
The molecular mechanism of the triplet state formation in bodipy-phenoxazine photosensitizer dyads confirmed by ab initio prediction of the spin polarization†
Maria
Kosaka
 *a,
Katsuki
Miyokawa
a and
Yuki
Kurashige
*abc
*a,
Katsuki
Miyokawa
a and
Yuki
Kurashige
*abc
aDepartment of Chemistry, Graduate School of Science, Kyoto University, Kitashirakawa Oiwake-cho, Sakyo-ku Kyoto 606-8502, Japan. E-mail: kosaka@theoc.kuchem.kyoto-u.ac.jp; kura@kuchem.kyoto-u.ac.jp
bFOREST, JST, Honcho 4-1-8, Kawaguchi, Saitama 332-0012, Japan
cCREST, JST, Honcho 4-1-8, Kawaguchi, Saitama 332-0012, Japan
First published on 30th October 2024
Abstract
Efficient formation of excited triplet states on metal-free photosensitizer dyads, bodipy-phenoxazine (BDP-PXZ) and tetramethylbodipy-phenoxazine (TMBDP-PXZ), was investigated using ab initio calculations. We revealed the reason why two different triplet transient species, 3CT and 3BDP, can co-exist only for BDP-PXZ as observed in the previous study with the TR-EPR measurements. It was found that the state mixing of 3CT enables the transition from 1CT to 3CT and 3BDP states only for BDP-PXZ. This mixing effect is commonly seen in the singlet states of twisted intermolecular charge transfer molecules, though the key factor that determines the mixing of the excited states of the dyes was found to be the electron-donating ability of the substituents rather than their steric hindrance. This mechanism was corroborated by comparing the spin polarization ratio of the triplet spin-sublevels measured by TR-EPR with the theoretical predictions. The spin polarization ratio of the triplets should contain information about the transition via intersystem crossing, e.g. the twisted angle of two chromophores of the dyad, and thus it can be a powerful tool to analyze the molecular mechanism of photochemical processes at the electronic structure level. These insights on the molecular structures’ effect provided by this theoretical study would be a compass to molecular design of metal-free triplet photosensitizers.
1 Introduction
Triplet excited states of organic chromophores are some of the most familiar paramagnetic species in chemistry. Because their radiative and non-radiative decays to the ground state are spin-forbidden processes, they usually have a long enough lifetime to collide with or transfer the energy to other molecules, which allows for a wide range of applications, e.g., photon upconversion,1–5 triplet sensitizers,6–10 dynamic nuclear polarization,11–14etc. Conversely, the generation of triplet excited states of metal-free organic chromophores by photo-irradiation is not so efficient because the spin–orbit coupling (SOC) which induces intersystem crossing (ISC) from the S1 state to the Tn state is usually small, except for a few cases. One such exception is the transition between n–π* and π–π* excited states, in which the transition between the in-plane and out-of-plane 2p orbitals on an atom generates angular momentum, but there is a limited variety of organic chromophores with low-lying n–π* excited states.The spin–orbit coupling induced by charge transfer (SOCT)15,16 in covalently linked aromatic molecules is a promising alternative mechanism that can generate triplet states of a wide range of chromophores. For example, unsubstituted boron-dipyrromethene (BDP) and its derivatives have attracted attention for their unique properties.17,18 They have high fluorescence quantum yields often approaching one, and thus it is difficult to obtain a sufficient quantity of triplets by photo-irradiation. Filatov and co-workers have successfully obtained the triplet excited state of BDP in their pioneering work using the SOCT-ISC mechanism.19 A necessary condition for the SOCT should be that the energy levels of the singlet and triplet charge transfer states, i.e.1CT and 3CT, lie between those of the singlet and triplet local excited states, 1BDP and 3BDP in this paper, and thus it should be valuable if we can predict energy levels of the excited states along the decay process using theoretical calculations.
To predict photochemical processes on covalently linked donor–accepter systems is still challenging because the characters of low-lying excited states drastically vary with the twist angle between the molecules, though they are hardly measurable with only a few exceptions20 despite their importance. The locally excited (LE) state responsible for the absorption/emission of photons is stabilized in the planar conformation, whereas the charge transfer (CT) state, which is sensitive to the polarity of the environment, is stabilized in the perpendicular conformation, and what complicates the situation is that these states can be mixed at the angle where they are close in energy. At the same time, it can be a strength if its character is well-designed. The sensitivity of the luminescence properties of the twisted intramolecular charge transfer (TICT) state is used for sensing environmental polarity, microenvironmental viscosity, and chemical species.21,22 For highly efficient thermally activated delayed fluorescence materials (TADFs) for organic light-emitting diodes (OLEDs), the CT character can make the S1–T1 gap small, which enhances the reverse ISC, and the LE character will compensate for the lack of fluorescence efficiency.23–26 For SOCT-ISC molecules, the subject of this study, the twist angle should also alter the SOC between the singlet and triplet excited states; thus, a detailed theoretical analysis based on the molecular structure is necessary.
Here, molecular mechanisms of the triplet state formation in the bodipy-phenoxazine (BDP-PXZ) dyads were investigated using quantum chemical calculations. Interestingly, it has been suggested that a small modification, an addition of the methyl groups, of the BDP moiety prevents the generation of the 3CT state, while it was generated together with the 3BDP state when the substituents were absent.27 It was also suggested that 3CT and 3BDP states exhibit different EPR patterns, i.e., different spin polarizations, which should contain the information of the spin-selective ISC that the molecules went through,28e.g. the twisted angle between the chromophores, and they can be used for the corroboration of the proposed mechanism by comparing with the theoretical predictions. However, the calculation method has not been established to predict the spin polarization in the magnetic axis for triplet states of organic chromophores, because quantitative calculations of the zero-field splitting (ZFS) D-tensor are necessary. Our recent developments in the prediction of the spin–spin coupling (SSC)29 based on the ab initio density-matrix renormalization group (DMRG) algorithm opened up the possibility of quantitative prediction of the magnetic properties of organic chromophores. By comparing the theoretical predictions with the TR-EPR experimental results, we can confirm the character of the transient species and the mechanism at the electronic structure level.
2 Computational details
Bodipy-phenoxazine dyads shown in Fig. 1 are adopted in this study, and the dyads synthesized experimentally are modeled by replacing the C4H9 alkyl-chain with a hydrogen atom.27 Geometry optimizations were performed with the def2-TZVP basis sets30–32 and a long-range corrected BLYP functional33 (LC-BLYP) in which the range separation parameter was tuned to μ = 0.15 was adopted (see the ESI†). The polarizable continuum model34 (PCM) was used to account for the solvent effect of toluene. All the excited states including the lowest triplet state were calculated by the time-dependent density functional theory (TD-DFT) using the ORCA5 program package.35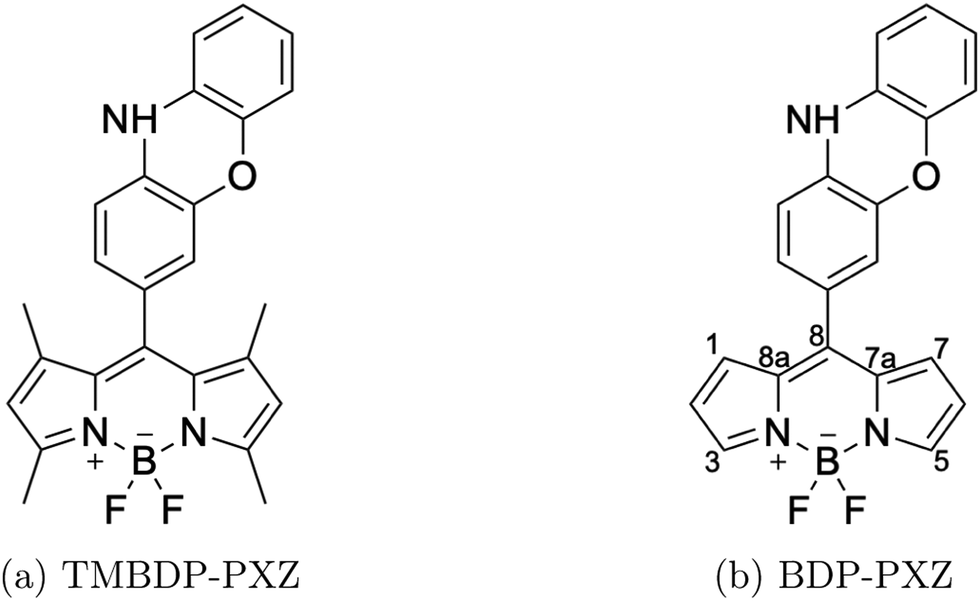 | ||
| Fig. 1 Chemical structures of BDP-PXZ and TMBDP-PXZ. | ||
The matrix elements of spin–orbit coupling between the 1CT and each spin sublevel (x, y, z axes of the molecular frame) of 3CT and 3BDP states were calculated by TD-DFT. The ZFS D-tensor was calculated using the method reported in ref. 29, in which the expected values of the spin–spin coupling operator in the Breit–Pauli Hamiltonian
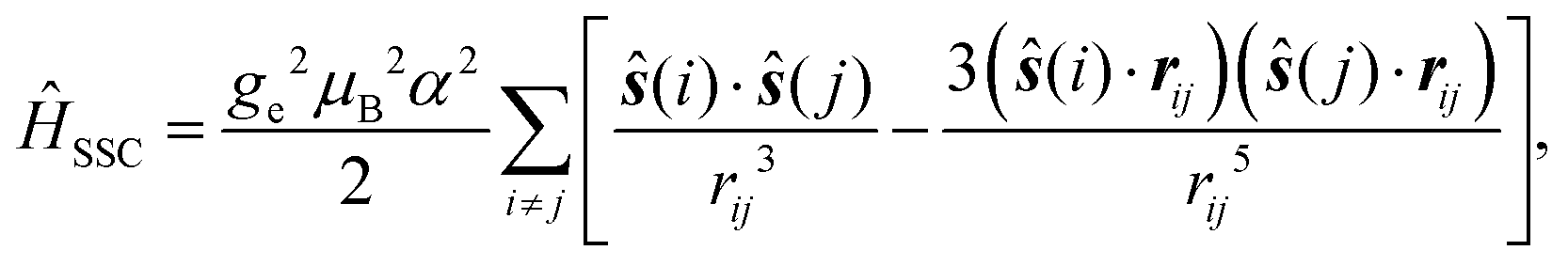 | (1) |
 | (2) |
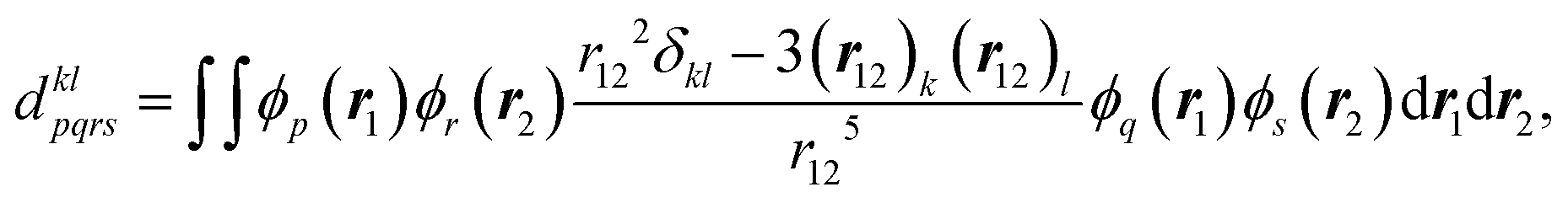 | (3) |
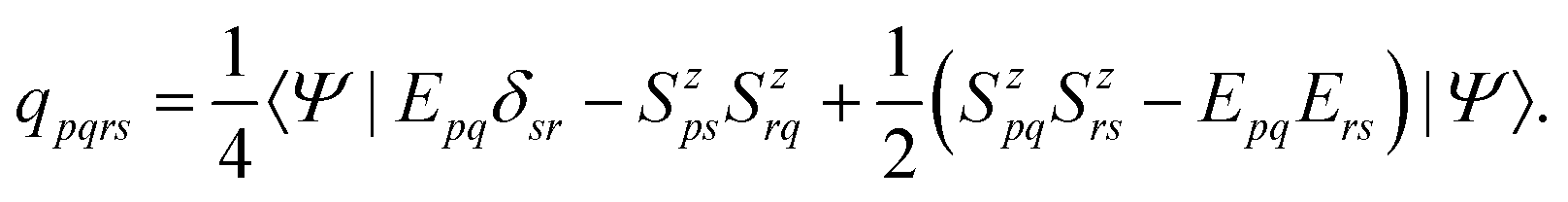 | (4) |
3 Results
3.1 Molecular mechanism of the formation of the triplet states
To elucidate the molecular mechanism of the formation of the triplet states, we performed the geometry optimization for 1CT, 3CT, and 3BDP states with constraints for the dihedral angle θ between BDP and PXZ moieties. Fig. 2 shows the potential energy curves (PECs) of TMBDP-PXZ and BDP-PXZ in toluene along the dihedral angle θ. The dihedral angle of TMBDP-PXZ at the equilibrium structure of the ground state was found to be θ = 80°, i.e., BDP and PXZ are almost perpendicular due to the steric hindrance of the CH3 groups, while that of BDP-PXZ was θ = 47°. In both systems, after the photo-irradiation θ will increase along the slope of the 1CT state towards θ = 90°, where the 1CT and 3CT states are energetically very close due to the small exchange coupling for the perpendicular conformation. The shapes of PECs of 1CT and 3CT are similar for TMBDP-PXZ and BDP-PXZ systems, but that of the 3BDP state is significantly different due to the presence and absence of the CH3 group; the perpendicular structure (θ ≃ 90°) is the energy minimum in TMBDP-PXZ but is unstable in BDP-PXZ. In fact, in the perpendicular structure, the energy level of 3BDP is lower by 0.4 eV than 1CT and 3CT states for TMBDP-PXZ, in contrast, it is very close in energy to 1CT and 3CT states for BDP-PXZ and as will be discussed 3BDP and 3CT are strongly mixed at around θ = 90°.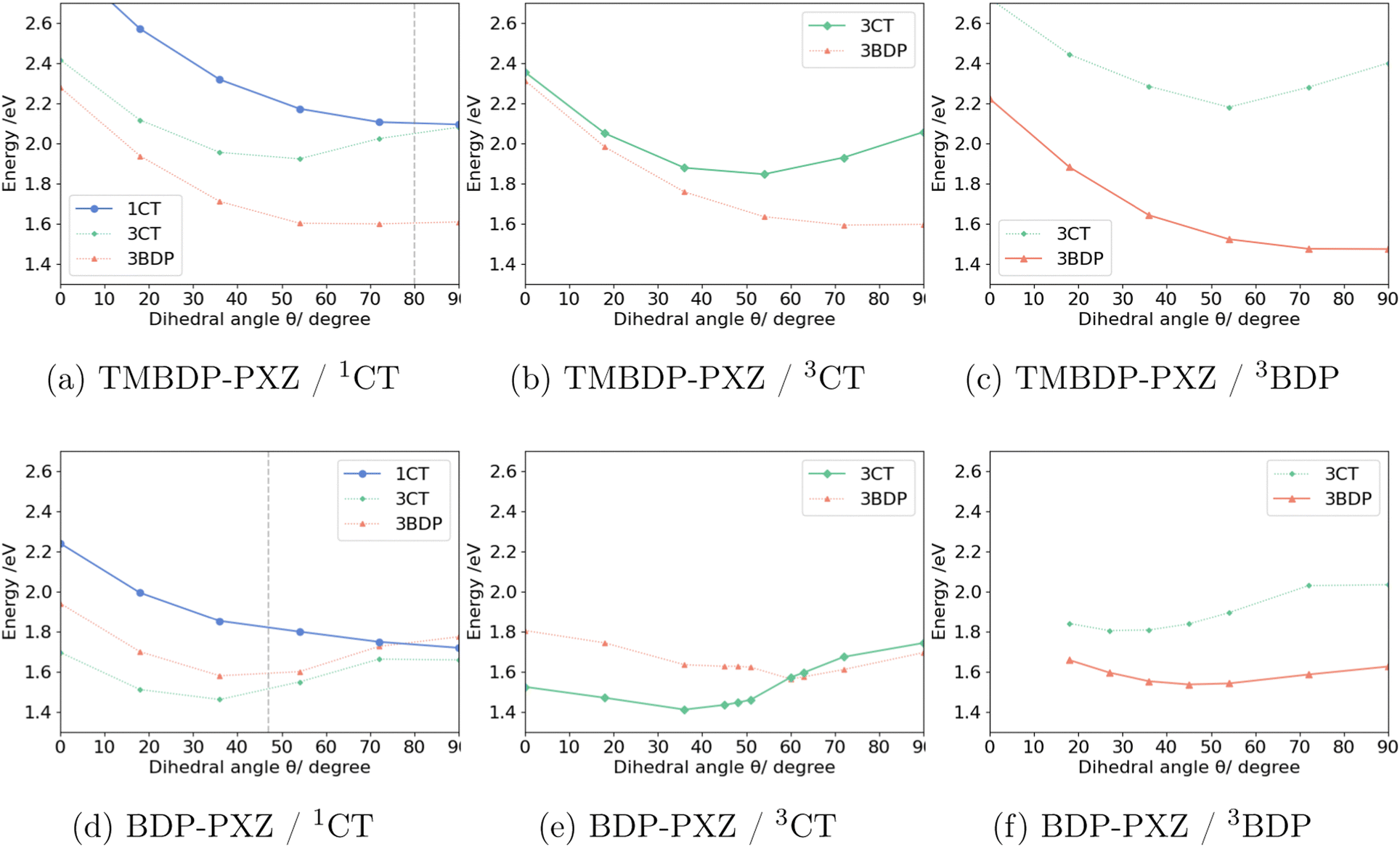 | ||
| Fig. 2 Potential energy curves of 1CT, 3CT, and 3BDP states along the dihedral angle between BDP and PXZ moieties, calculated with LC-BLYP/def2-TZVP under the solvent effect of toluene by LR-CPCM. The geometries were optimized for 1CT: (a) and (d), 3CT: (b) and (e), 3BDP: (c) and (f) states of BDP-PXZ and TMBDP-PXZ with constraints for the dihedral angle θ between BDP and PXZ moieties. The target state is depicted with solid lines, while others with dotted lines; the former was calculated with an equilibrium solvent and the latter with a non-equilibrium solvent. The gray dashed lines in (a) and (d) indicate θ for the ground state stabilized structure. | ||
In the TMBDP-PXZ system, after the transition to either the 3BDP or 3CT state the wavepackets will immediately decay to the 3BDP state, which lies at lower energy than the 3CT state even in the solvent environment that minimizes the energy level of 3CT as shown in Fig. 2b. In the BDP-PXZ system, the wavepackets that transit to the 3CT state can stay on the 3CT potential energy surface because 3CT has lower energy than 3BDP in the solvent reorganized for the 3CT state, and the wavepacket that directly transits to the 3BDP state from 1CT state will stay in the 3BDP state because it is lower in energy than 3CT in the solvent reorganized for the 3BDP state. This should be the reason why only the BDP-PXZ system exhibits the dual components in the TR-EPR spectra, while TMBDP-PXZ does not. Note that the state-specific effects, which can be evaluated by the corrected LR method41,42 for example, were neglected in the calculations based on the LR-CPCM method. It may stabilize the excited states with the CT character and make it easier for 3CT and 3BDP to coexist. The detailed analysis for the difference in energetics caused by the CH3 groups will be discussed in Section 3.4.
3.2 Spin polarization induced by intersystem crossing
To reveal the spin polarization axis in the molecular frame, which cannot be determined by the TR-EPR experiment for randomly oriented molecules, we computed the SOC matrix elements between 1CT and 3CT (〈1CT|Hsoc|3CTμ〉) and 1CT and 3BDP (〈1CT|Hsoc|3BDPμ〉), where μ = x, y, z at the structures on the PECs of 1CT in Fig. 2a and d. Table 1a and b show the SOC matrix elements of TMBDP-PXZ and BDP-PXZ, respectively, for each component 3CTx, 3CTy, and 3CTz where x, y, and z correspond to the molecular axis of the BDP moiety shown in Fig. 3; the long axis is x, the short axis is y, and the out-of-plane axis is z. As shown in Table 1a, the SOC of 〈1CT|Hsoc|3BDPμ〉 is x-polarized and significantly larger than that of 〈1CT|Hsoc|3CTμ〉 at any dihedral angle θ.| θ | 0° | 18° | 36° | 54° | 72° | 90° |
|---|---|---|---|---|---|---|
| (a) TMBDP-PXZ | ||||||
| |〈1CT|Hsoc|3CTμ〉|/cm−1 | ||||||
| x | 0.11 | 0.09 | 0.05 | 0.01 | 0.01 | 0.01 |
| y | 0.03 | 0.07 | 0.11 | 0.14 | 0.14 | 0.02 |
| z | 0.00 | 0.01 | 0.00 | 0.01 | 0.02 | 0.01 |
| ΔE/eV | ||||||
| 0.52 | 0.45 | 0.36 | 0.25 | 0.08 | 0.01 | |
| |〈1CT|Hsoc|3BDPμ〉|/cm−1 | ||||||
| x | 0.29 | 0.39 | 0.47 | 0.56 | 0.66 | 0.71 |
| y | 0.13 | 0.11 | 0.08 | 0.03 | 0.03 | 0.01 |
| z | 0.02 | 0.01 | 0.03 | 0.03 | 0.04 | 0.01 |
| ΔE/eV | ||||||
| 0.66 | 0.63 | 0.61 | 0.57 | 0.51 | 0.49 | |
| (b) BDP-PXZ | ||||||
| |〈1CT|Hsoc|3CTμ〉|/cm−1 | ||||||
| x | 0.00 | 0.03 | 0.03 | 0.11 | 0.44 | 0.48 |
| y | 0.07 | 0.13 | 0.15 | 0.15 | 0.06 | 0.00 |
| z | 0.01 | 0.01 | 0.00 | 0.02 | 0.01 | 0.00 |
| ΔE/eV | ||||||
| 0.54 | 0.48 | 0.39 | 0.25 | 0.09 | 0.06 | |
| |〈1CT|Hsoc|3BDPμ〉|/cm−1 | ||||||
| x | 0.22 | 0.33 | 0.40 | 0.52 | 0.52 | 0.56 |
| y | 0.04 | 0.02 | 0.01 | 0.03 | 0.05 | 0.00 |
| z | 0.02 | 0.02 | 0.00 | 0.03 | 0.01 | 0.00 |
| ΔE/eV | ||||||
| 0.30 | 0.29 | 0.27 | 0.20 | 0.02 | −0.06 | |
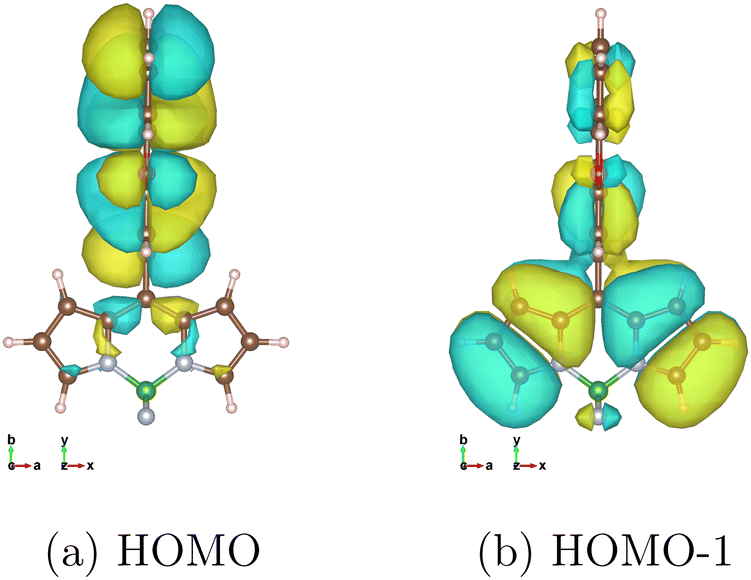 | ||
| Fig. 3 Surface plots of canonical MOs (isovalue = 0.008), calculated with LC-BLYP/def2-TZVP under the solvent effect of toluene by LR-CPCM, non-equilibrium condition. | ||
Theoretically, the SOC is usually accounted by the change in the occupation of the atomic orbitals that have different angular momentum, e.g., the change in the occupation of 2py and 2pz orbitals, such as a lone pair orbital and π orbital, of an atom will contribute x-polarized SOC. As far as we know, it has never been explicitly analyzed, but the SOC in linked π-conjugated systems should originate from the transition between the in-plane (σ orbital) and out-of-plane (π orbital) associated with a charge transfer excitation. For example, Fig. 3 shows the canonical molecular orbitals that represent most of the transitions between 1CT and 3BDP at the θ = 90° structure. The transition appears to involve the difference in occupations of 2px in the HOMO and 2pz in HOMO−1 of the C7a and C8a atoms. It causes the change in the angular momentum about the x-axis and is considered to be the origin of the x-polarized SOC of 3BDP shown in Table 1a and b. This is an explanation for the mechanism of SOCT-ISC at the electronic level, i.e., the reason why the SOC matrix elements between the charge transfer state and locally excited state in linked π-conjugated molecules are significant. The mechanism should be universal for other covalently linked π-conjugated systems, e.g., the efficient reverse ISC of TADF. Note that an atom that is directly linked to the other aromatic moiety, the C8 atom in this case, usually has the largest contribution to this type of the generation of SOC because the in-plane 2px or 2py of the linked atom, which has the same symmetry as the π orbitals of the other aromatic moiety, can strongly mix with those, but the C8 atom happens to be located at the node of the π orbital of BDP in HOMO−1 and cannot contribute at least to the Cs (θ = 90°) structure.
For the same reasons, it is reasonable that the SOC matrix element 〈1CT|Hsoc|3CTμ〉 for TMBDP-PXZ is weak because the transition between 1CT and 3CT does not involve the change in the electron configuration. It suggests that the direct transition from 1CT to 3BDP, which exhibits larger couplings than those with 3CT by one or two order magnitudes, can occur through the minimum energy crossing point (MECP), which should be displaced along other degrees-of-freedom that are orthogonal to the minimum energy path of 1CT shown in Fig. 2a. In fact, we found that the MECP is located at θ = 87° with less than 7 kcal mol−1 above the energy minimum of 1CT state (see the ESI†), so the direct transition to 3BDP should be quite feasible. Note that the energy gap between 3BDP and 3CT should be small at around the MECP, and two triplet states can be strongly mixed and an ISC transition to 3CT could be possible by borrowing the x-polarized SOC of 3BDP, although it should soon decay to the lower energy state 3BDP anyway without changing the polarization in the molecular frame conserving the angular momentum. For BDP-PXZ, in which the mixture of two components observed in the TR-EPR spectra,27 not only 3BDP but also 3CT should be generated although the transition from 1CT to 3CT is considered to be inefficient due to the weak coupling character as explained above. As can be seen in Table 1b, however, the SOC matrix element between 1CT and 3CT is large enough (∼0.5 cm−1) at around θ = 90° structure. This should be explained by the state mixing of 3CT and 3BDP; those are very close in energy at around θ = 90° structure (Fig. 2d). Indeed, it was found that the electron configurations of the 3CT state obtained by TDDFT consist of 57.6% HOMO of PXZ → LUMO of BDP (3CT configuration) and 38.7% HOMO of BDP → LUMO of BDP (3BDP configuration). The latter configuration should contribute to the large x-polarized SOC of 3CT in BDP-PXZ.
3.3 Prediction of the ZFS D-tensor in the molecular frame
The ZFS D-tensor in the molecular frame was calculated at the level of DMRG-CASSCF theory, in which the active space consists of the full valence π orbitals CAS(28e, 25o), and diagonalized to assign the magnetic axis in the molecular frame; the method is described in detail in ref. 29. Table 2 summarizes ZFS of the spin sublevels of 3BDP and 3CT for BDP-PXZ, and the polarization axis with the correspondence between the molecular axes x, y, and z and magnetic axes x′, y′, and z′ for 3BDP and x′′, y′′, z′′ for 3CT. The ZFS parameters, D and E values, were predicted to be −79.44 mT and 6.87 mT for 3BDP at the 3BDP energy minimum structure and −44.24 mT and 17.75 mT for 3CT at the 3CT energy minimum structure, respectively. The predicted D values are in good agreement with the values determined by the TR-EPR spectra in ref. 27. There are discrepancies in the trend between the theoretical and the experimental E values, of which the absolute value is often small and sensitive to the change in the D-tensor.29 In addition, the magnetic axis in the molecular frame was determined by diagonalizing the D tensor, and it was found that the x-axis, which is the axis of the electron polarization induced by the spin-selective ISC from 1CT (see Section 3.2), corresponds to the z′ and y′′ for 3BDP and 3CT, respectively. It is also consistent with the TR-EPR experiments, which strongly support the proposed molecular mechanism in Section 3.1.3.4 Insights into the molecular mechanism
As proposed using the theoretical calculations and confirmed by the consistency between the calculations and the experiments of the TR-EPR measurement, the transitions from 1CT to 3CT and 3BDP should occur at around the θ = 90° structure, hence the steric hindrance by CH3 groups of TMBDP-PXZ is not critical to the mechanism, rather the electron donor character of the groups, which should affect the energy levels of the HOMO and LUMO of the BDP moiety, seems to be more important for the mechanism.Regarding this point, the TD-DFT calculation for the BDP monomer was performed. As summarized in Table 3, when CH3 groups were added, both the HOMO and LUMO energies of the BDP monomer increase by 0.67 eV and 0.45 eV, respectively, and the excitation energy of 3BDP slightly decreases by 0.15 eV. It should have more impacts on 3CT because only the LUMO of BDP, which accepts the electrons from the HOMO of the donor PXZ that should not be altered by the substitution of CH3 to the BDP moiety, is involved in the charge transfer. Thus only the 3BDP is observed for TMBDP-PXZ, whereas the 3CT is observed together with 3BDP for BDP-PXZ in the TR-EPR measurements.
| HOMO | LUMO | T1 | |
|---|---|---|---|
| BDP | −7.44 | −2.00 | 1.71 |
| TMBDP | −6.76 | −1.54 | 1.56 |
To investigate the impact of the electron-withdrawing/donating groups on the BDP moiety, the potential energy curves were constructed for three other BDP-PXZ derivatives with different substituent groups X (X = Me, NO2, and NH2) on C3 and C5 atoms in BDP, where the groups bring an insignificant steric effect to the PXZ moiety (Fig. 4). The curves have similar shapes throughout θ, the dihedral angles between BDP and PXZ moieties as reaction coordinates; however quite different in energy levels of 3CT and 3BDP (see the ESI†).
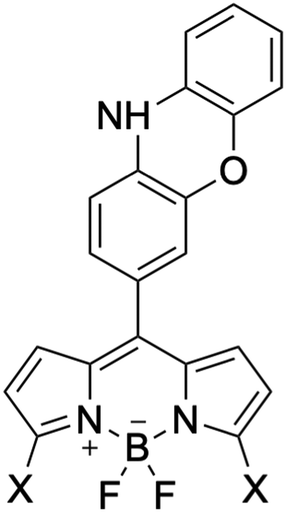 | ||
| Fig. 4 Chemical structures of BDP-PXZ derivatives with substituent groups X, as X = Me, NO2, and NH2 in potential energy curve analysis, and X = F, Cl, NH2, OMe, Me, H, CF3, Ac, CHO, CN, and NO2 in Hammett substituent constant analysis. | ||
Moreover, with a series of various substituent groups (X = F, Cl, NH2, OMe, Me, H, CF3, Ac, CHO, CN, and NO2) on the C3 and C5 atoms of BDP moiety, the scatter plots between the Hammett substituent constants and the energy levels of 1CT, 3CT, and 3BDP states were obtained, as the positive substituent constants correspond to electron-withdrawing groups while the negatives to electron-donating groups.43 The energy levels are calculated using the optimized structures under the constraints for the dihedral angle θ = 90° for clarity. As shown in Fig. 5, there were certain correlations between the substituent constant and 1CT and 3CT energy levels; the correlation coefficient was −0.973 for 1CT, and −0.967 for 3CT, respectively. This result makes it clear that the electron-donating groups on the C3 and C5 atoms of BDP raise the energy level of 1,3CT states, as the BDP-LUMO level gets higher, and vice versa. On the other hand, the 3BDP level is the highest when X = H, and it gets lower for both electron-donating and withdrawing groups. It is natural, considering that the 3BDP state involves not only BDP-LUMO but also BDP-HOMO. In this way, it was reasonably elucidated that the electron donor character of the groups, which should affect the energy levels of the HOMO and the LUMO of the BDP moiety, is more important for the formation mechanism of 3CT and 3BDP transient species, rather than their steric hindrance. The different dependencies of the CT and LE excited states on the substituent effects should be useful for designing molecules that meet the necessary condition of an efficient SOCT that the energy levels of the singlet and triplet charge transfer states lie between those of the singlet and triplet local excited states.
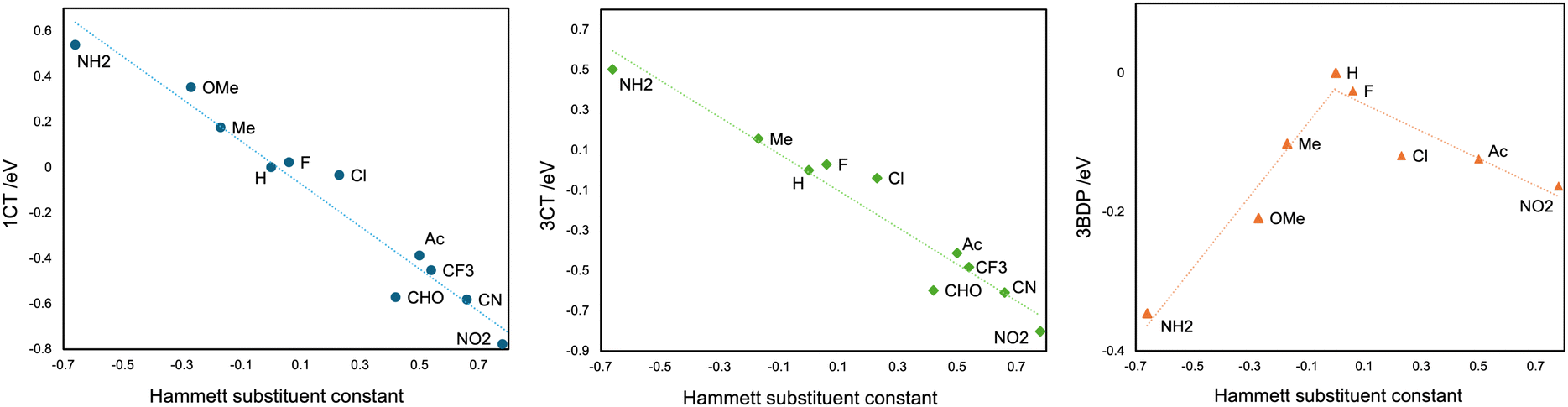 | ||
| Fig. 5 The scatter plot between the Hammett substituent constants and the energy levels of 1CT, 3CT, and 3BDP states for BDP-PXZ derivatives with a series of different substituent groups, calculated with LC-BLYP/def2-TZVP under the solvent effect of toluene by LR-CPCM, equilibrium condition. | ||
4 Summary
We revealed the details of the triplet formation process of BDP-PXZ and TMBDP-PXZ via the SOCT-ISC; the reason why the two different triplet transient species were experimentally observed for BDP-PXZ, and only one species for TMBDP-PXZ, even though the structures differ only by the substitution of the methyl groups. As shown in the analysis of the potential energy curves along the dihedral angles between BDP and PXZ moieties as the reaction coordinates, in both molecules the transition from 1CT to either the 3BDP or 3CT state occurs at about the orthogonal conformation, where the 1CT and 3CT states are close in energy.In BDP-PXZ, it was found that the wavepacket that transits to the 3CT and 3BDP states can remain on their adiabatic potential energy surface because both states are the lowest triplet states in their solvent environment. In TMBDP-PXZ, on the other hand, the 3BDP state is more stable than the 3CT state even in the solvent environment that is optimized for the 3CT state, and only the 3BDP state will be observed regardless of which triplet excited state surfaces the wavepacket has at first transited to. The key to this difference is therefore not the steric hindrance, but the electron donating effect of the CH3 groups on the BDP moiety, which increases the energy levels of the BDP's HOMO and LUMO to elevate the 3CT.
Utilizing the correlation analysis between the energy levels of the excited states and Hammett substituent constants, it was reasonably elucidated that the electron-donating/withdrawing substituent groups on the BDP moiety are strongly correlated with 1,3CT states; and with 3BDP, have an analogous trend with the CT state for electron-withdrawing groups, while the opposite trend for electron-donating ones. It should be universal for the 3CT and 3BDP states of dyad systems and can be exploited to control the formation process of the triplet state by the SOCT-ISC.
Furthermore, with the calculation of the spin–orbit coupling matrix element between 1CT and each triplet state, it was shown that the triplet states generated by the SOCT-ISC should be spin polarized to the component of the BDP's long axis (3CTx and 3BDPx). This result, together with the prediction of the ZFS tensor in the molecular frame, is quite consistent with the TR-EPR spectral results, which strongly supports the SOCT-ISC mechanism we propose. Our theoretical study has reasonably discovered the fundamental mechanisms of the triplet formation of BDP-PXZ systems and the effect of the molecular structures. These insights would be a compass to molecular design of metal-free triplet photosensitizers.
Author contributions
Y. K. conceived the project, M. K. carried out the calculations and analyzed the computational results, K. M. and Y. K. partly contributed to the calculations, and M. K. and Y. K. participated in the composition and revision of the manuscript.Data availability
The data supporting this study's findings are available from the corresponding authors upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partly supported by the JST-FOREST Program (JPMJFR221R), JST-CREST Program (JPMJCR23I6), MEXT Q-LEAP Program (JPMXS0120319794), JSPS KAKENHI (JP23K26614), and Tokyo Ohka Foundation for The Promotion of Science and Technology. The computation was partly performed at the Research Center for Computational Science, Okazaki, Japan (Project: 24-IMS-C028).References
- R. Pérez-Ruiz, Photon Upconversion Systems Based on Triplet–Triplet Annihilation as Photosensitizers for Chemical Transformations, Top. Curr. Chem., 2022, 380, 23 CrossRef.
- N. Yanai and N. Kimizuka, New Triplet Sensitization Routes for Photon Upconversion: Thermally Activated Delayed Fluorescence Molecules, Inorganic Nanocrystals, and Singlet-to-Triplet Absorption, Acc. Chem. Res., 2017, 50, 2487–2495 CrossRef CAS.
- T. N. Singh-Rachford and F. N. Castellano, Photon Upconversion Based on Sensitized Triplet–Triplet Annihilation, Coord. Chem. Rev., 2010, 254, 2560–2573 CrossRef CAS.
- J. Zhou, Q. Liu, W. Feng, Y. Sun and F. Li, Upconversion Luminescent Materials: Advances and Applications, Chem. Rev., 2015, 115, 395–465 CrossRef CAS.
- M. Zhong and Y. Sun, Recent Advancements in the Molecular Design of Deep-Red to near-Infrared Light-Absorbing Photocatalysts, Chem Catal., 2024, 100973 CrossRef CAS.
- V.-N. Nguyen, Y. Yan, J. Zhao and J. Yoon, Heavy-Atom-Free Photosensitizers: From Molecular Design to Applications in the Photodynamic Therapy of Cancer, Acc. Chem. Res., 2021, 54, 207–220 CrossRef CAS.
- X. Zhang, Z. Wang, Y. Hou, Y. Yan, J. Zhao and B. Dick, Recent Development of Heavy-Atom-Free Triplet Photosensitizers: Molecular Structure Design, Photophysics and Application, J. Mater. Chem. C, 2021, 9, 11944–11973 RSC.
- M. A. Filatov, Heavy-Atom-Free BODIPY Photosensitizers with Intersystem Crossing Mediated by Intramolecular Photoinduced Electron Transfer, Org. Biomol. Chem., 2020, 18, 10–27 RSC.
- J. Großkopf, T. Kratz, T. Rigotti and T. Bach, Enantioselective Photochemical Reactions Enabled by Triplet Energy Transfer, Chem. Rev., 2022, 122, 1626–1653 CrossRef PubMed.
- C.-C. Ko and V. W.-W. Yam, Coordination Compounds with Photochromic Ligands: Ready Tunability and Visible Light-Sensitized Photochromism, Acc. Chem. Res., 2018, 51, 149–159 CrossRef CAS PubMed.
- T. Hamachi and N. Yanai, Recent Developments in Materials and Applications of Triplet Dynamic Nuclear Polarization, Prog. Nucl. Magn. Reson. Spectrosc., 2024, 142–143, 55–68 CrossRef CAS.
- K. Nishimura, H. Kouno, Y. Kawashima, K. Orihashi, S. Fujiwara, K. Tateishi, T. Uesaka, N. Kimizuka and N. Yanai, Materials Chemistry of Triplet Dynamic Nuclear Polarization, Chem. Commun., 2020, 56, 7217–7232 RSC.
- A. Kagawa, M. Negoro, K. Takeda and M. Kitagawa, Magnetic-Field Cycling Instrumentation for Dynamic Nuclear Polarization-Nuclear Magnetic Resonance Using Photoexcited Triplets, Rev. Sci. Instrum., 2009, 80, 044705 CrossRef PubMed.
- K. Takeda, K. Takegoshi and T. Terao, Dynamic Nuclear Polarization by Electron Spins in the Photoexcited Triplet State: I. Attainment of Proton Polarization of 0.7 at 105 K in Naphthalene, J. Phys. Soc. Jpn., 2004, 73, 2313–2318 CrossRef CAS.
- Z. E. X. Dance, S. M. Mickley, T. M. Wilson, A. B. Ricks, A. M. Scott, M. A. Ratner and M. R. Wasielewski, Intersystem Crossing Mediated by Photoinduced Intramolecular Charge Transfer: Julolidine-Anthracene Molecules with Perpendicular π Systems, J. Phys. Chem. A, 2008, 112, 4194–4201 CrossRef CAS PubMed.
- C. V. Suneesh and K. R. Gopidas, Long-Lived Photoinduced Charge Separation Due to the Inverted Region Effect in 1,6-Bis(Phenylethynyl)pyrene-Phenothiazine Dyad, J. Phys. Chem. C, 2010, 114, 18725–18734 CrossRef CAS.
- G. Ulrich, R. Ziessel and A. Harriman, The Chemistry of Fluorescent Bodipy Dyes: Versatility Unsurpassed, Angew. Chem., Int. Ed., 2008, 47, 1184–1201 CrossRef CAS PubMed.
- N. Boens, V. Leen and W. Dehaen, Fluorescent Indicators Based on BODIPY, Chem. Soc. Rev., 2012, 41, 1130–1172 RSC.
- M. A. Filatov, S. Karuthedath, P. M. Polestshuk, H. Savoie, K. J. Flanagan, C. Sy, E. Sitte, M. Telitchko, F. Laquai, R. W. Boyle and M. O. Senge, Generation of Triplet Excited States via Photoinduced Electron Transfer in Meso -Anthra-BODIPY: Fluorogenic Response toward Singlet Oxygen in Solution and in Vitro, J. Am. Chem. Soc., 2017, 139, 6282–6285 CrossRef CAS PubMed.
- K. Suzuki and H. Kaji, Torsion Angle Analysis of a Thermally Activated Delayed Fluorescence Emitter in an Amorphous State Using Dynamic Nuclear Polarization Enhanced Solid-State NMR, J. Am. Chem. Soc., 2023, jacs.3c05204 Search PubMed.
- S. Sasaki, G. P. C. Drummen and G.-I. Konishi, Recent Advances in Twisted Intramolecular Charge Transfer (TICT) Fluorescence and Related Phenomena in Materials Chemistry, J. Mater. Chem. C, 2016, 4, 2731–2743 RSC.
- Z. R. Grabowski, K. Rotkiewicz and W. Rettig, Structural Changes Accompanying Intramolecular Electron Transfer: Focus on Twisted Intramolecular Charge-Transfer States and Structures, Chem. Rev., 2003, 103, 3899–4032 CrossRef PubMed.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Highly Efficient Organic Light-Emitting Diodes from Delayed Fluorescence, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- F. B. Dias, T. J. Penfold and A. P. Monkman, Photophysics of Thermally Activated Delayed Fluorescence Molecules, Methods Appl. Fluoresc., 2017, 5, 012001 CrossRef PubMed.
- Y. Tao, K. Yuan, T. Chen, P. Xu, H. Li, R. Chen, C. Zheng, L. Zhang and W. Huang, Thermally Activated Delayed Fluorescence Materials Towards the Breakthrough of Organoelectronics, Adv. Mater., 2014, 26, 7931–7958 CrossRef CAS PubMed.
- Z. Yang, Z. Mao, Z. Xie, Y. Zhang, S. Liu, J. Zhao, J. Xu, Z. Chi and M. P. Aldred, Recent Advances in Organic Thermally Activated Delayed Fluorescence Materials, Chem. Soc. Rev., 2017, 46, 915–1016 RSC.
- Y. Dong, A. A. Sukhanov, J. Zhao, A. Elmali, X. Li, B. Dick, A. Karatay and V. K. Voronkova, Spin–Orbit Charge-Transfer Intersystem Crossing (SOCT-ISC) in Bodipy-Phenoxazine Dyads: Effect of Chromophore Orientation and Conformation Restriction on the Photophysical Properties, J. Phys. Chem. C, 2019, 123, 22793–22811 CrossRef CAS.
- K. Sakamoto, T. Hamachi, K. Miyokawa, K. Tateishi, T. Uesaka, Y. Kurashige and N. Yanai, Polarizing agents beyond pentacene for efficient triplet dynamic nuclear polarization in glass matrices, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2307926120 CrossRef CAS.
- K. Miyokawa and Y. Kurashige, Zero-Field Splitting Tensor of the Triplet Excited States of Aromatic Molecules: A Valence Full-π Complete Active Space Self-Consistent Field Study, J. Phys. Chem. A, 2024, 128, 2349–2356 CrossRef CAS PubMed.
- F. Weigend and R. Ahlrichs, Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- F. Weigend, Accurate Coulomb-fitting Basis Sets for H to Rn, Phys. Chem. Chem. Phys., 2006, 8, 1057 RSC.
- A. Hellweg, C. Hättig, S. Höfener and W. Klopper, Optimized Accurate Auxiliary Basis Sets for RI-MP2 and RI-CC2 Calculations for the Atoms Rb to Rn, Theor. Chem. Acc., 2007, 117, 587–597 Search PubMed.
- Y. Tawada, T. Tsuneda, S. Yanagisawa, T. Yanai and K. Hirao, A Long-Range-Corrected Time-Dependent Density Functional Theory, J. Chem. Phys., 2004, 120, 8425–8433 CrossRef CAS PubMed.
- V. Barone and M. Cossi, Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- F. Neese, Software Update: The ORCA Program System—Version 5.0, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1606 Search PubMed.
- D. Zgid and M. Nooijen, The Density Matrix Renormalization Group Self-Consistent Field Method: Orbital Optimization with the Density Matrix Renormalization Group Method in the Active Space, J. Chem. Phys., 2008, 128, 144116 CrossRef PubMed.
- D. Ghosh, J. Hachmann, T. Yanai and G. K.-L. Chan, Orbital Optimization in the Density Matrix Renormalization Group, with Applications to Polyenes and \beta-Carotene, J. Chem. Phys., 2008, 128, 144117 CrossRef PubMed.
- T. Yanai, Y. Kurashige, D. Ghosh and G. K.-L. Chan, Accelerating Convergence in Iterative Solution for Large-Scale Complete Active Space Self-Consistent-Field Calculations, Int. J. Quantum Chem., 2009, 109, 2178–2190 CrossRef CAS.
- D. Ganyushin, N. Gilka, P. R. Taylor, C. M. Marian and F. Neese, The Resolution ofthe Identity Approximation for Calculations of Spin-Spin Contribution to Zero-Field Splitting Parameters, J. Chem. Phys., 2010, 132, 144111 CrossRef.
- Q. Sun, T. C. Berkelbach, N. S. Blunt, G. H. Booth, S. Guo, Z. Li, J. Liu, J. D. McClain, E. R. Sayfutyarova, S. Sharma, S. Wouters and G. K.-L. Chan, P y SCF: The Python-based Simulations of Chemistry Framework, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8 Search PubMed.
- M. Caricato, B. Mennucci, J. Tomasi, F. Ingrosso, R. Cammi, S. Corni and G. Scalmani, Formation and Relaxation of Excited States in Solution: A New Time Dependent Polarizable Continuum Model Based on Time Dependent Density Functional Theory, J. Chem. Phys., 2006, 124, 124520 CrossRef PubMed.
- S. Chibani, A. D. Laurent, B. Le Guennic and D. Jacquemin, Improving the Accuracy of Excited-State Simulations of BODIPY and Aza-BODIPY Dyes with a Joint SOSCIS(D) and TD-DFT Approach, J. Chem. Theory Comput., 2014, 10, 4574–4582 CrossRef CAS PubMed.
- C. Hansch, A. Leo and R. W. Taft, A Survey of Hammett Substituent Constants and Resonance and Field Parameters, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp03386h |
| This journal is © the Owner Societies 2024 |