
Computational insights into Diels–Alder reactions of paramagnetic endohedral metallofullerenes: M@C82 (M = Sc, Y, La) and La@C72†
Linfeng
Nie
b,
Yuanyuan
Sun
 *ab and
Yang
Wang
*b
*ab and
Yang
Wang
*b
aInstitute of Innovation Materials and Energy, Yangzhou University, Yangzhou 225002, P. R. China. E-mail: syy@yzu.edu.cn
bSchool of Chemistry and Chemical Engineering, Yangzhou University, Yangzhou 225002, P. R. China. E-mail: yangwang@yzu.edu.cn
First published on 23rd September 2024
Abstract
In fullerene chemistry, Diels–Alder cycloaddition is an essential reaction for exohedral modification of carbon cages. M@C2v(9)-C82 (M = Sc, Y, and La), incorporating one metal atom within the fullerene cage, are key compounds for understanding the impact of both endohedral and exohedral modifications on their electronic structures. In this work, the Diels–Alder (DA) cycloaddition of cyclopentadiene (Cp) to M@C2v(9)-C82 (M = Sc, Y, and La) and La@C2(10612)-C72 was systematically studied using density functional theory. The most reactive bonds were initially chosen for detailed mechanistic exploration, considering both concerted and stepwise mechanisms. Our findings revealed that DA cycloadditions for the three metals (Sc, Y, and La) consistently exhibit the same regioselectivity, favoring the concerted attack on the [5,6] bond. This observation is in agreement with previous experimental and theoretical studies on the regioselectivity of the Diels–Alder reaction between La@C2v(9)-C82 and Cp. In the case of La@C2(10612)-C72, the most favored pathway is the concerted attack on the [6,6] bond both kinetically and thermodynamically. In toluene and ortho-dichlorobenzene, while the energy barriers and the reaction free energies increased to different extents for most pathways, the regioselectivity largely mirrored that observed in the gas phase.
1. Introduction
Due to their large interior spaces, fullerenes exhibit remarkable ability to encapsulate different metal atoms or metallic clusters, which gives rise to a unique class of carbon materials known as endohedral metallofullerenes (EMFs).1–3 One of the key characteristics of EMFs is that a certain number of electrons (typically no more than 6e) transfer from the encaged entities to the fullerene framework, resulting in significantly strengthened interactions between the embedded species and the carbon cage.4 It is well-known that stable fullerenes must adhere to the isolated pentagon rule (IPR), wherein each pentagonal ring should only be adjacent to hexagonal rings.5 Despite this, the stabilization between the metal ions and the outer cage has led to the discovery of several EMFs that violate the IPR.6–15 In particular, recent theoretical studies on small EMFs and quasi-fullerenes have revealed that non-IPR carbon cages can be greatly stabilized by metal insertion, exhibiting a significant change in the bonding characteristics, stability, aromaticity, and reactivity.10–15Among various sizes of carbon cages, C82 demonstrates exceptional stability in encapsulating metal atoms (M) to form mono-metallofullerenes M@C82 which have different cage isomers (C2v and Cs) being isolated. Since the first experimental report on La@C82,16 various rare-earth metal atoms have been successfully encapsulated inside diverse fullerene cavities.17–22 In particular, when certain rare-earth metal atoms (such as Sc, Y, La, Ce or Gd) are encapsulated within C2v-C82, three electrons are transferred to the carbon framework. Therefore, the electronic structure can be expressed as M3+@C823−, resulting in unpaired electron spin on the carbon cage, which imparts paramagnetic properties to EMFs.
The Diels–Alder (DA) cycloaddition approach is widely employed to functionalize fullerenes and generate fullerene derivatives. The interaction between the interior metal atoms and the carbon framework endows EMFs with unique reactivity and regioselectivity compared to their empty counterparts. However, despite the high experimental yields and unique paramagnetic properties of M@C82 (where M represents a rare-earth metal), research studies on their DA reactions have been relatively limited, with an exception of La@C2v-C82.23–26 Although La@C2v-C82 has 24 non-equivalent carbon atoms and 35 non-equivalent bonds, its DA reaction with cyclopentadiene (Cp) displays high regioselectivity with only one adduct observed experimentally.23 It was presumed that the addition occurred by attacking a specific [6,6] bond (type B, as shown in Fig. 1) near the La atom, which was solely based on the shape of the singly occupied molecular orbital (SOMO) without further isolation of this monoadduct. Subsequent work by the same group demonstrated, through X-ray crystallographic analysis, that 1,2,3,4,5-pentamethylcyclopentadiene (Cp*) was proved to mainly attack a type D [5,6] bond C8–C15 (as shown in Fig. 2a), contradicting the high spin densities or the π-orbital axis vector (POAV) value at this addition site. However, it was speculated that this regioselectivity mainly stemmed from the high positive charge densities.26,27 Later, systematic theoretical calculations corrected the addition site of Cp with La@C2v-C82 in ref. 23 to bond C8–C15, indicating consistent regioselectivity between Cp and Cp* cycloaddition to La@C2v-C82.25
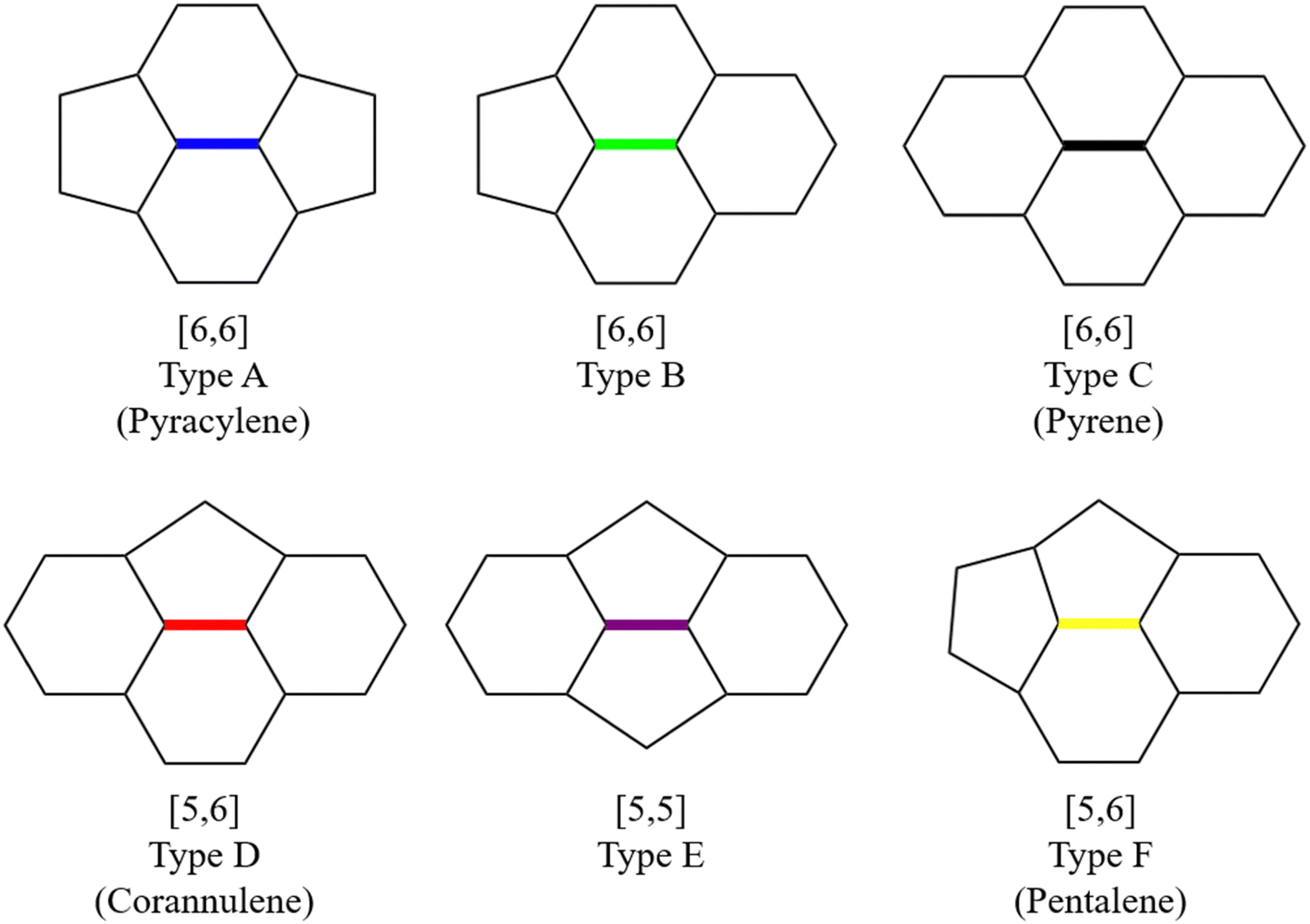 | ||
| Fig. 1 Representation of different bond types [5,5], [5,6], and [6,6] in fullerene structures.28 | ||
 | ||
| Fig. 2 Schlegel diagrams of (a) M@C2v-C82 (M = Sc, Y, and La) and (b) La@C2-C72 with labeling of cage C atoms used in this study. Adjacent pentagons in the C72 cage are depicted in green. | ||
Theoretical calculations play a significant role in unraveling the regioselectivity, feasibility, and reaction mechanisms of EMF-involved reactions. In our previous work, the cycloaddition of s-cis-1,3-butadiene (BD) to paramagnetic TiSc2N@Ih-C80 was systematically investigated by taking into account all the possible addition sites.29 Our findings revealed that the endohedral metal atom in EMFs significantly increases the energy barrier and reduces the reaction energy for Diels–Alder (DA) cycloadditions, making EMFs generally less reactive than their empty fullerene counterparts.29 This has been generally recognized across a wide variety of EMFs with different metalloclusters.30–32 Different from the traditional concerted mechanism, it was found that the reaction was kinetically favored and prone to occur through a [4+3] stepwise mechanism. To date, both experimental and theoretical investigations into the Diels–Alder reactions involving Sc@C2v-C82, Y@C2v-C82, and IPR-violating EMFs remain scarce. The exploration of DA reactions involving different rare-earth metal-embedded fullerenes offers a valuable platform for comparative analysis of reactivity and regioselectivity trends, as well as understanding the role of different metal species in influencing chemical reactions. Herein, a systematic theoretical exploration on the DA additions of M@C2v-C82 (M = Sc, Y, and La, C2v-C82 is abbreviated as C82 hereafter) is presented. Additionally, La@C2(10612)-C72 with a non-IPR carbon cage was also investigated (where C2(10612)-C72 is abbreviated as C72 hereafter), which has been synthesized and characterized experimentally.33 By delving into these reactions, we aim to not only fill the existing research gap but also contribute to a deeper understanding of the fundamental mechanisms governing these reactions.
2. Computational details
The BP86 functional34–36 was employed in all the DFT calculations, along with Grimme's DFT-D3 dispersion correction with Becke–Johnson damping,37 which is a commonly employed functional to explore the reactivities and thermodynamics of EMFs.38 The Def2-SVP39,40 basis set was adopted for C, H, and Sc elements, while the Stuttgart/Dresden SDD41,42 relativistic effective core potentials with the corresponding basis set were employed for Y and La elements. In order to reduce the high computational cost, the Def2-SVP basis set was combined with the W0639,40 density fitting auxiliary basis set. The use of the above functional and basis sets has been previously verified in our previous works.29,43 All the DFT calculations were conducted using the Gaussian 16 programs suite.44The initial geometric structures of the carbon cages C2v(9)-C82 and C2(10612)-C72 were generated using the FullFun software.45,46 Subsequently, full geometry optimizations were carried out for all relevant species. Following this, the vibrational frequency analyses were conducted to confirm the stability of each optimized configuration and to obtain the thermal corrections to Gibbs free energies at 298.15 K and 1 atm. Furthermore, intrinsic reaction coordinate (IRC) calculations47,48 were carried out to guarantee that the obtained transition state connected the proper reactant and product along the reaction pathway. To take into account the solvent effect, single point calculations were performed using the solvation model based on density (SMD).48
3. Results and discussion
3.1 Isomers of M@C82 (M = Sc, Y, and La) and La@C72
The geometric structures of different EMF isomers, M@C82 (M = Sc, Y, and La) as well as La@C72, were explored in the first step. For each EMF, the metal atom was encapsulated in the fullerene cage and located above the center of the rings, considering all nonequivalent rings within the fullerene structure. For M@C82, the number of isomers varies with the atomic size from Sc to La after full geometric optimization. Sc@C82 exhibits the highest diversity among the studied endohedral metallofullerenes, with six distinct isomers (labeled a to f) as shown in Fig. 3. Y@C82 has three isomers (labeled a, e and f), demonstrating a limited set of stable configurations. In contrast, La@C82 possesses four isomers, lacking configurations c and d. These results are consistent with our previous research findings on M@C82 (M = Sc, Y, and La), further validating the relationship between atomic size and isomeric diversity in these endohedral fullerenes.43 The relative energies of these isomers are illustrated in Table 1. Among these isomers, isomer a is particularly noteworthy because the metal atom is positioned off-center, adjacent to a hexagonal ring along the C2 axis. Remarkably, this arrangement aligns closely with experimental determinations and is identified as the most stable isomer.49 Therefore, isomer a serves as a focal point for systematically investigating the Diels–Alder (DA) reaction in the subsequent phases of our study. Following full optimization for La@C72, two different isomers with distinct La atom positions were identified, as shown in Fig. S1 (ESI†). The energy difference between these two isomers is 14.7 kcal mol−1. Notably, in the more stable isomer (isomer a), the La atom is positioned adjacent to the fused pentagons, which is consistent with the experimental observations33 and employed in the following calculations. As shown in Table S1 (ESI†), the Mulliken population and natural population analysis (NPA) reveal that the charge transfer between M and the fullerene cage remains relatively consistent across different isomers of the same EMF. This consistency suggests that the relative energies of isomers are not decisively determined by the charge transfer from the metal atom to the carbon cage, but are more likely influenced by other factors, such as the complex covalent and dispersion interactions between the metal and the carbon cage, and the specific distribution of electron density within the EMF.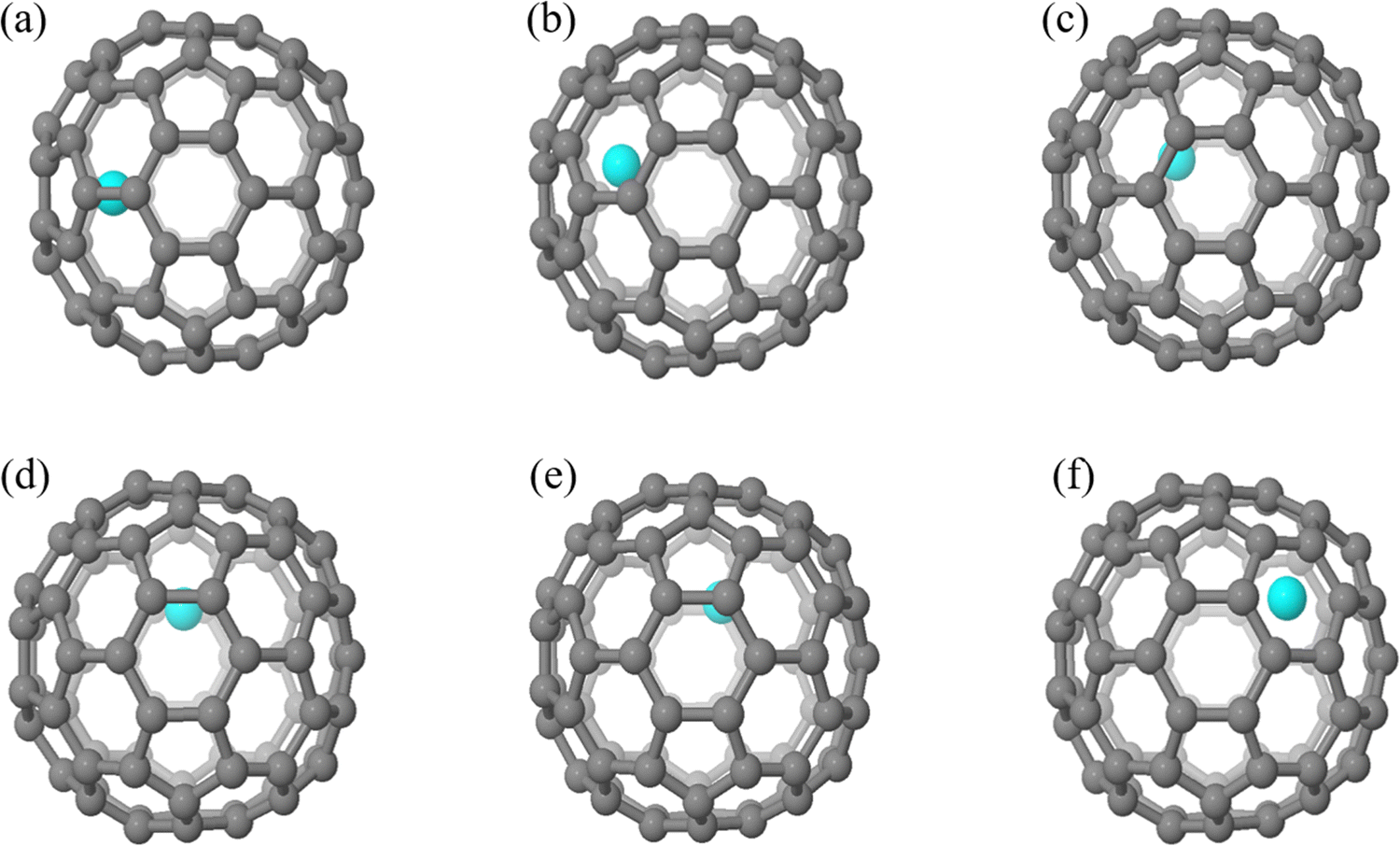 | ||
| Fig. 3 Geometrical structures of M@C82 (M = Sc, Y, and La) isomers with different metal atom positions. | ||
| Isomer | Sc@C82 | Y@C82 | La@C82 |
|---|---|---|---|
| The hyphen (-) indicates that no stable isomer was found. | |||
| a | 0.0 | 0.0 | 0.0 |
| b | 0.4 | - | 0.9 |
| c | 4.1 | - | - |
| d | 11.0 | - | - |
| e | 12.7 | 13.8 | 14.6 |
| f | 26.7 | 28.5 | 28.6 |
3.2 Simple hydride model
In our previous work, a mono-hydride model was developed to accurately predict the relative thermodynamic stability of regioisomers of EMF dimers.43 Based on this model, a basic dihydride model was proposed to alleviate the computational burden and to provide preliminary predictions of the regioselectivity of Diels–Alder (DA) adducts. Initially, two hydrogen atoms were positioned along the POAV direction of each C atom in the C–C bond, and each C–H bond distance was set to a typical value of 1.12 Å, consistent with that of fully optimized EMF hydrides. This method resulted in the generation of 35 distinct dihydrides for M@C82 (where M = Sc, Y, and La) and 55 dihydrides for La@C72. Single-point energy calculations were conducted for these dihydrides to evaluate their relative stabilities. The computed relative energies are depicted in Fig. S2 (ESI†), providing valuable insights into the regioselectivity of the DA cycloaddition. Based on the results of this model, dihydrides with relative energies less than 15 kcal mol−1 were chosen, as configurations with lower relative energies are more likely to be favored in the actual reaction. There are four different bond types in the C82 cage (labeled A, B, C, and D) and six bond types in the C72 cage (labeled A, B, C, D, E, and F). The relative energies of the dihydrides formed on type C bonds are notably higher, exceeding 26.0 kcal mol−1 for both M@C82 and La@C72. Given the high energies associated with these configurations, type C bonds were excluded from further consideration in the exploration of the concerted mechanism.For each of these chosen dihydrides, the corresponding DA adducts formed at the same addition sites were constructed. Full optimizations were carried out for both the selected dihydrides and their corresponding DA adducts. As illustrated in Fig. 4, there is a strong linear relationship between the relative energies of the dihydrides and their respective DA adducts. This relationship is quantitatively supported by a coefficient of determination (R2) consistently exceeding 0.91, which underscores the accuracy and predictive capability of the model. According to Hammond's postulate and the Evans–Polanyi principle,50–52 the activation energy barrier is anticipated to align with the overall energy trend of the reaction. Therefore, for further study, the addition sites corresponding to the dihydrides with the lowest relative energies were selected for each EMF (M@C82 and La@C72, M = Sc, Y, and La). These selected sites included different bond types within the fullerene cages. In Fig. 4, the solid circles highlight the specific addition sites that were chosen to investigate the concerted mechanism of the DA cycloaddition in this study.
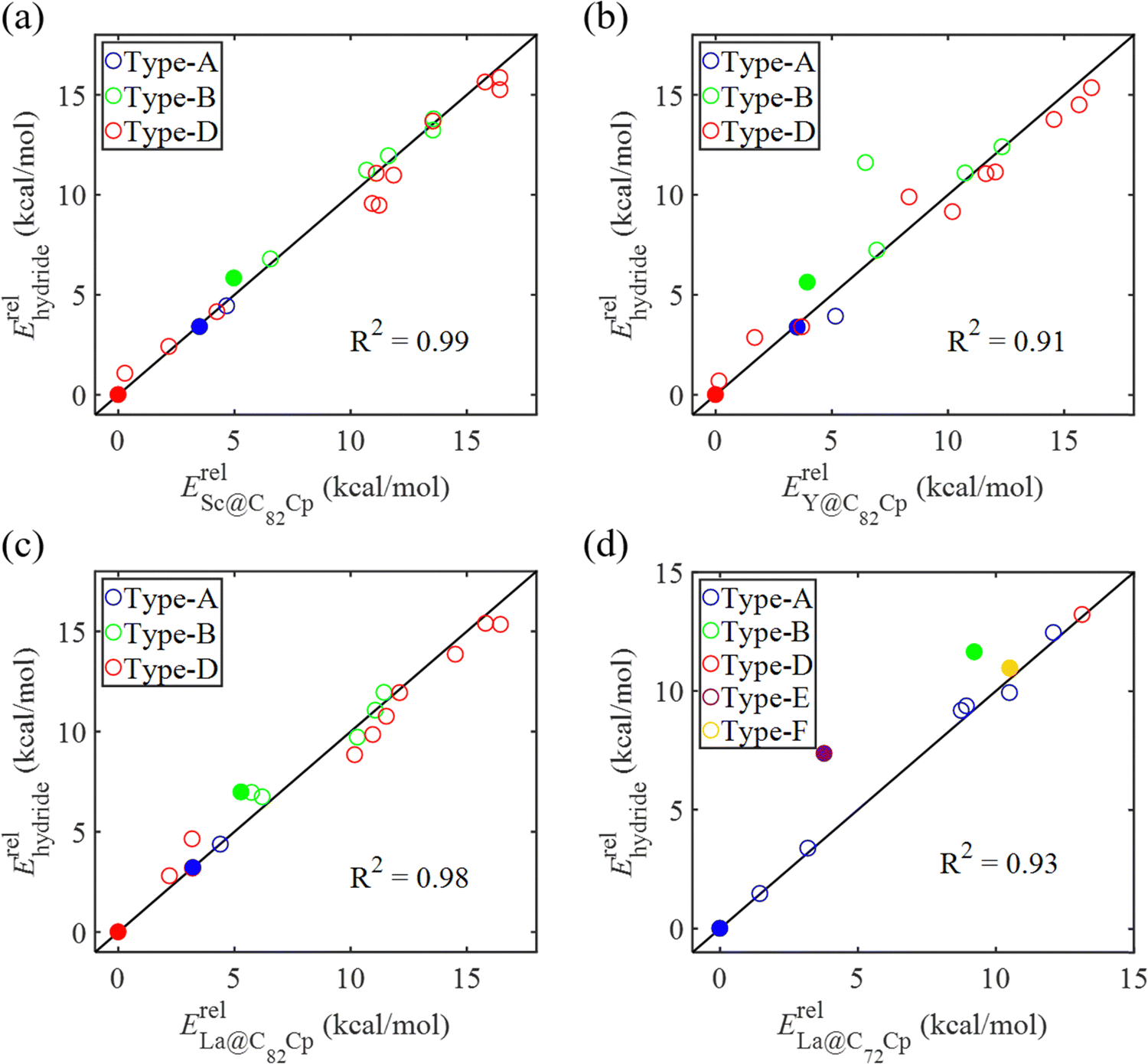 | ||
Fig. 4 Relative energies of EMF dihydrides Erelhydrideversus relative energies of Diels–Alder adducts (a) 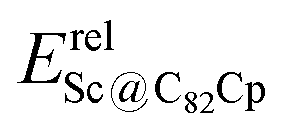 , (b) , (b)  , (c) , (c)  , and (d) , and (d) 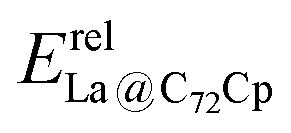 . ZPE corrections are not included. The relative energies are given in kcal mol−1. . ZPE corrections are not included. The relative energies are given in kcal mol−1. | ||
3.3 The intermediate model
When cycloaddition between EMFs and Cp molecules proceeds via a stepwise mechanism, the Cp moiety in the intermediate molecule exhibits the ability to rotate around the newly formed C–C single bond. This rotational flexibility can potentially lead to the formation of three different configurations. Therefore, all non-equivalent C atoms in the carbon cage (24 in C82 and 36 in C72) were considered as possible addition sites for the formation of intermediate species, denoted as Ca. Each addition site leads to the possibility of forming three distinct adducts, as shown in Fig. S3a (ESI†). Additionally, taking into account the two mirror orientations of the Cp moiety relative to the addition bond (as shown in Fig. S3b, ESI†), a combined total of 144 intermediate isomers for M@C82 and 216 for La@C72 were identified. These potential intermediates, when situated at certain addition sites, undergo direct transformation into either final products or reaction complexes after full optimization, which suggests that the DA reaction may not proceed through a stepwise mechanism at these particular addition sites. In each intermediate, the other adduct C atom in Cp spatially corresponds to an adduct C atom in fullerene, which was denoted as Cβ. Consequently, the intermediates were classified based on the bond types between Cα and Cβ in the fullerene cage as depicted in Fig. 1. Fig. 5 depicts the successfully optimized geometric structures for the potential intermediates. For Sc, Y, and La@C82, the most stable intermediate is formed at the C7 atom of the type B [6,6] bond, while the Cp moiety bonded with a C atom (C13 for Sc and La@C82, C22 for Y@C82) of the type C [6,6] bond exhibits the highest energy. Because of the failure optimization of the intermediate formed on type A bonds, it is inferred that the DA cycloaddition of M@C82 cannot proceed through the stepwise mechanism for type A bonds. In the case of La@C72, the most stable intermediate is formed on the C33 atom of the type D [5,6] bond, and the optimization of the intermediate formed on the [5,5] bond also failed. To comprehensively explore the stepwise mechanism, the most stable intermediate for each bond type was selected for detailed analysis (see solid legends shown in Fig. 5).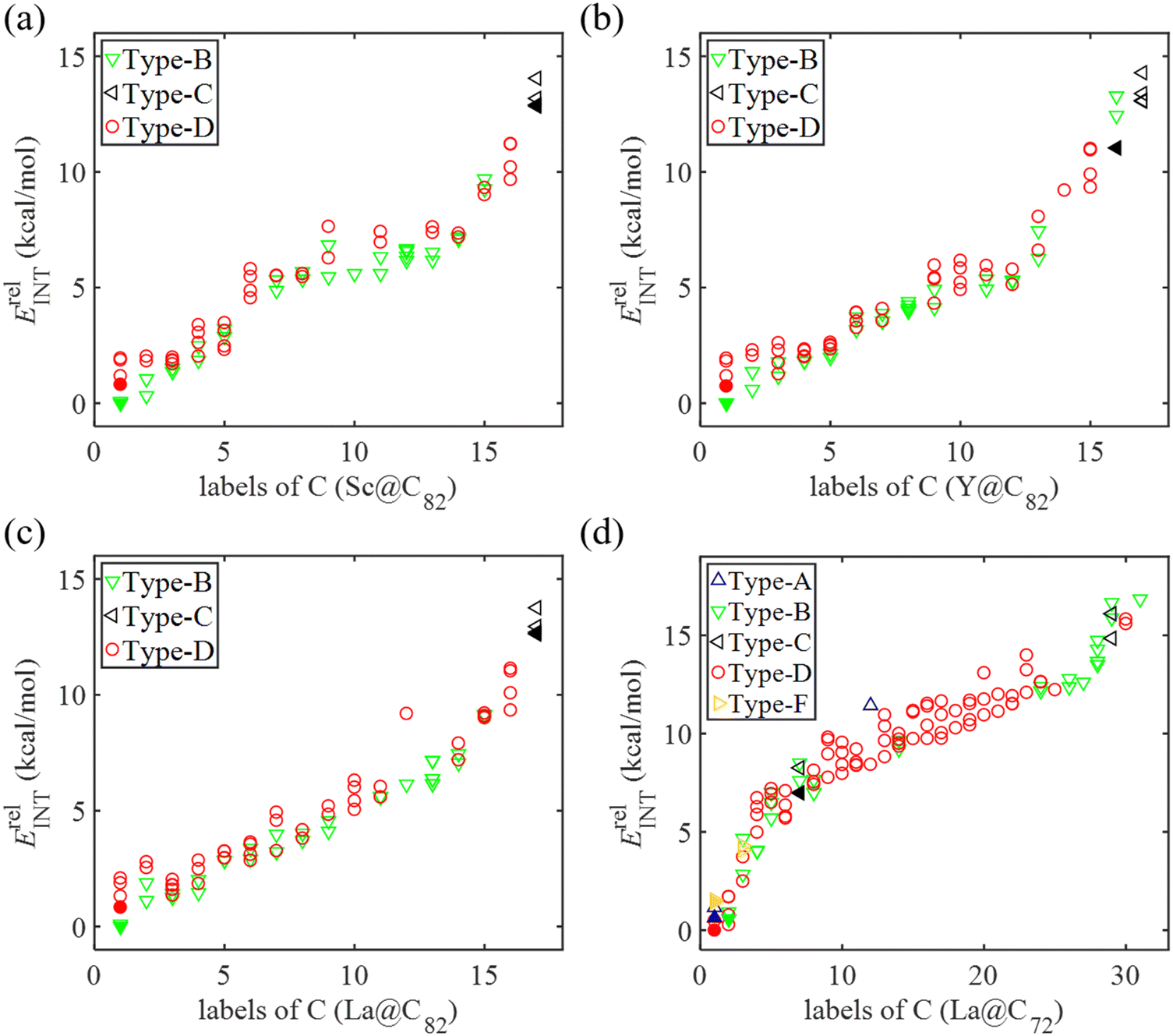 | ||
| Fig. 5 Relative energies of all potential intermediates (INTs) ErelINT for the Diels–Alder reaction between Cp and (a) Sc@C82, (b) Y@C82, (c) La@C82, and (d) La@C72 through the stepwise mechanism. The relative energies are given in kcal mol−1. | ||
3.4 Systematic exploration of the concerted and stepwise mechanisms
Fig. 6 and 7 depict the Gibbs free energy profiles for the cycloaddition of Cp to EMFs in this study, illustrating the lowest energy pathways for different bond types through both the concerted and the stepwise mechanisms, respectively. When Cp reacts with Sc, Y, and La@C82 through the concerted mechanism, the preferred addition sites for each bond type (A, B, and D) are identical, as shown in Fig. 6. This consistency implies that these metals exert a similar influence on the regioselectivity of the reactions, suggesting a predictable pattern in their chemical behavior. The results reveal that the activation energies for the addition reactions occurring at the type D bond C8–C15 are very close to those at the type A bond C1–C3 for Sc, Y, and La@C82, with differences being smaller than 0.4 kcal mol−1. This indicates that kinetically, the addition processes at these sites are nearly equivalent. However, the differences in reaction energies are considerably larger, with values of −3.4, −3.3, and −1.7 kcal mol−1 for Sc, Y, and La@C82, respectively. These significant differences in reaction energies highlight that thermodynamically, the additions at the type D bond are more favorable than those at the type A bond. Moreover, the additions to the type B bond C45–C56 exhibit the highest activation energies (15.8, 13.9, and 16.5 kcal mol−1 for Sc, Y, and La@C82, respectively), indicating that these reactions are the most kinetically hindered. Additionally, the reactions at the type B bond are also the least exothermic, making them less favorable from both kinetic and thermodynamic perspectives.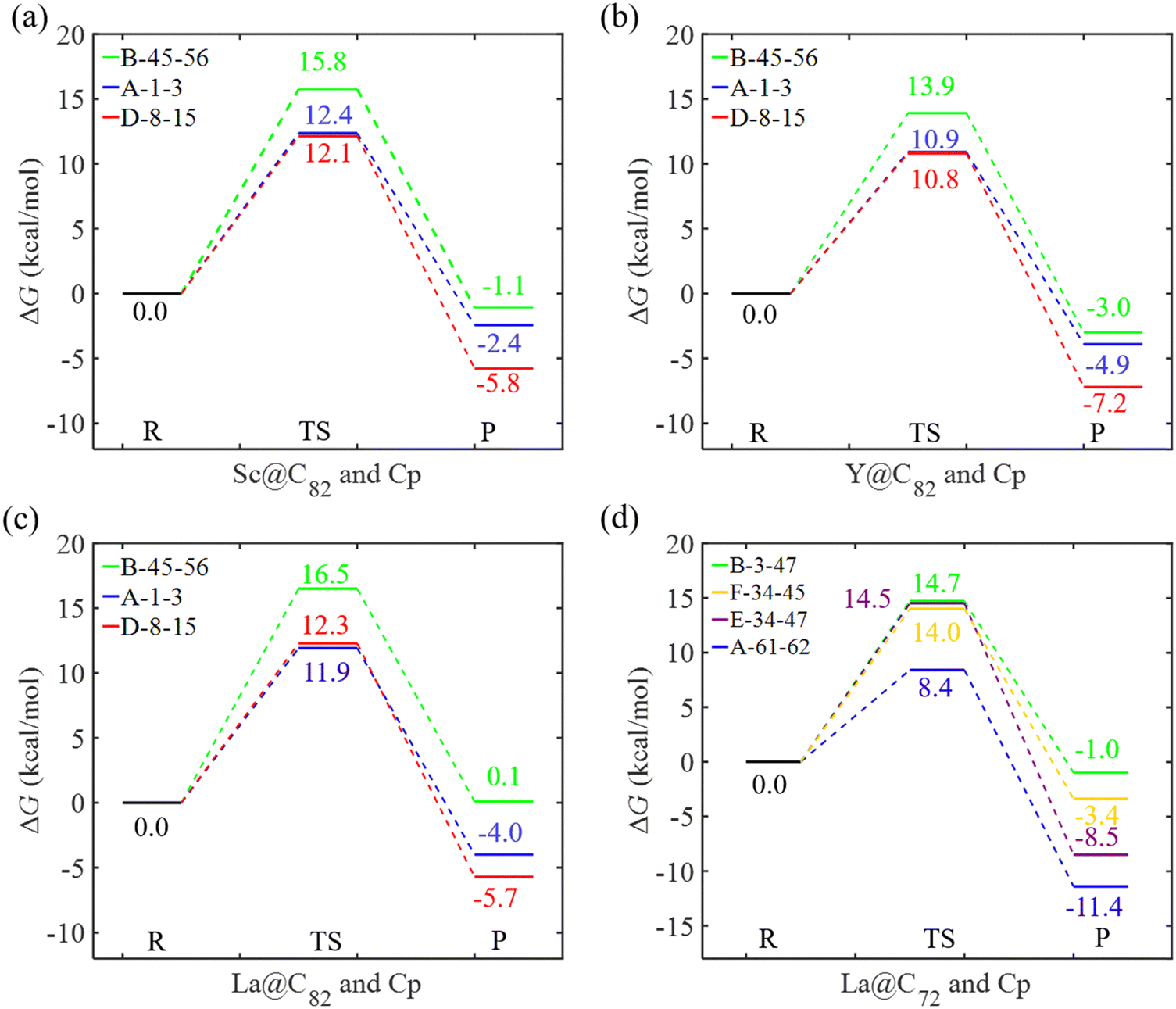 | ||
| Fig. 6 Gibbs free energy profile for the Diels–Alder reaction between Cp and (a) Sc@C82, (b) Y@C82, (c) La@C82, and (d) La@C72via the concerted mechanism in the gas phase. The relative energies are given in kcal mol−1. | ||
 | ||
| Fig. 7 Gibbs free energy profile for the Diels–Alder reaction between Cp and (a) Sc@C82, (b) Y@C82, (c) La@C82, and (d) La@C72via the stepwise mechanism in the gas phase. The relative energies are given in kcal mol−1. | ||
When the DA reaction proceeds through the stepwise mechanism, the free energy barriers and the reaction energies exhibit the following trends: type B bond < type D bond < type C bond. In particular, the energy barrier for the rate-determining step of the addition on the type B bond C7–C13 is 11.5, 10.0 and 11.7 kcal mol−1 for Sc, Y, and La@C82, respectively, which is slightly lower than the corresponding lowest free energy barrier for concerted pathways (12.1, 10.8, and 11.9 kcal mol−1). This indicates that the addition on the type B bond is slightly more favorable when proceeding via the stepwise mechanism compared to the concerted mechanism. The addition on the type C bond demonstrates the highest activation energies and reaction energies through the stepwise mechanism, specifically on C13–C6 for Sc@C82, and on C13–C22 for Y and La@C82. In contrast to the concerted mechanism, the reaction free energies following the stepwise mechanism are mostly positive, which indicates that the cycloadditions are endergonic through the stepwise mechanism. Therefore, accounting for both kinetic and thermodynamic results, the regioselectivity of the DA reaction between Cp and M@C82 (M = Sc, Y, and La) favors the attack on the [5,6] bond C8–C15 through the concerted mechanism. This preference aligns with the regioselectivity observed in the DA reaction between Cp* and La@C82,26 and is also consistent with the theoretical findings regarding the regioselectivity between Cp and La@C82.25
For the DA reactions of La@C72 with the non-IPR carbon cage, the transition state of the concerted addition to the selected type D bond could not be located. The relative energy of the related adduct on this bond was 13.2 kcal mol−1 (as shown in Fig. 4d), and the corresponding dihydride is the most stable among all the dihydrides added to the same type of bonds (see Fig. S2, ESI†). Thus, Cp cycloadditions on type D bonds were not explored through the concerted mechanism for La@C72. When examining the concerted mechanism for Cp's attack on various bonds in C72, the energy barrier for the attack on the type A bond (C61–C62) was found to be 8.4 kcal mol−1. This barrier is notably lower than those for attacks on types B, E, and F bonds, which have barriers of 14.7, 14.5, and 14.0 kcal mol−1 respectively, as shown in Fig. 6d. This pathway not only has the lowest energy barrier but is also the most exergonic, with a reaction free energy of −11.4 kcal mol−1. Through the stepwise mechanism, the kinetically most favored additions are on types D (C33–C32) and A (C33–C7) bonds with similar activation energies (12.3 and 12.6 kcal mol−1), followed in sequence by the additions on types F, B, and C bonds. The reactions of the cycloadditions on types D and F are exergonic, with reaction free energies −1.8 and −2.6 kcal mol−1, respectively. In contrast, the reactions involving types A, B, and C bonds are endergonic, with reaction energies ranging from 2.7 to 10.7 kcal mol−1. Therefore, when taking into account both kinetic and thermodynamic aspects, the most favored pathway is the concerted addition of Cp to the type A bond (C61–C62) of La@C72.
As primary solvents in DA reactions involving EMFs, toluene and ortho-dichlorobenzene (o-DCB) were also examined for their effects on the regioselectivity of the DA reaction, as outlined in Table 2 and Table S1 (ESI†). These results show that in both toluene and o-DCB, the activation energies and reaction energies increased to different extents for most of the selected pathways as shown in Table S2 (ESI†), which indicates that the solvent environment significantly influenced the energetics of the reaction. The concerted reactions even became thermodynamically unfavorable with reaction energies greater than 1.8 kcal mol−1, especially for types A and B bonds in Sc and Y@C82, as well as the type B bond in La@C82. For the stepwise addition, the activation energies on the type B bond (C7–C13) were slightly lower compared to the activation energies for the concerted addition on the D bond (C8–C15) for M@C82. However, the Gibbs reaction energies for the stepwise addition were all larger than 1.1 kcal mol−1, indicating that even though the activation barrier might be lower, the overall reaction remained less favorable energetically. An exception was observed for the stepwise addition on La@C82 in toluene, which had a Gibbs reaction energy of −0.5 kcal mol−1, suggesting a slight thermodynamic favorability in this specific case. For La@C72, the concerted attacks of Cp on types B and F bonds were found to be endergonic in both toluene and o-DCB. Conversely, the concerted attacks on types A and E bonds were exergonic. In o-DCB, the kinetically most favored pathway was the concerted addition on the bond C61–C62, with Gibbs energy barriers of 11.7 kcal mol−1, which was also the most exergonic reaction with a reaction energy of −7.4 kcal mol−1, indicating both kinetic and thermodynamic favorability. In toluene, the kinetically most favored pathway was also the concerted addition on the C61–C62 bond with Gibbs energy barriers of 11.6 kcal mol−1. However, the corresponding reaction energy was −7.1 kcal mol−1, highlighting strong thermodynamic favorability as well. Interestingly, the thermodynamically most favored pathway in toluene was the concerted addition on the C34–C47 bond, which suggests that different solvent environments can shift the balance between kinetic and thermodynamic control of the reaction.
| Reactants | Machanisma | Typeb | No. C–C | ΔG‡ (ΔGR) | ΔG‡ (ΔGR) | ΔG‡ (ΔGR) |
|---|---|---|---|---|---|---|
| Gas | Toluene | o–DCB | ||||
| a Concerted (C) or stepwise (S) mechanism for the DA cycloadditions. b Different bond types. The Gibbs free energy barriers and reaction energies are given in kcal mol−1. | ||||||
| Sc@C82 + Cp | C | D | 8–15 | 12.1 (−5.8) | 16.5 (−0.4) | 16.2 (−1.3) |
| S | B | 7–13 | 11.4, 11.3 (0.6) | 15.0, 14.9 (5.6) | 14.8, 14.7 (4.7) | |
| Y@C82 + Cp | C | D | 8–15 | 10.8 (−7.2) | 16.2 (−1.6) | 15.6 (−2.3) |
| S | B | 7–13 | 10.3, 10.2 (−0.9) | 15.3, 14.8 (5.5) | 14.9, 14.3 (4.2) | |
| La@C82 + Cp | C | D | 8–15 | 12.4 (−5.7) | 12.4 (−5.9) | 13.3 (−5.1) |
| S | B | 7–13 | 11.5, 11.7 (1.0) | 10.5, 9.0 (−0.5) | 11.9, 11.2 (1.1) | |
| La@C72 + Cp | C | A | 61–62 | 8.4 (−11.4) | 11.6 (−7.1) | 11.7 (−7.4) |
| S | D | 33–32 | 12.3, 10.6 (−2.6) | 16.9, 16.2 (3.1) | 16.6, 15.0 (2.0) | |
4. Conclusion
Systematic investigations of the DA cycloaddition reactions between cyclopentadiene (Cp) and the paramagnetic EMFs M@C82 (M = Sc, Y, and La) as well as La@C72 were conducted using density functional theory (DFT). Both the concerted and stepwise mechanisms were considered, providing comprehensive insights into the reaction pathways, regioselectivity, and solvent effects. The most reactive bonds for the concerted addition mechanism were identified using a fundamental hydride model, which allowed for the prediction of the most favorable sites based on the corresponding relative dihydride energies. For the stepwise addition mechanism, intermediates were screened according to their relative energies to determine the most likely addition sites. For M@C82, the concerted addition on the type D bond (C8–C15) was found to be both kinetically and thermodynamically favorable, with activation barriers smaller than 12.1 kcal mol−1 and reaction free energies smaller than −5.7 kcal mol−1. This suggests that the concerted mechanism is the predominant pathway for these reactions, despite the slightly higher activation barriers compared to the stepwise addition (difference less than 0.8 kcal mol−1). Moreover, most stepwise additions were found to be endergonic, further supporting the preference for the concerted pathway. In the case of La@C72, despite two of the selected pathways being exergonic via the stepwise addition (types D and F bonds), the concerted addition on the C61–C62 bond emerged as the main product due to its lowest activation energy (8.4 kcal mol−1) and highest exothermicity (reaction energy of −11.4 kcal mol−1). The interaction region indicator (IRI) distribution isosurfaces,53 mapped with the sign(λ2)ρ function, were computed using the Multiwfn program54 for the main adduct on C8–C15 in M@C82 and on C61–C62 in La@C72. Our calculations indicate that repulsive interactions between the metal atom and the carbon cage exist (see Fig. S4, ESI†), which are likely due to Pauli repulsion between the electron clouds. When the encapsulated metal atom loses its valence electrons, it becomes a positively charged atomic core, leading to a contraction of its electron cloud (with low polarizability). Since the encapsulation of metal atoms in EMFs is a strongly exothermic process, it can be concluded that the nature of the interaction is predominantly ionic, driven by electrostatic attraction, which exceeds Pauli repulsion.Additionally, this study examined the effects of solvents such as toluene and o-DCB, which were found to increase both activation energies and reaction energies for most pathways. Although the activation energy barriers were a little higher than those of the lowest stepwise pathways, the addition on the type D bond (C8–C15) still generated the major product for M@C82 thermodynamically. For La@C72, the regioselectivity observed in o-DCB remained consistent with that in the gas phase. However, in toluene, the thermodynamically most favored pathway became the concerted addition on bond C34–C47.
In summary, Sc, Y, and La do not significantly influence the regioselectivity of the reaction, as the regioisomers of the main product, whether formed through a concerted or stepwise mechanism, are identical for all three metals. Furthermore, while the barriers of the rate-determining step vary slightly with different metals, the relative trends depend on the reaction medium, showing variations in vacuum, toluene, and o-DCB.
Data availability
The data are available from the corresponding author upon reasonable request.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (22073080).References
- A. A. Popov, S. Yang and L. Dunsch, Chem. Rev., 2013, 113(8), 5989–6113 CrossRef PubMed.
- J. Zhao, X. Huang, P. Jin and Z. Chen, Coord. Chem. Rev., 2015, 289–290, 315–340 CrossRef.
- L. Bao, P. Peng and X. Lu, Acc. Chem. Res., 2018, 51(3), 810–815 CrossRef PubMed.
- A. Rodríguez-Fortea, A. L. Balch and J. M. Poblet, Chem. Soc. Rev., 2011, 40(7), 3551–3563 RSC.
- H. W. Kroto, Nature, 1987, 329(6139), 529–531 CrossRef.
- C.-R. Wang, T. Kai, T. Tomiyama, T. Yoshida, Y. Kobayashi, E. Nishibori, M. Takata, M. Sakata and H. Shinohara, Nature, 2000, 408(6811), 426–427 CrossRef.
- S. Stevenson, P. W. Fowler, T. Heine, J. C. Duchamp, G. Rice, T. Glass, K. Harich, E. Hajdu, R. Bible and H. C. Dorn, Nature, 2000, 408(6811), 427–428 CrossRef CAS PubMed.
- F. Liu, S. Wang, C.-L. Gao, Q. Deng, X. Zhu, A. Kostanyan, R. Westerström, F. Jin, S.-Y. Xie, A. A. Popov, T. Greber and S. Yang, Angew. Chem., Int. Ed., 2017, 56(7), 1830–1834 CrossRef CAS PubMed.
- N. Chen, C. M. Beavers, M. Mulet-Gas, A. Rodríguez-Fortea, E. J. Munoz, Y.-Y. Li, M. M. Olmstead, A. L. Balch, J. M. Poblet and L. Echegoyen, J. Am. Chem. Soc., 2012, 134(18), 7851–7860 CrossRef CAS.
- A. Muñoz-Castro and R. B. King, J. Comput. Chem., 2017, 38(19), 1661–1667 CrossRef.
- A. Miralrio and L. Enrique Sansores, Int. J. Quantum Chem., 2017, 117(6), e25335 CrossRef.
- A. Muñoz-Castro and R. Bruce King, J. Comput. Chem., 2017, 38(1), 44–50 CrossRef.
- C. A. Celaya, M. Reina, J. Muñiz and L. E. Sansores, ChemistrySelect, 2018, 3(24), 6791–6801 CrossRef CAS.
- A. Miralrio, A. Muñoz-Castro, R. B. King and L. E. Sansores, J. Phys. Chem. C, 2018, 122(1), 798–807 CrossRef CAS.
- C. A. Celaya, J. Muñiz and L. E. Sansores, Comput. Theor. Chem., 2019, 1152, 7–19 CrossRef CAS.
- Y. Chai, T. Guo, C. Jin, R. E. Haufler, L. P. F. Chibante, J. Fure, L. Wang, J. M. Alford and R. E. Smalley, J. Phys. Chem., 1991, 95(20), 7564–7568 CrossRef CAS.
- T. Suzuki, Y. Maruyama, T. Kato, K. Kikuchi and Y. Achiba, J. Am. Chem. Soc., 1993, 115(23), 11006–11007 CrossRef CAS.
- M. Takata, B. Umeda, E. Nishibori, M. Sakata, Y. Saitot, M. Ohno and H. Shinohara, Nature, 1995, 377(6544), 46–49 CrossRef CAS.
- J. Ding, L.-T. Weng and S. Yang, J. Phys. Chem., 1996, 100(26), 11120–11121 CrossRef CAS.
- J. Ding and S. Yang, J. Am. Chem. Soc., 1996, 118(45), 11254–11257 CrossRef CAS.
- J. Ding, N. Lin, L.-T. Weng, N. Cue and S. Yang, Chem. Phys. Lett., 1996, 261(1), 92–97 CrossRef CAS.
- E. Nishibori, M. Takata, M. Sakata, M. Inakuma and H. Shinohara, Chem. Phys. Lett., 1998, 298(1), 79–84 CrossRef CAS.
- Y. Maeda, J. Miyashita, T. Hasegawa, T. Wakahara, T. Tsuchiya, T. Nakahodo, T. Akasaka, N. Mizorogi, K. Kobayashi, S. Nagase, T. Kato, N. Ban, H. Nakajima and Y. Watanabe, J. Am. Chem. Soc., 2005, 127(35), 12190–12191 CrossRef PubMed.
- Y. Maeda, S. Sato, K. Inada, H. Nikawa, M. Yamada, N. Mizorogi, T. Hasegawa, T. Tsuchiya, T. Akasaka, T. Kato, Z. Slanina and S. Nagase, Chem. – Eur. J., 2010, 16(7), 2193–2197 CrossRef PubMed.
- M. Garcia-Borràs, J. M. Luis, M. Swart and M. Solà, Chem. – Eur. J., 2013, 19(14), 4468–4479 CrossRef PubMed.
- S. Sato, Y. Maeda, J.-D. Guo, M. Yamada, N. Mizorogi, S. Nagase and T. Akasaka, J. Am. Chem. Soc., 2013, 135(15), 5582–5587 CrossRef PubMed.
- L. Feng, T. Wakahara, T. Nakahodo, T. Tsuchiya, Q. Piao, Y. Maeda, Y. Lian, T. Akasaka, E. Horn, K. Yoza, T. Kato, N. Mizorogi and S. Nagase, Chem. – Eur. J., 2006, 12(21), 5578–5586 CrossRef PubMed.
- A. Rodríguez-Fortea, S. Irle and J. M. Poblet, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1(3), 350–367 Search PubMed.
- J. Sun and Y. Wang, Inorg. Chem., 2022, 61(48), 19183–19192 CrossRef CAS PubMed.
- M. Garcia-Borràs, S. Osuna, J. M. Luis, M. Swart and M. Solà, Chem. – Eur. J., 2013, 19(44), 14931–14940 CrossRef PubMed.
- P. Zhao, X. Zhao and M. Ehara, J. Org. Chem., 2016, 81(18), 8169–8174 CrossRef CAS.
- Z. Zhang, Y. Zhang, W. Xue, Z. Guo and P. Cui, ChemistrySelect, 2017, 2(28), 8880–8885 CrossRef CAS.
- T. Wakahara, H. Nikawa, T. Kikuchi, T. Nakahodo, G. M. A. Rahman, T. Tsuchiya, Y. Maeda, T. Akasaka, K. Yoza, E. Horn, K. Yamamoto, N. Mizorogi, Z. Slanina and S. Nagase, J. Am. Chem. Soc., 2006, 128(44), 14228–14229 CrossRef CAS.
- J. P. Perdew, Physical Review B: Condens. Matter Mater. Phys., 1986, 33(12), 8822–8824 CrossRef PubMed.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 34(10), 7406 CrossRef.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38(6), 3098–3100 CrossRef CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32(7), 1456–1465 CrossRef CAS PubMed.
- A. F. Sattarova, Y. N. Biglova and A. G. Mustafin, Int. J. Quantum Chem., 2022, 122(7), e26863 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7(18), 3297–3305 RSC.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8(9), 1057–1065 RSC.
- M. Dolg, U. Wedig, H. Stoll and H. Preuss, J. Chem. Phys., 1987, 86(2), 866–872 CrossRef CAS.
- P. Schwerdtfeger, M. Dolg, W. H. E. Schwarz, G. A. Bowmaker and P. D. W. Boyd, J. Chem. Phys., 1989, 91(3), 1762–1774 CrossRef CAS.
- L. Wang and Y. Wang, Inorg. Chem., 2020, 59(15), 10962–10975 CrossRef CAS PubMed.
- M. e Frisch; G. Trucks; H. B. Schlegel; G. Scuseria; M. Robb; J. Cheeseman; G. Scalmani; V. Barone; G. Petersson and H. Nakatsuji, Gaussian 16, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- Y. Wang, S. Díaz-Tendero, M. Alcamí and F. Martín, J. Am. Chem. Soc., 2017, 139(4), 1609–1617 CrossRef.
- Y. Wang, S. Díaz-Tendero, M. Alcamí and F. Martín, J. Chem. Theory Comput., 2018, 14(3), 1791–1810 CrossRef PubMed.
- C. Gonzalez and H. B. Schlegel, J. Chem. Phys., 1989, 90(4), 2154–2161 CrossRef.
- C. Gonzalez and H. B. Schlegel, J. Phys. Chem., 1990, 94(14), 5523–5527 CrossRef.
- E. Nishibori, M. Takata, M. Sakata, H. Tanaka, M. Hasegawa and H. Shinohara, Chem. Phys. Lett., 2000, 330(5), 497–502 CrossRef.
- G. S. Hammond, J. Am. Chem. Soc., 1955, 77(2), 334–338 CrossRef.
- M. G. Evans and M. Polanyi, Trans. Faraday Soc., 1938, 34, 11–24 RSC.
- P. Pla, Y. Wang and M. Alcamí, Phys. Chem. Chem. Phys., 2020, 22(16), 8846–8852 RSC.
- T. Lu and Q. Chen, Chem.: Methods, 2021, 1(5), 231–239 CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33(5), 580–592 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Geometrical structures of La@C2-C72 isomers, relative energies of dihydrides for M@C82 (M = Sc, Y, and La) and La@C72, intermediate conformers for the addition of Cp to M@C82 (M = Sc, Y, and La), Gibbs free energy barriers and reaction energies for the addition of Cp to M@C82 (M = Sc, Y, and La) and La@C72 in gas-phase, toluene, and o-DCB, Cartesian coordinates for most important structures. See DOI: https://doi.org/10.1039/d4cp02538e |
| This journal is © the Owner Societies 2024 |