
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
trans-Stilbene aggregates and crystallites in polystyrene films: microscopy and spectroscopy studies
Ivan
Halimski
 a,
Renata
Karpicz
a,
Andrej
Dementjev
a,
Marija
Jankunec
b,
Jevgenij
Chmeliov
ac,
Mindaugas
Macernis
c,
Darius
Abramavicius
c and
Leonas
Valkunas
*a
a,
Renata
Karpicz
a,
Andrej
Dementjev
a,
Marija
Jankunec
b,
Jevgenij
Chmeliov
ac,
Mindaugas
Macernis
c,
Darius
Abramavicius
c and
Leonas
Valkunas
*a
aCenter for Physical Sciences and Technology, Sauletekio ave. 3, LT-10257, Vilnius, Lithuania. E-mail: leonas.valkunas@ftmc.lt
bInstitute of Biochemistry, Life Sciences Center, Vilnius University, Sauletekio ave. 7, LT-10257, Vilnius, Lithuania
cInstitute of Chemical Physics, Faculty of Physics, Vilnius University, Sauletekio ave. 3, LT-10257, Vilnius, Lithuania
First published on 19th August 2024
Abstract
Solid and liquid stilbene forms were characterized using a range of techniques, including atomic force microscopy, coherent anti-Stokes Raman scattering microspectroscopy and optical spectroscopy. The obtained experimental results were analyzed by means of quantum chemical calculations and using a non-negative matrix factorization algorithm. It was confirmed that pure cis-stilbene formed a homogeneous fluid film on a glass substrate, whereas pure trans-stilbene formed crystals. Mixtures of trans-stilbene and polystyrene were shown to form stable solid films, which were non-homogeneous on the microscopic scale: stilbene molecules self-organized into microcrystals, which floated on the surface of polystyrene glass.
Introduction
Various photoinduced intra- and intermolecular processes in organic compounds can be employed to design diverse components of highly sensitive instruments. Nowadays, purely organic compounds are utilized for detection in a broad spectral range from IR rays1 to X-rays.2 One of the most fundamental light-induced reactions that plays an important role in photochemistry and different biological processes is photoisomerization. Stilbene molecules are classic examples demonstrating such type of behaviour.3 It is well known that under UV irradiation, trans–cis isomerization of stilbene can occur in liquid solutions.3–6 However, in solid glasses, such conversion is impeded due to steric constraints.7–9 Such functionality of stilbene has been utilized for practical purposes of sensing. For instance, polycrystals obtained via hot pressing10 of stilbene grains and single crystals grown via either melt Bridgman–Stockbarger11 or solution-based12–14 technique have been considered as candidates for ionizing radiation registration. Stilbene monocrystals are also well known for their UV-detection ability.15–17trans-Stilbene (Tstilbene; the molecular structure is shown in Fig. 1c) crystallizes in the monoclinic centrosymmetric point group 2/m. Its structure was first determined in 1937 by Robertson and Woodward.18 Later refinements by Finder et al.,19 Bernstein20 and Hoekstra et al.21 resulted in a good understanding of Tstilbene's properties in monocrystals. Mixtures of stilbene and polystyrene (PS) form technologically stable films, which can be utilized for specific applications such as sensing and screening; however, they show some specific properties that are not well understood. Stilbene-related spectroscopic data and variability of excitation relaxation rates in these films demonstrate unusual temperature dependence8 while the corresponding fluorescence spectral shifts are highly dependent on the concentration of stilbene.9
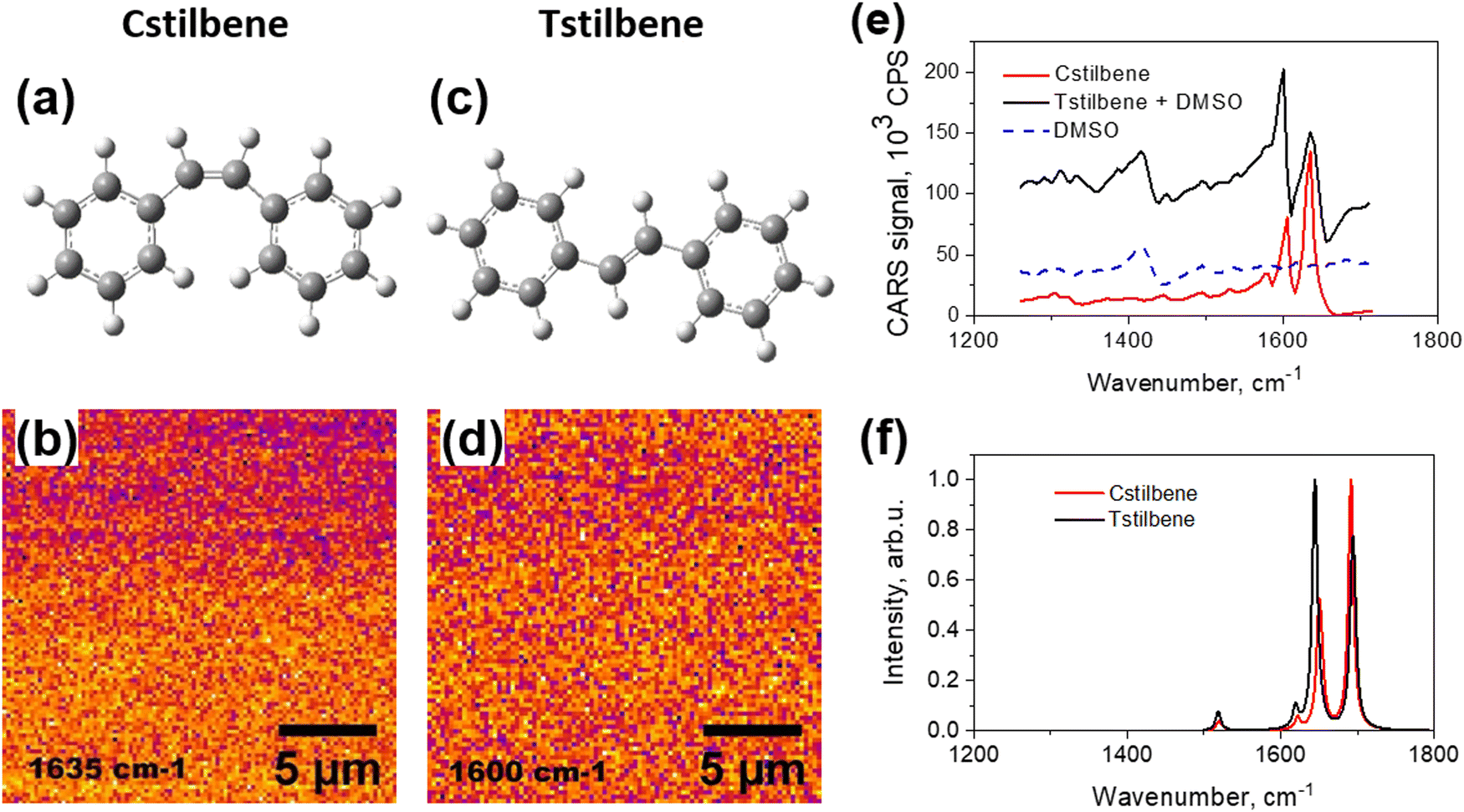 | ||
| Fig. 1 Chemical structures of Cstilbene (a) and Tstilbene (c). CARS images, captured at a resonance frequency of 1635 cm−1 for pure Cstilbene, (b) and diluted solution of Tstilbene in DMSO (d) with a resonance frequency of 1600 cm−1. Measured CARS spectra of pure Cstilbene and mono-molecular Tstilbene (e). CARS spectrum of DMSO is shown for reference. Calculated Raman spectra of Cstilbene and Tstilbene molecules (f). | ||
Increasing stilbene concentration in the PS mixture leads to clustering of stilbene molecules.9 This severely affects both the absorption and emission properties of the materials. Although clustering of stilbene was proposed in our previous publication,9 the precise 3D morphology of the films at high concentrations of stilbene molecules was not identified. Fundamentally, the self-assembly of stilbene into microcrystals results from the interplay of stilbene–stilbene intermolecular interactions and stilbene–PS intermolecular interactions. Atomic force microscopy (AFM) is one of the highest resolution approaches to directly study the surface morphology of samples, and thus, it can be efficiently used to detect the formation of molecular clusters on the surfaces of films. However, the inner molecular structure (either inside the clusters or in the bulk of films) can be better resolved by applying different approaches. For instance, second- or third-harmonic generation optical microscopy provides a possibility to improve both the spectral and spatial resolutions in 3D. Meanwhile, coherent anti-Stokes Raman scattering (CARS) microscopy utilizes third-order nonlinear spectroscopy effects and allows the detection of distinct vibrational spectra,22,23 so they can be utilized for molecular recognition in optically transparent regions, which is not possible in high-harmonic-generation microscopy. Compared to regular bulk spectroscopic methods, optical imaging provides the possibility to reveal structural heterogeneity on the length scale of the optical resolution. Thus, it allows distinguishing different molecules with high fidelity and sensitivity.
In this paper, we demonstrate how Tstilbene microcrystals are self-organized on polystyrene films. Previous studies using transmission electron microscopy (TEM) demonstrated the presence of small aggregates or even crystals, but it was not possible to determine the position of the crystals with respect to the PS matrix.9 We utilized CARS and AFM techniques to characterize the morphology of the stilbene clusters self-assembled in PS films of variable thickness using various stilbene concentrations. The results were supported by optical spectroscopy approaches as well as by quantum chemical calculations. To demonstrate the reliability of the obtained results, appropriate examination of cis-stilbene's (Cstilbene; chemical structure is shown in Fig. 1a) properties was also carried out. We show that when increasing the Tstilbene concentration during the sample preparation, the samples exhibited a smooth transition from mono-molecular properties to those of monocrystals. In the latter case, Tstilbene clusters were observed floating on the surface of the PS films. The sample morphology could therefore be easily controlled with high precision.
Experimental
Sample preparation
trans- and cis-stilbene, polystyrene, and chloroform were purchased from Sigma-Aldrich, USA. Thin films of Tstilbene (≥96% C14H12; powder, CAS: 103-30-0) in PS (neat powder; analytical standard, for GPC, 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) matrices with different mass ratios were used for steady-state spectroscopy, and AFM and CARS microscopy analyses. The studied films were prepared in the following way: PS powder was dissolved in chloroform (≥99.8%) with 30 mg ml−1 concentration. It dissolved immediately, and the stock solution was further diluted with chloroform to obtain solutions at varying concentrations of 15, 10, 5, and 3 mg ml−1. After that the Tstilbene was dissolved in the PS solution (with different Tstilbene mass concentrations with respect to PS of 0.5%, 10%, 20%, 60%, and 80%) and then thin films were prepared on quartz substrates (15 mm × 24 mm), pre-cleaned with chloroform, via a spin-coating technique with the Spin-Coater KW-4A (Chemat Technology, USA) device (70 rpm for the first 3 s and then 1200 rpm for 30 s). Since chloroform evaporated during the spin-coating procedure, solid glass-like thin films of Tstilbene in the PS matrix were obtained.
000) matrices with different mass ratios were used for steady-state spectroscopy, and AFM and CARS microscopy analyses. The studied films were prepared in the following way: PS powder was dissolved in chloroform (≥99.8%) with 30 mg ml−1 concentration. It dissolved immediately, and the stock solution was further diluted with chloroform to obtain solutions at varying concentrations of 15, 10, 5, and 3 mg ml−1. After that the Tstilbene was dissolved in the PS solution (with different Tstilbene mass concentrations with respect to PS of 0.5%, 10%, 20%, 60%, and 80%) and then thin films were prepared on quartz substrates (15 mm × 24 mm), pre-cleaned with chloroform, via a spin-coating technique with the Spin-Coater KW-4A (Chemat Technology, USA) device (70 rpm for the first 3 s and then 1200 rpm for 30 s). Since chloroform evaporated during the spin-coating procedure, solid glass-like thin films of Tstilbene in the PS matrix were obtained.
For CARS microscopy, Tstilbene thin films (of the same thickness and Tstilbene concentrations) were prepared on a 0.17-mm-thick cover borosilicate glass. Drops of Tstilbene in PS and pure Cstilbene (≥96% C14H12; liquid, CAS: 645-49-8) were prepared on the same glass for comparison.
Atomic force microscopy (AFM)
The surface topography was characterized in the tapping mode by TESP probes (with a nominal spring constant of 42 N m−1, Bruker, USA) with the AFM system DimensionIcon (Bruker, USA). The film thickness was determined by visualization of the scratched area and post-image analysis (Nanoscope Analysis 1.9, Bruker, USA). The image resolution was kept at 128 pixels or higher.Coherent anti-Stokes Raman microspectroscopy (CARS)
Microscopic characterization of the stilbene samples was performed by exploiting a polarization-resolved CARS technique24 using a custom-built single-wavelength CARS microspectrometer as described elsewhere.22 The CARS signal in the spectral range of 780–840 nm was generated by mixing picosecond pulses of fixed (1064 nm) and tunable (900–935 nm) laser beams and focusing it on the sample. The power of incident radiation on the sample plane was about 1–5 μW. Operation of the microscope in a polarization-resolved mode was implemented by placing an achromatic half-wave-plate (HWP) (AHWP05M-980, Thorlabs, Sweden) in the optical path in front of the sample.23 The direction of the linear polarization of the incident laser beam was changed by rotating the HWP in the range from −180° to +180° in increments of 10°. For each HWP position, a full image of the object was acquired. The imaging was performed using the commercial Olympus IX71 microscope in combination with the piezo scanning system P-517.3CL (Physik Instrumente, Germany) running in the raster-scanning regime. The oil immersion objective (NA = 1.42, spatial resolution ≈ 0.5 μm) was utilized for imaging.Optical spectroscopy
Absorption spectra were measured with a V670 spectrometer (Jasco Corporation, Japan). The baseline, i.e., absorption of the pure PS matrix on the substrate, was removed from all the recorded spectra. Fluorescence excitation spectra were measured with an FS5 spectrofluorometer (Edinburgh Instruments, UK). The excitation source was a xenon lamp, and the bandwidths for emission and excitation were 0.15 nm and 2 nm, respectively.Steady-state and time-resolved fluorescence (fluorescence decay kinetics) were recorded on an F920 spectrophotometer (Edinburgh Instruments, UK). All the samples were excited at 300 nm with a picosecond pulsed diode PLED-300 (the beam diameter was about 0.4 mm, resulting in a beam area of 0.13 mm2). While measuring the samples containing Tstilbene crystallites, the illuminating beam position on the film was focused to cover only a few of them. The 2-nm spectral slit was chosen, while the time resolution was 200 ps, excitation pulse duration was 850 ps, excitation power was 0.08 mW, and pulse repetition rate was 5 MHz. All the fluorescence (FL) spectra were corrected for the instrument sensitivity. Since for the highest concentrations, the optical densities at the excitation wavelength were around 1.0, some reabsorption effects could take place. However, this would lead to only small changes in the spectral behavior.8 All the FL decay kinetics were fitted as a convolution of the instrument response function (IRF, see Fig. 5b) with mono- and double-exponential decay kinetics. IRF for the F920 device was measured the same way as the FL decay kinetics.
Computational details
Quantum chemical calculations of the molecular structure and properties of separate Tstilbene and Cstilbene molecules were done at the density functional theory (DFT) level using the B3LYP and CAM-B3LYP functionals in conjunction with the cc-pVDZ basis set, as implemented in the Gaussian 16 package.25 Such an approach has been shown to demonstrate good agreement with the experimental data.8,9,26,27 These computational approaches align with those utilized in our earlier investigations of stilbene and analogous molecules.8,9 The vibrational properties were computed at the optimized electronic ground state of the molecule, while structures with imaginary frequencies were excluded from the computational study.Two-component analysis of the fluorescence spectra and decay kinetics data were performed using a multiplicative update algorithm28 for the non-negative matrix factorization.29 In short, all the collected FL spectra were simultaneously fitted as a two-component superposition as follows:
| F(λ,n) = S1(λ)·C1(n) + S2(λ)·C2(n), | (1) |
Results
CARS and AFM microscopy studies
The CARS spectra of Cstilbene and Tstilbene molecules were studied first for comparison. Pure Cstilbene (Fig. 1a) in ambient conditions was in a liquid phase. Thus, Cstilbene formed a thin liquid droplet on the substrate and could be scanned in the CARS microscope, which detects CARS signal at a specific frequency and at some specific spot on the sample. The scanned CARS signal intensity from the Cstilbene sample is presented in Fig. 1b and showed a homogeneous background, implying an ideal homogeneous liquid, as expected. The CARS spectrum presented in Fig. 1e exhibited characteristic single-molecular features: two main peaks at 1604 and 1634 cm−1. The bands were sharp, with the linewidth full-width at half maximum (FWHM) equal to 14 cm−1. Since the Cstilbene does not associate with other molecules, its signal was used as a reference, while further study was related to Tstilbene.The CARS measurements of pure molecular Tstilbene (Fig. 1c) were more cumbersome as Tstilbene tends to form large clusters or aggregates. Dissolving Tstilbene in DMSO at small concentrations prevented the clustering, so the Tstilbene-in-DMSO droplet was studied as a sample containing molecular non-aggregated Tstilbene. The CARS microscopy image of the droplet also showed a homogeneous profile with no structural patterns (Fig. 1d), which confirmed the homogeneous distribution of Tstilbene in the droplet. We additionally recorded the CARS spectrum of pure DMSO, which is shown in Fig. 1e with the dashed blue line. Notice that in the 1500–1700 cm−1 spectral region DMSO showed no fine structure of the spectrum. Hence, the CARS spectrum of Tstilbene showed sharp peaks at slightly shifted positions compared to Cstilbene: 1599 and 1635 cm−1. Additionally, the intensities of the two peaks were reversed compared to Cstilbene. The FWHM of the Tstilbene 1635 cm−1 band was 28 cm−1.
This intensity-reversal property was confirmed by the single-molecular B3LYP/cc-pVDZ DFT calculations shown in Fig. 1f. Since here we aimed just to understand the major qualitative differences in the Raman spectra of the trans and cis configurations of stilbenes and the origin of each observed band, the calculation result is presented without any scaling factors (though a better quantitative correspondence between the measured and calculated spectra could be obtained by rescaling the wavenumbers of the latter by 0.9685).26 Note that changes in the splitting and intensities between the two main peaks for Cstilbene and Tstilbene were also confirmed. As follows from our calculations, Cstilbene exhibited a narrower energy gap between the two Raman bands compared to Tstilbene. Both Raman bands were predominantly associated with C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching and C–H bending modes: the higher frequency bands at 1696 cm−1 for Tstilbene and at 1691 cm−1 for Cstilbene were primarily attributed to CC stretching vibrations, corresponding to the double bond structure inherent in the stilbene molecule. Conversely, the active Raman bands at lower frequencies (1646 cm−1 for Tstilbene and 1650 and 1653 cm−1 for Cstilbene) represent symmetric and antisymmetric vibrations and were dominated by C–H bending modes emanating from the phenyl rings.
C stretching and C–H bending modes: the higher frequency bands at 1696 cm−1 for Tstilbene and at 1691 cm−1 for Cstilbene were primarily attributed to CC stretching vibrations, corresponding to the double bond structure inherent in the stilbene molecule. Conversely, the active Raman bands at lower frequencies (1646 cm−1 for Tstilbene and 1650 and 1653 cm−1 for Cstilbene) represent symmetric and antisymmetric vibrations and were dominated by C–H bending modes emanating from the phenyl rings.
Tstilbene in PS films formed clusters of various sizes depending on the concentration, as has been revealed by TEM previously.9 Similar but much higher resolution images were obtained by scanning CARS microscopy at 1600 cm−1 resonant frequency, as shown in Fig. 2. The clear formation of μm-sized clusters could be observed at 60% or higher concentrations. Such clusters were observed in all the samples, except at the small 0.5% Tstilbene concentration.
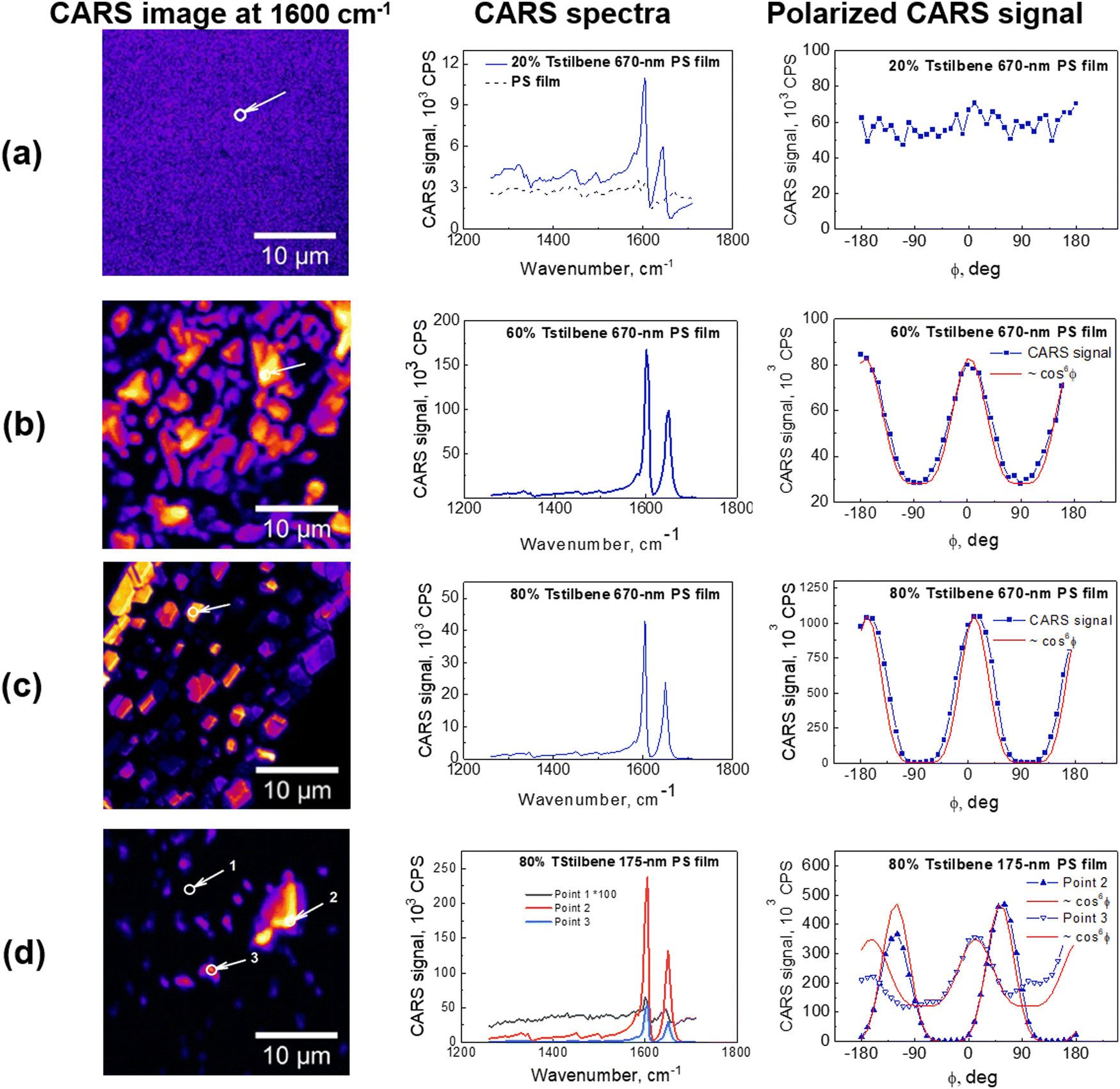 | ||
| Fig. 2 Left: CARS images of various films of Tstilbene and PS mixtures at 1600 cm−1. Middle: CARS spectra at the selected points indicated with arrows. Right: Polarized CARS signal measured at 1600 cm−1 at the same selected spatial point. Each data point is the result of averaging over an area of about 1 μm2 at the chosen position of the sample (a)–(d). The types of films are indicated in the middle panels. | ||
CARS spectra were recorded for all the samples at various spots of the obtained CARS images. Surprisingly, the characteristic Tstilbene peaks did not depend on the spot position. Hence, the CARS spectral shape was not affected by clusterization. Our previous calculations9 demonstrated that clusterization leads to a permanent twisting of the Tstilbene molecules. Apparently, this twisting does not affect the molecular vibrational characteristics. Also, similar spectra and model calculations were obtained in ref. 30 and 31 for similar stilbene-like dimers. While the dimers in those studies due to chemical constrains could not be exactly matched with our samples, similar ring twisting was observed as in our studies.
Next, we applied AFM to measure the thickness of the films and to survey the films’ surface landscape. The obtained AFM images, presented in Fig. 3a, demonstrated that the Tstilbene aggregates were rather bulky and were located on the surface of the PS. Essentially, the PS matrix acts as a glue, which keeps the relatively large Tstilbene aggregates attached, making the whole sample similar to crumbs scattered on a surface. Moreover, intermediate Tstilbene concentrations demonstrated the formation and growth of various sizes of clusters, which could be controlled by varying the thickness of the PS film and the concentration of Tstilbene in the initial solution.
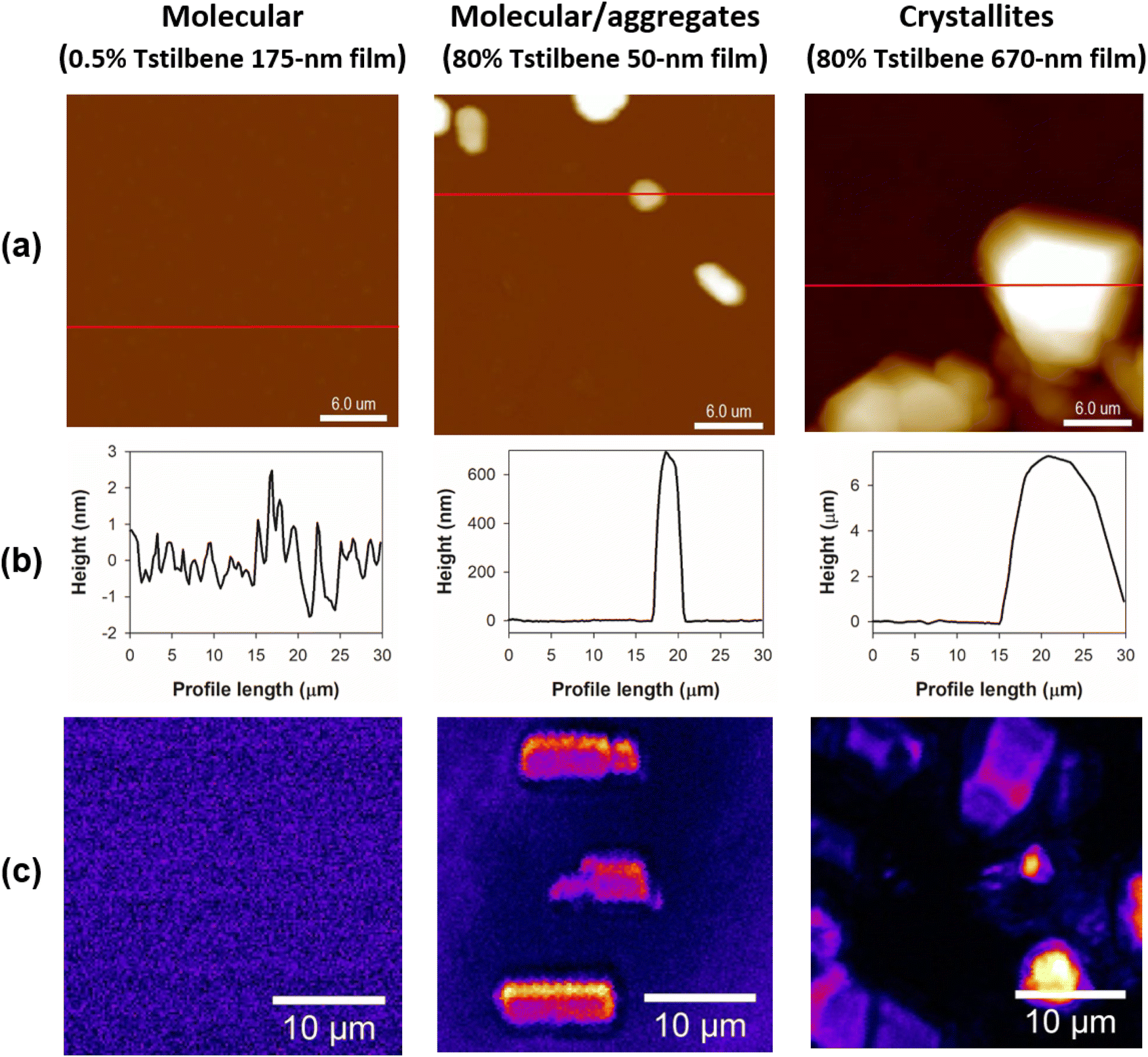 | ||
| Fig. 3 (a) AFM representative 30 μm × 30 μm topography images of Tstilbene-containing PS films. (b) Intensity profiles (the distance in the Z direction from the film surface) along the red lines indicated in (a). (c) CARS images of the same films at 1600 cm−1 in the similarly sized 30 μm × 30 μm range. | ||
CARS spectrometry has one additional advantage compared to AFM and TEM: the polarization of the excitation laser field can be employed. If the CARS signal intensity does not depend on the polarization, the sample is isotropic. However, that is not the case in the presence of molecular ordering. Polarization-resolved CARS images must be post processed in order to extract microscopic information about orientational ordering on the angular distribution of Raman scatterers inside the object.32 For this purpose, a region of approximately 1 μm2 was selected here in the image of the object, and the amplitude of the CARS signal was averaged over the area. This procedure was performed with images recorded for each direction of linear polarization of the incident laser beam. The obtained values were then plotted, revealing a dependence of the CARS response on the angle of the linear polarization direction of the incident beam.
CARS is essentially a third-order nonlinear optical response, where the material interacts with the excitation optical field three times, and the fourth interaction generates the output field. In this terminology, the CARS field amplitude, defined by E(CARS)δ = χ(CARS)αβγδEγEβEα, can be described by the following equation,
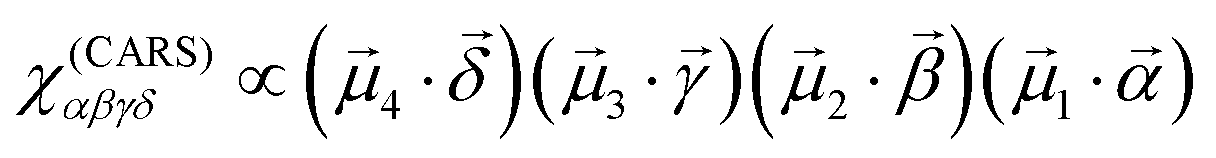 | (2) |
![[small mu, Greek, vector]](https://www.rsc.org/images/entities/i_char_e0e9.gif) i is the i-th interacting transition electric dipole moment, while
i is the i-th interacting transition electric dipole moment, while 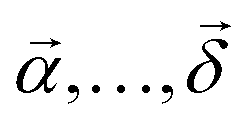 are unit vectors indicating optical field polarization directions. Assuming that polarization of the detection (last interaction) is fixed, the excitation field corresponds to three polarizations
are unit vectors indicating optical field polarization directions. Assuming that polarization of the detection (last interaction) is fixed, the excitation field corresponds to three polarizations 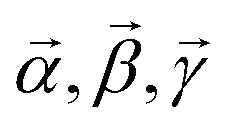 , and the dot product is written in the form (1·
, and the dot product is written in the form (1·![[small alpha, Greek, vector]](https://www.rsc.org/images/entities/i_char_e118.gif) ) = |1|cosθ, where θ is the angle between the excitation field and the transition dipole moment. Thus we immediately come to the result that the amplitude depends on the third power of a harmonic function:
) = |1|cosθ, where θ is the angle between the excitation field and the transition dipole moment. Thus we immediately come to the result that the amplitude depends on the third power of a harmonic function:| χ(CARS)αβγδ ∝ cos3θ | (3) |
The spectrometer essentially measures not the amplitude, but rather the intensity, which is given by the amplitude squared. The net CARS intensity as a function of excitation polarization direction thus should behave as
| I(CARS) ∝ cos6θ | (4) |
For a completely disordered stilbene, as expected, the modulation of the amplitude of the polarization curve disappears, as we observed in the films with a lower Tstilbene concentration (see Fig. 2a). On the other hand, the polarization-resolved measurements of the 80% Tstilbene 670-nm film revealed a strong dependence of the CARS signal on the polarization direction of the incident laser beam (see the right panel in Fig. 2c). The obtained dependence is typical of highly oriented molecular structures. The modulation of the CARS amplitude of the polarization curve had a contrast of about 1:250, and could be well approximated by a cos6θ function, in accordance with theory. Thus, it can be concluded that the specific stilbene μm-sized clusters were indeed Tstilbene monocrystals.
The 175-nm film with 80% Tstilbene concentration (see points 1 and 3 in Fig. 2d) did not contain properly shaped microcrystals, compared to the 670-nm film (note the non-vanishing constant background for the polarized CARS measurements, typical for the completely disordered molecular stilbene, as shown in Fig. 2a). This data indicated the extensive presence of mostly the glassy form of stilbene, with the separate molecules or their small aggregates exhibiting very weak ordering. On the other hand, polarization measurements at the point corresponding to the very strong CARS signal (point 2 in Fig. 2d) showed a completely modulated response and the curves were well approximated by cos6θ, suggesting also the presence of some micrometer-sized monocrystals of stilbene in the polymer film.
Finally, for the matrix of the 670-nm film of polystyrene and a 60% concentration of stilbene (see Fig. 2b), the polymer film was composed of unstructured stilbene unevenly distributed within the polymer. The polarization curves were also modulated but contained a steady component of about 1/3 of the full amplitude. As already mentioned, this steady component occurred due to the non-resonance contribution of the molecular orientationally disordered stilbene, polystyrene, or/and the glass substrate at the frequency of stilbene resonance. Moreover, the fitting result (over the alternating component) showed some deviations from the cos6θ function, indicating that the CARS response originated not from a single crystal but arose from the polycrystalline stilbene with a partially disordered structure.
Spectroscopy studies
Tstilbene molecules efficiently absorb light in the near-UV spectral region. The blue lines in Fig. 4 show the absorption spectra of various films recorded by transmission measurements. Notice that isomerization into Cstilbene was not possible due to the solid state of the film. The absorption spectrum contained a broad band at around 300 nm [the measured optical density (OD) of this band for various samples is presented in Table 1] and barely depended on the sample. This is surprising since the morphology of the samples varied significantly. It is well known that upon aggregation molecules often form new intense bands, usually denoted as J or H bands depending on their position with respect to the mono-molecular band. Here the sample with microcrystals had the same absorption spectrum as the one containing only molecular stilbene. One possibility for such a situation is that molecules in the crystal were resonantly non-interacting, thus intermolecular excitons were not formed and, therefore, the absorption spectrum was not affected by molecular crystallization. The other possibility is that this measurement does not collect signals from the crystals.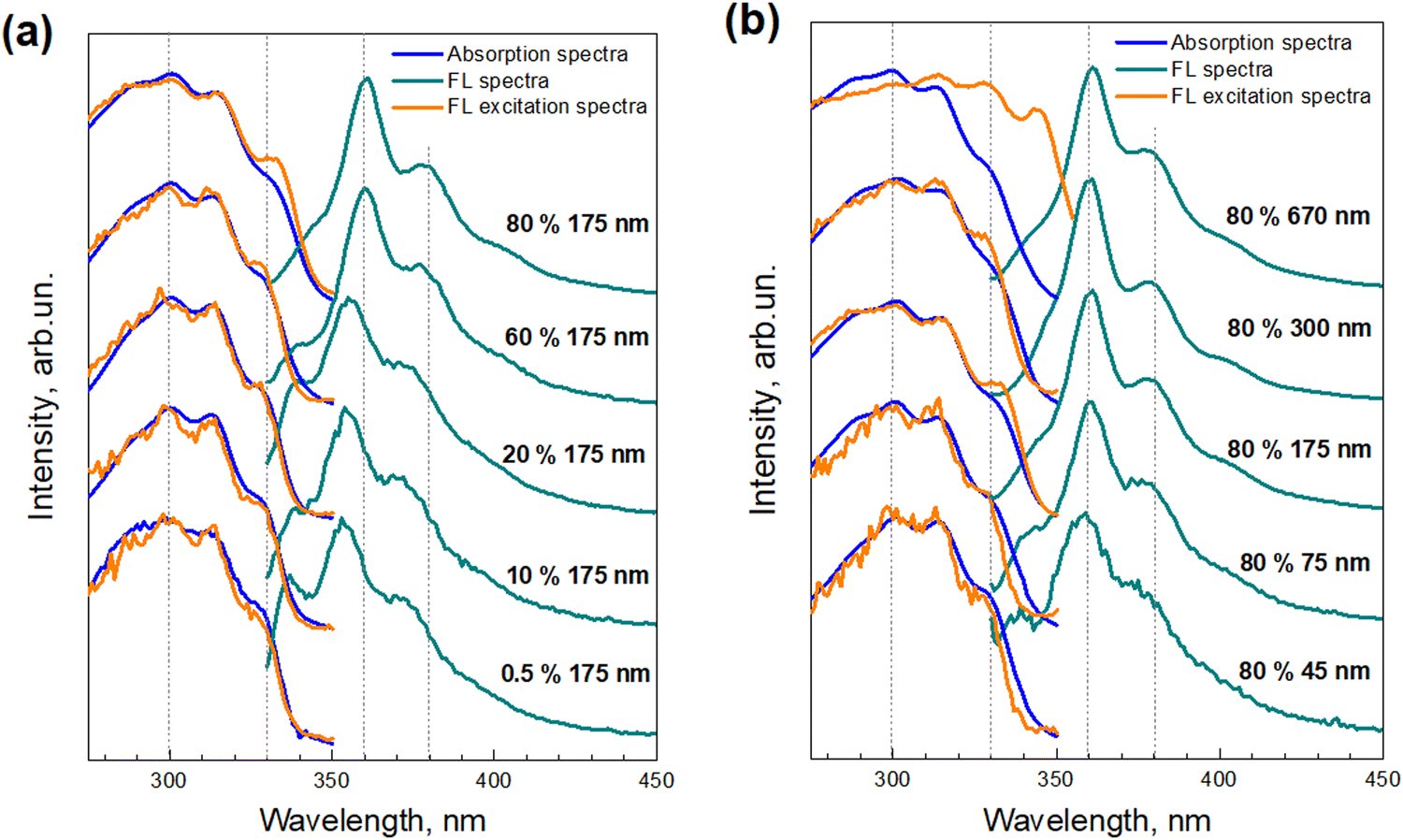 | ||
| Fig. 4 Steady-state absorption (blue lines), FL excitation (orange lines), and FL (dark cyan lines) spectra of Tstilbene films, dependent on the concentration of Tstilbene in the PS matrix of the 175-nm film thickness (a), and on the film thickness for 80% Tstilbene (b). All the spectra were normalized and shifted along the linear vertical axis for clarity. | ||
| Tstilbene concentration in PS, wt% | Film thickness, nm | OD at 300 nm | λ FL, nm | τ 1, ns (%) | τ 2, ns (%) | τ ave, ns |
|---|---|---|---|---|---|---|
| 0.5% | 175 | 0.02 | 353 | 0.71 (100%) | — | 0.71 |
| 10% | 175 | 0.17 | 355 | 0.26 (100%) | — | 0.26 |
| 20% | 175 | 0.30 | 357 | 0.28 (100%) | — | 0.28 |
| 60% | 175 | 0.38 | 361 | 0.19 (80%) | 1.65 (20%) | 0.48 |
| 80% | 45 | 0.11 | 360 | 0.30 (87%) | 1.68 (13%) | 0.48 |
| 80% | 75 | 0.29 | 361 | 0.38 (75%) | 1.62 (25%) | 0.69 |
| 80% | 175 | 0.68 | 361 | 0.51 (61%) | 1.50 (39%) | 0.90 |
| 80% | 300 | 0.76 | 361 | 0.51 (36%) | 1.48 (63%) | 1.12 |
| 80% | 670 | 1.53 | 361 | — | 1.60 (100%) | 1.60 |
Contrarily to the absorption spectra, the fluorescence spectra of the samples, also presented in Fig. 4, exhibited a strong dependence on the Tstilbene concentration and the film thickness. At a low Tstilbene concentration, there were three distinct bands at around 337, 353, and 370 nm. As the Tstilbene concentration or film thickness was increased, the bluest band disappeared while the two remaining bands shifted to longer wavelengths. Since these changes occurred monotonically, they should represent the increasing amount of the Tstilbene crystallites in the films. To check this assumption, we performed a simultaneous fit by decomposing the measured FL spectra of all the samples into two components: one representing the FL spectrum of the molecular Tstilbene, and the other corresponding to the Tstilbene crystallites, with varying only the relative amplitudes of both components. To maximize the unambiguity of such decomposition, the FL spectrum of the molecular Tstilbene was fixed to the one obtained for the 175-nm films containing just 0.5% of Tstilbene, when no crystallites were formed. However, no such simple direct attribution for the crystallite spectrum was possible, since the spectra of all the samples contained a smaller or larger contribution from the monomeric Tstilbene – hence more detailed mathematical analysis based on the non-negative matrix factorization algorithm was needed. Using this method, the FL spectrum of the Tstilbene crystallites and the weighting amplitudes of both components were then determined simultaneously for all the measured samples (see the Experimental section for the mathematical details).
The results of this spectral decomposition are shown in Fig. 5a and b and clearly demonstrate that the fluorescence signal in the Tstilbene films was indeed a superposition of two components: one originating from separate Tstilbene molecules, and another related to the crystallites. The “pure” FL spectra of these two Tstilbene forms are shown in Fig. 5a with dark yellow and green lines, respectively.
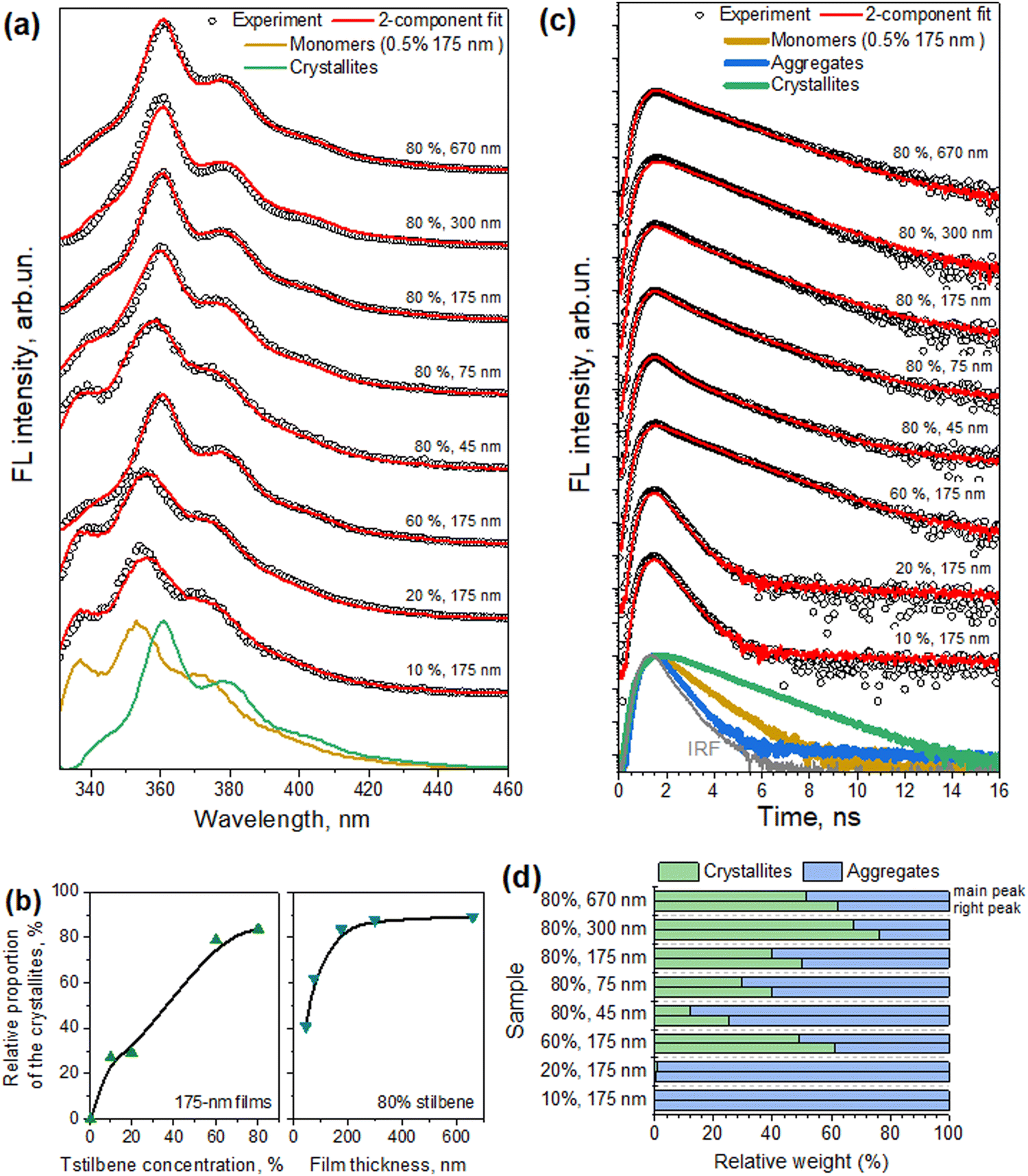 | ||
| Fig. 5 Results from the two-component analysis of the measured fluorescence spectra and kinetics. (a) The measured FL spectra and their fits are shown with black circles and red lines, respectively. For clarity, all the spectra are shifted along the linear vertical axis. Decomposed normalized FL spectra of the molecular and crystallite forms of Tstilbene are also shown (dark yellow and green lines, respectively). (b) Relative weighting factors of crystallite Tstilbene in different samples, corresponding to the 2-component fits of the FL spectra shown in panel (a). (c) The determined FL decay kinetics of the monomeric stilbene (dark yellow line, obtained from the measurements of the 175-nm films prepared at 0.5% stilbene concentration), small molecular aggregates (blue line), and crystallites (green lines). For clarity, all the measured FL decay kinetics at the main peak of the FL spectrum (circles) and their fits (red lines) are shifted along the logarithmic vertical axis; the gray line represents IRF. (d) Relative contributions of crystallite and aggregate FL decay kinetics in various samples shown in panel (c). Upper bars correspond to the kinetics measured at the main spectral peak (around 360 nm) and lower bars represent the results for the kinetics measured at the secondary right peak (around 375 nm). | ||
As already mentioned, such a superposition was not observed in the absorption measurements, which were obtained by transmission detection. One possibility for explaining this discrepancy is that Tstilbene crystals efficiently absorb the whole field, so that detection of the optical transmission signal is available only from the PS matrix with a single-molecular Tstilbene and small aggregates of Tstilbene in it. The absorption characteristics of Tstilbene crystals can be then accessed from the FL excitation spectrum, which additionally takes into account photons absorbed by the crystals. The measured FL excitation spectra for various samples are shown in Fig. 4 with orange lines. In this case, additional spectral lines at 330 and 340 nm appeared at large concentrations of Tstilbene (especially for the 670-nm sample, where the crystals dominate the spectra). The spectra suggest that the crystals show a sharp absorption edge at ∼340–350 nm with the lowest energy exciton peaking at 340 nm.
For completeness, we additionally present the fluorescence decay kinetics in Fig. 5c, measured at the main peak of the corresponding fluorescence spectrum. For clarity, all the kinetics were shifted vertically by factors of 10. At the smallest Tstilbene concentration (0.5%), the FL kinetics exhibited a mono-exponential decay with a lifetime of ∼710 ps (see Table 1). At a slightly larger Tstilbene concentration, the FL lifetime dropped down to ∼260 ps, indicating the formation of small Tstilbene aggregates and hence the manifestation of the concentration quenching effect.33–35 It is noteworthy to mention, however, that this timescale was at the detection limit of our spectrometer, thus the true FL decay kinetics might be even faster. Further increasing the Tstilbene concentration, however, resulted in bi-exponential FL decay, with a new slow decay component of ∼1.6 ns. The relative amplitude of this slow component gradually rose with increasing the Tstilbene concentration and film thickness. Then the whole decay kinetics became almost single-exponential for the 670-nm films and 80% Tstilbene. Such a behavior again suggests additive contributions of the fast and slow components originating from the small aggregates and crystallites forms of Tstilbene present in all the samples (except for the 0.5% Tstilbene-containing film, representing only molecular non-aggregated species). Again, the assumption was validated by global decomposition of all the FL decay kinetics, measured in various samples at several distinct wavelengths representing the corresponding peaks in the FL spectra, into the weighted sum of two components, similar to the spectral decomposition performed earlier. The results of such a decomposition are shown in Fig. 5c and d. Note that no strict one-to-one correspondence between the weighting factors for crystallites in the spectral (Fig. 5b) and temporal (Fig. 5d) plots was expected, since various spots on the films, containing various amount of crystallites, were illuminated in different measurements. Since no third spectral component was needed for the spectral decomposition described above, we can conclude that the FL spectrum of molecular stilbene – both purely monomeric and arranged into small molecular aggregates – remained the same, but severely changed when larger crystallites were formed.
In summary, all the measured kinetics could be described as originating from completely distinct fluorescing species of Tstilbene: pure molecular contribution (mean lifetime τave = 0.71 ns), contribution of small aggregates exhibiting concentration quenching (τave = 0.26 ns), and long-lived contribution from the crystallites (τave = 1.64 ns), shown with dark yellow, blue, and green lines in Fig. 5c, respectively. It is also interesting to mention that the ratio of the crystallite contributions in the same sample at two different wavelengths (cf. upper and lower bars in Fig. 5d) approximately scaled with the ratio of the intensities of the normalized FL spectra of the crystallites and aggregates (shown in Fig. 5a) at the corresponding wavelengths. Such a scaling, of course, should be expected, thus it indirectly supports the feasibility of our two-component analysis, applied independently to the FL spectra and FL decay kinetics. All the decay times and amplitudes are listed in Table 1.
Discussion
A complete broad-spectrum characterization of stilbene molecules in various compositions was described in the previous section, covering the vibrational and electronic transition properties and how are they affected by aggregation and crystallization. Both the CARS and AFM data (Fig. 2 and 3) indicated the formation of ordered crystal structures when the relative concentration of Tstilbene molecules in the PS films increased. However, it has to be stressed that CARS and AFM provide different information: AFM visualizes the surface roughness without referring to the chemical nature of “mountains” and “valleys” (they could be properties of polystyrene as well, and AFM would not make such a distinction). On the other hand, in the present study, the CARS microscope was tuned specifically to be sensitive to one type of molecule – stilbenes. Additionally, our studies using polarized CARS measurements showed the formation of Tstilbene microcrystals spontaneously in the PS films, which was not possible using solely AFM. It is also important to point out that the CARS microscope detects the signal from the whole volume, while AFM detects only the surface profile.Additionally, CARS spectroscopy directly provided a possibility to discriminate between Cstilbene and Tstilbene molecules. This analysis allows a definite attribution of the molecular composition to either the cis- or trans-form. Moreover, as follows from our CARS studies shown in Fig. 1, Cstilbene was not present in the aggregate formation of Tstilbene.
The sensitivity of CARS microscopy depends on the interplay of the CARS spectra of different components. Surprisingly, the Raman spectra of Tstilbene in the mono-molecular and crystal forms were almost identical. The detected signal variation came from the differences in the sample thickness and macroscopic ordering of the molecules, the latter being utilized for the detection of crystals.
Absorption and fluorescence spectroscopy were employed only for the trans-stilbene-based samples. We found an extremely efficient absorbance of the Tstilbene microcrystals, which seemed to absorb nearly all the incident field. Therefore, their contribution was not reflected in the absorption measurements, while it was supported by the fluorescence excitation spectrum. Such type of absorption is characteristic of crystals. The crystal spectra were described by a specific edge in the absorption spectrum, above which all the photons with shorter wavelengths are absorbed due to the crystal band structure. Although we made no experiments for the absorption measurement of Tstilbene monocrystal, our fluorescence excitation measurements showed this tendency.
The fluorescence spectrum could be clearly assigned to the superposition of the two components, which we attributed to the mono-molecular and crystalline components. The mono-molecular component disappeared at a high Tstilbene concentration (80%) and the fluorescence decayed mono-exponentially. Keeping in mind that the optical field was still absorbed by independent Tstilbene molecules and by microcrystals, excitation transport between single molecules in the film and crystals was very efficient, as was proposed in our previous paper.8 Two types of fluorescence centers dominated at high concentrations and all the emission events took place in these types of centers. One type primarily could be assigned to single isolated molecules, while the other was related to crystals, although their assignment to specific structural motifs remains obscure.
Overall, specific conditions of the sample preparation led to variations in the density of the microcrystals and a variation of their sizes. Most importantly, we found that stilbene crystals were created on the surface of the polystyrene film. Consequently, this became the main reason why stilbene–styrene interactions were not observed and, thus, did not play a major role.
Changing the sample preparation conditions allowed a fine control of the sample properties, as reflected in the optical data shown in Fig. 4 and 5. Evidently, the film thickness affected the sizes of the crystals. The thickness as a geometrical parameter may be the primary cause leading to the larger microcrystals (and heterogeneity of their distribution) on the film surface (Fig. 2 and 3). Thicker samples contain much more Tstilbene molecules within their diffusion length, hence larger microcrystals can form. This could be utilized for the precise preparation of specific Tstilbene microcrystals. The controlled microfabrication of Tstilbene nano- and microcrystals with their unique properties is a promising field for device fabrication.
The fluorescence decay dependence on the stilbene concentration and on the sample thickness (Table 1 and Fig. 5) supported the conclusion that microcrystals were gradually formed with increasing the stilbene concentration containing sinks of excitations, while the signal corresponding to the crystals became dominating as soon as crystals appeared. An additional third component of the fluorescence decay could be attributed to the Tstilbene aggregates. While the decay component of the crystals was the slowest, the kinetics corresponding to the aggregates was the fastest one. Most probably some type of fast decaying fluorescent traps were inherent for the aggregates and were not connected to the crystals.
The molecular skeleton of Tstilbenes and similar molecules is known to create orientational disorder in their crystals.36 Dynamic disorder with the pedal motion was proposed to occur at higher temperature, even in the molecules that showed no traces of the disorder at room temperature. However, this dynamics is not expected at room temperature, while it is a natural source of the static disorder at room temperature conditions. This causes a localization of the crystal states, and this may be a reason why the fluorescence spectra at high Tstilbene concentrations were broad and structureless and correlated with the fluorescence spectra of the aggregates.
Conclusions
The packing of functional blends of guest molecules in host molecular films is still poorly understood. It is well recognized that molecular packing and the overall film morphology are responsible for variations in the quantum efficiency of organic solar cells, while a slow relaxation of the morphology is usually related to device degradation. We demonstrated that the application of CARS microscopy not only provides the possibility to distinguish between different molecular conformers, but, together with AFM as well as absorption/fluorescence spectroscopy, allows a very precise description of the bulk morphology, and determination of the microcrystalline domains; thus, allowing assigning the characteristic spectroscopic properties of the crystalline domains and of the medium among such microstructures.Author contributions
All authors agreed to the final version of the manuscript. IH: investigation, data curation, formal analysis, visualization, and writing – original draft. RK: conceptualization, methodology, data curation, formal analysis, visualization, funding acquisition, writing – original draft, writing – review & editing. AD: methodology, data curation, visualization, and writing – original draft. MJ: methodology, data curation, visualization and writing – original draft. JC: methodology, software, writing – review & editing. MM: methodology, formal analysis, software, and writing – review & editing. DA: conceptualization, writing – review & editing, and funding acquisition. LV: supervision, conceptualization, project administration, writing – review & editing.Data availability
All research results are present in the manuscript and can be reproduced using the methodology described in the article.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Experiments were supported by Horizon Europe FLORIN (Project No. 101086142). Theoretical analysis and calculations were supported by the Research Council of Lithuania (Grant No. S-MIP-23-48). Computations were performed using high performance supercomputer ‘‘VU HPC’’ Saulėtekis of Vilnius University at Faculty of Physics.Notes and references
- P. Jacoutot, A. D. Scaccabarozzi, T. Zhang, Z. Qiao, F. Aniés, M. Neophytou, H. Bristow, R. Kumar, M. Moser, A. D. Nega, A. Schiza, A. Dimitrakopoulou-Strauss, V. G. Gregoriou, T. D. Anthopoulos, M. Heeney, I. McCulloch, A. A. Bakulin, C. L. Chochos and N. Gasparini, Small, 2022, 18, 2200580 CrossRef CAS.
- R. Dobužinskas, A. Poškus, V. Jankauskas, M. Viliūnas, E. Kamarauskas, M. Daškevičienė, V. Getautis, K. Arlauskas and D. Abramavičius, Adv. Sens. Res., 2024, 2400018 CrossRef.
- D. H. Waldeck, J. Mol. Liq., 1993, 57, 127–148 CrossRef CAS.
- G. Likhtenshtein, Stilbenes: applications in chemistry life sciences and materials sciences, Wiley-VCH, 2010 Search PubMed.
- T. Teka, L. Zhang, X. Ge, Y. Li, L. Han and X. Yan, Phytochemistry, 2022, 197, 113128 CrossRef CAS PubMed.
- C. Wang, M. D. J. Waters, P. Zhang, J. Suchan, V. Svoboda, T. T. Luu, C. Perry, Z. Yin, P. Slavíček and H. J. Wörner, Nat. Chem., 2022, 14, 1126–1132 CrossRef CAS PubMed.
- M. R. Ams, D. Ajami, L. C. Stephen, J. S. Yang and J. Rebek Jr., Beilstein J. Org. Chem., 2009, 79, 5–8 Search PubMed.
- R. Karpicz, N. Ostapenko, Y. Ostapenko, Y. Polupan, I. Lazarev, N. Galunov, M. Macernis, D. Abramavicius and L. Valkunas, Phys. Chem. Chem. Phys., 2021, 23, 3447–3454 RSC.
- R. Karpicz, G. Kareivaite, M. Macernis, D. Abramavicius and L. Valkunas, Phys. Chem. Chem. Phys., 2023, 25, 21183–21190 RSC.
- S. V. Budakovsky, N. Z. Galunov, A. Y. Rybalkooleg, A. Tarasenko and V. V. Yarychkin, Mol. Cryst. Liq. Cryst., 2002, 385, 71–77 CrossRef CAS.
- S. K. Lee, Y. H. Cho, B. H. Kang, W. G. Lee, J. K. Kim, G. D. Kim, N. Z. Galunov and Y. K. Kim, Prog. Nucl. Sci. Technol., 2011, 1, 292–295 CrossRef.
- L. Carman, N. Zaitseva, H. P. Martinez, B. Rupert, I. Pawelczak, A. Glenn, H. Mulcahy, R. Leif, K. Lewis and S. Payne, J. Cryst. Growth, 2013, 368, 56–61 CrossRef CAS.
- N. Zaitseva, A. Glenn, L. Carman, H. Paul Martinez, R. Hatarik, H. Klapper and S. Payne, Nucl. Instrum. Methods Phys. Res., Sect. A, 2015, 789, 8–15 CrossRef CAS.
- H. Klapper, N. Zaitseva and L. Carman, J. Cryst. Growth, 2015, 429, 74–81 CrossRef CAS.
- G. F. Knoll, Radiation Detection and Measurement, John Wiley & Sons, Inc., 4th edn, 2010 Search PubMed.
- N. Zaitseva, L. Carman, A. Glenn, J. Newby, M. Faust, S. Hamel, N. Cherepy and S. Payne, J. Cryst. Growth, 2011, 314, 163–170 CrossRef CAS.
- J. M. Verbeke, P. L. Kerr and M. K. Prasad, Characterization of a stilbene detector array for safeguards, LLNL-PROC-776637, Lawrence Livermore National Laboratory, USA, 2019, pp. 1–10.
- J. M. Robertson and I. Woodward, Proc. R. Soc. London, Ser. A, 1937, 162, 568–583 CAS.
- C. J. Finder, M. G. Newton and N. L. Allinger, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1974, 30, 411–415 CrossRef CAS.
- J. Bernstein, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1975, 31, 1268–1271 CrossRef.
- A. Hoekstra, P. Meertens and A. Vos, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1975, 31, 2813–2817 CrossRef.
- A. Dementjev, G. Mordas, V. Ulevičius and V. Gulbinas, J. Microsc., 2015, 257, 217–225 CrossRef CAS PubMed.
- A. Dementjev, D. Rutkauskas, I. Polovy, M. Macernis, D. Abramavicius, L. Valkunas and G. Dovbeshko, Sci. Rep., 2020, 10, 17097 CrossRef CAS PubMed.
- S. Brasselet, Adv. Opt. Photonics, 2011, 3, 205 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2019 Search PubMed.
- T. Egawa, K. Shinashi, T. Ueda, E. J. Ocola, W. Y. Chiang and J. Laane, J. Phys. Chem. A, 2014, 118, 1103–1112 CrossRef CAS PubMed.
- A. L. Dobryakov, I. Ioffe, A. A. Granovsky, N. P. Ernsting and S. A. Kovalenko, J. Chem. Phys., 2012, 137, 244505 CrossRef CAS.
- M. W. Berry, M. Browne, A. N. Langville, V. P. Pauca and R. J. Plemmons, Comput. Stat. Data Anal., 2007, 52, 155–173 CrossRef.
- W. H. Lawton and E. A. Sylvestre, Technometrics, 1971, 13, 617–633 CrossRef.
- G. C. Bazan, W. J. Oldham, R. J. Lachicotte, S. Tretiak, V. Chernyak and S. Mukamel, J. Am. Chem. Soc., 1998, 120, 9188–9204 CrossRef CAS.
- A. Ruseckas, E. B. Namdas, J. Y. Lee, S. Mukamel, S. Wang, G. C. Bazan and V. Sundström, J. Phys. Chem. A, 2003, 107, 8029–8034 CrossRef CAS.
- B. C. Chen and S. H. Lim, Appl. Phys. Lett., 2009, 94, 171911 CrossRef.
- B. Zhang, H. Soleimaninejad, D. J. Jones, J. M. White, K. P. Ghiggino, T. A. Smith and W. W. H. Wong, Chem. Mater., 2017, 29, 8395–8403 CrossRef CAS.
- G. Fanciullo, I. Conti, P. Didier, A. Klymchenko, J. Léonard, M. Garavelli and I. Rivalta, Phys. Chem. Chem. Phys., 2022, 24, 1787–1794 RSC.
- S. Barysaitė, J. Chmeliov, L. Valkunas and A. Gelzinis, J. Phys. Chem. B, 2024, 128, 4887–4897 CrossRef.
- J. Harada and K. Ogawa, J. Am. Chem. Soc., 2001, 123, 10884–10888 CrossRef CAS.
| This journal is © the Owner Societies 2024 |