
Modeling the ionization mechanism of amorphous solid particles without an external energy source coupled to mass spectrometry†
Styliani
Consta‡
 *,
Lisa M.
Wingen
,
Yiming
Qin§
,
Veronique
Perraud
and
Barbara J.
Finlayson-Pitts
*,
Lisa M.
Wingen
,
Yiming
Qin§
,
Veronique
Perraud
and
Barbara J.
Finlayson-Pitts
Department of Chemistry, University of California, Irvine, CA 92697-2025, USA. E-mail: sconstas@uwo.ca
First published on 23rd October 2024
Abstract
Ionization via desorption of charged analytes from the surface of solid amorphous glutaric acid particles, without the assistance of an external energy source, has been shown to be a promising method that can be coupled to mass spectrometry. We conduct mechanistic studies of the later stages of this ionization process using atomistic molecular dynamics. Our analysis focuses on the hydrogen bonding, diffusion, and ion desorption from nano-aggregates of glutaric acid. These nano-aggregates exhibit an extended H-bonded network, often comprising H-bonded chains, linear dimeric assemblies, and occasionally cyclic trimeric assemblies. These local structures serve as centers for proton transfer reactions. The intermediate hydrocarbon chain between the proton-carrying oxygen sites prevents proton diffusion over a long distance unless there is significant translational or rotational movement of the proton-carrying diacid molecule. Our calculations show that diffusion on the surface is an order of magnitude faster than in the core of the nano-aggregate, which aids effective proton transfer on the particle's exterior. We find that ionic species desorb from the aggregate's surface through independent evaporation events of small clusters, where the ion is coordinated by only a few glutaric acid molecules. Near the nano-aggregate's Rayleigh limit, jets capable of releasing multiple ions were not observed. These observations suggest a more general ion-evaporation mechanism that applies to low-dielectric particles of various sizes, complementing the original ion-evaporation mechanism proposed for aqueous droplets with an approximate radius of 10–15 nm. The combined evidence from molecular modeling presented here and the thermodynamic properties of solid and supercooled liquid glutaric acid indicates that the stronger signals of glutaric acid observed in the mass spectra, relative to other experimentally tested diacids, can be attributed to its significantly lower melting point and the reduced enthalpy of vaporization of its amorphous state compared to other tested diacids.
1 Introduction
Mass spectrometry (MS) is a major experimental technique used in the analysis of atmospheric aerosol particles. In most ionization methods coupled to MS, a sample of aerosol particles is completely dissolved in a solvent or is vaporized at high temperatures, and their composition is detected without distinguishing between the surface and bulk interior constituents. However, a coveted goal is the development of surface-sensitive methods that can detect surface molecules1–3 and even profile the particle composition as a function of depth from the surface. The surface composition is of particular interest because it plays a direct and decisive role in chemical reactivity and interaction with living tissues, as well as particle growth mechanisms. In this direction, a few ionization methods have emerged4–6 such as extractive electrospray ionization MS for heterogeneous solid particles,7 and ionization without an external energy source.1,8–12 Even though ionization methods without an external energy source have been successful in detection of analytes,1,12 their molecular mechanism is still unexplored. Molecular modeling is an important method that allows us to explore the molecular details of these mechanisms. Knowledge of the mechanistic details can facilitate optimization of the ionization process by systematically searching for effective substrates and optimal conditions. Moreover, molecular modeling may assist in unraveling the relation between the desorbed species detected in MS and their depth from the surface of the carrier particles.In Qin et al.,1 solid dicarboxylic acid particles comprised of malonic acid (C3, MA), succinic acid (C4, SA), glutaric acid (C5, GA), and adipic acid (C6, AA) were formed from direct atomization of their aqueous solutions. Although there was no ionization source or any external energy source present, diacid molecules were observed as charged species by a triple quadrupole mass analyzer. It was found that among the diacid series, GA provided the strongest analyte signal even at room temperature.1 It is noted that the formation of ions without an external source of energy is similar to processes and results reported by Trimpin et al.8–13
In this manuscript we present mechanistic studies of the later stages of this ionization process1 using molecular dynamics. Specifically, the questions we address here are: (a) What is the H-bonded network in these systems that is responsible for proton transfer reactions? (b) What is the mechanism of the analyte desorption from the diacid particles? (c) Why does GA give a stronger signal in the mass spectra than the other experimentally tested diacids? Our hypothesis is that, under the conditions of the later stages of the ionization process, the GA particle's external layers may be amorphous and softened to a higher degree than that of the other experimentally tested diacids. A softened, disordered surface will facilitate analyte adsorption and charging of the analyte.
Specifically, the experimental steps for the generation and ionization of diacid particles are as follows:
1. An aqueous solution of a diacid was prepared in ∼20–30 mM concentrations. The initial solution had pH ∼ 3.
2. Droplets were formed by atomizing the solution and then dried with diffusion dryers.
3. As the H2O evaporated, the droplets underwent a phase transformation at 295 K, forming solid particles. Atomic force microscopy measurements of the GA particles after drying showed that many had oblong shapes with edges and corners as shown in Fig. S1 in ESI.†
4. Then, the diacid particles were sampled at the inlet of the mass spectrometer without any ionization source, where the initial pressure was of the order of mbar and the temperature of the inlet was set to 30–150 °C but the measured highest temperature was ∼60 °C. In this region, the particles shrank through evaporation and ionic species were desorbed that were subsequently detected by the mass analyzer. Fig. S2 in ESI,† shows the temperatures that particles were exposed to at atmospheric pressure just before entering the MS over the range of source temperature set points used in Qin et al.1 Ions were detected in positive ion mode as [GA + H]+ (protonated GA) or [GA + NH4]+ (due to trace NH3 in air) and in negative ion mode as [GA–H]−.
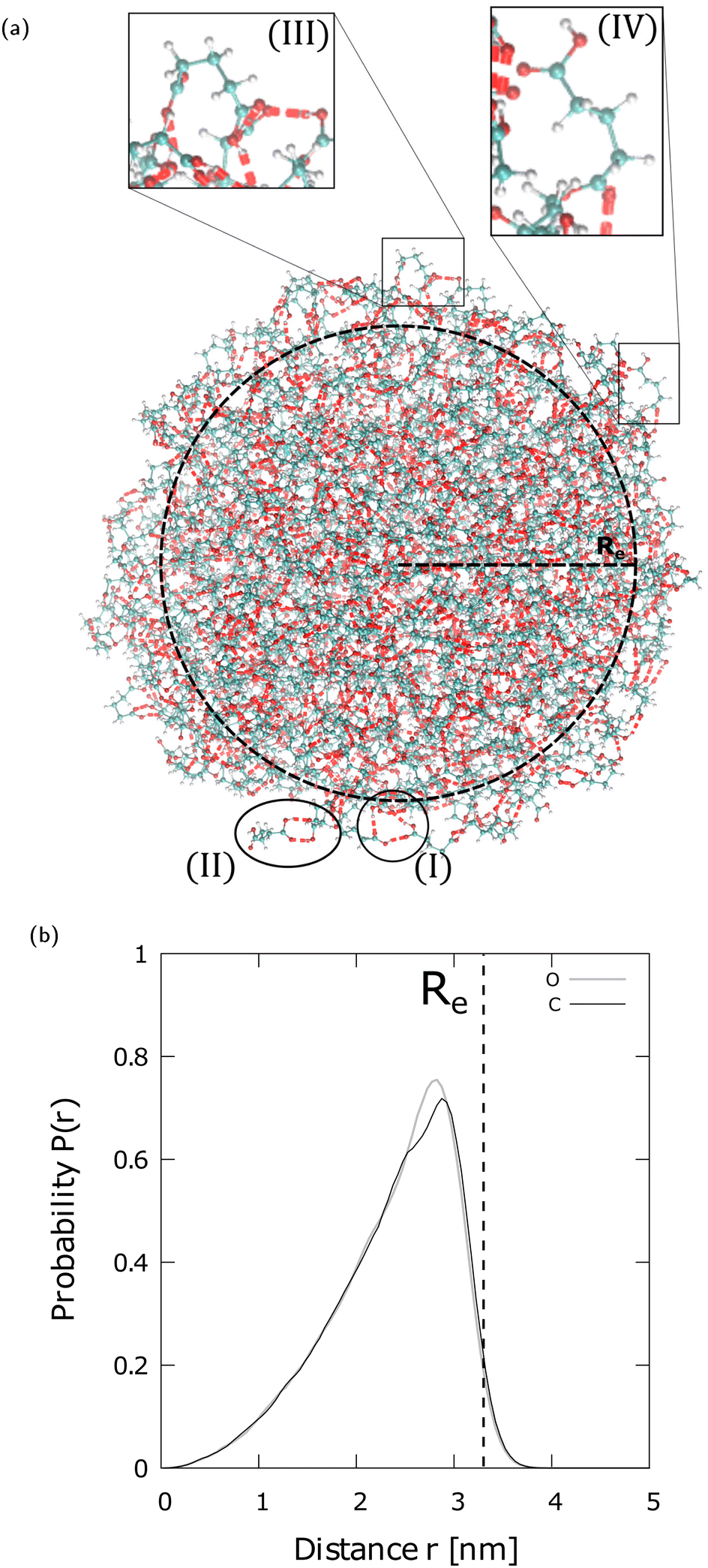
In this study, we investigate neutral and charged GA nano-aggregates composed of up to several hundreds of molecules (Re ∼ 3.3 nm, where Re denotes the equimolar radius), and a few thousands of GA molecules (Re ∼ 6.3 nm), respectively. For the first time the H-bond network in diacid nano-aggregates, diffusion coefficients and the desorption mechanism of charged species from the parent aggregates are examined. To compute a radially dependent diffusion coefficient in finite sized systems, where Einstein's equation and velocity autocorrelation function approaches cannot be applied,14 a general method is presented, that is not limited to the specific systems studied here.
Previous research investigating the structure of GA nano-aggregates in the context of atmospheric aerosols15,16 has pointed to the importance of intermolecular H-bonds using molecular modeling17 and the strength of gas phase dimer formation with four H-bonds of the odd diacids, such as GA and pimelic acid (C7), using quantum chemistry methods.18
One of the questions addressed in the simulations performed here is the selection of the system sizes that we model atomistically so as on the one hand they are computationally feasible, and on the other hand they are representative of the relevant processes to ionization in the larger particles with diameters of ∼100–200 nm generated in the experiments. In the experimental diacid particles, the bulk portion dominates, however, the relevant chemistry in MS experiments such as charging and ion desorption occur on the surface. Therefore, in atomistic modeling, which is often computationally limited by the system size, the size selection should include both bulk-like and surface regions, to be representative of the relevant processes observed in the experiments. To choose the system size we accumulate evidence from a number of studies of a variety of systems where the transition of properties from small clusters to bulk have been examined.19 For example, experiments and models on the melting of Au and Al nanoparticles20,21 have reported that nanoparticles with a radius of approximately less than or equal to 2.5 nm show a rapid change with an almost infinite slope of the melting temperature as a function of radius. For larger radii, the slope becomes smaller and then slowly plateaus as the melting temperature converges to its bulk value. The rapid change for radius <2.5 nm is attributed to the fact that in these smaller system sizes the surface determines their physical properties. As the radius increases, a bulk-like interior that gradually increases plays an ever more important role in the melting, until it dominates over the surface contribution. Our previous study on the structure of H2O droplets of various sizes22 (see Table 1 in Kwan et al.22) has also shown a non-negligible bulk-like interior for droplets of radius ∼3 nm. Drawing experience from these different systems, we infer that diacid aggregates of Re ∼ 3.3 nm to 6.3 nm that are studied here, contain a non-negligible bulk-like interior in addition to the surface and subsurface region. Regardless of the precise structure of the particle's interior, it is the subsurface and surface that play the key chemical role in the ionization process.
2 Models and simulation methods
2.1 Pristine neutral diacid nano-aggregates
Molecular dynamics (MD) simulations of atomistically modeled pristine diacid aggregates were performed. The majority of the simulations were performed for GA nano-aggregates with N = 700, where N denotes the number of molecules in the nano-aggregate. By visual inspection of the MD trajectories we estimate that a N = 700 GA aggregate has Re ∼ 3.3–3.5 nm. For comparison of H-bonding patterns, simulations were also performed for oxalic acid (C2, OA), MA, GA, SA, and AA nano-aggregates with N = 3, 5, 8, 13, 21 diacid molecules at temperature (T) equal to 300 K.The MD simulations were performed using the software NAMD version 2.12.23 VMD 1.9.4a47 was used for the visualization of the trajectories.24 The molecular models were prepared using the CHARMM CGenFF molecular parametrization.25–27 CHARMM CGenFF includes in its parametrization experimental heats of vaporization and molecular volumes of the pure liquids, which are important physical properties for the systems that we investigate. Additional validation of the force field parameters arises from comparisons of the radial distribution functions (rdf) of the nano-aggregates with previous studies of gaseous phase monomeric conformers,28,29 and dimeric structures18 modeled by high level quantum chemistry methods. These comparisons are presented in the Results and discussion section.
Newton's equation of motion for each atomic site was integrated using the velocity-Verlet algorithm with a time step of 1 fs. The electrostatic interactions were treated using the multilevel summation method.30 In order for the multilevel summation method to be applied, the nano-aggregate was placed at the center of a spherical volume with a radius of 10 nm, where the spherical boundary condition was applied. For GA the temperature range of 360–420 K was examined. The temperature was controlled by using the Langevin thermostat with the damping coefficient set to 1 ps−1. Simulations were also performed at T < 360 K. At this lower temperature the molecules were almost immobile within the simulation time. The particles studied in the experiments1 are considered to be amorphous solids. The modeled GA nano-aggregates at T = 360–420 K have an amorphous mobile interface and a much less mobile amorphous interior.
For the molecular model used here, the temperature at which mobility within the nano-aggregate is observed, is near the experimental31 melting point of GA (95–98 °C) and is within the temperature range of the experiments (see Fig. S2 in ESI†). This is not expected considering the delicate relationship between phase diagram and molecular models. Here, we elaborate on this interesting coincidence. Nanoparticles with a radius of a few nanometers are expected to melt at a lower temperature than the bulk because of the large ratio of surface to volume number of molecules. Indeed, the studies of Buffat and de With showed that the melting temperature of Au nanoparticles for diameters <5 nm exhibit a dramatic decrease relative to bulk.19,20 For diameters 5–6 nm they also show19,20 that the decrease of the melting temperature is ∼14–10% relative to the bulk. We deduce from the above studies that the modeled N = 700 GA nano-aggregate with diameter of ∼7 nm will show a decrease approximately 10% in the melting temperature relative to the bulk. Based on these qualitative predictions, the melting temperature of the modeled N = 700 GA is predicted to be at 350 K. Interestingly, our simulations show an unexpected agreement with the above predictions on the mobility of the GA molecules at T > 350 K.
In the study of the diffusion coefficient, we carried out simulations of an aggregate comprised 700 GA molecules. The aggregates were set to temperatures of 360 K, 380 K, 400 K, and 420 K. The coupling with the Langevin thermostat was reduced to a damping coefficient set of 0.1 ps−1. The simulation time for an equilibrium run was 5 ns. The configurations were reoriented every 10 ps to remove spurious rotations.
2.2 Charged GA nano-aggregates
Charged GA aggregates composed of N = 4096 GA molecules and 32 GA− ions, where the negative charge is carried by a carboxylate group, were simulated at T = 420 K. This system has Re ∼ 6.3 nm. The simulation protocol was the same as that for the pristine GA nano-aggregates as described above. The relaxation time lasted several nanoseconds. Test simulations were performed at a range of T > 420 K where rapid ion release was observed. At T = 420 K single ion release after simulation time of several hundreds of picoseconds was observed that was selected for analysis.3 Results and discussion
3.1 H-Bonding in diacid nano-aggregates
To examine the radial distribution function (rdf) between various oxygen sites of diacid molecules within aggregates we label the O sites as shown in Fig. 1(a). To interpret the rdfs it is reminded that the diacid molecules may attain many conformations. The presence of several conformers for a single gaseous phase diacid molecule has been reported in the literature.28,29,32 The lowest energy conformers28,29,32 of an isolated single molecule will be present at room temperature because their energy difference is near the thermal energy at this temperature (∼2.4 kJ mol−1). In aggregates comprised of even a few diacid molecules, higher energy conformers may also appear at room temperature because of stabilization provided in the condensed phase. Fig. S3 in ESI,† shows that the rdfs between O1 and O2 up to a distance of 6.0 Å for N = 3–21 SA molecules converge when N = 21. For this reason we use rdfs for OA to AA nano-aggregates with N = 21 molecules (Fig. 1(b)) to examine their local structure.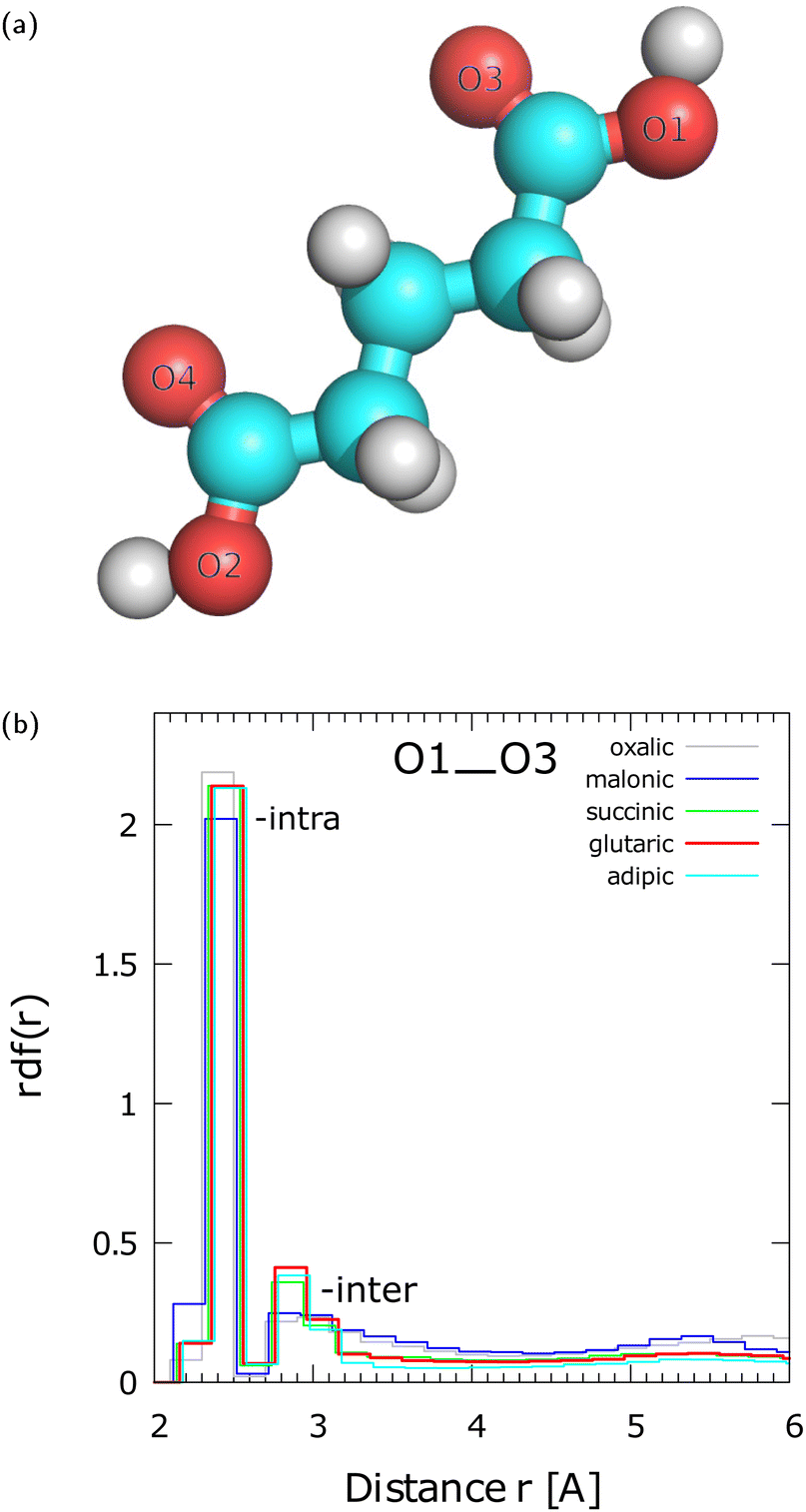 | ||
| Fig. 1 (a) Numbering of O sites in GA to be used in radial distribution functions (rdf). The same numbering is also followed for all the other diacids examined here. (b) O1–O3 rdf at T = 300 K for various diacids nano-aggregates from OA (C2) to AA (C6) with N = 21 molecules. | ||
Fig. 1(b) shows the rdfs between O1 and O3 for different pristine diacid nano-aggregates with N = 21 molecules. The first peak at ∼2.3 Å arises from the intramolecular O1–O3 interaction within the same carboxyl group. The O1–O3 distance of the first peak, which is almost the same for all the diacids, is in agreement with the value of 2.25–2.26 Å for the conformers of a single OA molecule calculated by Weber et al.32 as shown in their Fig. 1, using quantum density functional theory. The second broader peak is centered at ∼2.85 Å and arises from intermolecular H-bonded interactions. The O1–O3 rdf indicates that GA, SA, and AA show similar local structure of H-bonding. Among the three diacids the peak of GA is marginally higher than that of SA and AA. Interestingly, the order in the height of the 2nd peak for GA, SA, and AA follows the trend for intermolecular H-bond strength found by Elm et al.18 for gaseous phase dimers. The second peak of OA (C2) and MA (C3) is broader than of the other diacids and they show an incipient third broad peak between 4.5–6.0 Å. The differences between OA and MA and the other diacids in the formation of gaseous phase dimers have been discussed by Elm et al.18 OA and MA are different from the other diacids in that they cannot form curved conformers that can lead to four H-bonds with another diacid in gaseous phase dimers.18 Moreover, previous research18 has shown that OA prefers to form intramolecular H-bonds instead of intermolecular bonds in gaseous phase dimers. Therefore, at r > 2.7 Å, we attribute the differences between OA and MA, and the C4–C6 diacids to the longer hydrocarbon chains of C4–C6 that lead to a larger number of conformers, which in turn can readily form H-bonds with neighboring molecules. The O1–O4 rdf is presented in Fig. S4(a) in ESI† and the interpretation of the peaks is discussed in Section S4 in ESI† using the example of a MA trimer (Fig. S4(b) in ESI†). The rdfs between various oxygen sites of N = 700 GA are given in Fig. S5 in ESI.†
Analysis of the H-bonding within OA to AA nano-aggregates of various N reveals a variety of H-bonded patterns. The H-bonds are defined here with a donor–acceptor cutoff distance of 3.0 Å and an acceptor–donor-H angle of 30°. For N ≥ 5, a carboxyl group of a diacid molecule may utilize both its H-donor and acceptor sites in a carboxyl group to form dimeric assemblies (region (II) in Fig. 2(a) and in Fig. S6 in ESI†), and occasionally, cyclic trimeric assemblies (region (I) in Fig. 2(a) and Fig. S6 in ESI†). We will call the dimeric assemblies with two H-bonds “2-dimeric” in order to distinguish them from dimers with one H-bond. Visual inspection showed frequent formation of a single intermolecular H-bond linking two diacid molecules. We observed that the structure of the amorphous diacid nano-aggregates is dominated by long H-bonded chains as shown in Fig. 2(b) for the example of a nano-aggregate with N = 13 SA molecules. The H-bonded chain length distribution in various diacid types is an interesting quantity to compute in future research. The single intermolecular H-bonds between two carboxyl groups, diacid chains, 2-dimeric and cyclic trimeric assemblies can be found on the surface as well as in the interior of the aggregates. Elm et al.18 have calculated using quantum density functional theory, that GA (C5) and pimelic acid (C7) form the strongest H-bonds in gas phase dimers relative to the other C2–C8 diacids. However, these diacid conformers were bent and they formed four intermolecular H-bonds at zero temperature. We did not detect such dimeric structures in the nano-aggregates studied here.
 | ||
| Fig. 2 (a) Typical snapshots of dimeric and cyclic trimeric assemblies formed within the diacid aggregates. The N = 5 oxalic acid nano-aggregate is selected for clarity. (b) Typical example of H-bonded chains in N = 13 SA nano-aggregates. The SA molecules that participate in the chain are enlarged. The chain is traced by an orange-colored line. The white spheres represent H sites, the red O and the blue C. The red dotted lines indicate the H-bonds. The definition of the H- bond is discussed in the text. | ||
Fig. 3(a) shows a typical snapshot of an N = 700 GA nano-aggregate. Examples of cyclic trimeric and dimeric assemblies on its surface are encircled (regions (I) and (II) in Fig. 3(a)). The common H-bonded patterns of dimers, trimers, and H-bonded chains, encountered in different types of diacids and in different nano-aggregate sizes suggest that their presence is also expected in the larger amorphous particles observed in experiments.
 | ||
| Fig. 3 (a) Same as in Fig. 2(a) but for an N = 700 GA nano-aggregate with Re 3.3 Å. The encircled regions show (I) a trimer, and (II) a dimer on the nano-aggregate's surface. The enlarged regions (III) and (IV) show “bent” GA conformers at the surface. The color coding of the atomic sites and the definition of the H-bond is the same as in Fig. 2. (b) Probability profile (P(r)) of the O sites and the C sites in the –CH2– groups of an N = 700 GA nano-aggregate, estimated in spherical shells centered at the center of mass (COM), located at zero on the x-axis. The bin size is 0.5 Å and the curves are normalized to 1. | ||
Fig. 3(b) shows the probability of O sites and C sites in the –CH2– groups estimated in spherical shells centered around the center of mass (COM) of a N = 700 GA. The probability profiles show that there is a small preference for the exposure of –CH2– groups toward the vacuo (or vapor). The slight preference for the –CH2– groups toward the vacuo (or vapor) is attributed to GA conformations that are “bent” as shown in regions (III) and (IV) in Fig. 3(a). These bent structures are also consistent with those shown in Fig. 1 of Elm et al.,18 where gaseous phase diacid dimers are modeled using high level quantum chemistry methods. Within the approximations of the molecular model used here, the lack of a strong orientation preference indicates that both polar (–COOH) and non-polar (–CH2–) sites of GA are accessible for interactions with analytes.
The local dimeric and cyclic trimeric assemblies are the centers for proton transfer reactions. Because of the significance of proton transfer in ionization processes coupled to mass spectrometry, we analyze the H-bond network33,34 in a N = 700 GA nano-aggregate. Fig. 4(a) shows the probability density of the H-bond occupancy. Using a distance cutoff of a H-bond of 3 Å between the donor and acceptor site and an angle of 30° the occupancy is defined as the average number of H-bonds counted in the MD trajectory over the total number of possible H-bonds. The distribution is well approximated by the binomial distribution with M = 1400 and p = 0.68 as
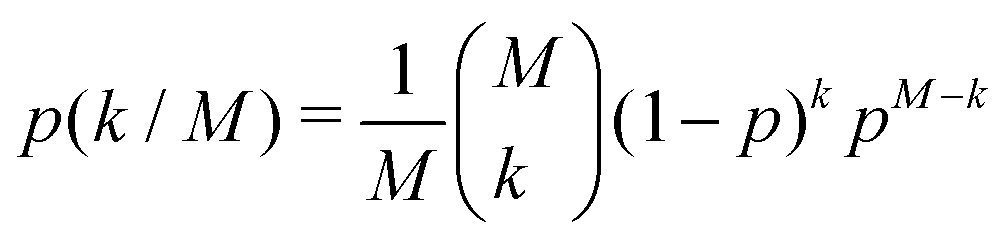 | (1) |
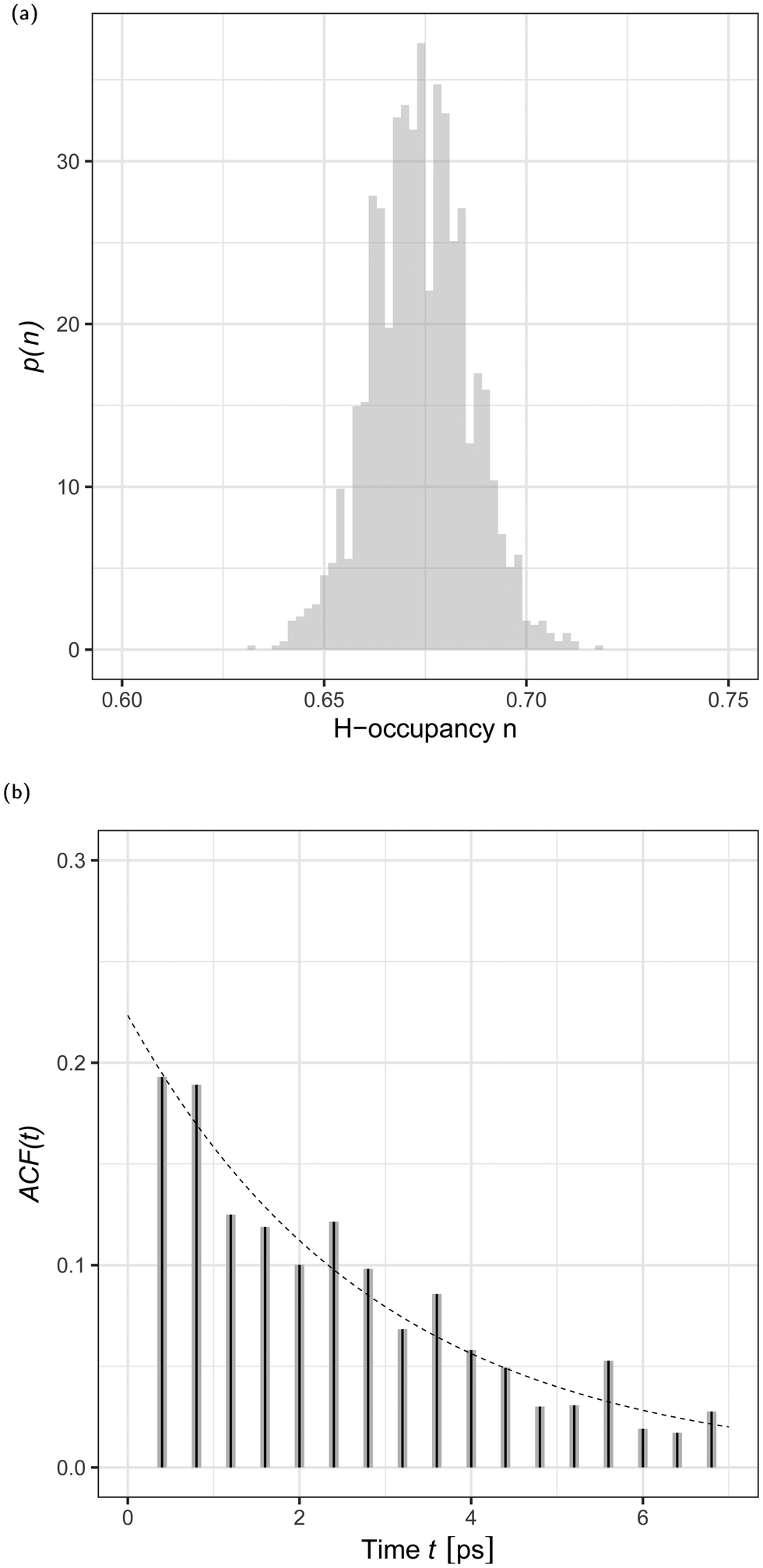 | ||
| Fig. 4 (a) Probability density, p(n), of H-bond occupancy, n, for an N = 700 GA nano-aggregate at 360 K. (b) H-Bond time autocorrelation function, ACF(t), as a function of time, t. | ||
In a sample run of 1 ns at T = 360 K we analyzed the dynamic structure of the H-bond network. We observed that the pristine GA network is subject to continuous formation and disruption (total or partial) of the H-bonded assemblies. Fig. 4(b) shows the H-bond number correlation function (ACF(τ)),
| ACF(τ) = 〈δNH(t)δNH(t + τ)〉 | (2) |
![[N with combining macron]](https://www.rsc.org/images/entities/i_char_004e_0304.gif) H is the difference between the number of H-bonds (NH) at time t and the average number of H-bonds (H) computed from Fig. 4(a). The angular brackets 〈⋯〉 denote an ensemble average. We analysed the time series by fitting the data to the stationary autoregressive moving average processes35 using the R statistical analysis software.36 The H-bond number autocorrelation function, shown in Fig. 4(b), is well approximated, after the initial relaxation time37 by a single exponential decay. The best fit is achieved by the autoregressive moving average (1,1) model shown in eqn (4.4.2) in ref. 35 and the parameters of the dashed line are given by eqn (4.4.5) in the same reference. The corresponding relaxation time is trelax = 2.8 ± 0.6 ps. This time corresponds to 1 Å displacement in the surface layer of the aggregate due to diffusion. This relaxation time will depend on the molecular model used, as for example simulations of the H-bond lifetime in H2O have demonstrated significant variability depending on the H2O model.38
H is the difference between the number of H-bonds (NH) at time t and the average number of H-bonds (H) computed from Fig. 4(a). The angular brackets 〈⋯〉 denote an ensemble average. We analysed the time series by fitting the data to the stationary autoregressive moving average processes35 using the R statistical analysis software.36 The H-bond number autocorrelation function, shown in Fig. 4(b), is well approximated, after the initial relaxation time37 by a single exponential decay. The best fit is achieved by the autoregressive moving average (1,1) model shown in eqn (4.4.2) in ref. 35 and the parameters of the dashed line are given by eqn (4.4.5) in the same reference. The corresponding relaxation time is trelax = 2.8 ± 0.6 ps. This time corresponds to 1 Å displacement in the surface layer of the aggregate due to diffusion. This relaxation time will depend on the molecular model used, as for example simulations of the H-bond lifetime in H2O have demonstrated significant variability depending on the H2O model.38
3.2 Diffusion
The rate of the proton diffusion in an aggregate depends on the dominant mechanism responsible for displacement of the proton. The two candidates are: the Grotthuss mechanism46,47 of proton transfer by exchanging the hydrogen bonds, and the diffusion mechanism of the molecule that carries the proton. The hydrogen bonding patterns were discussed in the previous section. In the following discussion we address diffusion in an aggregate of diacids, using as a typical example the N = 700 GA nano-aggregate.From the direct observation of the magnitude of the displacement, where the magnitude is defined as the distance between two locations at two different points in time, of a molecule in the aggregate, we concluded that the diffusion coefficient is position dependent. This has a simple explanation. The Stokes–Einstein relationship shows the diffusion coefficient to be proportional to the inverse of the drag friction. We expect the drag friction to be lower near the surface as fewer number of molecules will participate in the momentum transfer responsible for the friction.
In the course of the simulations of a N = 700 GA nano-aggregate at T = 360 K, we recorded the distance of each molecule from the COM of the aggregate and its displacement squared, denoted hereafter as Δ2, in 10 ps intervals. Fig. 5 shows the distribution of Δ2 as a function of r (distance from the aggregate's COM) using the equilibrium simulation run. The displacements squared reveal marked variability depending on the location of the molecule. As expected a pronounced increase in the magnitude of the displacement, Δ, with r is evident in the figure. A conventional technique of calculating diffusion coefficients using either the velocity auto-correlation function, or calculating the ratio of the displacements squared over time is not directly applicable for this system, because firstly, the system is finite and, secondly, the diffusion coefficient depends on the location of the molecule.14
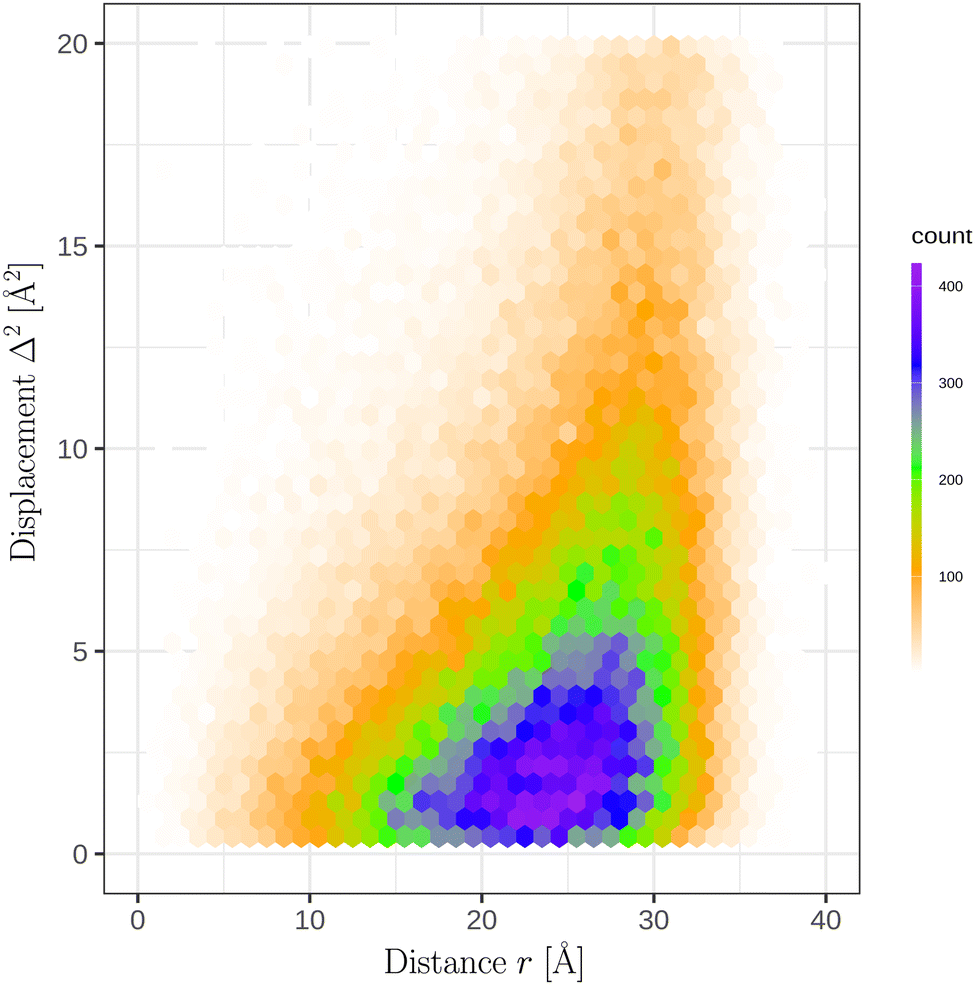 | ||
| Fig. 5 Distribution of the squared displacements, Δ2, as a function of the distance r from the COM of a N = 700 GA nano-aggregate. The COM is located at zero on the x-axis. The plot comprises of 1.4 × 105 observations that were collected in the equilibrium simulation run. | ||
We devised a new technique to investigate position dependent diffusion coefficients in a finite-size system. The steps we performed are described below. Firstly, an ensemble of molecule displacements squared, shown in Fig. 5, was analyzed over a finite interval of time when the position of the molecule does not change significantly. By experimentation, a 10 ps time interval was chosen to be sufficiently brief to preserve the location of a molecule in the aggregate (e.g. within this time interval a molecule in the interior does not reach the surface boundary). Secondly, a simple functional model of the diffusion coefficient dependence on r (distance from the aggregate's COM) was proposed. Thirdly, the ensemble of displacements was analyzed using the maximum likelihood estimate and the optimal parameters for the r dependence of the model of the diffusion coefficient was selected. Lastly, the model was verified by comparing the computed probability distribution of the displacements squared with the theoretical prediction.
For a diffusive process the probability, denoted as Pr, of Δ2 for a fixed value of position r, is given by the gamma distribution (Γ) with the shape parameter α = 3/2:
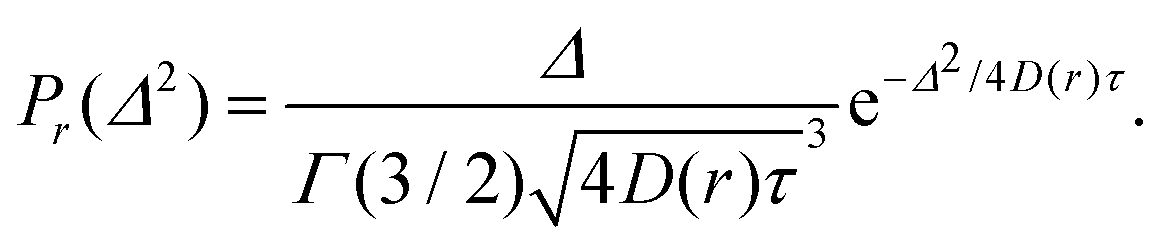 | (3) |
Using the maximum likelihood estimate method, a search for a relationship of D(r) (radially dependent diffusion coefficient) on the molecule position relative to the COM of the aggregate was performed using the following equation
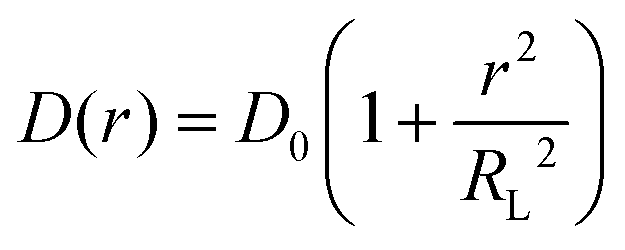 | (4) |
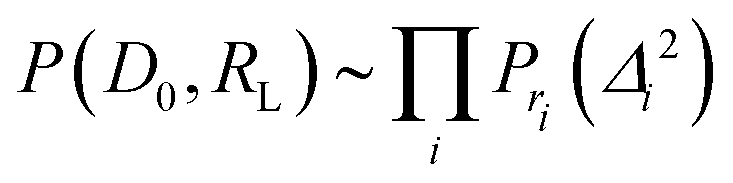 | (5) |
| T [K] | D 0 [nm2 ns−1] | R L [Å] |
|---|---|---|
| 360 | 0.184 ± 0.002 | 15.0 ± 0.1 |
| 380 | 0.230 ± 0.002 | 14.3 ± 0.1 |
| 400 | 0.309 ± 0.003 | 14.3 ± 0.1 |
| 420 | 0.405 ± 0.004 | 14.4 ± 0.1 |
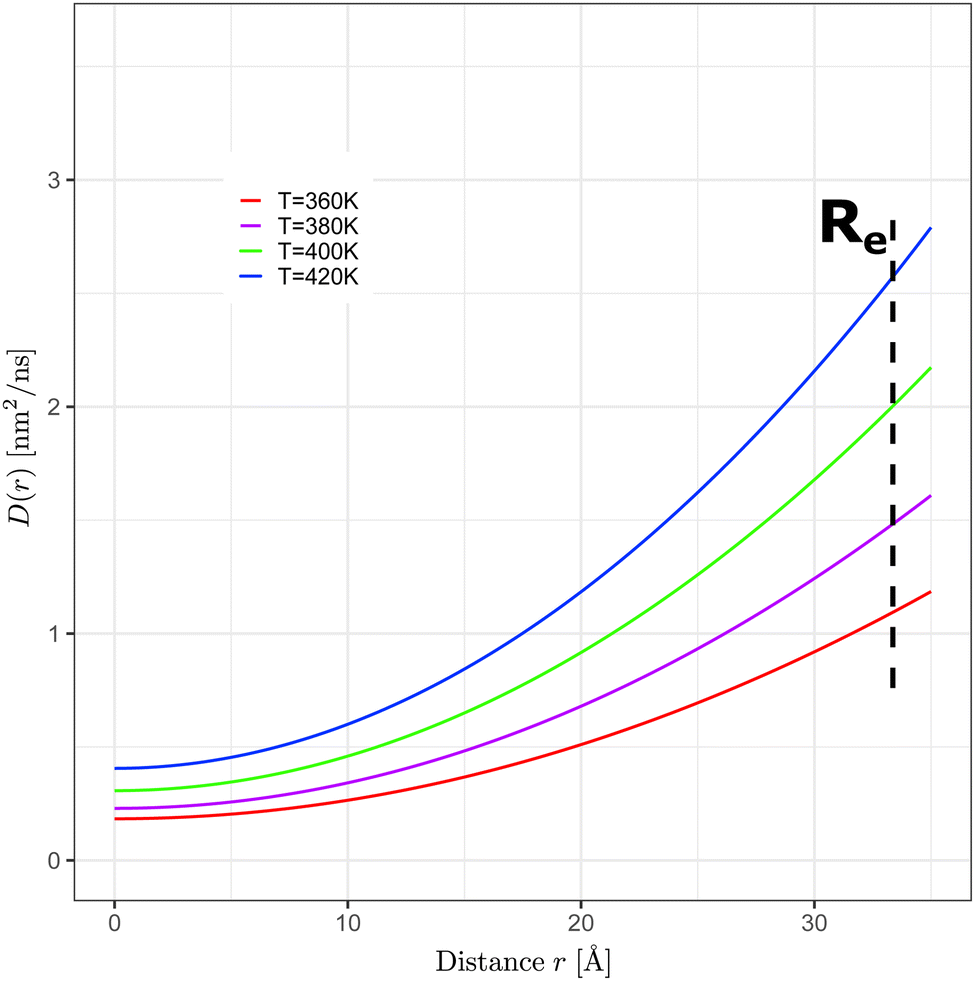 | ||
| Fig. 6 Radially dependent diffusion coefficients, D(r), as a function of the distance r from the N = 700 GA nano-aggregate's COM (COM is located at zero on the x-axis) at T = 360 K, 380 K, 400 K, and 420 K. D(r) was computed using eqn (4) with the parameters shown in Table 1. | ||
To test the reliability of our method, in Fig. 7, we compared the histogram of the random variable Xi = Δi2/4τD(ri) using the calculated diffusion coefficient (gray histogram) with that of the gamma distribution with shape 3/2 (solid line). Fig. 7 shows that the histogram from the calculated diffusion coefficient follows closely the theoretical curve of the gamma distribution. The possible sources of any discrepancy are: the diffusion coefficient dependence has more complex dependence on r than that we assumed here (eqn (4)), and, secondly, the diffusion can be anisotropic close to the surface due to pronounced droplet shape fluctuations.
 | ||
| Fig. 7 Normalized probability distribution (P) of the scaled squared displacement (Xi). The solid line represents the gamma probability distribution with the shape parameter 3/2. | ||
To analyze the temperature dependence of the diffusion coefficient, the logarithm of the diffusion coefficient in the aggregate's interior was plotted as a function of the inverse temperature as shown in Fig. 8. A modified Arrhenius behavior of the diffusion coefficient was assumed given by
| D(T) ∼ Te−ΔE‡/kBT | (6) |
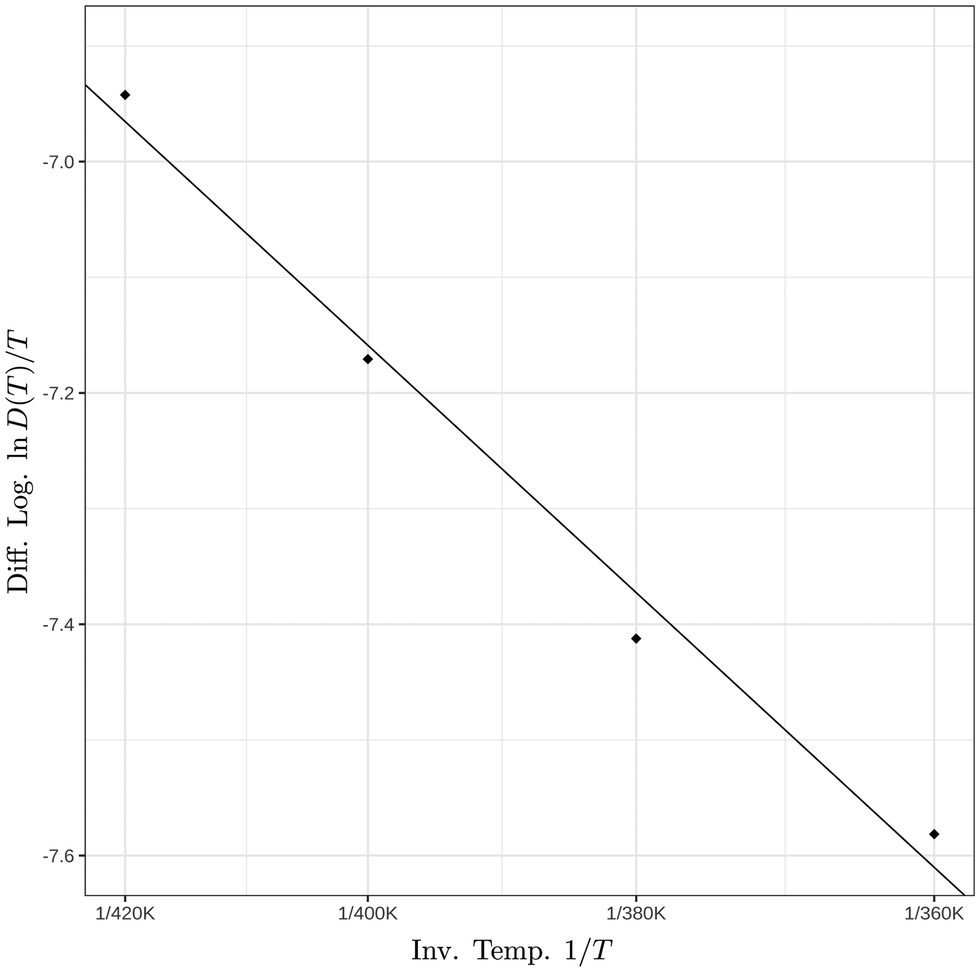 | ||
| Fig. 8 Natural logarithm of the ratio of the interior diffusion coefficient over temperature as a function of the inverse temperature. The corresponding activation barrier is ΔE‡ = 3.3 ± 0.3 kcal mol−1. | ||
At this point, we relate the information that the simulations provided for the N = 700 GA nano-aggregate to the surface mobility of the larger particles observed in the experiments. Similarly to N = 700 GA, the larger particles may also have a more mobile surface relative to the interior because of the lower drag friction on the surface. The susceptibility of the larger particles to surface softening will be further enhanced by the combination of a number of factors that are all present in the particles. These factors are the inherent amorphous nature of the systems, surface irregularities,19,48 and presence of impurities such as ionic species on the surface and interior, residual water, and adsorbed analytes. The crystalline GA has a distinctly lower melting temperature31,49 than the other experimentally tested diacids, which may imply that the combination of all these factors may lead to surface softening at a much lower temperature than that of the other diacids. A more mobile surface layer than the interior is expected to enhance proton transfer and promote ion desorption from the surface. The manner in which the diffusion coefficient changes from the surface to the interior in the large particles may be different from the quadratic law found here for the N = 700 GA nano-aggregates.
3.3 Ion desorption from multiply charged GA nano-aggregates
We examined the manner in which ions are released from the diacid aggregates by simulating a system composed of N = 4096 GA molecules and 32 GA− ions at T = 420 K (see MD Movie in ESI†). The discussion that follows applies to liquid droplets and to aggregates that have a solid or highly viscous interior surrounded by a softened or partially melted surface.The stability of viscous finite-sized systems with unbalanced charge is determined by the competition of two opposing forces: the Coulomb repulsion among the unbalanced charge carriers that leads to emission of excess charge and the surface tension force that holds the system connected. This competition of forces is captured in the Rayleigh model50 that gives rise to the Rayleigh limit. The Rayleigh limit yields the maximum amount of unbalanced charge that a viscous finite-sized system can hold just before the system becomes unstable.50 Lord Rayleigh conjectured that at the instability point a significant amount of charge is rapidly emitted via jets.50–52 The Rayleigh limit is defined by
| Q2 = 64π2γε0R3 | (7) |
By performing several test simulations with various numbers of GA− and at various temperatures, we found that a nano-aggregate comprised N = 4096 GA molecules and 32 GA− ions at T = 420 K emits a single cluster composed of a single GA− ion coordinated with 2–3 GA molecules in approximately several hundreds of picoseconds. The fast but not explosive rate of ion desorption indicated that the system is found near its Rayleigh limit. The charge of 32 GA− is lower than the initial estimate of 42e–50e at T = 298.15 K. Two reasons contribute to the lower value, the lower surface tension at the higher temperature (T = 420 K), and the fact that the experimental surface tension may be different from that of the molecular model. Eqn (7) with the input variables of radius and surface tension provides an initial estimate of the Rayleigh limit, but it is the simulation testing that pinpoints the aggregates' Rayleigh limit more accurately.
We observed that the ion release takes place via independent ion-cluster desorption events (here “ion-cluster” refers to cluster composed of the ion coordinated by diacid molecules). A typical sequence of desorption events is shown in Fig. 9. The events take place in ∼100 ps intervals from (a) to (b) to (c) and in ∼50 ps from (c) to (d). The entire ion-cluster desorption is shown in the MD Movie in ESI.† In the specific simulation, a cluster is desorbed comprising of a single GA− coordinated with three GA molecules. We note that the N = 4096 GA nano-aggregate is sufficiently large to be able to form jets, because it is approximately equal in volume to a droplet of water with 2.85 × 104 H2O molecules for which we have found the formation of Rayleigh jets that emit a train of multiply charged clusters.51
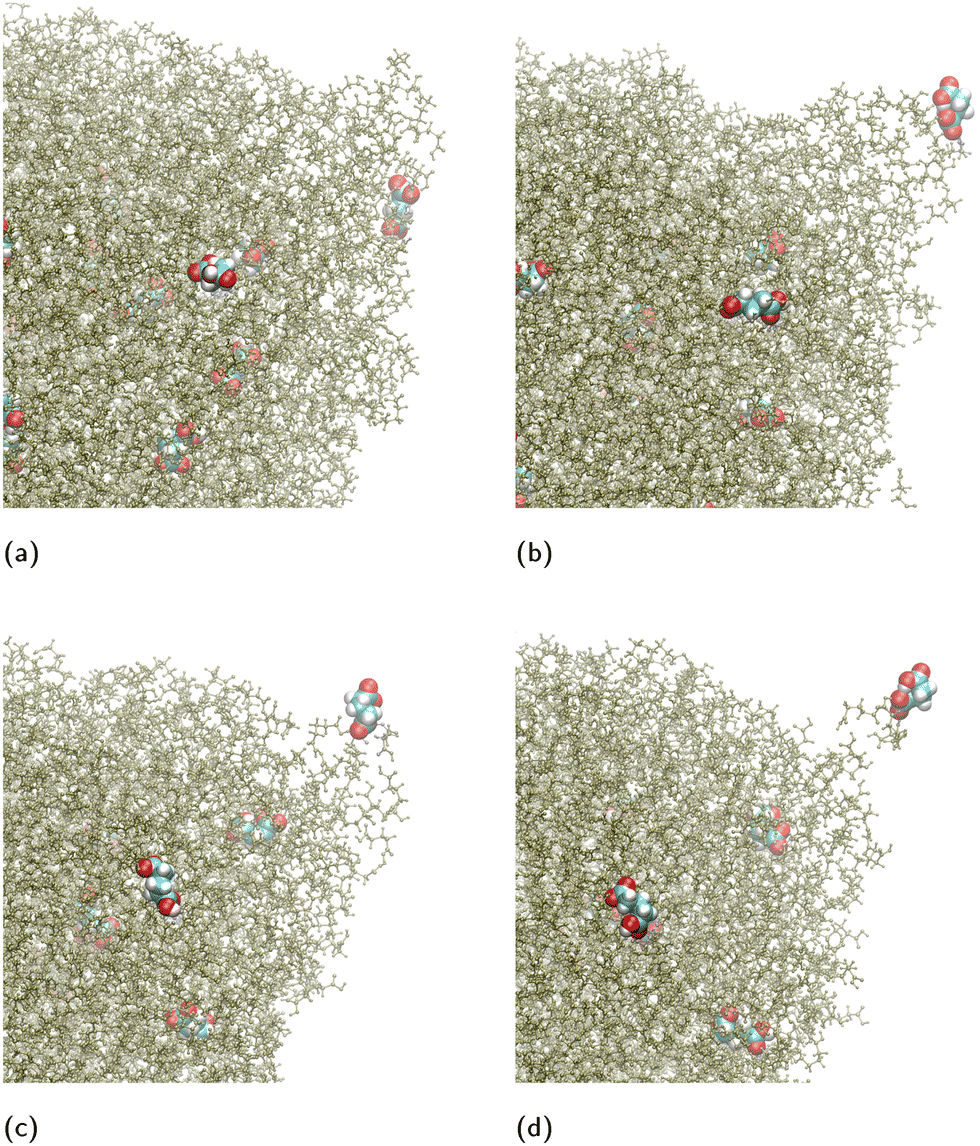 | ||
| Fig. 9 Typical snapshots of ion-cluster desorption from a nano-aggregate composed of N = 4096 GA molecules and 32 GA− ions. For clarity only the region of the nano-aggregate near the location of desorption is shown. The GA− ions has been enlarged and colored differently from the rest of the GA molecules for visualization purposes. (a) A GA− ion approaches an existing surface protrusion, which has a trapezoidal prism shape. (b) The ion goes to one of the corners of the prism and a cone is formed because of the pulling force as the ion escapes. (c) The ion continues to be at the tip of the cone. (d) The ion, coordinated with three GA molecules, detaches from the nano-aggregate. The entire process is shown in a MD Movie in ESI.† | ||
Now we contrast the ion emission mechanism from aggregates with ε < 18, such as the diacids, with that from droplets (liquid or solid surrounded by a mobile surface) with ε > 18. Ramos and Castellanos extended54 the theory of G. I. Taylor for the cone angle55 by finding its dependence on ε. For ε < 18, there are no theoretical predictions for the cone angle.54,55 All the diacids from MA to AA, have a dielectric constant44,45 less than 18. Therefore, according to the theoretical predictions, for the diacid aggregates ion emission will take place via single-ion emission events, where the ion carries with it some of the parent particle's molecules, or via a new mechanism that still is to be discovered. The former mechanism has the features of the ion-evaporation mechanism as it was formulated by Iribarne and Thomson.56–62
Droplets of dielectric constant >18 emit ions from conical fluctuations51,61 as it has been shown in simulations of charged aqueous droplets. Simulations have revealed that conical fluctuations, as shown in shape (II) in Fig. 10(a), appear randomly on the droplet surface at the Rayleigh limit or below but very near to it. When a cone appears, initially it contains no ions. Ions may be found in the vicinity of the cone, or in its base or they may be far away, in which case the cone, which is void of ions, will retract without emitting any ions. If ions are found in the vicinity of the cone, they may enter the conical region as shown in (II) in Fig. 10(a) and be emitted from the tip of the cone51,61 as shown in (III) in Fig. 10(a). For large droplets, the conical fluctuations at the Rayleigh limit form jets that emit multiply charged progeny droplets ((III) in Fig. 10(a)). We note a distinction between the conical shapes on the droplet surface (void of ions), that may provide the path for ion emission and cones that may appear because of the pulling of the ion from the surface, even when a droplet or a planar interface contains no unbalanced charge.63 In the latter case, a “neck” commonly appears as the intermediate configuration when an ion is pulled from a liquid surface and it will not originate from initially formed cones. Once ions are emitted, the Rayleigh jet retracts ((IV) in Fig. 10(a)). In minute nanodroplets, with Re approximately less than 2.5 nm, the conical shapes may emit only one or two ions.61 Therefore, for these minute systems the ion-evaporation mechanism56–58 (IEM) of Iribarne and Thomson becomes indistinguishable from a fission via a Rayleigh jet.61 Recall that the experiments of Iribarne and Thomson have shown that the IEM mechanism is the fastest ion-emission pathway for droplets of radius up to ∼10–15 nm, where the droplet's largest size depends on the nature of the ion.56
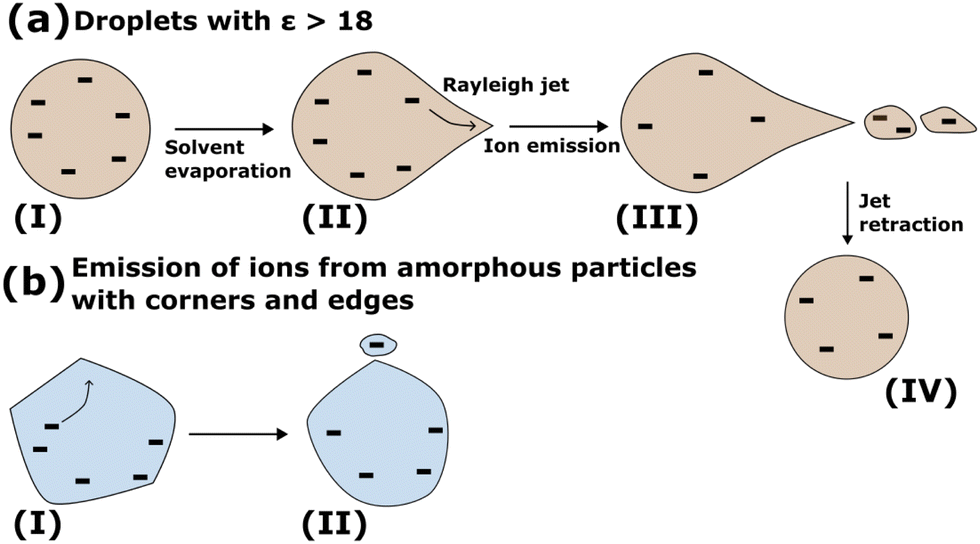 | ||
| Fig. 10 Schematic of ion emission (a) from droplets of ε > 18, and (b) from amorphous particles. The droplet (a) and particle (b) are shown to be charged by negative (−) unbalanced ions because simulations for the N = 4096 GA nano-aggregate have been performed with anions. The same mechanistic steps will also hold for positive ions. Details of the steps are described in the text. | ||
For aggregates with ε < 18, the emission of a single ion coordinated by a few substrate molecules is indistinguishable from the IEM mechanism of Iribarne and Thomson. Because of this similarity, we suggest a broader IEM mechanism that extends in the regime of low ε < 18 where there is no definition of a Taylor cone angle according to analytical theory54,55 and in particles of any size. In this mechanism, ions can be emitted by independent events from any place on the surface without the formation of long jets. The amorphous micro-aggregates may have irregular morphology that can have long-living corners and edges (Fig. S1 in ESI†). These corners may play the role of a Taylor cone. Ions that are in the vicinity of these irregularities may diffuse to the corners from where they can be emitted as shown schematically in Fig. 10(b). We point out a difference between the emission of ions from Rayleigh jets (Fig. 10(a)) and the mechanism described for the ion-desorption from the diacid nano-aggregates (Fig. 10(b)). In the Rayleigh jet (Fig. 10(a)), ions may not completely reside on the surface and they are still pulled into the conical fluctuation. In the desorption of diacids (Fig. 10(b)) ions are found on the outer surface and they may transfer toward a surface irregularity. This difference is important to be further explored in additional studies because it provides insight into the relation of the depth of the ions from the surface of the aggregate and their desorption mechanism.
Here, simulations of GA aggregates containing a small number of GA− were presented. The mechanism of ion desorption when protonated GA replaces GA− is expected to be the same because it is the low dielectric constant of the diacids that plays a key role in the mechanism and not the sign of the charge. Because of the low dielectric constant of diacids, the ion desorption mechanism shares similarities with that from liquid helium nanodroplets59–62 in that ions are emitted via single ion-release events regardless of the droplet size. It is noted that the charge generation in the liquid helium droplets is different from the ionization without an external energy source, but regardless of the origin of the charge, the low dielectric constant of the parent aggregate plays a key role in the ion emission mechanism.
In summary, the experimental data1 and presented simulations suggest a broader IEM mechanism that extends in the regime of ε < 18 where there is no definition of a Taylor cone angle according to analytical theory54,55 and for particles of any size. In this mechanism ions are released by independent desorption events without the formation of long jets. In amorphous particles, surface irregularities may provide corners that may also serve as places for ion desorption.
The exact location of the charging of the analytes cannot be readily detected in experiments or simulations. The charging may occur on the substrate's surface or by charge transfer between protonated GA molecules ([GA + H]+) and the analyte in the small desorbed clusters. Here we simulated multiply charged GA nano-aggregates, but we expect the same mechanism to hold for the other diacids as well because of their low dielectric constant.
3.4 Why do the GA aggregates give a stronger MS signal?
Some of the qualities of the substrate (if not all) to produce high intensity signals in the mass spectra even near room temperature are abundance of charge donor sites, and mobility of the surface molecules which will allow on the one hand, efficient charge transfer and on the other hand, facile release of clusters containing the ion coordinated by substrate molecules. A moderate to lower dielectric constant value of the substrate may also facilitate the proton transfer when a donor and acceptor site are within the range that proton transfer can occur.Here we compare physical properties of diacids that are relevant to the ionization process. The odd and even numbered diacids show significant differences in physical properties as has been discussed by Thalladi31 and Bilde et al.64 For example, the odd and even number of carbons results in alternating lower and higher melting points, respectively.31,64 Some physical properties relevant to this study are summarized in Table 2.
| Diacid type (C#) | MP (°C) | p 0l (Pa) | pKa1, pKa2 |
|---|---|---|---|
| MA (C3) | 135 | (6.2+3.2−2.1) × 10−4 | 2.83, 5.09 |
| SA (C4) | 184–190 | (1.3+1.5−0.7) × 10−3 | 4.2, 5.6 |
| GA (C5) | 95–98 | (1.0+0.3−0.2) × 10−3 | 4.34, 5.22 |
| AA (C6) | 151–154 | (1.8+1.0−0.7) × 10−4 | 4.43, 5.41 |
Solid diacids from MA to AA are subject to polymorphism,31,66,67 which indicates that they are prone to formation of amorphous68,69 solids. Regardless of the precise structure of the particle's interior, it is the structure and mobility of subsurface and surface that is of interest in the ionization process. Among the diacids we examine here, GA has the lowest melting point (Table 2). The lower melting point of GA may indicate higher tendency to surface-melting70–73 relative to the other diacids in the inlet of the mass spectrometer where the temperature of the particles may reach 60 °C (Section 1, Step 4). Additional factors that lower the melting point and will facilitate surface melting in the experimentally produced particles are, surface irregularities19,48 such as edges and corners (Fig. S1 in ESI†) where molecules may be more mobile because of a lower coordination number than other surface molecules, and the presence of foreign species in the surface such as ions, adsorbates as well as traces of H2O.
The difference between the boiling (302–304 °C) and melting point (95–98 °C) of GA is significantly higher than that of the other tested diacids, which indicates that the liquid GA has the strongest H-bonding among MA, SA and AA. The stronger H-bonding of GA will reduce its volatility at room temperature relative to the other diacids.
The enthalpy of sublimation of solid MA to AA reported by Bilde et al.64 increases from 111 ± 15 kJ mol−1 to 131 ± 18 kJ mol−1 for MA to AA, respectively, as shown in Table 2 in ref. 64. The saturation vapor pressures49,64,74–76 of the solid MA to GA accumulate around the value of 10−9 bar at 298 K and do not differ significantly as shown in Table 2 in ref. 64. Differently, AA has an order of magnitude lower saturation vapor pressure.64 Differences in the enthalpies of sublimation of solid diacids and the saturation vapor pressures cannot explain the higher volatility of GA molecules. However, the fact that evaporated charged GA molecules have been detected in MS is a strong indication that GA molecules evaporate easier than the other diacids.
The structure of an amorphous softened surface layer of a particle may be closer to that of a supercooled diacid liquid. The saturation vapor pressures of supercooled diacid liquid64 at 298 K (Table 2) show that the values for SA and GA are almost the same. The enthalpies of vaporization of supercooled diacid liquids shown in Table 2 in ref. 64 indicate that supercooled liquid GA has the lowest enthalpy of vaporization in the series of MA to AA. It is noted, however, that the reported enthalpies of vaporization of MA to AA are not clearly distinguishable because they are within their error bar values. Similarity in the values of the enthalpy of vaporization for supercooled liquid SA and GA indicates that the low enthalpy of vaporization alone cannot be responsible for the distinct GA ionization capacity.
Since the values of the thermodynamic variables presented by Bilde et al.64 for the single phase bulk diacid do not provide an explanation for the volatility of GA, we infer that there is a different phase on the surface of GA particles. This difference in volatility may arise from the local interactions of the adsorbed species and the GA nanoparticle. Higher mobility on the surface layer combined with a lower enthalpy of vaporization will satisfy the conditions for a substrate that has available proton donor sites to yield a strong MS signal. Softening of the surface of the diacid particles will increase the diffusion ability of the diacid molecules, and thus the proton transfer, in the particles at the temperatures at which experiments are performed.
Another aspect to be considered in the charging mechanism is the role of water. AA (C6) and SA (C4) have much lower solubility77,78 in H2O relative to MA and GA, which may favour the formation of crystals during the drying process. MA and GA (both with odd numbers of carbons) have high solubility77 in H2O (the solubility of MA exceeds that of GA by a few times) that may favour the inclusion of H2O mainly in the particle's interior. The H2O impurities may facilitate MA and GA melting relative to AA and SA. However, experiments1 with MA show signal intensities over an order of magnitude lower than GA. This may indicate that the trapping of H2O alone may not be the decisive factor in the signal intensity.
Experiments have also been performed with self-nucleated GA particles.1 This process does not involve an atomizer and aqueous solutions. The signals of GA are still present in the mass spectra, even though weaker. This observation also supports the fact that H2O alone will not play a key role in the presence of a signal, but still it is not known whether it plays a role in enhancing it. The hygroscopicity78 of the diacid particles and the role of H2O in the structure and mobility of amorphous diacid aggregates are topics to be examined in future studies.
We also argue that the pK values of the examined di-acids do not differentiate the signal intensity between GA and the other experimentally tested diacids. With the exception of the pKa1 value of MA, all the other pKa values of the diacids in aqueous solution are similar (Table 2), thus the pKa values in the parent solution are expected to affect similarly the chemistry in the later stages of ionization if their values are maintained. However, within the amorphous diacid particles, different structure of the micro-environment from that in the bulk solution, variability between surface and interior, and finite-sized effects due to possible H2O inclusions in the amorphous matrix may alter the pKa values relative to those in the initial aqueous solution.79 We speculate that the randomness in the amorphous state will smooth out differences among the experimentally tested diacids, which may lead to similar pKa values for all of the diacids, but different from their values in the initial aqueous solution.
4 Conclusion
By using molecular modeling of nano-aggregates composed of diacids, insights into the H-bonded network that is responsible for proton transfer, surface mobility and desorption of ions were gained. These insights are transferable to aggregates with diameters of 100–200 nm observed in experiments.To analyze H-bonding and diffusion, we atomistically modeled GA nano-aggregates of size that is large enough to encompass both surface and bulk-like interior. For comparison, smaller pristine systems composed of up to a few tens of malonic, adipic and succinic acid molecules were also studied. We found that all the amorphous diacid nano-aggregates are characterized by substantial H-bonding where the molecules form chains of several molecules connected to one another with one and two H-bonds, linear dimeric assemblies with one or two H-bonds, and occasionally cyclic trimeric assemblies. The universality of these H-bonded patterns in nano-aggregates of different size and composition indicates that they will also appear in the large particles that were tested experimentally. A dense H-bonding network similar to that of H2O cannot be formed in these systems because of the hydrocarbon chains between the carboxyl groups. Proton transfer is facilitated during the lifetime of the H-bond because of the diacids' low dielectric constants.44,45 A successful proton transfer involves the breaking of the H-bond once the transfer has occurred. Proton diffusion throughout the system will be achieved by the translational and the rotational diffusion of the proton-carrier molecules.
A general method for computing a radially dependent diffusion coefficient in nanoparticles was developed. Throughout the temperature range of 360 K to 420 K, which is near the bulk melting point of crystalline GA, GA nano-aggregates show an order of magnitude faster diffusion in the surface layer of thickness of ∼1 nm than near the center, where the molecules are almost immobile within the simulation time. The mobility on the surface leads to the facile formation of dimeric and cyclic trimeric H-bonded assemblies, which in turn, will allow an efficient proton transfer through the surface molecules via translational and rotational diffusion. The details of the rotational diffusion are still to be studied in future research. The simulations of the mobility on the surface are presented for nanoparticles of a radius of a few nanometers. There are several other factors that increase the mobility of the surface in the experimentally produced nanoparticles and the simulated ones. These factors include surface irregularities (e.g. edges and corners) that assist in the softening of the materials,19,48 the presence of foreign species such as ions and adsorbed analytes in their outer layers that lower the melting point, as well as the amorphous nature of the particles themselves due to their inherent polymorphism.
The final step of the ionization process is the desorption of analytes from the diacid substrates. We found that the desorption occurs via small clusters where a GA− ion may be coordinated by less than a handful of GA molecules. In simulations of charged N = 4096 GA aggregates containing 32 GA− ions, a long Rayleigh jet formation,50 in which ions can enter and eject from its tip as it was previously reported51 for H2O, was not observed. It is noted that the Rayleigh jet is not unique to H2O, as theory54,55 predicts a conical angle for dielectric liquids with dielectric constant >18. The lack of long conical deformations on the surface of GA is consistent with the fact that for materials with dielectric constant <18, such as the diacids, analytical theory54,55 does not predict a cone angle for a jet. However, similarly to the Rayleigh jets, in GA nanoparticles long-living conical surface irregularities can attract neighboring ions, which may travel to the tip of the cone and desorb. The simulations suggest a broader ion-evaporation mechanism56–58 that extends in the regime of low dielectric constant (<18) for particles of any size. Studies of the desorption mechanism and the depth of the ionic species in aerosol particles will assist in establishing a relationship between the mass spectra signals and the profiling of the composition of aerosol particles.
We addressed the question of why GA aggregates give a significantly stronger signal intensities in the mass spectra than malonic, succinic, and adipic acids, which were compared in the experiments. Thermodynamic data that already existed in the literature64 complemented with the molecular studies performed here suggest that the combination of the significantly lower melting point of GA than that of the other tested diacids with a reduced enthalpy of vaporization of the amorphous state may be the reason that leads to the distinctly stronger signals of GA in the mass spectra. The reduced enthalpy of vaporization may arise from the local interactions between the ionic species or analytes and surrounding GA molecules.
There are still a number of molecular questions to investigate in future studies but this study highlights the intricate nature of viscous GA particles and provides a good framework to modeling the spontaneous ionization of GA particles in a mass spectrometer without an external energy source. Future studies may examine the possibility of strengthening the GA signal by lowering its melting point using adipic acid as an additive.80 Synergy of experiments and molecular modeling can examine the ability of GA aggregates to adsorb analytes of different size and polarity relative to the other diacids. Local disorder and conformational changes of the diacid substrate induced by the adsorption of analytes and how this local environment affects the desorption of small clusters containing ionic species should be further examined. Moreover, the factors that affect the minimal adsorbed concentration required to provide MS signals await exploration. The depth profiling of the analytes from the surface is another critical question to be examined by combining experiments and molecular simulations. This question is intimately related to the desorption mechanism of the ionic species and the composition of the desorbed clusters.
Data availability
Data supporting this article have been included as part of the ESI.† Data for this article, including MD trajectories are available at https://doi.org/10.7910/DVN/G52HGM.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. C. acknowledges a Fulbright Canada Research Chair Climate Change, Air Quality and Atmospheric Chemistry fellowship held at the Department of Chemistry, University of California, Irvine for funding this research. BJFP acknowledges NSF grant no. 2030175 and LMW acknowledges NSF grant no. 2331523 for funding this research. S. C. acknowledges insightful discussions on experimental methods used in atmospheric aerosol studies with Prof. J. Hemminger, Prof. J. Smith, Prof. S. Nizkorodov and members of their groups, and discussions on computational methods for liquid–vapor interfaces with Prof. D. Tobias. The discussions took place at the Chemistry Department, University of California, Irvine. S. C. expresses gratitude for the hospitality offered by Prof. B. J. Finlayson-Pitts and by members of all the atmospheric chemistry groups. S. C. is grateful to Dr. Anatoly Malevanets for very insightful discussions on charged systems. Digital Research Alliance of Canada is acknowledged for providing the computing facilities.Notes and references
- Y. Qin, L. Wingen and B. Finlayson-Pitts, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2209134119 CrossRef CAS.
- B. J. Finlayson-Pitts, Phys. Chem. Chem. Phys., 2009, 11, 7760–7779 RSC.
- B. J. Finlayson-Pitts, L. M. Wingen, V. Perraud and M. J. Ezell, Commun. Chem., 2020, 3, 108 CrossRef PubMed.
- L. Wingen and B. Finlayson-Pitts, Chem. Sci., 2019, 10, 884–897 RSC.
- T. Nah, M. Chan, S. R. Leone and K. R. Wilson, Anal. Chem., 2013, 85, 2087–2095 CrossRef CAS PubMed.
- M. N. Chan, T. Nah and K. R. Wilson, Analyst, 2013, 138, 3749–3757 RSC.
- S. Kumbhani, T. Longin, L. Wingen, C. Kidd, V. Perraud and B. Finlayson-Pitts, Anal. Chem., 2018, 90, 2055–2062 CrossRef CAS PubMed.
- S. Trimpin, J. Am. Soc. Mass Spectrom., 2015, 27, 4–21 CrossRef.
- S. Trimpin and E. D. Inutan, J. Am. Soc. Mass Spectrom., 2013, 24, 722–732 CrossRef CAS.
- S. Trimpin, B. Wang, C. B. Lietz, D. D. Marshall, A. L. Richards and E. D. Inutan, Crit. Rev. Biochem. Mol. Biol., 2013, 48, 409–429 CrossRef CAS PubMed.
- S. Trimpin, I.-C. Lu, S. Rauschenbach, K. Hoang, B. Wang, N. D. Chubatyi, W.-J. Zhang, E. D. Inutan, M. Pophristic and A. Sidorenko, et al. , J. Am. Soc. Mass Spectrom., 2017, 29, 304–315 CrossRef PubMed.
- S. Trimpin, M. Pophristic, A. Adeniji-Adele, J. W. Tomsho and C. N. McEwen, Anal. Chem., 2018, 90, 11188–11192 CrossRef CAS PubMed.
- S. Trimpin, B. Wang, E. D. Inutan, J. Li, C. B. Lietz, A. Harron, V. S. Pagnotti, D. Sardelis and C. N. McEwen, J. Am. Soc. Mass Spectrom., 2012, 23, 1644–1660 CrossRef CAS PubMed.
- P. Liu, E. Harder and B. Berne, J. Phys. Chem. B, 2004, 108, 6595–6602 CrossRef CAS.
- K. Kawamura and S. Bikkina, Atmos. Res., 2016, 170, 140–160 CrossRef CAS.
- A. Chebbi and P. Carlier, Atmos. Environ., 1996, 30, 4233–4249 CrossRef CAS.
- A. Roose, C. Toubin, S. Dusanter, V. Riffault and D. Duflot, ACS Earth Space Chem., 2019, 3, 380–389 CrossRef CAS.
- J. Elm, N. Hyttinen, J. J. Lin, T. Kurtén and N. L. Prisle, J. Phys. Chem. A, 2019, 123, 9594–9599 CrossRef CAS.
- G. de With, Chem. Rev., 2023, 123, 13713–13795 CrossRef CAS PubMed.
- P. Buffat and J. P. Borel, Phys. Rev. A: At., Mol., Opt. Phys., 1976, 13, 2287 CrossRef CAS.
- V. I. Levitas and K. Samani, Nat. Commun., 2011, 2, 284 CrossRef PubMed.
- V. Kwan, S. Consta and S. M. Malek, J. Phys. Chem. B, 2023, 128, 193–207 CrossRef PubMed.
- J. C. Phillips, R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kalé and K. Schulten, J. Comput. Chem., 2005, 26, 1781–1802 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- B. R. Brooks, C. L. Brooks III, A. D. Mackerell Jr, L. Nilsson, R. J. Petrella, B. Roux, Y. Won, G. Archontis, C. Bartels and S. Boresch, et al. , J. Comput. Chem., 2009, 30, 1545–1614 CrossRef CAS PubMed.
- K. Vanommeslaeghe and A. D. MacKerell Jr, J. Chem. Inf. Model., 2012, 52, 3144–3154 CrossRef CAS PubMed.
- K. Vanommeslaeghe, E. P. Raman and A. D. MacKerell Jr, J. Chem. Inf. Model., 2012, 52, 3155–3168 CrossRef CAS.
- T. H. Nguyen, D. E. Hibbs and S. T. Howard, J. Comput. Chem., 2005, 26, 1233–1241 CrossRef CAS PubMed.
- X. Sheng, X. Song, H. Zhu, C. A. Ngwenya and H. Zhao, Chem. Phys., 2019, 524, 14–20 CrossRef CAS.
- D. J. Hardy, Z. Wu, J. C. Phillips, J. E. Stone, R. D. Skeel and K. Schulten, J. Chem. Theory Comput., 2015, 11, 766–779 CrossRef CAS.
- V. R. Thalladi, M. Nüsse and R. Boese, J. Am. Chem. Soc., 2000, 122, 9227–9236 CrossRef CAS.
- K. H. Weber, F. J. Morales and F.-M. Tao, J. Phys. Chem. A, 2012, 116, 11601–11617 CrossRef CAS PubMed.
- A. Luzar, J. Chem. Phys., 2000, 113, 10663–10675 CrossRef CAS.
- A. Luzar and D. Chandler, Nature, 1996, 379, 55–57 CrossRef CAS.
- J. D. Cryer and K.-S. Chan, Time series regression models, Springer, 2008 Search PubMed.
- R Core Team, R: A language and environment for statistical computing, R Foundation for Statistical Computing, Vienna, Austria, 2021 Search PubMed.
- D. Chandler, Introduction to modern statistical mechanics, Oxford University Press, New York, 1987 Search PubMed.
- J. Liu, X. He, J. Z. Zhang and L.-W. Qi, Chem. Sci., 2018, 9, 2065–2073 RSC.
- C.-J. Tsai, J.-S. Lin, C. Deshpande and L.-C. Liu, Part. Part. Syst. Charact., 2005, 22, 293–298 CrossRef.
- B. Forsyth, B. Y. Liu and F. J. Romay, Aerosol Sci. Technol., 1998, 28, 489–501 CrossRef CAS.
- V. V. Pervukhin, D. G. Sheven’ and Y. N. Kolomiets, Tech. Phys., 2016, 61, 1262–1268 CrossRef CAS.
- S. Consta and R. Kapral, J. Chem. Phys., 1994, 101, 10908–10914 CrossRef CAS.
- P. L. Geissler, C. Dellago, D. Chandler, J. Hutter and M. Parrinello, Science, 2001, 291, 2121–2124 CrossRef CAS PubMed.
- S. Ponomarenko, Y. Y. Borovikov, T. Sivachek and D. Vovk, Russ. J. Gen. Chem., 2003, 73, 1769–1773 CrossRef CAS.
- V. Madhurima, K. Sudheendran and K. James Raju, Mol. Simul., 2006, 32, 331–337 CrossRef CAS.
- N. Agmon, Chem. Phys. Lett., 1995, 244, 456–462 CrossRef CAS.
- D. Marx, ChemPhysChem, 2006, 7, 1848–1870 CrossRef CAS PubMed.
- R. Shidpour, H. H. Delavari and M. Vossoughi, Chem. Phys., 2010, 378, 14–18 CrossRef CAS.
- M. Bilde, B. Svenningsson, J. Mønster and T. Rosenørn, Environ. Sci. Technol., 2003, 37, 1371–1378 CrossRef CAS.
- L. Rayleigh, Philos. Mag., 1882, 14, 184–186 Search PubMed.
- S. Consta, J. Phys. Chem. B, 2022, 126, 8350–8357 CrossRef CAS.
- V. Kwan and S. Consta, Chem. Phys. Lett., 2020, 137238 CrossRef CAS.
- S. Vepsäläinen, S. M. Calderón, J. Malila and N. L. Prisle, Atmos. Chem. Phys., 2022, 22, 2669–2687 CrossRef.
- A. Ramos and A. Castellanos, Phys. Lett. A, 1994, 184, 268–272 CrossRef CAS.
- G. Taylor, Proc. R. Soc. London, Ser. A, 1964, 280, 383–397 Search PubMed.
- J. Iribarne and B. Thomson, J. Chem. Phys., 1976, 64, 2287–2294 CrossRef CAS.
- M. Labowsky, J. Fenn and J. F. De La Mora, Anal. Chim. Acta, 2000, 406, 105–118 CrossRef CAS.
- I. G. Loscertales and J. Fernández De La Mora, J. Chem. Phys., 1995, 103, 5041–5060 CrossRef CAS.
- S. Kollotzek, O. V. Lushchikova, L. Tiefenthaler, F. Zappa and P. Scheier, Int. J. Mol. Sci., 2022, 23, 3613 CrossRef CAS.
- F. Laimer, L. Kranabetter, L. Tiefenthaler, S. Albertini, F. Zappa, A. M. Ellis, M. Gatchell and P. Scheier, Phys. Rev. Lett., 2019, 123, 165301 CrossRef CAS.
- V. Kwan, R. O’Dwyer, D. Laur, J. Tan and S. Consta, J. Phys. Chem. A, 2021, 125, 2954–2966 CrossRef.
- V. Kwan and S. Consta, J. Phys. Chem. A, 2022, 126, 3229–3238 CrossRef CAS.
- P. Loche, D. J. Bonthuis and R. R. Netz, Commun. Chem., 2022, 5, 55 CrossRef CAS.
- M. Bilde, K. Barsanti, M. Booth, C. D. Cappa, N. M. Donahue, E. U. Emanuelsson, G. McFiggans, U. K. Krieger, C. Marcolli and D. Topping, et al. , Chem. Rev., 2015, 115, 4115–4156 CrossRef CAS PubMed.
- J. D. Roberts and M. C. Caserio, Basic Principles of Organic Chemistry, W. A. Benjamin, Inc., Menlo Park, CA, 1977 Search PubMed.
- P. C. Belser, H. R. Timsina, T. M. Raymond and D. D. Dutcher, Aerosol Sci. Technol., 2019, 53, 464–472 CrossRef CAS.
- P. Espeau, P. Négrier and Y. Corvis, Cryst. Growth Des., 2013, 13, 723–730 CrossRef CAS.
- Y.-T. Cheng and W. L. Johnson, Science, 1987, 235, 997–1002 CrossRef CAS.
- S. Alexander, Phys. Rep., 1998, 296, 65–236 CrossRef CAS.
- U. Tartaglino, T. Zykova-Timan, F. Ercolessi and E. Tosatti, Phys. Rep., 2005, 411, 291–321 CrossRef CAS.
- H.-P. Cheng and R. S. Berry, Phys. Rev. A: At., Mol., Opt. Phys., 1992, 45, 7969 CrossRef CAS.
- D. W. Oxtoby, Nature, 1990, 347, 725–730 CrossRef CAS.
- J. Israelachvili, Intermolecular & Surface Forces, Academic Press, 2011 Search PubMed.
- M. Bilde and S. N. Pandis, Environ. Sci. Technol., 2001, 35, 3344–3349 CrossRef CAS PubMed.
- S. Chattopadhyay, H. J. Tobias and P. J. Ziemann, Anal. Chem., 2001, 73, 3797–3803 CrossRef CAS PubMed.
- Y. Tao and P. H. McMurry, Environ. Sci. Technol., 1989, 23, 1519–1523 CrossRef CAS.
- P. Saxena and L. M. Hildemann, J. Atmos. Chem., 1996, 24, 57–109 CrossRef CAS.
- C. Peng, M. N. Chan and C. K. Chan, Environ. Sci. Technol., 2001, 35, 4495–4501 CrossRef CAS PubMed.
- E. Mehler, M. Fuxreiter, I. Simon and B. Garcia-Moreno E, Proteins: Struct., Funct., Bioinf., 2002, 48, 283–292 CrossRef CAS PubMed.
- A. Kolyado, S. Alenova and I. Garkushin, Russ. J. Phys. Chem. A, 2016, 90, 1293–1297 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: (S1) Atomic force microscopy images of glutaric acid particles. (S2) Experimentally measured temperatures of the inlet elbow through which particles travel in the mass spectrometer as as function of the source temperature set points. (S3) Radial distribution functions of succinic acid as a function of diacid nano-aggregate size. (S4) Radial distribution functions of oxalic to adipic diacid nano-aggregates. (S5) Typical snapshots of H-bonding patterns in N = 21 succinic acid nano-aggregate. (S6) Molecular dynamics movie of evaporation of GA− from a GA nano-aggregate composed of 4096 GA molecules and 32 GA− ions. See DOI: https://doi.org/10.1039/d4cp03597f |
| ‡ Present address: Department of Chemistry, The University of Western Ontario, London, Ontario, N6A 5B7, Canada. Tel: +1 519 661 2111. |
| § Present address: School of Energy and Environment, City University of Hong Kong, Hong Kong, China. |
| This journal is © the Owner Societies 2024 |