
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Double-boron heterocyclic carbenes: a computational study of Diels–Alder reactions†
Changyu
Cao
a,
Congjie
Zhang
*a,
Junjing
Gu
b and
Yirong
Mo
 *c
*c
aKey Laboratory of Macromolecular Science of Shaanxi Province, School of Chemistry & Chemical Engineering, Shaanxi Normal University, Xi’an, 710062, China. E-mail: zcjwh@snnu.edu.cn
bDepartment of Chemistry, Center for Theoretical Chemistry Xiamen University, Xiamen, 361005, China
cDepartment of Nanoscience, Joint School of Nanoscience and Nanoengineering University of North Carolina at Greensboro, Greensboro, NC 27401, USA. E-mail: y_mo3@uncg.edu
First published on 22nd October 2024
Abstract
An aromatic boron-containing organic compound, C2B2H2, with an unusual C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond was experimentally synthesized in 2017. Here we investigate the structure and bonding nature of C2B2H2 and its derivatives C2B2R2 using DFT and VB theory. Although the CC bond in C2B2R2 consists of a π bond and a charge-shift (CS) bond, C2B2F2 has the lowest LUMO energy and its LUMO is similar to that of ethylene, suggesting that C2B2F2 can be an ideal dienophile for the Diels–Alder reaction. Subsequently, the mechanism and stereoselectivity of the Diels–Alder reaction of C2B2F2 with 5-substituted cyclopentadienes are studied. Computations demonstrate that these Diels–Alder reactions are feasible thermodynamically and kinetically. The stereoselectivity and distortion angles of C2B2R2 exhibit linear correlations with the electronegativity difference between the two substituents bonded to the C(sp3) of cyclopentadiene, suggesting that the stereoselectivity of related Diels–Alder reaction products can be modulated by the substitution of cyclopentadiene. Considering the current interest in boron neutron capture therapy (BNCT), we design six BNCT drugs through the Diels–Alder reaction of C2B2F2 with dienes containing peptide fragments. Thus, we demonstrate a new method for designing three-in-one BNCT drugs via the facile Diels–Alder reaction.
C bond was experimentally synthesized in 2017. Here we investigate the structure and bonding nature of C2B2H2 and its derivatives C2B2R2 using DFT and VB theory. Although the CC bond in C2B2R2 consists of a π bond and a charge-shift (CS) bond, C2B2F2 has the lowest LUMO energy and its LUMO is similar to that of ethylene, suggesting that C2B2F2 can be an ideal dienophile for the Diels–Alder reaction. Subsequently, the mechanism and stereoselectivity of the Diels–Alder reaction of C2B2F2 with 5-substituted cyclopentadienes are studied. Computations demonstrate that these Diels–Alder reactions are feasible thermodynamically and kinetically. The stereoselectivity and distortion angles of C2B2R2 exhibit linear correlations with the electronegativity difference between the two substituents bonded to the C(sp3) of cyclopentadiene, suggesting that the stereoselectivity of related Diels–Alder reaction products can be modulated by the substitution of cyclopentadiene. Considering the current interest in boron neutron capture therapy (BNCT), we design six BNCT drugs through the Diels–Alder reaction of C2B2F2 with dienes containing peptide fragments. Thus, we demonstrate a new method for designing three-in-one BNCT drugs via the facile Diels–Alder reaction.
Introduction
In 1928, Diels and his student Alder first synthesized an unsaturated six-membered ring from a dienophile and a diene and the reaction has been recognized as the Diels–Alder reaction or [4+2] cycloaddition.1 The Diels–Alder reaction can efficiently and economically produce unsaturated six-membered rings under relatively simple reaction conditions, and has been widely applied in organic chemistry,2,3 drug synthesis,4–6 and materials science.4,7,8 The simplest Diels–Alder reaction is the reaction of ethylene with butadiene to form cyclohexene. However, when a dienophile or diene with a complex structure exhibits stereo- and spatial selectivity, the accompanying Diels–Alder reaction can result in a diversity of products.9–12 For example, reactions of cyclopentadiene with cyclopropene and substituted cyclopropenes can give endo- or exo-type products due to the stereoselectivity.10 To better understand the stereoselectivity of Diels–Alder reaction, Houk and co-workers identified hyperconjugative aromaticity or antiaromaticity as the main element of stereoselectivity,13 though electrostatic,9,14 secondary orbital interaction15,16 and steric effect14,17 can also influence the stereoselectivity. In Diels–Alder reactions, the dienophile contains a classical double bond, such as CC in olefin, C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C in alkyne, and so on. In contrast to the classical dienophile, we theoretically found two types of novel dienophiles with an inverted CC bond composed of a π bond and a charge-shift (CS) bond,18i.e., B-heterocyclic carbene (BHC)19 and (Si and B)-heterocyclic carbene (SiBHC)20 as shown in Scheme 1a and b. Because the LUMOs of BHCs and SiBHCs are similar to that of ethylene, the Diels–Alder reactions of both BHCs and SiBHCs with diene can be expected.19–21 These Diels–Alder reactions are favourable both thermodynamically and kinetically due to their low energy barriers and exergonic outcomes. However, neither BHC nor SiBHC has been isolated experimentally. Nevertheless, in 2017, the Zhou group reported a novel molecule C2B2H2 which was experimentally obtained via the C–H bond activation reaction of acetylene by boron atoms, and confirmed that C2B2H2 has a rhombus structure with D2h symmetry as shown in Scheme 1c.22 Clearly, C2B2H2 is structurally similar to BHC and SiBHC predicted in our previous computational work.
C in alkyne, and so on. In contrast to the classical dienophile, we theoretically found two types of novel dienophiles with an inverted CC bond composed of a π bond and a charge-shift (CS) bond,18i.e., B-heterocyclic carbene (BHC)19 and (Si and B)-heterocyclic carbene (SiBHC)20 as shown in Scheme 1a and b. Because the LUMOs of BHCs and SiBHCs are similar to that of ethylene, the Diels–Alder reactions of both BHCs and SiBHCs with diene can be expected.19–21 These Diels–Alder reactions are favourable both thermodynamically and kinetically due to their low energy barriers and exergonic outcomes. However, neither BHC nor SiBHC has been isolated experimentally. Nevertheless, in 2017, the Zhou group reported a novel molecule C2B2H2 which was experimentally obtained via the C–H bond activation reaction of acetylene by boron atoms, and confirmed that C2B2H2 has a rhombus structure with D2h symmetry as shown in Scheme 1c.22 Clearly, C2B2H2 is structurally similar to BHC and SiBHC predicted in our previous computational work.
 | ||
| Scheme 1 Structures of (a) BHC, (b) SiBHC and (c) C2B2H2. | ||
Earlier calculations showed that BHC, SiBHC and their derivatives can be used to design novel molecules with planar tetracoordinate carbon23 or silicon24 (ptC or ptSi) and porous organic molecules (POMs).25 They can also react with dienes via the Diels–Alder reaction.20,21,26 In this work, we intended to study the structures and bonding nature of C2B2H2 and its derivatives C2B2R2 where R is a substituent group. Interestingly, we found that C2B2F2 is an ideal dienophile due to the similarity of its LUMO with that of ethylene and has the lowest LUMO energy level among the studied derivatives. Subsequently, the mechanism and stereoselectivity of the Diels–Alder reaction of C2B2F2 with 5-substituted cyclopentadienes were investigated.
Recent studies in the literature have shown that boron neutron capture therapy (BNCT) drugs might have potential application in tumor treatment.27,28 Because C2B2F2 contains both key elements of boron and fluorine, a family of three-in-one BNCT drugs might be obtained by the Diels–Alder reaction of C2B2F2 with dienes containing peptide fragments. Accordingly, the mechanism of such Diels–Alder reactions and the structural optimizations of the BNCT drugs were also computationally investigated.
Computational methods
Geometry optimizations and frequency calculations of C2B2H2 and its derivatives C2B2R2 (R = Me, SiH3, NH2, PH2, OH, SH, F, Cl, Br, Ph, tBu, CN and NO2) were carried out at the M06-2X/6-311++G** theoretical level.29–34 Similarly, the structures of reactants, transition states and products of the Diels–Alder reaction of C2B2F2 with substituted cyclopropenes were optimized and their vibrational frequencies were calculated at the same level of theory to confirm the true minima and transition states. The distortion/interaction model was used to describe the relationship between the activation energies vs. the distortion and interaction energies between dienes and dienophiles. Single-point solvation energies were computed using the solvation model based on density (SMD) for all species in the above Diels–Alder reactions, in which CH2Cl2 and water were used as the solvents. The Diels–Alder reaction mechanisms of C2B2F2 with six dienes containing peptide fragments were investigated using same approach in aqueous solution. The convergence criteria in geometry optimizations were 10−8 for energy, 0.000450 for maximum force, 0.000300 for root-mean-square (RMS) force, 0.001800 for maximum displacement, and 0.001200 for RMS displacement with the grid of 75302. The Wiberg bond indices (WBIs) for bonds were calculated using natural bond orbital (NBO) analysis35 to investigate the character of the inverted CC bond in C2B2R2.
To justify the reliability of the M06-2X/6-311++G** method, we used CBS-QB336 and DSD-PBEP86-D3(BJ)37,38/6-311++G** methods to optimize the geometries and calculate the WBIs of both A1 and A8, two key molecules in this work. The bond lengths and WBIs are compiled in Fig. 1. Although the central CC bond lengths were elongated in the order of M06-2X < CBS-QB3 < DSD-PBEP86-D3(BJ), all other bond lengths and the WBIs were consistent at all three theoretical levels. The Diels–Adler reaction of A8 with B10 was also investigated using the CBS-QB3 and DSD-PBEP86-D3(BJ) methods. The energy barriers and optimized geometries of the transition states are illustrated in Fig. S1 (ESI†). While there were insignificant differences for the optimal geometries of the transition states obtained at the three theoretical levels, the CBS-QB3 method generated the highest barriers and the DSD-PBEP86-D3(BJ) method the lowest barriers. Importantly, all three methods resulted in the same reaction mechanism, i.e., the Diels–Alder reaction prefers the anti-type product via the anti-type transition state. Considering that the M06-2X results were between the data from the CBS-QB3 and DSD-PBEP86-D3(BJ) methods, in the following we used the M06-2X results for discussion.
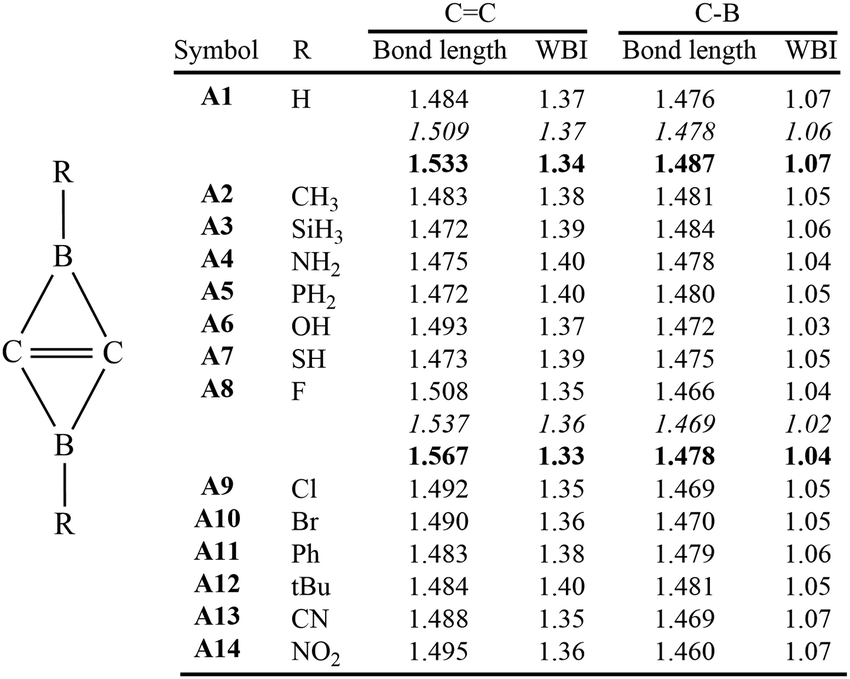 | ||
| Fig. 1 Lengths (in angstroms) and WBI values of the CC and C–B bonds in Ai (i = 1–14) at the M06-2X/6-311++G** level, with additional data for A1 and A8 at the CBS-QB3 (in italic) and DSD-PBEP86-D3(BJ)/6-311++G** (in bold) levels. | ||
Nucleus independent chemical shifts NICS(0) and NICS(1) at the centers of the three-membered rings from two C atoms and one B atom of C2B2R2 were calculated using the gauge-independent atomic orbital (GIAO) method.39 All above calculations were performed using Gaussian09 and Gaussian16.40,41 The valence bond (VB) analyses of the inverted CC bond in C2B2H2 and C2B2F2 were conducted using the breathing orbital VB (BOVB) method and the 6-31G* basis set.42,43 The BOVB calculations were carried out within the Xiamen valence bond (XMVB) package.44
Results and discussion
Based on our previous studies of BHCs and SiBHCs, we designed thirteen derivatives of C2B2H2, which were C2B2R2 (R = Me, SiH3, NH2, PH2, OH, SH, F, Cl, Br, Ph, tBu, CN and NO2). Optimized geometries and WBIs are summarized in Fig. 1. For C2B2H2 (A1), the CC and C–B bond lengths are 1.484 and 1.476 Å, respectively, well consistent with the literature data.22 The lengths of CC and C–B bonds of its derivatives (A2–A14) fluctuate in the ranges of 1.472–1.508 Å and 1.460–1.484 Å, respectively, indicating that the substituted group R bonded to the boron atoms influence their geometries slightly. The WBIs of CC and C–B bonds are close to 1.40 and 1.05 respectively. Thus, the central CC bonds are between single and double bonds, while C–B bonds are clearly single.
The occupied π-type and frontier molecular orbitals (MOs) of C2B2R2 are displayed in Fig. S2 (ESI†) and Fig. 2, respectively. As seen from Fig. S2 (ESI†), each C2B2R2 has an occupied π-type MO that extends the whole C2B2 unit, such as the HOMO−2 of C2B2H2 (A1) and C2B2F2 (A8). Thus, C2B2R2 contains a delocalized Π24 bond. Fig. 2 shows the HOMOs of all substituted C2B2R2 which are similar to those of BHCs or SiBHCs,19,20 and C2B2R2 can be similarly described in terms of resonance structures as shown in Scheme 2, where the covalent structure is weakly bonded with the characteristics of a singlet biradical. We were curious whether the inverted CC bond in C2B2R2 was a charge-shift (CS) bond. To answer this question, we calculated the contributions of both covalent (1) and ionic (2 and 3) resonance structures to the ground state, the bond energy, the resonance energy, and the ratio of the resonance energy to the bond energy for the inverted CC bond in C2B2H2 and C2B2F2 at the BOVB/6-31G* level. The results are compiled in Table S1 (ESI†). The contributions of the covalent and ionic resonance structures of C2B2H2 and C2B2F2 are about 60% and 40%, respectively, while the ratios of the resonance energy to the CC bond energy are 40% and 43%, respectively, meeting the definition of a CS bond.45 Thus, the inverted CC bond in C2B2R2 contains a π bond and a charge-shift (CS) bond, and is unsurprisingly analogous to the CC bonds in BHC and SiBHC.19,20 Accordingly, C2B2H2 and its derivatives C2B2R2 can be labelled as double boron heterocyclic carbenes (DBHCs).
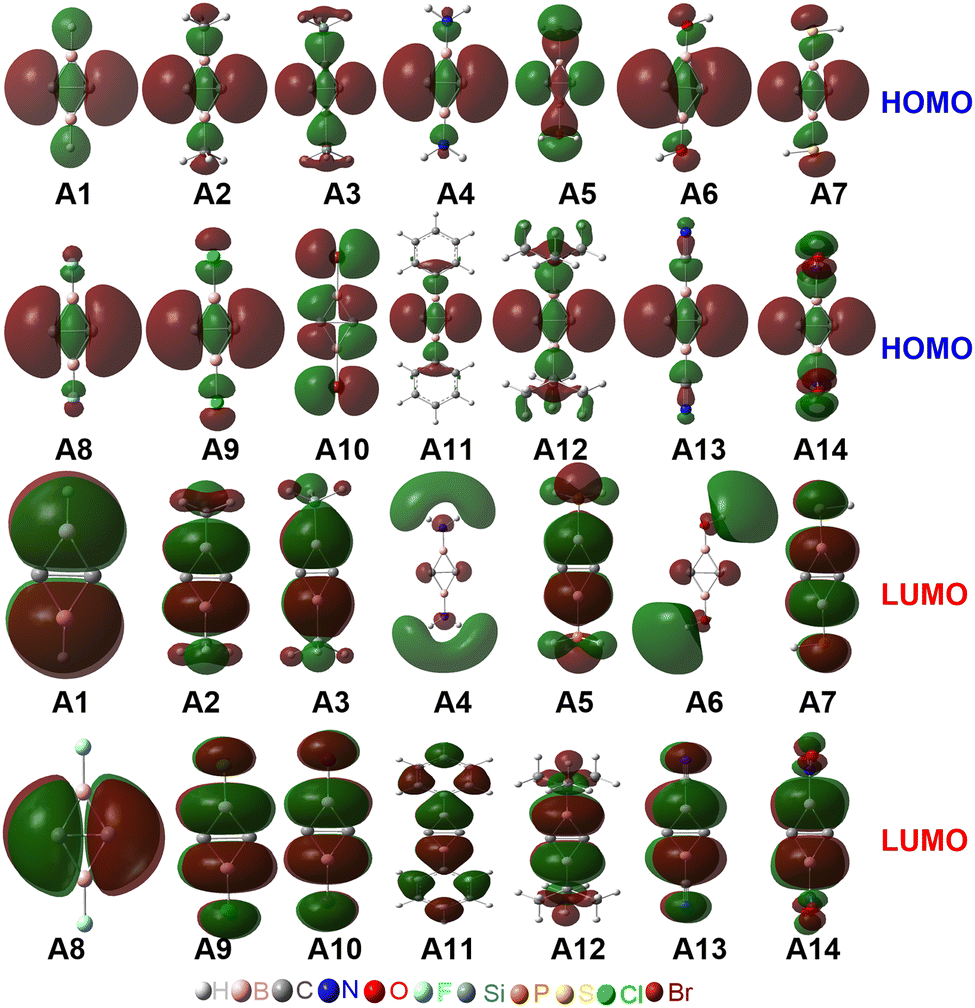 | ||
| Fig. 2 The HOMOs and LUMOs of Ai (i = 1–14). | ||
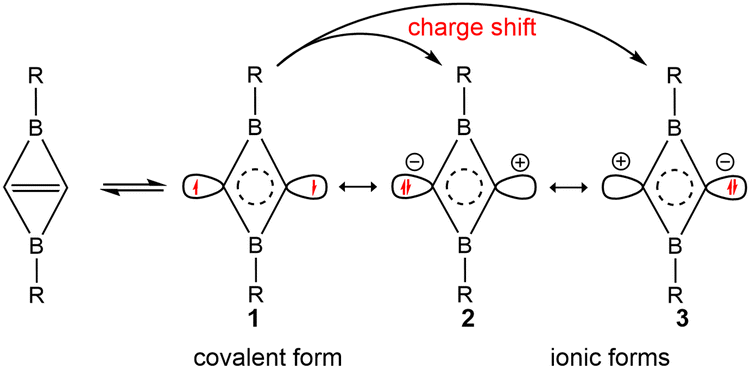 | ||
| Scheme 2 One covalent structure (1) and two ionic resonance structures (2 and 3) arising from charge shifting over the inverted CC bond in C2B2R2. | ||
Since C2B2H2 is an aromatic system,22 the values of NICS(0) and NICS(1) at the center of three-membered rings of C2B2R2 were calculated and are listed in Table S2 (ESI†). With the values of NICS(0) and NICS(1) ranging from −23.90 to −12.81 ppm, it can be confirmed that C2B2R2 are also aromatic and consistent with previous findings for C2B2H2.22
Fig. 2 shows that the LUMOs of A1–A3, A5, A7 and A9–A14 are the anti-bond MOs formed by the π orbitals of the two boron atoms; the LUMOs of A4 and A6 are a kind of σ orbital, but the LUMO of A8 is the anti-bond MO formed by the π orbitals of the two carbon atoms. Obviously, the LUMO of A8 is consistent with that of ethylene. In addition, the LUMO energy level of A8 is low (−0.85 eV). Following the frontier MO theory, A8 (C2B2F2) thus would be the most suitable dienophile due to its low LUMO energy level. To confirm this hypothesis, we studied the reaction mechanism and stereoselectivity of the Diels–Alder reaction of C2B2F2 with ten 5-substituted cyclopentadienes Bi (i = 1–10 as shown in Scheme 3), which can be sorted according to the order of van der Waals radius of the two substituent groups bonded to the C(sp3) atom. Between the two substituent groups R1 and R2, the van der Waals radius of R1 (top) is smaller than that of R2 (bottom). Because the two groups R1 and R2 are different, the Diels–Alder reaction of C2B2F2 with these substituted cyclopentadienes undergoes anti- and syn-type transition states to produce isomers as shown in Scheme 4, where the anti- and syn-type structures of transition states and products are named in terms of the position of R1 relative to the substituted cyclopentadienes and the CC bond. If the CC bond and R1 are situated on the same side, the transition states and products are anti-type structures; otherwise, transition states and products are syn-type structures.
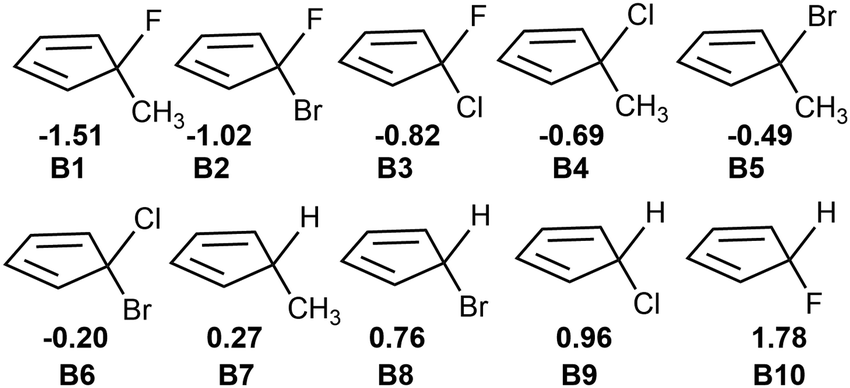 | ||
| Scheme 3 5-substituted cyclopentadienes Bi (i = 1–10) with the electronegativity difference between the two substituent groups. | ||
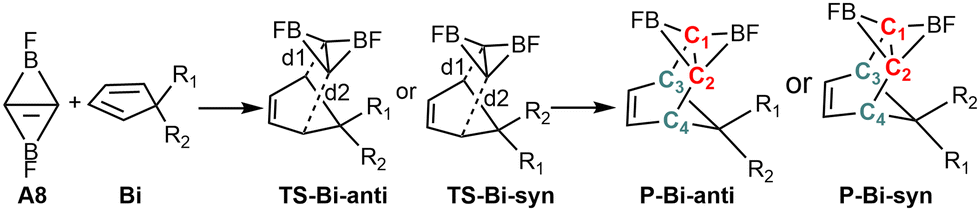 | ||
| Scheme 4 The reactants (C2B2F2 and Bi), transition states (TS-Bi-anti and TS-Bi-syn) and products (P-Bi-anti and P-Bi-syn) in the Diels–Alder reactions of C2B2F2 with (Bi, i = 1–10). | ||
The optimized geometries of the anti- and syn-type transition states of the Diels–Alder reactions of C2B2F2 with substituted cyclopentadienes are displayed in Fig. 3 and labelled as TS-Bi-anti and TS-Bi-syn (i = 1–10), respectively, in which the optimal distances (d1 and d2) between the carbon atoms of C2B2F2 and Bi are defined in Scheme 4. The structures of products undergoing TS-Bi-anti and TS-Bi-syn are P-Bi-anti and P-Bi-syn as shown in Fig. S3 (ESI†), respectively. The distances of d1 and d2 in TS-Bi-anti and TS-Bi-syn show no significant difference and are close to 2.26 Å, except for TS-B4-syn where d1 and d2 are 2.47 and 2.11 Å. These distances correspond well with those observed in the Diels–Alder reactions of BHCs with cyclopentadienes.21
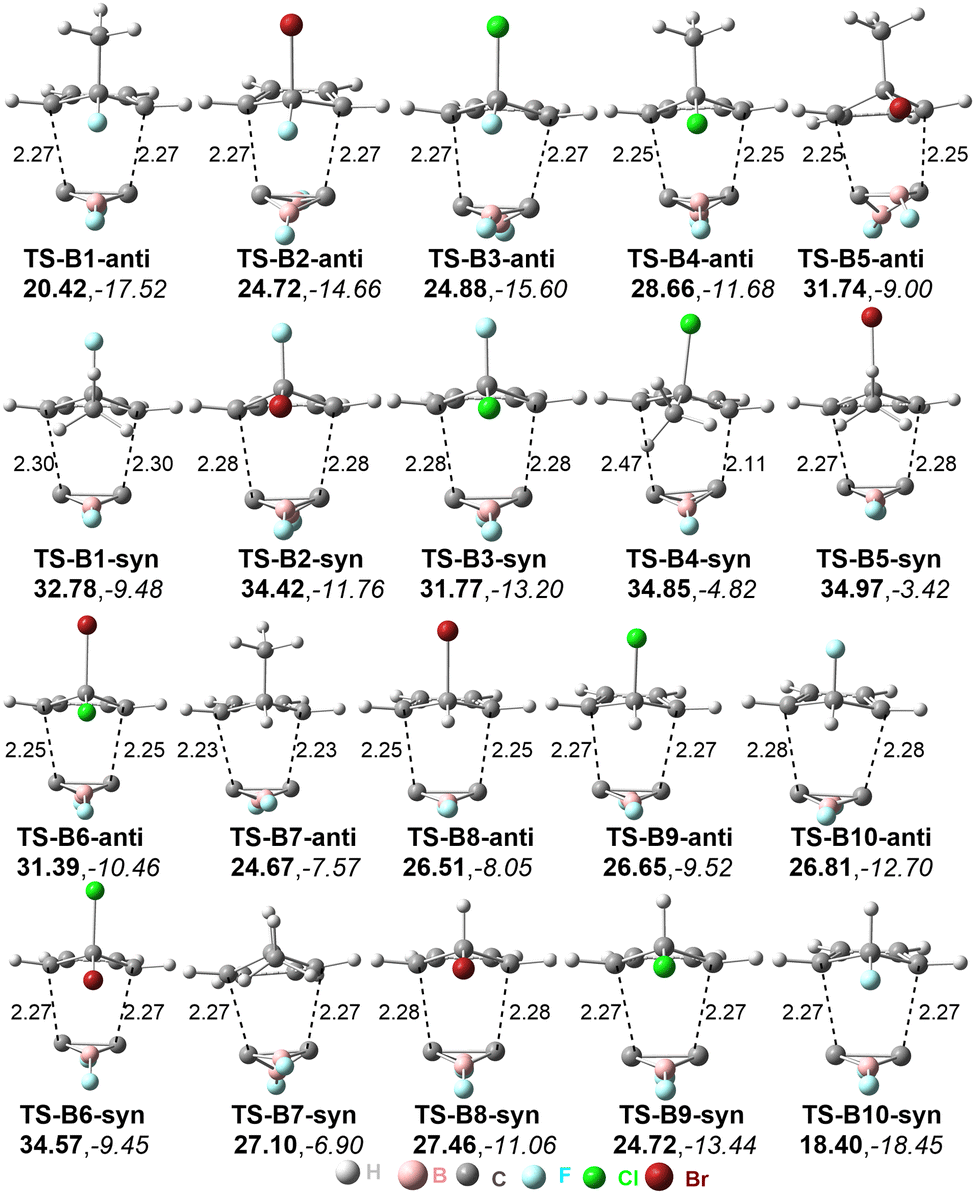 | ||
| Fig. 3 Optimized distances between C atoms in transition states (in Å), energy barriers ΔGTS (in bold), and the reaction free energy ΔGre (in italic). Energies are given in kcal mol−1. | ||
The reaction barriers and Gibbs free energies of the ten Diels–Alder reactions are given in Fig. 3. The barriers of the reactions via anti-transition states are mostly lower than those of syn-transition states, except for B9 and B10. In detail, the barriers undergoing anti-transition states range from 20.4 to 31.7 kcal mol−1 for Bi (i = 1–8), while for B9 and B10, the barriers via syn-transition states TS-Bi-syn (i = 9 and 10) are 24.72 and 18.40 kcal mol−1, respectively. The Diels–Alder reaction with the lowest barrier occurs between C2B2F2 and C5H5F (B10). All these ten Diels–Alder reactions are exergonic. Thus, these Diels–Alder reactions are both thermodynamically and kinetically feasible. As seen from Scheme 4, the products can be considered as derivatives of [1.2.2]-propellane. The lengths and WBIs of the inverted C–C bond in P-Bi-anti and P-Bi-syn are summarized in Table S2 (ESI†). For the inverted C–C bond, its distances and WBIs are close to 1.710 Å and 0.74 in all products, indicating that the inverted C–C bond is weak. Similar findings have been shown in the products of the Diels–Alder reactions of BHCs.26
To understand the different energy barriers in the Diels–Alder reactions via syn- and anti-type transition states, we examined the electronegativity difference (Δχ = χR2 − χR1) between the two substituent groups (R1 and R2) bonded to the C(sp3) of cyclopentadiene as shown in Scheme 4. We used Δχ to compare with the difference of the energy barriers via syn- and anti-type transition states (ΔΔGTS(s–a) = ΔGTSs − ΔGTSa). Fig. 4a shows the linear correlation between Δχ and ΔΔGTS(s–a) with the correlation coefficient of 0.94. Thus, the stereoselectivity of the Diels–Alder reaction of C2B2F2 with substituted cyclopentadienes can be modulated by the electronegativity difference between the two substituted groups bonded to the C(sp3) of cyclopentadiene.
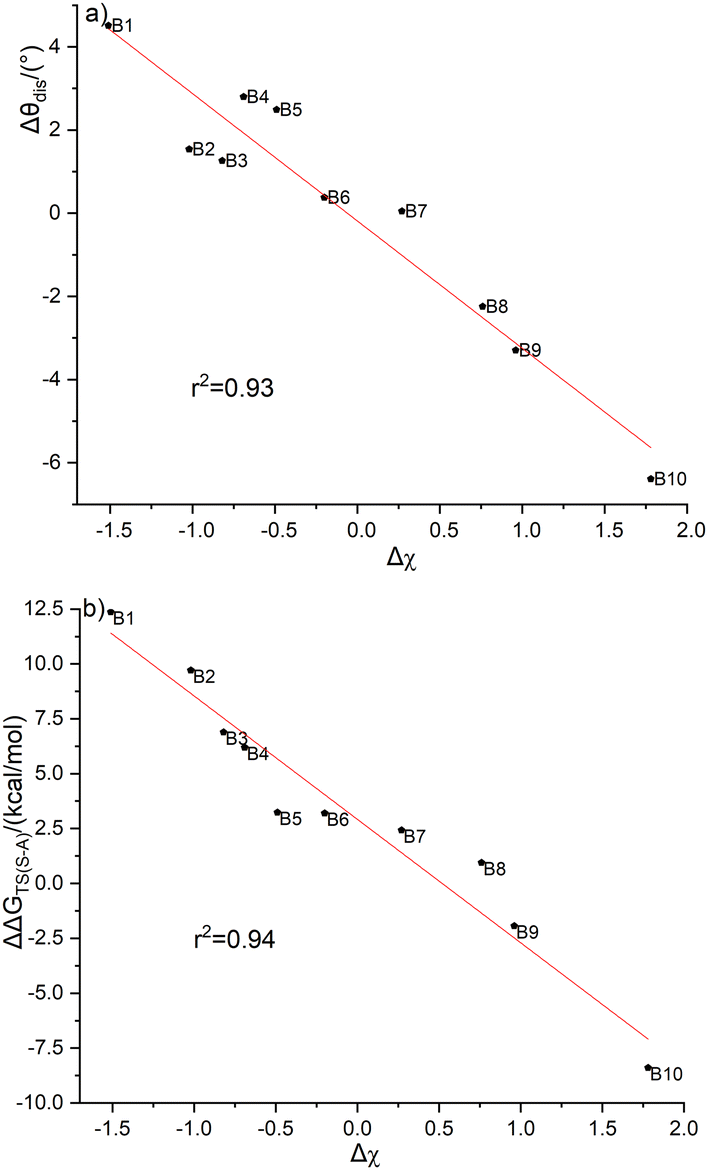 | ||
| Fig. 4 Correlations of the electronegativity difference with (a) reaction barriers (ΔΔGTS(s-a) = −5.62Δχ + 2.92, r2 = 0.94); (b) distortion angles of the ground-state 5-substituted cyclopentadienes (θdis = −0.31Δχ − 0.18, r2 = 0.93). | ||
Unlike cyclopentadiene whose five-membered ring is planar, the inequivalent substituent groups R1 and R2 drive the C(sp3) atom in substituted cyclopentadienes out of the planarity. We calculated the distortion angles (θdis) of substituted cyclopentadienes as defined in Fig. S4 (ESI†) to quantify such deviation. Interestingly, there is also a good linear correlation between Δχ and θdis as shown in Fig. 4b. Thus, the stereoselectivity of the Diels–Alder reactions of C2B2F2 with substituted cyclopentadienes is expected to correlate with the distortion angles of substituted cyclopentadienes. To elucidate such a correlation, the variations between the distortion angles of substituted cyclopentadienes from their isolated optimal states to their respective syn- and anti-transition states were calculated and are displayed in Fig. S5 (ESI†). Fig. S5 (ESI†) shows that a smaller variation of the distortion angle would result in a lower energy barrier. For example, the changes in the distortion angles of TS-B1-anti and TS-B1-syn are 24.4° and −33.0°, respectively. Accordingly, the energy barrier of TS-B1-anti is lower than that of TS-B1-syn. Thus, the selectivity of Diels–Alder reactions of C2B2F2 with substituted cyclopentadienes is largely ruled by the electronegativity difference between the two substituents, with the more electronegative substituent leading to a smaller distortion from the planar structure of cyclopentadiene and a lower reaction barrier.
In contrast to classical dienes, C2B2F2 contains two electron-deficient boron atoms which might exhibit interactions with substituted groups R1 or R2 in substituted cyclopentadienes. Therefore, we used the distortion/interaction model proposed by the Houk group to investigate the distortion energies (Edis-A8 and Edis-Bi) of C2B2F2 and substituted cyclopentadienes, as well as the activation energy (Eact) and the interaction energies (Eint) in their reactions. Calculated distortion, interaction, and activation energies of the anti- and syn-type transition states (TS-Bi-anti and TS-Bi-syn) are depicted in Fig. 5 which shows that Eact can be decomposed into three components, including Edis-Bi, Edis-A8 and Eint. All these three components may make significant contributions to the reaction barrier. For the example of TS-B1-anti, Edis-Bi, Edis-A8 and Eint are 12.60, 8.84 and −14.91 kcal mol−1, respectively, leading to the reaction barrier of 6.53 kcal mol−1.
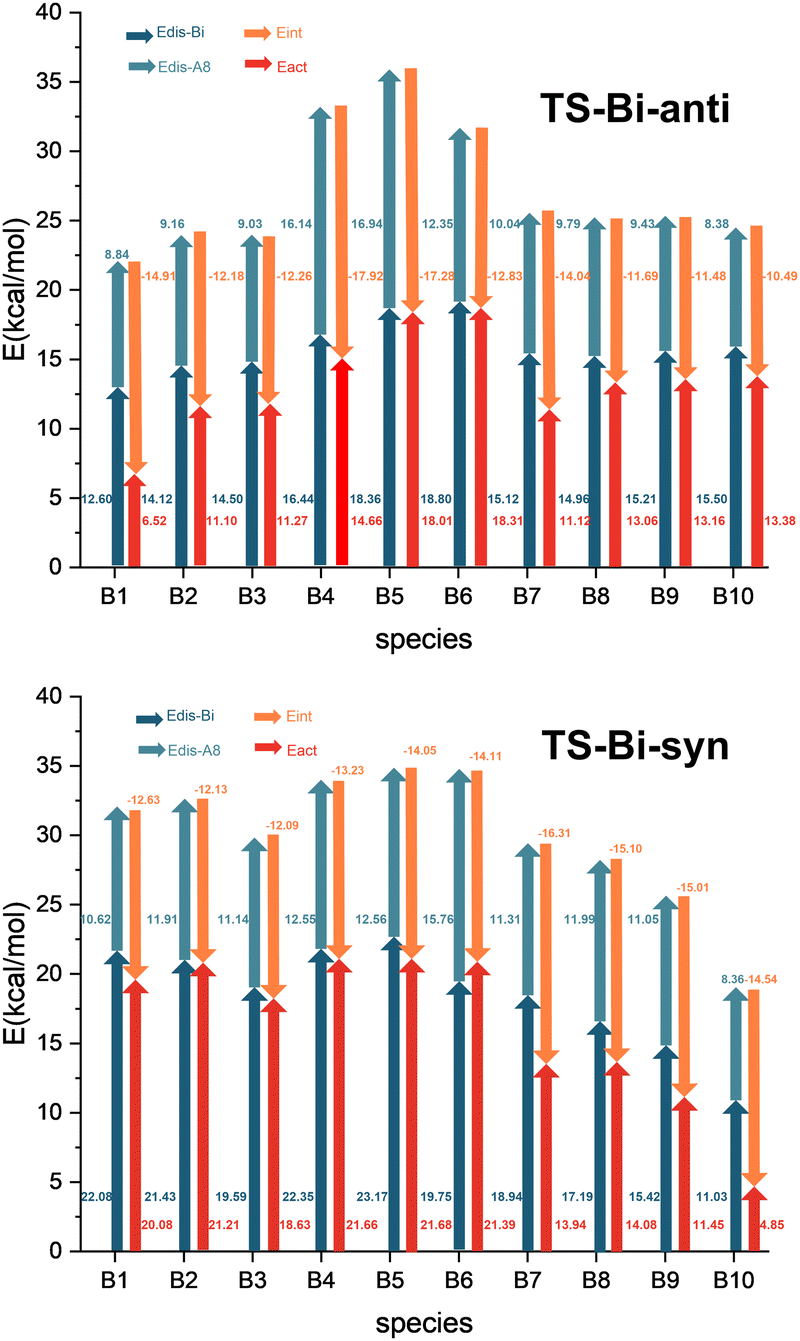 | ||
| Fig. 5 Energy analysis of the barriers (Eact, in red) in the Diels–Alder reactions of C2B2F2 with substituted cyclopentadienes based on the distortion/interaction model. | ||
The variation of the energy barriers of the Diels–Alder reactions of C2B2F2 with substituted cyclopentadienes in solvents including water and dichloromethane was further investigated, and the calculated results are illustrated in Fig. 6. Computations showed that the energy barriers slightly decrease by 0.03–1.86 kcal mol−1 in water, while they slightly increase by 0.09–1.42 kcal mol−1 in dichloromethane. Thus, such Diels–Alder reactions are more favourable to occur in water. Moreover, Fig. 6 demonstrates that the trends of the changes of barriers via anti- and syn-type transition states in solvents are consistent with that in gas, indicating that solvation effect has limited influence on the stereoselectivity of such Diels–Alder reactions.
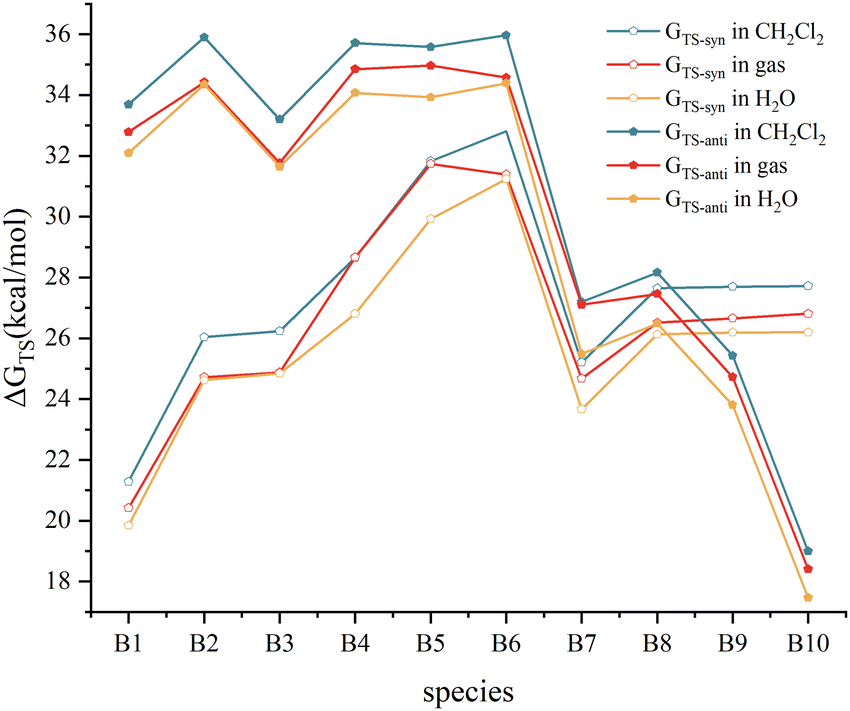 | ||
| Fig. 6 The energy barrier in gas and solvents (H2O and CH2Cl2). | ||
Boron neutron capture therapy (BNCT) is a radiation treatment designed to improve tumour control while reducing damage to surrounding tissues. The key to BNCT is the use of boronated compounds. Yan's group obtained a type of three-in-one BNCT drugs by photocatalytic B–C coupling via a carboranyl cage radical in 2022.46 Additionally, five classes of boron-containing drugs, bortezomib,47 ixazomib,48 vaborbactam,49 tavaborole50 and crisaborole,51 have been marketed during the past thirty years, while many boron-containing drugs, such as Dutogliptin52 and Acoziborole53 are in clinical trials. Based on the feasibility of the Diels–Alder reactions of C2B2F2 with 5-substituted cyclopentadienes, as well as the fact that C2B2F2 involves boron and fluorine atoms, we propose the synthesis of a family of three-in-one BNCT drugs through the Diels–Alder reactions of C2B2F2 with dienes containing peptide fragments. Here we computationally designed six dienes (Ci, i = 1–6) with peptide fragments as shown in Scheme 5 by modifying the unit bound to B(OH)2 of boron-containing drugs with a diene structure. The optimized geometries of Ci (i = 1–6) are displayed in Fig. S6 (ESI†), and their HOMOs and LUMOs are illustrated in Fig. S7 (ESI†). It should be noted that their HOMOs are all located at the modified diene unit. Due to the unsymmetrical groups bound to the diene unit in these dienes Ci (i = 1–6), C2B2F2 can attack them via both anti- and syn-forms. Accordingly, the subsequent Diels–Alder reactions can go through anti- and syn-type transition states (TS-Ci-anti, TS-Ci-syn, i = 1–6) and generate the corresponding products (P-Ci-anti and P-Ci-syn, i = 1–6). Optimized geometries of the transition states and products are shown in Fig. S8 and S9 (ESI†), respectively. The optimal distances (d1 and d2) between the carbon atoms of C2B2F2 and Ci in TS-Ci-anti and TS-Ci-syn for i = 1–3 are close in the range of 2.18–2.36 Å with a gap less than 0.17 Å, and consistent with those of TS-Bi-anti and TS-Bi-syn shown in Fig. 3. But for i = 4–6, the distances of d1 and d2 in TS-Ci-anti and TS-Ci-syn (i = 2, 4–6) are quite different with a gap between 0.23 and 0.59 Å, consistent with those of the Diels–Alder reactions of BHCs with butadiene.26
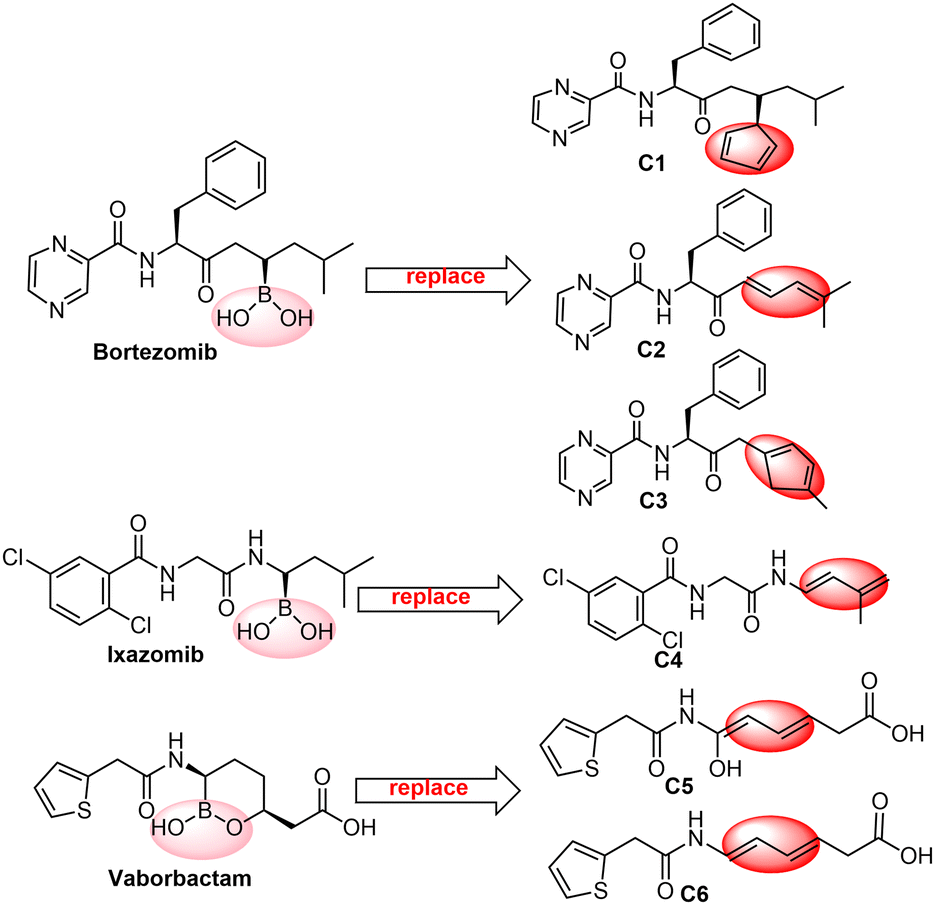 | ||
| Scheme 5 Six dienes (Ci, i = 1–6) with peptide fragments by modifying the unit bound to B(OH)2 of boron-containing drugs. | ||
The energy barriers and reaction free energies in water for the six Diels–Alder reactions were shown in Fig. 7. Fig. 7a shows that the energy barriers viaTS-Ci-anti (i = 1, 4–6) are lower than those of TS-Ci-syn, while the barriers viaTS-Ci-syn (i = 2 and 3) are lower than those of TS-Ci-anti. The barriers undergoing TS-Ci-anti (i = 1, 4–6) and TS-Ci-syn (i = 2 and 3) range from 18.71 to 26.30 kcal mol−1, indicating these Diels–Alder reactions are feasible kinetically. In particular, the barrier in the reaction of C2B2F2 with C4viaTS-C4-anti is the lowest. The reaction free energies (in Fig. 7b) are all negative, suggesting that these Diels–Alder reactions are also thermodynamically feasible. Furthermore, the lower the energy barrier of formation of P-Ci-anti or P-Ci-syn, the more negative the reaction free energy. Thus, a new method for designing BNCT drugs by the simple Diels–Alder reaction was anticipated. Moreover, such BNCT drugs contain boron, fluorine, and peptide fragments, i.e., three-in-one BNCT. If C2B2F2 were synthesized, it could be used to synthesize such BNCT drugs. These BNCT drugs might have potential applications in tumor treatment because of their special structures.
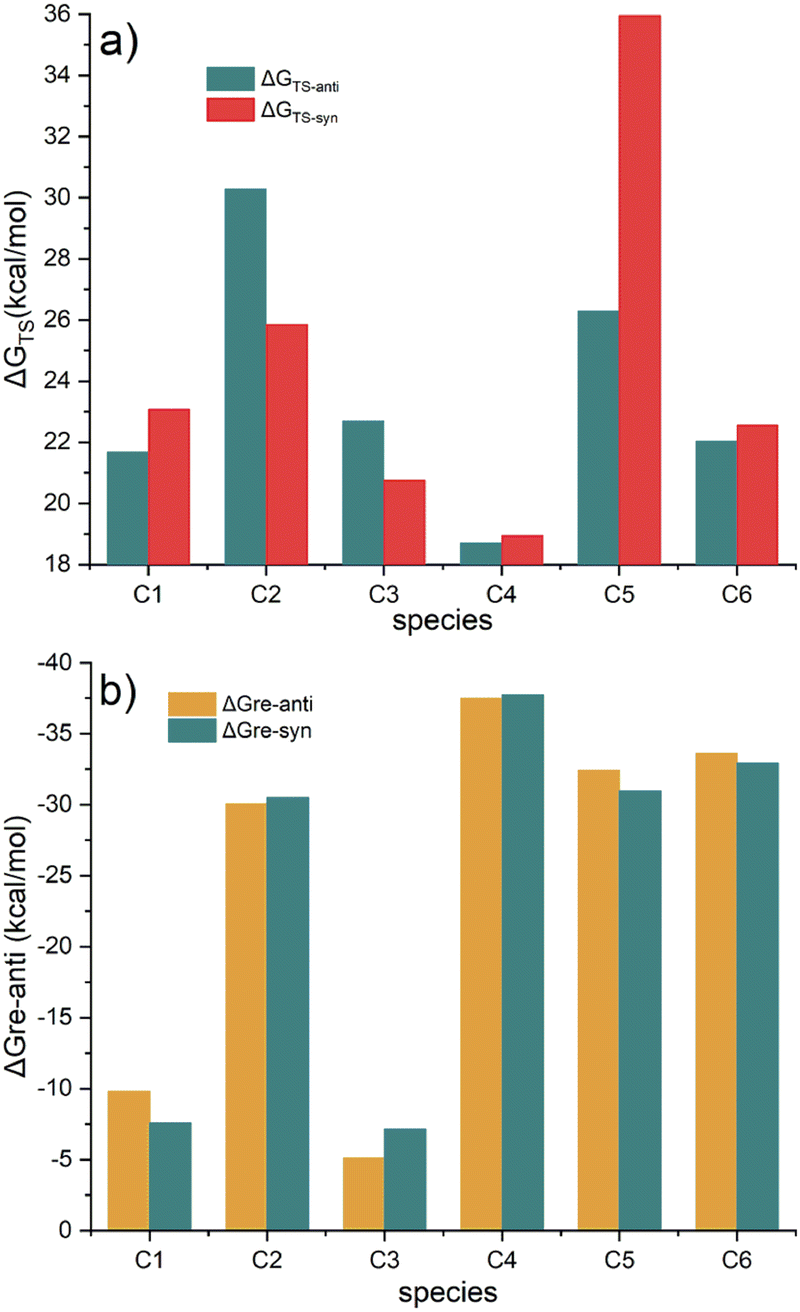 | ||
| Fig. 7 (a) The energy barriers (energy changes from reactants to the transition states); (b) reaction free energies (energy changes from reactants to products) of the Diels–Alder reactions of C2B2F2 with Ci (i = 1–6) in water. | ||
Conclusions
Using density-functional theory (DFT) and valence bond theory (VBT), the structure, stability, and bonding of the rhombus molecule C2B2H2 with D2h symmetry and its derivatives (C2B2R2) have been investigated in this work. We also explored the mechanism and stereoselectivity of the Diels–Alder reactions of C2B2F2 with ten kinds of 5-substituted cyclopentadienes at the M06-2X/6-311++G** level. Besides, six BNCT drugs were designed and it was demonstrated that they can be produced via Diels–Alder reactions of C2B2F2 with six dienes containing peptide fragments.Although our VBT computations confirmed that the inverted CC bond in the derivatives of C2B2H2 similarly contains a π bond and a charge-shift (CS) bond, C2B2F2 stands out as a preferable dienophile to proceed the Diels–Alder reaction due to its low LUMO energy level and the similarity of its LUMO to that of ethylene. DFT computations showed that the Diels–Alder reactions of C2B2F2 with 5-substituted cyclopentadienes are feasible both thermodynamically and kinetically, and the stereoselectivity of products is controlled by the substituent groups bonded to C(sp3) of cyclopentadiene. Finally, we demonstrated a new method for designing BNCT drugs by the Diels–Alder reactions of C2B2F2 with dienes containing peptide fragments. We anticipate the experimental synthesis of C2B2F2 and expect its promising potential for application in organic chemistry and in the pharmaceutical industry.
Author contributions
Changyu Cao: performing DFT computations and drafting the initial version of the manuscript. Junjing Gu: performing VBT calculations and summarizing the results. Congjie Zhang: initializing and supervising the project and writing the final version of the manuscript. Yirong Mo: supervising the project and editing the final version of the manuscript.Data availability
All data related to this work including structural information and energies are available in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the National Natural Science Foundation of China for financial support (No. 21373133). This work was performed in part at the Joint School of Nanoscience and Nanoengineering, a member of the Southeastern Nanotechnology Infrastructure Corridor (SENIC) and National Nanotechnology Coordinated Infrastructure (NNCI), which is supported by the National Science Foundation (Grant ECCS-2025462).Notes and references
- O. Diels and K. Alder, Synthesen in der hydroaromatischen Reihe, Justus Liebigs Ann. Chem., 1928, 460, 98–122 CrossRef.
- M. H. Cao, N. J. Green and S. Z. Xu, Application of the aza-Diels-Alder reaction in the synthesis of natural products, Org. Biomol. Chem., 2017, 15, 3105–3129 RSC.
- S. M. Morozova, Recent Advances in Hydrogels via Diels-Alder Crosslinking: Design and Applications, Gels, 2023, 9, 102 CrossRef CAS PubMed.
- T. N. Gevrek and A. Sanyal, Furan-containing polymeric Materials: Harnessing the Diels-Alder chemistry for biomedical applications, Eur. Polym. J., 2021, 153, 110514 CrossRef CAS.
- D.-Q. Li, S.-Y. Wang, Y.-J. Meng, Z.-W. Guo, M.-M. Cheng and J. Li, Fabrication of self-healing pectin/chitosan hybrid hydrogel via Diels-Alder reactions for drug delivery with high swelling property, pH-responsiveness, and cytocompatibility, Carbohydr. Polym., 2021, 268, 118244 CrossRef CAS PubMed.
- F. Cadamuro, L. Russo and F. Nicotra, Biomedical Hydrogels Fabricated Using Diels–Alder Crosslinking, Eur. J. Org. Chem., 2021, 374–382 CrossRef.
- X. Zhang, X. Chen, Z. Ye, W. Liu, X. Liu and X. Wang, Conductive hydrogels for bioelectronics: molecular structures, design principles, and operation mechanisms, J. Mater. Chem. C, 2023, 11, 10785–10808 RSC.
- G. Griffini, B. Rigatelli and S. Turri, Diels-Alder Macromolecular Networks in Recyclable, Repairable and Reprocessable Polymer Composites for the Circular Economy – A Review, Macromol. Mater. Eng., 2023, 308, 2300133 CrossRef.
- M. Imade, H. Hirao, K. Omoto and H. Fujimoto, Theoretical Study of Endo Selectivity in the Diels-Alder Reactions between Butadienes and Cyclopropene, J. Org. Chem., 1999, 64, 6697–6701 CrossRef.
- J. D. Xidos, T. L. Gosse, E. D. Burke, R. A. Poirier and D. J. Burnell, Endo—Exo and Facial Stereoselectivity in the Diels—Alder Reactions of 3-Substituted Cyclopropenes(I) with Butadiene, ChemInform, 2001, 123, 5482–5488 Search PubMed.
- M. Ramirez, D. Svatunek, F. Liu, N. K. Garg and K. N. Houk, Origins of Endo Selectivity in Diels–Alder Reactions of Cyclic Allene Dienophiles, Angew. Chem., Int. Ed., 2021, 60, 14989–14997 CrossRef.
- J. E. Baldwin and V. P. Reddy, Stereochemistry of the Diels-Alder reaction of butadiene with cyclopropene, J. Org. Chem., 1989, 54, 5264–5267 CrossRef.
- B. J. Levandowski, L. Zou and K. N. Houk, Hyperconjugative Aromaticity and Antiaromaticity Control the Reactivities and π-Facial Stereoselectivities of 5-Substituted Cyclopentadiene Diels–Alder Cycloadditions, J. Org. Chem., 2018, 83, 14658–14666 CrossRef PubMed.
- B. J. Levandowski and K. N. Houk, Hyperconjugative, Secondary Orbital, Electrostatic, and Steric Effects on the Reactivities and Endo and Exo Stereoselectivities of Cyclopropene Diels–Alder Reactions, J. Am. Chem. Soc., 2016, 138, 16731–16736 CrossRef.
- Y. Apeloig and E. Matzner, Evidence for the Dominant Role of Secondary Orbital Interactions in Determining the Stereochemistry of the Diels-Alder Reaction: The Case of Cyclopropene, J. Am. Chem. Soc., 1995, 117, 5375–5376 CrossRef.
- B. J. Levandowski, D. Svatunek, B. Sohr, H. Mikula and K. N. Houk, Secondary Orbital Interactions Enhance the Reactivity of Alkynes in Diels–Alder Cycloadditions, J. Am. Chem. Soc., 2019, 141, 2224–2227 CrossRef PubMed.
- J. M. Medina, J. L. Mackey, N. K. Garg and K. N. Houk, The Role of Aryne Distortions, Steric Effects, and Charges in Regioselectivities of Aryne Reactions, J. Am. Chem. Soc., 2014, 136, 15798–15805 CrossRef PubMed.
- S. Shaik, D. Danovich, W. Wu and P. C. Hiberty, Charge-shift bonding and its manifestations in chemistry, Nat. Chem., 2009, 1, 443–449 CrossRef PubMed.
- C. J. Zhang, F. Fan, Z. M. Wang, J. S. Song, C. S. Li and Y. R. Mo, B-Heterocyclic Carbene Arising from Charge Shift: A Computational Verification, Chem. – Eur. J., 2018, 24, 10216–10223 CrossRef PubMed.
- Z. Q. Tian, C. J. Zhang, Z. P. Pei, J. X. Liang and Y. R. Mo, (Si and B)-heterocyclic carbenes and theoretical design of new molecules, Mol. Syst. Design Eng., 2023, 8, 85–91 RSC.
- C. Zhang, H. Jiao and W. Jia, Theoretical predication of Diels-Alder reactions of highly strained dienophiles, Comput. Theor. Chem., 2020, 1175, 112734 CrossRef.
- J. W. Jian, W. Li, X. Wu and M. F. Zhou, Double C–H bond activation of acetylene by atomic boron in forming aromatic cyclic-HBC2BH in solid neon, Chem. Sci., 2017, 8, 4443–4449 RSC.
- C. Zhang, D. Ma, S. Yang and J. Liang, Theoretical Investigation of Promising Molecules for Obtaining Complexes with Planar Tetracoordinate Carbon, ACS Omega, 2016, 1, 620–625 CrossRef.
- C. J. Zhang, Z. Q. Tian and W. H. Jia, Elusive Magnetic Compounds Originated from Planar Tetracoordinate Silicon, a Theoretical Prediction, J. Phys. Chem. A, 2021, 125, 843–847 CrossRef PubMed.
- C. J. Zhang, Z. Q. Tian, W. H. Jia and Y. R. Mo, Rational design of porous organic molecules (POMs) based on B-heterocyclic carbenes, Mol. Syst. Design Eng., 2021, 6, 132–138 RSC.
- C. J. Zhang, Z. M. Wang, J. S. Song, C. S. Li and Y. R. Mo, Bonding and Diels–Alder reactions of substituted 2-borabicyclo(1.1.0)but-1(3)-enes: a theoretical study, Theor. Chem. Acc., 2019, 138, 106 Search PubMed.
- R. J. Grams, W. L. Santos, I. R. Scorei, A. Abad-García, C. A. Rosenblum, A. Bita, H. Cerecetto, C. Viñas and M. A. Soriano-Ursúa, The Rise of Boron-Containing Compounds: Advancements in Synthesis, Medicinal Chemistry, and Emerging Pharmacology, Chem. Rev., 2024, 124, 2441–2511 CrossRef.
- P. Coghi, J. Li, N. S. Hosmane and Y. Zhu, Next generation of boron neutron capture therapy (BNCT) agents for cancer treatment, Med. Res. Rev., 2023, 43, 1809–1830 CrossRef PubMed.
- Y. Zhao and D. G. Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions, J. Chem. Phys., 1980, 72, 650–654 CrossRef.
- A. D. McLean and G. S. Chandler, Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z = 11–18, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef CAS.
- M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. Defrees and J. A. Pople, Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second-row elements, 1982773654–3665.
- T. Clark, J. Chandrasekhar, G. W. Spitznagel and P. V. R. Schleyer, Efficient diffuse function-augmented basis sets for anion calculations. III. The 3-21+G basis set for first-row elements, Li–F, J. Comput. Chem., 1983, 4, 294–301 CrossRef.
- G. W. Spitznagel, T. Clark, P. von Ragué Schleyer and W. J. Hehre, An evaluation of the performance of diffuse function-augmented basis sets for second row elements, Na-Cl, J. Comput. Chem., 1987, 8, 1109–1116 CrossRef.
- E. D. Glendening, C. R. Landis and F. Weinhold, NBO 7.0: New vistas in localized and delocalized chemical bonding theory, J. Comput. Chem., 2019, 40, 2234–2241 Search PubMed.
- Y. Zhang, X.-R. Zeng and X.-Z. You, A new complete basis set model (CBS-QB3) study on the possible intermediates in chemiluminescence, J. Chem. Phys., 2000, 113, 7731–7734 CrossRef.
- S. Kozuch and J. M. L. Martin, DSD-PBEP86: in search of the best double-hybrid DFT with spin-component scaled MP2 and dispersion corrections, Phys. Chem. Chem. Phys., 2011, 13, 20104–20107 RSC.
- J. M. L. Martin and G. Santra, Empirical Double-Hybrid Density Functional Theory: A ‘Third Way’ in Between WFT and DFT, Isr. J. Chem., 2020, 60, 787–804 CrossRef.
- P. V. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. van Eikema Hommes, Nucleus-Independent Chemical Shifts:
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) A Simple and Efficient Aromaticity Probe, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef.
A Simple and Efficient Aromaticity Probe, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef. - G. W. T. M. J. Frisch, H. B. Schlegel, G. E. Scuseria, J. R. C. M. A. Robb, G. Scalmani, V. Barone, B. Mennucci, H. N. G. A. Petersson, M. Caricato, X. Li, H. P. Hratchian, J. B. A. F. Izmaylov, G. Zheng, J. L. Sonnenberg, M. Hada, K. T. M. Ehara, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, O. K. Y. Honda, H. Nakai, T. Vreven, J. A. Montgomery Jr., F. O. J. E. Peralta, M. Bearpark, J. J. Heyd, E. Brothers, V. N. S. K. N. Kudin, T. Keith, R. Kobayashi, J. Normand, A. R. K. Raghavachari, J. C. Burant, S. S. Iyengar, J. Tomasi, N. R. M. Cossi, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, C. A. V. Bakken, J. Jaramillo, R. Gomperts, R. E. Stratmann, A. J. A. O. Yazyev, R. Cammi, C. Pomelli, J. W. Ochterski, K. M. R. L. Martin, V. G. Zakrzewski, G. A. Voth, J. J. D. P. Salvador, S. Dapprich, A. D. Daniels, J. B. F. O. Farkas, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- G. W. T. M. J. Frisch, H. B. Schlegel, G. E. Scuseria, J. R. C. M. A. Robb, G. Scalmani, V. Barone, H. N. G. A. Petersson, X. Li, M. Caricato, A. V. Marenich, B. G. J. J. Bloino, R. Gomperts, B. Mennucci, H. P. Hratchian, A. F. I. J. V. Ortiz, J. L. Sonnenberg, D. Williams-Young, F. L. F. Ding, F. Egidi, J. Goings, B. Peng, A. Petrone, D. R. T. Henderson, V. G. Zakrzewski, J. Gao, N. Rega, W. L. G. Zheng, M. Hada, M. Ehara, K. Toyota, R. Fukuda, M. I. J. Hasegawa, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, K. T. T. Vreven, J. A. Montgomery Jr., J. E. Peralta, M. J. B. F. Ogliaro, J. J. Heyd, E. N. Brothers, K. N. Kudin, T. A. K. V. N. Staroverov, R. Kobayashi, J. Normand, A. P. R. K. Raghavachari, J. C. Burant, S. S. Iyengar, M. C. J. Tomasi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, R. L. M. J. W. Ochterski, K. Morokuma, O. Farkas, and A. D. J and F. J. B. Foresman, Gaussian 16, Revision C.02, Gaussian, Inc., Wallingford CT, 2019 Search PubMed.
- P. C. Hiberty and S. Shaik, Breathing-orbital valence bond method – a modern valence bond method that includes dynamic correlation, Theor. Chem. Acc., 2002, 108, 255–272 Search PubMed.
- P. Su and W. Wu, Ab initio nonorthogonal valence bond methods, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2013, 3, 56–68 Search PubMed.
- Z. Chen and W. Wu, Ab initio valence bond theory: A brief history, recent developments, and near future, J. Chem. Phys., 2020, 153, 090902 CrossRef PubMed.
- S. Shaik, D. Danovich, J. M. Galbraith, B. Braïda, W. Wu and P. C. Hiberty, Charge-Shift Bonding: A New and Unique Form of Bonding, Angew. Chem., Int. Ed., 2020, 59, 984–1001 CrossRef CAS PubMed.
- M. Chen, J. Xu, D. Zhao, F. Sun, S. Tian, D. Tu, C. Lu and H. Yan, Site-Selective Functionalization of Carboranes at the Electron-Rich Boron Vertex: Photocatalytic B−C Coupling via a Carboranyl Cage Radical, Angew. Chem., Int. Ed., 2022, 61, e202205672 CrossRef CAS PubMed.
- R. C. Kane, P. F. Bross, A. T. Farrell and R. Pazdur, Velcade: U.S. FDA approval for the treatment of multiple myeloma progressing on prior therapy, Oncologist, 2003, 8, 508–513 CrossRef.
- M. Hušák, A. Jegorov, J. Rohlíček, A. Fitch, J. Czernek, L. Kobera and J. Brus, Determining the Crystal Structures of Peptide Analogs of Boronic Acid in the Absence of Single Crystals: Intricate Motifs of Ixazomib Citrate Revealed by XRPD Guided by ss-NMR, Cryst. Growth Des., 2018, 18, 3616–3625 CrossRef.
- K. Bush and P. A. Bradford, Interplay between β-lactamases and new β-lactamase inhibitors, Nat. Rev. Microbiol., 2019, 17, 295–306 CrossRef CAS PubMed.
- S. J. Baker, T. Akama, Y.-K. Zhang, V. Sauro, C. Pandit, R. Singh, M. Kully, J. Khan, J. J. Plattner, S. J. Benkovic, V. Lee and K. R. Maples, Identification of a novel boron-containing antibacterial agent (AN0128) with anti-inflammatory activity, for the potential treatment of cutaneous diseases, Bioorg. Med. Chem. Lett., 2006, 16, 5963–5967 CrossRef CAS PubMed.
- S. J. Baker, C. Z. Ding, T. Akama, Y.-K. Zhang, V. Hernandez and Y. Xia, Therapeutic Potential of Boron-Containing Compounds, Future Med. Chem., 2009, 1, 1275–1288 CrossRef CAS.
- A. M. O'Farrell, A. van Vliet, K. Abou Farha, J. M. Cherrington, D. A. Campbell, X. Li, D. Hanway, J. Li and H.-P. Guler, Pharmacokinetic and Pharmacodynamic Assessments of the Dipeptidyl Peptidase-4 Inhibitor PHX1149: Double-Blind, Placebo-Controlled, Single- and Multiple-Dose Studies in Healthy Subjects, Clin. Ther., 2007, 29, 1692–1705 CrossRef.
- C. H. Baker and S. C. Welburn, The Long Wait for a New Drug for Human African Trypanosomiasis, Trends Parasitol., 2018, 34, 818–827 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp03615h |
| This journal is © the Owner Societies 2024 |