
On the importance of low-frequency modes in predicting pressure-induced phase transitions†
Anna
Hoser‡
 ,
Aleksandra
Zwolenik
and
Anna
Makal‡
*
,
Aleksandra
Zwolenik
and
Anna
Makal‡
*
Faculty of Chemistry, University of Warsaw, Żwirki i Wigury 101, 02-089, Warszawa, Poland. E-mail: am.makal@uw.edu.pl
First published on 25th July 2024
Abstract
The occurrence of ultra-low frequency oscillation mode as observed by means of periodic DFT calculations at Γ point in a molecular crystal at ambient conditions can be a valuable predictor of imminent pressure-induced phase transition. We illustrate it with a series of polymorphs of diacetyl-pyrenes, for two of which such soft-modes indicated the trajectory of pressure-induced structural reorganization at ≈0.8 GPa. Notably, susceptibility to pressure-induced phase transformations could not be predicted based on material's thermodynamic characteristics, and there were no unequivocal clues in their crystal packing.
Determination of the physicochemical properties of solid materials, such as their stability, remains one of the hottest objects in modern materials sciences.1,2 Predicting the occurrence and outcome of a phase transition appears particularly attractive. The appearance of a low-frequency vibrational mode can be a valuable indicator of the imminent phase transformation3,4 and has been understood and exploited as such in the field of inorganic, in particular magnetic, materials. In the field of molecular materials, however, the assessment of vibrational properties in order to predict phase transformation has been utilized but rarely. A recent successful study of lattice dynamics of polymorphs of quinacridone showed that such an assessment may indeed have predictive power as to the probability and modes of temperature-induced phase transitions in functional organic materials.5 On cue, an experimentally determined lowest-frequency vibration in 1,2,4,5-tetrabromobenzene (TBB) represented a rotational ‘gateway’ mode for inducing the temperature-dependent phase transition from β to γ-TBB.6 Importantly, the presence of this crucial mode was reproduced by periodic DFT calculations.
The appearance of a low-frequency mode has also been known to indicate the onset and trajectory of a pressure-induced phase transition in certain minerals. The calcite I to calcite II symmetry-lowering transformation, observed experimentally at ≈1.5 GPa, has been shown to be forecast by theoretically predicted negative-frequency mode at 0.8 GPa.7 It must be stressed that in contrast to the multi-temperature studies, experimental search of ultra-low frequency modes by means of terahertz IR/Raman spectroscopy at variable pressure is wrought in difficulties due to the necessity of sample placement in a specialized container and only available at a limited number of facilities worldwide. Hence, the ability to predict structural transformations by means of theoretical computations may be the only reasonably accessible option.
Similarly to TBB, the isomers of diacetylpyrene represent a family of rigid, flat molecules which readily crystallize under a variety of conditions. Contrary to the initial assumptions,8 they yield multiple polymorphs under standard conditions.9,10 Originally reported diacetyl-pyrene polymorphs, further marked as α, display crystal structures based on ‘tapes’ of flat molecules and dominated by C–H⋯π interactions. The new (β) polymorphs more recently described by our group display packing based on π⋯π stacking of the flat molecules in the main crystallographic direction and hence distinct morphologies and luminescent properties. Most importantly, while apparently stable in a wide temperature range, certain α-polymorphs of diacetyl-pyrene (2′′AP-α, 2°AP-α) have been found to undergo displacive single-crystal-to-single-crystal (SCSC) phase transitions at increased but relatively low pressure of 0.8 GPa. On the other hand, the β polymorphs stabilized predominantly by π-stacking did not show any indication of SCSC phase transitions up to and beyond 2.0 GPa.
Having recently characterized new polymorphs of the 1,3-diacetyl pyrene11 and 1,6-diacetyl pyrene (described in ESI†), we were in position to perform a comparative study on a whole set of diacetyl-pyrene isomers. Our goal was to determine whether the propensity for pressure-induced SCSCs could be linked to, and predicted by, the crystal packing features, relative polymorph stability, the vibrational enthropy or the presence of low-frequency vibrational modes.
Crystal structures of all considered polymorphs have been redetermined at 100 K based on relatively high-resolution data for the sake of consistency (Table 1). In order to verify the occurrence/absence of pressure-induced phase transformations in 2′AP polymorphs, we conducted a series of single-crystal high-pressure X-ray diffraction experiments. A total of two datasets per each 2′AP polymorph were collected and crystal structures determined at pressures of ≈0.5 GPa and 2.0 GPa, showing no indication of SCSC phase transitions (Section S2.1 in ESI†).
|
|
|
|
||||
|---|---|---|---|---|---|---|
| a Melting points in italics were determined from DSC by Rajagopal et al.8 b No soft modes -no non-translational vibrational modes with ν < 18 cm−1. c Properties obtained with 5/12 lowest frequency modes refined accordingly. | ||||||
| Form | α9 | β9 | α10 | β11 | α | β |
| Crystal system | Monoclinic | Orthorhombic | Orthorhombic | Monoclinic | Monoclinic | Monoclinic |
| Space group | P21/c | Fdd2 | Pnma | P21/c | P21/n | P21/c |
| Z | 4 | 8 | 4 | 4 | 2 | 4 |
| Z′ | 1 | 0.5 | 0.5 | 1 | 0.5 | 2 × 0.5 |
| a [Å] | 8.13770(10) | 4.69175(9) | 7.2159(2) | 7.15110(13) | 9.2009(4) | 8.02950(10) |
| b [Å] | 22.9058(3) | 35.5546(5) | 16.4984(4) | 10.90793(19) | 7.3090(3) | 23.1438(4) |
| c [Å] | 7.23590(10) | 16.2326(3) | 11.2994(2) | 17.7744(3) | 10.0763(5) | 7.45590(10) |
| β [°] | 93.7010(10) | 90 | 90 | 102.4944(19) | 98.993(5) | 95.865(2) |
| V [3] | 1345.96(3) | 2707.81(8) | 1345.20(5) | 1353.63(4) | 669.30(5) | 1378.30(4) |
| Density [g cm−3] | 1.413 | 1.405 | 1.418 | 1.405 | 1.421 | 1.379 |
| Melting point [°C] | 162/161.78a | 157 | 181/179.08a | 174 | 206/205.78a | 196 |
| HP phase transition | Yes | Yes | No | |||
| CRYSTAL17 calculations at (Γ point) | ||||||
| Soft mode ν [cm−1] | 3 | Noneb | −19 | Noneb | Noneb | Noneb |
| E latt [kJ mol−1] | −177.6 | −178.0 | −174.3 | −172.6 | −177.9 | −172.9 |
| ΔElatt [kJ mol−1] | 0 | −0.4 | 0 | 1.7 | 0 | 5.0 |
| ΔEele [kJ mol−1] | 0 | −2.3 | 0 | −0.9 | 0 | 1.7 |
| ΔTSvib [kJ mol−1] | 0 | −4.7 | 0 | 0.9 | 0 | 4.8 |
| ΔGΓ [kJ mol−1] | 0 | 2.1 | 0 | −1.2 | 0 | 0.4 |
| Normal modes refinement | ||||||
| Soft mode ν [cm−1] | 18/18c | 17/39c | ||||
| ΔTSvib [kJ mol−1] | 0 | 1.1/1.1c | 0 | 1.0/2.0c | 0 | −0.3/0.1c |
| ΔGNoMoRe [kJ mol−1] | 0 | −2.4/−2.4c | 0 | −1.8/−2.8c | 0 | 3.5/3.2c |
In order to systematically analyze relative stabilities of the polymorphs and gain insight into lattice dynamics of the studied materials, we optimized the initial experimental crystal geometry for all systems (Table 1) with unit cell parameters fixed at X-ray determined values (functional: B3LYP-D3, basis set: 6-31G(d,p), using CRYSTAL17,12 details in ESI†). Based on optimized structures, we calculated: (a) lattice Elatt and total electronic Eele energies, (b) vibrational frequencies and their normal mode vectors at Γ point of the Brillouin zone (BZ). Corresponding Gibbs free energies G were derived according to the formula:
| G = Eele + ZPE + Hvib + pV − TSvib | (1) |
In many instances relative polymorph stability can be gauged from their lattice energies Elatt alone.13,14 The polymorphs of diacetyl-pyrene isomers however show very comparable lattice energies (Table 1), and already proved challenging from the crystal structure prediction point of view.9 This prompted us to investigate all components of Gibbs free energy between polymorphs. It was shown that entropy (S),13 ZPE,15 or Hvib16 can alter the stability order of polymorphs at a given temperature. However, accurate determination of these thermodynamic properties solely through computations remains challenging, requiring calculation of vibrational frequencies and their normal mode vectors at multiple points in the BZ. Such an approach has very high computational costs. We turned instead to dynamic quantum crystallography methods, which postulate, that certain information about the vibrations in crystal structures can be gained from the diffraction data and atomic displacement parameters (ADPs). We applied normal mode refinement (NoMoRe),17,18 based on a direct adjusting of selected vibrational frequencies from DFT calculations at Γ point of BZ against single crystal X-ray diffraction data, thus obtaining a much more comprehensive description of the lattice vibrations (see ESI† for details). We then calculated the more exact vibrational contributions to the Gibbs free energy, namely ZPE, Hvib and Svib based on the refined frequencies. Such an approach can yield thermodynamic properties in excellent agreement with calorimetric measurements.18 We validated our computational approaches by how well they reproduced the relative polymorph stability based on experimentally established melting points.
All the originally discovered α polymorphs of diacetyl-pyrene isomers emerge uniformly as those characterized by the higher melting point (Table 1). This trend was not reproduced by either lattice energies or Gibbs free energies computed for analyzed systems. Only the 2′AP polymorphic system appears as monotropic,19 with the 2′AP-α displaying the lowest lattice energy Elatt, electronic energy Eele and Gibbs free energy G irrespective of the calculation method. The vibrational entropy contribution in this system is small and indeed negligible after the NoMoRe refinement, varying between −0.3 kJ mol−1 to 0.1 kJ mol−1, for different approaches.
In the case of the 2′′AP and 2°AP polymorphic systems, namely the systems where pressure-induced phase transitions could occur, the results were much more ambiguous. The differences in Elatt between α and β forms are very small (<1 kJ mol−1 for 2′′AP and <2 kJ mol−1 for 2°AP), approaching the threshold of accuracy achievable with density functional theory (DFT).20,21 The differences in Gibbs free energy at Γ-point in these cases were also very similar, with ΔGΓ not exceeding 2, and reproduced the order of the melting points only in the case of 2°AP. Dynamic quantum crystallography approach (NoMoRe) yielded lower Gibbs free energies and indicated as more stable the 2′′AP-β and 2°AP-β polymorphs, in disagreement with experimentally determined melting points. Notably, 2°AP-β shows significant anharmonic vibrations above 200 K,11 only superficially accounted for in our calculations (see Section S1.7 of ESI† for details).
All β polymorphs, stabilized predominantly by the π-stacking of molecules in one of the crystallographic directions, displayed more pronounced atomic displacement parameters even at 100 K (Fig. 1) for at least one of the independent molecules, in agreement with their lower densities. Such a feature should correlate with more stabilizing vibrational contributions to G, in particular with TSvib. Considering the results from DFT calculations at Γ point, no such correlation can be observed. However, this approach does not take into account acoustic modes, which also contribute to entropy and should be better accounted for in the NoMoRe approach. Indeed, in the case of 2′′AP and 2°AP isomers, the β polymorphs showed a non-negligible stabilizing enthropic component to their Gibbs free energy after selected low-frequencies were refined against X-ray diffraction data. Somewhat surprisingly, the vibrational enthropic component TSvib obtained in NoMoRe appears smaller for the exact α polymorphs which showed the presence of soft- or ‘gateway’ vibrational modes and propensity for pressure-induced phase transitions. However, since the relative differences in TSvib do not exceed 2 kJ mol−1, the noted trend is within the accuracy limit of the current computational approach.
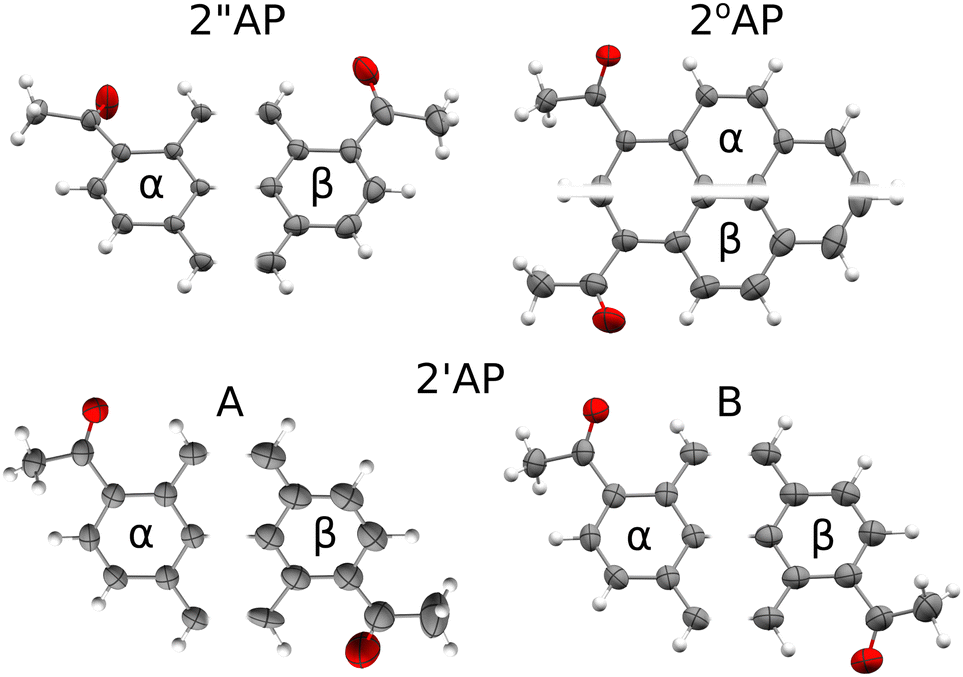 | ||
| Fig. 1 Comparison of the atomic displacement parameters between the α–β polymorphs of diacetyl-pyrene as determined at 100 K. Only half of each molecule has been drawn for easier comparison. All ADP-s are drawn at 90% probability level. Notably, all β polymorphs display more prominent atomic displacement parameters involving in-plane oscillations of the pyrene moieties. | ||
Apparently neither Elatt nor ΔG or ΔTSvib as computed here can be an indicator of the imminent pressure-induced phase transformation. Our dynamic quantum crystallography models turned out to be insufficient even to describe minute discrepancies in Gibbs free energies for the polymorphic systems of diacetyl-pyrene isomers. Reasons for this insufficiency may stem from refinements performed against diffraction data at 100 K only or from conducting frequency calculations essentially within harmonic approximation. Last but not least, models from NoMoRe based on adjusting of only a few frequencies appear too simplified even for such relatively rigid compounds. Improving the accuracy of the NoMoRe approach will be the object of future studies.
Nor can the propensity for pressure-induced phase transition be easily inferred from the comparative investigation of the intermolecular interaction networks (Fig. 2). While the very characteristic stacks of molecules in the β polymorphs with their dominant interaction energies seem to prevent pressure-induced phase transitions, there are no clear clues among the α polymorphs. Intermolecular interaction networks of 2′′AP-α and 2°AP-α the materials prone to pressure-induced phase transformations appear less 'interaction dense’ than of 2′AP-α. It is conceivable to descry ‘voids’ in which there are no strong interactions, allowing for tilting of the molecules under external stimuli. It would appear that a molecule in 2′AP-α is more consistently stabilized than molecules in 2′′AP-α or 2°AP-α and has literally no space to change its orientation. However, this kind of consideration is rather ambiguous and not easily transferable to a wider variety of organic materials.
 | ||
| Fig. 2 Crystal packing of diacetyl-pyrene polymorphs seen from one arbitrary direction. Energy frameworks calculated with CrystalExplorer17 are overlaid on the crystal packing motifs. Blue tubes represent the total intermolecular interaction energies more negative than −10 kJ mol−1 and their thickness is scaled by the |E|. | ||
Most importantly and conclusively, for the very crystal forms which were prone to pressure-induced phase transitions, we observed ultra-low frequency modes, which in the case of 2′′AP-α amounted to only 3 cm−1 at Γ point while in the case of 2°AP-α turned out to be an imaginary mode (−19 cm−1). Upon applying the NoMoRe approach, providing a more generalized view, both frequencies refined to well-defined positive values (Table 1), confirming the meaningfulness of these vibrations. Careful inspection of the trajectories of these 'soft' modes proved that these vibrations in each instance coincided exactly with the reorientation of the molecules occurring in the course of the pressure-induced phase transitions (Fig. 3).
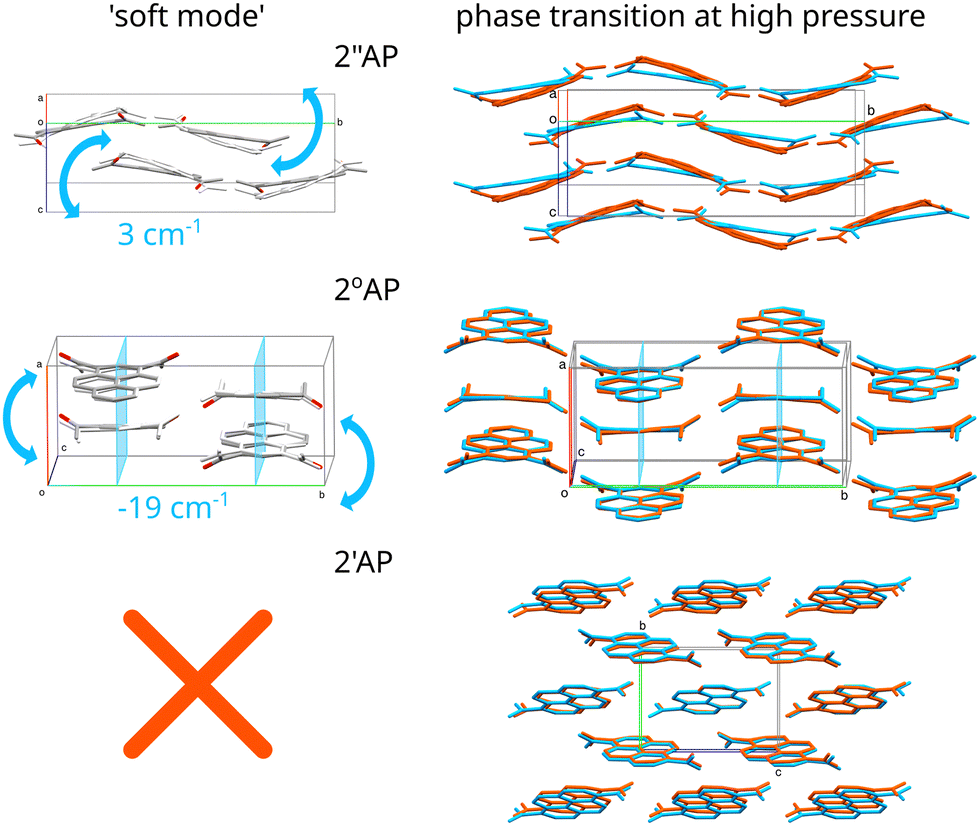 | ||
| Fig. 3 Representations of the soft modes detected in the case of 2′′AP-α and 2°AP-α polymorphs (on the left) and the overlays of the crystal structures from 1 atm (blue) and ≈1 GPa (orange) illustrating the molecular shifts induced by increased pressure (right). Notably in the case of 2′AP-α the crystal structure undergoes compression without showing a tendency for molecules to tilt. | ||
In the case of 2′′AP-α the transformation involved tilting a molecule along its main axis, in this instance parallel to and out of the existing molecular tape, thus reorganizing a network of intermolecular interactions.9 The exact same tilting motion of the molecule constituted the soft-mode in question. In the case of 2°AP-α the transformation also involved tilting the whole molecule along its main axis, coinciding with the crystallographic [001] direction, and out of the crystallographic m[010] plane. As the tilt became more prominent, the m[010] symmetry would no longer be present in the resulting new phase.
In the case of the third isomer of diacetyl-pyrene, neither of the polymorphs showed any indication of an oncoming phase transition up to 2.0 GPa. The only noticeable structural changes occurring with pressure were molecules shifting steadily closer towards each other. There were no indications of tilting or rotating of molecules (see Section 2.1.1 of ESI† for details). Likewise, no low-frequency modes other than the usual translational ones could be found among the calculated crystal vibrational modes for both 2′AP-α and 2′AP-β.
The occurrence of a single low-frequency oscillation mode in the course of theoretical calculations can thus be shown as a predictor of the possible pressure-induced phase transition. Moreover, it can indicate the direction in which such a transformation would proceed, allowing to forecast certain properties of a new phase, such as symmetry lowering. Computational approach proposed here for detecting low-frequency modes is relatively accessible to a wide community of users and for a larger variety of molecular systems and comes with a bonus in the estimates of all components of Gibbs free energy.
While there were a few notable cases of quantitative phase transition characterization in molecular crystals at non-ambient conditions by computational means, these involved exclusively simple compounds such as ammonia22 or urea23 with a very small (<20) number of atoms and electrons per unit cell. More importantly, reliable modeling of properties for such materials (e.g.: relative stabilities, Raman frequencies) required methods such as Møller–Plesset perturbation (MP2) or even coupled clusters (CCSD(T)),2,22 absolutely prohibitive for larger molecules in terms of computational cost and convergence success rate.
Our simplified approach to qualitatively analyze the material's oscillation modes and interpret them might in that context be the only viable option for larger (>40 atoms) molecular systems in the solid state.
Author contributions
AZ: investigation and analysis (crystallography and periodic DFT calculations), visualization; AM: conceptualization, funding and resources, investigation and analysis (crystallography and periodic DFT calculations), visualization, supervision of AZ. AH: quantum crystallography (NoMoRe) methodology and software, investigation and analysis (periodic DFT calculations and NoMoRe approach). All Authors contributed to data validation, writing initial draft and reviewed the final version of the manuscript.Data availability
Crystallographic data for all presented compounds has been deposited at the CCDC under numbers 2332696–2332704† and can be obtained from https://www.ccdc.cam.ac.uk/structures/. Further data supporting this manuscript have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was financially supported by the National Science Centre Poland (NCN) based on decision DEC-2015/17/B/ST4/04216. Theoretical DFT calculations in atom-centered approach were preformed using resources provided by PLGrid grants plgamakalhp5 thorough plgamakalhp7a.References
- J. Hoja, A. M. Reilly and A. Tkatchenko, WIREs Comput. Mol. Biosci., 2017, 7, e1294 CrossRef.
- D. Firaha, Y. M. Liu, J. van de Streek, K. Sasikumar, H. Dietrich, J. Helfferich, L. Aerts, D. E. Braun, A. Broo, A. G. DiPasquale, A. Y. Lee, S. Le Meur, S. O. Nilsson Lill, W. J. Lunsmann, A. Mattei, P. Muglia, O. D. Putra, M. Raoui, S. M. Reutzel-Edens, S. Rome, A. Y. Sheikh, A. Tkatchenko, G. R. Woollam and M. A. Neumann, Nature, 2023, 623, 324–328 CrossRef CAS PubMed.
- G. Venkataraman, Bull. Mater. Sci., 1979, 1, 129–170 CrossRef CAS.
- V. Heine, X. Chen, S. Dattagupta, M. T. Dove, A. Evans, A. P. Giddy, S. Marais, S. Padlewski, E. Salje and F. S. Tautz, Ferroelectrics, 1992, 128, 255–264 CrossRef CAS.
- A. Giunchi, L. Pandolfi, R. G. Della Valle, T. Salzillo, E. Venuti and A. Girlando, Cryst. Growth Des., 2023, 23, 6765–6773 CrossRef CAS PubMed.
- A. J. Zaczek, L. Catalano, P. Naumov and T. M. Korter, Chem. Sci., 2019, 10, 1332–1341 RSC.
- M. Ukita, K. Toyoura, A. Nakamura and K. Matsunaga, J. Appl. Phys., 2016, 120, 142118 CrossRef.
- S. K. Rajagopal, A. M. Philip, K. Nagarajan and M. Hariharan, Chem. Commun., 2014, 50, 8644–8647 RSC.
- D. Tchon, D. Bowskill, I. Sugden, P. Piotrowski and A. Makal, J. Mater. Chem. C, 2021, 9, 2491–2503 RSC.
- D. Tchon and A. Makal, IUCrJ, 2021, 8, 1006–1017 CrossRef CAS PubMed.
- A. Zwolenik, D. Tchon and A. Makal, IUCrJ, 2024, 11, 519–527 CrossRef CAS PubMed.
- R. Dovesi, A. Erba, R. Orlando, C. M. Zicovich-Wilson, B. Civalleri, L. Maschio, M. Rérat, S. Casassa, J. Baima, S. Salustro and B. Kirtman, WIREs Comput. Mol. Biosci., 2018, 8, e1360 CrossRef.
- J. Nyman and G. M. Day, CrystEngComm, 2015, 17, 5154–5165 RSC.
- A. J. Cruz-Cabeza, S. M. Reutzel-Edens and J. Bernstein, Chem. Soc. Rev., 2015, 44, 8619–8635 RSC.
- S. A. Rivera, D. G. Allis and B. S. Hudson, Cryst. Growth Des., 2008, 8, 3905–3907 CrossRef CAS.
- J. Nyman and G. M. Day, Phys. Chem. Chem. Phys., 2016, 18, 31132–31143 RSC.
- A. A. Hoser and A. O. Madsen, Acta Crystallogr., Sect. A: Found. Adv., 2016, 72, 206–214 CrossRef CAS PubMed.
- A. A. Hoser, M. Sztylko, D. Trzybinski and A. O. Madsen, Chem. Commun., 2021, 57, 9370–9373 RSC.
- J. Bernstein, Polymorphism in molecular crystals, Oxford University Press, Oxford; New York, 2 edn, 2020 Search PubMed.
- F. D. Pia, A. Zen, D. Alfè and A. Michaelides, How accurate are simulations and experiments for the lattice energies of molecular crystals, 2024.
- A. E. Phillips and H. C. Walker, J. Phys.: Energy, 2023, 6, 011001 Search PubMed.
- L. Huang, Y. Han, J. Liu, X. He and J. Li, Sci. Rep., 2020, 10, 7546 CrossRef CAS PubMed.
- A. Mazurek, L. Szeleszczuk and D. M. Pisklak, Molecules, 2020, 25, 1584 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Details concerning used methodology, additional tables and figures describing new investigated structures and intermolecular interactions in 2'AP polymorphs, movies illustrating the crucial soft-modes. CCDC 2332696–2332704. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cp02368d |
| ‡ These authors contributed equally to this work. |
| This journal is © the Owner Societies 2024 |