
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInsights into the electronic structure of non-steroidal anti-inflammatory drugs: soft X-ray study of fenoprofen, ketoprofen and methyl salicylate in the gas phase†
Hanan
Sa’adeh
 *ab,
Assimo
Maris
*c,
Kevin C.
Prince
bd,
Oksana
Plekan
b,
Cesare
Grazioli
e,
Marcello
Coreno
f and
Robert
Richter
b
*ab,
Assimo
Maris
*c,
Kevin C.
Prince
bd,
Oksana
Plekan
b,
Cesare
Grazioli
e,
Marcello
Coreno
f and
Robert
Richter
b
aDepartment of Physics, The University of Jordan, Amman, 11942, Jordan. E-mail: Hanan.Saadeh@ju.edu.jo
bElettra Sincrotrone Trieste, in Area Science Park, 34149 Basovizza, Trieste, Italy
cDipartimento di Chimica G. Ciamician, Università di Bologna, 40139 Bologna, Italy. E-mail: assimo.maris@unibo.it
dCharles University, Faculty of Mathematics and Physics, Department of Surface and Plasma Science, V Holešovičkách 2, Prague, 18000, Czech Republic
eCNR – Istituto Officina dei Materiali (IOM), Area Science Park - Basovizza, S.S. 14 Km 163.5, Trieste 34149, Italy
fCNR – Istituto di Struttura della Materia (ISM), Area Science Park - Basovizza, S.S. 14 Km 163.5, Trieste 34149, Italy
First published on 14th November 2024
Abstract
The valence and core electronic structure of three non-steroidal anti-inflammatory drugs (methyl salicylate, fenoprofen and ketoprofen) have been studied by photoelectron and soft X-ray absorption spectroscopy, supported by theoretical calculations of the molecular and electronic structure. The conformational landscape has been explored for sixteen low-energy conformers of fenoprofen and ketoprofen, and the energies of both compounds fall into two groups with steric similarities, separated by about 3 kJ mol−1. Valence band photoelectron spectra agree with previous results, and the spectra have been calculated using two approaches. We find the outer valence Green's function method gives good results, but the P3+ method is a little better, particularly for outer valence ionic states. Carbon and oxygen 1s photoemission spectra are reported and are in acceptable agreement with the theory. The C and O K near-edge X-ray absorption fine structure spectra are reported and interpreted by comparison with reference compounds. We analyse the data to provide rough estimates of the energies of the unoccupied orbitals in methyl salicylate.
1. Introduction
Non-steroidal anti-inflammatory drugs (NSAIDs) are commonly used medications to relieve pain and reduce inflammation. They reduce the body's production of prostaglandins, which trigger inflammation, fever, and pain. NSAIDs block the enzyme cyclooxygenase (COX) used by the body to make prostaglandins.1 By reducing the production of prostaglandins, the drugs help relieve the discomfort of fever and reduce inflammation and associated pain. The motivation for studying NSAIDs is mainly based on their pharmaceutical applications1 but can also be extended to their environmental impact.2 In a previous study, we investigated the most widely used compounds in this group of drugs (aspirin, paracetamol and ibuprofen) through gas-phase X-ray photoelectron spectroscopy (XPS).3 Gas-phase studies, where extrinsic effects are eliminated, provide unique advantages in exploring and characterizing the intrinsic electronic structure of the isolated molecules. In the present work, we extend our gas-phase investigation to three other NSAIDs: methyl salicylate (MS), fenoprofen (FP), and ketoprofen (KP), with the long-term goal of identifying spectroscopic properties that are indicative of pharmaceutical efficacy.MS (C8H8O3, also known as methyl 2-hydroxybenzoate) is an organic ester naturally produced by plants to defend against predators or pathogens. It can be synthesized from salicylic acid (which we have previously studied in the gas phase4). Among other uses, MS is used in high concentrations to treat joint and muscular pain, and in low concentrations as a flavoring agent. Similarly to ibuprofen, FP (C15H14O3, 2-(3-phenoxyphenyl)propanoic acid) and KP (C16H14O3, 2-(3-benzoylphenyl)propanoic acid) are 2-phenylpropanoic acids and have analgesic and antipyretic effects. They are related to the NSAID ibuprofen, but their molecular structure is different because of the aromatic ring substituents: ibuprofen has an isobutyl in the para position with respect to the carboxylic chain, while FP and KP have a phenoxy and a benzoyl group respectively in the meta position. KP is generally prescribed for the treatment of acute pain, chronic arthritis, and severe toothaches. It is also known as an important water pollutant.2 The schematic structures of the three compounds are shown in Fig. 1.
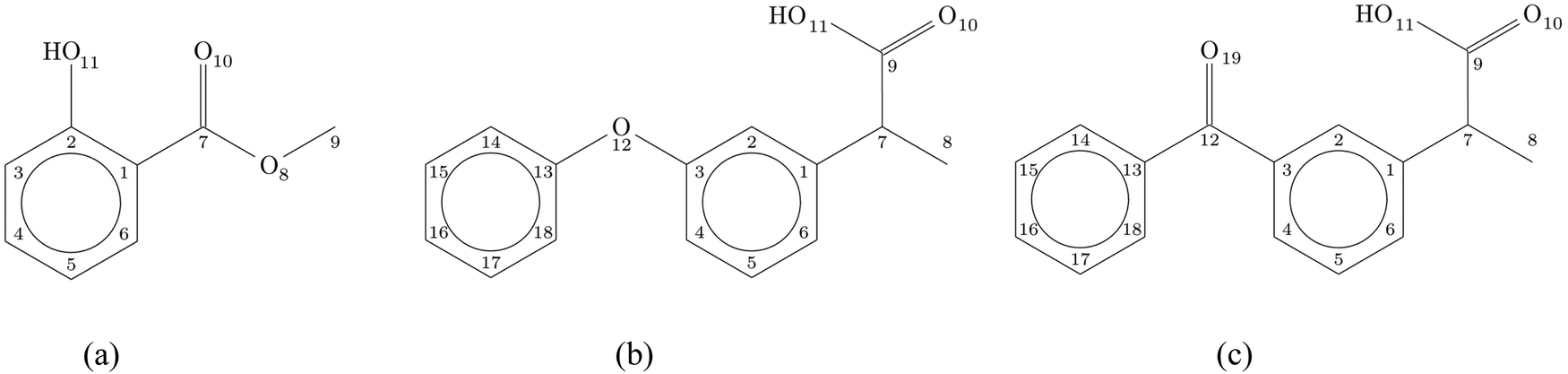 | ||
| Fig. 1 Schematic structure and numbering of the atoms of (a) methyl salicylate (MS), (b) fenoprofen (FP) and (c) ketoprofen (KP). | ||
KP and FP have very similar structures as they both have two benzene rings (see Fig. 1) connected by a carbonyl group and ether oxygen atom, respectively. The question arises as to how this small change impacts the electronic structure of these two molecules.
MS has a simpler geometric structure (only one benzene ring). Its intramolecular hydrogen bond permits proton transfer in its excited states and the dynamics have been the subject of a few recent studies.5–7 The rotational spectra have been reported and the observed structures of this molecule have been explained using quantum chemical calculations.8
The valence band photoelectron spectra of the three proposed molecules were reported by Novak et al.9 using He I radiation and kinetic energies in the 8–16 eV energy range, and the results were in good agreement with their calculations. To the best of our knowledge, no gas phase core level ionization and X-ray absorption spectra have been reported. In the present work, core-level X-ray photoemission spectra (XPS) and near-edge X-ray absorption fine structure (NEXAFS) spectra have been measured, as well as the valence band spectra. We present new calculations of the valence spectra and compare two methods, Outer Valence Green's Function and the renormalized Partial Third Order (P3+) self-energy approximations. The experimental XPS results are compared to predictions based on quantum mechanical calculations.
2. Experimental methods
MS and KP were purchased from Sigma-Aldrich (now Merck), with purity >99% and >98%, respectively, and used without further purification. FP was purchased from abcr GmbH with specified purity >95% and used without further purification. At room temperature, MS is liquid, FP is a viscous oily liquid, and KP is solid. Vapour from the samples was admitted to the vacuum chamber via an effusive needle source at room temperature (MS) or evaporated from a crucible at temperatures of 323 K (FP) or 333 K (KP), with a background pressure in the measurement chamber of 1–3 × 10−8 mbar.The experiments were performed using the VG analyzer end-station at the GasPhase (GAPH) Photoemission beamline of Elettra-Sincrotrone Trieste, Italy.10 The photoemission spectra were measured with a combined photon and analyzer energy resolution of: 0.2 eV (or better) at photon energies 60 and 100 eV (valence); 0.4 eV (or better) at photon energy 382 eV (C 1s); and 0.7 eV (or better) at photon energies 588 and 628 eV (O 1s). The energy scale of the valence band spectrum was calibrated by observing the first ionization potential of H2O (present in the residual gas), binding energy 12.62 eV,11 while for core-level photoemission, the spectra were referred to the following core levels of CO2: 297.6 eV, C 1s,12 and 541.3 eV, O 1s.13,14 The NEXAFS spectra at the C and O K edges were measured by scanning the photon energy and measuring the total ion yield. The ion yield was normalised to the photon flux measured by a photodiode behind the sample. The photon energy scale was calibrated by reference to the resonances of CO2 at 290.77 eV (C 1s → π*)15 and 535.4 eV (O 1s → π*).16
3. Computational methods
Minimum energy structures and related Hessian matrices were determined using the valence triple-zeta quality Karlsruhe polarized type basis set (Def2-TZVP17) in combination with the Becke-three-parameters Lee–Yang–Parr hybrid density functional theory (B3LYP18,19) corrected by the D3 version of Grimme's empirical dispersion with Becke–Johnson damping (B3LYP-D3(BJ)20,21). The obtained geometries were used for subsequent calculations to estimate the vertical ionization potentials (vIPs) using the same basis set. Outer valence band IPs were calculated using electron propagator theory (EPT22,23) and by applying the Outer Valence Green's Function (OVGF24–26) or alternatively the renormalized Partial Third Order (P3+27) self-energy approximations. The inner shell vIPs were estimated by means of the symmetry-adapted cluster/configuration interaction method (SAC-CI28) with the MIDI! basis set.29 All the mentioned calculations were done using the Gaussian16™‡ software package (G16, Rev. A.03).4. Results and discussion
4.1. Computational results
The most stable conformer of MS in the gas phase, as determined experimentally by rotational spectroscopy,8 is characterized by Cs symmetry and the carbonyl oxygen atom forming two hydrogen bonds with the hydroxyl and methyl groups, respectively. Our calculated rotational constants, at the B3LYP-D3(BJ)/Def2-TZVP level, (A = 2183, B = 834, C = 606 MHz) are close to the experimental values (A = 2169, B = 833, C = 604 MHz8) and confirm the reliability of the calculated structure, therefore justifying its use for the simulation of the ionization spectra.To our knowledge, no experimental information on the structures of FP and KP in the gas phase is available. However rotational spectroscopy data on the unsubstituted ring systems, that is, diphenyl ether30 and benzophenone,31–33 respectively, show that the two phenyl rings do not lie in the same plane, but are staggered, being rotated in opposite directions with respect to the C3–O12–C13 and C3–C12(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O19)–C13 planes in FP and KP, respectively. Two orientations of the propanoic acid group are possible, with it facing toward the phenyl group (#A) or the oxygen atom (#B), as shown in Fig. 2. However, depending on the rotation of the phenyl groups, for each of the #A and #B species, two non-equivalent conformers exist, namely #A′/#A′′ and #B′/#B′′. They are represented in Fig. 2 using blue and red sticks which indicate whether the bond is directed in front of or behind the plane, respectively. Concerning the side chain, since C7 is a chiral carbon atom, two enantiomers (R and S) are possible for each conformer. Here we refer to the S-species, which in the case of KP is also called dexketoprofen. The hydrogen atom bound to the chiral atom is almost coplanar with the phenyl ring. Therefore, two orientations (#1 and #2) are possible as shown in Fig. 2. An additional degree of freedom is provided by the orientation of the carboxyl group. It is perpendicular to the C1–C7 bond connecting the phenyl and the propyl chain allowing two possible arrangements, with the carbonyl oxygen atom being syn or anti with respect to the hydrogen atom bound to the chiral atom. Overall, there are 24 = 16 non-equivalent skeletal conformers. Here we considered the lower energy arrangement of the carboxyl group, with the hydroxyl interacting with the carbonyl oxygen through a hydrogen bond. Another sixteen conformers refer to the hydroxyl pointing in the opposite direction, but we do not consider this arrangement because it is far less stable. For instance, in formic acid, the energy difference between cis- and trans-rotamers determined by microwave relative intensity measurements is 16.3(4) kJ mol−1.34
O19)–C13 planes in FP and KP, respectively. Two orientations of the propanoic acid group are possible, with it facing toward the phenyl group (#A) or the oxygen atom (#B), as shown in Fig. 2. However, depending on the rotation of the phenyl groups, for each of the #A and #B species, two non-equivalent conformers exist, namely #A′/#A′′ and #B′/#B′′. They are represented in Fig. 2 using blue and red sticks which indicate whether the bond is directed in front of or behind the plane, respectively. Concerning the side chain, since C7 is a chiral carbon atom, two enantiomers (R and S) are possible for each conformer. Here we refer to the S-species, which in the case of KP is also called dexketoprofen. The hydrogen atom bound to the chiral atom is almost coplanar with the phenyl ring. Therefore, two orientations (#1 and #2) are possible as shown in Fig. 2. An additional degree of freedom is provided by the orientation of the carboxyl group. It is perpendicular to the C1–C7 bond connecting the phenyl and the propyl chain allowing two possible arrangements, with the carbonyl oxygen atom being syn or anti with respect to the hydrogen atom bound to the chiral atom. Overall, there are 24 = 16 non-equivalent skeletal conformers. Here we considered the lower energy arrangement of the carboxyl group, with the hydroxyl interacting with the carbonyl oxygen through a hydrogen bond. Another sixteen conformers refer to the hydroxyl pointing in the opposite direction, but we do not consider this arrangement because it is far less stable. For instance, in formic acid, the energy difference between cis- and trans-rotamers determined by microwave relative intensity measurements is 16.3(4) kJ mol−1.34
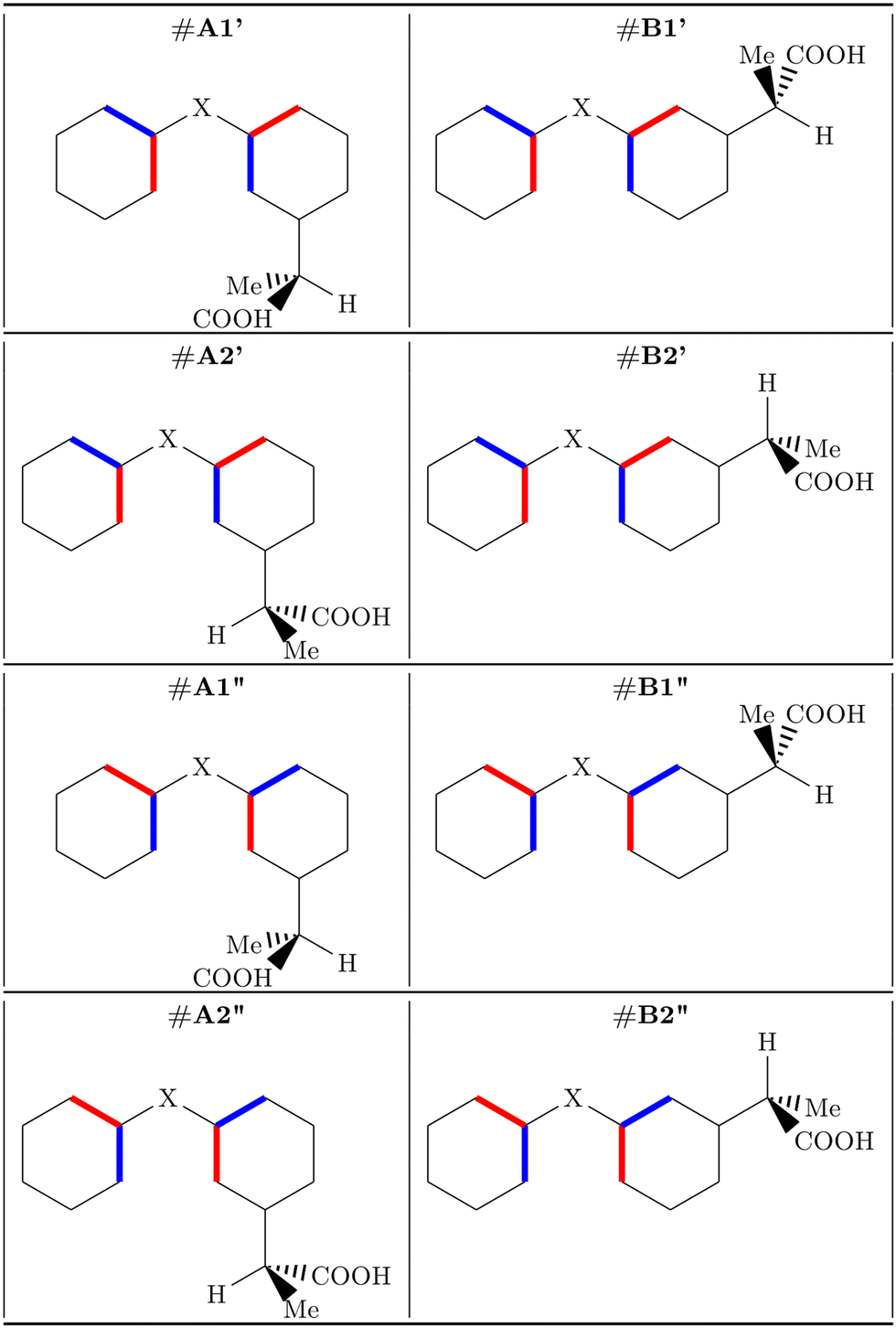 | ||
| Fig. 2 S-conformers of fenoprofen (X = O) and ketoprofen (X = CO). Blue and red sticks indicate bonds directed in front of or behind the plane, respectively. | ||
Geometry optimization followed by evaluation of the Hessian matrix was performed for each of the sixteen more stable conformers of FP and KP. The relative energy values are listed in Table S1 and represented in Fig. S1 (ESI†), while the atomic coordinates are reported in Tables S2 and S3 in the ESI.† All these conformers lie in the 0–7 kJ mol−1 energy region. In both molecules, the anti species are more stable than the syn ones, the relative energy lying in the 0–1.3 kJ mol−1 range for anti-FP, 3.8–6.3 kJ mol−1 for syn-FP, 0–2.9 kJ mol−1 for anti-KP, and 4.2–7.0 kJ mol−1 for syn-KP.
In each of these subgroups, the conformers with the propanoic acid substituent on the same side of the unsubstituted ring (#A) are in general more stable than those with the substituent on the opposite side (#B), except for the case of the #A2′′ and #B2′′ forms.
As regards the alkyl chain, it can assume two orientations, corresponding to the two eclipsed positions of the hydrogen atom bound to the chiral carbon atom with respect to the phenyl ring. In general, these two orientations have a similar energy, however, it is worth noting that the preferred one is different for the two molecules, as shown in Fig. 3. Conformer #S-A1′′-anti was chosen for subsequent calculations. In the crystalline phase, #KP-R-B2′-anti has been identified in the pure compound (CCDC: KEMRUP35) while as a macromolecular ligand both #KP-A (Protein Data Bank36 codes: 6QS9,375ZWR,386UQR39) and #KP-B (Protein Data Bank36 codes: 7JWN,406UQR,396OCK41) forms were determined. Only #FP-A (codes: 4NED, 2X7H) conformers have been reported.
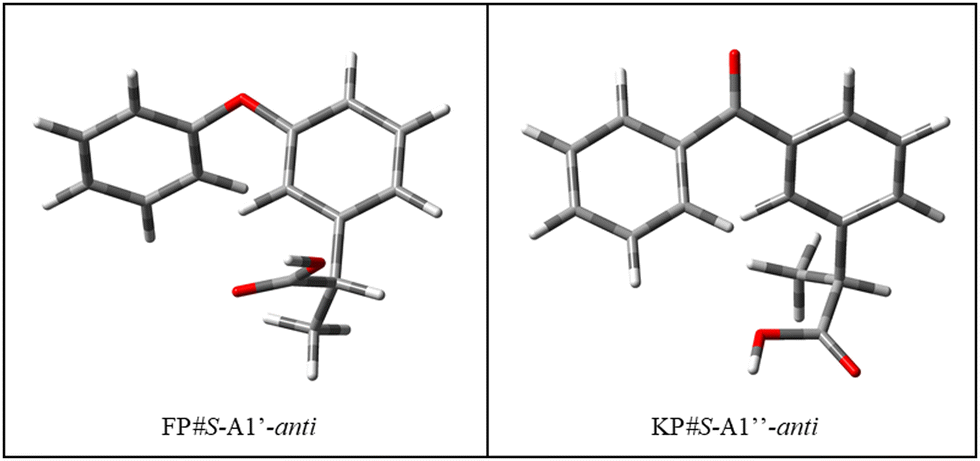 | ||
| Fig. 3 Structure of the most stable conformers of fenoprofen (FP) and ketoprofen (KP) according to B3LYP-D3(BJ)/Def2-TZVP calculations. | ||
4.2. Results: valence band
The experimental valence band photoelectron spectra of MS, FP and KP are shown in Fig. 4, while the peak assignments of the outer valence bands, based on the theoretical vIPs are given in Tables 1–3. The experimental ionization energies obtained by Novak et al.9 are also listed and are in reasonable agreement with the present data. The complete list of calculated energies and experimental peaks is given in the ESI† (Tables S4–S6). The calculated pole strengths of the valence band molecular orbitals vary between 0.84 and 0.89, indicating that the single electron approximation is valid. The experimental spectra are compared to the simulated ones in Fig. S2a–S4a in the ESI,† together with illustrations of all molecular orbital charge densities in Fig. S2b–S4b (ESI†). Fig. 5 and 6 show charge density maps of the lowest energy molecular orbitals. | ||
| Fig. 4 Valence photoelectron spectra of methyl salicylate (red), fenoprofen (green) and ketoprofen (blue). Photon energy: 100 eV for methyl salicylate and ketoprofen, and 60 eV for fenoprofen. The fenoprofen and ketoprofen spectra have been offset vertically for clarity. The features are numbered so that peaks approximately correspond. | ||
| No. | C s | Koopmans | OVGF | P3+ | Exp. | Exp.9 | Label |
|---|---|---|---|---|---|---|---|
| MOE/eV | vIP/eV | vIP/eV | IP/eV | IP/eV | |||
| a Values obtained by fitting three Gaussian functions to the experimental peak. | |||||||
| 31 | σ | −15.60 | 13.80 | 13.50 | 13.4a | 13.4 | VII |
| 32 | π | −15.20 | 13.96 | 13.69 | |||
| 33 | σ | −14.93 | 13.37 | 13.27 | 13.1a | 13.0 | |
| 34 | σ | −14.61 | 13.15 | 12.93 | 12.9a | 12.61 | |
| 35 | σ | −13.91 | 12.30 | 12.30 | 12.0 | 12.1 | VI |
| 36 | π | −13.32 | 11.90 | 11.87 | 11.58 | 11.35 | V |
| 37 | π–COOCH3 | −13.04 | 11.64 | 11.37 | 11.20 | IV | |
| 38 | σ (nO) | −12.51 | 10.89 | 10.54 | 10.47 | 10.40 | III |
| 39 | πPh | −9.78 | 9.48 | 9.68 | 9.57 | 9.45 | II |
| 40 | πPh/OH | −8.75 | 8.55 | 8.73 | 8.64 | 8.60 | I |
| No. | Character | Koopmans | OVGF | P3+ | Exp. | Exp.9 | Label |
|---|---|---|---|---|---|---|---|
| MOE/eV | vIP/eV | vIP/eV | IP/eV | IP/eV | |||
| 54 | −13.82 | 12.34 | 12.29 | 11.80 | 12.1 | V | |
| 55 | −13.77 | 12.29 | 12.24 | ||||
| 56 | −13.71 | 12.26 | 12.22 | ||||
| 57 | −13.34 | 11.88 | 11.76 | ||||
| 58 | −13.17 | 11.90 | 11.77 | ||||
| 59 | −12.85 | 11.36 | 11.28 | 11.02 | 11.0 | IV | |
| 60 | σ (nO) propanoic acid moiety | −12.24 | 10.69 | 10.45 | 10.44 | 10.45 | III |
| 61 | π Ph2, Ph1 | −9.49 | 9.30 | 9.40 | 9.20 | 9.15 | II |
| 62 | π Ph1, Ph2 | −9.25 | 9.13 | 9.22 | |||
| 63 | π Ph2, Ph1 | −9.21 | 8.97 | 9.11 | |||
| 64 | π Ph1, Ph2 | −8.48 | 8.06 | 8.32 | 8.27 | 8.25 | I |
| No. | Character | Koopmans | OVGF | P3+ | Exp. | Exp.9 | Label |
|---|---|---|---|---|---|---|---|
| MOE/eV | vIP/eV | vIP/eV | IP/eV | IP/eV | |||
| 55 | −14.17 | 12.56 | 12.49 | 11.88 | 12.1 | V | |
| 56 | −14.02 | 12.47 | 12.40 | ||||
| 57 | −13.94 | 12.42 | 12.43 | ||||
| 58 | −13.73 | 12.23 | 12.21 | ||||
| 59 | −13.58 | 12.04 | 12.05 | ||||
| 60 | −13.37 | 12.13 | 11.99 | ||||
| 61 | −13.24 | 11.90 | 11.86 | ||||
| 62 | σ (nO) propanoic acid moiety | −12.43 | 10.86 | 10.63 | 10.58 | 10.55 | III |
| 63 | σ (nO), O19 | −11.49 | 9.65 | 9.29 | 8.98 | I | |
| 64 | π Ph2, Ph2 | −9.60 | 9.33 | 9.51 | 9.41 | 9.4 | II |
| 65 | π Ph1, Ph2 | −9.43 | 9.22 | 9.37 | |||
| 66 | π Ph1, Ph2 | −9.38 | 9.18 | 9.36 | |||
| 67 | π Ph1, Ph2 | −9.31 | 9.14 | 9.29 | 8.98 | 9.0 | I |
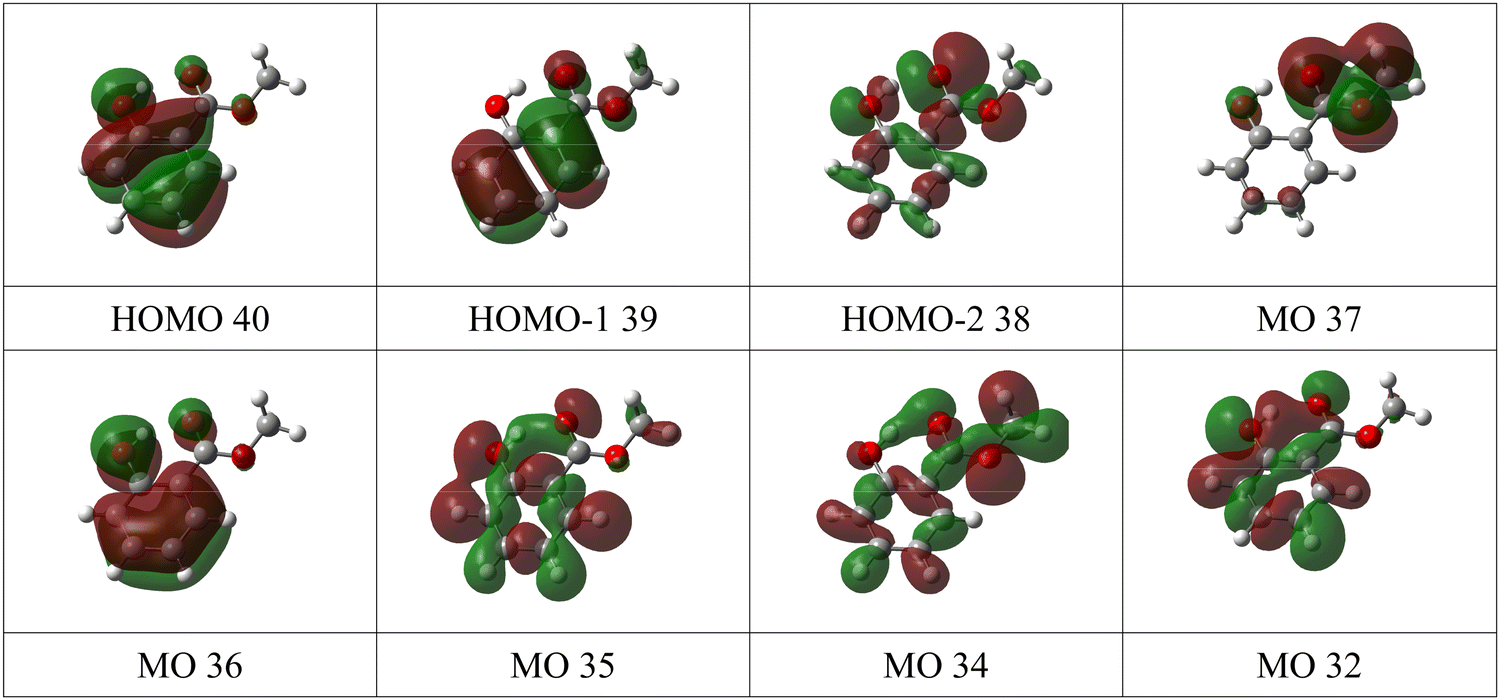 | ||
| Fig. 5 Charge density maps of the eight lowest energy molecular orbitals of methyl salicylate. | ||
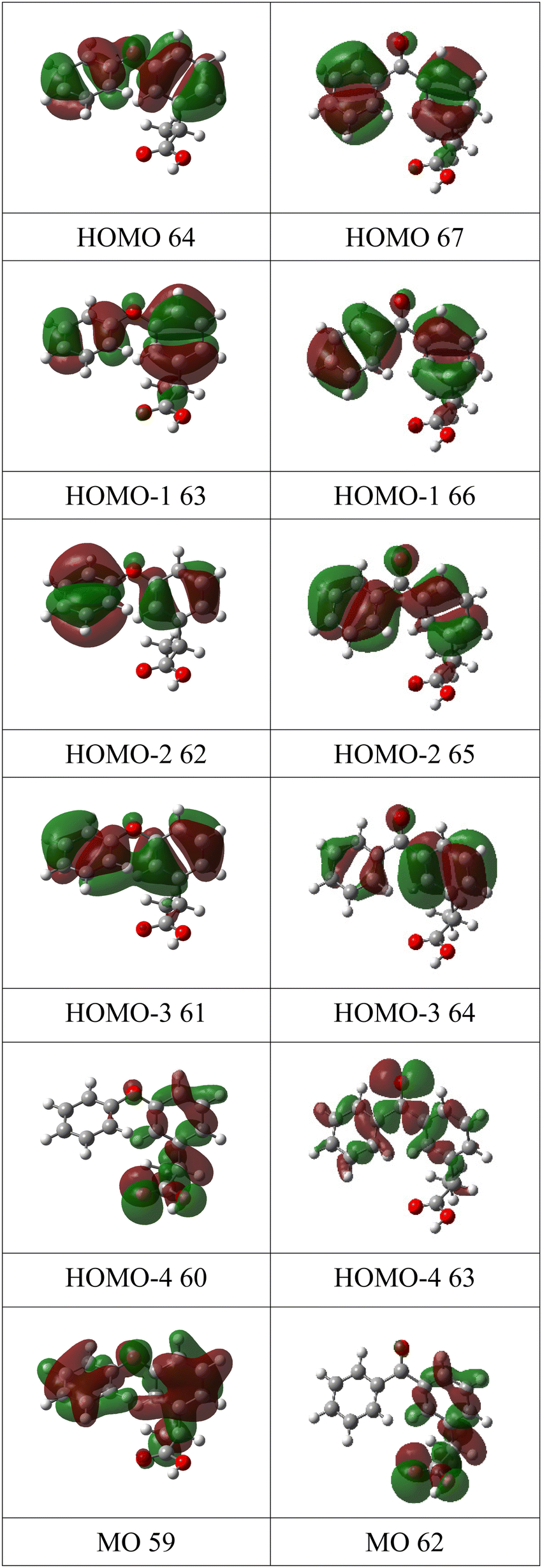 | ||
| Fig. 6 Charge density maps of the six lowest energy molecular orbitals of fenoprofen (left column) and ketoprofen (right column). | ||
The valence band spectrum of MS is broadly similar in shape to that of the parent molecule, salicylic acid, Fig. S5 of the ESI,† with some shifts in energy, and some additional structure. The experimental peak I at 8.65 and II at 9.57 eV (Fig. 4 and Fig. S2a, ESI†) agree well with the theoretical binding energies of the π-type highest occupied molecular orbital (HOMO) and second highest occupied molecular orbital (HOMO−1), with the calculated values slightly underestimating (OVGF) or overestimating (P3+) the experimental values (Table 1). The charge density maps (Fig. 5) indicate that these peaks correspond to ionization of two π bonds of the aromatic ring, with a contribution from the OH group to the HOMO. Band II shows vibronic splitting with a vibrational quantum of about 140 meV; for neutral MS, the strongest IR absorption band with this energy is the O–CH3 stretching vibration.42 Excitation of this vibration on ionization of the HOMO−1 orbital is consistent with the partial localization of the orbital on the O8 atom, bonded to the CH3 group.
The strong experimental peak III at 10.47 eV matches the vIP of a σ-type orbital (HOMO−2) mainly described by the non-bonding oxygen atom orbitals (nO), with the carbonyl oxygen character being strongest. The orbital charge density is however widely distributed over the molecule. Band IV, of π symmetry, is strongly localised on the ester group, with some OH character. Band V is assigned to an orbital of π symmetry and localized mainly on the phenyl ring with a contribution from the carbonyl group. Band VI is assigned to an orbital of σ symmetry, while band VII is assigned to three σ orbitals and one π orbital. These are not resolved, and the structure of the peak suggests that at least three peaks are contributing, so the peak was fitted with three Gaussian functions. At higher binding energy, most experimental bands correspond to two or more predicted ionic states.
The measured first ionization potential of KP (Fig. 4 and Fig. S4a, ESI†) is in good agreement with calculations; the theoretical values slightly overestimate the experimental values. FP (Fig. 4 and Fig. S3a, ESI†) has the lowest first ionization potential (8.27 eV) among the three compounds, with only slight differences between the theoretical and experimental HOMO ionization energies (about +0.04 eV using P3+, and −0.21 eV using OVGF). In general, the agreement between the calculated and measured energies for the outer valence molecular orbitals of these compounds is good. The calculated P3+ ionization energies agree slightly better with the experiment than those obtained by the OVGF, particularly for the outermost states. The root mean square (rms) differences between the theoretical (OVGF, P3+) and all experimental energies are (0.31, 0.25), (0.38, 0.30), and (0.39, 0.32) eV for MS, FP, and KP, respectively. For the first three ionic states, the rms difference between P3+ theory and experiment is less than 0.2 eV, namely, 0.09 eV (MS), 0.11 eV (FP), and 0.18 eV (KP), which is quite satisfactory. We conclude that the P3+ calculation is more reliable in predicting the outer valence ionizations for the three compounds.
FP and KP are structurally similar: both have two benzene rings, compared to one ring in MS, and both contain propanoic acid groups. However, the shape of their first three bands (I, II, and III) differs, while at binding energies greater than 11 eV, the spectral features of FP and KP are reasonably similar. To understand why the outer valence spectra are so different despite the close structural similarities, we again consider the charge density maps of the orbitals, Fig. 6. Band I of FP has phenyl π bond character, is distributed over both rings with approximately equal weight, and is assigned to ionization of the HOMO. The second band II is assigned to three unresolved ionic states, all with phenyl π bond character localised on both rings, but with varying relative contributions from the substituted and unsubstituted rings. Band III is assigned to ionization of the HOMO−4 and is mostly localised on the propanoic acid moiety.
For KP, the lowest energy band I is assigned to two ionic states, one of which is due to ionization of the HOMO, has phenyl π bond character and is delocalised over both rings. In contrast to the HOMO of FP, the charge distributions on the phenyl rings are elongated in the direction of the keto bridge. According to the P3+ calculations, this band overlaps molecular orbital 63, which has lone pair oxygen character, and whose dominant contribution is localised on the carbonyl group. The OVGF calculations place this orbital at higher energy, but the distribution of intensities favours the P3+ assignment. Band II of KP is assigned to three states of phenyl π character distributed over both rings. The first is similar to the HOMO−1 of FP, with the charge densities more equally balanced on the two rings; for FP the substituted ring provides more π bond character. The calculated ionization energies are similar, 9.36 eV for KP and 9.11 eV for FP. The third orbital contributing to band II has a much stronger contribution from the phenyl π orbitals from the substituted ring than from the unsubstituted ring. As in the case of FP, they have varying weights on each of the rings, and the charge distributions show some similarities with those of FP.
Band III of KP is localised mainly on the propanoic acid group, as in the case of FP, and has a similar energy. Overall, the change from the ether bridge in FP to the keto bridge in KP significantly perturbs the outer orbitals, and increases the first ionization potential. An orbital with strong carbonyl π bond character, which has no counterpart in FP, appears and is accidentally degenerate in energy with the HOMO.
4.3. Results: core levels
The carbon and oxygen 1s spectra of MS, FP and KP are shown in Fig. 7–12, and the theoretical and experimental core electron binding energies are summarised in Table 4. The calculated pole strengths of the core-level molecular orbitals vary between 0.74 and 0.77 for C 1s and between 0.77 and 0.79 for O 1s, indicating that the single-electron approximation is valid. The C 1s spectra were fitted with Gaussian profiles, and the relative intensity ratios of the C 1s peaks for the three compounds (Table 5) are very consistent with the expected stoichiometric ratios.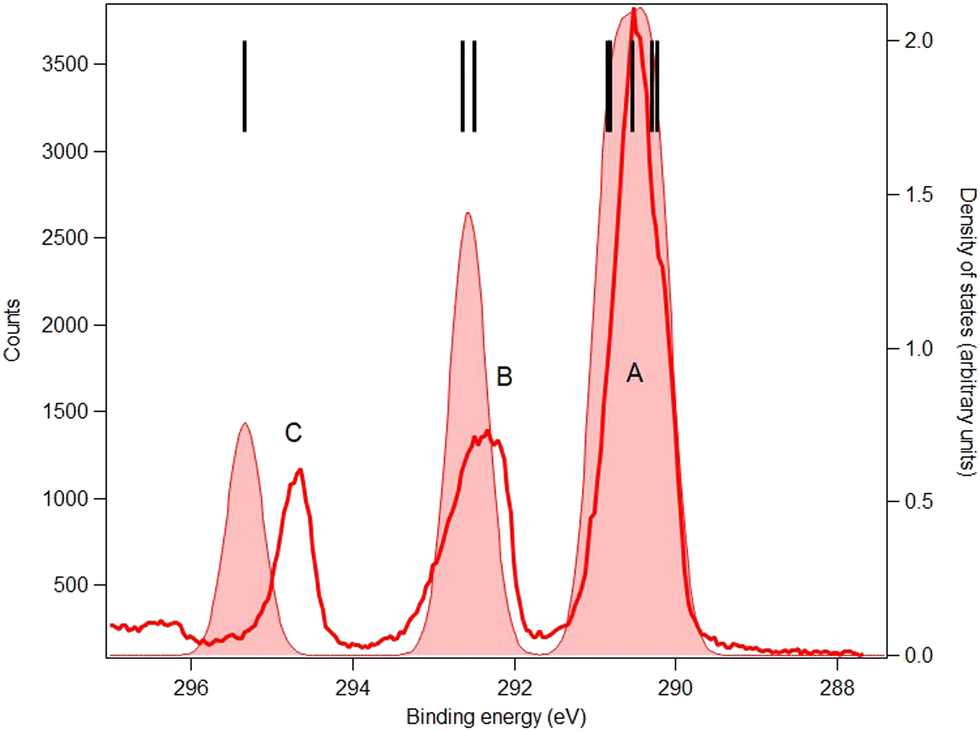 | ||
| Fig. 7 C 1s core-level spectrum (red line) and simulation (red full area) of methyl salicylate. Photon energy: 382 eV. Black bars: theoretical vertical ionization potentials (at the SAC-CI/MIDI! level of calculation). The theoretical curve has been shifted to lower binding energy by 0.58 eV. | ||
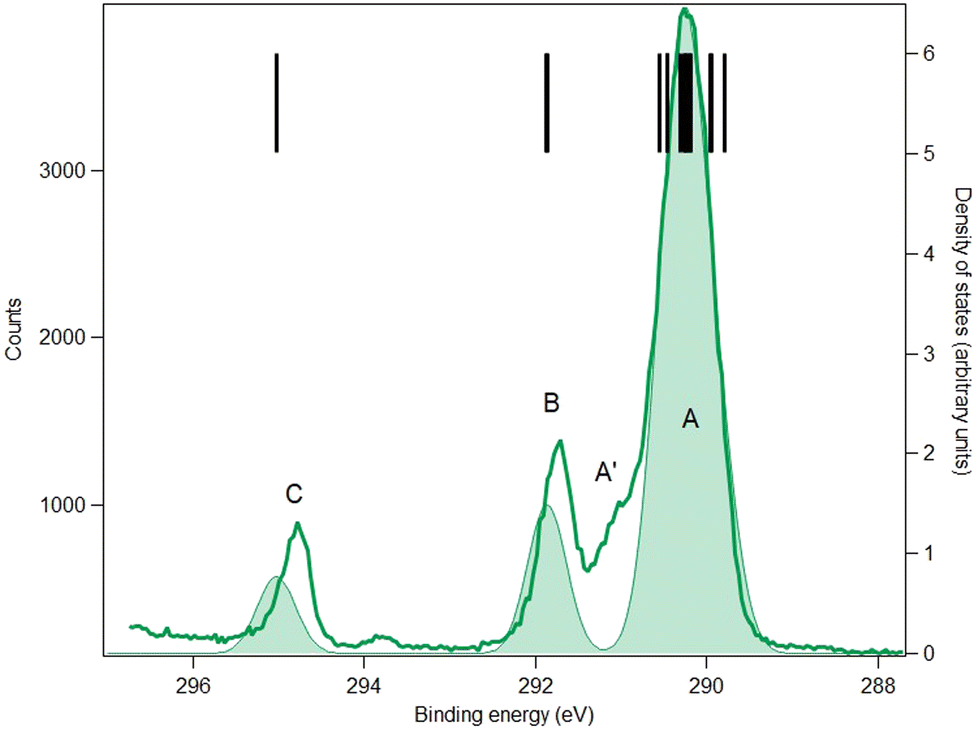 | ||
| Fig. 8 C 1s core-level spectrum (green line) and simulation (green full area) of fenoprofen. Photon energy: 382 eV. Black bars: theoretical vertical ionization potentials (at the SAC-CI/MIDI! level of calculation). The theoretical curve has been shifted to lower binding energy by 0.13 eV. | ||
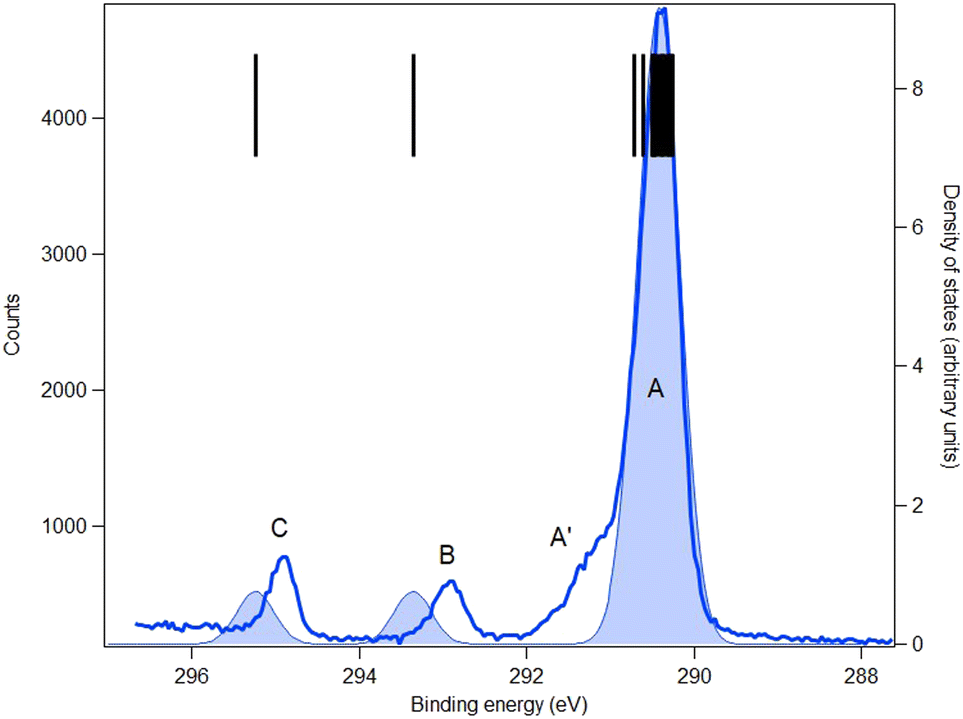 | ||
| Fig. 9 C 1s core-level spectrum (blue line) and simulation (blue full area) of ketoprofen. Photon energy: 382 eV. Black bars: theoretical vertical ionization potentials (at the SAC-CI/MIDI! level of calculation). | ||
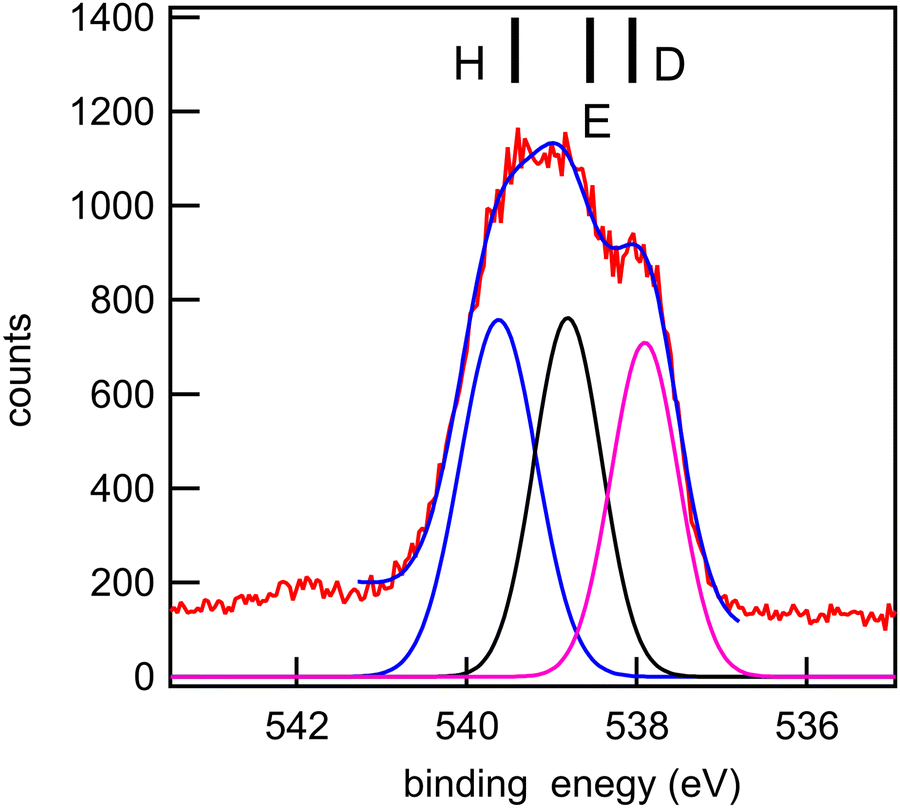 | ||
| Fig. 10 O 1s core-level spectrum (red line: data; blue line: smoothed data) of methyl salicylate and a fit with three Gaussian functions, whose integrated intensity variations were limited to ±10% of the mean intensity, and whose energies and widths were allowed to vary freely. Photon energy: 628 eV. Black bars: theoretical vertical ionization potentials (at the SAC-CI/MIDI! level of calculation). | ||
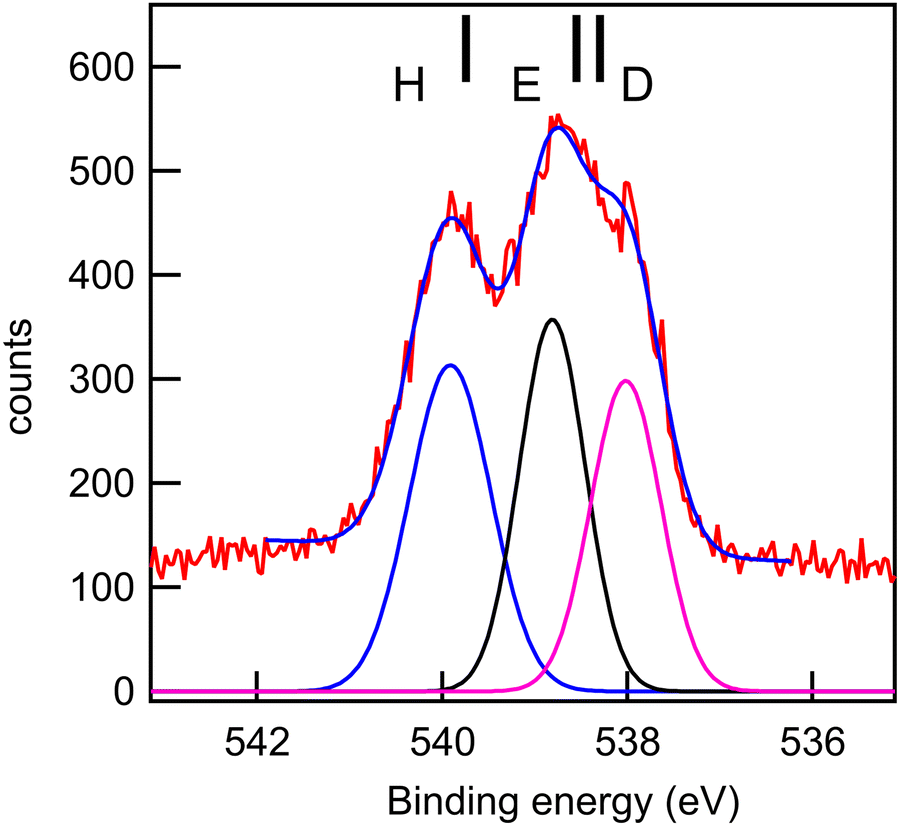 | ||
| Fig. 11 O 1s core-level spectrum (red line: data; blue line: smoothed data) of fenoprofen and a fit with three Gaussian functions, whose integrated intensity variations were limited to ±10% of the mean intensity, and whose energies and widths were allowed to vary freely. Photon energy: 628 eV. Black bars: theoretical vertical ionization potentials (at the SAC-CI/MIDI! level of calculation). | ||
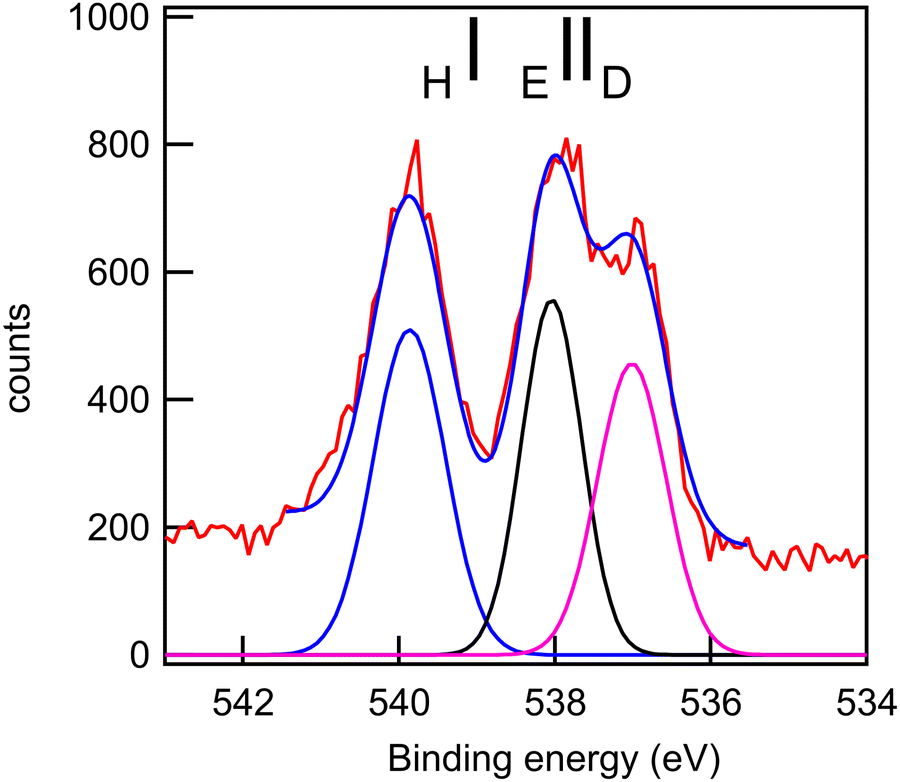 | ||
| Fig. 12 O 1s core-level spectrum (red line: data; blue line: smoothed data) of ketoprofen and a fit with three Gaussian functions, whose integrated intensity variations were limited to ±10% of the mean intensity, and whose energies and widths were allowed to vary freely. Photon energy: 588 eV. Black bars: theoretical vertical ionization potentials (at the SAC-CI/MIDI! level of calculation). | ||
| Core | No. | Assignment | MOE/eV | vIP/eV | IP/eV | Label |
|---|---|---|---|---|---|---|
| Methyl salicylate | ||||||
| C 1s | 11 | C3 (Ph) | 304.00 | 290.80 | 290.48 | A |
| 10 | C5 (Ph) | 304.05 | 290.88 | |||
| 9 | C1 (Ph) | 304.48 | 291.11 | |||
| 8 | C4 (Ph) | 304.67 | 291.42 | |||
| 7 | C6 (Ph) | 304.72 | 291.39 | |||
| 6 | C9 (OCH3) | 305.62 | 293.21 | 292.44 | B | |
| 5 | C2 (C–OH) | 306.48 | 293.08 | |||
| 4 | C7 (CO) |
308.59 | 295.91 | 294.71 | C | |
| O 1s | 3 | O10 (CO) |
556.24 | 538.05 | 537.87 | D |
| 2 | O11 (OH) | 556.57 | 538.55 | 538.60 | E | |
| 1 | O8 (OCH3) | 557.77 | 539.43 | 539.37 | H | |
| Fenoprofen | ||||||
| C 1s | 18 | C8 (CH3) | 303.71 | 290.37 | 290.23 291.02 | A A′ |
| 17 | C16 (Ph) | 303.95 | 290.33 | |||
| 16 | C4 (Ph) | 303.95 | 290.09 | |||
| 15 | C2 (Ph) | 303.96 | 289.92 | |||
| 14 | C6 (Ph) | 303.97 | 290.08 | |||
| 13 | C14 (Ph) | 303.98 | 290.36 | |||
| 12 | C18 (Ph) | 304.02 | 290.32 | |||
| 11 | C15 (Ph) | 304.15 | 290.59 | |||
| 10 | C17 (Ph) | 304.16 | 290.59 | |||
| 9 | C5 (Ph) | 304.23 | 290.43 | |||
| 8 | C7 (*C) | 304.62 | 290.68 | |||
| 7 | C1 (Ph) | 304.65 | 290.39 | |||
| 6 | C13 (Ph) | 305.89 | 292.00 | 291.75 | B | |
| 5 | C3 (Ph) | 306.04 | 291.99 | |||
| 4 | C9 (COOH) | 308.00 | 295.16 | 294.80 | C | |
| O 1s | 3 | O10 (CO) |
555.87 | 537.59 | 537.90 | D |
| 2 | O12 (PhOPh) | 557.26 | 537.85 | 538.70 | E | |
| 1 | O11 (OH) | 557.39 | 539.04 | 539.90 | H | |
| Ketoprofen | ||||||
| C 1s | 19 | C8 (CH3) | 303.85 | 290.49 | 290.40 290.87 | A A′ |
| 18 | C15 (Ph) | 304.08 | 290.49 | |||
| 17 | C17 (Ph) | 304.10 | 290.51 | |||
| 16 | C14 (Ph) | 304.20 | 290.47 | |||
| 15 | C5 (Ph) | 304.23 | 290.37 | |||
| 14 | C16 (Ph) | 304.26 | 290.60 | |||
| 13 | C4 (Ph) | 304.30 | 290.30 | |||
| 12 | C13 [C18] (Ph) | 304.31 | 290.31 | |||
| 11 | C18 [C13] (Ph) | 304.33 | 290.45 | |||
| 10 | C2 [C3] (Ph) | 304.42 | 290.25 | |||
| 9 | C6 [C2, C03] (Ph) | 304.43 | 290.40 | |||
| 8 | C3 [C2, C06] (Ph) | 304.44 | 290.28 | |||
| 7 | C1 (Ph) | 304.69 | 290.38 | |||
| 6 | C7 (*C) | 304.76 | 290.71 | |||
| 5 | C12 (CO) |
306.71 | 293.36 | 292.93 | B | |
| 4 | C9 (COOH) | 308.14 | 295.24 | 294.93 | C | |
| O 1s | 3 | O19 (CO) |
555.86 | 537.03 | 536.87 | D |
| 2 | O10 (CO) |
556.00 | 537.68 | 537.93 | E | |
| 1 | O11 (OH) | 557.53 | 539.12 | 539.89 | H | |
| Sample | C 1s intensity ratios | O 1s intensity ratios |
|---|---|---|
| Methyl salicylate | A![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :B:C = 5.1:2.0:0.9 :B:C = 5.1:2.0:0.9 |
D:E:H = 0.91:0.98:1.10 |
| Fenoprofen | A:A′:B:C = 10.2:1.8:2.0:1.0 |
D:E:H = 0.94:0.98:1.08 |
| Ketoprofen | A:A′:B:C = 9.9:4.3:0.8:1.0 |
D:E:H = 0.90:1.01:1.09 |
The C 1s spectrum of MS (Fig. 7) is the simplest of the three compounds, and shows three well-resolved peaks A, B and C. Peak C is assigned to the carbonyl carbon atom (C7), peak B is assigned to carbon bonded to the hydroxyl moiety (C2) and methyl (C9) carbon atom, and the broad peak A is assigned to the other five benzene ring carbon atoms. The theory overestimates the binding energy of the carbonyl carbon atom in MS by about 1.2 eV, but for the remaining seven carbon atoms the average energy difference between theory and experiment is less than 0.7 eV.
The C 1s spectra of FP (Fig. 8) and KP (Fig. 9) show three main peaks A, B and C, in addition to a minor peak/shoulder A′ that is not present in the spectrum of MS. The peaks C are assigned to the ionization of the carboxylic carbon atoms (C9) in both compounds. Peaks B are assigned to the two ring carbon atoms (C3 and C13) bonded to the ether oxygen atom in FP, and to the carbonyl carbon atom (C12) in ketoprofen. The broad peak A is assigned to the remaining carbon atoms (methyl carbon C8 and other benzene ring carbon atoms), while the shoulder A′ may be a vibrational structure (not included in the theory). Studies on the influence of vibrations on the C 1s signal of some organic molecules (see for example ref. 43 for benzene rings and ref. 44 for a series of linear alkanes) have highlighted that the vibrational tail can extend up to 0.8 eV above the main lines (i.e., the vertical ionization energies) in agreement with this interpretation. The theoretical binding energies agree within 0.3 eV with the experimental values in both compounds, which is satisfactory, and the rms difference between the fitted and theoretical energies is 0.25 eV in FP and 0.18 in KP, which also indicates good agreement.
The experimental and theoretical oxygen 1s core spectra of the three compounds (Fig. 10–12) are not fully resolved, but fitted with three peaks D, E, and H, corresponding to the three oxygen atoms. For MS, fitting with Gaussian functions, whose binding energy, intensity and peak widths are free parameters, does not yield a physically meaningful result, so we modified our fitting procedure. It is known that intensities in core photoemission of molecules may vary from their stoichiometric ratio, due to a number of phenomena such as variations of pole strength, and scattering, but usually the variation is no more than about 10%.45 We therefore performed a constrained fit, with 3 peaks, whose integrated intensity variations were limited to ±10% of the mean intensity, and whose energies and widths were allowed to vary freely. The results are shown in Table 5, and the binding energies extracted are stable for small variations in assumed width and intensities. The ratio of integrated intensities for MS is 0.91:0.98:1.10, and this is close to the expected stoichiometric ratio. FP and KP spectra were analysed in the same way.
The rms energy difference between theory and experiment is 0.2, 0.75, and 0.4 eV for MS, FP, and KP, respectively. The calculations (Table 4) and the shape of the spectra (Fig. 10–12) indicate that the differences in binding energies D and E are very small (0.3–0.7 eV). The highest binding energy peak H is assigned to the hydroxyl oxygen atom (O11) in FP and KP, and to the oxygen atom attached to the methyl group (O8) in MS, and we note that this is the most intense peak in all cases. The lowest binding energy peak D is assigned to the oxygen atom of the carbonyl group in the three compounds: O10 in MS and FP, and O19 in KP. The remaining oxygen atoms, O11 in MS, O12 in FP, and O10 in KP are assigned to the peak E.
4.4. Results: near-edge X-ray absorption fine structure spectra
The C and O K edge NEXAFS spectra of MS, FP, and KP are shown in Fig. 13 and 14, and the resonance energies are summarised in Tables 6 and 7.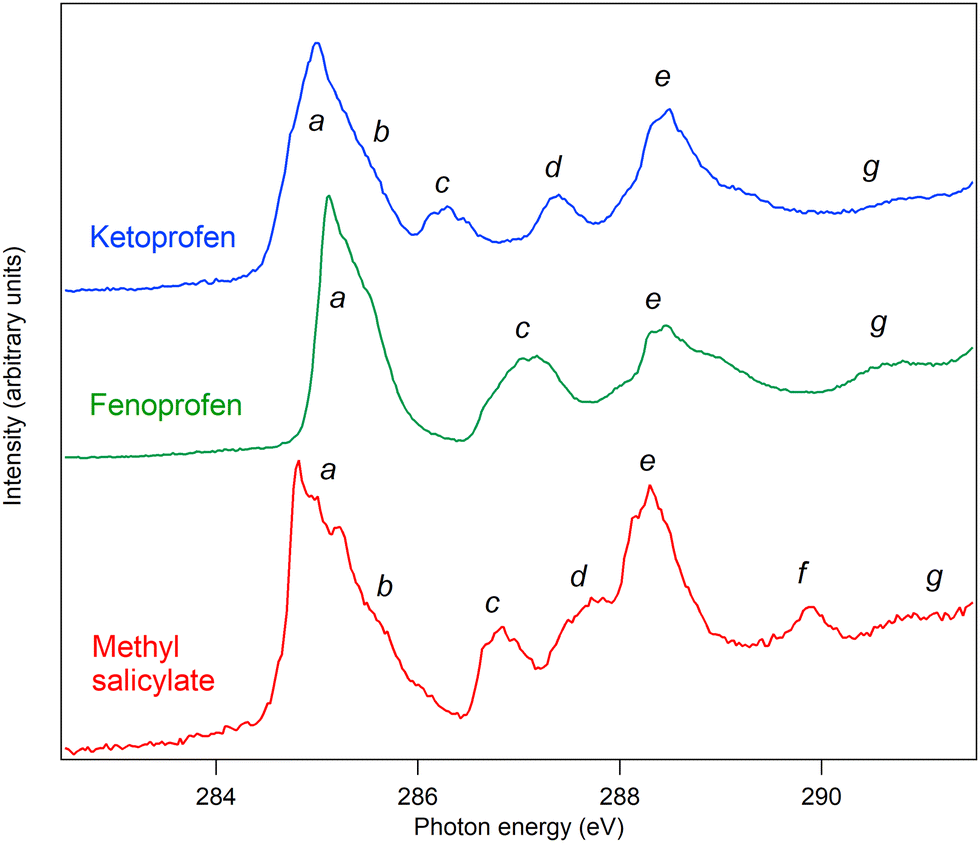 | ||
| Fig. 13 Carbon near-edge X-ray absorption fine structure spectra of methyl salicylate (red), fenoprofen (green) and ketoprofen (blue). The fenoprofen and ketoprofen spectra have been offset vertically for clarity. | ||
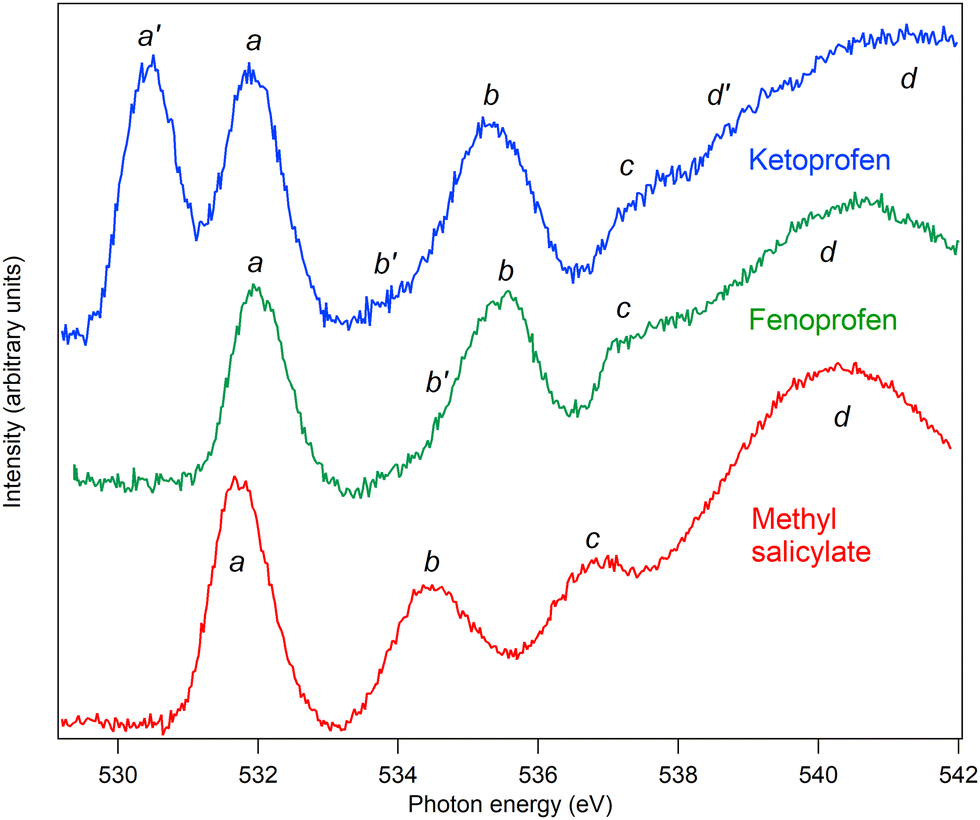 | ||
| Fig. 14 Oxygen near-edge X-ray absorption fine structure spectra of methyl salicylate (red), fenoprofen (green) and ketoprofen (blue). The fenoprofen and ketoprofen spectra have been offset vertically for clarity. | ||
| Label | Methyl salicylate | Fenoprofen | Ketoprofen | |||||
|---|---|---|---|---|---|---|---|---|
| Energy (eV) | Term value (eV) | Energy (eV) salicylic acid4 | Assignment | Energy (eV) | Assignment | Energy (eV) | Assignment | |
| a | 284.8 | 5.7 | 284.6 | C3, C1, C6, C4 1s → π1* | 285.1 | C1, C5, C15, C17 1s → π1* | 284.8 | C1, C5, C15, C17 1s → π1* |
| 284.9 | 5.6 | 284.7 | 285.3 | 285.0 | ||||
| 285.2 | 5.3 | 284.9 | 285.5 | 285.4 | ||||
| b | 285.4 | 5.1 | 285.6 | C5, C3 1s →π2* | 285.6 | C1, C5, C15, C17 1s → π2* | ||
| c | 286.7 | 5.9 | 286.6 | C2 1s → π1* | 286.8 | C13, C3 1s → π1*, C1, C2, C4–C6 1s → 3s | 286.3 | C13, C3 1s → π1*, C1, C2, C4–C6 1s → 3s |
| 287.0 | 5.7 | 286.7 | 287.2 | C14–C18 1s→ 3s | C14–C18 1s→ 3s | |||
| d | 287.7 | 4.8 (C2) | 287.7 | C2 1s → π2*, C1 1s → π3*/3p | 287.4 | |||
| 2.8 (C1) | ||||||||
| e | 288.2 | 6.5 (C7) | 288.1 | C7 1s → π1* (C9, 1s → 3s) | 287.9 | C7, C8 1s → 3s, 3p Rydberg C9 1s → π* | 288.1 | C7, C8 1s → 3s, 3p Rydberg C9 1s → π* |
| 4.3 (C9) | ||||||||
| 288.4 | 6.3 (C7) | 288.2 | 288.4 | 288.4 | ||||
| 4.1 (C9) | ||||||||
| 288.8 | 5.9 (C7) | 288.4 | 288.8 | 288.6 | ||||
| 3.7 (C9) | ||||||||
| 289.7 | 289.1 | |||||||
| f | 289.9 | 0.6 (C3, C5, C4) | 289.6 | C3, C5, C4 1s → π3*, π3*/3p (C9 1s → 4p) | ||||
| 2.5 (C9) | ||||||||
| g | 291.2 | 1.2 (C9) | σ shape resonance | 290.6 | 290.5 | |||
| Label | Methyl salicylate | Fenoprofen | Ketoprofen | |||
|---|---|---|---|---|---|---|
| Energy (eV) | Assignment of corresponding feature in salicylic acid (energy, eV)4 | Energy (eV) | Assignment | Energy (eV) | Assignment | |
| a′ | 530.4 | O19 1s to πc* | ||||
| a | 531.8 | O10 1s → π1* (531.9) | 532.0 | O10 1s → πa* | 531.9 | O10 1s → πa* |
| b′ | 534.8 | 533.9 | ||||
| b | 534.5 | O11 1s → π1* O8 1s → π1*, 3s (535.2) | 535.5 | O11 1s → πa*, 3s | 535.3 | O11 1s → πa*, 3s |
| c | 536.6 | O8, O11 1s → Rydberg, O2 1s → π2* (537.7) | 537.3 | Rydberg transitions | 537.4 | Rydberg transitions |
| d′ | 538.7 | |||||
| IP | 539.4 | 539.9 | 539.9 | |||
| d | 540.4 | σ shape resonance | 540.1 | σ shape resonance | 541.4 | σ shape resonance |
The C K edge spectrum of MS can be assigned by comparison with the spectrum of the closely related compound salicylic acid,4 for which detailed calculations were carried out. The spectra are very similar (see Fig. S6, ESI†), and in Table 6, their peak energies are compared: the energies differ by an average of 0.19 eV, and for most features, MS has a slightly higher excitation energy. We therefore assign the spectrum to the same transitions.
Table 6 also lists the term values of the excitations of MS, calculated using the experimental binding energies. Due to overlap and spectral broadening, the true binding energies of different core levels contributing to a given experimental peak may vary by about ±0.2 eV. In an independent electron model, the term values correspond to the energies of the unoccupied orbitals with respect to the vacuum level. This model is a crude approximation as the orbital character changes between the neutral, core ionized and core excited states due to different relaxation, and may depend on the spatial overlap of the core holes and unoccupied orbitals, but the data provide some qualitative information. Peaks a, c and e are due to transitions from several core levels to the π1* orbital, and the term values are 5.7, 5.9 and 6.5 eV. Peaks b and d correspond to transitions from core levels to the π2* orbital, with term values of 5.1 and 4.8 eV. Peaks d and f contain contributions from transitions to the π3* orbital, with term values of 2.8 and 0.6 eV. These results suggest that the model is sufficiently accurate to predict the energetic order of the unoccupied orbitals, with π1* at 5.7–6.5 eV, π2* at 4.8–5.1 eV, and π3* at 0.6–2.8 eV below the vacuum level.
MS differs from salicylic acid by the substitution of a methyl group, which may give rise to additional resonance structure. In methanol and dimethyl ether, which contain O–CH3 groups, the lowest energy C 1s excitation is a transition to a 3s Rydberg state at about 288 eV, and the most prominent excitation is to a 4p Rydberg state at about 289.4 eV.46 We therefore assign peak e (at 288.2 eV) to a C9 1s → 3s transition (as well as other transitions), and peak f (289.9 eV) to a C9 1s → 4p transition. Peak g is assigned to a σ resonance.
The structures of FP and KP have phenyl rings and carboxylic acid chromophores in common with MS, but display a number of structural differences. The acid groups are propanoic acid, so that the COOH motif is separated from the phenyl ring by the methanetriyl carbon, C7, rather than being directly bonded to the phenyl ring. There are also two phenyl rings instead of one, linked by an ether group (FP) or a keto group (KP). We assign the NEXAFS spectra using the building block principle, and with reference to related compounds.
The spectral features of the two phenyl groups joined by an ether bridge are expected to be similar to those of phenol.4 The features a and c at 285.1 and 286.8 eV correspond closely to similar features in phenol at 285.1 and 287.04 eV, and are therefore assigned to corresponding transitions. Peak a is assigned to C1, C5, C15, C17 1s → π1*, in other words, to C 1s transitions from carbon atoms in the meta position in the two rings, to the π1* orbital of each ring, which is derived from the degenerate benzene ring π* system on substitution of one hydrogen atom by an oxygen atom.
Peak c is assigned to C13, C3 1s → π1*, C1, C2, C4–C6 1s → 3s, that is to C 1s transitions to the same unoccupied orbitals from the phenyl carbon atoms bonded to oxygen, as well as transitions from all unsubstituted carbon atoms to 3s Rydberg states. We do not expect a strong effect due to the substitution at C1 by propanoic acid, as substitution of hydrogen by a saturated carbon atom does not generally have a strong effect.
Peak d is present for KP but not for FP: we therefore assign it to an excitation localized at the keto group linking the two phenyl rings, that is, C12 1s transitions to the π* unoccupied orbital of the C12O19 moiety.
At energy higher than 288 eV, phenol does not show strong structures, so we assign peak e at 287.9–288.8 eV to a C9 1s to π* transition within the carboxylic acid group. A suitable model compound is butanoic acid, which has excitations at about 286.9–287.8 eV (methyl and methylene C 1s to 3s and 3p Rydberg) and 288.74 eV (carboxylic C 1s to π*).47 We assign peak e to overlapping transitions of C7 and C8 1s to Rydberg orbitals, and C9 1s to π*.
For the O K edge spectra, a similar approach can be followed, and the spectrum of MS can be assigned by comparison with the spectrum of salicylic acid.4 The energy of the first absorption peak a is within 0.15 eV of the first resonance of salicylic acid, and so is assigned to the same transition, namely, carbonyl O10 1s to π1*. The second peak, b, is at 0.7 eV lower energy than the corresponding peak of salicylic acid and is a little narrower. In salicylic acid, this feature is assigned to two main electronic transitions, namely the OH of the carboxylic acid, O8 1s to π1* and 3s at higher energy. At still higher energy, there is a weak transition from the hydroxy O11 1s to π2* [4, ESI†]. Since the resonance is narrower, we suggest that the presence of the methyl group bonded to O8 quenches the higher energy 3s Rydberg state transition, so that the peak of spectral intensity appears at lower energy. This is illustrated in Fig. S7 in the ESI,† where the spectra of MS and salicylic acid have been normalised to the intensity of the first peak and superposed.
Peak c is at 0.7 eV lower than the corresponding feature in salicylic acid, where it was assigned to transitions from the OH 1s groups to Rydberg states, as well as a transition from O8 1s to π2*.
As noted above, the spectra of FP can be compared with the spectra of appropriate building blocks. The O K edge spectrum of the propanoic acid moiety is comparable to that of the short-chain fatty acid butanoic acid,47 where resonances occur at 532.0 and 535.3 eV. We assign peak a at 532.0 eV to the O10 1s to πa* transition, where πa* refers to the antibonding orbital localised on the CO bond, and peak b at 535.5 to O11 1s to πa* and 3s transitions, as in benzoic acid, where the resonance occurs at 535.0 eV.4 The spectra of the phenyl rings of FP bridged by an ether group can be compared, as above, with the spectrum of phenol, which has broad resonances at 534.8 and 535.7 eV. These overlap the excitations of O10, so we conclude that there are also contributions to peak b from O12 1s to 3s and πb* orbitals, where the πb* orbitals are localised on the phenyl rings and ether oxygen. Because the peak is broad, we cannot determine the energetic ordering of πa* and πb*. Peak c is assigned to transitions to Rydberg states, while peak d is assigned to σ shape resonances.
Structurally, KP differs from FP by the substitution of the ether bridge by a keto bridge, and spectrally, it differs mainly by the appearance of an additional peak a′. It is straightforward to assign this to O19 1s to πc* transitions, where πc* indicates the antibonding π* orbital localised on the keto bridge, and possibly hybridised with the phenyl π* orbitals. The resonance energy of 530.4 eV is lower than that of acetone, 531.4 eV, and closer to that of benzaldehyde, 531.0 eV48 suggesting that hybridisation may indeed occur, even though the phenyl rings are not coplanar with the keto group. Other features of the spectrum are assigned as for FP. By overlapping the spectra normalised to the intensity of peak a, it can be seen that there is a decrease in relative intensity of peak b, particularly on the higher energy flank, see ESI,† Fig. S8. This confirms that there is a contribution to this peak of excitations from the ether oxygen O12 in FP, which is not present in KP.
5. Summary, conclusions and perspectives
Valence, C 1s and O 1s core photoelectron spectra and NEXAFS spectra of the three NSAIDs methyl salicylate, fenoprofen and ketoprofen were measured. The valence spectra agree with the published He I spectra of the outer valence region, and we have extended the data to include the inner valence region. New calculations were performed using two methods, and we found that the P3+ method was somewhat more accurate than the OVGF method, in agreement with the fact that the P3+ method includes third-order self-energy contributions, which provide a more accurate description of electron correlation effects compared to the second-order approximations used in OVGF.49The relative energies of sixteen conformers of FP and KP have been calculated. Both compounds show two groups of similar energy: eight syn conformers, in which the hydrogen bonded to the chiral C7 atom is syn with respect to the carbonyl group of the carboxylic acid group, and eight anti conformers. The syn conformers have an average energy of about 3 kJ mol−1 higher than the anti-conformers. This energy difference could have significant consequences in the biological environment. However, in the condensed phase, the effect of the intermolecular interactions could become dominant and modify the relative stability of the conformers. For this reason, further investigation of the solvent effect by either implicit or explicit models would provide important information.
The C 1s and O 1s XPS spectra are in good agreement with the theory. The O 1s spectra are less resolved due to vibrational and lifetime broadening and were fitted assuming approximately stoichiometric intensities of the three oxygen atoms.
The C and O K edge NEXAFS spectra of all three compounds were measured and assigned by reference to similar molecules. By calculating term values, we make predictions of the energies of the unoccupied molecular orbitals in methyl salicylate.
The obtained results extend our previous core and valence level studies3,4 and add to the available electronic structure data9 regarding NSAIDs. Novak et al.9 have summarised the data regarding the docking of these compounds with the COX-2 enzyme, where the drug blocks access to arachidonic acid, preventing the formation of prostaglandins. Arachidonic acid consists of a carboxylic acid moiety and a polyunsaturated chain, so it is not surprising that many NSAIDs also contain a carboxylic acid group. Frequently they also have aromatic groups which occupy the same hydrophobic site as the polyunsaturated chain. However, as pointed out by Novak et al., there does not appear to be a simple correlation between biological activity and the particular building blocks of drugs; rather the interaction is synergistic in nature, with many factors playing a role. We intend to continue our studies of these drugs to build up a more complete picture of their basic properties.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully thank our colleagues at Elettra for providing high-quality synchrotron light. Part of this work was carried out during a sabbatical leave (academic year 2022–2023) granted to H. S. from the University of Jordan, Amman, Jordan. H. S. acknowledges the TRIL fellowship awarded by the Abdus Salam International Centre for Theoretical Physics (ICTP), Trieste, Italy. A. M. acknowledges the CINECA award under the ISCRA initiative, for the availability of high-performance computing resources and support. We thank Ms Roberta Totani for assistance during some of the measurements of one of the compounds.References
- J. R. Vane and R. M. Botting, Am. J. Med., 1998, 104, 2S–8S, DOI:10.1016/S0002-9343(97)00203-9.
- L. Hykrdová, O. Bajt and J. Jirkovský, J. Hazard. Mater., 2018, 353, 70–79, DOI:10.1016/j.jhazmat.2018.03.048.
- H. Sa'adeh, K. C. Prince, R. Richter, V. Vasilyev, D. P. Chong and F. Wang, Phys. Chem. Chem. Phys., 2023, 5, 10946–10955, 10.1039/D2CP05810C.
- A. Hill, H. Sa'adeh, D. Cameron, F. Wang, A. B. Trofimov, E. Y. Larionova, R. Richter and K. C. Prince, J. Phys. Chem. A, 2021, 125(45), 9877–9891, DOI:10.1021/acs.jpca.1c07523.
- F. Ling, D. Liu, S. Li, W. Li, B. Zhang and P. Wang, J. Chem. Phys., 2019, 151, 094302, DOI:10.1063/1.5115307.
- J. Catalan, Phys. Chem. Chem. Phys., 2012, 14, 8903–8909, 10.1039/C2CP23742C.
- R. D. Massaro and E. Blaisten-Barojas, J. Chem. Phys., 2011, 135, 164306, DOI:10.1063/1.3653969.
- S. Melandri, B. M. Giuliano, A. Maris, L. B. Favero, P. Ottaviani, B. Velino and W. Caminati, J. Phys. Chem. A, 2007, 111, 9076–9079, DOI:10.1021/jp0723970.
- I. Novak, L. Klasinc, D. P. Chong and S. P. McGlynn, Spectrochim. Acta, Part A, 2013, 112, 110–115, DOI:10.1016/j.saa.2013.04.012.
- R. R. Blyth, R. Delaunay, M. Zitnik, J. Krempasky, R. Krempaska, J. Slezak, K. C. Prince, R. Richter, M. Vondracek, R. Camilloni, L. Avaldi, M. Coreno, G. Stefani, C. Furlani, M. de Simone, S. Stranges and M.-Y. Adam, J. Electron Spectrosc. Relat. Phenom., 1999, 101–103, 959–964, DOI:10.1016/S0368-2048(98)00381-8.
- K. Kimura, S. Katsumata, Y. Achiba, T. Yamazaki and S. Iwata, Handbook of He(I) Photoelectron Spectra of Fundamental Organic Molecules, Japan Scientific Societies Press, Tokyo, Japan, 1981 Search PubMed.
- V. Myrseth, J. D. Bozek, E. Kukk, L. J. Sæthre and T. D. Thomas, J. Electron Spectrosc. Relat. Phenom., 2002, 122, 57–63, DOI:10.1016/S0368-2048(01)00321-8.
- T. D. Thomas and R. W. Shaw Jr., J. Electron Spectrosc. Relat. Phenom., 1974, 5, 1081–1094, DOI:10.1016/0368-2048(74)85066-8.
- T. Hatamoto, M. Matsumoto, X.-J. Liu, K. Ueda, M. Hoshino, K. Nakagawa, T. Tanaka, H. Tanaka, M. Ehara, R. Tamaki and H. Nakatsuji, J. Electron Spectrosc. Relat. Phenom., 2007, 155, 54–57, DOI:10.1016/j.elspec.2006.10.002.
- M. Tronc, G. C. King and F. H. Read, J. Phys. B: At. Mol. Phys., 1979, 12, 137–157, DOI:10.1088/0022-3700/12/1/020.
- G. R. Wight and C. E. Brion, J. Electron Spectrosc. Relat. Phenom., 1974, 3, 191–205, DOI:10.1016/0368-2048(74)80010-1.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305, 10.1039/B508541A.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652, DOI:10.1063/1.464913.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789, DOI:10.1103/PhysRevB.37.785.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104, DOI:10.1063/1.3382344.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465, DOI:10.1002/jcc.21759.
- J. Linderberg and Y. Öhrn, Propagators in Quantum Chemistry, John Wiley and Sons, Inc., Hoboken, NJ, 2nd edn, 2004 Search PubMed.
- D. Danovich, WIREs Comput. Mol. Sci., 2011, 1(3), 377–387, DOI:10.1002/wcms.38.
- L. S. Cederbaum, J. Phys. B: At. Mol. Phys., 1975, 8(2), 290–303 CrossRef CAS.
- W. von Niessen, J. Schirmer and L. S. Cederbaum, Comput. Phys. Rep., 1984, 1(2), 57–125, DOI:10.1016/0167-7977(84)90002-9.
- V. G. Zakrzewski, J. V. Ortiz, J. A. Nichols, D. Heryadi, D. L. Yeager and J. T. Golab, Int. J. Quant. Chem., 1996, 60, 29–36 CrossRef.
- J. V. Ortiz, J. Chem. Phys., 1996, 104(19), 7599–7605, DOI:10.1063/1.471468.
- M. Ehara, J. Hasegawa and H. Nakatsuji, “SAC-CI method applied to molecular spectroscopy” in “Theory and Applications of Computational Chemistry, The First Forty Years”, Elsevier, Amsterdam, 2005, ch. 39, pp. 1099–1141 DOI:10.1016/B978-044451719-7/50082-2.
- R. E. Easton, D. J. Giesen, A. Welch, C. J. Cramer and D. G. Truhlar, Theor. Chim. Acta, 1996, 93, 281–301, DOI:10.1007/BF01127507.
- M. Fatima, D. Maué, C. Pérez, D. S. Tikhonov, D. Bernhard, A. Stamm, C. Medcraft, M. Gerhards and M. Schnell, Phys. Chem. Chem. Phys., 2020, 22, 27966–27978, 10.1039/D0CP04104A.
- A. Maris, S. Melandri, W. Caminati and P. G. Favero, Chem. Phys. Lett., 1996, 256(4), 509–512, DOI:10.1016/0009-2614(96)00467-8.
- C. West, G. Sedo and J. van Wijngaarden, J. Mol. Spectrosc., 2017, 335, 43–48, DOI:10.1016/j.jms.2016.12.002.
- H. Tan, M. Yang, C. Huang, S. Duan, M. Sun, Q. Chen, C. Jiao and Y. Wu, Appl. Sci., 2020, 10(23), 8471, DOI:10.3390/app10238471.
- W. H. Hocking, Z. Naturforsch., A: Phys. Sci., 1976, 31(9), 1113–1121, DOI:10.1515/zna-1976-0919.
- P. P. Briard and J. C. Rossi, Acta Crystallogr., 1990, C46, 1036–1038, DOI:10.1107/S0108270189004968.
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov and P. E. Bourne, Nucleic Acids Res., 2000, 28, 235–242, DOI:10.1093/nar/28.1.235.
- R. Castagna, S. Donini, P. Colnago, A. Serafini, E. Parisini and C. Bertarelli, ACS Omega, 2019, 4, 13270–13278, DOI:10.1021/acsomega.9b01442.
- T. D. Ngo, C. Oh, P. Mizar, M. Baek, K. Park, L. Nguyen, H. Byeon, S. Yoon, Y. Ryu, B. H. Ryu, T. D. Kim, J. W. Yang, C. Seok, S. S. Lee and K. K. Kim, ACS Catal., 2019, 9, 755–767, DOI:10.1021/acscatal.8b02797.
- P. Gasser, S. S. Tarchevskaya, P. Guntern, D. Brigger, R. Ruppli, N. Zbären, S. Kleinboelting, C. Heusser, T. S. Jardetzky and A. Eggel, Nat. Commun., 2020, 11, 165, DOI:10.1038/s41467-019-13815-w.
- M. P. Czub, A. J. Stewart, I. G. Shabalin and W. Minor, IUCrJ, 2022, 9, 551–561, DOI:10.1107/S2052252522006820.
- K. Zielinski, B. Sekula, A. Bujacz and I. Szymczak, Chirality, 2020, 32, 334–344, DOI:10.1002/chir.23162.
- R. D. Massaro, Y. Dai and E. Blaisten-Barojas, J. Phys. Chem. A, 2009, 113, 10385–10390, DOI:10.1021/jp905887m.
- V. Myrseth, K. J. Børve, K. Wiesner, M. Bässler, S. Svensson and L. J. Sæthre, Phys. Chem. Chem. Phys., 2002, 4, 5937–5943, 10.1039/B208160A.
- T. Karlsen, K. J. Børve, L. J. Sæthre, K. Wiesner, M. Bässler and S. Svensson, J. Am. Chem. Soc., 2002, 124, 7866–7873, DOI:10.1021/ja010649j.
- O. Travnikova, M. Patanen, J. Soderstrom, A. Lindblad, J. J. Kas, F. D. Vila, D. Ceolin, T. Marchenko, G. Goldsztejn, R. Guillemin, L. Journel, T. X. Carroll, K. J. Borve, P. Decleva, J. J. Rehr, N. Martensson, M. Simon, S. Svensson and L. J. Saethre, J. Phys. Chem. A, 2019, 123, 7619–7636, DOI:10.1021/acs.jpca.9b05063.
- K. C. Prince, R. Richter, M. de Simone, M. Alagia and M. Coreno, J. Phys. Chem. A, 2003, 107, 147172, DOI:10.1016/j.elspec.2022.147172.
- H. Sa'adeh, M. Masič, P. Bolognesi, R. Richter and K. C. Prince, J. Electron Spectrosc. Relat. Phenom., 2022, 256, 10946–10955, 10.1039/D2CP05810C.
- A. P. Hitchcock, S. G. Urquhart and E. G. Rightor, J. Phys. Chem., 1992, 96, 8736–8750, DOI:10.1021/j100201a015.
- R. Flores-Moreno and J. V. Ortiz, in Efficient and Accurate Electron Propagator Methods and Algorithms, Practical Aspects of Computational Chemistry, ed. J. Leszczynski, M. Shukla, Springer, Dordrecht, 2009 DOI:10.1007/978-90-481-2687-3_1.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp03600j |
| ‡ Gaussian is a registered trademark of Gaussian, Inc. 340 Quinnipiac St. Bldg. 40 Wallingford, CT 06492 USA. |
| This journal is © the Owner Societies 2024 |