. Hölsä, T. Laamanen, M. Lastusaari, M. Malkamäki and L. C. V. Rodrigues, Opt. Mater. Express, 2012, 2, 371 CrossRef CAS.
- J. Xu and S. Tanabe, J. Lumin., 2019, 205, 581–620 CrossRef CAS.
- N. Basavaraju, K. R. Priolkar, D. Gourier, S. K. Sharma, A. Bessière and B. Viana, Phys. Chem. Chem. Phys., 2015, 17(3), 1790–1799 RSC.
- D. Gourier, A. Bessière, S. K. Sharma, L. Binet, B. Viana, N. Nasavaraju and K. R. Priolkar, J. Phys. Chem. Solids, 2014, 75(7), 826–837 CrossRef CAS.
- C. Lee, Y. Liu, B. Hulme, L. Chang, S. Ke, E. Wang, Y. Wu, B. Lin, Y. Jiang and L. Liu, ChemPhotoChem, 2023, 7(12), e202300143 CrossRef CAS.
- M. K. Hussen and F. B. Dejene, J. Sol-Gel Sci. Technol., 2018, 88, 454–464 CrossRef CAS.
- R. E. Rojas-Hernandez, F. Rubio-Marcos, M. A. Rodriguez and J. F. Fernandez, Sustainable Energy Rev., 2018, 81, 2759–2770 CrossRef CAS.
- M. Montalti, L. Prodi, E. Rampazzo and N. Zaccheroni, Chem. Soc. Rev., 2014, 43(12), 4243–4268 RSC.
- K. Sztandera, M. Gorzkiewicz and B. Klajnert-Maculewicz, Mol. Pharmaceutics, 2019, 16(1), 1–23 CrossRef CAS PubMed.
- Q. Le Masne De Chermont, C. Chanéac, J. Seguin, F. Pellé, S. Maîtrejean, J. P. Jolivet, D. Gourier, M. Bessodes and D. Scherman, Proc. Natl. Acad. Sci. U S A, 2007, 104(22), 9266–9271 CrossRef CAS PubMed.
- Y. Liu, J. A. McLeod, L. Y. Chang, C. K. Chang, Y. Jiang, Z. Wang, A. Lefebvre, X. Chen and L. Liu, Mater. Today Commun., 2024, 38, 108080 CrossRef CAS.
- M. Jin, F. Li, J. Xiahou, L. Zhu, Q. Zhu and J.-G. Li, J. Alloys Compd., 2023, b, 167491 CrossRef.
- H. Jiang, L. Liu, K. Yu, Z. Yin, S. Zheng, L. Song, J. Shi and Y. Zhang, J. Rare Earths, 2022, 40(9), 1389–1398 CrossRef CAS.
- A. Bessière, S. Jacquart, K. Priolkar, A. Lecointre, B. Viana and D. Gourier, Opt. Express, 2011, 19(11), 10131–10137 CrossRef PubMed.
- Y. Jiang, Y. Li, C. Richard, D. Scherman and Y. Liu, J. Mater. Chem. B, 2023, 7(24), 3796–3803 RSC.
- T. Lécuyer, M.-A. Durand, J. Volatron, M. Desmau, T. Lai-Kuen, Y. Corvis, J. Seguin, G. Wang, D. Alloyeau, D. Scherman, N. Mignet, G. Gazeau and C. Richard, Nanoscale, 2020, 12, 1967–1974 RSC.
- S. M. Moghimi, A. C. Hunter and T. L. Andresen, Annu. Rev. Pharmacol. Toxicol., 2012, 52, 481–503 CrossRef CAS PubMed.
- X. Sun, L. Song, N. Liu, J. Shi and Y. Zhang, ACS Appl. Nano Mater., 2021, 4(7), 6497–6514 CrossRef CAS.
- W.-T. Shiu, X. Li, L.-Y. Chang, J.-L. Chen, Y.-Y. Lin, B.-H. Lin, G. E. Sterbinsky, T. Wu, J. A. McLeod and L. Liu, J. Lumin., 2023, 263, 120113 CrossRef CAS.
- Y. Wang, C.-X. Yang and X.-P. Yan, Nanoscale, 2017, 9(26), 9049–9055 RSC.
- B. B. Srivastava, S. K. Gupta and Y. Mao, J. Mater. Chem. C, 2020, 8(19), 6370–6379 RSC.
- H.-X. Zhao, X.-X. Yang and X. P. Yan, Nanoscale, 2016, 8(45), 18987–18994 RSC.
- H. Chen, B. Zheng, C. Liang, L. Zhao, Y. Zhang, H. Pan, W. Ji, X. Gong, H. Wang and J. Chang, Mater. Sci. Eng., C, 2017, 79, 372–381 CrossRef CAS PubMed.
- W.-T. Shiu, L.-Y. Chang, Y. Jiang, M. Shakouri, Y.-H. Wu, B.-H. Lin and L. Liu, Phys. Chem. Chem. Phys., 2022, 24(35), 21131–21140 RSC.
- C. Bharti, N. Gulati, U. Nagaich and A. Pal, Int. J. Pharm. Invest., 2015, 5(3), 124 CrossRef CAS PubMed.
- C. Fu, T. Liu, L. Li, H. Liu, D. Chen and F. Tang, Biomaterials, 2013, 34(10), 2565–2575 CrossRef CAS PubMed.
- B. Xu, S. Li, R. Shi and H. Liu, Signal Transduction Targeted Ther., 2023, 8(1), 435 CrossRef CAS PubMed.
- M. Manzano and M. Vallet-Regí, Adv Funct Mater., 2020, 30(2), 1902634 CrossRef CAS.
- J. Shi, X. Sun, S. Zheng, J. Li, X. Fu and H. Zhang, Biomaterials, 2018, 152, 15–23 CrossRef CAS PubMed.
- L. Zhan-Jun, Z. Hong-Wu, S. Meng, S. Jiang-Shan and F. Hai-Xia, J. Mater. Chem., 2012, 22, 24713–24720 RSC.
- A. Satpathy, W. Huang, T. Liu, T. Su, W. Zhang, M. Kamiński, M. Grzegorczyk, J. Chen, D. Cherng, K. Lu, X. Chen, S. Mahlik and R. Liu, Adv. Opt. Mater., 2024 DOI:10.1002/adom.202400130.
- Z. Li, Y. Zhang, X. Wu, X. Wu, R. Maudgal, H. Zhang and G. Han, Adv. Sci., 2015, 2(3), 1500001 CrossRef PubMed.
- Y. Lin, J. Hu, L. Wu, Q. Zou, D. Chen, D. Huang, H. Lu, S.-B. Wang and H. Zhu, J. Mater. Chem. B, 2021, 9(4), 1131–1137 RSC.
- Y. F. Gao, R. Zou, G.-F. Chen, B.-M. Liu, Y. Zhang, J. Jiao, K.-L. Wong and J. Wang, Chem. Eng. J., 2021, 420, 130021 CrossRef CAS.
- W. Huang, T. Su, M. Chan, J. Tsai, Y. Do, P. Huang, M. Hsiao and R. Liu, Angew. Chem., 2021, 133(13), 7031–7035 CrossRef.
- R. Zhou, J. Huang, J. Shi, L. Huang, X. Zhang, K. L. Wong, H. Zhang, D. Jin, J. Wang and Q. Su, Nano Res., 2017, 10, 2070–2082 CrossRef.
- X. H. Lin, L. Song, S. Chen, X. F. Chen, J. J. Wei, J. Li, G. Huang and H. H. Yang, ACS Appl. Mater. Interfaces, 2017, 9(47), 41181–41187 CrossRef CAS PubMed.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12(4), 537–541, DOI:10.1107/S0909049505012719.
- P. M. Aneesh, K. M. Krishna and M. K. Jayaraj, J. Electrochem. Soc., 2009, 156(3), K33 CrossRef CAS.
- Y. Liang, J. Ouyang, H. Wang, W. Wang, P. Chui and K. Sun, Appl. Surf. Sci., 2012, 258(8), 3689–3694 CrossRef CAS.
- L. Zu, X. Cui, Y. Jiang, Z. Hu, H. Lian, Y. Liu, Y. Jin, Y. Li and Z. Wang, Mater, 2015, 8(4), 1369–1383 CrossRef CAS PubMed.
- M. Shakeri, Z. Khatami Shal and P. Van Der Voort, Mater, 2021, 14(17), 5082 CrossRef CAS PubMed.
- W. Nie, F. M. Michel-Calendini, C. Linarès, G. Boulon and C. Daul, J. Lumin., 1990, 46(3), 177–190 CrossRef CAS.
- J. Su, S. Ye, X. Yi, F. Q. Lu, X. B. Yang and Q. Y. Zhang, Opt. Mater. Express, 2017, 7(3), 734–743 CrossRef CAS.
- Z. Hu, D. Ye, X. Lan, W. Zhang, L. Luo and Y. Wang, Opt. Mater. Express, 2016, 6, 1329H–1338H CrossRef.
- S. H. Choi, S. B. Kwon, B. K. Kang, J. H. Yoo, Y. H. Song and D. H. Yoon, J. Lumin., 2021, 234, 117945 CrossRef CAS.
- J. S. Kim, J. S. Kim and H. L. Park, Solid State Commun., 2004, 131(12), 735–738 CrossRef CAS.
- X. Bai, C. Lin, Y. Wang, J. Ma, X. Wang, X. Yao and B. Tang, Dent. Mater., 2020, 36(6), 794–807 CrossRef CAS PubMed.
- Z. Neščáková, K. Zheng, L. Liverani, Q. Nawaz, D. Galusková, H. Kaňková, M. Michálek, D. Galusek and A. R. Boccaccini, Bioact. Mater., 2019, 4, 312–321 Search PubMed.
- M. Cui, M. Li, J. Wang, R. Chen, Z. Xu, J. Wang, J. Han, G. Hu, R. Sun, X. Jiang, B. Song and T. He, Angew. Chem. Int. Ed., 2021, 60(28), 15490–15496 CrossRef CAS PubMed.
- A. Anedda, C. M. Carbonaro, F. Clemente, R. Corpino, S. Grandi, P. Musarelli and A. Magistris, J. Non-Cryst. Solids, 2003, 322, 68–72 CrossRef CAS.
- A. Anedda, C. M. Carbonaro, F. Clemente, R. Corpino and P. C. Ricci, J. Phys. Chem. B, 2005, 109, 1239–1242 CrossRef CAS PubMed.
- Q. He, J. Shi, X. Cui, C. Wei, L. Zhang, W. Wu, W. Bu, H. Chen and H. Wu, Chem. Commun., 2011, 47, 7947–7949 RSC.
- K. Huang, W. Wu, S. Xu, P. Yan, Z. Wei and Q. Xu, Energy Environ. Mater., 2023 DOI:10.1002/eem2.12655.
- J. Yang, Y. Zhou, H. Ming, E. Song and Q. Zhang, ACS Appl. Electron. Mater., 2022, 4(2), 831–841 CrossRef CAS.
- A. Fernández-Osorio, M. Tapia, A. R. Vázquez-Olmos and J. Chávez, J. Solid State Chem., 2019, 269, 328–335 CrossRef.
- C. Liu, G. Che, Z. Xu and Q. Wang, J. Alloys Compd., 2009, 474, 250–253 CrossRef CAS.
- T. Zhang, A. Du, C. Chen, X. Ji, B. Zhou, J. Shen and Z. Zhang, RSC Adv., 2019, 9, 33883 RSC.
- E. K. Griffith, E. D. Ingall, P. L. Morton, D. A. Tavakoli and B. Lai, J. Synchrotron Radiat., 2019, 26, 1302–1309 CrossRef PubMed.
- L. Liu and T. Sham, Small, 2012, 8(15), 2371–2380 CrossRef CAS PubMed.
| This journal is © the Owner Societies 2024 |