
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploring the phase behavior of C8-BTBT-C8 at ambient and high temperatures: insights and challenges from molecular dynamics simulations†
Mosè
Casalegno
 *a,
Simone
Provenzano
b,
Guido
Raos
a and
Massimo
Moret
*c
*a,
Simone
Provenzano
b,
Guido
Raos
a and
Massimo
Moret
*c
aDepartment of Chemistry, Materials and Chemical Engineering “G. Natta”, Politecnico di Milano, via L. Mancinelli 7, 20131 Milano, Italy. E-mail: mose.casalegno@polimi.it
bApplied Materials Inc., Reggio Emilia, Italy
cDepartment of Materials Science, Università degli Studi di Milano-Bicocca, Via R. Cozzi 55, 20125 Milano, Italy. E-mail: massimo.moret@unimib.it
First published on 29th July 2024
Abstract
C8-BTBT-C8 is one promising candidate for the development of high-performance electronic devices based on thin-film technologies. Its monoclinic polymorph has a well-established role in thin-film growth. Yet, quite little information is available about its dynamics on the molecular scale, and the structures of the mesophases which form at high temperature (about 100 K above ambient temperature). The present study is devoted to the analysis of such phases, with the ultimate goal of developing molecular models. Already at ambient temperature, our molecular dynamics simulations reveal a rich conformational behavior of the alkyl side chains, with gauche conformations as leading structural defects. Heating promotes the formation of a stacking faulted mesophase (380 K), and a smectic phase, at 385 K, upon side chain melting. Although more disordered, this phase bears several analogies with the smectic A phase, experimentally observed at 382.5 K. At higher temperatures, the increase in configurational disorder is brought by molecular diffusion and other phenomena, finally leading to an isotropic molten phase. Our in-depth analysis, complemented by hot-stage polarizing microscopy data, provides interesting insights into this material, highlighting the challenges associated with the modeling of soft semiconducting systems.
Introduction
Organic semiconductors are a wide class of materials with a broad spectrum of applications, ranging from solar energy conversion to sensing.1–4 The success of many applications requires highly ordered, large area crystalline structures, to maximize the efficiency of charge transport along the available intra and intermolecular channels.5–8 Molecular assemblies suitable for this purpose are often obtained by means of thin film technologies. Here, the active material is deposited onto an inert substrate and subsequently engineered during device manufacture. The precise control of thin film morphology represents a formidable issue, due to the many factors that affect molecular organization.9,10 The development of ordered crystalline phases results from the competition between weak molecular interactions and thermal motion. The interactions with the substrate, in combination with kinetic effects, may heavily interfere with this process, triggering the formation of poorly ordered molecular assemblies, or crystal polymorphs different from those observed in the bulk.11 At the same time, the precise control of such interactions may eventually lead to structures with new and interesting properties.Although examples of surface-induced polymorphism have occasionally been reported12–14 several active materials, once deposited, develop crystalline thin phases closely resembling bulk ones. This especially occurs when film–substrate interactions are weak,15 so that the interactions between adsorbed molecules play a dominant role in thin film assembling. In such cases, the characterization of bulk polymorphs and their thermal behavior represents a preliminary step toward a better understanding of on-surface dynamics. Removing the complexity associated with surface interactions can be expected to simplify the investigations; nonetheless the detailed characterization of the different polymorphs of a given compound remains challenging in many cases.
One specific issue concerns the availability of structural models for materials for which some phases are elusive and difficult to detail at the atomistic level. One meaningful example is provided by liquid crystals (LCs)16,17 for which partially disordered mesophases are often observed. LC materials often result from the addition of long alkyl side chains to π-conjugated aromatic cores.18 This design guarantees high solubility in common organic solvents while preserving the desired film-forming properties. At the same time, it brings some degree of molecular “softness” which may allow for the formation of stable phases characterized by conformational and/or configurational disorder. Quite a large variety of such mesophases have now been recognized,18–20 including the smectic phases, where the molecules are organized in layers, the translational symmetry being broken in at least one direction in space.21–23
In contrast to highly ordered crystalline phases, the characterization of smectic and other disordered phases cannot rely on experimental techniques alone but requires the integration of experimental data and computational modeling techniques, such as molecular dynamics.24–26 So far, this method has proven valuable in the characterization of the bulk properties of “soft” semiconducting materials,26–31 including polymers.32
Herein, molecular dynamics is applied to the study of the thermal behavior of a solution-processable semiconductor, 2,7-dioctyl-[1]benzothieno[3,2-b][1]benzothiophene (C8-BTBT-C8).33,34 C8-BTBT-C8 is well-known among BTBT derivatives,35 thanks to the high charge-carrier mobility displayed in organic field-effect transistor (OFET) device architectures.36–38 C8-BTBT-C8 is characterized by two distinct, reversible phase transitions at high temperature: from the ambient temperature monoclinic crystal polymorph (space group P21/a) to a smectic A phase (SmA) at 382.5 K, followed by melting to the isotropic phase at 398 K.33,39 A second, triclinic polymorph has recently been discovered at low temperature.31
A number of studies have highlighted the importance of the monoclinic polymorph in C8-BTBT-C8 thin film growth.15,40–46 Yet, little information is available on the dynamics of this polymorph on the molecular scale. One concrete issue involves the characterization of structural defects playing a role at ambient and high temperatures. In this respect, gauche conformations, characterized by the synclinal alignment of vicinal methylene groups in alkyl side chains, are of special interest, as testified by recent reports on asymmetric BTBT derivatives.47–49 Besides, the development of high temperature mesophases calls for detailed investigations, due to the role of such phases in the dewetting/rewetting processes taking place during thermal treatments.43,44
In contrast to another study28 which has primarily focused on C8-BTBT-C8 equilibrium structures, here we investigate the thermal behavior of C8-BTBT-C8 in a wide range of temperatures (from ambient to high temperatures). Special emphasis is put on the analysis of the phase transition mechanisms and the resulting phases. Our goal is twofold: to gain some insights into the structure of C8-BTBT-C8, and develop atomistic models which may supplement the available experimental information.
Computational methods
Molecular dynamics simulations
The monoclinic crystal structure of C8-BTBT-C8 was used as a starting point for the MD simulations. Fig. 1A shows two molecules within the monoclinic unit cell (space group P21/a) with the following lattice parameters: a = 5.927(7) Å, b = 7.88(1) Å, c = 29.18(4) Å, β = 92.443(4)°.34 The structure is characterized by a herringbone arrangement of BTBT cores in the ab plane. | ||
| Fig. 1 (A) C8-BTBT-C8 unit cell. The unit cell lengths are reported, along with the A and B labels used to distinguish the two alkyl side chains. (B) and (C) Views of the supercell input structure along the a (A) and c axes (C). For better clarity, hydrogen atoms (top and bottom views) and alkyl side chains (bottom view) have been omitted. Carbon and sulfur atoms are highlighted in orange and yellow, respectively. | ||
A supercell consisting of 15 × 15 × 3 unit cells along a, b, and c axes (1350 molecules, 97![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 atoms overall) was constructed to simulate a C8-BTBT-C8 bulk crystal. Views of this input structure are provided in Fig. 1B.
200 atoms overall) was constructed to simulate a C8-BTBT-C8 bulk crystal. Views of this input structure are provided in Fig. 1B.
Full atomistic simulations were performed using the GROMACS 2021.2 package.50,51 To model intra and intermolecular interactions we adopted an all-atom force field (FF) based on the CHARMM36 parametrization,52–56 within the CHARMM general force field framework.57–59 This choice was justified by the need to model the thermal behavior of C8-BTBT-C8 on silica surfaces in an ongoing study,60 for which a force field compatible with CHARMM has recently been made available.61
A straightforward protocol was then used for force field generation and implementation. A single C8-BTBT-C8 molecule was first submitted to the CGNEFF web utility,62 in order to define atom types and charges. The atomic charges were subsequently refined at the HF/6-31G(d)//HF/6-31G(d) level, following the RESP-A1 approach,63via the PyRED utility.64,65 Files suitable for use in GROMACS were finally obtained via porting utilities.66 The force field parameters and the MD input files in GROMACS format are supplied with the ESI.†
The leapfrog algorithm with a time step of 1 fs was used to integrate the equations of motion. Temperature was controlled by coupling with a velocity rescaling thermostat,67 with a coupling constant of 1.0 ps.
All simulations were performed at ambient pressure. During equilibration and all the production runs involving phase transitions, the pressure was controlled via the Berendsen barostat (BE).68 Due to its sensitivity to large box deformations, the Parrinello–Rahman (PR) barostat69 was only used to equilibrate the structures obtained from BE runs at ambient temperature (see below). For both barostats, the coupling time constant was set at 5.0 ps and isothermal compressibility at 1 × 10−6 atm−1.
No constraints were applied to the simulation box lengths and angles, thus allowing for the development of triclinic P1 structures. Periodic boundary conditions were applied along all dimensions in all systems. Electrostatic interactions were treated via the particle-mesh-Ewald method70 with default settings (Fourier grid spacing of 0.12 nm and PME order of 4).
Following CHARMM specifications for GROMACS,71 the cutoff for non-bonded interactions was set at 1.2 nm. Non-bonded forces were switched in the range between 1.0 to 1.2 nm via a force-switch modifier. All simulations were carried out under the NPT ensemble.
During force field validation (see below), the starting input structure was equilibrated via a short run (5 ns) under the BE barostat, then a longer (10 ns) production run was performed under the PR barostat. A short (0.5 ns), dense NPT-PR run was also carried out, saving the trajectories at each ps, to be used for analysis purposes. All these simulations were performed at 293 K and 1 atm, so as to compare with experimental XRD data.
To study the behavior of C8-BTBT-C8 at high temperatures, independent 100-ns NPT runs were performed at different temperatures. The choice of the simulation time of the production runs was made on the basis of preliminary simulations and previous calculations on a similar system.30
In all cases, the aforementioned equilibrated structure obtained at 293 K was used as the starting point. To better handle the large box deformations expected during phase transitions, all these runs were performed under the BE barostat. In some cases, these simulations were extended beyond 100 ns.
To further check our results, we performed two additional simulations, gradually cooling the corresponding phases (see below) from 460 to 385 K and from 385 to 293 K. In both cases, a linear temperature gradient was applied. Each simulation was performed under the BE barostat, with an overall duration of 100 ns.
MD trajectories were collected every 50 ps, and processed mostly using in-house programs, in order to extract relevant physical and structural information, including powder XRD patterns. Additional software packages, including Mercury,72 Discovery Studio,73 VMD,74 Gnuplot,75 Mathematica,76 and OriginPro,77 were also used in the data analysis and figure production.
Structural descriptors
Fig. 2 summarizes the different structural descriptors used in the present work to characterize the molecular structures. Molecular orientation was quantified by means of the core tilt angle, φ (see Fig. 2A) between the vector connecting the two farthest atoms of the BTBT aromatic core (ui for the i-th molecule), and the phase director, q. The latter, corresponding to the average orientation of all tilt angles, was chosen as the eigenvector associated with the largest eigenvalue of the Cartesian ordering matrix, Q, namely:78 | (1) |
| φi(t) = ±cos−1(ui(t)·q(t)) | (2) |
 | (3) |
 | ||
Fig. 2 Definition of structural descriptors used in the present work. (A) Definition of the core tilt angle, φ, calculated as the angle between the vector connecting the two farthest C atoms (dashed red line connecting the green atoms) and the phase director, q. The core normal was defined as the vector product between the vectors connecting selected core atoms (blue and orange). The molecular centroid (small red circle) is also shown. (B) Intramolecular side chain parameters: torsion angles τ1–τ7, C8 chain length, ![[small script l]](https://www.rsc.org/images/entities/i_char_e146.gif) , measured between the red carbon atoms (dashed red line). The tilt angle δ was considered only at 293 K and calculated with respect to the c* axis, except for Table S3 (ESI†). For clarity only chain A is shown. The same labels were also adopted for chain B. , measured between the red carbon atoms (dashed red line). The tilt angle δ was considered only at 293 K and calculated with respect to the c* axis, except for Table S3 (ESI†). For clarity only chain A is shown. The same labels were also adopted for chain B. | ||
At any given time during the simulation, the value of P2(t) provides indications about the overall molecular orientation. Its value can be expected to approach unity when the molecules are, on average, aligned along the phase director. Values approaching zero, conversely, are associated with the loss of molecular ordering, which occurs, for example, in isotropic molten phases. We also considered 〈P2(t)〉, namely the average of P2(t) over all the MD frames.
Fig. 2B collects the side chain intramolecular parameters for chains A and B. These include seven C–C–C–C torsion angles labeled τ1–τ7, the C8 chain length, ![[small script l]](https://www.rsc.org/images/entities/char_e146.gif) , and the side chain tilt angle, δ.
, and the side chain tilt angle, δ.
Results and discussion
Dynamics at ambient temperature and force field validation
In the early stages of our investigation, we performed MD simulations at ambient temperature (293 K) aimed at providing a basis for a proper discussion of simulations at higher temperatures and assessing FF reliability by comparison with XRD data at the same temperature.34To this end, the bulk supercell detailed in Fig. 1 was shortly equilibrated (5 ns) via the BE barostat at 293 K. Subsequently, a longer (10 ns) run was performed using the PR barostat at the same temperature, followed by a 500-ps dense NPT-PR run.
The MD trajectories extracted from this simulation showed a rich conformational behavior of the C8 chains embedded in the crystal structure. Analysis of 500 frames 1 ps apart provided more than 1 million different observations for each parameter describing the chain conformations. The seven C–C–C–C torsion angles τ1–τ7 defined in Fig. 2 show several interesting trends. The first torsion along the C8 chain, τ1, describes the rotation of the whole chain with respect to the central BTBT core (Fig. 2). Chains A and B (defined in Fig. 1) show identical distributions, with a global two maxima distribution centered at ca. ±65° arising from the superposition of the two independent distributions (Fig. 3A). Merging the data for chains A and B, according to centro-symmetry, gives the distribution reported in Fig. 3B for the 500 ps trajectory. The merged τ1 distribution centered at positive values is left tailed with the peak mode at +67.3° in agreement with the experimental value of 65.7(7)°.34 Values of τ1 around 65° allow relieving of non-bonded repulsive contacts between the first two methylene units of the octyl chains and the BTBT core.79 In Fig. 3B, the left tail from τ1 = +30° down to negative values accounts for ca. 13% of all conformations, including τ1 = 0°, an arrangement with the first two methylene groups closest to BTBT C–H moieties.
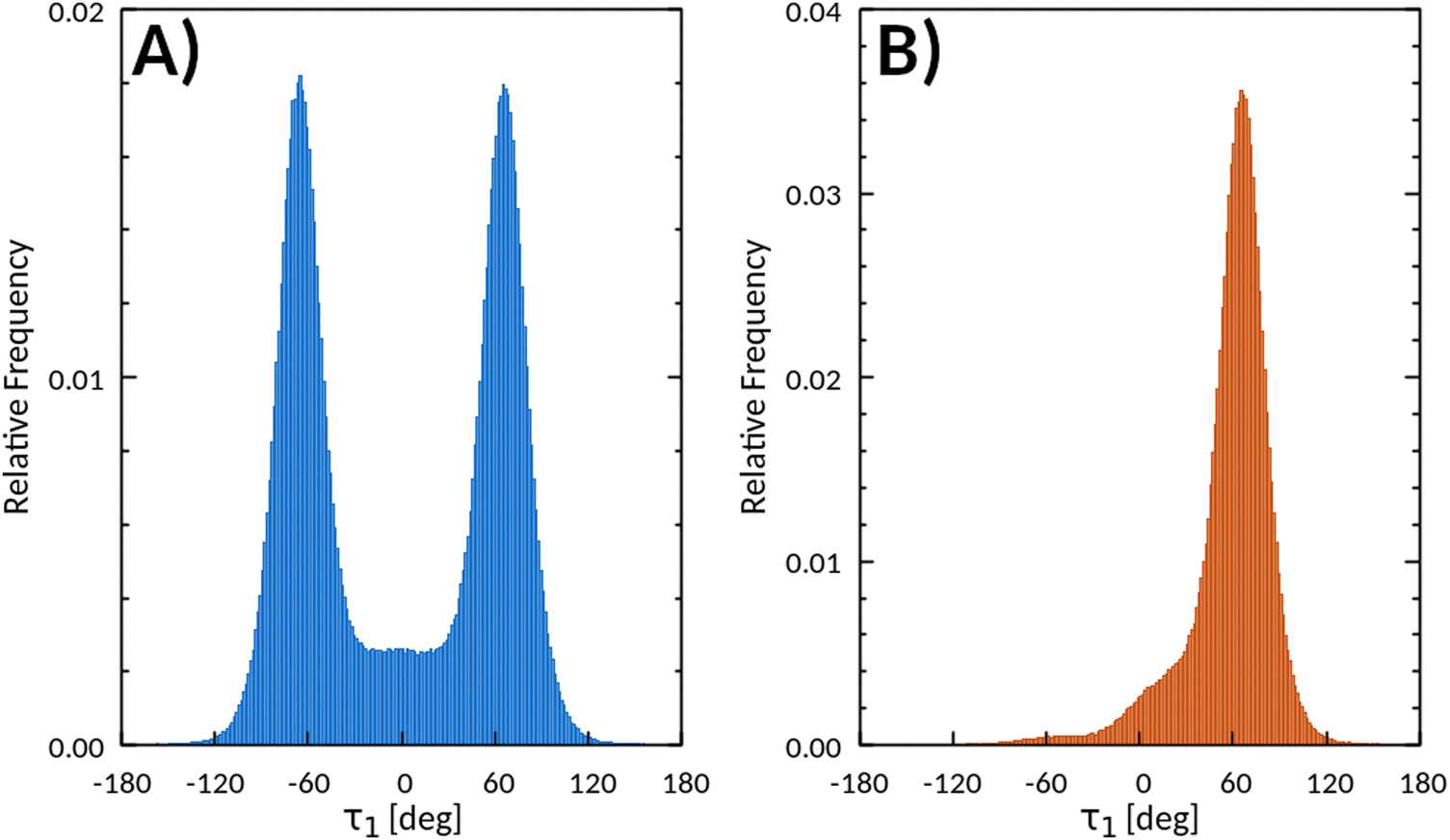 | ||
| Fig. 3 (A) Cumulative distributions for chains A and B that show identical distributions with opposite sign of τ1; (B) distribution obtained after merging data for A and B side chains according to centro-symmetry. The distribution of τ1 for a single chain emerges, arbitrarily chosen as that with positive τ1. | ||
In Fig. S1A (ESI†) the trajectories of two randomly chosen molecules (adjacent in the simulation box) show different behaviors for their alkyl chains. Both chains of molecule #329 have oscillations of τ1 around the mean value with few short trips toward 0°. In contrast, the chain of molecule #330 with positive τ1 at 325 ps suddenly jumps into the region of negative torsions, lying around −67° for about 65 ps. In the same time interval, increased fluctuations appear also for the other chain but without stabilizing at positive values of τ1. For a given molecule, conformations with both chains having τ1 of the same sign are unlikely, as evidenced by the density plot of Fig. S1B (ESI†) with sparingly occupied regions with this feature (white circles). These regions correspond to heavily non-centrosymmetric molecules whose intramolecular potential energy (due to stretching, bending, van der Waals terms) is not particularly unfavorable. However, when both τ1 are around +67° (or −67°) (top molecule in Fig. S1C, ESI†) clashing of the chains with neighbour almost centrosymmetric molecules (bottom molecule in Fig. S1C, ESI†) is produced. Therefore, random cavities can appear from time to time due to thermal fluctuations of the crystal structure allowing existence of these chain conformations for short times.
Torsion angles τ2–τ7, defined in Fig. 2, are all characterized by sharp distributions centered at 180°, i.e. anti conformations (Fig. S2, ESI†). However, gauche populations at ca. ±67° (data are normalized in the range 0–360° so that −67° corresponds to +293°) are always present with varying populations for different positions along the chain. This gives rise to two distinct subgroups of torsions: the first subset comprises torsion angles τ2, τ4 and τ6 exhibiting only tiny gauche satellites, evidenced by the histograms in Fig. S2 and data in Table S1 (ESI†). While these secondary peaks are hardly visible for τ2 and τ4, their relevance grows slightly but steadily on moving toward the free end of the chain. A similar trend for gauche conformations is observed also for torsions τ3, τ5 and τ7. In this subgroup, the gauche populations are easily observable already in τ3 (closest to the BTBT core), grow slightly with τ5 and reach a 2/1 anti/gauche ratio for τ7 at the end of the alkyl chain (Fig. S2, ESI†).
Although the increasing relevance of the gauche maxima on moving away from the BTBT core toward the end of the alkyl chains can be expected, due to an increased freedom of motion at the end of the alkyl chains, the origin of the even/odd τ pattern here reported is unclear and, to the best of our knowledge, has never been explicitly reported in previous experimental or computational studies. Indeed, MD simulations of polyethylene reveal no or very few gauche defects for infinite chains.80,81
For n-alkanes, infrared spectroscopy on deuterated molecules evidenced the presence of gauche defects and gauche–anti–gauche kinks, with gauche defects concentrated at the end of chains, especially in the rotator phases.82,83 Compared to n-alkanes, the present system possesses an increased freedom for the conformational dynamics of chains due to the presence of the BTBT core which acts as a spacer within the ab plane, consistently distancing alkyl chains from each other (Fig. S3 (ESI†) qualitatively compares the steric hindrance of C8-BTBT-C8 side chains with n-octane ones).
Eclipsed conformations are practically absent (see Fig. S2 (ESI†) at 0, 120 and 240°) as they represent high intramolecular potential energy states and appear only for short times during conformational interchange. Therefore, anti conformations dominate the chain regions described by τ2–τ4–τ6 but become less relevant along the sequence τ3–τ5–τ7. This overall behavior allows the adoption of a simple description of the conformational space of the C8 chains with negligible loss of information. By using only τ3, τ5 and τ7 as descriptors of the chains dynamics, the reduced conformational space comprises 27 clusters of torsions arising from the three peaks in each of the τ3, τ5, τ7 distributions (Fig. S4A–C, ESI†). Owing to differences in the relevance of the anti and gauche populations, the clusters are strongly scattered in size. Fig. S4D (ESI†) summarizes these clusters as spheres whose diameter is proportional to the number of observations. The central cluster at (τ3, τ5, τ7) = (180°, 180°, 180°) collects 57.3% of all data points, followed by two clusters (180°, 180°, 68°) and (180°, 180°, 295°) each comprising 9.4% of points. More than half of the chain dynamics comprises zig-zag fully extended chains with six anti conformations (all-trans), providing the longest chains (see Fig. 2). With other combinations of the τ2–τ7 angles the number of gauche defects increases, including double or multiple gauche defects, and kinked chains arise, more sterically demanding for accommodation within the surrounding neighbors. As an example, τ3, τ5 and τ7 all close to ±67° coupled to the almost monomodal distributions of τ2–τ4–τ6 give τ2–τ7 sequences of the type agagag (where a = anti and g = gauche; in the subsequent discussion g+ will represent τ around +67°, g–τ around −67°, i.e. +293°) including ca. 6% of all conformations. Fig. S4E (ESI†) shows the observed agagag conformations which include the other four enantiomeric cases. When considering the presence of gauche defects independently of their sign the conformations exceeding 1% of cases are reported in Table S2 (ESI†).
At 293 K 18% of chains bear only one terminal gauche defect, aaagag and agagag, accounting for 6% each, while agaaaa and aaaaga, with only one g defect, are less likely than agagaa and agaaag with two gauche defects. These eight most frequent cases account for 98% of conformations. Interestingly, the aaaaga case (gauche close to the end of chain) is less frequent than agaaaa where the defect is close to the BTBT core, in contrast with other reports.82,83
Out of 36 = 729 possible main cases (a, g+, g−) for six torsions τ2–τ7, in molecules comprising two or more gauche conformations, the sign (same or opposite) and the position of the gauche defects within the chains determine only a moderate increase of the intramolecular potential energy compared to the all-trans conformation but, more importantly, increase steric crowding and non-bonded interactions with the surrounding molecules.84 According to our calculations, the average non-bonded contribution to the potential energy at 293 K is about 12.5 kJ mol−1 (per molecule) greater than that evaluated for the starting configuration with all-trans conformation (as from the XRD data34). Most of this energy is associated with the electrostatic interactions, thus highlighting the role of partial atomic charges in side chain dynamics.
The octyl chain length, , exhibits a bimodal distribution (Fig. S5A, ESI†) with peaks at 8.12 Å and 8.95 Å, with full widths at half maximum (FWHM) of 0.631° and 0.257°, respectively. The length of C8 chains (defined in Fig. 2B) is the outcome of the overall ensemble of local conformations τ2–τ7; the longest alkyl chains represent 56% of all data points (threshold at 8.7 Å) and implies fully stretched chains with all-trans conformations (Fig. S5B–G, ESI†). All other combinations of local conformations give rise to a broad distribution due to the many different sequences of torsions with shorter chains. On the contrary, the molecular centroids fluctuate around the average without strong correlations with conformational changes of the alkyl chains.
The value of in the monoclinic ambient temperature structure of C8-BTBT-C834 is 8.90 Å, close to the sharpest peak of the simulation. However, at 293 K the MD simulation affords an average cell c axis of 28.20(3) Å, thus one Å shorter than the experimental value of 29.18(4) Å. The deviation from the experimental value of the d001 thickness can arise from the force field parameters that influence the length and tilt angle of the chains, the tilt of the BTBT core (see Fig. 2), molecular motion along the c axis, and the interlocking pattern of methyl groups at the interface between adjacent (001) layers (see below). As to the effect of chain length, the statistical distribution in single MD frames mirrors the 500 ps temporal distribution reported in Fig. S5A (ESI†). Therefore, there are constantly more than 50% of molecules with fully extended chains able to sustain the thickness of the (001) monolayer. Fig. 4 provides a color-coded representation of the temporal evolution of torsion angles τ2–τ7 for four selected molecules. The bright green areas evidence predominance of torsions with τ = 180° ± 10°, completed by anti conformations more deviated from the ideal angle (olive green areas). The different behavior of “even” and “odd” torsions is easily appreciated by noting that τ2–τ4–τ6 plots are green-colored most of the time while τ3–τ5–τ7 plots experience frequent conformational changes. The gauche defects (blue-coded for positive values, red coded for negative values translated by +360°), appear and disappear several times during the 500 ps trajectory, including sudden changes from g+ to g− or vice versa.
 | ||
| Fig. 4 Color-coded representation of the temporal evolution of torsion angles τ2–τ7 for four selected molecules during a 500-ps run at 293 K. According to the look up table at the right of the plots anti conformations are represented by bright green and olive-green areas, gauche conformations are evidenced by blue or red regions, and very rare eclipsed conformations by white or black areas. | ||
The experimental value of δ (see Fig. 2) is 35.6° (ref. 34) while the mean tilt of the two molecules in the unit cell averaged during the 500 ps trajectory is 37.0°. The values of δ in Fig. S6A (ESI†) (after merging the data of both chains) and Table S3 (ESI†) show a monomodal distribution without correlation between the tilt of chain A and chain B. The distribution is slightly skewed toward the c* axis with a mean tilt of 37.3(4.3)°. The tilt of chains referred to as the mean tilt direction (see the discussion at higher temperatures below) gives the sharper distribution of Fig. S6B (ESI†) (with an average value of 7.02(3.6)°), with deviations becoming less and less likely as they become larger.
The herringbone angle between adjacent molecules, often used to describe the packing features of polycyclic molecules, including organic semiconductors, is 52.27° for the average unit cell of the MD simulation, to be compared with the experimental value at ambient temperature of 56.42°. To further characterize the dynamics of C8-BTBT-C8, Fig. S7A (ESI†) shows the distribution of the BTBT core tilt angle φ and the angle between the projection onto the ab plane of the normal to the BTBT core and the a axis (Fig. S7B, ESI†). Two symmetric peaks centered at 64.0(4.7)° and 116.0(4.7)° (each for one of the independent molecules in the unit cell in the NPT simulation not constrained to P21/a symmetry) confirm the 21 axis.
The angle of the BTBT core normal with the reciprocal vector c* (Fig. S7C, ESI†) indicates that on average the BTBT cores are orthogonal to the ab plane with a maximum excursion of 10° below and above the plane.85,86 The influence of static and dynamic features in semiconducting molecular crystals upon their physical properties and performance has been deeply analyzed in terms of alkyl chain length,48,87 static and dynamic disorder and thermal motions.86,88
The monomolecular layers contacting each other at the (001) interface represents a crucial structural aspect of monoclinic C8-BTBT-C8. As mentioned above, the terminal methyl groups are the most mobile region of the C8-BTBT-C8 molecules due to molecular librations about the mass center and the conformational flexibility of the octyl chains. The cumulative effects are included in the X-ray structural model as time- and space-averaged coordinates and anisotropic displacement parameters (ADP) (Fig. S8A, ESI†). The usual trend in the average mean square displacement of atoms along the alkyl chains is evident in Fig. S8B (ESI†) (for one selected molecule) with increasing amplitudes for the terminal atoms.
The systematic effect of librational disorder modeled with ADP is the apparent shortening of bonds on moving from the center to the periphery of molecules as observed in the C8-BTBT-C8 crystal structure. The same trend emerges in our simulations with an anomalously short value of 1.12 Å for the CH2–CH3 distance, to be compared with the experimental value of 1.46 Å. Another difference between simulations and experiment appears in the separation between (001) adjacent monomolecular layers; planes passing through the last methylene units along the chains are 4.45 Å apart in the X-ray structure and 4.28 Å for the average MD structure, obtained by averaging the atomic coordinates over 500 ps, whilst keeping two independent molecules (see Fig. S9, ESI†). The interlocking of methyl groups is also slightly different on going from simulation to experiment (see Fig. S10, ESI†).
The series of Cn-BTBT-Cn structures with n = 2–589 and n = 10, 1239 show different (001) interface structures depending on the value of n. Moreover, the methyl groups from the present MD simulations are more mobile and disordered than what emerges from the experimental crystal structure at ambient temperature (and because of that they have not been used to set the interface reference plane).
The results of the above analysis are summarized in Table 1, where experimental and calculated unit cell parameters and densities are compared. Overall, the present force field exhibits a slight increase along the a and b-axis, and a moderate decrease along the c-axis. Nonetheless, the space group and molecular symmetry are neatly reproduced by the MD simulations, all parameters agreeing on a statistical basis with the P21/a space group symmetry and −1 molecular point group symmetry of the monoclinic structure.34
Furthermore, the average MD crystal structure (Fig. S9, ESI†) provides a good match to the experimental one, as evidenced by the agreement of the corresponding powder XRD patterns reported in Fig. 5, with differences mainly depending on the different cell axes length.
 | ||
| Fig. 5 Calculated XRD powder spectra for the experimental34 and the average unit cell at 293 K. | ||
The minimal differences in geometrical parameters, preserving de facto the published monoclinic symmetry are an indication of the good performance of the chosen force field. This outcome is quite remarkable considering that the present force field was not specifically designed to describe compound dynamics in the solid state. Different factors may be responsible for the above results, the most significant being the recalculation of atomic charges. Indeed, short test simulations performed prior to atomic charge optimization showed worse performance in terms of unit cell parameters (not shown). Following our previous work,30 we have recently started to test other force fields, which gave acceptable results at 293 K, but failed modeling the phase transition to the smectic phase.
Thermal behavior at high temperatures
As a further step in our investigation, we attempted at modelling the dynamics of bulk C8-BTBT-C8 at different temperatures. This material is characterized by a transition from crystalline to the smectic (SmA) phase at 382.5 K, and a transition to the isotropic melt at 398 K.43 More recently, a triclinic, low-temperature phase has also been discovered, upon cooling the monoclinic polymorph between 140 and 150 K.31 Hereafter, we report on the results obtained at high temperatures.Based on the above experimental data, different temperatures were investigated, in the range between 380 K and 500 K, so as to account for possible deviations between calculated and experimental phase transition temperatures. In the following, we shall report the results obtained at a few representative temperatures, namely 380, 385, and 460 K.
Fig. 6 collects snapshots of the final structures extracted from NPT-BE simulations at the different temperatures. For comparison purposes, the structure at 293 K is also reported. Unit cell parameters, densities, and average order parameters associated with these simulations are collected in Table 2. A comparison with experimental data has been performed by measuring the thermal expansion of mm sized crystals of C8-BTBT-C8 (grown from tetrachloroethane solutions) with a hot stage in the range 303–373 K. The expansion along the a axis was ca. 1% whereas along the b the crystals enlarged by 4% with a linear trend.
 | ||
| Fig. 6 Views of the structures obtained from the NPT simulations along the a axis, at (A) 293 K; (B) 380 K; (C) 385 K; (D) 460 K. The structure at 293 K refers to the 500-ps NPT-PR run (see above). | ||
| T [K] | a [Å] | b [Å] | c [Å] | α [deg] | β [deg] | γ [deg] | Density [g L−1] | 〈P2(t)〉 |
|---|---|---|---|---|---|---|---|---|
| 380 | 5.941(37) | 8.95(16) | 28.02(15) | 87.96(81) | 87.51(28) | 90.023(42) | 1034.5(4) | 0.841(3) |
| 385 | 5.82(12) | 9.37(33) | 28.29(14) | 88.10(55) | 87.19(31) | 90.02(18) | 991.1(7) | 0.86(3) |
| 460 | 6.452(18) | 9.02(18) | 28.73(46) | 90.040(45) | 90.47(11) | 89.929(55) | 920.6(4) | 0.004(31) |
At 380 K, a phase transition was observed, with the starting structure evolving to a smectic phase with stacking faults. The time-resolved supercell density profiles, reported in Fig. 7A, and the evolution of box parameters (see Fig. S11, ESI†) suggest an overall process consisting of two subsequent stages.
 | ||
| Fig. 7 Time-resolved profiles of (A) density, and (B) order parameter P2(t), extracted from the NPT-BE run at 380, 385 and 460 K. | ||
The first stage was a fast (4 ns) supercell relaxation, characterized by a quick drop in density, due to the increase in supercell volume. The supercell size increased along b and c, whereas it decreased along a. A modest change in supercell shape was related to the change in the β angle, from 91.03° to 87.52° (at 4 ns). No relevant changes were detected in the two remaining angles. This initial stage was followed by a more gradual and slower structural rearrangement, lasting about 30 ns overall.
Fig. 7B reports the evolution of P2(t) at all the investigated temperatures. As mentioned in the Methods section, P2(t) quantifies the instantaneous change of the average overall molecular orientation with respect to the phase director (see eqn (1)–(3) and Fig. 2A). Its starting value was close to unity at the beginning of each simulation, since the equilibrated structure at 293 K was used in all cases as the starting point. Then, different outcomes were observed, depending on the simulation temperature.
At 380 K, the evolution of P2(t) is consistent with the hypothesis of two consecutive stages proposed above. During the former, the molecular orientation along the phase director, essentially aligned with c*, changed only slightly. After about 4 ns, a more significant rearrangement occurred, with P2(t) gradually approaching 0.841(3) after 30 ns (see Table 2).
To investigate this outcome further, we considered the φ angle behavior in more detail. Based on the definition of φ given in Fig. 2, two equal groups of molecules were recognized in the starting crystal structure,34 one with φ = +2.52°, the other with φ = −2.51°. After relaxation at 293 K, these values settled at φ = +4(2)° and φ = −4(2)°.
Fig. 8A follows the evolution of φ at 380 K. For better clarity, the original distributions, collected every 50 ps, were window-averaged over a time interval of 3 ns. At the beginning of the simulation, the molecular orientations were evenly distributed in two main ensembles, resembling those recognized at 293 K. After about 4 ns, all molecules changed their orientations gradually and simultaneously, with two/thirds of the molecules tilting toward positive φ values. The tilt angle distributions look asymmetrical with respect to φ = 0 due to the phase director orientation. This was shifted by about 6.7° with respect to the c* axis, as a consequence of different tilt angle populations.
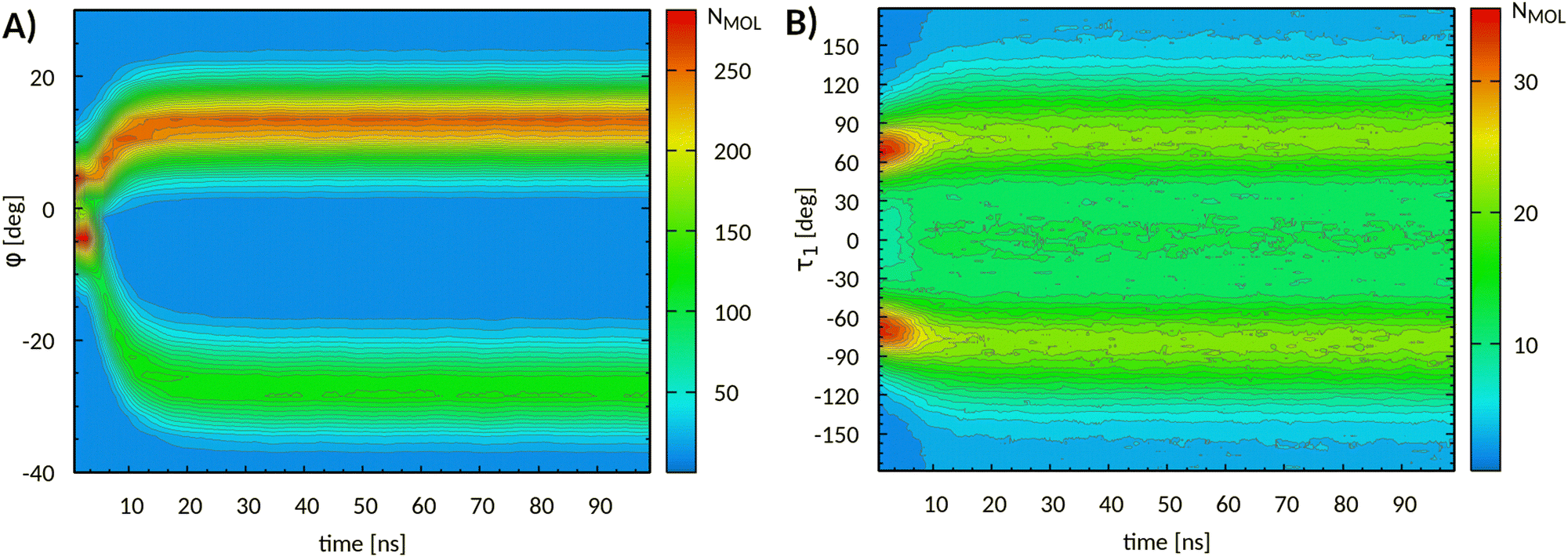 | ||
| Fig. 8 (A) Evolution of core tilt angle φ and (B) of torsional angle τ1 obtained from MD simulations at 380 K. The absolute molecular populations are also reported. For better clarity, the data have been window-averaged over time intervals of 3 ns. | ||
By the end of the simulation, core tilt angles attained stable averages, namely 12(4)° (900 molecules) and −27(4)° (450 molecules), respectively (first 30 ns discarded). The effect of this structural rearrangement is clearly visible in the tilted molecular layers that appear in Fig. 6B. The resulting system is partially disordered and the original symmetry lost. The faulted stacking of the layers of Fig. 6B could be an artifact due to the small size of the simulation box. However, stacking faults are commonly observed in other layered organic (and inorganic) crystals,90 including semiconductors (e.g. quaterthiophene91). In fact, the faulted interfaces require only a modest cost (a few %) in terms of stabilizing energy compared to the correct stacking sequence and lead to polysynthetic twinning and polytypes.
Next, we monitored the conformational changes associated with the alkyl side chain torsions. Fig. 8B shows the evolution of τ1 over time (chain A), the syn conformation being at 0°. The two local maxima at the early stage of the simulation are representative of the gauche configurations, g+ (+67°) and g− (−67°), already present at 293 K (see above). During the phase transition, both distributions broadened to include previously unsampled syn conformations. After about 30 ns, the maxima of the distribution attained constant values, e.g. 76.5° and 106.5°, with slightly different populations (see Fig. S12, ESI†).
As shown in the same figure, the gauche peak positions of τ2–τ7 angles were essentially unaltered with respect to those observed at 293 K (±67°). The distributions for the τ2–τ7 angles showed the even/odd τ pattern already recognized at lower temperature, albeit with a relative increase of gauche populations. Such an increase was quite significant, as testified by the anti/gauche ratio for the torsions τ3, τ5, and τ7, which were 1.4, 1.1, and 0.7, respectively. The same analysis performed on chain conformations at 293 K was extended here, in order to identify the most frequently occurring patterns. Table S4 (ESI†) reports the most representative conformations (with populations above 1%). Compared to 293 K, the contributions of individual conformations were more evenly distributed. The all-trans conformation, aaaaaa, previously accounting for the largest fraction (56%) of cases, turned out to be less representative (about 11%) than conformations with a terminal gauche defect, namely aaaaag, agagag, and aaagag. These conformations accounted for more than 40% of cases, overall. The remaining conformations featured at least one pattern of the form aga, whereas gg and ggg defects were only occasionally present.
Increasing the temperature to 385 K provided the smectic phase reported in Fig. 6C. This temperature is close to that experimentally observed for the phase transition to the smectic A phase,43,44 and the lowest temperature at which this transition was observed within 100 ns.
The inspection of density and order parameter profiles (see Fig. 7) suggests a process made of three consecutive stages. The former two stages closely resembled those at 380 K, namely a short supercell relaxation (1 ns), followed by the development of a partially disordered structure. The third stage started after about 27 ns, when the density (1030 g L−1) and the order parameter (close to 0.8) changed suddenly. The transition, triggered by a significant increase in side-chain conformational disorder, extended from a group of molecules to the whole structure. Notably, while the density quickly settled at a constant value (991.1(7) g L−1, see Table 2), the box lengths a and b were still evolving after 100 ns (see Fig. S13, ESI†). The P2(t) profile did not attain an asymptotic value over the simulation time span, thus suggesting the presence of orientational disorder in the newly formed structure.
This conclusion is consistent with the evolution of the core tilt angle reported in Fig. 9A. The development of the mesophase obtained at 380 K is clearly visible up to 27 ns, when the transition to the new phase settled in. Molecular orientations were, on average, distributed in two groups centered at about ±10° with respect to the phase director. Such an alignment is visible in the structure reported in Fig. 6C. At about 36 ns, a major change in core tilt orientations occurred, from positive to negative values, which appeared to be an irreversible process. The factors triggering such a process are not known at present and will be the subject of further investigations.
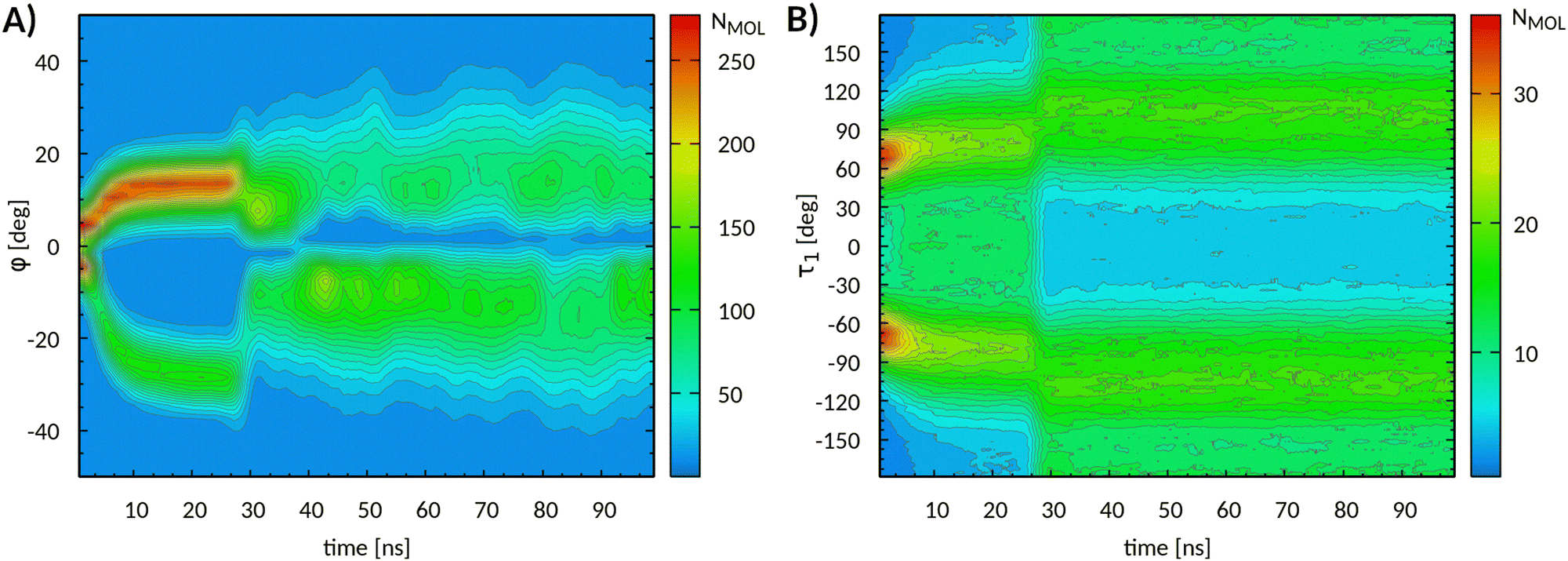 | ||
| Fig. 9 (A) Evolution of core tilt angle φ and (B) of torsional angle τ1 obtained from MD simulations at 385 K. The absolute molecular populations are also reported. For better clarity, the data have been window-averaged over time intervals of 3 ns. | ||
Concurrently with the aforementioned molecular rearrangements, the development of the new phase was characterized by conformational changes mostly related to τ1 torsions, as highlighted by Fig. 9B. The most notable change was represented by the increase of anti conformations at the expenses of syn ones, starting at 27 ns. Meanwhile, the maxima of the distribution gradually shifted at ± 110°. The distributions of the remaining angles (see Fig. S14, ESI†) were essentially unaltered, except for a slight increase in gauche populations along with the temperature increase. Also in this case, as shown by the conformational analysis, reported in Table S5 (ESI†), at least two gauche conformations were more representative than the all-trans one. This result is consistent with recent reports on similar compounds.47,48
The thermal behavior so far reported bears strong analogies to the “side-chain melting” also observed in other small molecule semiconductors,92 as well as π-conjugated polymers93 and oligomers.94 As mentioned above, the presence of the aromatic core in BTBT derivatives increases the conformational freedom of n-alkyl side chains, compared to that corresponding to n-alkanes.
A variety of molecular diffusion phenomena took place in the alkyl molten regions after the phase transition. As shown in the supplementary movie (see Movie S1, ESI†), where the molecules have been colored according to their orientation (red for φ < 0°, blue for φ > 0°), these events included the transfer between the adjacent layer and/or the orientation reversal with respect to the layer plane. The transfer between adjacent layers was detected for 23 molecules. Among them, 11 completely reversed their orientation (e.g. |φ| > 90°). Others (11), conversely, having reversed their orientation, returned to their starting layer after a brief excursion into the molten region. This behavior was, in fact, experimentally observed in polymorphic transitions of asymmetrical BTBT derivatives.20,49,95,96
All of the above mentioned phenomena did not disrupt the layered structure. We may thus classify the structure obtained at this temperature (Fig. 6C) as a smectic phase. Experimentally, the transition from the monoclinic to a smectic A phase is observed at about 382.5 K.43,44 The phase we have obtained at 385 K is more disordered than a smectic A phase, due to molecular diffusion. We note, nonetheless, that this phase has not yet been precisely detailed on the molecular scale, and according to the above mentioned reports, diffusion phenomena are not uncommon in BTBT derivatives.
Among the different factors responsible for our results, we believe that the delicate balance between inter- and intra-molecular forces underlying the process had a major role. An accurate description of such forces is known to be a real challenge for classical force fields, as demonstrated in a number of works.28,30 To further support this consideration, we add that, so far, our attempts at modeling this phase transition using other parameterizations, such as GAFF/AMBER,97 did not succeeded.
The difficulties of such simulations are further exacerbated by the complex and sluggish dynamic behavior of this smectic mesophase. During several heating/cooling runs performed on C8-BTBT-C8 with hot-stage polarizing microscopy several patterns typical of smectic systems appeared (see Fig. 10 and Fig. S15–S17 (ESI†) for selected snapshots of structural transformations of C8-BTBT-C8 samples). Even while keeping the temperature constant the analyzed samples remained dynamic for several minutes while reducing their potential energy by coarsening/ripening, a symptom of slow kinetics during these structural changes.
 | ||
| Fig. 10 (A)–(I) Focal conic domains in rapid motion/rearrangement after the crystal to mesophase transformation happened at 383 K (heating rate +1 K min−1, images taken with Linkam THMS-600 heating stage and metallographic microscope Olympus BX51). Images are 360 μm along the horizontal axis. The dark regions in images from D to I correspond to regions of isotropic melt as viewed under a crossed polarizer/analyzer. | ||
To complement the previous results and check for the presence of additional phases, not detected by NPT simulations, we performed a simulation gradually cooling the system from 385 to 293 K. A linear temperature gradient was applied, over a time span of 100 ns. During the simulation a partial reordering of the molecular layer was observed, as testified by a modest increase in the order parameter (not shown). The side chain conformational disorder did not change appreciably, and no other mesophases were formed, according to our previous results. No sign of highly ordered domains resembling the crystal phase was detected upon cooling. This result was expected, considering the slow kinetics of crystallization (see Fig. 10), far beyond the time scale accessible with molecular dynamics.
Further NPT simulations were performed at higher temperatures up to 460 K. The results obtained at 390 and 400 K turned out qualitatively similar to those at 385 K, in terms of mechanism and final structures. Also in this case, three consecutive stages were required to describe the overall process. As expected, increasing the temperature shortened the duration of the former two stages compared to 385 K. As shown in Fig. S18 (ESI†), at 400 K the starting structure turned smectic after about 7 ns. Again, the molecular orientations were dynamically swapping between two main groups. In analogy with the previous case, the increase of side chain conformational disorder during the transition was mostly related to the τ1 torsional (see Fig. S19, ESI†), with the remaining distributions less affected by the temperature increase. In fact, the angles τ2–τ7 explore most of their conformational space at ambient temperature, whereas τ1 only a small fraction, due to the steric contacts between the first methylene group and BTBT core hydrogens. Increasing the temperature allows this angle to gain full access to its conformational space. Finally, molecular diffusion phenomena at 390 and 400 K were closely similar to those observed at 385 K, albeit more pronounced.
The phase transition to the isotropic melt was detected starting at 460 K. As shown in Fig. 7A, after an initial drop, the density followed by a more gradual decrease, while approaching the value of 920.6(4) g L−1 (see Table 2). According to the P2(t) profile (see Fig. 7B), the orientational order was completely lost after about 30 ns, as testified by its average value, 0.004(31) (see Table 2). The visual inspection of the MD trajectories confirmed the development of an increasingly disordered structure from the layered one, finally leading to the isotropic molten phase reported in Fig. 6D.
Also in this case, we performed an additional simulation by gradually cooling the molten phase from 460 to 385 K over 100 ns. A significant increase in the order parameter, from 0.1 to about 0.7, was detected, corresponding to the development of large smectic domains within the isotropic phase. A preliminary analysis of these domains revealed significant analogies with the mesophase observed at 385 K (see Fig. S20, ESI†). No other phase transitions were detected within this temperature range, in line with the results reported above.
Conclusions
In this work, we have systematically investigated the dynamics and the thermal behavior of C8-BTBT-C8 at ambient and high temperatures. Our choice of its monoclinic polymorph as a reference structure was mainly dictated by two reasons: the availability of a fully solved XRD structure, and the fact this same structure is similar to that developed in thin film forming processes of technological relevance.33,42The MD simulations performed at ambient temperature revealed quite a significant freedom in side chain conformational dynamics, when compared to n-alkanes. This outcome is due to the presence of BTBT cores, whose steric hindrance limits the potential energy increase associated with the accessed conformations. Among them, gauche conformations appear to play a leading role as structural defects. This observation is in line with recent experimental findings on asymmetric BTBT derivatives.47–49 Furthermore, our results suggest that a similar behavior may also be extended to species isomorphic with C8-BTBT-C8, namely Cn-BTBT-Cn, with n = 7, 10, 12.34,39,98 Besides, our simulations revealed other interesting structural features, such as the even/odd alternating pattern in the side chain torsion distributions, which do not have a clear explanation at present, and may be the subject of future studies.
Increasing the temperature, two phase transitions were observed. The first one, occurring at 380 K, resulted in a stacking faulted structure with molecular layers tilted differently with respect to the c* axis. Currently, no experimental evidence supports the existence of this structure for BTBT derivatives. Nonetheless, its appearance is not unlikely, taking into account its modest energy cost and the fact stacking faults are not uncommon in organic semiconductors.91 Simulations on larger crystal assemblies will eventually help assess whether this phase is an artifact associated with the box size.
According to our simulations, the second phase transition observed at 385 K was triggered by side chain melting. The in-depth analysis of the resulting phase revealed a number of interesting structural features, such as the diffusion of molecules across neighbor molecular layers, sometimes accompanied by their complete reversal. An entirely similar phenomenon, leading to a change in molecular packing features has been experimentally observed for Ph-BTBT-C10 by different groups.49,95 Interestingly, in our previous work on Ph-BTBT-C10,30 we were not able to detect diffusion processes, plausibly due to a much slower kinetics compared to C8-BTBT-C8.
The phase we have detected at 385 K can be classified as smectic, yet not as the smectic A phase, owing to the presence of diffusing molecules and the distribution of core tilt orientations. These features also characterize the mesophases at 390 and 400 K, and reliably all the mesophases up to the isotropic melting point (460 K). This suggests that the phase at 385 K might already be a partially molten phase. The existence of this phase is difficult to prove experimentally, also due to the limited structural information that can be extracted from XRD experiments at this temperature.99 Nonetheless, we note that the coexistence of difference phases is not unlikely, as testified by the hot stage experiments (see Fig. 10 and Fig. S15–S17, ESI†).
The force field used for the simulations was chosen in the need to conduct a parallel study on silica surfaces.60 According to our analysis, the description of side chain dynamics should be improved to better describe the crystal structure. Meanwhile, we note that attempts at modeling this particular phase transition using other force fields were, so far, unsuccessful. Based on our previous study,30 this suggests that large differences likely exist among the members of the BTBT family in terms of force field requirements and sensitivity.
Data availability
All the open access software used in this work has been explicitly cited in the main manuscript. All main simulation input files used in the present work have been included as ESI.† Some minor codes used for structure pre-processing and editing, developed by our group, have not been included in the present work. The data supporting this article have been included in the manuscript, and the remaining as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the CINECA award under the ISCRA initiative (project SOX2BT-2, no. HP10CXOGKQ) for the availability of high-performance computing resources and support.References
- P. Hu, X. He and H. Jiang, InfoMat, 2021, 3, 613–630 CrossRef CAS.
- Y. Diao, B. C. K. Tee, G. Giri, J. Xu, D. H. Kim, H. A. Becerril, R. M. Stoltenberg, T. H. Lee, G. Xue, S. C. B. Mannsfeld and Z. Bao, Nat. Mater., 2013, 12, 665–671 CrossRef CAS PubMed.
- Y. Yuan, G. Giri, A. L. Ayzner, A. P. Zoombelt, S. C. B. Mannsfeld, J. Chen, D. Nordlund, M. F. Toney, J. Huang and Z. Bao, Nat. Commun., 2014, 5, 1–9 Search PubMed.
- C. Wang, H. Dong, L. Jiang and W. Hu, Chem. Soc. Rev., 2018, 47, 422–500 RSC.
- G. Schweicher, V. Lemaur, C. Niebel, C. Ruzié, Y. Diao, O. Goto, W. Y. Lee, Y. Kim, J. B. Arlin, J. Karpinska, A. R. Kennedy, S. R. Parkin, Y. Olivier, S. C. B. Mannsfeld, J. Cornil, Y. H. Geerts and Z. Bao, Adv. Mater., 2015, 27, 3066–3072 CrossRef CAS PubMed.
- H. Minemawari, T. Yamada, H. Matsui, J. Tsutsumi, S. Haas, R. Chiba, R. Kumai and T. Hasegawa, Nature, 2011, 475, 364–367 CrossRef CAS PubMed.
- S. Kwon, J. J. Kim, G. Kim, K. Yu, Y.-R. R. Jo, B.-J. J. Kim, J. J. Kim, H. Kang, B. Park and K. Lee, Adv. Mater., 2015, 27, 6870–6877 CrossRef CAS PubMed.
- R. Akai, K. Oka, S. Dekura, K. Yoshimi, H. Mori, R. Nishikubo, A. Saeki and N. Tohnai, J. Phys. Chem. Lett., 2023, 14, 3461–3467 CrossRef CAS PubMed.
- C. H. Lee, T. Schiros, E. J. G. Santos, B. Kim, K. G. Yager, S. J. Kang, S. Lee, J. Yu, K. Watanabe, T. Taniguchi, J. Hone, E. Kaxiras, C. Nuckolls and P. Kim, Adv. Mater., 2014, 26, 2812–2817 CrossRef CAS PubMed.
- Y. Diao, L. Shaw, Z. Bao and S. C. B. Mannsfeld, Energy Environ. Sci., 2014, 7, 2145–2159 RSC.
- A. O. F. Jones, Y. H. Geerts, J. Karpinska, A. R. Kennedy, R. Resel, C. Röthel, C. Ruzié, O. Werzer and M. Sferrazza, ACS Appl. Mater. Interfaces, 2015, 7, 1868–1873 CrossRef CAS PubMed.
- R. G. D. Valle, E. Venuti, A. Brillante and A. Girlando, ChemPhysChem, 2009, 10, 1783–1788 CrossRef PubMed.
- M. Yoneya, M. Kawasaki and M. Ando, J. Phys. Chem. C, 2012, 116, 791–795 CrossRef CAS.
- L. Viani, C. Risko, M. F. Toney, D. W. Breiby and J. L. Brédas, ACS Nano, 2014, 8, 690–700 CrossRef CAS PubMed.
- L. Lyu, D. Niu, H. Xie, Y. Zhao, N. Cao, H. Zhang, Y. Zhang, P. Liu and Y. Gao, Phys. Chem. Chem. Phys., 2017, 19, 1669–1676 RSC.
- H. Iino and J. I. Hanna, Adv. Mater., 2011, 23, 1748–1751 CrossRef CAS PubMed.
- H. Iino and J. I. Hanna, Polym. J., 2017, 49, 23–30 CrossRef CAS.
- W. Park, C. Yun, S. Yun, J. J. Lee, S. Bae, D. Ho, T. Earmme, C. Kim and S. Y. Seo, J. Ind. Eng. Chem., 2022, 114, 161–170 CrossRef CAS.
- S. Inoue, K. Nikaido, T. Higashino, S. Arai, M. Tanaka, R. Kumai, S. Tsuzuki, S. Horiuchi, H. Sugiyama, Y. Segawa, K. Takaba, S. Maki-Yonekura, K. Yonekura and T. Hasegawa, Chem. Mater., 2022, 34, 72–83 CrossRef CAS.
- S. Hofer, J. Unterkofler, M. Kaltenegger, G. Schweicher, C. Ruzié, A. Tamayo, T. Salzillo, M. Mas-Torrent, A. Sanzone, L. Beverina, Y. H. Geerts and R. Resel, Chem. Mater., 2021, 33, 1455–1461 CrossRef CAS PubMed.
- P. Ostwald and P. Pieranski, Smectic and Columnar Liquid Crystals: Concepts and Physical Properties Illustrated by Experiments, CRC Press, Boca Raton, US, 2005 Search PubMed.
- P. Ostwald and P. Pieranski, Nematic and Cholesteric Liquid Crystals: Concepts and Physical Properties Illustrated by Experiments, CRC Press, Boca Raton, US, 2005 Search PubMed.
- C. Zannoni, Liquid Crystals and their Computer Simulations, Cambridge University Press, Cambridge, UK, 2022 Search PubMed.
- V. Bhat, C. P. Callaway and C. Risko, Chem. Rev., 2023, 123, 7498–7547 CrossRef CAS PubMed.
- W. Wang, C. Yang, H. Fan, J. Zhang and X. Wang, Appl. Surf. Sci., 2022, 579, 152203 CrossRef CAS.
- G. Han, X. Shen and Y. Yi, Adv. Mater. Interfaces, 2015, 2, 1–8 Search PubMed.
- M. Ando, T. B. Kehoe, M. Yoneya, H. Ishii, M. Kawasaki, C. M. Duffy, T. Minakata, R. T. Phillips and H. Sirringhaus, Adv. Mater., 2015, 27, 122–129 CrossRef CAS.
- M. Yoneya, H. Minemawari, T. Yamada and T. Hasegawa, J. Phys. Chem. C, 2017, 121, 8796–8803 CrossRef CAS.
- M. Casalegno, M. Moret, R. Resel and G. Raos, Cryst. Growth Des., 2016, 16, 412–422 CrossRef CAS PubMed.
- A. Baggioli, M. Casalegno, G. Raos, L. Muccioli, S. Orlandi and C. Zannoni, Chem. Mater., 2019, 31, 7092–7103 CrossRef CAS.
- M. Asher, R. Jouclas, M. Bardini, Y. Diskin-Posner, N. Kahn, R. Korobko, A. R. Kennedy, L. Silva De Moraes, G. Schweicher, J. Liu, D. Beljonne, Y. Geerts and O. Yaffe, ACS Mater. Au, 2022, 2, 699–708 CrossRef CAS PubMed.
- M. Casalegno, T. Nicolini, A. Famulari, G. Raos, R. Po and S. V. Meille, Phys. Chem. Chem. Phys., 2018, 20, 28984–28989 RSC.
- H. Ebata, T. Izawa, E. Miyazaki, K. Takimiya, M. Ikeda, H. Kuwabara and T. Yui, J. Am. Chem. Soc., 2007, 129, 15732–15733 CrossRef CAS PubMed.
- T. Izawa, E. Miyazaki and K. Takimiya, Adv. Mater., 2008, 20, 3388–3392 CrossRef CAS.
- P. Xie, T. Liu, J. Sun and J. Yang, Adv. Funct. Mater., 2022, 32, 1–27 Search PubMed.
- D. He, Y. Zhang, Q. Wu, R. Xu, H. Nan, J. Liu, J. Yao, Z. Wang, S. Yuan, Y. Li, Y. Shi, J. Wang, Z. Ni, L. He, F. Miao, F. Song, H. Xu, K. Watanabe, T. Taniguchi, J. Bin Xu and X. Wang, Nat. Commun., 2014, 5, 1–7 Search PubMed.
- Z. Chai, S. A. Abbasi and A. A. Busnaina, ACS Appl. Mater. Interfaces, 2018, 10, 18123–18130 CrossRef CAS PubMed.
- Y. Huang, J. Sun, J. Zhang, S. Wang, H. Huang, J. Zhang, D. Yan, Y. Gao and J. Yang, Org. Electron., 2016, 36, 73–81 CrossRef CAS.
- T. Izawa, E. Miyazaki and K. Takimiya, Adv. Mater., 2008, 20, 3388–3392 CrossRef CAS.
- T. Matsuzaki, Y. Shibata, R. Takeda, T. Ishinabe and H. Fujikake, Jpn. J. Appl. Phys., 2017, 56, 11601 CrossRef.
- R. Janneck, N. Pilet, S. P. Bommanaboyena, B. Watts, P. Heremans, J. Genoe and C. Rolin, Adv. Mater., 2017, 29, 1703864 CrossRef PubMed.
- T. Hawly, M. Johnson, A. Späth, H. Nickles Jäkel, M. Wu, E. Spiecker, B. Watts, A. Nefedov and R. H. Fink, ACS Appl. Mater. Interfaces, 2022, 14, 16830–16838 CrossRef CAS PubMed.
- M. Dohr, H. M. A. Ehmann, A. O. F. Jones, I. Salzmann, Q. Shen, C. Teichert, C. Ruzié, G. Schweicher, Y. H. Geerts, R. Resel, M. Sferrazza and O. Werzer, Soft Matter, 2017, 13, 2322–2329 RSC.
- M. Dohr, O. Werzer, Q. Shen, I. Salzmann, C. Teichert, C. Ruzié, G. Schweicher, Y. H. Geerts, M. Sferrazza and R. Resel, ChemPhysChem, 2013, 14, 2554–2559 CrossRef CAS PubMed.
- A. M. Moh, K. Sasaki, T. Shinagawa, S. Watase and M. Izaki, Jpn. J. Appl. Phys., 2018, 57, 4–8 CrossRef.
- C. Grigoriadis, C. Niebel, C. Ruzié, Y. H. Geerts and G. Floudas, J. Phys. Chem. B, 2014, 118, 1443–1451 CrossRef CAS PubMed.
- N. Shioya, M. Yoshida, M. Fujii, T. Shimoaka, R. Miura, S. Maruyama and T. Hasegawa, J. Phys. Chem. Lett., 2022, 13, 11918–11924 CrossRef CAS PubMed.
- S. Arai, S. Inoue, M. Tanaka, S. Tsuzuki, R. Kondo, R. Kumai and T. Hasegawa, Phys. Rev. Mater., 2023, 7, 25602 CrossRef CAS.
- T. Oka, N. Shioya, T. Shimoaka and T. Hasegawa, J. Phys. Chem. C, 2023, 127, 7560–7564 CrossRef CAS.
- D. van der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. C. Berendsen, J. Comput. Chem., 2005, 26, 1701–1718 CrossRef CAS PubMed.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- Y. Huang, J. Sun, J. Zhang, S. Wang, H. Huang, J. Zhang, D. Yan, Y. Gao and J. Yang, Org. Electron., 2016, 36, 73–81 CrossRef CAS.
- R. B. Best, X. Zhu, J. Shim, P. E. M. Lopes, J. Mittal, M. Feig and A. D. MacKerell, J. Chem. Theory Comput., 2012, 8, 3257–3273 CrossRef CAS PubMed.
- A. D. Mackerell, M. Feig and C. L. Brooks, J. Comput. Chem., 2004, 25, 1400–1415 CrossRef CAS PubMed.
- A. D. MacKerell, D. Bashford, M. Bellott, R. L. Dunbrack, J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, W. E. Reiher, B. Roux, M. Schlenkrich, J. C. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiórkiewicz-Kuczera, D. Yin and M. Karplus, J. Phys. Chem. B, 1998, 102, 3586–3616 CrossRef CAS PubMed.
- J. B. Klauda, R. M. Venable, J. A. Freites, J. W. O’Connor, D. J. Tobias, C. Mondragon-Ramirez, I. Vorobyov, A. D. MacKerell and R. W. Pastor, J. Phys. Chem. B, 2010, 114, 7830–7843 CrossRef CAS PubMed.
- K. Vanommeslaeghe, E. Hatcher, C. Acharya, S. Kundu, S. Zhong, J. Shim, E. Darian, O. Guvench, P. Lopes, I. Vorobyov and A. D. MacKerell, J. Comput. Chem., 2010, 31, 671–690 CrossRef CAS PubMed.
- K. Vanommeslaeghe and A. D. MacKerell, J. Chem. Inf. Model., 2012, 52, 3144–3154 CrossRef CAS PubMed.
- K. Vanommeslaeghe, E. P. Raman and A. D. MacKerell, J. Chem. Inf. Model., 2012, 52, 3155–3168 CrossRef CAS PubMed.
- S. Provenzano and M. Casalegno, Characterization of thermal behavior and surface properties of C8-BTBT-C8 by means of molecular dynamics simulations, Master Degree thesis, Politecnico di Milano, 2022.
- F. S. Emami, V. Puddu, R. J. Berry, V. Varshney, S. V. Patwardhan, C. C. Perry and H. Heinz, Chem. Mater., 2014, 26, 2647–2658 CrossRef CAS.
- CGENFF website. [Online]. Available: https://cgenff.umaryland.edu/ (accessed: 2023-05-15).
- C. I. Bayly, P. Cieplak, W. D. Cornell and P. A. Kollman, J. Phys. Chem., 1993, 97, 10269–10280 CrossRef CAS.
- F. Y. Dupradeau, A. Pigache, T. Zaffran, C. Savineau, R. Lelong, N. Grivel, D. Lelong, W. Rosanski and P. Cieplak, Phys. Chem. Chem. Phys., 2010, 12, 7821–7839 RSC.
- E. Vanquelef, S. Simon, G. Marquant, E. Garcia, G. Klimerak, J. C. Delepine, P. Cieplak and F. Y. Dupradeau, Nucleic Acids Res., 2011, 39, 511–517 CrossRef PubMed.
- P. Bjelkmar, E. Lindahl, P. Larsson, M. A. Cuendet and B. Hess, J. Chem. Theory Comput., 2010, 6, 459–466 CrossRef CAS PubMed.
- G. Bussi, D. Donadio and M. J. Parrinello, Chem. Phys., 2007, 126, 014101 Search PubMed.
- H. J. C. Berendsen, J. P. M. Postma, W. F. Van Gunsteren, A. Dinola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- M. Parrinello and A. Rahman, J. Appl. Phys., 1981, 52, 7182–7190 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- GROMACS website. [Online]. Available: https://www.GROMACS.org (accessed 2023-05-15).
- C. F. MacRae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, J. Appl. Crystallogr., 2020, 53, 226–235 CrossRef CAS PubMed.
- Biovia Discovery Studio 2021, Dassault Systemes: 1233 Lujiazui Ring Road, Pudong, 200120 Shanghai, China. https://www.3ds.com/products-services/biovia (accessed 2023-05-15).
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- Gnuplot release 5.2. Thomas Williams and Colin Kelley. Gnuplot 4.6: An Interactive Plotting Program. https://sourceforge.net/projects/gnuplot (accessed 2023-05-15).
- Mathematica, Version 13.3, Wolfram Research, Inc., Champaign, IL, 2023 Search PubMed.
- OriginPro, Version 2021, OriginLab Corporation, Northampton, MA, USA Search PubMed.
- G. Tiberio, L. Muccioli, R. Berardi and C. Zannoni, ChemPhysChem, 2009, 10, 125–136 CrossRef CAS PubMed.
- A. Baggioli, C. A. Cavallotti and A. Famulari, Phys. Chem. Chem. Phys., 2016, 18, 29616–29628 RSC.
- I. E. Mavrantza, D. Prentzas, V. G. Mavrantzas and C. Galiotis, J. Chem. Phys., 2001, 115, 3937–3950 CrossRef CAS.
- T. L. Phillips and S. Hanna, Polymer, 2005, 46, 11035–11050 CrossRef CAS.
- M. Maroncelli, H. L. Strauss and R. G. Snyder, J. Chem. Phys., 1985, 82, 2811–2824 CrossRef CAS.
- N. Wentzel and S. T. Milner, J. Chem. Phys., 2010, 132, 044901 CrossRef PubMed.
- J. B. Klauda, R. W. Pastor and B. R. Brooks, J. Phys. Chem. B, 2005, 109, 15684–15686 CrossRef CAS PubMed.
- A. Troisi and G. Orlandi, J. Phys. Chem. A, 2006, 110, 4065–4070 CrossRef CAS PubMed.
- T. Vehoff, Y. S. Chung, K. Johnston, A. Troisi, D. Y. Yoon and D. Andrienko, J. Phys. Chem. C, 2010, 114, 10592–10597 CrossRef CAS.
- M. Alkan and I. Yavuz, Phys. Chem. Chem. Phys., 2018, 20, 15970–15979 RSC.
- S. Illig, A. S. Eggeman, A. Troisi, L. Jiang, C. Warwick, M. Nikolka, G. Schweicher, S. G. Yeates, Y. Henri Geerts, J. E. Anthony and H. Sirringhaus, Nat. Commun., 2016, 7, 1–10 Search PubMed.
- H. Minemawari, M. Tanaka, S. Tsuzuki, S. Inoue, T. Yamada, R. Kumai, Y. Shimoi and T. Hasegawa, Chem. Mater., 2017, 29, 1245–1254 CrossRef CAS.
- R. Boistelle, Defect structures and growth mechanisms of long-chain alkanes, in Current Topics in Materials Sciences, ed. Kaldis, E., North Holland Publ. Co, Amsterdam, 1980, vol. 413 Search PubMed.
- M. Moret, M. Campione, S. Caprioli, L. Raimondo, A. Sassella, S. Tavazzi and D. Aquilano, J. Phys. Conf. Ser., 2007, 61, 831 CrossRef CAS.
- D. W. Davies, G. Graziano, C. Hwang, S. K. Park, W. Liu, D. Yuan, S. C. B. Mannsfeld, S. Y. G. Wang, Y. S. Chen, D. L. Gray, X. Zhu and Y. Diao, Cryst. Growth Des., 2023, 23, 719–728 CrossRef CAS.
- V. A. Pozdin, D. M. Smilgies, H. H. Fong, M. Sorensen and M. He, J. Mater. Chem. C, 2016, 4, 5255–5262 RSC.
- M. Casalegno, A. Famulari and S. V. Meille, Macromolecules, 2022, 55, 2398–2412 CrossRef CAS.
- S. Hofer, A. Hofer, J. Simbrunner, M. Ramsey, M. Sterrer, A. Sanzone, L. Beverina, Y. Geerts and R. Resel, J. Phys. Chem. C, 2021, 125, 28039–28047 CrossRef CAS PubMed.
- M. Yoneya, Jpn. J. Appl. Phys., 2020, 59, 90909 CrossRef CAS.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- P. Pandey, L. Fijahi, N. McIntosh, N. Turetta, M. Bardini, S. Giannini, C. Ruzié, G. Schweicher, D. Beljonne, J. Cornil, P. Samorì, M. Mas-Torrent, Y. H. Geerts, E. Modena and L. Maini, J. Mater. Chem. C, 2023, 11, 7345–7355 RSC.
- G. Schweicher, G. Liu, P. Fastré, R. Resel, M. Abbas, G. Wantz and Y. H. Geerts, Mater. Chem. Front., 2021, 5, 249–258 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary figures and tables (PDF); supplementary movie showing the evolution of core tilt angles at 385 K (AVI); input structures, force field parameters, topology, and example MD input file for C8-BTBT-C8 (in GROMACS format) (ZIP). See DOI: https://doi.org/10.1039/d4cp01884b |
| This journal is © the Owner Societies 2024 |