
Theoretical study of covalent organic frameworks and heterojunctions for the oxygen reduction reaction†
Xiao-Xiao
Guo
a,
Jing-Hua
Guo
 *a,
Dong
Hou
a and
Gang
Chen
*b
*a,
Dong
Hou
a and
Gang
Chen
*b
aLaboratory of Advanced Materials Physics and Nanodevices, School of Physics and Technology, University of Jinan, Jinan, Shandong 250022, China. E-mail: sps_guojh@ujn.edu.cn
bSchool of Physics and Electronics, Shandong Normal University, Jinan, Shandong 250358, China. E-mail: phdgchen@sdnu.edu.cn
First published on 21st November 2024
Abstract
In this study, the catalytic potential of two representative covalent organic frameworks (COFs), COF-366 and Pc-PBBA COF, was investigated in the oxygen reduction reaction (ORR). Transition metal atoms (Fe or Co) were incorporated into the center of porphyrin or phthalocyanine rings to form Fe(Co)–N–C single-atom catalysts, which can catalyze the ORR with overpotentials between 0.49 V and 0.69 V. In order to improve the catalytic activity, we introduced graphene or C3N4 layers to build heterojunctions with the COFs. Charge density difference and Bader charge analysis demonstrated that the introduction of a graphene (or C3N4) layer increases the population of electrons on the Fe and Co ions, which facilitates the flow of electrons to the intermediates and promotes the ORR catalytic reaction. For COFs@graphene heterojunctions, the overpotential decreases by 10.9–20.3% for Fe-based active sites and 30.3–36.4% for Co-based active sites. For COFs@C3N4 heterojunctions, the overpotential decreases by 7.3–39.1% for Fe-based active sites and 15.116.3% for Co-based active sites. The analysis of the volcano plot reveals that the COF-366_Co@graphene heterojunction is positioned at the apex of the volcano plot with an overpotential of 0.31 V.
1. Introduction
With the increase in social needs, the excessive consumption of fossil fuels has triggered an energy crisis, climate change, and environmental pollution. Hydrogen is a clean energy source, and its combustion product is only water. Fuel cells are the best way to utilize hydrogen energy. The oxygen reduction reaction (ORR) occurring at the cathode is a sluggish reaction, which is one of the critical bottlenecks in fuel cells.1,2 Platinum group metals (e.g., platinum and palladium) are widely used as catalysts in the ORR due to their excellent catalytic performance. The high cost has prompted researchers to actively seek more cost-effective alternatives.3,4Covalent organic frameworks (COFs) are a type of crystalline porous polymer designed and synthesized by condensation reactions in 2005.5 In recent years, it has been reported that COFs have certain electrocatalytic ability due to their ligands containing N–C sites or Fe/Co–N–C sites.6,7 For instance, Hu et al. conducted a systematic study on the catalytic performance of Fe/Co–N–C with various active sites (FeCoN6, FeCoN7, and FeCoN8) for the ORR. They found that the top side of the Co atom is the primary active center for the ORR. By adjusting the atomic structure of the active sites, an extremely low overpotential of only 0.22 V can be achieved.8 Liu et al. developed two COF-based catalysts, CoTAPP-PATA-COF and CoTAPP-BDTA-COF, which both exhibited good ORR performance in basic media.9 Fang et al. prepared a large-area, self-supporting CoP-TOB COF, which showed excellent performance with a half-wave potential of 0.82 V in the ORR process.10 You et al. constructed two additional types of COFs (MBD-COF and DAF-COF) by introducing methyl groups (MGs). They confirmed that the carbon adjacent to the MGs serves as the active center for the ORR, which improves the kinetics of oxygen reduction.11
In recent years, many theoretical studies have been carried out to explore the electrocatalytic properties of COFs. Jin et al. performed first-principle calculations to investigate the electrocatalytic performance of two-dimensional M-COFs (M = Sc–Zn, Ru, Rh, Pd, Ag, Ir). They reported that Co-COF is a perfect ORR catalyst with a low overpotential (ηORR) of 0.23 V.12 The binding energy of the key intermediate species on the Co-porphyrin active center is optimal due to the suitable metal d bands, resulting in good ORR catalytic activity. Six homonuclear (2TM-COF) and fifteen heteronuclear (TM1TM2-COF) dual-atom catalysts (DACs) are designed by the Zhou group, among which RhIr-COF exhibited the best OER catalytic activity with an overpotential (ηOER) value of 0.29 V.13 It also revealed that the charge of the embedded dual-metal atoms is the most important factor governing the catalytic activity.
Combining density functional theory (DFT) with machine learning (ML) approaches, several TM atoms embedded on a 2D COF as single-atom catalysts are investigated, and the results demonstrated that the geometric configuration and the electronic properties mainly controlled the catalytic activity.14,15 Hu et al. found that the spin state of the active sites has a significant impact on catalytic performance. By appropriately adjusting the external field (such as adsorbed species) to change the spin state of transition metals, catalytic performance can be optimized, achieving the maximization of catalytic efficiency.16
However, pure COFs face some limitations in catalysis, such as active site deactivation, instability problems in aqueous solutions, and simple functionality, which may not meet the requirements of applications. Heterojunctions17,18 provide the advantages of synergistic effects, enhanced catalytic activity, and tailored electronic properties, which may address the limitations of pure COFs. Liu et al. designed a catalyst composed of core–shell carbon frameworks derived from COFs and metal–organic (MOFs). The synthetic COF@ZIF800 exhibited excellent bifunctional catalytic performance, and the half-wave potential for the ORR was 0.85 V in a 0.1 M KOH solution.19 Chen et al. utilized carbon nanotubes (CNTs) as the core and two-dimensional COFs as the shell to synthesize a novel one-dimensional van der Waals heterostructure, with an optimal half-wave potential of 0.83 V in the ORR.20
Herein, we selected two typical COFs (COF-366 and Pc-PBBA COF) to explore their potential for ORR catalytic applications.21,22 COF-366 is a porphyrin-derived COF that features a two-dimensional layered architecture with a pore diameter of about 20 Å and a Langmuir surface area of 506 m2 g−1. The Pc-PBBA COF has a square lattice made up of phthalocyanine macrocycles that are interconnected by phenylene bis(boronic acid) linkers. The pore width is approximately 20 Å, and the interlayer distance is 3.34 Å. Its Langmuir surface area and pore volume are 506 m2 g−1 and 0.258 cm3 g−1, respectively. The structural diagrams of COF-366 and PC-PBBA COF are shown in Fig. 1. A transition metal (Fe or Co) atom was embedded in the center of the porphyrin or phthalocyanine ring to form a Fe–N–C single-atom catalyst, and its ORR catalytic mechanism has been systematically investigated. Then we constructed the COFs_Fe(Co) heterostructures with graphene or C3N4 layers and explored their impact on the catalytic performance.
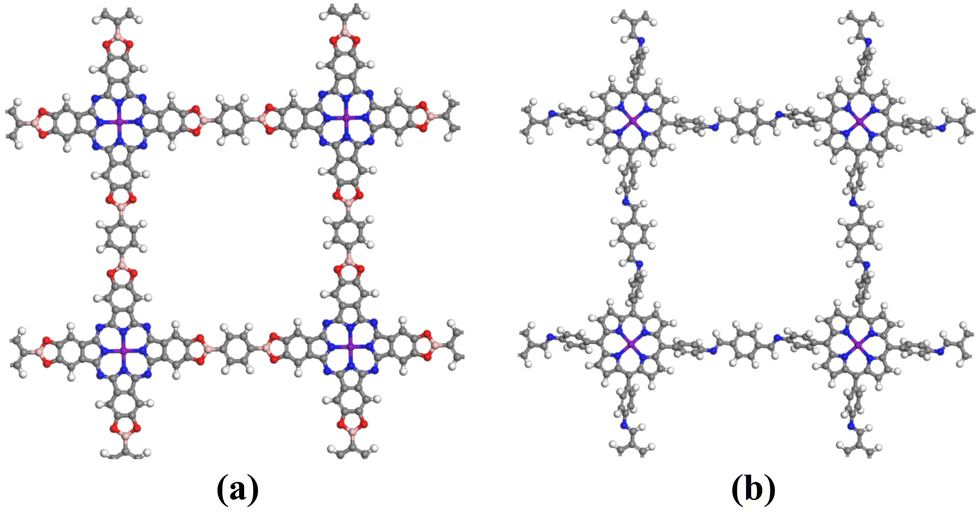 | ||
| Fig. 1 The structural diagrams of (a) Pc-PBBA COF and (b) COF-366. The gray, white, blue, red, pink, and purple spheres represent C, H, N, O, B, and X (X = Fe, Co) atoms, respectively. | ||
2. Method
In this study, we performed all computational analyses utilizing spin-polarized DFT implemented within the Vienna Ab initio Simulation Package (VASP).23 The electron–nucleus interactions are precisely described using the projector augmented wave (PAW) method.24 For the exchange and correlation functional, we employed the Perdew–Burke–Ernzerhof (PBE) functional within the framework of the generalized gradient approximation (GGA).25,26 Regarding the description of valence electrons, we employed the plane wave basis set with an energy cutoff of 500 eV. For geometric optimization, the Brillouin zone integration is performed with a 1 × 1 × 1 gamma K-point27 mesh. The convergence criteria of electronic and geometric optimization were determined as 1 × 10−5 eV for energy and 0.03 eV Å−1 for force, respectively. The computational details of the adsorption-free energies and reaction-free energies can be seen in the ESI.† The DFT-D3 method27 of Grimme with Becke–Jonson damping28 was employed to accurately describe the strong vdW interaction between the two counterparts of the heterostructure and the long-range electrostatic interactions of ORR species with catalysts. Meanwhile, the solvation effect was considered in the study, which was described by the implicit solvation model embedded in VASPsol, and the dielectric constant of water was set to 80.29 The projected density of states (PDOS) was calculated using the HSE06 hybrid functional,30,31 and a 3 × 3 × 1 k-point sampling was used. In the calculations of the spin moment, the DFT+U method was applied to the 3d orbital of Fe and Co to describe the strong Coulomb interaction.32 For iron and cobalt, we use U = 4 eV, J = 1 eV, and U = 3.3 eV, J = 0 eV, respectively, which is consistent with previous studies.33,343. Results and discussion
3.1 Exploration of the ORR Mechanism on COFs
In this work, the ORR mechanism was explored on Pc-PBBA COF and COF-366 embedded with Fe and Co atoms, which were labeled as Pc-PBBA COF_Fe, Pc-PBBA COF_Co, COF-366_Fe and COF-366_Co. To evaluate the stability of the materials, we calculated the binding energy of Fe and Co embedded in COF-366 and Pc-PBBA COF by the formula:| Eb = EM_X − EM − EX | (1) |
M denotes Pc-PBBA COF or COF-366. The symbol X represents transition metals of Fe and Co. EM_X, EM, and EX refer to the total energy of these structures in their most stable configurations. The calculated binding energies of the four materials Pc-PBBA COF_Fe, COF-366_Fe, Pc-PBBA COF_Co, and COF-366_Co are −4.62 eV, −3.50 eV, −8.68 eV and −8.50 eV, respectively. The binding energies are negative, indicating that the structures are stable. The total density of states (TDOS) of Pc-PBBA COF_Fe, COF-366_Fe, Pc-PBBA COF_Co, and COF-366_Co as shown in Fig. S1 (ESI†), and their bandgaps were calculated to be 1.20 eV, 1.79 eV, 1.44 eV, and 1.90 eV, respectively. These values are within the semiconductor range (0.1–3 eV), indicating that these materials have electrical conductivity and can be utilized as catalysts for fuel cells.
In the ORR reaction, it is acknowledged that there are two main pathways: a 2-electron path and a 4-electron path. The 2-electron path involves the reduction of O2 into H2O2 and its further reduction to H2O, while the 4-electron path directly reduces O2 into H2O. We studied the H2O2 adsorption on the COF surface, and the most stable adsorption configurations are shown in Fig. 2. Interestingly, it was found that H2O2 could only be adsorbed on Pc-PBBA COF_Co (Fig. 2b), which indicates that the ORR may occur along a 2-electron pathway. When loaded on the other three materials, it will be decomposed into an oxygen and water molecule, indicating that the formation of H2O2 can be inhibited. ORR is performed via the 4-electron pathway.
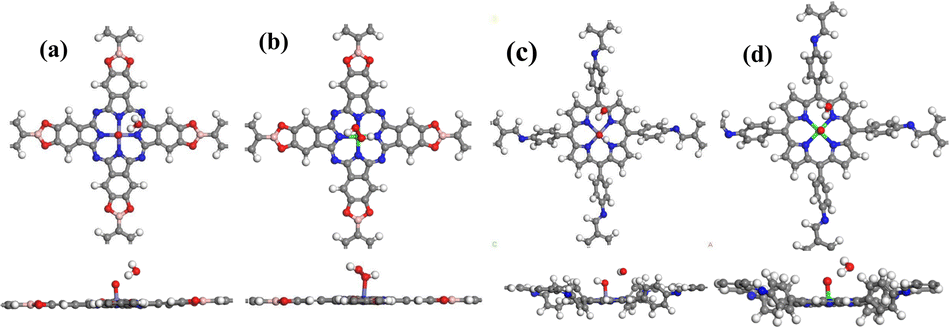 | ||
| Fig. 2 The adsorption model of H2O2 on (a) Pc-PBBA COF_Fe, (b) Pc-PBBA COF_Co, (c) COF-366_Fe and (d) COF-366_Co. The gray, white, blue, red, pink, purple and green spheres represent C, H, N, O, B, Fe and Co atoms, respectively. The top one is the top view, and the bottom one is the side view. | ||
Because the four-electron reduction is widely regarded as the most efficient route for the ORR, we focused on exploring the feasibility of ORR reactions occurring in this pathway. Within a stepwise hydrogenation mechanism, an in-depth assessment of the adsorption characteristics of the key intermediates (*O2, *OOH, *OH, and *O) has been conducted. The adsorption configurations and the adsorption enthalpies (ΔH) are depicted in Fig. 3, which confirms the structural stability and provides valuable insights into the catalytic process. The adsorption enthalpy (ΔH) at T = 0 K was calculated from
| ΔH = ΔE +ΔZPE | (2) |
| ΔE = EM_X*n − EM_X − En | (3) |
| ΔZPE = ZPEM_X*n − ZPEM_X − ZPE*n | (4) |
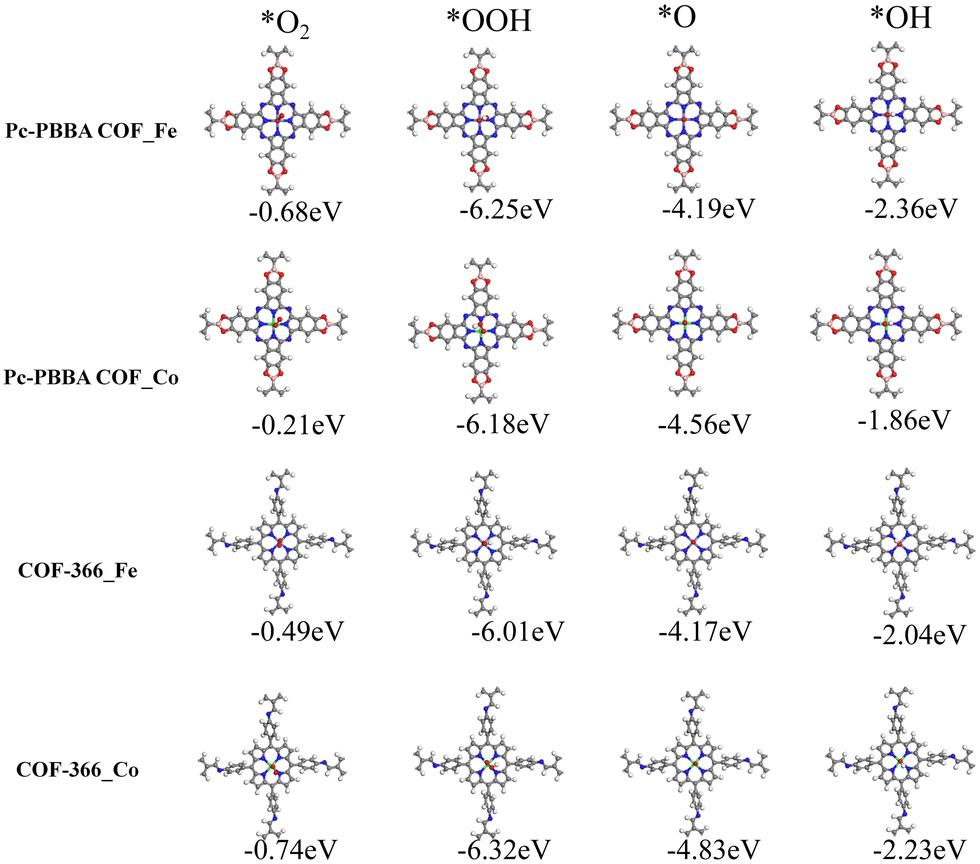 | ||
| Fig. 3 The stable adsorption configurations of four intermediate (*O2, *OOH, *O, *OH) on Pc-PBBA COF_Fe (Co) and COF-366_Fe (Co), respectively. The gray, white, blue, red, pink, purple and green spheres represent C, H, N, O, B, Fe and Co atoms, respectively. The numbers below the structure indicate the adsorption enthalpies. | ||
The adsorption free energies of the four pivotal intermediates are shown in Fig. 4. The reaction steps are downhill at the electrode potential of U = 0 V, indicating the spontaneity of the ORR. At U = 1.23 V, the initial electron and proton transfer step for the Pc-PBBA COF_Co, COF-366_Fe, and COF-366_Co sites is notably sluggish, which indicates that the formation of *OOH intermediates predominantly constrains the ORR process. At these three active sites, the first reaction step is identified as the rate-determining step, which significantly influences the catalytic efficacy of the electrocatalyst. For Pc-PBBA COF_Fe site, the third step exhibits the most pronounced deceleration, which is the rate-determining step for this catalyst. The overpotential values for Pc-PBBA COF_Fe, COF-366_Fe, Pc-PBBA COF_Co, and COF-366_Co are 0.55, 0.69, 0.66, and 0.49 V, respectively. These values are still higher than the previous results reported for platinum-based catalysts, which are approximately 0.25 V.35
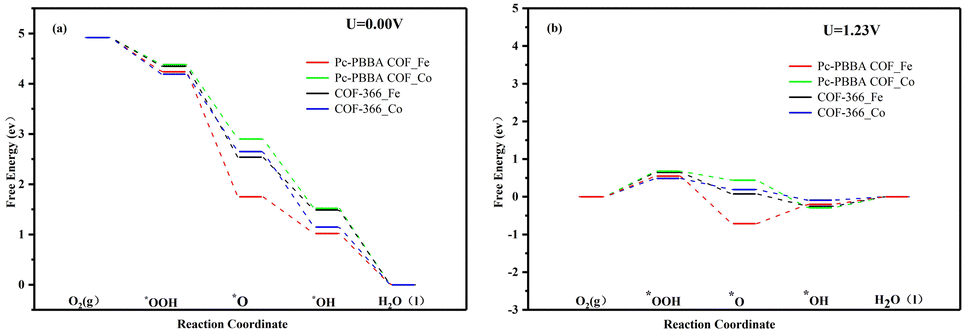 | ||
| Fig. 4 The free energy diagrams of the ORR pathways on Pc-PBBA COF_Fe (Co) and COF-366_Fe (Co) sites were calculated at (a) U = 0 V and (b) U = 1.23 V. | ||
The ORR catalytic activity mainly derives from electrons close to the Fermi level (Ef), which can be efficiently transported from the active site to the intermediate product.36Fig. 5 shows the PDOS of these four materials and demonstrates that the electronic states near Ef are mainly contributed by the 3d orbital of Fe or Co atoms. This indicates that the catalytic activity mainly originates from the central Fe or Co element.
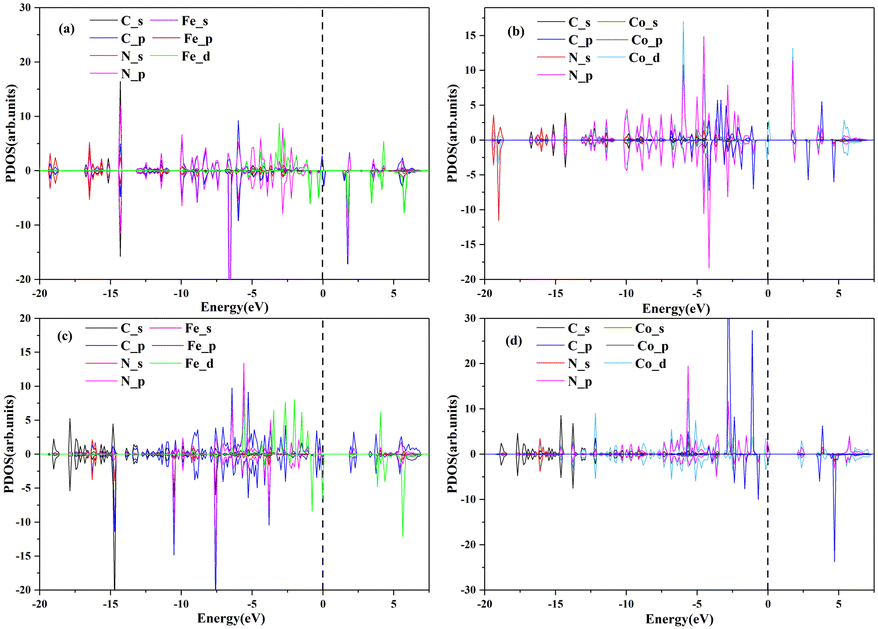 | ||
| Fig. 5 The PDOS of (a) Pc-PBBA COF_Fe, (b) Pc-PBBA COF_Co, (c) COF-366_Fe and (d) COF-366_Co structures. The dashed line indicates the Fermi level. | ||
3.2 Heterostructures comprised of the COFs_Fe(Co) with graphene
Graphene (GP), a versatile two-dimensional carbon nanomaterial, is composed of carbon's sp2 hybrid orbitals in a hexagonal honeycomb lattice. Remarkably, it serves as an exemplary catalyst carrier.37 Given that the catalytic ability of COFs_Fe(Co) is not yet up to the level of Pt-based catalysts, heterostructures were constructed by combining with graphene to explore the design method of catalysts with higher activity.Based on the primary cell of graphene, we first constructed a tetragonal supercell structure with a cell parameter around 22 A, and then compounded it with the COF structure to construct a bilayer heterojunction structure. The lattice mismatch rates for the heterojunctions Pc-PBBA COF_Fe@GP, Pc-PBBA COF_Co@GP, COF-366_Fe@GP, and COF-366_Co@GP are 2.63%, 1.56%, 2.51%, and 1.97%, respectively. These all fall within the range of low mismatch rates, which are considered favorable for forming stable heterostructures, helping to reduce lattice defects at the interfaces and thereby enhancing the catalytic performance of the materials. A vacuum layer of 15 Å was added to remove the influence of periodic boundary conditions on the surface model. The stable heterojunction structures were then obtained based on DFT optimization without any symmetry constrains. The heterostructures comprised of COFs_Fe(Co) with graphene are shown in Fig. 6.
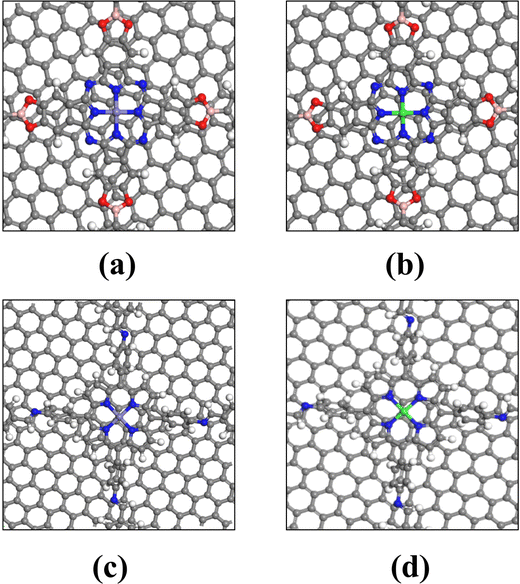 | ||
| Fig. 6 The heterojunction structure diagram of (a) Pc-PBBA COF_Fe@GP, (b) Pc-PBBA COF_Co@GP, (c) COF-366_Fe@GP, and (d) COF-366_Co@GP. The gray, white, blue, red, pink, purple and green spheres represent C, H, N, O, B, Fe and Co atoms, respectively. | ||
Table 2 lists the adsorption free energies of the intermediates on each heterojunction, and the free energy diagrams of the ORR pathways are shown in Fig. 7. All reaction steps display a downward trend at U = 0 V, indicating spontaneous ORR reactions. Comparison with Table 1 reveals that the adsorption free energy is reduced at Pc-PBBA COF_Fe and Pc-PBBA COF_Co active sites. In particular, the reaction energy of the first electron transfer step is reduced, which results in a much lower overpotential for the reaction.
| Structures | ΔG*O2 | ΔG*OOH | ΔG*O | ΔG*OH | η ORR |
|---|---|---|---|---|---|
| Pc-PBBA COF_Fe | 4.49 | 4.25 | 1.75 | 1.02 | 0.55 |
| COF-366_Fe | 4.59 | 4.35 | 2.53 | 1.48 | 0.69 |
| Pc-PBBA COF_Co | 4.70 | 4.37 | 2.89 | 1.52 | 0.66 |
| COF-366_Co | 4.39 | 4.18 | 2.65 | 1.15 | 0.49 |
| Structures | ΔG*O2 | ΔG*OOH | ΔG*O | ΔG*OH | η ORR |
|---|---|---|---|---|---|
| Pc-PBBA COF_Fe@GP | 4.39 | 3.99 | 1.58 | 0.84 | 0.49 |
| COF-366_Fe@GP | 4.47 | 3.97 | 1.54 | 0.87 | 0.55 |
| Pc-PBBA COF_Co@GP | 4.62 | 4.15 | 2.67 | 1.25 | 0.46 |
| COF-366_Co@GP | 4.49 | 4.28 | 2.68 | 1.36 | 0.31 |
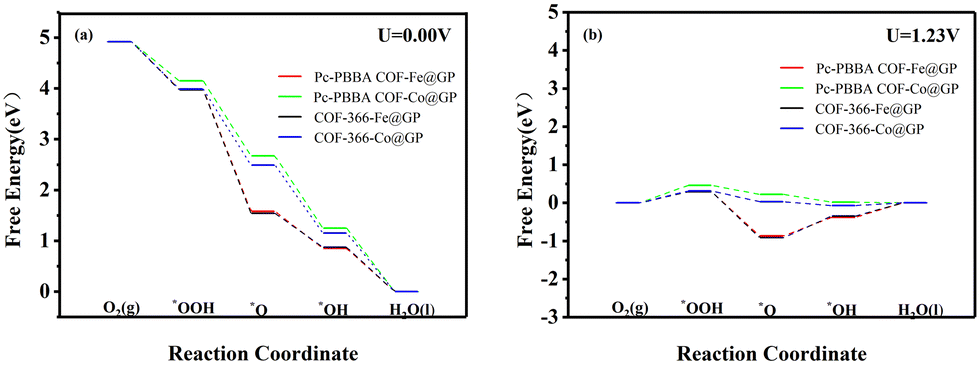 | ||
| Fig. 7 The free energy diagrams of the ORR pathways on Pc-PBBA COF_X@GP and COF-366_X@GP (X = Fe, Co) sites were calculated at (a) U = 0 V and (b) U = 1.23 V. | ||
Subsequently, we explored the effect of graphene layers on the charge distribution in the vicinity of the Fe (Co) active sites. Fig. 8 represents the calculated charge density difference on the heterojunction. It can be seen that the electrons are transferred from graphene to the Fe- (or Co-) active site. Bader charge analysis38 reveals that about 1.05–1.29 electrons were transferred to Fe ions, resulting in the conversion of the active site from Fe2+ to Fe+. About 0.18–0.26 electrons are transferred to Co ions, so the valence state of Co+ remains essentially unchanged. The introduction of a graphene layer increases the population of electrons on the Fe and Co ions, which facilitates the flow of electrons to the intermediates and can promote the ORR catalytic reaction. Meanwhile, Bader charge analysis shows that only 0.01–0.13 electrons were transferred from graphene to the COF layer after the heterojunction was constructed, which demonstrates that some electrons are also transferred from the COF layer to the graphene layer. Charge density difference also reveals that, at the periphery of the active sites, electrons are transferred from the COF layer to the graphene layer.
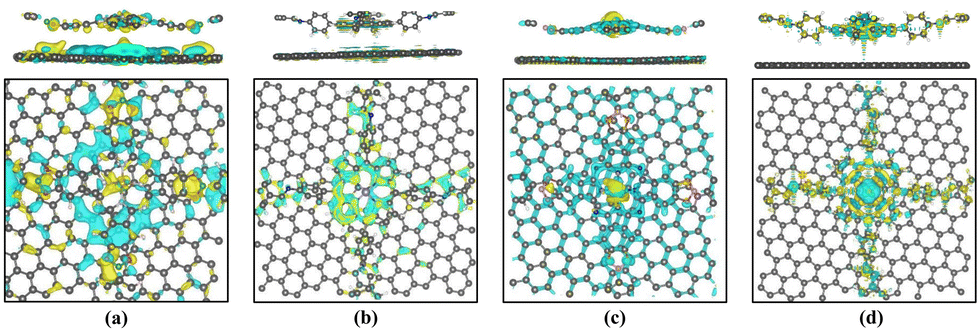 | ||
| Fig. 8 Charge density difference of (a) Pc-PBBA COF_Fe@GP, (b) COF-366_Fe@GP, (c) Pc-PBBA COF_Co@GP, and (d) COF-366_Co@GP. The yellow and cyan colors represent electron accumulation and depletion regions, respectively. The isosurface level is set to 0.007 e Bohr−3. | ||
Due to the transfer of charge between the bilayer materials, a built-in electric field is formed between the heterojunctions. We calculated the electrostatic field in the vicinity of the active site of Fe(Co) based on the distribution of electrostatic potentials. Since the intermediate species of the ORR adsorbed at 1.5–3.5 Å from the active site, Fig. 9 shows the electric field distribution profile in this region. The electric field strength is between 2.0 × 109 V m−1–7.0 × 1010 V m−1. The formation of a built-in electric field promotes the transfer ability of electrons and facilitates the catalytic reaction, which is consistent with the past study.39
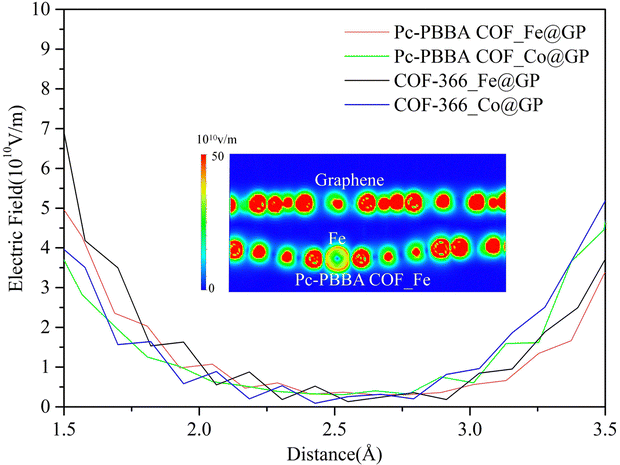 | ||
| Fig. 9 The electric field distribution profile in the vicinity of the active site of Fe(Co). The inset shows an isosurface map of the electric field distribution inside the Pc-PBBA COF_Fe@GP heterojunction. | ||
3.3 Heterostructures composed of the COFs_Fe(Co) with C3N4
Carbon nitride (C3N4) has shown significant potential in photocatalysts, fuel cell electrodes, light-emitting devices, and chemical sensors, which has sparked our interest. We jointly constructed a heterojunction with COFs_Fe(Co) and C3N4, and its structural models are shown in Fig. 10. The lattice mismatch rates for Pc-PBBA COF_Fe@C3N4, Pc-PBBA COF_Co@C3N4, COF-366_Fe@C3N4, and COF-366_Co@C3N4 are 2.17%, 1.78%, 2.05%, and 1.73%, respectively. The adsorption-free energies of the intermediates are shown in Table 3, and the free energy diagrams of the ORR pathways are depicted in Fig. 11. It's evident from the table that all the activation-free energies of the intermediates are positive, indicating endothermic reactions. All the reaction steps exhibit a decreased trend at U = 0 V, revealing the spontaneous nature of the ORR reactions for these structures. At U = 1.23 V, for COF-366_Co@C3N4, the notable fourth electron transfer step influences the catalyst's performance the most. This step directly determines the electrocatalysts' catalytic ability. Compared to the pure COF-366_Co, the overpotential decreases from 0.49 to 0.41 V. Similarly, for the Pc-PBBA COF_Fe, Pc-PBBA COF_Co and COF-366_Fe materials, the formation of the C3N4 heterojunction makes the occurrence of the rate-limiting step easier and the overpotential is decreased to 0.51, 0.56 and 0.42 V, respectively.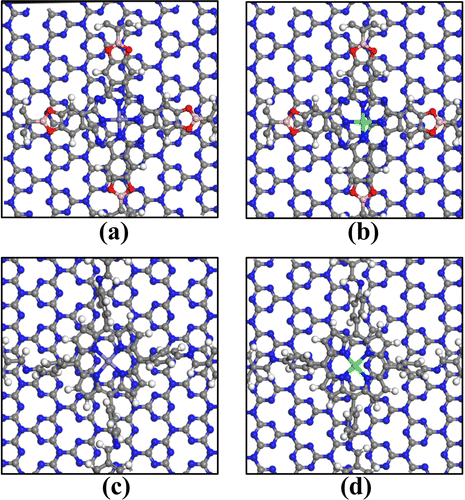 | ||
| Fig. 10 The heterojunction structure diagram of (a) Pc-PBBA COF_Fe@C3N4, (b) Pc-PBBA COF_Co@C3N4, (c) COF-366_Fe@C3N4 and (d) COF-366_Co@C3N4. The gray, white, blue, red, pink, purple and green spheres represent C, H, N, O, B, Fe and Co atoms, respectively. | ||
| Structures | ΔG*O2 | ΔG*OOH | ΔG*O | ΔG*OH | η ORR |
|---|---|---|---|---|---|
| Pc-PBBA COF_Fe@C3N4 | 4.32 | 4.20 | 3.16 | 1.49 | 0.51 |
| COF-366_Fe@C3N4 | 4.50 | 4.11 | 2.65 | 1.38 | 0.42 |
| Pc-PBBA COF_Co@C3N4 | 4.37 | 4.25 | 3.30 | 1.46 | 0.56 |
| COF-366_Co@C3N4 | 4.27 | 4.10 | 2.65 | 1.39 | 0.41 |
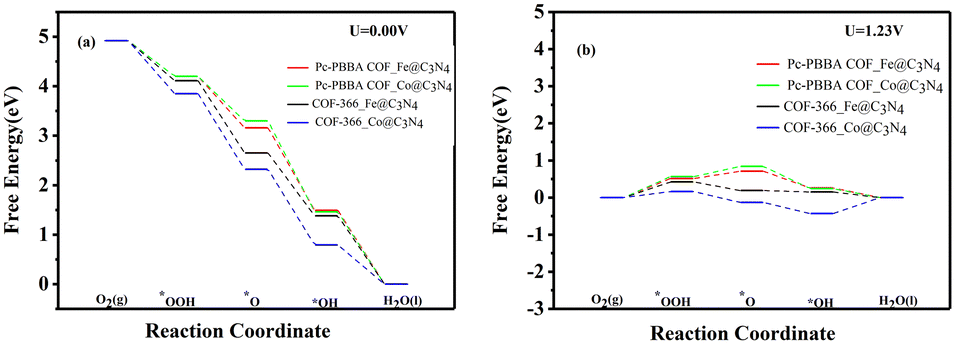 | ||
| Fig. 11 The free energy diagrams of the ORR pathways on Pc-PBBA COF_X@C3N4 and COF-366_X@C3N4(X = Fe, Co) sites were calculated at (a) U = 0 V and (b) U = 1.23 V. | ||
In Fig. 12, the differential charge density map indicates charge transfer at the active center site. Through Bader charge analysis of the active site in the C3N4 heterojunction, it is observed that electrons transfer from C3N4 to the transition metal at the active site. About 0.88–1.32 electrons are transferred to the Fe ions, causing the conversion of Fe2+ to Fe+ at the active center. Only 0.14–0.29 electrons are transferred to the Co ions, so the valence state of Co+ remains essentially unchanged. The introduction of a C3N4 layer increases the population of electrons on the Fe and Co ions, which can promote the ORR catalytic reaction. Charge density difference, as shown in Fig. 12, also reveals the transfer of electrons between the bilayer structure of the heterojunctions.
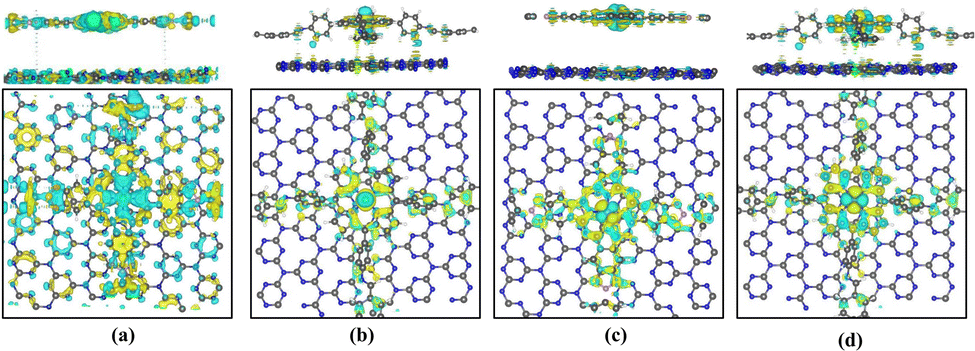 | ||
| Fig. 12 Charge density difference of (a) Pc-PBBA COF_Fe@C3N4, (b) COF-366_Fe@C3N4, (c) Pc-PBBA COF_Co@C3N4, and (d) COF-366_Co@C3N4. The yellow and cyan colors represent electron accumulation and depletion regions, respectively. The isosurface level is set to 0.004 e Bohr−3. | ||
3.4 The scaling relation and volcano plot
The scaling relationship between adsorption free energies is shown in Fig. 13(a). It indicates a linear correlation between ΔG*OOH and ΔG*OH, with ΔG*OOH = 0.40 ΔG*OH + 3.68 and R-squared at 0.08. Similarly, the ΔG*O is correlated with ΔG*OH by ΔG*O = 2.08ΔG*OH + 0.36 with the R-squared of 0.80. The *OOH and *OH species exhibit a strong linear correlation in their adsorption free energies due to their binding to the surface via the oxygen atom. Conversely, the weaker linear correlation between ΔG*OH and ΔG*O is due to the more intricate binding mechanism of the *O. The volcanic relationship between the ηORR and ΔG*OH is plotted in Fig. 13(b). The system located at the summit of the volcano is COF-366_Co@GP, which has an overpotential of 0.31 V and an OH adsorption free energy of 1.36 eV.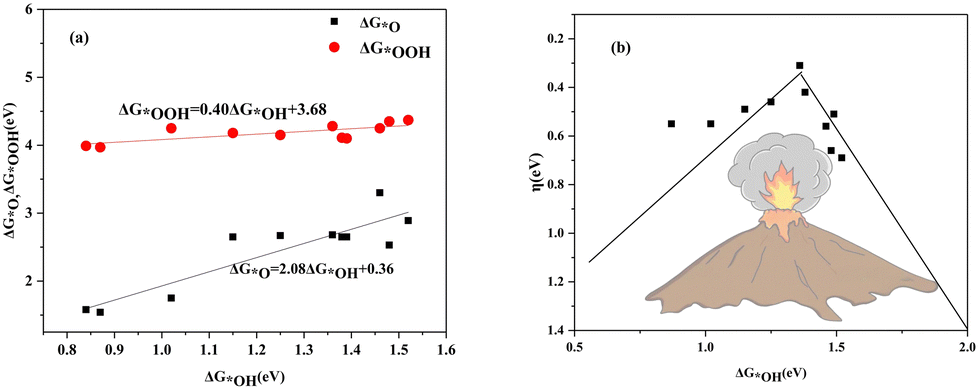 | ||
| Fig. 13 (a) The scaling relationship of ΔG*OOH and ΔG*OH associated with ΔG*OH. (b) The volcano curve of the calculated ηORR associated with ΔG*OH. | ||
In view of past studies showing that the spin states of active sites have a significant impact on catalytic performance, we calculated the spin moments of the Fe- and Co-active sites using the DFT+U method (as shown in Table S2, ESI†). The results show that the spin magnetic moment changes significantly during the ORR reaction, which may be one of the reasons why the ORR reaction always has an overpotential.
4. Conclusions
In this work, an Fe (Co) atom was embedded in the center of Pc-PBBA COF and COF-366 to form an Fe (Co)–N–C single atom catalyst, and its ORR catalytic mechanism has been systematically investigated. The analysis of the ORR reaction pathways indicates that Pc-PBBA COF_Fe, Pc-PBBA COF_Co,COF-366_Fe and COF-366_Co exhibit relatively good catalytic activity with ηORR of 0.55, 0.69, 0.66 and 0.49 V, respectively. To explore the design method of catalysts with higher activity, we introduced a graphene or C3N4 layer to form a heterostructure with the COFs.Based on the analysis of the above discussion, the following main conclusions can be drawn:
1. Electron transfer between heterojunction layers: Electron transfer between the COFs and graphene (C3N4) is bidirectional. In the vicinity of the Fe- (Co-) active sites, electrons are transferred from graphene (C3N4) to Fe (Co) ions of the COFs. At the periphery of the active sites, electrons are transferred from the COF layer to the graphene (C3N4) layer.
2. Built-in electric field between heterojunctions: A built-in electric field ranging from 2.0 × 109 V m−1 to 7.0 × 1010 V m−1 is formed between the heterojunctions, which can promote the transfer ability of electrons and facilitates the catalytic reaction.
3. Enhancement of catalytic activity: The construction of heterojunctions significantly enhanced the ORR catalytic activity. For COFs@graphene heterojunctions, the overpotential decreases by 10.9–20.3% for Fe-based active sites and 30.3–36.4% for Co-based active sites, respectively. For COFs@C3N4 heterojunctions, the overpotential decreases by 7.3–39.1% for Fe-based active sites and 15.1–16.3% for Co-based active sites, respectively.
4. Volcano plot analysis: COF-366_Co@GP positioned at the summit of the volcanic plot with an overpotential of 0.31 V, shows high activity but still lags behind Pt-based catalysts. Further research is needed to reduce the ORR overpotential and enhance the catalytic activity of COF heterojunctions.
Data availability
The authors declare that the data supporting the findings of this study are available within the paper and its ESI† files. Should any raw data files be needed in another format they are available from the corresponding author upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was jointly supported by the Shandong Provincial Natural Science Foundation (No. ZR2020MA076), the National Natural Science Foundation of China (Grant No. 12474275), the Major Scientific and Technological Innovation Project (MSTIP) of Shandong Province (No. 2019JZZY010209), and the High performance Computing Platform at University of Jinan.Notes and references
- S. Zaman, L. Huang, I. Douka, H. Yang and B. Y. Xia, Angew. Chem., Int. Ed., 2021, 60, 2–23 CrossRef PubMed.
- N. Thomas, D. D. Dionysiou and S. C. Pillai, J. Hazard. Mater., 2021, 404, 124082 CrossRef CAS PubMed.
- M. B. Vukmirovic, J. Zhang, K. Sasaki, A. U. Nilekar, F. Uribe, M. Mavrikakis and R. R. Adzic, Electrochim. Acta, 2007, 52, 2257–2263 CrossRef CAS.
- Z. Ma, Z. P. Cano, A. Yu, Z. Chen, G. Jiang, X. Fu, L. Yang, T. Wu, Z. Bai and J. Lu, Angew. Chem., Int. Ed., 2020, 132, 18490–18504 CrossRef.
- A. P. Cote, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef CAS PubMed.
- S. Huang, D. Tranca, F. Rodríguez-Hernández, J. Zhang, C. Lu, J. Zhu, H. W. Liang and X. Zhuang, Angew. Chem., Int. Ed., 2024, 63, e202314833 CrossRef CAS PubMed.
- Y. Zhang, X. Zhang, L. Jiao, Z. Meng and H.-L. Jiang, J. Am. Chem. Soc., 2023, 145, 24230–24239 CrossRef CAS PubMed.
- R. Hu, Y. Li, Q. Zeng and J. Shang, Appl. Surf. Sci., 2020, 525, 146588 CrossRef CAS.
- M. Liu, S. Liu, C.-X. Cui, Q. Miao, Y. He, X. Li, Q. Xu and G. Zeng, Angew. Chem., Int. Ed., 2022, 61, e202213522 CrossRef CAS PubMed.
- J. Tang, Z. Liang, H. Qin, X. Liu, B. Zhai, Z. Su, Q. Liu, H. Lei, K. Liu and C. Zhao, Angew. Chem., Int. Ed., 2023, 135, e202214449 CrossRef.
- Z. You, B. Wang, Z. Zhao, Q. Zhang, W. Song, C. Zhang, X. Long and Y. Xia, Adv. Mater., 2023, 35, 2209129 CrossRef CAS PubMed.
- J. Ji, C. Zhang, S. Qin and P. Jin, Sustainable Energy Fuels, 2021, 5, 5615–5626 RSC.
- Y. Zhou, L. Chen, L. Sheng, Q. Luo, W. Zhang and J. Yang, Nano Res., 2022, 15, 7994–8000 CrossRef CAS.
- J. Wang, Z. Zhang, Y. Li, Y. Qu, Y. Li, W. Li and M. Zhao, ACS Appl. Mater. Interfaces, 2021, 14, 1024–1033 CrossRef PubMed.
- Y. Wang, X. Huang, H. Fu and J. Shang, J. Mater. Chem. A, 2022, 10, 24362–24372 RSC.
- X. Hu and N. Q. Su, J. Phys. Chem. Lett., 2023, 14, 9872–9882 CrossRef CAS PubMed.
- W. Zhou, W. Q. Deng and X. Lu, Interdiscip. Mater., 2024, 3, 87–112 CAS.
- Y. Yang, Y. Li, X. Ma, L. Xie, D. Lv, L. Jiang, J. He, D. Chen and J. Wang, Catal. Sci. Technol., 2023, 13, 5599–5609 RSC.
- M. Liu, Q. Xu, Q. Miao, S. Yang, P. Wu, G. Liu, J. He, C. Yu and G. Zeng, J. Mater. Chem. A, 2022, 10, 228–233 RSC.
- C. Liu, F. Liu, H. Li, J. Chen, J. Fei, Z. Yu, Z. Yuan, C. Wang, H. Zheng and Z. Liu, ACS Nano, 2021, 15, 3309–3319 CrossRef CAS PubMed.
- S. Wan, F. Gándara, A. Asano, H. Furukawa, A. Saeki, S. K. Dey, L. Liao, M. W. Ambrogio, Y. Y. Botros and X. Duan, Chem. Mater., 2011, 23, 4094–4097 CrossRef CAS.
- E. L. Spitler and W. R. Dichtel, Nat. Chem., 2010, 2, 672–677 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B., 1976, 13, 5188–5192 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich and S. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456 CrossRef CAS PubMed.
- K. Mathew, R. Sundararaman, K. Letchworth-Weaver, T. A. Arias and R. G. Hennig, J. Chem. Phys., 2014, 140, 084106 CrossRef PubMed.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207–8215 CrossRef CAS.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2006, 124, 219906 CrossRef.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505–1509 CrossRef CAS.
- Z. Duan and G. Henkelman, ACS Catal., 2020, 10, 12148–12155 CrossRef CAS.
- H. Zhou, X. Hu, W. H. Fang and N. Q. Su, Phys. Chem. Chem. Phys., 2022, 24, 16300–16309 RSC.
- W. Chen, J. Huang, J. Wei, D. Zhou, J. Cai, Z. D. He and Y. X. Chen, Electrochem. Commun., 2018, 96, 71–76 CrossRef CAS.
- W. Xiao, H. Sheng-Xiang, L. Heng, D. Lian-Wen, W. Hao, X. Yun-Chao, H. Jun and H. Long-Hui, Acta Phys. Sin., 2019, 68, 187301 CrossRef.
- A. K. Geim, Science, 2009, 324, 1530–1534 CrossRef CAS PubMed.
- R. F. W. Bader, J. Mol. Struct. THEOCHEM, 1994, 360, 167–174 Search PubMed.
- J. Wu, X. Gao and Z. Chen, Chem. Eng. J., 2024, 492, 152241 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp02749c |
| This journal is © the Owner Societies 2024 |