
Revealing the enhancement of Li plating/stripping efficiency in TEGDME-based low-concentration electrolytes for anode-free lithium metal batteries†
Yushen
Wang
 ab and
Hidenori
Noguchi
*ab
ab and
Hidenori
Noguchi
*ab
aGraduate School of Science and Engineering, Hokkaido University, Sapporo 065-8628, Japan
bCenter for Green Research on Energy and Environmental Materials, National Institute for Materials Science, Tsukuba 305-0044, Japan. E-mail: NOGUCHI.Hidenori@nims.go.jp
First published on 24th September 2024
Abstract
Anode-free lithium metal batteries (AFLMBs) have attracted great attention owing to their higher energy density compared to conventional lithium metal batteries. Unfortunately, AFLMBs still suffer from poor Coulombic efficiency (CE) due to severe dendrite growth and the unstable solid–electrolyte interface (SEI) at the anode. Therefore, we explored the effect of concentration and LiNO3 additive on the SEI layer and on Li plating/stripping efficiency using a low-concentration tetraethylene glycol dimethyl ether (TEGDME)-based electrolyte in an AFLMB anode half-cell configuration. It was found that the formation of protective Li2O-based SEI layers when blocking the oxidative subsequent SEI formation (called “OSS”) above 2.2 V vs. Li/Li+ improved Li plating/stripping stability, while the LiNO3 additive suppressed the oxidation of Li2O into Li2O2 and sustained the existence of Li2O without blocking OSS, therefore improving CE performance. Although increasing the concentration (from 0.4 M to 2.0 M) did not have a major effect, the 2.0 M electrolyte with LiNO3 additive shifted the dominant SEI species from Li2O to LiF, further enhancing CE performance.
1. Introduction
Li metal batteries (LMBs) have been considered as a promising alternative to Li-ion batteries, as the replacement of the conventional graphite anode with the Li metal anode can greatly enhance the energy density owing to the low redox potential (−3.04 V vs. SHE) and high theoretical capacity (3860 mA h g−1) of Li metal.1,2 However, the most fundamental obstacle that hinders the practical application of LMBs lies in the highly reactive nature of Li metal, which leads to the occurrence of parasitic reactions and dendrite growth during charge–discharge, resulting in the irreversible loss of both Li metal and electrolyte. To compensate the loss in the Li reservoir, LMBs require the usage of an excess amount of Li metal in the anode side, which however increases the weight and volume of batteries, the cost, as well as the risk during battery operation.3Under this circumstance, anode-free Li–metal batteries (AFLMBs) have attracted growing attention as an alternative to LMBs.3–6 In the configuration of AFLMBs, the anode active material is absent in the as-fabricated state, and a bare anode current collector (e.g. Cu) is used as a substrate for Li deposition to create a Li anode after initial charging. It is worth noting that in AFLMBs, the negative to positive electrode capacity (N/P) ratio is 0 and 1 after cell assembly and after initial charging, respectively, meaning that all Li resources in the anode come from the cathode during initial charging. Therefore, the use of Li metal can be fully minimized, rendering greatly enhanced energy density and safety compared with LMBs. Furthermore, based on the above properties, the performance of AFLMBs is exclusively dependent on Li plating/stripping efficiency, making the analysis of capacity loss more practical.7
Despite the above advantages, the research bottleneck in AFLMBs mainly originates from the immense volume expansion and quick capacity drop during the initial state.8 In particular, due to the limited Li inventory (low N/P) in AFLMBs, Li plating on the anode usually takes place less evenly;9 hence the mechanical breakage of the solid electrolyte interphase (SEI) in stripping and further plating, which often occurs in LMBs, can be more easily found in AFLMBs. Depending on different electrochemical stages, the SEI can be classified into the “native-SEI (or N-SEI)” prior to applying bias voltage,10 the “pre-SEI” prior to initial Li plating, and the “subsequent-SEI (or s-SEI)” forming after the breakage of pre-SEI.11 An ideal SEI layer should facilitate rapid Li+ conduction while being electrically insulated to suppress electrolyte decomposition, and currently the most important thing in designing the SEI layer concerns the modification of electrolytes.
There are diverse strategies for modifying electrolytes applied in AFLMBs. In general, the selection of solvent is confined to carbonate-based or ether-based solvents, or the “co-solvent”. Compared with carbonate-based electrolytes, ether-based electrolytes have better Li metal compatibility (i.e. less dendrite formation), which ensures a higher Li plating and stripping efficiency. However, the oxidation stability (>4 V) of ether-based electrolytes usually cannot surpass that of carbonate-based electrolytes.12 As an ether-based solvent, tetraethylene glycol dimethyl ether (TEGDME) can tolerate high potential to ensure a wide operating window.13 Moreover, considering the unstable nature of TEGDME solvent-based electrolytes in the presence of Li metal,14,15 TEGDME was selected in this study in view of studying the performance degradation and enhancement in a broad potential range for AFLMBs. Meanwhile, the modification of Li salts in the electrolyte involves several technical routes, such as dual-salt electrolytes,16 high-concentration electrolytes (HCEs),4,17 and localized high-concentration electrolytes (LHCEs).18 Typically, the composition of the SEI layer is highly dependent on the concentration of the electrolyte which is related to the solvation structure of Li+.19 For low-concentration electrolytes (<3 M), the free solvent dominated solvation structure (i.e., more interaction between Li+ and solvent molecules) leads to the formation of fragile organic-rich SEI layers, while HCEs (>3 M) can usually assure an anion-involved solvation structure, giving rise to the formation of a more rigid inorganic-rich SEI and the enhancement of anti-oxidation ability.20 However, HCEs generally possess higher viscosity and lower conductivity compared to low-concentration electrolytes, and the use of higher amount of Li salt in HCEs not only reduces the energy density but also raises the cost since Li salt accounts for more than half of the total cost in electrolytes.21,22 Therefore, using low-concentration electrolytes while ensuring their high-voltage stability is considered as a promising electrolyte modification approach in the future.
Another common technique in electrolyte modification is using electrolyte additives. Among them, LiNO3 has been studied by many groups as a powerful additive applied in LMBs,15,23–25 and can be also applied in AFLMBs.26 It can generate N-rich components in the inner SEI layer including Li3N, LiNxOy, LiNO2, and R-NO2, most of which are known as good Li+ conductors that can promote efficient cycling of Li plating/stripping.15,23,25 Furthermore, it was reported that adding LiNO3 as an additive can regulate the morphology of plated Li from dendritic Li to spherical Li to suppress the undesired reaction area between Li and the free solvent.26 In the case of using LiTFSI with the LiNO3 additive, the coordination of TFSI− anions with Li+ can be strengthened, and the decomposition of TFSI− anions can be facilitated to generate low electronic conductive components such as LiF or Li2CO3.24
In the present study, the effects of electrolyte concentration and LiNO3 additive on SEI layer structure and Li plating/stripping efficiency were investigated using the Li‖Cu half-cell in TEGDME-based low-concentration electrolytes (0.4 M and 2.0 M) dissolving the LiTFSI salt. In total, four kinds of electrolytes were prepared prior to measurements. For convenience, these electrolytes are hereafter abbreviated as 0.4LT, 0.4LTLN, 2.0LT, and 2.0LTLN, where LT means LiTFSI and LN means 0.1 M LiNO3 as an additive. Then the properties of formed SEI layers during electrochemical cycling were elucidated based on their Li-ion/electron conduction ability, chemical composition, and surface morphology.
2. Results and discussion
2.1. Identification of electrochemical reactions during cycling
The first- and second-cycle CV curves measured in 0.4LT are shown in Fig. 1a, in which all the observed peaks are named by using abbreviated symbols. During the first cycle without (hereafter, w/o) Li plating/stripping, three redox peak couples were observed, corresponding to the reduction and oxidation of the electrolyte (REL/OEL),24,27,28 the reduction and oxidation of copper oxides (CuxO) remaining on the Cu substrate before measurement (RCO/OCO),29 and the reduction and oxidation reactions related to pre-SEI formation (RPS/(OPS + OPS′)).10,29,30 However, when Li plating/stripping was involved, a strong oxidative current started to flow from 2.2 V, which was related to the s-SEI layer construction process occurring on the surface of unstripped Li metal. This process was named “OSS” which paired with the corresponding reduction process RSS of the next cycle,29 whereas this redox couple was only partially reversible since the peak RSS was apparently weaker. From the second cycle, one can observe another redox couple relevant to s-SEI construction (RSS′/(OSS′ + OSS′′)) involved in the commonly accepted potential area where the SEI layer usually forms.30 Consequently, it is evident that there are two sets of s-SEI related redox reactions in this electrolyte: the OSS/RSS and the RSS′/(OSS′ + OSS′′), with the former occurring at a higher potential. Notably, the abovementioned redox couples were observed in all tested electrolytes in this study, except for artificially blocking OSS by scanning up to 2.2 V which presented only the RSS′/(OSS′ + OSS′′) couple from the second cycle, as summarized in Fig. 1b. | ||
| Fig. 1 (a) Enlarged view of the first- and second-cycle CV curves measured in 0.4LT; (b) enlarged view of the first- and second-cycle CV curves measured in 0.4LT, 0.4LT w/o OSS, 0.4LTLN, 2.0LT and 2.0LTLN with Li plating/stripping. The prime symbols ′ and ′′ on the capitals indicate that the corresponding peaks locate at a lower potential compared to those without the prime symbol. The arrows represent the direction of scanning. Potential range with Li plating/stripping: −0.5 to 3.2 V, and potential range w/o Li plating/stripping: 0.2–3.2 V. Scan rate: 10 mV s−1. | ||
2.2. Comparison of CE performances
Fig. 2a shows the calculated CE values up to 50 cycles in different electrolytes based on their CV curves shown in Fig. S1 (ESI†) by taking the influence of concentration, LiNO3 additive and reaction OSS into consideration. Overall, increasing the concentration from 0.4 M to 2.0 M enhanced the CE values. The CE in 2.0LT displayed a 4-fold increase at the beginning compared with 0.4LT, while both showed a gradual decrease afterwards, revealing a very unstable Li plating/stripping process during cycling. In the meantime, blocking reaction OSS by scanning up to 2.2 V can effectively stabilize the CE performance in prolonged cycles in both 0.4LT and 2.0LT. This indicates that reaction OSS must be one of the major causes for CE degradation, which may promote unstable SEI layer formation. Furthermore, adding 0.1 M LiNO3 as an additive in either 0.4LT or 2.0LT can not only enhance but also stabilize the CE performance. Interestingly, this remains true when adding LiNO3 based on blocking OSS, while the opposite operation (blocking OSS based on adding LiNO3) did not show any positive effect. Therefore, it is concluded that adding LiNO3 could significantly compensate for the degradation caused by the reaction OSS, apart from its effect on enhancing the CE values. Besides, the current densities of Li plating/stripping in 0.4LT w/o OSS, 0.4LTLN, and 2.0LTLN were lower than those in 0.4LT and 2.0LT (Fig. S1, ESI†), which could result in a smoother and denser Li morphology.10 | ||
| Fig. 2 (a) 50-Cycle CE performances in 0.4LT, 0.4LTLN, 2.0LT and 2.0LTLN (with and w/o the occurrence of OSS); (b) cycling dependence of the peak potential and peak current density of OSS, RSS′ and REL obtained from CV measured in 0.4LT, 0.4LT w/o OSS, 0.4LTLN, 2.0LT and 2.0LTLN. | ||
2.3. Li-ion/electronic conductivity studies of the SEI layer
Since the CE performances should be deeply affected by the stability of electrolytes which can induce corresponding electrochemical reactions as cycling proceeds, a detailed analysis of the CV peaks was further conducted within the range of 0 to 3.2 V throughout the cycling process. Fig. 2b shows the potential and current variations of the peak OSS, RSS′ and REL during cycling obtained from the 50-cycle CV results measured in 0.4LT, 0.4LT w/o OSS, 0.4LTLN, 2.0LT and 2.0LTLN (Fig. S2, ESI†), which implies the reconstruction of the SEI layer. Peak OSS and RSS′, belonging to the two aforementioned s-SEI related redox reactions, were selected for discussion together with peak REL. Here, the OSS and RSS′ currents can be regarded as the indicator of the ionic conduction ability of the s-SEI layer (i.e. the larger or smaller currents represent the higher or lower efficiency of Li+ transport through the s-SEI layer), and the REL currents reflect the electronic conductivity of s-SEI which influences the reaction kinetics between the electrolyte and plated Li. For 0.4LT, the largely increased REL currents with a severe polarization, together with the decreased OSS and RSS′ currents demonstrate the formation of thick s-SEI layers with poor Li+ conduction and rapid electronic tunneling. However, blocking reaction OSS remarkably reduced the electronic conductivity to suppress the decomposition of the electrolyte to a certain extent, whereas the Li+ conductivity still decreased as the RSS′ currents almost exhausted within 10 cycles. This could explain why CE became more stable but was not enhanced when blocking reaction OSS in 0.4LT. By increasing the concentration to 2.0LT, the Li+ conductivity substantially increased as the OSS and RSS′ currents were much larger than those in 0.4LT after several cycles, but the electrolyte decomposition still proceeded as indicated by the increased REL current. In the case of both 0.4LTLN and 2.0LTLN, one can observe a cathodic peak at 1.70 V belonging to the reduction of NO3− from the second cycle (marked by the triangle symbol in Fig. S2, ESI†), a similar value as that reported by Fu et al. (1.66 V in the first cycle),24 while it was not observed in the first cycle in this study probably due to the overlap from the strong REL peak. This demonstrates that LiNO3 will be reduced prior to the reduction of LiTFSI, which is in agreement with Fu et al.'s report. Notably, it was found that adding 0.1 M LiNO3 not only increased Li+ conductivity but also greatly suppressed the electrolyte decomposition, as supported by the peak current density comparison between the electrolytes with and w/o LiNO3 additive (Fig. 2b). However, it should be mentioned that the REL currents in 2.0LTLN gradually increased during cycling and surpassed the REL currents in 0.4LTLN. This might be due to the decomposition of LiNO3, which caused a decrease of the LiNO3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :LiTFSI ratio leading to the weakened ability to suppress further electrolyte decomposition.
:LiTFSI ratio leading to the weakened ability to suppress further electrolyte decomposition.
2.4. Composition and morphology studies of the SEI layer
The first-cycle QCM test was performed in 0.4LT, 0.4LTLN, 2.0LT and 2.0LTLN, by which the real-time change of mass and surface roughness of the SEI layer were obtained. Prior to the first Li plating, the increase of mass on the Cu surface measured was induced by electrolyte reduction, CuxO reduction, and the pre-SEI formation (Fig. 3a). The average MPE (mass change per mol e−) values were then calculated from the slope of the plot with Δm as a function of the passed charge, showing that the deposits in 2.0LTLN were the heaviest among the four electrolytes (Fig. 3b). The relatively small MPE values in the REL + RCO area could be attributed to (1) the generation of soluble products or predominant inorganic deposits from TFSI− reduction,31,32 and (2) the reduction of CuxO causing a decrease of deposit mass. Then, pre-SEI formation (RPS) reactions occurred based on the products during REL + RCO reactions. Considering the formation of potential pre-SEI species in the LiTFSI/TEGDME based electrolytes, e.g. Li2O (MPE = 15), LiF (MPE = 26) and Li2CO3 (MPE = 37),33,34 as well as the corresponding reductions among them (for example, Li2CO3 reducing to Li2O, MPE = 7),35 it can be inferred that the major species in the pre-SEI layer was likely to be LiF and Li2O for all electrolytes, while the relative ratio of LiF was higher for 2.0LTLN than that for other 3 electrolytes. Fig. 3c shows the first-cycle CV curves as well as potential dependent Δm and ΔR (the change of resonant resistance, which is positively correlated with the change of surface roughness)36 measured in the four electrolytes. It can be found that the surface layer in the 2.0 M electrolytes displayed an elevated roughness before electrochemical measurement compared with 0.4 M electrolytes mainly due to the increased density and viscosity by increasing the concentration,37 whereas the effect of the N-SEI layer should not be ignored. Markedly, both Δm and ΔR increased to various extents after the initial potential scan for all electrolytes, while the overall increase of mass was smaller in the electrolytes with LiNO3 additive. Moreover, the considerably reduced ΔR during Li plating/stripping for the electrolytes with LiNO3 additive can be regarded as a greatly mitigated roughening process.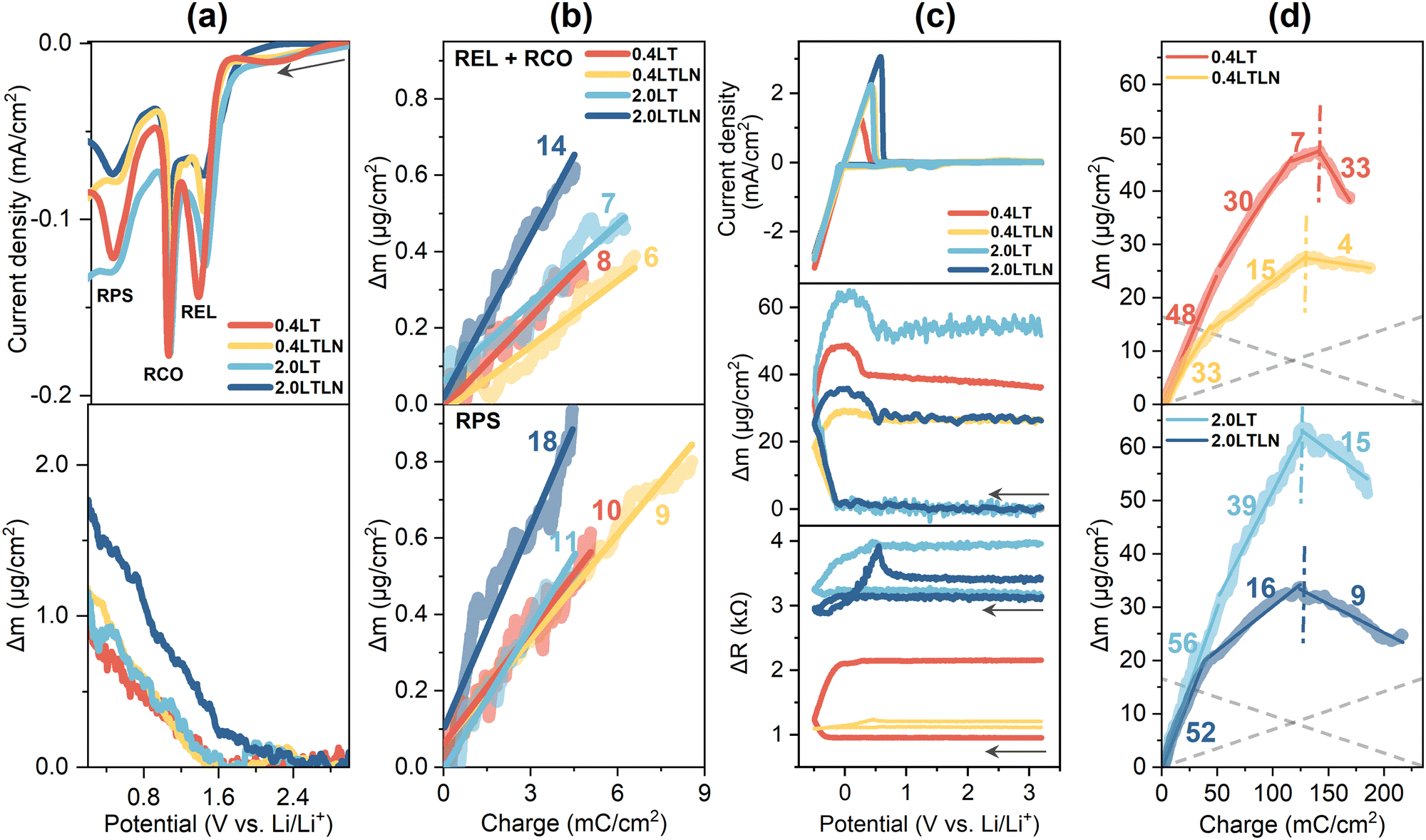 | ||
| Fig. 3 (a) The cathodic process prior to initial Li plating and the simultaneous mass change measured in 0.4LT, 0.4LTLN, 2.0LT and 2.0LTLN; (b) calculated MPE values during the reaction REL + RCO and reaction RPS, respectively, in the above four electrolytes; (c) first-cycle CV curves as well as the potential dependence of Δm and ΔR measured in 0.4LT, 0.4LTLN, 2.0LT and 2.0LTLN. Scan rate: 10 mV s−1. The arrows represent the direction of scanning; (d) calculated MPE values during the initial Li plating/stripping in the above four electrolytes. The vertical dash-dotted lines function as borderlines to separate the Li plating region and the Li stripping region; the gray dashed lines were manually added as a reference showing only Li plating and stripping (MPE = 7).38 | ||
The MPE values during the initial Li plating/stripping in these electrolytes were also calculated for the prediction of possible dominant SEI components generated in this process, which were exhibited in the Δm vs. charge profile (Fig. 3d). Here, multiple MPE values were confirmed during Li plating reflecting complex electrochemical processes, where an extra SEI layer was formed presumably due to the reaction of plated Li metal with electrolytes as verified by MPE values much larger than 7 (ideally, it can be regarded as Li plating/stripping exclusively taking place when MPE ≈ 7, i.e., 5 to 9).39 The major species of the SEI layer are more likely to be the organic Li salts (MPE around 50–80) from decomposed TEGDME solvent since these electrolytes are low-concentrated (<3 M) and free-solvent dominated.33 It should be noted that the total charge consumed during Li plating should include the charge from associated electrolyte reactions in the case of the MPE value larger than 7; thus the actual MPE related to electrolyte reactions is normally larger than the observed one which is dependent on the ratio of the charge from electrolyte reactions. Nevertheless, in this study the electrolytes with LiNO3 additive presented smaller MPE values during Li plating, demonstrating the possibility of more inorganic species generated with the help of LiNO3 (Fig. 3d). During Li stripping, the extra mass loss except for the stripped Li was observed for 0.4LT and 2.0LT probably due to the concurrent oxidation of SEI species and/or the crack of the SEI layer. In contrast, the MPE closer to 7 for 0.4LTLN and 2.0LTLN indicates that a relatively protective inorganic-based SEI was formed during Li plating, with which the reaction of Li metal with the electrolyte can be efficiently suppressed.
The analyses of the cycling-dependent MPE and ΔR were conducted for the 10th and 20th cycles. As shown in Fig. 4a, the MPE value of −7 during Li stripping markedly deviated from 7 in the 20th cycle for 0.4LT, showing that the simultaneous increase of surface mass originated from the more undesired side reactions. Upon increasing the concentration to 2.0LT, the continuously decreased mass except for that contributed by stripped Li may imply the mechanical disconnection of unreactive Li and/or SEI components from the surface, giving rise to a substantial decrease of surface mass from the 10th to the 20th cycle. These behaviors also reflected on the prominent rising of surface roughness in 0.4LT and 2.0LT (Fig. 4b). In contrast, by either blocking OSS or adding LiNO3 to 0.4LT, a more stable Li plating/stripping process was achieved as indicated by their MPE close to 7. Moreover, reaction OSS kept on increasing the surface roughness upon cycling, while blocking OSS was not as effective as adding LiNO3 in suppressing the increased roughness. The above discussions were evidenced by the SEM and optical images of the electrode surface after cycling among different electrolytes (Fig. S3, ESI†). The whisker-like lithium accumulates in 0.4LT, whereas some film-like products with a relatively smooth surface morphology can be found when blocking OSS. However, a dark grey deposit indicating the formation of a thick layer on the Cu surface was visually observed regardless of the OSS process.40,41 With the help of LiNO3 additive, smoother morphology and the silver white color of Li deposits prove the formation of denser and dendrite-free SEI layers, and further blocking reaction OSS in 0.4LTLN did not hugely change the surface morphology. Although both blocking OSS and adding LiNO3 additive can result in a better Li stripping reversibility, the lower CE values in 0.4LT compared with those in 0.4LTLN should be associated with the inferior Li+ conductivity as well as higher surface roughness. However, for 2.0LTLN cycled to the 10th and 20th cycles, the increase of surface roughness was not effectively suppressed (Fig. 4b), and the MPE value of 1 or 2 during Li stripping indicates the simultaneous occurrence of electrolyte decomposition in spite of the best CE performance among the tested electrolytes (Fig. 4a). This agrees well with the CV results where the REL currents in 2.0LTLN gradually increased upon cycling (see Fig. 2b).
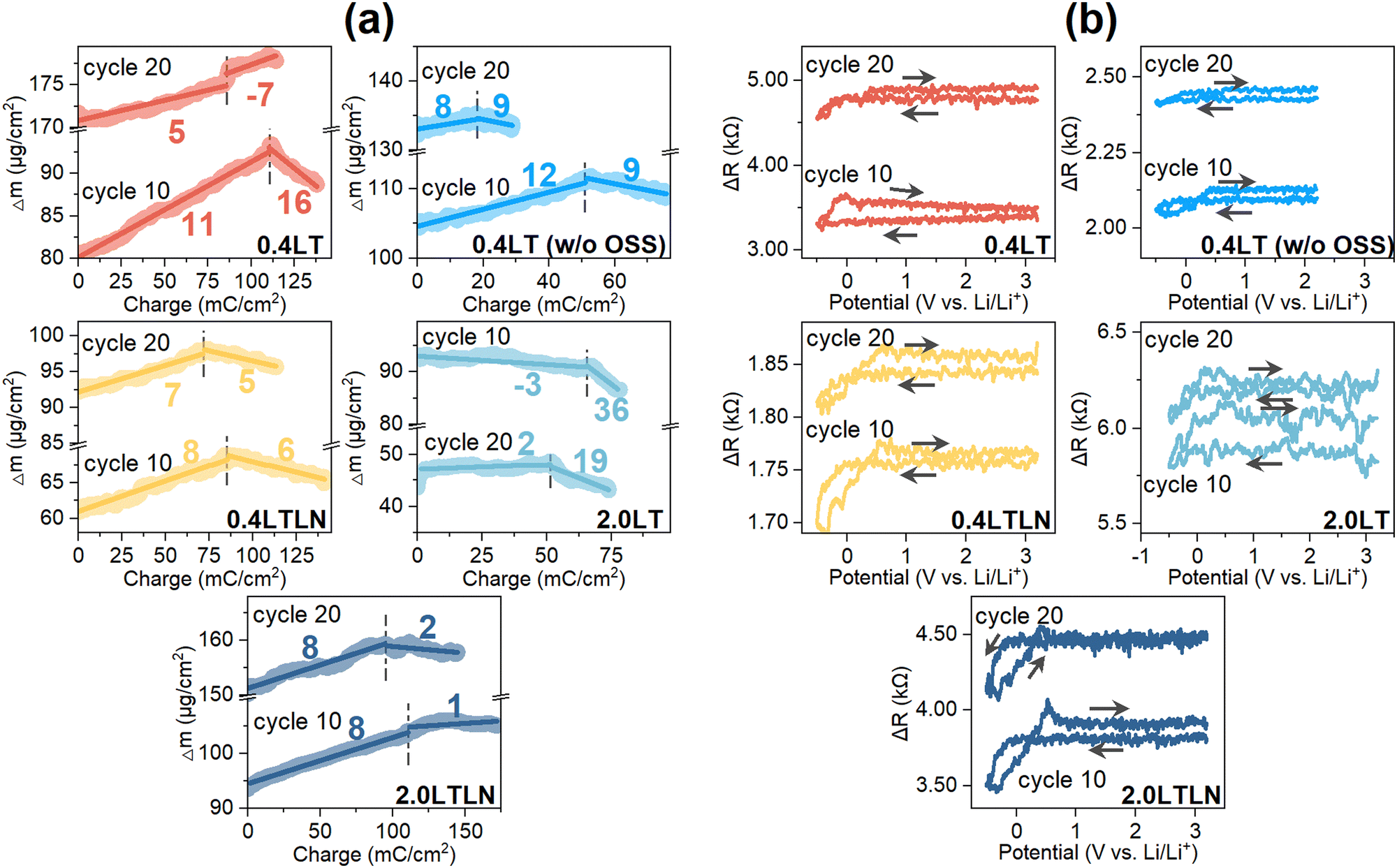 | ||
| Fig. 4 (a) Calculated MPE values during Li plating/stripping in the 10th and 20th cycles measured in 0.4LT, 0.4LT w/o OSS, 0.4LTLN, 2.0LT and 2.0LTLN. The negative MPE indicates the loss (or growth) of mass during Li plating (or stripping). The vertical dash-dotted lines function as borderlines to separate the Li plating region and the Li stripping region; (b) potential-dependent ΔR in the10th and 20th cycles measured in 0.4LT, 0.4LT w/o OSS, 0.4LTLN, 2.0LT and 2.0LTLN. | ||
To better understand the detailed variation of SEI composition at different stages of cycling, the SEI layer formed in 0.4LT, 0.4LT w/o OSS, 0.4LTLN and 2.0LTLN after cycling 50 times was characterized using in-depth XPS analysis by Ar+ sputtering. Fig. 5 shows the Li 1s, N 1s, O 1s, S 2p, F 1s, and C 1s XPS spectra of the SEI layers formed at different periods of cycles on the Cu substrate in these electrolytes. In either O 1s or C 1s spectra, two peaks were observed in all electrolytes: one corresponding to the –C–O–C– bonding of poly(ethylene oxide) ((CH2–CH2–O)n−) or ROCO2Li from the decomposition of TEGDME molecules, and the other corresponding to Li2CO3.15,42 Typically, the LiNO3 decomposition products, such as Li3N and LiNxOy that reportedly enable high Li+ conductivity,15,23 were confirmed for 0.4LTLN and 2.0LTLN (Fig. 5a and b). Moreover, the spectral intensity displayed a descending order of 0.4LT > 0.4LTLN > 2.0LTLN, showing demonstrating the improved stability of s-SEI layers, which suppress electrolyte decomposition. In addition, a down-shift of binding energy from the O 1s and C 1s spectra was found for the two 0.4LT electrolytes upon cycling, which was due to the presence of polar decomposed species (such as carbonates, which could involve in the further degradation process) on the electrode surface that caused the formation of an electric potential gradient at the interface.43 However, the downshift of binding energy was not observed for 0.4LTLN and 2.0LTLN during cycling; instead the decrease of the inorganic constituent (e.g. Li2CO3) and the increase of the organic constituent (e.g. ROCO2Li) could be the proof of the formation of a heterogeneous SEI layer structure.42,44
 | ||
| Fig. 5 (a) Li 1s, (b) N 1s, (c) O 1s, (d) S 2p, (e) F 1s, and (f) C 1s depth profiling XPS spectra of the SEI layers for 0.4LT, 0.4LT w/o OSS, 0.4LTLN and 2.0LTLN at different etching times; (g) the atomic concentration percentage of the s-SEI composition after different etching times cycled in 0.4LT, 0.4LT w/o OSS, 0.4LTLN and 2.0LTLN. | ||
Furthermore, Li2O was detected (Fig. 5a and c) for 0.4LT w/o OSS, which as discussed above could generate from the SEI layer related reactions including the reactions between the plated Li metal and the electrolyte. However, Li2O was absent in the O 1s spectra for 0.4LT (Fig. 5c). Instead, lithium peroxide (Li2O2) was detected,45 suggesting that Li2O could be oxidized to Li2O2 during OSS in 0.4LT (the co-existence of Li2O and Li2O2 detected in the Li 1s spectra may indicate that this oxidation is an incomplete process).29 Interestingly, Li2O was detected for 0.4LTLN at the occurrence of OSS but was hardly found for 2.0LTLN. It was previously reported that the decomposition of LiNO3 could contribute to the formation of Li2O together with Li3N.23 The formed insoluble Li2O at the anode side contributes to the construction of the SEI layer.46 However, the existence of Li2O2 cannot be excluded since it is difficult to unambiguously distinguish Li2O2 and Li2O in the spectra due to their close binding energy. To this end, in situ SERS measurement was conducted before and after reaction OSS (Fig. S4, ESI†) to clarify whether the oxidation of Li2O occurred or not via OSS in 0.4LTLN and 2.0LTLN. It should be mentioned that the SERS band of Li2O (near 530 cm−1)47 cannot be confirmed due to the broad peak from the Cu substrate at 500–600 cm−1. However, in both 0.4LT and 2.0LT, lithium superoxide (LiO2) and Li2O2 were detected after reaction OSS.48–50 This indicates that the SEI component Li2O was oxidized to Li2O2 and insoluble LiO2 on the electrode surface during reaction OSS when using low-donor-number TEGDME as solvent.51–53 However, neither Li2O2 nor LiO2 was confirmed for 0.4LTLN and 2.0LTLN regardless of reaction OSS, which implies that on the one hand the LiNO3 additive strongly suppressed the oxidation of Li2O via reaction OSS in 0.4LTLN, and on the other hand Li2O was not the major SEI component in 2.0LTLN since both Li2O and its oxidation products were hardly found.
The function of LiNO3 additive in suppressing the oxidation of Li2O was further proved by a more careful analysis of the OSS process in 0.4LT and 0.4LTLN via QCM. When repeating the QCM test with a focus on the OSS process in 0.4LT, a sudden change of MPE around 2.7 V was found in all three tests, which was highly likely to be caused by the oxidation of Li2O (Fig. S5a, ESI†),54,55 irrespective of the distinct MPE values among these tests probably due to the complicated physical and chemical phenomena caused by SEI crack and Li dendrites during OSS. In contrast, the sudden change of MPE was not observed in 0.4LTLN, meaning that the oxidation of Li2O hardly occurred during OSS (Fig. S5b, ESI†). Sharon et al. reported that Li2O2 can be oxidized by the reaction product (NO2) originated from LiNO3 which can act as a redox mediator in the Li–oxygen battery.56 However, this process was thought to occur at the cathode side above 3.6 V vs. Li/Li+. Therefore, the possibility of LiNO3 further oxidizing Li2O2 that was generated from Li2O oxidation during OSS can be excluded in our study. It is worth noting that the possible reaction showing the oxidation of Li2O to Li2O2 is considered as
| 2Li2O − 2e− → Li2O2 + 2Li+ | (R1) |
The correspondent MPE of reaction (R1) equals −7. Assuming that the change of MPE was mostly due to Li2O oxidation, then the ratio of the charge released by electrochemical Li2O oxidation over the total charge during OSS can be calculated. As a result, similar ratios (see Table S1, ESI†) were confirmed in all tests using 0.4LT, further proving the above assumption. This demonstrates that the oxidation of Li2O was the major OSS reaction that transferred 80–90% of the total charge, which can be effectively suppressed by LiNO3.
Since we have demonstrated that the addition of LiNO3 can effectively inhibit Li2O oxidation, it was expected that the total charge transferred during the OSS process should be less for the electrolytes with LiNO3 than for the electrolytes w/o LiNO3. However, if we look more carefully at Fig. S5 (ESI†), the transferred charges from 2.6 V to 3.2 V for 0.4LT and for 0.4LTLN were very close to each other (5–6 mC cm−2), meaning that when LiNO3 was added, some extra electrochemical oxidation reactions occurred during OSS that compensated for the reduced charge from Li2O oxidation. Note that the XPS spectra for 0.4LTLN exhibit three major differences with respect to that of 0.4LT regarding the species (Fig. 5): (1) the disappearance of Li2O2; (2) the generation of nitrogen-containing species including Li3N, LiNO2 and a series of LiNxOy components; and (3) the disappearance of lithium carbide (Li2C2) during cycling. Since Li2C2 was only related to the deposited Li metal which will be discussed in the next section, the only difference created by LiNO3 during the OSS process (except for the suppression of Li2O oxidation) was the electrochemical oxidation of specific nitrogen-containing species (equivalent to peak a in Fig. S6, ESI†). The possible reactions can be expressed as
| LixNOy − e− → Li(x−1)NOy + Li+ (x > 1, y ≥ 0) | (R2) |
As an example, the oxidation of Li3N to LiNOx (x = 2, 3) can possibly occur, whose reverse reaction occurs around 1.7 V during negative scanning (equivalent to peak a′′ in Fig. S6, ESI†).24,57 Considering the fact that the extra reactions instead of Li2O oxidation occurred in the OSS process for 0.4LTLN, such reactions are therefore highly possible to be related to reaction (R2). However, the detailed reaction scheme is still unclear and requires a further study.
The comparison among the four electrolytes regarding the S 2p spectra (Fig. 5d) leads to the conclusion that the oxidation of short-chain lithium polysulfides (e.g. Li2S/Li2S2), which were from the reduction of LiTFSI, towards Li2SO4 occurred via OSS.29 This could be related to the degradation of CE performance in 0.4LT.58 It also suggests that the LiNO3 additive cannot prevent the oxidation of these short-chain lithium polysulfides which were from TFSI− decomposition, whereas the lack of these species did not degrade the CE performance in the presence of LiNO3 additive, showing the importance of the presence of Li2O rather than Li2S/Li2S2. Notably, the peak intensity of LiF in 0.4LTLN is much weaker than that in 0.4LT (Fig. 5e). This observation may, on the contrary, imply that the very-low CE in 0.4LT strongly correlates with the severe decomposition of LiTFSI (thus, more LiF). It was previously reported that the LiNO3 additive can facilitate the decomposition of LiTFSI to generate more LiF.24 The different results in our study might be because we used a low-concentration electrolyte while they adopted a concentrated electrolyte (3.25 M). Meanwhile, the atomic concentration percentage of the s-SEI composition demonstrates a higher F ratio for 2.0LTLN compared with that for 0.4LTLN (Fig. 5g). This also demonstrates that LiF was the major SEI component for 2.0LTLN which played a decisive role in enhancing and stabilizing the CE performance, in line with the above discussions on the pre-SEI species. For the more dilute 0.4 M electrolytes, however, the existence of Li2O (and the existence of short-chain lithium polysulfides when blocking OSS) was rather regarded as the major contributor than LiF to the improved Li plating/stripping efficiency.29
2.5. Studies of Li inventory loss
As shown in Fig. 5f, Li2C2 was only found in the inner s-SEI layer measured in 0.4LT with the occurrence of OSS, whereas it was observed throughout all s-SEI layers when blocking OSS. On the other hand, it was not detected at any depth of the s-SEI layers formed in 0.4LTLN and 2.0LTLN. It was reported that the formation of Li2C2 is prone to occur on the massively plated Li metal surface.59 These differences therefore indicate that with the occurrence of OSS a poor reversibility of Li plating/stripping will lead to the accumulation of Li2C2, and ultimately a severe loss of the Li inventory fundamentally prohibits the further formation of Li2C2. In sharp contrast, a greatly improved reversibility of the dissolution of formed Li2C2 together with an enhanced Li stripping efficiency can be achieved by the LiNO3 additive. The in situ SERS also proves that Li2C2 is the major component of the formed s-SEI layers, which was detected around 1853 cm−1 on the plated Li metal surface (Fig. S7, ESI†), similar to Schmitz et al.'s report.59 During the initial 10 cycles for 0.4LT, the amount of deposited Li2C2 drastically decreased even at −0.5 V where Li was deeply plated, showing the severe loss of the Li inventory. A similar tendency was also observed in 2.0LT, while for 0.4LT w/o OSS, 0.4LTLN and 2.0LTLN, the intensity of the Li2C2 band almost did not change during cycling, meaning that a more stable SEI layer was formed to suppress Li loss via reacting with electrolyte and reconstructing s-SEI layers. These results were well in accordance with the observed C 1s spectral behaviors. Meanwhile, the depth-dependent atomic concentration percentage also demonstrates the Li inventory change during cycling, as shown in Fig. 5g. In the case of 0.4LT, the apparent drop of Li content in the outer SEI layer proves the severe decomposition of the electrolyte associated with Li exhaustion. By contrast, Li deposits were more evenly distributed across the s-SEI layers during cycling by blocking OSS, and a homogeneous distribution of Li was almost achieved with the help of LiNO3 additive.2.6. Proposed model of major SEI components with morphology variation via OSS
In our previous study, a model showing the possible major SEI components at each stage of the first CV cycle in the dilute LiTFSI–TEGDME electrolyte was proposed.29 The N-SEI layer contains species such as LiF, Li2CO3 and Cu oxides.10 After electrolyte reductive decomposition, some inorganic species like Li2O mainly constituted the pre-SEI layer. During reaction OSS, these species were thought to be oxidized to construct the s-SEI layer. In this study, a new model was proposed based on the above results in order to show the variation of major species via OSS in more detail, with the emphasis on the effect of LiNO3 (Fig. 6). The major SEI components in the 0.4LT electrolyte prior to reaction OSS include Li2O and Li2Sx. When adding LiNO3, nitrogen-containing species appear to be another major species. When increasing the concentration to 2.0 M, LiF replaces Li2Ox and becomes the major species. In the case of w/o LiNO3 additive, Li dendrite growth proceeds more heavily via OSS due to the change of major SEI components (Li2O → Li2O2, Li2Sx → Li2SOx), resulting in the roughening of the electrode surface. By adding LiNO3 additive, the problem can be solved through the prevention of Li2O oxidation, and the resultant electrode surface after OSS will retain rather lower roughness. | ||
| Fig. 6 The proposed model of the change of major SEI components via OSS under the influence of electrolyte concentration and LiNO3 additive in the LiTFSI/TEGDME electrolytes. | ||
3. Conclusions
In conclusion, by adopting the AFLMB anode half-cell (Li‖Cu) configuration, this study provided a viewpoint in evaluating the positive and negative impacts of the electrochemical reactions in the potential range of 0–3.2 V vs. Li/Li+, with an emphasis on the effect of the “pre-SEI” layer and “s-SEI” layer modified by concentration and the LiNO3 additive. Specifically, two low-concentration (0.4 M and 2.0 M) LiTFSI/TEGDME electrolytes with and w/o LiNO3 additive were investigated and the performance of Li plating/stripping under different modification conditions was discussed in detail. It was found that purely increasing salt concentration from 0.4 M to 2.0 M was not beneficial in improving the stability of s-SEI layers on the surface under a low-concentration condition due to the unavoidable electrolyte decomposition, while either blocking the reaction OSS at above 2.2 V or adding 0.1 M LiNO3 as an additive could result in the formation of Li2O-based SEI layers with smaller surface roughness (i.e., less amount of dendrite) by blocking the oxidation of Li2O to Li2O2, suppressing the decomposition of the electrolyte as well as enabling a more stable and efficient Li plating/stripping. Meanwhile, the effect of increasing the concentration (to 2.0 M) in the presence of 0.1 M LiNO3 additive shifted the dominant SEI species from Li2O to LiF, further assuring the enhanced Li plating/stripping performance. However, in consideration of the gradual decomposition of LiNO3 additive during cycling, the optimization of the LiNO3:LiTFSI ratio should be further considered to achieve better performances based on the concept of low-concentration. In addition, other countermeasures targeting up-regulation of the composition and morphology caused by reaction OSS will be investigated in the future. These results suggest that adding LiNO3 as an additive could be of great help in sustaining the SEI layer stability when the operating potential range of AFLMBs is considered to be broadened. In addition, this study provides a promising strategy to optimize the compositional and morphological conditions that could be degraded by reaction OSS and similar reactions occurring in a higher potential region.
Author contributions
H. N.: provided the conception and design of the study. W. Y.: acquired data analysis. H. N., W. Y.: interpreted the results. W. Y.: wrote the draft of the manuscript. H. N.: provided the study materials, laboratory samples, instrumentation, computing resources, and other analysis tools. H. N.: acquired the financial support. H. N., W. Y.: analyzed and interpreted the data, and revised the manuscript critically for important intellectual content. H. N.: managed and coordinated responsibility for the research activity planning and execution.Data availability
The data that support the findings of this study are available from the corresponding author (H. N.) upon reasonable request.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This research was funded by the “Fundamental Research on Materials for Energy Conversion and Storage” project at the Research Center of Energy and Environmental Materials (GREEN) of NIMS. The authors gratefully acknowledge the NIMS Battery Research Platform.References
- X.-B. Cheng, R. Zhang, C.-Z. Zhao and Q. Zhang, Toward safe lithium metal anode in rechargeable batteries: a review, Chem. Rev., 2017, 117, 10403–10473 CrossRef CAS PubMed.
- W. Xu, J. Wang, F. Ding, X. Chen, E. Nasybulin, Y. Zhang and J.-G. Zhang, Lithium metal anodes for rechargeable batteries, Energy Environ. Sci., 2014, 7, 513–537 RSC.
- S. Nanda, A. Gupta and A. Manthiram, Anode-free full cells: a pathway to high-energy density lithium–metal batteries, Adv. Energy Mater., 2021, 11, 2000804 CrossRef CAS.
- J. Qian, B. D. Adams, J. Zheng, W. Xu, W. A. Henderson, J. Wang, M. E. Bowden, S. Xu, J. Hu and J.-G. Zhang, Anode-free rechargeable lithium metal batteries, Adv. Funct. Mater., 2016, 26, 7094–7102 CrossRef CAS.
- R. Weber, M. Genovese, A. J. Louli, S. Hames, C. Martin, I. G. Hill and J. R. Dahn, Long cycle life and dendrite-free lithium morphology in anode-free lithium pouch cells enabled by a dual-salt liquid electrolyte, Nat. Energy, 2019, 4, 683–689 CrossRef CAS.
- R. V. Salvatierra, W. Chen and J. M. Tour, What Can be Expected from “Anode-Free” Lithium Metal Batteries?, Adv. Energy Sustainable Res., 2021, 2, 2000110 CrossRef CAS.
- R. Rodriguez, K. E. Loeffler, R. A. Edison, R. M. Stephens, A. Dolocan, A. Heller and C. B. Mullins, Effect of the electrolyte on the cycling efficiency of lithium-limited cells and their morphology studied through in situ optical imaging, ACS Appl. Energy Mater., 2018, 1, 5830–5835 CrossRef CAS.
- A. P. Cohn, N. Muralidharan, R. Carter, K. Share and C. L. Pint, Anode-free sodium battery through in situ plating of sodium metal, Nano Lett., 2017, 17, 1296–1301 CrossRef CAS PubMed.
- A. M. Tripathi, W.-N. Su and B. J. Hwang, In situ analytical techniques for battery interface analysis, Chem. Soc. Rev., 2018, 47, 736–851 RSC.
- S. Menkin, C. A. O’Keefe, A. B. Gunnarsdóttir, S. Dey, F. M. Pesci, Z. Shen, A. Aguadero and C. P. Grey, Toward an understanding of SEI formation and lithium plating on copper in anode-free batteries, J. Phys. Chem. C, 2021, 125, 16719–16732 CrossRef CAS PubMed.
- C.-J. Huang, B. Thirumalraj, H.-C. Tao, K. N. Shitaw, H. Sutiono, T. T. Hagos, T. T. Beyene, L.-M. Kuo, C.-C. Wang and S.-H. Wu, Decoupling the origins of irreversible coulombic efficiency in anode-free lithium metal batteries, Nat. Commun., 2021, 12, 1452 CrossRef CAS PubMed.
- C.-H. Jo, K.-S. Sohn and S.-T. Myung, Feasible Approaches for Anode-Free Lithium-Metal Batteries as Next Generation Energy Storage Systems, Energy Storage Mater., 2023, 57, 471–496 CrossRef.
- X. Gao, Y. Chen, L. R. Johnson, Z. P. Jovanov and P. G. Bruce, A rechargeable lithium–oxygen battery with dual mediators stabilizing the carbon cathode, Nat. Energy, 2017, 2, 1–7 Search PubMed.
- J. Heine, P. Hilbig, X. Qi, P. Niehoff, M. Winter and P. Bieker, Fluoroethylene carbonate as electrolyte additive in tetraethylene glycol dimethyl ether based electrolytes for application in lithium ion and lithium metal batteries, J. Electrochem. Soc., 2015, 162, A1094 CrossRef CAS.
- S.-J. Zhang, Z.-W. Yin, Z.-Y. Wu, D. Luo, Y.-Y. Hu, J.-H. You, B. Zhang, K.-X. Li, J.-W. Yan and X.-R. Yang, Achievement of high-cyclability and high-voltage Li–metal batteries by heterogeneous SEI film with internal ionic conductivity/external electronic insulativity hybrid structure, Energy Storage Mater., 2021, 40, 337–346 CrossRef.
- T. T. Beyene, H. K. Bezabh, M. A. Weret, T. M. Hagos, C.-J. Huang, C.-H. Wang, W.-N. Su, H. Dai and B.-J. Hwang, Concentrated dual-salt electrolyte to stabilize Li metal and increase cycle life of anode free Li–metal batteries, J. Electrochem. Soc., 2019, 166, A1501–A1509 CrossRef CAS.
- T. T. Beyene, B. A. Jote, Z. T. Wondimkun, B. W. Olbassa, C.-J. Huang, B. Thirumalraj, C.-H. Wang, W.-N. Su, H. Dai and B.-J. Hwang, Effects of concentrated salt and resting protocol on solid electrolyte interface formation for improved cycle stability of anode-free lithium metal batteries, ACS Appl. Mater. Interfaces, 2019, 11, 31962–31971 CrossRef CAS PubMed.
- T. T. Hagos, B. Thirumalraj, C.-J. Huang, L. H. Abrha, T. M. Hagos, G. B. Berhe, H. K. Bezabh, J. Cherng, S.-F. Chiu and W.-N. Su, Locally concentrated LiPF6 in a carbonate-based electrolyte with fluoroethylene carbonate as a diluent for anode-free lithium metal batteries, ACS Appl. Mater. Interfaces, 2019, 11, 9955–9963 CrossRef CAS PubMed.
- J. Zheng, J. A. Lochala, A. Kwok, Z. D. Deng and J. Xiao, Research progress towards understanding the unique interfaces between concentrated electrolytes and electrodes for energy storage applications, Adv. Sci., 2017, 4, 1700032 CrossRef PubMed.
- B. Wu, C. Chen, L. H. Raijmakers, J. Liu, D. L. Danilov, R.-A. Eichel and P. H. Notten, Li-growth and SEI engineering for anode-free Li–metal rechargeable batteries: A review of current advances, Energy Storage Mater., 2023, 57, 508–539 CrossRef.
- Z. Li, H. Rao, R. Atwi, B. M. Sivakumar, B. Gwalani, S. Gray, K. S. Han, T. A. Everett, T. A. Ajantiwalay and V. Murugesan, Non-polar ether-based electrolyte solutions for stable high-voltage non-aqueous lithium metal batteries, Nat. Commun., 2023, 14, 868 CrossRef CAS PubMed.
- T. D. Pham, A. Bin Faheem, J. Kim, H. M. Oh and K.-K. Lee, Practical High-Voltage Lithium Metal Batteries Enabled by Tuning the Solvation Structure in Weakly Solvating Electrolyte, Small, 2022, 18, 2107492 CrossRef CAS PubMed.
- Q. Shi, Y. Zhong, M. Wu, H. Wang and H. Wang, High-capacity rechargeable batteries based on deeply cyclable lithium metal anodes, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 5676–5680 CrossRef CAS PubMed.
- J. Fu, X. Ji, J. Chen, L. Chen, X. Fan, D. Mu and C. Wang, Lithium nitrate regulated sulfone electrolytes for lithium metal batteries, Angew. Chem., 2020, 132, 22378–22385 CrossRef.
- Z. L. Brown, S. Heiskanen and B. L. Lucht, Using triethyl phosphate to increase the solubility of LiNO3 in carbonate electrolytes for improving the performance of the lithium metal anode, J. Electrochem. Soc., 2019, 166, A2523 CrossRef CAS.
- D. W. Kang, J. Moon, H.-Y. Choi, H.-C. Shin and B. G. Kim, Stable cycling and uniform lithium deposition in anode-free lithium-metal batteries enabled by a high-concentration dual-salt electrolyte with high LiNO3 content, J. Power Sources, 2021, 490, 229504 CrossRef CAS.
- M. Tułodziecki, J.-M. Tarascon, P.-L. Taberna and C. Guéry, Catalytic reduction of TFSI-containing ionic liquid in the presence of lithium cations, Electrochem. Commun., 2017, 77, 128–132 CrossRef.
- F. S. Gittleson, R. C. Sekol, G. Doubek, M. Linardi and A. D. Taylor, Catalyst and electrolyte synergy in Li–O2 batteries, Phys. Chem. Chem. Phys., 2014, 16, 3230–3237 RSC.
- Y. Wang and H. Noguchi, Li Plating/Stripping Efficiency in Ether-based Dilute Electrolyte for Anode-free Lithium–metal Batteries: Effect of Operating Potential Range on Subsequent SEI Layer Structure, Batteries Supercaps, 2023, 6, e202300359 CrossRef CAS.
- S. J. An, J. Li, C. Daniel, D. Mohanty, S. Nagpure and D. L. Wood III, The state of understanding of the lithium-ion-battery graphite solid electrolyte interphase (SEI) and its relationship to formation cycling, Carbon, 2016, 105, 52–76 CrossRef CAS.
- I. Shterenberg, M. Salama, H. D. Yoo, Y. Gofer, J.-B. Park, Y.-K. Sun and D. Aurbach, Evaluation of (CF3SO2)2N–(TFSI) based electrolyte solutions for Mg batteries, J. Electrochem. Soc., 2015, 162, A7118 CrossRef CAS.
- G. G. Eshetu, X. Judez, C. Li, M. Martinez-Ibañez, I. Gracia, O. Bondarchuk, J. Carrasco, L. M. Rodriguez-Martinez, H. Zhang and M. Armand, Ultrahigh performance all solid-state lithium sulfur batteries: salt anion's chemistry-induced anomalous synergistic effect, J. Am. Chem. Soc., 2018, 140, 9921–9933 CrossRef CAS PubMed.
- D. Aurbach and M. Moshkovich, A Study of Lithium Deposition–Dissolution Processes in a Few Selected Electrolyte Solutions by Electrochemical Quartz Crystal Microbalance, J. Electrochem. Soc., 1998, 145, 2629 CrossRef CAS.
- S. Koike, T. Fujieda, N. Wakabayashi and S. Higuchi, Electrochemical and quartz microbalance technique studies of anode material for secondary lithium batteries, J. Power Sources, 1997, 68, 480–482 CrossRef CAS.
- N. Tian, C. Hua, Z. Wang and L. Chen, Reversible reduction of Li2CO3, J. Mater. Chem. A, 2015, 3, 14173–14177 RSC.
- K. Naoi, M. Mori and Y. Shinagawa, Study of Deposition and Dissolution Processes of Lithium in Carbonate-Based Solutions by Means of the Quartz-Crystal Microbalance, J. Electrochem. Soc., 1996, 143, 2517–2522 CrossRef CAS.
- H. Muramatsu, E. Tamiya and I. Karube, Computation of equivalent circuit parameters of quartz crystals in contact with liquids and study of liquid properties, Anal. Chem., 1988, 60, 2142–2146 CrossRef CAS.
- K. Kanamura, S. Shiraishi and Z. Takehara, Quartz crystal microbalance study for lithium deposition and dissolution in nonaqueous electrolyte with HF, Electrochemistry, 1999, 67, 1264–1267 CrossRef CAS.
- K. S. Smaran, S. Shibata, A. Omachi, A. Ohama, E. Tomizawa and T. Kondo, Anion-dependent potential precycling effects on lithium deposition/dissolution reaction studied by an electrochemical quartz crystal microbalance, J. Phys. Chem. Lett., 2017, 8, 5203–5208 CrossRef CAS PubMed.
- F. Qiu, X. Zhang, Y. Qiao, X. Zhang, H. Deng, T. Shi, P. He and H. Zhou, An ultra-stable and enhanced reversibility lithium metal anode with a sufficient O2 design for Li–O2 battery, Energy Storage Mater., 2018, 12, 176–182 CrossRef.
- F. Qiu, S. Ren, X. Mu, Y. Liu, X. Zhang, P. He and H. Zhou, Towards a stable Li–CO2 battery: the effects of CO2 to the Li metal anode, Energy Storage Mater., 2020, 26, 443–447 CrossRef.
- P. Verma, P. Maire and P. Novák, A review of the features and analyses of the solid electrolyte interphase in Li–ion batteries, Electrochim. Acta, 2010, 55, 6332–6341 CrossRef CAS.
- D. Giacco, M. Carboni, S. Brutti and A. G. Marrani, Noticeable Role of TFSI− Anion in the Carbon Cathode Degradation of Li–O2 Cells, ACS Appl. Mater. Interfaces, 2017, 9, 31710–31720 CrossRef CAS PubMed.
- S. K. Heiskanen, J. Kim and B. L. Lucht, Generation and evolution of the solid electrolyte interphase of lithium–ion batteries, Joule, 2019, 3, 2322–2333 CrossRef CAS.
- K. P. Yao, D. G. Kwabi, R. A. Quinlan, A. N. Mansour, A. Grimaud, Y.-L. Lee, Y.-C. Lu and Y. Shao-Horn, Thermal stability of Li2O2 and Li2O for Li–air batteries: in situ XRD and XPS studies, J. Electrochem. Soc., 2013, 160, A824 CrossRef CAS.
- Y. Hayashi, S. Yamada, T. Ishikawa, Y. Takamuki, M. Sohmiya, H. Otsuska, K. Ito, Y. Kubo and M. Saito, Enhancement of bifunctional effect for LiNO3/glyme electrolyte by using dual solvent system for Li–O2 batteries, J. Electrochem. Soc., 2020, 167, 020542 CrossRef CAS.
- S. Hy, F. Felix, J. Rick, W.-N. Su and B. J. Hwang, Direct in situ observation of Li2O evolution on Li-rich high-capacity cathode material, Li[NixLi(1−2x)/3Mn(2−x)/3]O2 (0 ≤ x ≤ 0.5), J. Am. Chem. Soc., 2014, 136, 999–1007 CrossRef CAS PubMed.
- L. Andrews, Matrix infrared spectrum and bonding in the lithium superoxide molecule, LiO2, J. Am. Chem. Soc., 1968, 90, 7368–7370 CrossRef CAS.
- Y. Qiao and S. Ye, Spectroscopic investigation for oxygen reduction and evolution reactions with tetrathiafulvalene as a redox mediator in Li–O2 battery, J. Phys. Chem. C, 2016, 120, 15830–15845 CrossRef CAS.
- Z. Peng, S. A. Freunberger, L. J. Hardwick, Y. Chen, V. Giordani, F. Bardé, P. Novák, D. Graham, J.-M. Tarascon and P. G. Bruce, Oxygen reactions in a non-aqueous Li+ electrolyte, Angew. Chem., Int. Ed., 2011, 50, 6351–6355 CrossRef CAS PubMed.
- Y. Wang, N.-C. Lai, Y.-R. Lu, Y. Zhou, C.-L. Dong and Y.-C. Lu, A solvent-controlled oxidation mechanism of Li2O2 in lithium–oxygen batteries, Joule, 2018, 2, 2364–2380 CrossRef CAS.
- L. Luo, B. Liu, S. Song, W. Xu, J.-G. Zhang and C. Wang, Revealing the reaction mechanisms of Li–O2 batteries using environmental transmission electron microscopy, Nat. Nanotechnol., 2017, 12, 535–539 CrossRef CAS PubMed.
- Z. Zhu, A. Kushima, Z. Yin, L. Qi, K. Amine, J. Lu and J. Li, Anion-redox nanolithia cathodes for Li–ion batteries, Nat. Energy, 2016, 1, 1–7 Search PubMed.
- R. S. Assary and L. A. Curtiss, Oxidative decomposition mechanisms of lithium peroxide clusters: an Ab Initio study, Mol. Phys., 2019, 117, 1459–1468 CrossRef CAS.
- Y. Qiao, K. Jiang, H. Deng and H. Zhou, A high-energy-density and long-life lithium–ion battery via reversible oxide–peroxide conversion, Nat. Catal., 2019, 2, 1035–1044 CrossRef CAS.
- D. Sharon, D. Hirsberg, M. Afri, F. Chesneau, R. Lavi, A. A. Frimer, Y.-K. Sun and D. Aurbach, Catalytic behavior of lithium nitrate in Li–O2 cells, ACS Appl. Mater. Interfaces, 2015, 7, 16590–16600 CrossRef CAS PubMed.
- B. N. Olana, S. D. Lin and B.-J. Hwang, In situ diffuse reflectance infrared Fourier-transformed spectroscopy study of solid electrolyte interphase formation from lithium bis (trifluoromethanesulfonyl) imide in 1,2-dimethoxyethane and 1,3-dioxolane with and without lithium nitrate additive over lithium and copper metal anodes, Electrochim. Acta, 2022, 416, 140266 CrossRef CAS.
- W. Li, H. Yao, K. Yan, G. Zheng, Z. Liang, Y.-M. Chiang and Y. Cui, The synergetic effect of lithium polysulfide and lithium nitrate to prevent lithium dendrite growth, Nat. Commun., 2015, 6, 7436 CrossRef CAS PubMed.
- R. Schmitz, R. A. Mueller, R. W. Schmitz, C. Schreiner, M. Kunze, A. Lex-Balducci, S. Passerini and M. Winter, SEI investigations on copper electrodes after lithium plating with Raman spectroscopy and mass spectrometry, J. Power Sources, 2013, 233, 110–114 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp02755h |
| This journal is © the Owner Societies 2024 |