
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSubstituent effects on the photophysics of the kaede chromophore†
Anam
Fatima
,
Giovanni
Bressan
 ,
Eleanor K.
Ashworth
,
Philip C. B.
Page
,
James N.
Bull
* and
Stephen R.
Meech
*
,
Eleanor K.
Ashworth
,
Philip C. B.
Page
,
James N.
Bull
* and
Stephen R.
Meech
*
School of Chemistry, University of East Anglia, Norwich NR4 7TJ, UK. E-mail: s.meech@uea.ac.uk; james.bull@uea.ac.uk
First published on 6th November 2024
Abstract
Kaede is the prototype of the optical highlighter proteins, which are an important subclass of the fluorescent proteins that can be permanently switched from green to red emitting forms by UV irradiation. This transformation has important applications in bioimaging. Optimising brightness, i.e. enhancing fluorescence characteristics, in these proteins is an important objective. At room temperature, the excited state dynamics of the red form of the kaede chromophore are dominated by a broad distribution of conformers with distinct excited state kinetics. Here, we investigate substituent effects on the photophysics of this form of the kaede chromophore. While an electron withdrawing substituent (nitro) red shifts the electronic spectra, the modified chromophores showed no significant solvatochromism. The lack of solvatochromism suggests small changes in permanent dipole moment between ground and excited electronic states, which is consistent with quantum chemical calculations. Ultrafast fluorescence and transient absorption spectroscopy reveal correlations between radiative and nonradiative decay rates of different conformers in the chromophores. The most significant effect of the substituents is to modify the distribution of conformers. The results are discussed in the context of enhancing brightness of optical highlighter proteins.
1. Introduction
Isolation of fluorescent proteins (FPs) from marine jellyfish and corals, and the subsequent creation of numerous mutants, drove a revolution in bioimaging by permitting the labelling of specific proteins in live cells with a variety of colours.1–5 The kaede FP, isolated from the stony coral Trachyphyllia geoffroyi, is the original example of an “optical highlighter”.6,7 These FPs undergo irreversible photoconversion between a green and red emitting chromophore upon UV irradiation. This property has been harnessed to allow permanent in vivo labelling of a sub-population of a sample already specifically labelled with the unconverted FP.8,9 The subsequent temporal-spatial evolution of that sub-population is readily monitored by fluorescence microscopy. The permanent colour change of these photoconvertible FPs distinguishes them from reversibly switchable (rsFPs) examples, which undergo a reversible photochromic conversion between fluorescent and nonfluorescent forms, usually by a sequential photoisomerisation and deprotonation mechanism.1 The rsFPs are widely exploited in super-resolution bioimaging.The mechanism of photoswitching in kaede FP has been described previously.6,8–12 Initially, a green form of the kaede FP exists, in which the chromophore familiar from the original green FP is formed from the tripeptide sequence, His–Tyr–Gly. Irradiation in the UV initiates photochemistry that leads to a β-elimination reaction, ultimately incorporating the His sidechain into the conjugated π system. This extended conjugation gives rise to the red-shifted spectrum of the chromophore (1 in Fig. 1). The red-absorbing chromophore is photostable and yields a quite strongly emissive protein (ϕf ≈ 0.37) with an emission spectrum that is easily distinguished from the original green fluorescence.
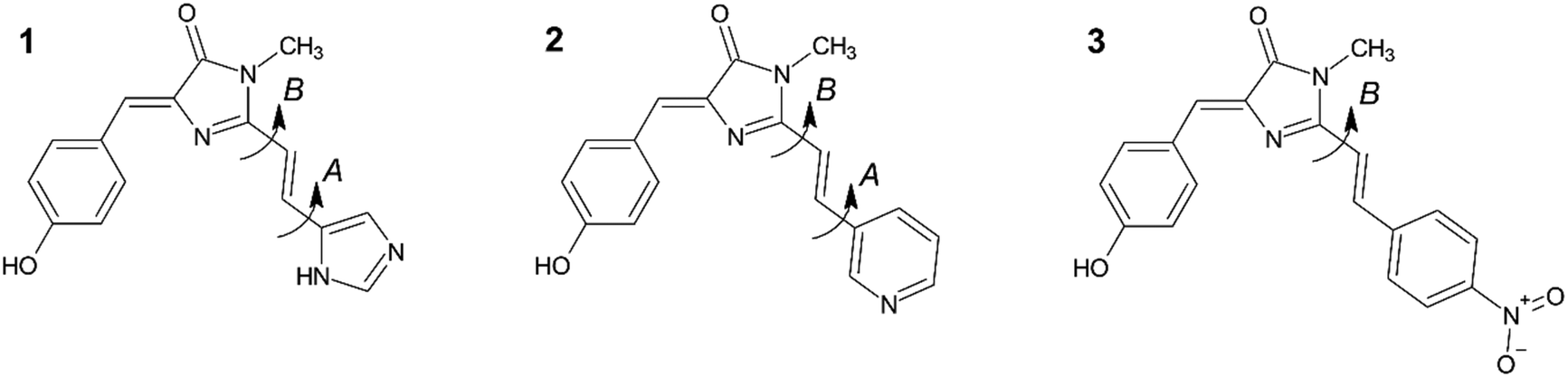 | ||
| Fig. 1 The red kaede FP chromophore analogues considered in this work: 1 – wild-type, 2 – pyridine (meta), 3 – nitrobenzene (para). 1 and 2 both have four possible ground state conformers linked with rotation about the single bonds denoted A and B. On the other hand, 3 possess two conformers linked with rotation about B. Potential energy surfaces (see ESI,† Fig. S1) indicate that the gas phase (i.e. no solvent) conformations lie within 5 kJ mol−1 of each other with interconversion barriers of 25–30 kJ mol−1, suggesting population of multiple conformers at T = 300 K. | ||
Since the discovery of kaede FP, several other optical highlighter proteins have been reported, which together constitute an important and distinct class of FPs.8,9,13 The desirable optical highlighter properties of these FPs has prompted wide interest in the photophysics of the red form of the chromophore, with the ultimate goal of providing a template for the rational design of improved derivatives.12,14–18 Studies on GFP variants have shown that substitutions such as halogenation or nitration on the phenoxy ring or incorporating non-canonical amino acid (ncAAs) into the GFP-like chromophores can significantly shift the absorption and emission spectra to the red, revealing how molecular modifications influence photophysics.19,20 In this work, red form of kaede chromophore 1, and two of its derivatives 2 and 3, have been synthesised (Fig. 1) and their spectroscopy in solution investigated. For 1, the photophysics have been studied in both gas and solution phase.14,16–18,21,22 Significantly, 1 is observed to have very different photophysics to those of the original green fluorescent protein (GFP) chromophore due to the additional conjugated functionality.18 For example, the absorption and emission spectra of both the neutral and anionic forms of 1 are red shifted from the GFP chromophore. In addition to this red-shift, 1 has a much higher quantum yield for fluorescence than the GFP chromophore (ca. 5 × 10−3 and <10−4, respectively, in aqueous solution) and exhibits markedly more complex excited-state decay kinetics, showing high non-exponentiality with sub-picosecond to tens of picosecond decay times.18 Thus, extending the conjugation of the GFP chromophore (HBDI) by a double bond plus single aromatic ring unit has a profound effect on the chromophore photophysics. This contrasts with previous studies where extension of conjugation in HBDI by a single double bond only modified the photophysics slightly (e.g. in the chromophore of the kindling fluorescent protein23).
In this work, we probe the origin of the photophysical changes to the red form of the kaede chromophore and discuss the potential for them to be chemically altered by contrasting the spectroscopy and photophysics of 1 with those of two derivatives having different aromatic substituents. Specifically, we compare solvent- and temperature-dependent spectroscopy and excited state dynamics of 1–3 (Fig. 1) and discuss the results in light of quantum chemical calculations.
2. Methods
2.1 Experimental
2.2 Computational
Quantum chemical calculations were performed using the Gaussian 16 software package.26 Geometries were optimised at the ωB97X-D/cc-pVTZ level of theory27,28 with and without the SMD (solvation model based on density) implicit solvation model assuming EtOH or THF,29 followed by harmonic vibrational frequency analysis. Absorption and fluorescence spectra were modelled in the Franck–Condon-Herzberg–Teller framework as implemented in Gaussian 16.303. Results and analysis
3.1 Steady-state electronic spectroscopy
Absorption spectra of 1–3 in polar hydrogen-bonding ethanol (EtOH, εr = 24.3 at 298 K) and weakly polar THF (εr = 7.6 at 298 K) are shown in Fig. 2; these chromophores are not sufficiently soluble for study in less polar solvents. Absorption maxima are summarised in Table 1. In EtOH, the absorption spectrum of 1 exhibits a broad featureless profile with a maximum at ca. 436 nm. The absorption spectrum of 2 is essentially identical to that of 1. For 3, the absorption spectrum is also broad and featureless, but the maximum is red shifted to 455 nm, indicating a significant effect of the electron withdrawing NO2 group. The absorption spectra of 1 and 2 in THF are again near identical and are blue shifted by ca. 4 nm compared to EtOH. The absorption spectrum of 3 in THF closely matches that in EtOH (i.e. red shifted by ≈19 nm relative to 1 and 2).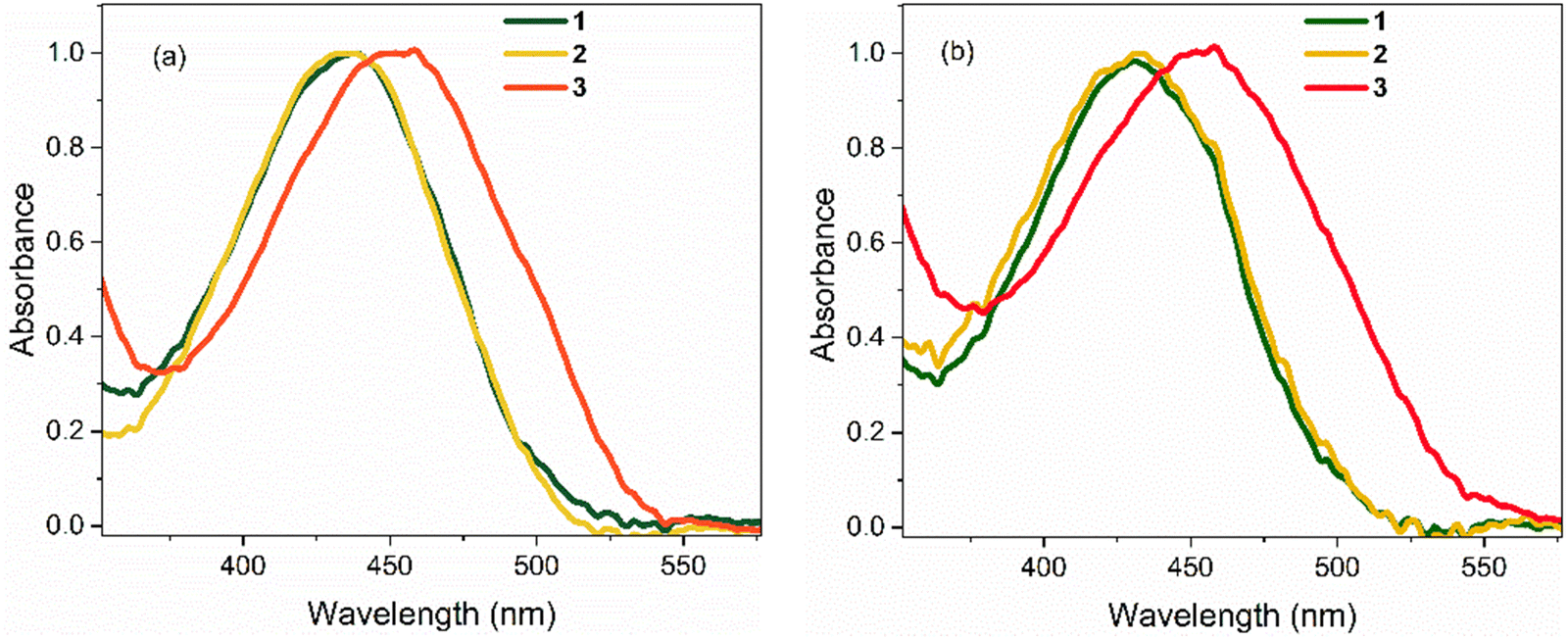 | ||
| Fig. 2 Steady-state absorption spectra of 1–3 at T = 298 K in: (a) EtOH, and (b) THF. In both solvents, the spectra for 1 and 2 are nearly identical. All spectra are normalised to unit intensity. | ||
| Solvent | Molecule | λ abs,max/nm | λ em,max (λav)/nm | Δλ (Δλ)a/cm−1 | Φ f |
|---|---|---|---|---|---|
| a Stokes shift calculated from the average (first moment) fluorescence wavelength, λav. b Reported relative to 1 in THF by integrating the area under the emission spectrum normalised for absorbance. The absolute quantum yield in EtOH was measured as ≈ 5 × 10−3.18 Uncertainties in relative quantum yields are ±0.02. | |||||
| EtOH | 1 | 436 | 525 (539) | 3888 (4383) | 0.12 |
| 2 | 436 | 525 (550) | 3888 (4757) | 0.14 | |
| 3 | 455 | 585 (620) | 4884 (5849) | 0.08 | |
| THF | 1 | 432 | 535 (552) | 4457 (5033) | 1.00 |
| 2 | 432 | 545 (557) | 4800 (5195) | 0.98 | |
| 3 | 455 | 602 (622) | 6537 (7071) | 1.07 | |
Emission spectra for the three chromophores are shown in Fig. 3 (normalised according to absorbance at the excitation wavelength as to display their relative quantum yields); spectral peak maxima, mean fluorescence wavelength, and Stokes shifts are given in Table 1. In both solvents, the emission spectra of 1 and 2 show modest vibronic structure, more obviously in THF, which is not evident in the absorption spectra. This lack of mirror symmetry between absorption and emission spectra has been discussed for 1 in terms of a broad distribution of conformers, which have different distributions in the ground and excited states. The mirror image relation is restored at low temperature (discussed in Section 3.2). In contrast to 1 and 2, the room temperature emission spectra of 3 are broad and essentially featureless. From Fig. 1 and the conformer energetics summarised in the ESI,† we expect 1 and 2 to have four conformers in solution at room temperature. On the other hand, 3 has only two conformers. The broadening of the emission spectra for 3 compared with 1 and 2 is thus unlikely to be associated with conformers, rather our TD-DFT calculations indicate that the rocking motion about bonds A and B sampling geometries accessible at room temperature (T = 300 K ≈ 3 kJ mol−1) produces a broader distribution of vertical transition energies for 3 than for 1 or 2. The same effect occurs on the excited state for fluorescence. This effect is presumably amplified in hydrogen-bonding solvents such as ethanol due to the strong hydrogen bonds with the nitro group.
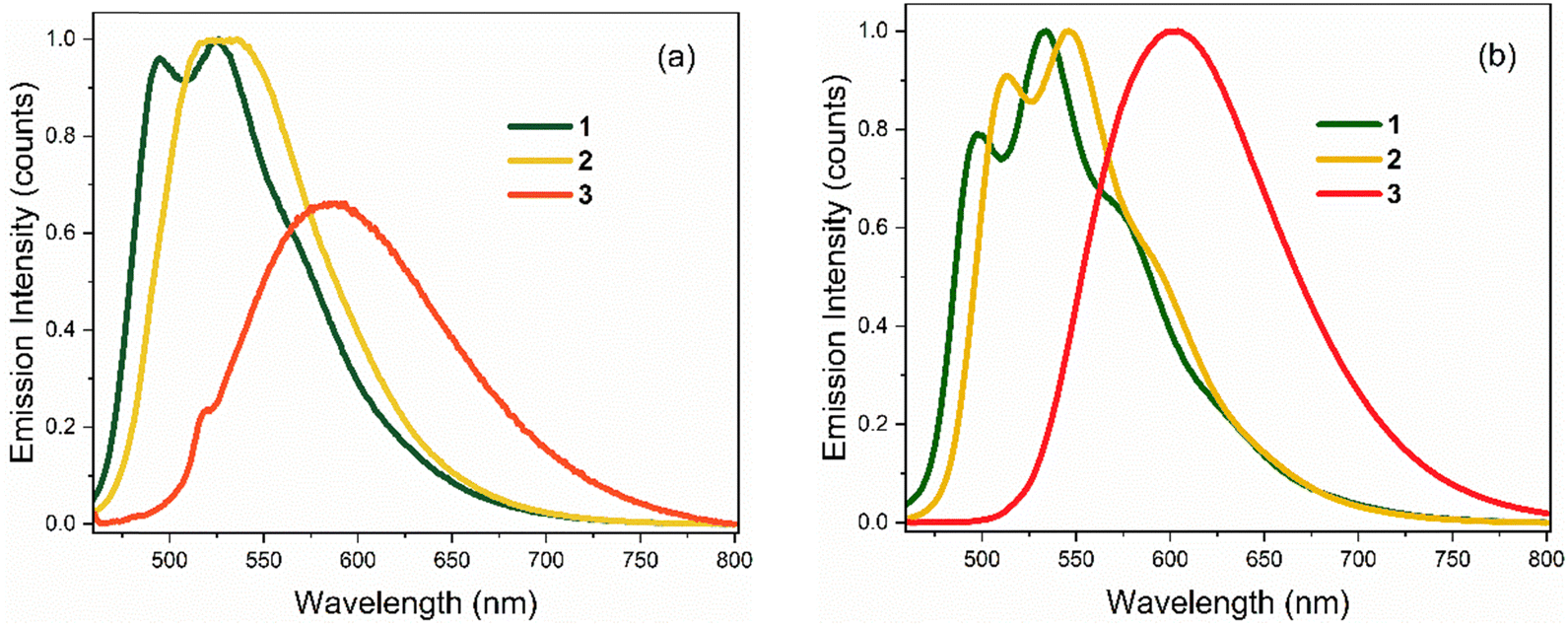 | ||
| Fig. 3 Steady-state emission spectra of 1–3 at T = 298 K in: (a) EtOH, and (b) THF. Spectra are normalised to reflect relative quantum yields (in THF, all three chromophores have similar quantum yields). | ||
The solvent-dependent Stokes shift data (Table 1), which includes data calculated from the first moment of the emission rather than peak maximum to remove the effect of the solvent-dependent vibronic structure, do not suggest strong solvatochromism in these derivatives. The Stokes shift is slightly larger for 3 than for 1 and 2, perhaps suggesting a larger dipole moment change on excitation associated with the electron withdrawing substituent. However, for all three chromophores the Stokes shift is larger in THF than in the more polar EtOH. This result is at odds with expectations from the continuum model of solvatochromism developed by Lippert and Mataga, which estimates the Stokes shift as:
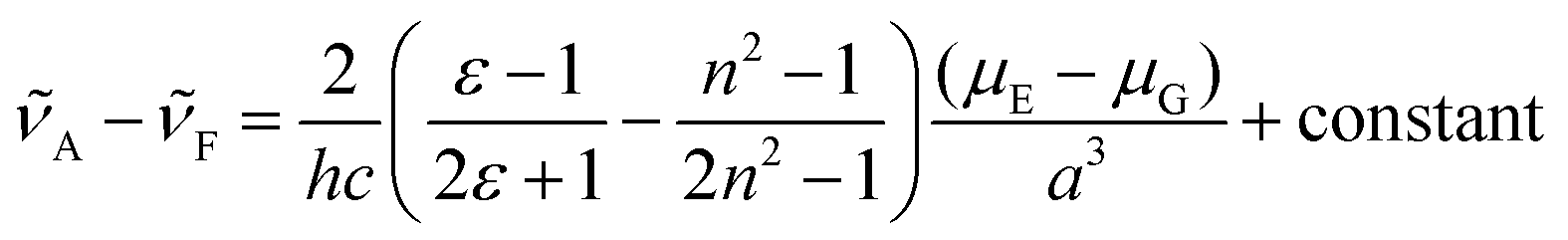
From the Lippert–Mataga equation, polar solvents should result in an increase in Stokes shift whenever there is a significant change in permanent electric-dipole moment between ground and excited state, Δμ = μE − μG. Based on data for 1–3, we conclude that Δμ is small in these chromophores in solution. This conclusion is supported from quantum chemical calculations of the S0 and S1 electronic states (Table 2). The small blue shift observed in THF might then reflect the influence of higher-order terms in the model, or a specific solvent effect not included in continuum models, such as hydrogen bonding. It is significant that incorporation of a strongly electron accepting NO2 group into the conjugated kaede backbone (3) leads to a large spectral red shift but no increase in solvatochromism – i.e., the substituent has not led to a large contribution of charge transfer (CT) configurations in the emitting state. Again, this conclusion is consistent with quantum chemical calculations (Table 2), which predict a modest Δμ for 3.
| μ G/D | μ E/D | |
|---|---|---|
| 1 | 5.8/4.4 | 6.5/5.4 |
| 2 | 4.9/4.5 | 4.8/4.6 |
| 3 | 1.3/0.8 | 2.8/1.8 |
The most remarkable feature of the calculations is that the absolute value of the dipole moments for 3 are calculated to be significantly below those of 1 and 2, indicating a substituent effect on electronic structure underpinning the observed red shift in absorption. This is rationalised from a consideration of the calculated natural type orbitals (NTOs, Fig. 4). Briefly, NTOs are a visualisation of the total orbital changes on excitation in terms of an electron and hole, and account for multiconfigurational character of a transition. For 1 and 2, the S1 ← S0 transitions are dominated by the HOMO → LUMO configurations (>95%), meaning that the NTOs resemble the canonical molecular orbitals. Within in our computational framework (isolated molecule with SMD continuum solvation but no specific solute–solvent interactions), the transition in 3 is delocalised across the molecule with little charge-transfer character. For 3, the transition is still predominantly HOMO → LUMO in character (≈80%) but has gained some HOMO → LUMO+1 character (≈15%). However, because the HOMO has less electron density on the phenyl-nitro moiety, and because the LUMO+1 is more localised on the phenyl-nitro moiety, the transition has a small extent of charge-transfer character. While not as large as might be anticipated for molecule with a strongly electron withdrawing functional group, the charge-transfer character is still larger for 3 than for 1 and 2.
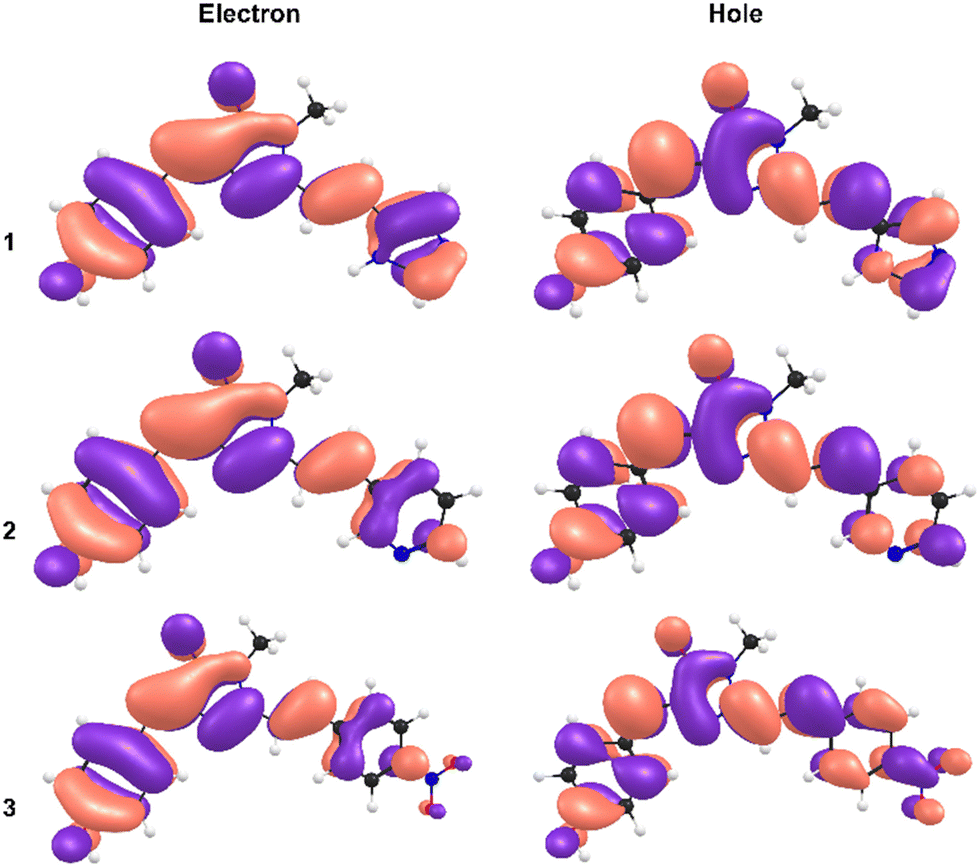 | ||
| Fig. 4 Natural transition orbitals (NTOs) for the electron (left) and the hole (right), for 1 (upper), 2 (middle), and 3 (lower). | ||
In contrast to the electronic spectra, the fluorescence quantum yields for the three chromophores show some solvent polarity dependence (Table 1). While all three chromophores have similar quantum yields in THF, in EtOH (more polar and hydrogen-bonding in character) the quantum yields are reduced by an order of magnitude. The fluorescence quenching in EtOH is particularly marked for 3, which has a 13-fold lower quantum yield than in THF. A similar solvent-dependent fluorescence yield was already noted for 1,18 however the effect is significantly enhanced for 3. This polar solvent quenching in 3 was further investigated in THF/EtOH mixtures, where relative fluorescence intensity and peak wavelength were monitored as a function of EtOH mole fraction (ESI,† Fig. S2). The fluorescence yield decreases rapidly with increasing mole fraction of EtOH (XEtOH); >75% of the total extent of the quenching 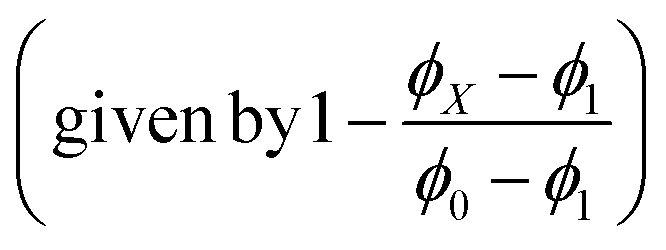 has occurred by XEtOH = 0.25. Concurrently, the spectrum shows a small (ca. 4 nm) red shift. The decrease in fluorescence quantum yield and red shift continues until XEtOH = 0.36, where quenching is 90% complete. At this mole fraction, the emission spectrum starts to shift back to the blue, reaching 585 nm at XEtOH = 1.0 with only a further 10% decrease in emission intensity. From the rapid quenching of fluorescence at low XEtOH, it is concluded that a specific solvent–solute interaction, probably hydrogen bonding, accelerates radiationless decay. The accompaniment of this solvent–solute interaction by a spectral red shift indicates that formation of the complex leads to increased stabilisation of the excited state relative to the ground state. The red shift reverses as the polar medium begins to dominate the chromophore environment, suggesting preferential stabilisation of the ground state by the polar environment. The obvious structural difference between 3 and the other two chromophores is the electron withdrawing nitro group, which introduces a site of charge separation. We expect stronger hydrogen-bonding interactions between the nitro group in 3 compared with the functional groups in 1 and 2, stabilising the charge separation.
has occurred by XEtOH = 0.25. Concurrently, the spectrum shows a small (ca. 4 nm) red shift. The decrease in fluorescence quantum yield and red shift continues until XEtOH = 0.36, where quenching is 90% complete. At this mole fraction, the emission spectrum starts to shift back to the blue, reaching 585 nm at XEtOH = 1.0 with only a further 10% decrease in emission intensity. From the rapid quenching of fluorescence at low XEtOH, it is concluded that a specific solvent–solute interaction, probably hydrogen bonding, accelerates radiationless decay. The accompaniment of this solvent–solute interaction by a spectral red shift indicates that formation of the complex leads to increased stabilisation of the excited state relative to the ground state. The red shift reverses as the polar medium begins to dominate the chromophore environment, suggesting preferential stabilisation of the ground state by the polar environment. The obvious structural difference between 3 and the other two chromophores is the electron withdrawing nitro group, which introduces a site of charge separation. We expect stronger hydrogen-bonding interactions between the nitro group in 3 compared with the functional groups in 1 and 2, stabilising the charge separation.
3.2 Low temperature measurements
The temperature dependence of the emission spectra for 3 are shown in Fig. 5a in EtOH; corresponding data for 1–2 in EtOH and for 1 and 3 in 2-methyl-THF (2MTHF) are shown in the ESI† (Fig. S3). 2MTHF is favoured over THF for low temperature measurements because it forms a more uniform glassy matrix on rapid freezing. As previously reported for 1,18 a >100-fold increase in fluorescence intensity was observed in both solvents when cooling from T = 298 K to 100 K, accompanied by emergence of distinct, well-resolved vibronic structure in the emission spectrum at temperatures close to the glass transition. In both solvents, the fluorescence spectra of 3 exhibit an initial bathochromic shift as the temperature decreases to ca. 210 K, followed by a shift towards the blue, accompanied by the appearance of vibronic structure as the temperature decreases further. The initial red shift was only seen for 3; both 1 and 2 show a monotonic blue shift with decreasing temperature. The red followed by blue shift with decreasing temperature would be consistent with an initial thermochromism for 3 as the solvent polarity increases with decreasing temperature until,33 at the lower temperature, the solvent reorientation time becomes slow compared with the fluorescence lifetime, preventing full solvation from occurring so that the spectrum blue shifts. However, solvent-dependent data for 3 (Fig. 2 and Table 1) did not show large solvatochromism, so thermochromism is also expected to be modest. Thus, the initial red shift more likely reflects preferential population of a red shifted conformer of 3 as temperature is reduced. The absence of this effect for 1 and 2 suggests that the NO2 substituent has modified the excited-state conformer distribution.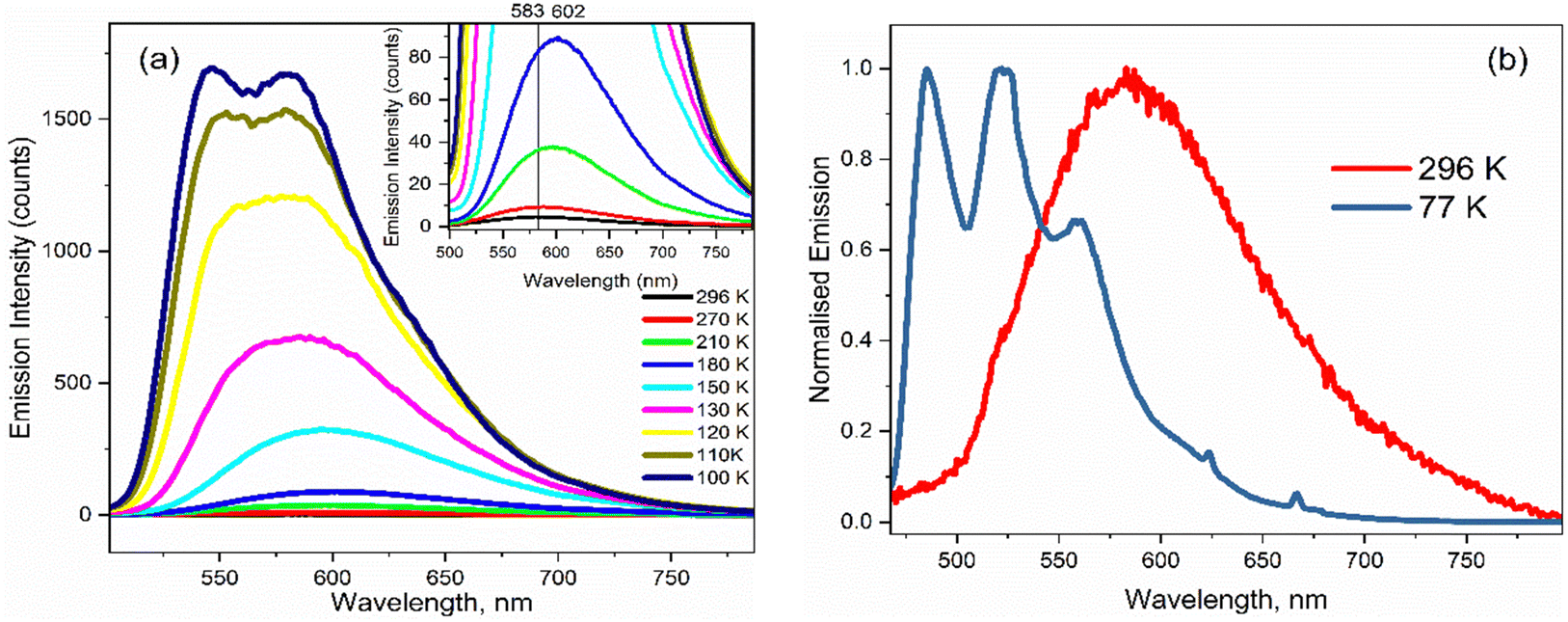 | ||
| Fig. 5 (a) Emission spectra for 3 in EtOH as a function of temperature. The inset highlights the initial red shift in the emission maximum on decreasing from T = 296 K to 180 K. (b) Normalised emission spectra at room temperature and at T = 77 K showing the emergence of vibronic structure at low temperature. | ||
The most striking feature of Fig. 5 is the large increase in fluorescence quantum yield with decreasing temperature. This was observed for all three chromophores in EtOH, and for 1 (see ref. 18) and 3 in 2MTHF, but the temperature dependence is qualitatively different in the two solvents (2 was not studied in 2MTHF because of its similarity to 1). For 3 in 2MTHF, the initial increase in emission quantum yield with decreasing temperature was slower but accelerated strongly at lower temperatures, leading to a biphasic temperature dependence (ESI,† Fig. S4); a similar observation was made for 1. In contrast, the temperature-dependent increase in fluorescence yield in EtOH was continuous for all derivatives, and the data could be analysed by an Arrhenius plot, under the assumption of a single dominant non-radiative decay rate, knr, in which case knr ∝ ϕ−1f. The Arrhenius analysis (ESI,† Fig. S5) yielded an activation energy for radiationless decay at Ea = 11 ± 2 kJ mol−1 for 1–3 in EtOH, in good agreement with our earlier study of 1.18 A probable source of radiationless decay in 1–3 is the presence of a conical intersection along an excited state isomerisation coordinate, which has been well characterised for the GFP chromophore.34–38 In the GFP chromophore, the conical intersection is accessed along a barrierless (albeit complex) pathway leading to ultrafast decay. The longer mean lifetime and higher fluorescence yields for 1–3 suggest a barrier in that coordinate. The measured activation energy contains contributions from both the intrinsic barrier height along the reaction coordinate and the activation energy for viscous flow in the solvent. The latter arises because solvent friction opposes large scale nuclear reorganisation, such as an excited state isomerisation (as was established for 1).18 Solvent friction for a given solvent will make the same contribution in the measured Ea of 1–3, so the common overall Ea observed suggests that the intrinsic barrier to the conical intersection is not a strong function of substituent.
As the glass transition temperature of the solvent is approached, the increase in fluorescence intensity is accompanied by the emergence of distinct, well resolved vibronic structure in the emission spectra (Fig. 5b). Also, the fluorescence excitation spectra for the three variants in EtOH at T = 77 K revealed corresponding vibronic structures in the S0 ← S1 transition, indicating the emergence of a mirror-image relation between fluorescence and absorption at low temperature (ESI,† Fig. S6). The manifestation of clear vibronic structures at low temperature points to an inhomogeneous distribution of solute–solvent structures around the equilibrium configuration at room temperature, with this conformational distribution being coupled to the transition energy, thus leading to the broadened spectrum observed (Fig. 5a and b). As the temperature decreases, the thermal distribution narrows, suppressing inhomogeneous broadening, in turn enabling the recovery of vibronic structures in the emission spectra. The vibronic structure is largely reproduced using Franck–Condon-Herzberg–Teller simulations (see Fig. S7, ESI†) and is consistent with groups of transitions as well as combination bands involving stretching modes on the two terminal ring systems (1600–1700 cm−1 depending on the functional group) and in-plane rocking modes.
3.3 Time-resolved spectroscopy
The three chromophores in EtOH and THF were further studied by ultrafast spectroscopy. Time resolved fluorescence (TRF) measurements were used to probe the decay kinetics of the emissive states with ≈75 fs time resolution, while transient absorption (TA) with ≈120 fs resolution was used to probe ground state recovery (and any non-emissive excited states). The decay kinetics of 1 were studied earlier in several solvents and were shown to exhibit an unusually broad distribution of excited-state decay times.18 This was discussed in terms of distributions of conformers with widths that differ in the ground and excited states.| Solvent | Molecule | α 1 (wt%) | τ 1/ps | α 2 (wt%) | τ 2/ps | α 3 (wt%) | τ 3/ps | α 4 (wt%) | τ 4/ps | 〈τm〉/ps |
|---|---|---|---|---|---|---|---|---|---|---|
| a 3 required a small risetime component to suitably fit the data (as assessed by fitting residuals), with a component weighting of ≈0.1%. Interpretations for these risetime components are described later in the text. | ||||||||||
| EtOH | 1 | 55 (49.5) | 0.40 | 40 (36) | 3.0 | 16 (14.4) | 32.2 | — | — | 6 |
| 2 | 138 (57.5) | 0.40 | 69 (28.7) | 3.7 | 33 (13.7) | 52.3 | — | — | 8 | |
| 3 | −0.2 (0.10)a | 0.08 | 118 (57.0) | 4.6 | 89 (42.9) | 19.0 | — | — | 10 | |
| THF | 1 | 30 (36.5) | 0.30 | 24 (29.2) | 5.0 | 12 (14.6) | 31.1 | 16 (19.5) | 320 | 68 |
| 2 | 30 (36.5) | 0.15 | 34 (41.4) | 3.0 | 8 (9.0) | 30.2 | 10 (12) | 500 | 65 | |
| 3 | −0.05 (0.07)a | 0.12 | 36 (52.1) | 1.7 | 13 (18.8) | 29.8 | 20 (28.9) | 450 | 137 | |
In both solvents, 3 is distinct from 1 and 2 in that a sub-picosecond rising component was identified and the sub-picosecond fluorescence decay time is absent; thus, the decay, while still non-single exponential, could be fit with one fewer decaying components than 1 and 2. The risetime was recorded as a function of wavelength (ESI,† Fig. S8 and Table S1), and was found to occur principally on the red edge of the emission. In many heteroaromatic systems a red edge risetime is indicative of polar solvation dynamics, i.e. the time-dependent polar solvent stabilisation of a permanent dipole moment that changes on electronic excitation. In such a case, the risetime observed is principally a function of the solvent dynamics (i.e. a function of the dielectric relaxation time).39 Solvation times for EtOH and THF are different to one another, being several picoseconds for EtOH and sub-picosecond for THF. These solvent dynamics data contrast with the data in Table 3, where the risetimes are ca. 100 fs and similar in both solvents. Considering these data, along with the observed weak solvatochromism in the steady-state measurements, and the absence of a risetime for 1 and 2, we conclude that the risetime in 3 reflects intramolecular nuclear relaxation and not solvent dynamics. Thus, uniquely, 3 has a risetime but lacks the sub-picosecond decay of 1 and 2. This suggests that some conformers in the excited state of 1 and 2 can rapidly (<1 ps) access a conical intersection with the ground state, leading to the sub-picosecond decay time. In contrast, for 3, a blue-emitting state relaxes rapidly on the excited state surface to populate red-emitting conformers rather than the ground state (Fig. 7).
A second distinctive feature of 3 is the remarkable narrowing of the distribution of excited-state decay times in EtOH compared to both 1 and 2, and also 3 in THF. This is evident in Fig. 6b and Table 3, where the distribution is seen to be a few to tens of picoseconds, rather than sub-picosecond to hundreds of picoseconds seen in the other solutes and solvent. This result is ascribed to a narrower distribution of conformers being populated in the excited state upon electronic excitation, which may arise from a narrower initial ground state distribution (see Fig. 7) by virtue of only two conformers (compared with four conformers for 1 and 2, see above) coupled with role of hydrogen-bonding in EtOH stabilising the nitro group compared with in THF.
 | ||
| Fig. 6 Ultrafast fluorescence up-conversion of 1–3 excited at ≈400 nm in (a) EtOH and (b) THF. Time-resolved emission decays were recorded at the wavelength maximum of the respective emission spectrum at T = 298 K. The emission counts are presented on a log scale after background subtraction to emphasise the multi-exponential nature of the decays. Insets show a magnified view of the sub-picosecond decay and rising components. The grey curve in the insets show the instrument response function. | ||
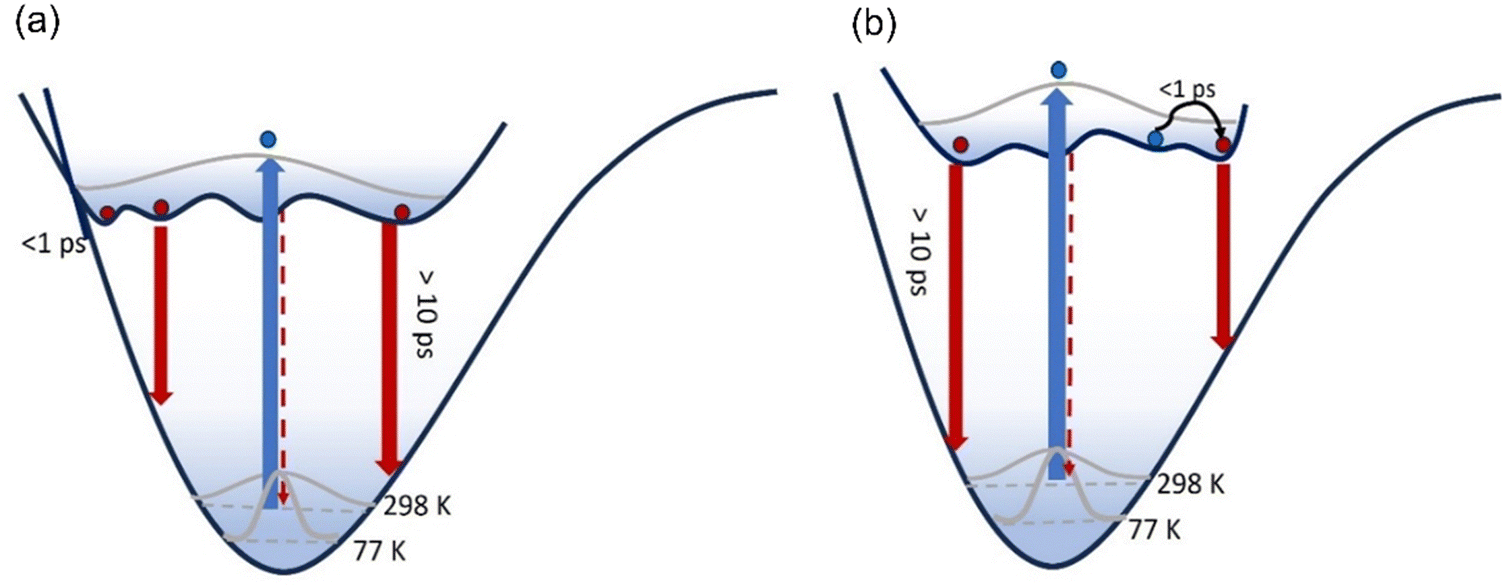 | ||
| Fig. 7 Excitation of a broad ground state conformer distribution populates an excited state PES characterised by multiple minima with low energy barriers which are populated within 100 fs. These conformers then relax to the ground state, each emitting fluorescence with distinct spectra transition moments and lifetimes. For 3, the excited state PES is modified (b) compared to 1 and 2 (a). Initially the blue-emitting state rapidly relaxes over a low barrier to populate a red-emitting state with large transition moment within 1 ps giving the risetime observed. | ||
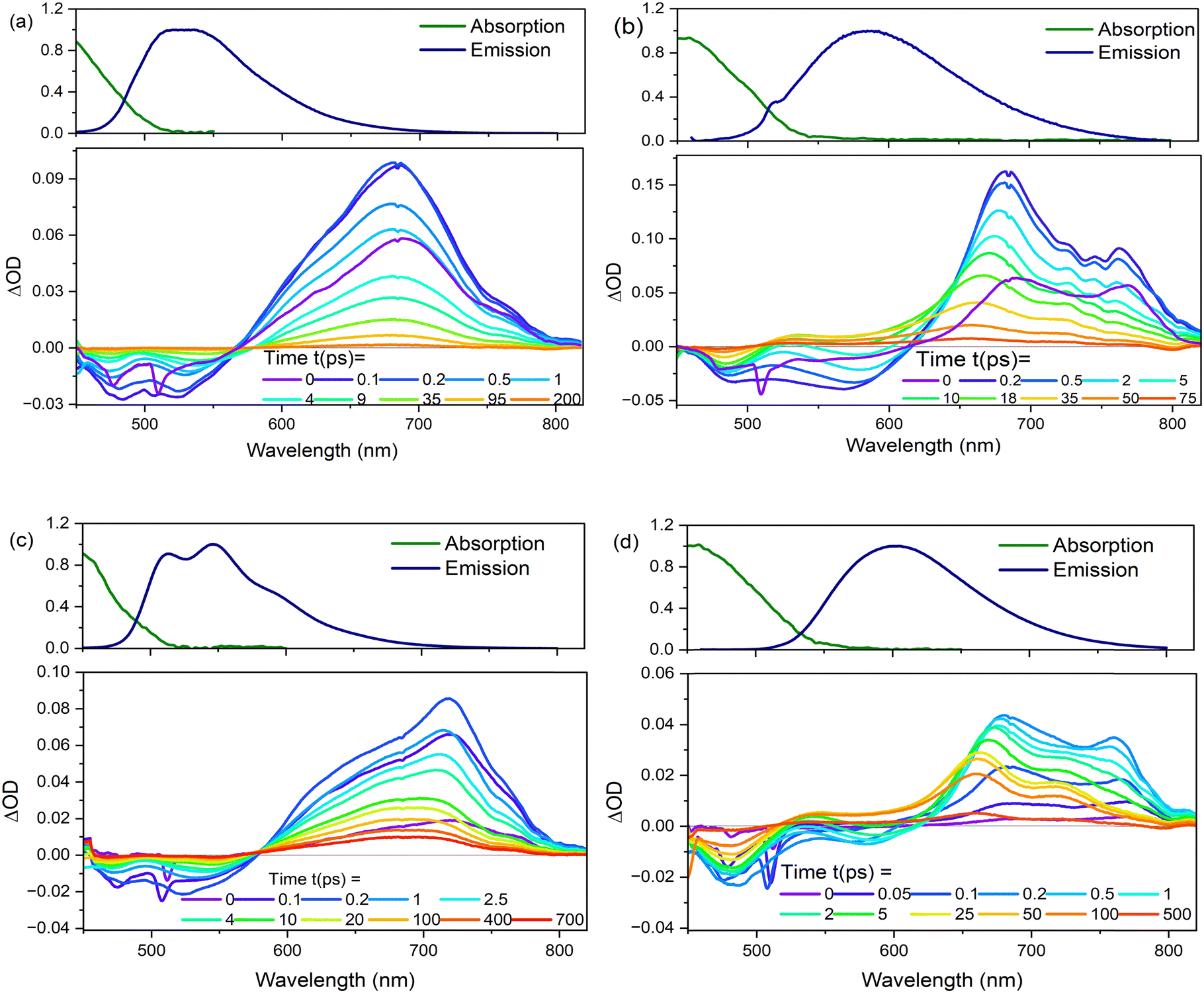 | ||
| Fig. 8 Transient absorption spectra plotted as a function of different delays after the pump pulse for (a) 2 and (b) 3 in EtOH, and (c) 2, and (d) 3 in THF at T = 298 K. Steady-state absorption and emission spectra are shown for comparison of GSB and SE. | ||
| Solvent | 1 | 2 | 3 | |
|---|---|---|---|---|
| Ethanol | τ 1/fs | 326 | 143 | 687 |
| τ 2/ps | 3 | 1.6 | 8 | |
| τ 3/ps | 50 | 45 | 31 | |
| THF | τ 1/fs | 333 | 265 | 288 |
| τ 2/ps | 8 | 2.5 | 3 | |
| τ 3/ps | 58 | 20 | 40 | |
| τ 4/ps | 497 | 904 | 293 |
The TA data were globally analysed using GloTarAn40 in terms of the minimum number of exponential components, three for EtOH and four for THF (quality of fit data are presented in the ESI† for key wavelengths, Fig. S10). The additional component for THF is consistent with the observations of more complex fluorescence decay observed in that solvent (see Section 3.3.1). Previously, the TA spectra of 1 were analysed with only two decaying components,18 but the signal-to-noise has been improved here permitting this more detailed analysis of all three chromophores. Analysis in terms of decay-associated difference spectra (DADS, i.e. under the assumption of a parallel decay model, which is consistent with the TRF data) are shown in Fig. 9 with the corresponding decay constants in Table 4. The complete TA data and full analyses (including the evolution ADS (EADS) that result from a sequential kinetic model) are presented in the ESI† (Fig. S11 and S12). Overall, there is qualitative agreement between the TA time constants from DADS and time constants from TRF (compare Tables 3 and 4), considering that the TRF were recorded at a single wavelength (when the data are known to be somewhat wavelength dependent18). However, the ≈100 fs risetime for 3 was not clearly resolved in TA, which may be associated with the lower time resolution in our measurements.
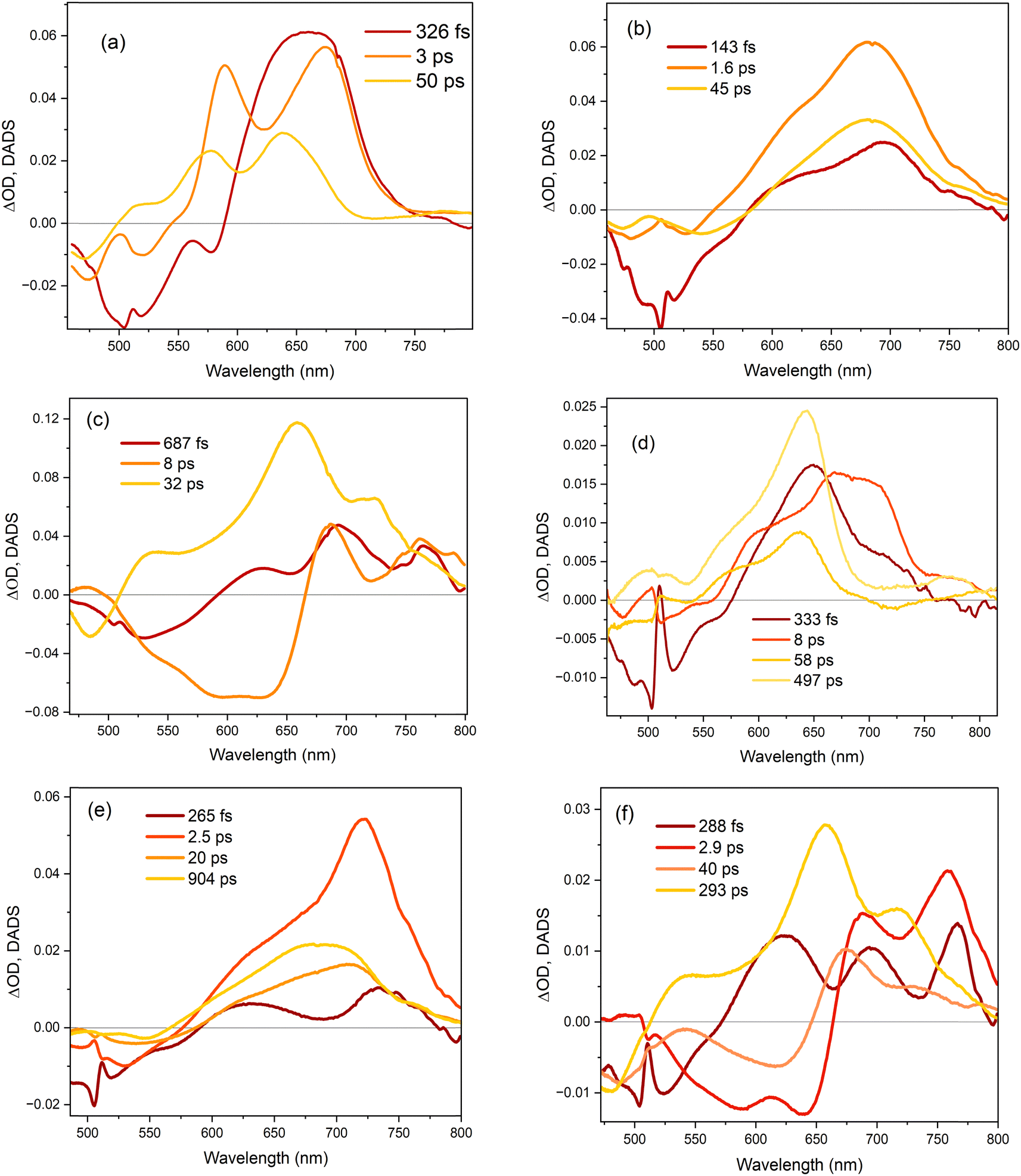 | ||
| Fig. 9 Decay associated difference spectra (DADS) for (a) 1, (b) 2 and (c) 3 in ethanol, and (d) 1, (e) 2 and (f) 3 in THF. The DADS were obtained from global fits of the kinetic traces to the transient absorption spectra. | ||
For 1 in EtOH, the EADS (ESI† Fig. S11) and DADS (Fig. 9a and d) are similar, with the fastest resolved component showing a single broad ESA and major contributions to the SE. The biphasic contribution to the ESA appears in the longer-lived components, and both ESA and SE shift to shorter wavelength. In THF, the sub-picosecond SE remains prominent. The relative prominence of the SE in the shortest-lived DADS can arise either because the shorter-lived conformers have the largest weight or the largest absorption cross section. The latter explanation (larger cross section) is favoured since SE is weaker in subsequent DADS but the ESA is not greatly reduced. The DADS resolve the red and blue shifted contributions of the ESA into picosecond (red shifted) and tens of picosecond (blue shifted) contributions. The EADS shows a similar picture of faster decay of the red shifted ESA (Fig. S12c and d, ESI†). Thus, the conformers with different lifetimes also have distinct ESA, pointing to differences in their electronic structure. Chromophore 2 shows qualitatively similar behaviour to 1 (Fig. 9b and e), but the two components in the ESA (ESI† Fig. S11) are less well resolved. Once again, 3 shows qualitatively different behaviour to 1 and 2. In both EtOH and THF, the maximum SE appears in the second (picosecond time constant) DADS, recalling the sub-picosecond risetime and absence of sub-picosecond decay in the TRF. Evidently, in 3, the fast relaxation populated the conformer with the largest SE cross section. The ESA is again resolved into two components, as for 1, except the shorter-lived component is more strongly red shifted, to wavelengths longer than 750 nm.
4 Discussion
The extension of the GFP chromophore by an additional double bond and imidazole ring in kaede had a dramatic effect on the chromophore photophysics, not only in shifting the spectrum markedly to the red but also increasing the quantum yield of fluorescence and making the decay kinetics markedly more complex, as described elsewhere.18 We speculated that kaede photophysics might also be sensitive to the nature of the group extending the conjugation. In fact, the effect of modifying the nature of the histidine ring is shown here to be much more modest. A detailed investigation of steady state and time resolved photophysics showed that exchanging the imidazole in 1 for a pyridine in 2 does not lead to a large perturbation in either spectra or dynamics, neither does it change the effect of solvent polarity. In neither case was significant solvatochromism observed, consistent with the absence of a large contribution of charge transfer configurations in the excited state. This result was in good agreement with quantum chemical calculations. When the ring substituent in 1 was replaced with a strong electron withdrawing nitrobenzene group (3) the substituent effect was greater than for 2, leading to a significant red shift in absorption and emission. Furthermore, the emission is broader and did not show the weak vibronic structure seen for 1 and 2. However, despite the electron withdrawing character of the NO2 substituent, 3 also does not show significant solvatochromism (consistent with the electronic structure calculations).While the emission of 1–3 is much stronger than for the GFP chromophore, it remains below 1% at room temperature. The fluorescence yield is enhanced at low temperature, showing that the radiationless pathway that dominates excited state decay at room temperature is thermally activated, and, at least in EtOH, all three derivatives have very similar activation energies for radiationless decay. In all three cases, vibronic structure is resolved at low temperature, consistent with thermally populated distribution of conformers at higher temperature. Further analysis revealed the Franck–Condon active vibrational modes as ring stretching and rocking modes.
TRF and TA data showed that the excited state decay kinetics departed dramatically from a single exponential model, but that the data could be understood based on a broad ground state distribution of conformers that, upon electronic excitation, rapidly populate distinct minima on the excited state surface. These excited state conformers were shown to have distinct emission lifetimes, transition moments and transient absorption spectra. The most significant substituent effect was on the conformer distribution. While 1 and 2 have very similar dynamics, the electron withdrawing substituent on 3 gave a narrower distribution with longer lifetimes.
In the context of the behaviour of the chromophore in the protein matrix, there are only two ultrafast studies of the red form of kaede, the protein in which 1 is the chromophore.41,42 In the protein at physiological pH, 1 exists in both neutral and anion forms. The dominant photophysical process observed is Förster resonance energy transfer (FRET) from the neutral to the red-shifted anion form on a 10–20 ps timescale. Inter-protein FRET is possible because kaede exists as a tetramer, placing donor and acceptor in close proximity. The picosecond timescale FRET is of course only possible because of the much greater mean lifetime of 1 in kaede compared to in solution, which is reported as several nanoseconds.42 This is consistent with the enhanced fluorescence quantum yield in the protein (0.3 compared to 0.005).
A key question is the mechanism of fluorescence enhancement in the protein, and whether the substituent effects revealed here suggest a route to further improving the overall brightness of the optical highlighter FPs. The most striking feature of the photophysics of 1–3 is the complex multi-exponential decay kinetics. This was assigned to multiple excited state conformers, and the comparison for TRF and TA data suggests each conformer has a distinct transition moment and radiationless decay rate. For optimum brightness, it is necessary to optimise for large transition moment and small radiationless decay. Unfortunately, the present data show that these two scales together (Fig. 8), so that relationship must be overcome in the protein. The observation that the electron withdrawing nitrobenzene ring modifies the conformer distribution suggests a route forward, using unnatural amino acid substitution to modify the histidine side group to be more electron withdrawing. For FPs derived from avGFP, the enhancement is >104 and the mechanism has been the topic of much research. The most recent work suggests that the twisting reaction, which dominates excited state dynamics in solution, is suppressed in the protein by means of electrostatic interaction with the surrounding matrix of amino acid residues.43–46 Thus, modifying the electrostatic environment of the chromophore may be a route to improving brightness in kaede. Although, neither experiment nor calculation pointed to large changes in electrostatic interaction for 1–3, comparison of THF and ethanol suggested that suppression of hydrogen-bonding interactions may improve fluorescence yield.
5. Conclusions
The photophysics of the red form chromophore of the optical highlighter FP kaede have been studied by steady state and ultrafast spectroscopy, and quantum chemical calculations. It was known that the addition of a group extending the conjugation of the GFP chromophore had a dramatic effect on the chromophore photophysics. Here, we probed whether the nature of the extending moiety had a similarly dramatic effect. To this end 1–3 were compared, and in general changes in the nature of the group had only a modest effect. The photophysics are complex, which is assigned to the existence of multiple excited-state conformers. The principal substituent effect was a change in the distributions of conformers. Possible ways in which these results might inform the design of brighter optical highlighter proteins was discussed, including genetic modification of the chromophore and its electrostatic environment.Data availability
Additional data and coordinates from calculations are available in ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to EPSRC for financial support to SRM (EP/X011410/1, EP/V00817X/1, EP/J009148/1) and JNB (EP/W018691/1). EKA thanks UEA for the award of a studentship.References
- R. Nifosì, B. Storti and R. Bizzarri, La Rivista del Nuovo Cimento, 2024, 47, 91–178 CrossRef.
- V. Sample, R. H. Newman and J. Zhang, Chem. Soc. Rev., 2009, 38, 2852–2864 RSC.
- R. N. Day and M. W. Davidson, Chem. Soc. Rev., 2009, 38, 2887–2921 RSC.
- N. C. Shaner, P. A. Steinbach and R. Y. Tsien, Nat. Methods, 2005, 2, 905–909 CrossRef CAS PubMed.
- R. Y. Tsien, Annu. Rev. Biochem., 1998, 67, 509–544 CrossRef CAS PubMed.
- H. Mizuno, T. K. Mal, K. I. Tong, R. Ando, T. Furuta, M. Ikura and A. Miyawakil, Mol. Cell, 2003, 12, 1051–1058 CrossRef CAS PubMed.
- R. Ando, H. Hama, M. Yamamoto-Hino, H. Mizuno and A. Miyawaki, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 12651–12656 CrossRef CAS PubMed.
- V. Adam, M. Lelimousin, S. Boehme, G. Desfonds, K. Nienhaus, M. J. Field, J. Wiedenmann, S. McSweeney, G. U. Nienhaus and D. Bourgeois, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 18343–18348 CrossRef CAS.
- J. Wiedenmann, S. Ivanchenko, F. Ostwald, F. Schmitt, C. Rocker, A. Salih, K. D. Spindler and G. U. Nienhaus, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 15905–15910 CrossRef CAS.
- I. Hayashi, H. Mizuno, K. I. Tong, T. Furuta, F. Tanaka, M. Yoshimura, A. Miyawaki and M. Ikura, J. Mol. Biol., 2007, 372, 918–926 CrossRef CAS.
- R. M. Wachter, J. L. Watkins and H. Kim, Biochemistry, 2010, 49, 7417–7427 CrossRef CAS.
- T. D. Krueger, J. N. Henderson, I. L. Breen, L. Zhu, R. M. Wachter, J. H. Mills and C. Fang, Front. Chem, 2023, 11, 1328081 CrossRef CAS PubMed.
- S. Habuchi, H. Tsutsui, A. B. Kochaniak, A. Miyawaki and A. M. van Oijen, PLoS One, 2008, 3, e3944 CrossRef PubMed.
- N. S. Baleeva, K. A. Myannik, I. V. Yampolsky and M. S. Baranov, Eur. J. Org. Chem., 2015, 5716–5721 CrossRef CAS.
- S. O. Zaitseva, E. R. Zaitseva, A. Y. Smirnov, N. S. Baleeva and M. S. Baranov, J. Bioorg. Chem., 2020, 46, 120–123 CrossRef CAS.
- E. K. Ashworth, M. H. Stockett, C. Kjaer, P. C. B. Page, S. R. Meech, S. B. Nielsen and J. N. Bull, J. Phys. Chem. A, 2022, 126, 1158–1167 CrossRef CAS.
- N. J. A. Coughlan, M. H. Stockett, C. Kjaer, E. K. Ashworth, P. B. C. Page, S. R. Meech, S. B. Nielsen, L. Blancafort, W. S. Hopkins and J. N. Bull, J. Chem. Phys., 2021, 155, 124324 CrossRef PubMed.
- K. Addison, P. Roy, G. Bressan, K. Skudaite, J. Robb, P. C. B. Page, E. K. Ashworth, J. N. Bull and S. R. Meech, Chem. Sci., 2023, 14, 3763–3775 RSC.
- S. Zhang and H.-w Ai, Nat. Chem. Biol., 2020, 16, 1434–1439 CrossRef CAS.
- B. G. Oscar, L. Zhu, H. Wolfendeen, N. D. Rozanov, A. Chang, K. T. Stout, J. W. Sandwisch, J. J. Porter, R. A. Mehl and C. Fang, Front. Mol. Biosci., 2020, 7, 131 CrossRef CAS.
- J. Tay, M. A. Parkes, K. Addison, Y. Chan, L. Zhang, H. C. Hailes, P. C. Bulman Page, S. R. Meech, L. Blancafort and H. H. Fielding, J. Phys. Chem. Lett., 2017, 765–771 CrossRef CAS PubMed.
- I. V. Yampolsky, A. A. Kislukhin, T. T. Amatov, D. Shcherbo, V. K. Potapov, S. Lukyanov and K. A. Lukyanov, Bioorg. Chem., 2008, 36, 96–104 CrossRef CAS PubMed.
- K. Addison, I. A. Heisler, J. Conyard, T. Dixon, P. C. B. Page and S. R. Meech, Faraday Discuss., 2013, 163, 277–296 RSC.
- I. A. Heisler, M. Kondo and S. R. Meech, J. Phys. Chem. B, 2009, 113, 1623–1631 CrossRef CAS.
- C. R. Hall, J. Conyard, I. A. Heisler, G. Jones, J. Frost, W. R. Browne, B. L. Feringa and S. R. Meech, J. Am. Chem. Soc., 2017, 139, 7408–7414 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Rev. B.01, Gaussian, Inc., Wallingford CT, 2016.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- T. H. Dunning, Jr., J. Chem. Phys., 1989, 90, 1007–1023 CrossRef.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- F. Santoro, A. Lami, R. Improta, J. Bloino and V. Barone, J. Chem. Phys., 2008, 128, 224311 CrossRef PubMed.
- A. Kawski, Z. Naturforsch., A: Phys. Sci., 2002, 57, 255–262 CrossRef CAS.
- N. Bakhshiev, Opt. Spectrosc., 1959, 7, 29 Search PubMed.
- N. H. Damrauer and J. K. McCusker, Inorg. Chem., 1999, 38, 4268–4277 CrossRef CAS.
- C. M. Jones, N. H. List and T. J. Martinez, Chem. Sci., 2021, 12, 11347–11363 RSC.
- N. H. List, C. M. Jones and T. J. Martinez, Chem. Sci., 2022, 13, 373–385 RSC.
- A. Toniolo, S. Olsen, L. Manohar and T. J. Martinez, Faraday Discuss., 2004, 127, 149–163 RSC.
- M. E. Martin, F. Negri and M. Olivucci, J. Am. Chem. Soc., 2004, 126, 5452–5464 CrossRef CAS PubMed.
- A. Sinicropi, T. Andruniow, N. Ferre, R. Basosi and M. Olivucci, J. Am. Chem. Soc., 2005, 127, 11534–11535 CrossRef CAS.
- M. L. Horng, J. A. Gardecki, A. Papazyan and M. Maroncelli, J. Phys. Chem., 1995, 99, 17311–17337 CrossRef CAS.
- J. J. Snellenburg, S. P. Laptenok, R. Seger, K. M. Mullen and I. H. M. van Stokkum, J. Stat. Soft., 2012, 49, 1–22 Search PubMed.
- H. Hosoi, H. Mizuno, A. Miyawaki and T. Tahara, J. Phys. Chem. B, 2006, 110, 22853–22860 CrossRef CAS.
- E. Fron, M. Sliwa, V. Adam, J. Michiels, S. Rocha, P. Dedecker, J. Hofkens and H. Mizuno, Photochem. Photobiol. Sci., 2014, 13, 867–874 CrossRef CAS PubMed.
- J. W. Park and Y. M. Rhee, J. Am. Chem. Soc., 2016, 138, 13619–13629 CrossRef CAS.
- M. G. Romei, C. Y. Lin, I. I. Mathews and S. G. Boxer, Science, 2020, 367, 76 CrossRef CAS.
- J. Chang, M. G. Romei and S. G. Boxer, J. Am. Chem. Soc., 2019, 141, 15504–15508 CrossRef CAS PubMed.
- C. Y. Lin, M. G. Romei, L. M. Oltrogge, I. I. Mathews and S. G. Boxer, J. Am. Chem. Soc., 2019, 141, 15250–15265 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp03272a |
| This journal is © the Owner Societies 2024 |