
Early-stage oxidation and subsequent damage of the used nuclear fuel extractant TODGA; electron pulse radiolysis and theoretical insights†
Rupali G.
Deokar
 and
Andrew R.
Cook
*
and
Andrew R.
Cook
*
Brookhaven National Laboratory, Upton, NY 11973, USA. E-mail: rupalishinde171991@gmail.com; acook@bnl.gov; Tel: +1-(631) 344-4782
First published on 14th November 2024
Abstract
Radiation induced damage of extractant molecules is a well-known phenomenon responsible for reducing efficiency and increasing the waste and cost of reprocessing used nuclear fuel (UNF). As such, understanding early-stage (pico- to nanoseconds) radiation-induced reaction mechanisms is essential for informing the design of next generation extractants with enhanced radiation robustness. Here we utilized picosecond and nanosecond electron pulse radiolysis experiments to probe the early-stage radioactive environment experienced by the organic phase extractant N,N,N′,N′-tetraoctyldiglycolamide (TODGA), proposed for separating highly radioactive trivalent minor actinides (specifically americium and curium) from the trivalent lanthanides. Using comparisons to the similar ionization potential (IP) solute p-xylene, this work determined the mechanism of reaction with the ionized diluent (i.e., n-dodecane radical cation, DD˙+) is hole transfer to produce TODGA˙+. At high TODGA concentrations (>100 mM), the majority of this transfer occurs faster than 10 ps via the capture of DD˙+ holes prior to their solvation with a C37 = 300 mM. The surviving solvated holes were captured with k = (2.38 ± 0.15) × 1010 M−1 s−1. Attempts at subsequent hole transfer to lower IP solutes found that only 10% of holes were transferred, indicating bond rupture of TODGA˙+ occurs within 2.6 ns at 200 mM TODGA. Possible reaction pathways for the rapid decomposition of TODGA˙+ were explored using a combination of experiments and density functional theory (DFT) calculations.
1. Introduction
The most common way to separate radioactive metals from used nuclear fuel (UNF) is by biphasic liquid–liquid solvent extraction. For the liquid–liquid extractions and separation of the trivalent minor actinides (MA) from the lanthanides (Ln), diglycolamide (DGA) derivatives are considered one of the most important classes of extractants in both aqueous and organic media. Much research has focused on using N,N,N′,N′-tetraethyldiglycolamide (TEDGA) in the aqueous phase1,2 and N,N,N′,N′-tetraoctyldiglycolamide (TODGA, Fig. 1) in the organic phase.3–9 Different extraction schemes have been developed using these DGA extractants. For example: the actinide lanthanide separation process (ALSEP)10 uses a mixture of tetra-2-ethylhexyldiglycolamide (TEHDGA) and 2-ethylhexylphosphonic acid mono-2-ethylhexyl ester (HEH[EHP]) in n-dodecane (DD) for MA/Ln separations; the innovative selective actinide extraction (i-SANEX)3,7 and grouped actinide extraction (EURO-GANEX)8,9 processes use 0.2 M TODGA and 5 vol% 1-octanol or malonamide as a phase modifier in DD diluent; the amide-based radio-resources treatment with interim storage of transuranics (ARTIST)4,5 process incorporates TODGA in DD along with the phase modifier N,N-dihexyloctylamide (DHOA). A method that uses TODGA as an extractant for MA has been developed by Sasaki et al.6 where they reported that TODGA is a tridentate ligand and has good efficiency to extract MA such as americium and curium from aqueous nitric acid (HNO3) into DD. | ||
| Fig. 1 Molecular structure of N,N,N′,N′-tetraoctyldiglycolamide (TODGA). | ||
Due to the strong radioactive environment experienced during the separation process, the radiation chemistry of all diluents and extractants will play a major role in determining extraction efficiency, separation factors and solvent-recycle longevity.11–17 Zarzana et al.16 studied the degradation of DGAs in DD by using γ-radiolysis and picosecond electron pulse radiolysis, with and without contact with an aqueous HNO3 phase. They reported that the nature of degradation products of DGAs was not affected by the acidic aqueous phase or by variations in the alkyl sidechains. More recently, Kimberlin et al.17 reported additional dealkylation and nitrogen-carbonyl rupture products with nitric acid contact. Both reports suggested degradation originated from charge transfer from the ionized diluent, DD˙+, to TODGA. Identification of radiolysis products further suggested that the central part of the DGA was more susceptible to damage than the alkyl side chains. While many degradation products due to the radiolysis of TODGA in DD have been reported, the most common are N,N-dioctylacetamide and 2-hydroxy-N,N-dioctylacetamide from breakage of an etheric C–O bond near the center of the molecule.16–22
While degradation product studies under various conditions have implied various damage mechanisms,16–18,21,23–25 directly observed chemistry using time-resolved pulse radiolysis can provide more certainty, but is only beginning to be applied to such problems. Sugo et al.11,12,26 reported considerable work on the radiation chemistry of TODGA in both the organic phase and the organic phase contacted with aqueous acid. They used pulse radiolysis to suggest that a variety of amides including TODGA were all oxidized by charge transfer from ionized solvent molecules (DD˙+). Such a charge/hole transfer is supported by the ionization potential (IP) difference between DD and TODGA. Using nanosecond pulse radiolysis, Mezyk et al.27 studied the reaction kinetics between DD˙+ and TODGA, where they reported a rate constant of 9.7 × 109 M−1 s−1, later revised to (1.57 ± 0.28) × 1010 M−1 s−1.28,29 Following Sugo, they assumed the hole transfer (HT) mechanism between DD˙+ and TODGA (reaction (1)), but in a later report suggested that a proton transfer (PT) mechanism may also be involved13 (reaction (2)).
| DD˙+ + TODGA → DD + TODGA˙+ HT, ΔG = −1.02 eV | (1) |
| DD˙+ + TODGA → DD(−H)˙ + TODGA(+H)+ PT, ΔG = −1.07 eV | (2) |
Using computed free energies,28 they reported both reactions are equally energetically favourable, although experimental confirmation for which reaction mechanism dominates was not performed.
Mincher et al.30 examined a series of solvents including 2,2′,4,6,6′-pentamethylheptane (a branched dodecane), 1-dodecene, cyclohexane and DD with varied IPs in which to measure reaction kinetics between the solvent radial cation and TODGA. They found a better correlation between the reaction rate constant and differences in IP for PT than HT, suggesting PT rather than HT may be responsible for the initial step of TODGA degradation.
Clearly the nature of the reaction of DD˙+ formed by diluent radiolysis is not well known. In addition, while product studies and calculations might infer the mechanism of this reaction and subsequent damage to extractants like TODGA, the initial steps where damage begins are likewise not well established. The present paper uses picosecond time-resolved electron pulse radiolysis to examine these early-stage processes. Initial measurements described below explored the reaction of DD˙+ with TODGA at various concentrations (0–500 mM), as well as subsequent HT to indicator molecules such as tri-p-tolylamine (TTA) and 10-methylphenothiazine (MePTZ). Unlike previous reports using very low concentrations of extractant, picosecond pulse radiolysis enabled making observations with up to 500 mM TODGA, which is more relevant to proposed separations processes, enabling greater mechanistic understanding of radiation effects under real-word conditions. The current work also examines the impact of pre-solvated hole capture that has been reported when concentrations of solutes are similarly high.31–35 This mechanism can enable hole transfer far faster than expected. It is unique to radiolysis but of unknown importance in the context of UNF extraction. Building on observations, this study finally examines the mechanism of initial TODGA damage, with the aid of quantum chemical calculations.
2. Materials and methods
TODGA (99%) was used as received from Technocomm, Ltd. Solvents n-dodecane (DD, >99%) and dichloromethane (DCM, >99%) from Sigma-Aldrich were dried over 3A molecular sieves. 10-Methylphenothiazine (MePTZ, 98%) and p-xylene (99+%) were used as received from Millipore Sigma, as was tri-p-tolyl amine (TTA, 98%) from TCI. Samples were sealed in custom made 0.5 cm pathlength Suprasil cells and degassed using argon or ethene gas (Matheson Tri-gas). 0.3 M DCM was added to samples to scavenge electrons formed by ionization to reduce the rate of recombination in alkanes.Picosecond electron pulse radiolysis experiments were performed at the Brookhaven National Laboratory (BNL) Laser Electron Accelerator Facility (LEAF).36 9 MeV electron pulses of 5–10 ps duration were generated using UV photons from an ultrafast laser incident on a magnesium photocathode in an RF cavity. The output 3–5 nC electron pulses gave a dose of 10–20 Gy in samples, estimated by comparison to solvated electron absorption in a water sample. Data were normalized to the same dose using a Faraday cup. Experiments with ∼10 ps time resolution used the Optical Fiber Single-Shot (OFSS) transient absorption experiment, described previously.37 Briefly, the OFSS system utilizes a bundle of different length optical fibers that act as independent optical delay lines to collect complete transients within a 5 ns time window using a single electron pulse. 64 pulses were typically averaged to reduce noise. 1 ns time-resolved experiments utilized a pulsed xenon arc lamp and an FND-100 silicon photodiode, digitized using a LeCroy WaveRunner HRO 66Zi oscilloscope (12 bit, 600 MHz). Data were collected and processed with LabView (National Instruments) and Igor Pro (Wavemetrics) software programs.
To support the experimental observations, quantum chemical calculations were performed using the Gaussian1638 and GaussView39 software packages. Calculations used the B3LYP functional40,41 and 6-31G(d,p) basis set,42 with solvation provided by the polarizable continuum model (IEFPCM) self-consistent reaction field (SCRF), using n-dodecane as the solvent (ε = 2.0060).43 Geometries were shown to be stable to a local minima using frequency analysis. IPs were calculated using enthalpies from frequency calculations. Free energies of reactions likewise used free energies from frequency calculations, and were further corrected for standard states.44,45 Time dependent density functional theory (TD-DFT)46,47 calculations were used to estimate the absorbance maximum (λmax) of transient species.
3. Results and discussion
3.1. Oxidation of TODGA by DD˙+
Radiolysis of alkanes, such as DD used in this study, generates electrons (e−), radical cations (DD˙+), carbon centered radicals (DD(−H)˙), hydrogen atoms (H˙), and excited states (DD*):48DD ![[long arrow, wavy then straight]](https://www.rsc.org/images/entities/char_e0f6.gif) DD˙+, e−, DD(−H)˙, H˙, DD* DD˙+, e−, DD(−H)˙, H˙, DD* | (3) |
Radiolytically generated e− were scavenged by the well-known electron scavenger DCM (0.3 M)49,50 which also inhibits subsequent DD* formation. H˙ will mostly seek out or abstract another H˙ from the solvent, producing additional DD(-H)˙, and under process conditions DD(−H)˙ will react with oxygen to form largely inert peroxyl radicals, making DD˙+ the most important radiolytically-induced oxidizing organic species.12,13,16,17 The calculated IP in DD of DD and TODGA are 8.11 and 6.92 eV respectively (Table S1, ESI†), suggesting that DD˙+ should readily oxidize TODGA by HT (reaction (1)). Note that at high concentration, a significant fraction of TODGA˙+ can also be produced by direct ionization of TODGA, because it is a large fraction of the sample composition. The yield of directly ionized solute is typically estimated by the fraction of the total sample electron density due to the solute, further discussed below. As noted in the introduction, DD˙+ can also react with TODGA by PT (reaction (2)). PT is an often-reported fate of alkane radical cations,51,52 and Lewis Bases like TODGA have ethers and carbonyls that are good proton acceptors. According to calculated free energies in DD, both reactions (1) and (2) are energetically favourable, −1.02 eV vs. −1.07 eV, respectively.
A difficulty in determining whether DD˙+ reacts with TODGA by HT or PT is that TODGA˙+ does not have a readily observable absorption in DD. The spectrum of 200 mM TODGA in DD recorded as a function of time after radiolysis is seen in Fig. 2. Sugo et al.12 reported that a peak at 370 nm was due to TODGA˙+. In this work we observed a 375 nm band and also a weaker and broad band near 600 nm. Consistent with previous results,53 we found that the peak at 375 nm was largely quenched by O2 at a near diffusion-controlled rate and is thus dominated by the TODGA triplet absorption (Fig. S1(a), ESI†). The peak at 600 nm was not quenched significantly by the presence of O2 (Fig. S1(b), ESI†). The nature of the 600 nm band is unclear. Geminate ion recombination in alkanes is found to happen in about 5 ns, but the 600 nm band is too long lived (∼60 ns) to be geminately recombining TODGA+˙ and further does not appear to decay in the presence of hole scavengers discussed below. It seems quite likely that this band is due to a degradation product of TODGA or an impurity. One possibility might be the products of the PT reaction (reaction (2)), DD(−H)˙ and TODGA(+H)+, however TDDFT predicts these species absorb below 250 nm (Table S2, ESI†). While not reported previously, a dimer radical cation, (TODGA)2+˙, might form and possibly absorb at 600 nm, but the spectrum of 20 mM TODGA indicates this peak was unaffected by concentration (Fig. S2, ESI†), so this assignment is unlikely.
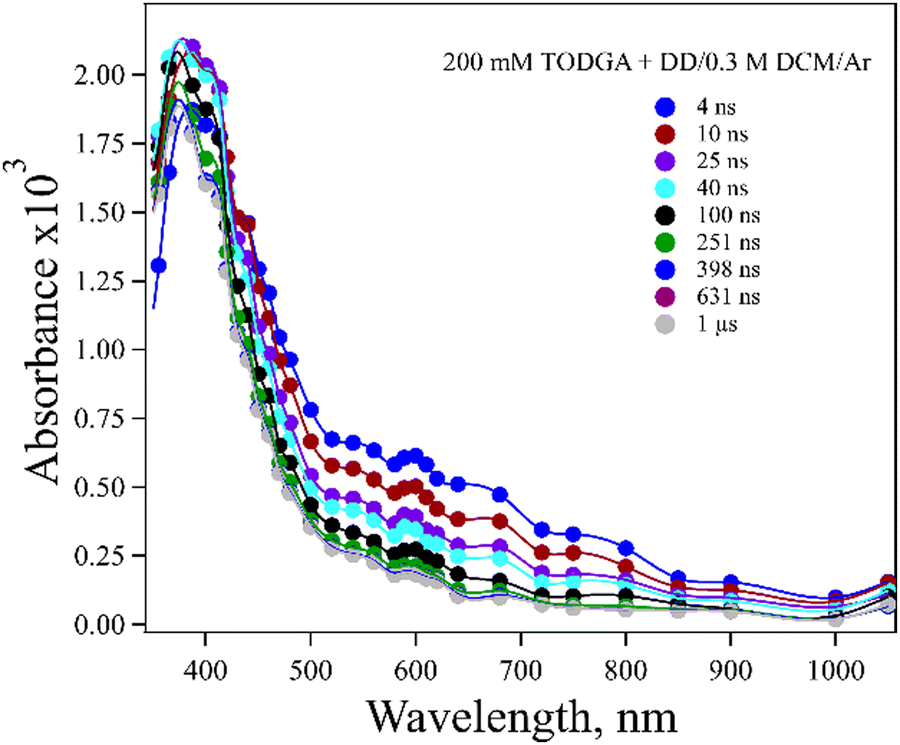 | ||
| Fig. 2 Transient spectra obtained for electron pulse-irradiated, Ar saturated 200 mM TODGA in DD/0.3 M DCM, from 4 ns to 1 μs. The peak at 375 nm is identified primarily as a triplet excited state of TODGA, and the peak at 600 nm is likely due to a degradation product of TODGA or an impurity. | ||
As it was not possible to directly observe the production of TODGA˙+, the loss of DD˙+ was monitored instead. The DD˙+ has an easily observed broad and strong absorption spectrum peaking at 850 nm, with an extinction coefficient (ε) = 1.2 × 104 M−1 cm−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 54 (Fig. S3, ESI†). Confidence in the results was gained by comparing to data collected using p-xylene instead of TODGA, also probing the decay of DD˙+. Arenes such as p-xylene are well known hole scavengers, are expected to make relatively stable radical cations, and have no proton-acceptor so only HT is possible. Both TODGA and p-xylene have similar computed IPs, 6.92 and 7.18 eV respectively, so have similar driving force for HT. Fig. 3 shows kinetic traces for both solutes at 0–500 mM recorded at 800 nm, near the DD˙+ absorption peak, with ∼10 ps resolution. The data have two key concentration dependent features: a resolved decay and a sudden loss of DD˙+ faster than experimental time-resolution. The decay of DD˙+ absorption seen with p-xylene (Fig. 3(a)) can only be attributed to HT from solvated DD˙+, with a rate determined with exponential fits to data, giving k = (2.55 ± 0.34) × 1010 M−1 s−1 (Fig. S4(a), ESI†). Likewise, the decay with TODGA (Fig. 3(b)) gives k = (2.38 ± 0.15) × 1010 M−1 s−1 (Fig. S4(b), ESI†). Previous work28,29 found the latter rate to be (1.57 ± 0.28) × 1010 M−1 s−1, but were unable to establish the mechanism of reaction. The ∼50% faster rates determined here may be attributable to a combination of better time-resolution and early-time enhancement of the apparent rate due to non-equilibrium effects when DD˙+ is suddenly created, as reported previously.55,56 While the observed DD˙+ decay with TODGA might be due to either HT or PT, the nearly identical rate to p-xylene suggests that the mechanism is HT. One might also expect slower rates for PT where a bond must be broken, however a few reports give PT rates of alkane˙+ to alcohols of 1 × 1010 M−1 s−1.57,58
54 (Fig. S3, ESI†). Confidence in the results was gained by comparing to data collected using p-xylene instead of TODGA, also probing the decay of DD˙+. Arenes such as p-xylene are well known hole scavengers, are expected to make relatively stable radical cations, and have no proton-acceptor so only HT is possible. Both TODGA and p-xylene have similar computed IPs, 6.92 and 7.18 eV respectively, so have similar driving force for HT. Fig. 3 shows kinetic traces for both solutes at 0–500 mM recorded at 800 nm, near the DD˙+ absorption peak, with ∼10 ps resolution. The data have two key concentration dependent features: a resolved decay and a sudden loss of DD˙+ faster than experimental time-resolution. The decay of DD˙+ absorption seen with p-xylene (Fig. 3(a)) can only be attributed to HT from solvated DD˙+, with a rate determined with exponential fits to data, giving k = (2.55 ± 0.34) × 1010 M−1 s−1 (Fig. S4(a), ESI†). Likewise, the decay with TODGA (Fig. 3(b)) gives k = (2.38 ± 0.15) × 1010 M−1 s−1 (Fig. S4(b), ESI†). Previous work28,29 found the latter rate to be (1.57 ± 0.28) × 1010 M−1 s−1, but were unable to establish the mechanism of reaction. The ∼50% faster rates determined here may be attributable to a combination of better time-resolution and early-time enhancement of the apparent rate due to non-equilibrium effects when DD˙+ is suddenly created, as reported previously.55,56 While the observed DD˙+ decay with TODGA might be due to either HT or PT, the nearly identical rate to p-xylene suggests that the mechanism is HT. One might also expect slower rates for PT where a bond must be broken, however a few reports give PT rates of alkane˙+ to alcohols of 1 × 1010 M−1 s−1.57,58
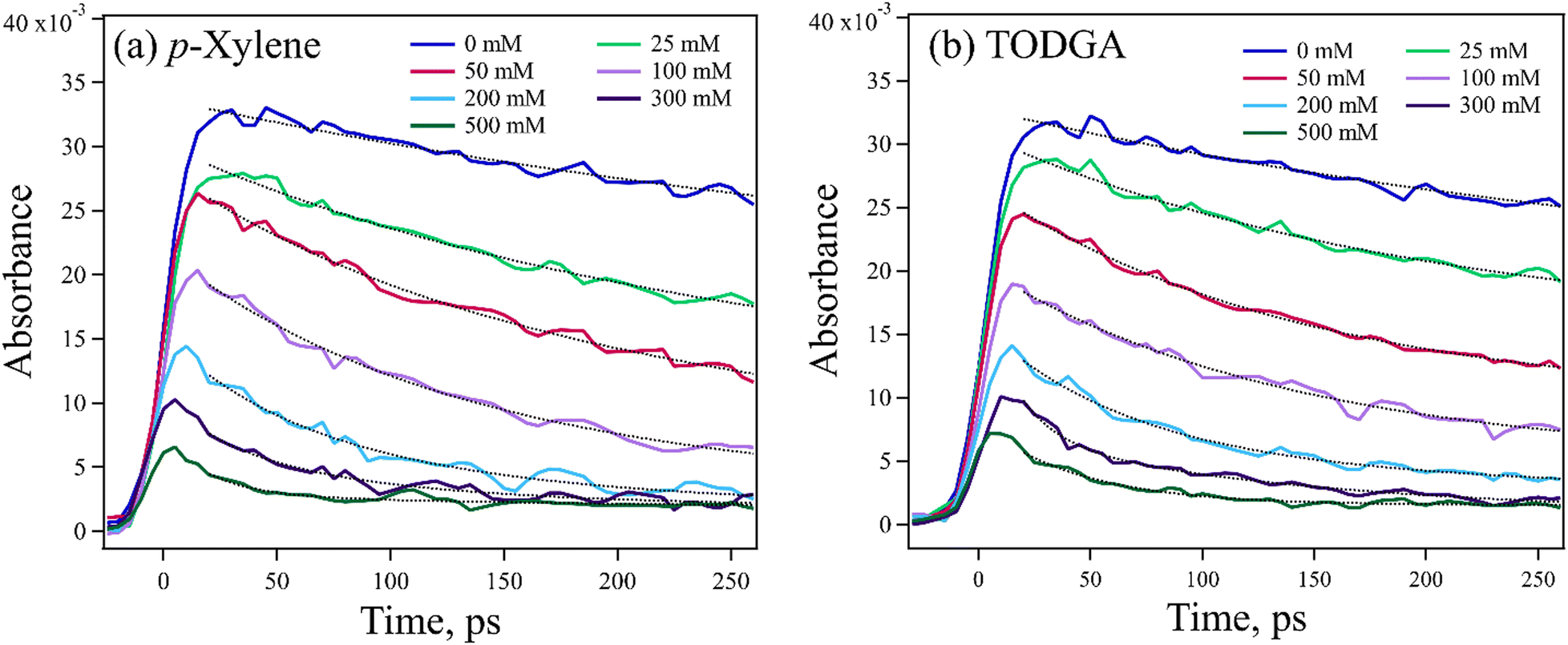 | ||
| Fig. 3 Kinetic traces at 800 nm where only DD˙+ absorbs were obtained by OFSS after electron pulse irradiation, for 0–500 mM (a) p-xylene or (b) TODGA in DD with 0.3 M DCM. Dashed lines are exponential fits to the data. HT from DD˙+ to p-xylene and TODGA after pulse radiolysis are described with a sudden component (<10 ps) followed by a resolved decay component, giving rate constants of (2.55 ± 0.34) × 1010 M−1 s−1 and (2.38 ± 0.15) × 1010 M−1 s−1 respectively. | ||
An important feature of the data in Fig. 3 is the sub-10 ps loss of DD˙+. With 500 mM of either solute, very little DD˙+ survives to transfer a hole to the solutes. Part of this loss is due to direct ionization of the solute, rather than the solvent. At 500 mM, this corresponds to a decrease in the DD˙+ signal by 35% and 6.5% for TODGA and p-xylene respectively (Table S3, ESI†). The remainder of the missing DD˙+ before 10 ps is due to ultrafast HT to solutes prior to the solvation of the initial DD˙+, as has been reported previously.31–35 On this timescale bimolecular PT is not likely. This feature in alkanes like DD will be further explored in a future publication. Fig. 4 plots the t = 0 extrapolated absorbance from the curves in Fig. 3vs. solute concentration. This data can be described by fitting the amount missing at early time to an exponential dependence, originally used by Hunt for describing pre-solvated electron capture in water:59,60
| f = exp(−[S]/C37) | (4) |
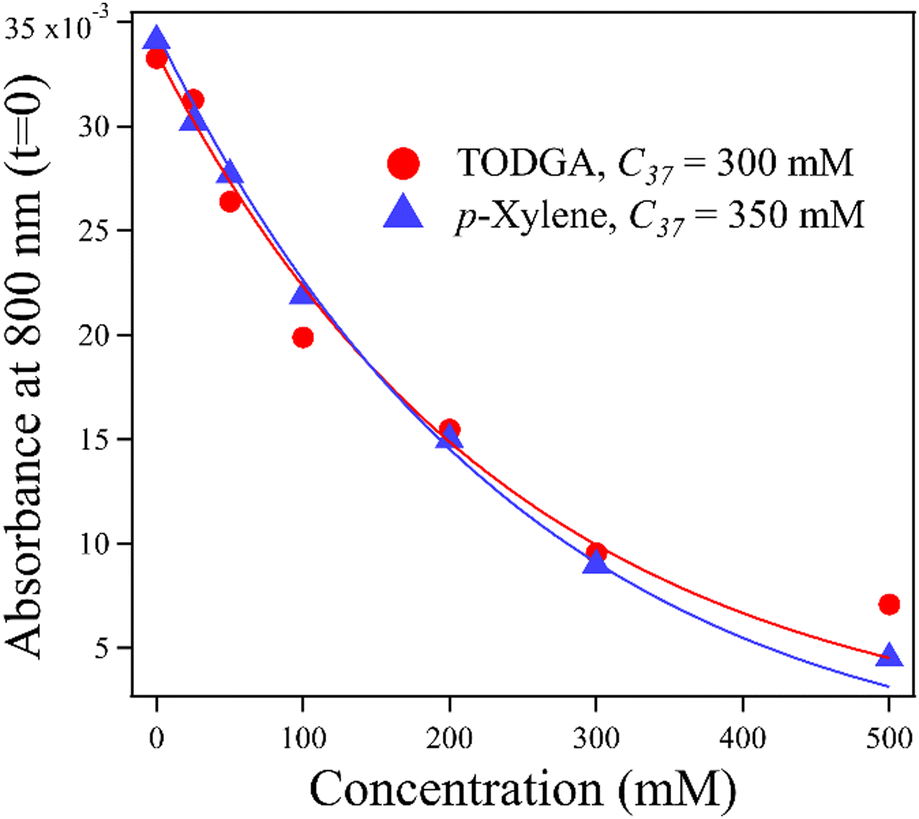 | ||
| Fig. 4 The loss of DD˙+ in the presence of p-xylene or TODGA is described as a function of solute concentration by both direct solute ionization and hole capture prior to solvation. The concentration at which 37% of solvent holes escape pre-solvated hole capture and become solvated is 350 and 300 mM for p-xylene and TODGA respectively. | ||
3.2. Can TODGA•+ transfer its hole to low IP solutes?
To quantify the oxidation of TODGA by reaction (1), rather than accepting a proton from DD˙+ in reaction (2), experiments attempted to transfer any captured holes to the lower IP (5.55 eV, Table S1, ESI†) solute tri-p-tolylamine (TTA). For clarity, results were compared to the equivalent process using p-xylene. These reactions are similarly energetically favourable:| p-xylene˙+ + TTA → p-xylene + TTA˙+ HT, ΔG = −1.59 eV | (5) |
| TODGA˙+ + TTA → TODGA + TTA˙+ HT, ΔG = −1.56 eV | (6) |
TTA was chosen as a hole indicator because it's radical cation is easily observed at 670 nm (Fig. S5, ESI†) with a large ε = 26200 M−1 cm−1.61 A 2 mM concentration of TTA was selected to avoid indicator oxidation by DD˙+. Measurements are further simplified because at this low concentration, most geminate ions (DD˙+, TODGA˙+) recombine before holes can be transferred to TTA, so results predominantly probe the chemistry of the slowly decaying free or homogeneous ions62 (free ion yield (Gfi) of alkanes is 0.1–0.2 per 100 eV63). A reasonable assumption however is that these measurements inform on the entire population of holes, as the nature of HT and PT events is the same for both the geminate and homogeneous ions. Kinetic measurements for 2 mM TTA in absence and presence of 200 mM p-xylene or TODGA in DD with 0.3 M DCM are shown in Fig. 5(a). Results show that the final absorbance of the sample with p-xylene is the same as the one with TTA only, confirming that all holes captured by p-xylene are transferred to TTA. By contrast, we find that by 100 ns the absorbance of TTA˙+ in the TODGA sample is at most only 10% of the TTA only sample. Results are tabulated in Table S4(a) (ESI†); note that the final determination of HT was made after subtracting off the signals from the p-xylene or TODGA only samples, where an unknown absorption (likely subsequent products or due to impurities, seen in Fig. 2) persisted to long times as can be seen in Fig. 5(a) as well as time-resolved spectra in Fig. S6 (ESI†).
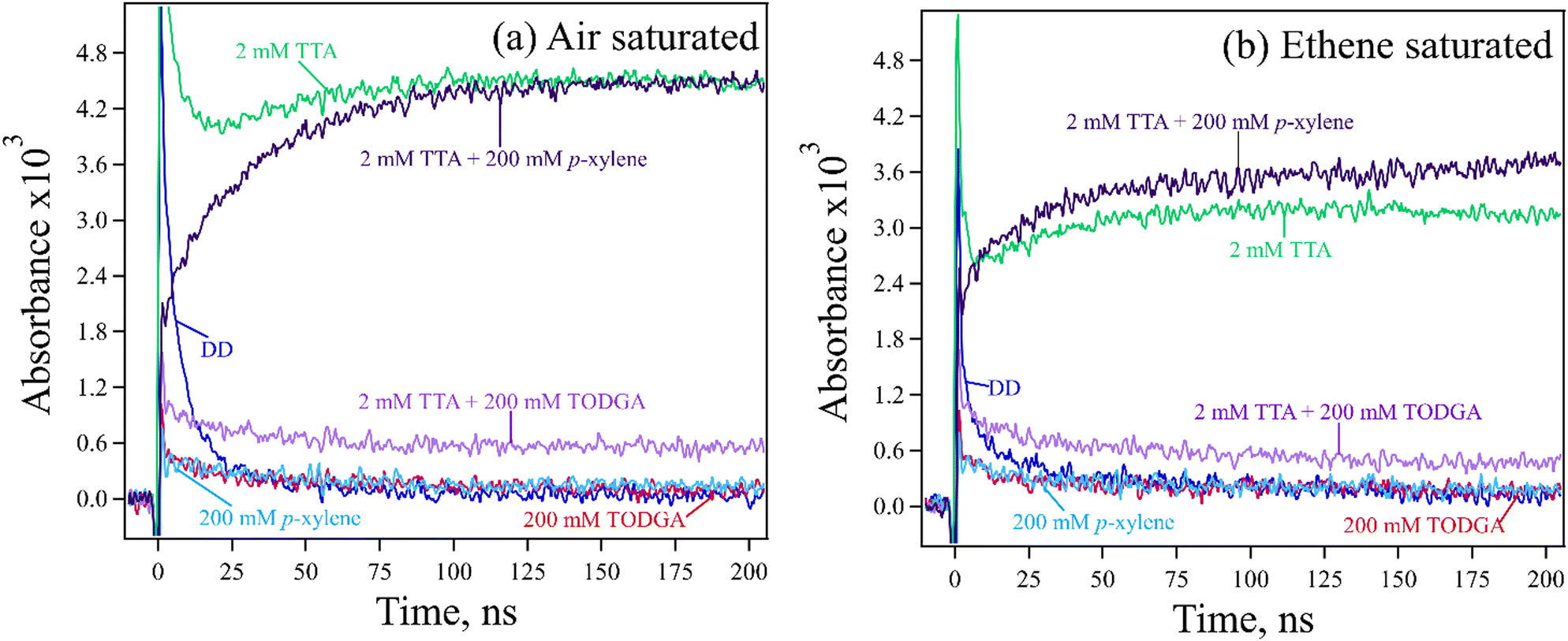 | ||
| Fig. 5 Kinetic traces at 670 nm obtained for electron pulse-irradiated 2 mM TTA in the absence and presence of 200 mM TODGA or p-xylene in DD/0.3 M DCM, where samples were saturated with (a) air or (b) ethene. From (a) and (b) it was found that 30% of TTA oxidation resulted from Cl˙. Results also show that all holes attached to p-xylene are transferred to TTA, while only 10-11% of holes attached to TODGA were transferred to TTA. | ||
While addition of DCM to samples is advantageous for increasing the lifetime of DD˙+, it also adds to TTA oxidation due to the production of Cl˙ atoms, as evidenced by the slow growths of TTA˙+ with both the TTA only and p-xylene containing samples. Cl˙ are formed primarily a result of dissociative electron capture by DCM followed by recombination:64
| CH2Cl2 + e− → CH2Cl˙ + Cl− (dissociative capture) | (7) |
| CH2Cl2˙+ + Cl− → CH2Cl2 + Cl˙ (recombination) | (8) |
As the chloride ion (Cl−) has a modest gas phase IP of 7.6 eV, Cl˙ can oxidize low IP molecules like TTA. To remove the impact of Cl˙ atom oxidation, solutions were saturated with ethene gas (solubility in DD is ∼95 mM65), which reacts with Cl˙ to make relatively inert carbon centered radicals before they can oxidize TTA.66Fig. 5(b) shows the results with all samples after ethene purging. We find that the absorbance of TTA˙+ decreased about 30%, indicating that this amount came from oxidation by Cl˙ atoms. Note that in the sample with p-xylene the amount of TTA˙+ is slightly higher than the TTA only sample. This is a result of the well-known ability of Cl˙ to form complexes with arenes, stabilizing them and increasing their lifetime, though also making them slower to oxidize solutes.67,68 We conclude that results with ethene purging still show that all holes captured by p-xylene are still transferred to TTA. When TODGA data are analysed as above, we find that up to 11% of holes captured by TODGA are transferred to TTA, essentially the same as without ethene.
A possible reason for the low transfer efficiency of holes from TODGA˙+ to TTA might be due to the exoergicity of the HT reaction, placing it in the Marcus inverted regime. However, given that the energetics of reactions (5) and (6) are essentially the same, we might expect them to have similar rates. To test whether energetics might play a role, analogous experiments to the above were performed using 10-methylphenothiazine (MePTZ, IP = 5.78 eV, MePTZ˙+λmax = ∼520 nm69,70) in place of TTA, which reduced the free energy of the reactions by 0.23 eV. Results were similar to TTA, finding 11.5% of holes captured by TODGA were transferred to MePTZ, as seen in Fig. S7 and Table S4(b) (ESI†).
While results indicate that TODGA transferred only ∼10% of its holes to indicators, here we ask if TTA˙+ might be made in other ways, suggesting even less transfer from TODGA˙+. These include direct ionization of TTA, and capture of geminate holes by TTA, forming some TTA˙+ without originating from TODGA˙+. The low concentration of TTA and high concentration of TODGA were expected to mitigate against these possibilities. We find indeed that this is the case, and the sum of these processes at most might contribute ∼0.3% of the observed TTA˙+ signal in the sample with both TODGA and TTA, well within the possible errors in our measurements (details in ESI,† Table S5 and, Section S1 and S2).
3.3. Why doesn’t TODGA˙+ transfer many holes to indicators?
The results above found that when 200 mM TODGA captured holes from DD˙+, it only transferred 10–11% to an indicator, TTA or MePTZ. The only likely explanation is that TODGA˙+ degraded faster than the holes could be transferred. As noted in the introduction, radiochemical degradation of extractants like TODGA is a well-known problem which depends on reaction conditions;12,14,16,22,24,25,71–74 here we find that a significant fraction of this degradation occurs quite rapidly after forming TODGA˙+. A rough estimate for the rate of degradation was made from the fraction of holes that are transferred to TTA, based on following logic. There are three possible fates for TODGA˙+: (i) recombination with anions; (ii) transfer of the holes to TTA (reaction (6)), and (iii) degradation/fragmentation:| TODGA˙+ → Degradation/fragmentation products | (9) |
As these measurements report on the chemistry of homogeneous ions, we expect their recombination will be slow. This is evident from our results with p-xylene and TTA (Fig. 5), where all holes are transferred to TTA and essentially no p-xylene˙+ is lost to recombination. The fraction of TODGA˙+ that transferred to TTA (fTTA) is thus given by the ratio of rate constants for reaction (6) (kHT) and reaction (9) (kdeg):
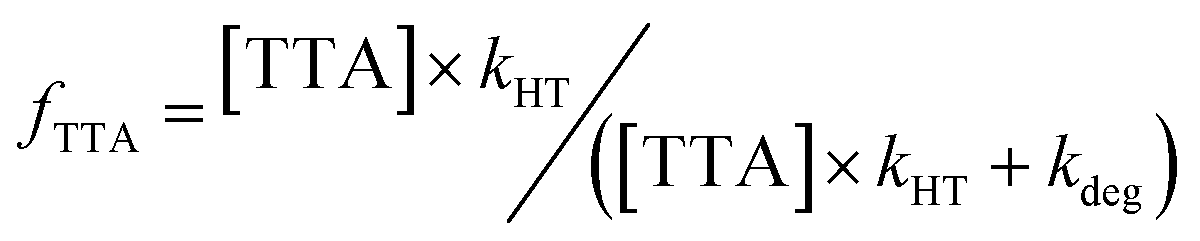 | (10) |
If TODGA˙+ degrades in within 2.6 ns, what is its fate? Experimentally identifying the correct pathway has proven difficult, in no small part because the products above have no easily observable absorption bands in the visible-nIR. Experiments and computations have suggested possibilities however, discussed below. It should be noted that more than one degradation pathway may be happening concurrently.
Firstly, a TODGA concentration study (0–400 mM) was performed with a fixed concentration of TTA (2 mM) in the presence of ethene (Fig. 6). A decrease in the amount of TTA˙+ absorption at 670 nm with increasing TODGA concentration was observed. The magnitude of the decrease in TTA˙+ is not consistent with simply assuming competition between TODGA and TTA for holes from DD˙+; to explain the observed production of TTA˙+, the rate constant for HT from DD˙+ to TTA would need to be in excess of 1011 M−1 s−1, which is not physically reasonable, and far in excess of the measured rate, 2.1 × 1010 M−1 s−1 (Fig. S8, ESI†). We further note that not only are the rates incompatible with our results, but such an analysis ignores the large fraction of DD˙+ holes that are captured by TODGA in <10 ps, leaving few DD˙+ to oxidize TTA in competition with TODGA by diffusion. A more likely explanation is that the TODGA concentration dependence indicates a reaction between TODGA˙+ and neutral TODGA, which removes the ability to transfer the hole to TTA. A plausible reaction for this is PT from TODGA˙+ to the oxygens of a neutral TODGA molecule:
| TODGA˙+ + TODGA → TODGA(−H)˙ + TODGA(+H)+ ΔG = −0.67 eV | (11) |
| TODGA˙+ → TODGA(−H)(+H)˙+ ΔG = −0.70 eV | (12) |
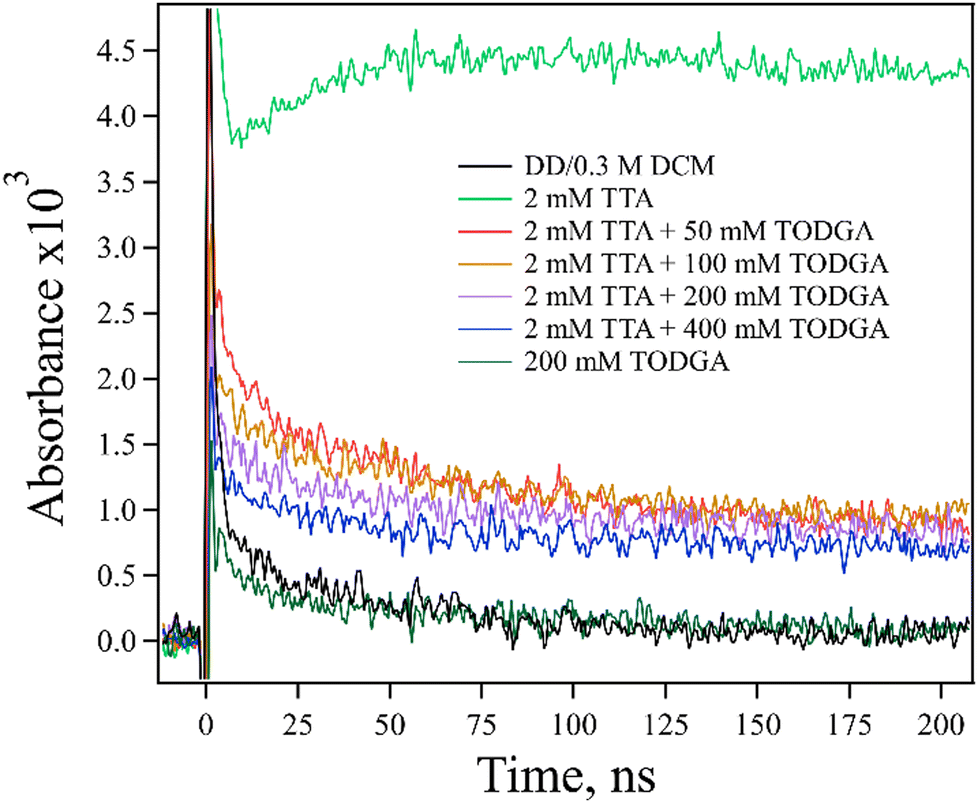 | ||
| Fig. 6 Kinetic traces obtained at 670 nm for electron pulse-irradiated, 2 mM TTA with 0–400 mM TODGA in DD/0.3 M DCM where samples were saturated with ethene. The decrease in TTA˙+ absorption with increasing TODGA concentration was observed, likely indicating a PT reaction between TODGA˙+ and TODGA (reaction (11)). | ||
However, this unimolecular reaction would not be expected to result in the concentration dependence seen in Fig. 6.
DFT calculations suggest that after PT, the TODGA(−H)˙ radical formed by removing a proton from a carbon next to the ether may further fragment by rupture of the etheric C–O bond, the products of which have been reported.16–20 Calculations predict this reaction is energetically favourable, with a free energy of −0.30 eV, resulting in the formation of N,N-dioctyl-2-oxoacetamide and the N,N-dioctylacetamide radical (Fig. S10, ESI†). Subsequent H abstraction by the N,N-dioctylacetamide radical can form N,N-dioctylacetamide, which is also a commonly observed degradation product of TODGA. TD-DFT calculations suggest the N,N-dioctylacetamide radical absorbs at 572 nm (Table S6, ESI†), which may be responsible for the observed 600 nm band in the TODGA spectrum (Fig. 2). To examine such a C–O bond rupture in TODGA(−H)˙, Galán et al. measured the degradation of methylated24,25 and dimethylated25 TODGA to test whether replacing protons on the carbons next to the ether with methyl groups inhibits TODGA decomposition. They reported that the addition of one methyl group increased the rate of degradation, while the addition of a second methyl group did in fact reduce TODGA degradation in alkane diluents. These results support the supposition above that PT to produce the etheric radical product may be an important vector for TODGA degradation.
A final possibility for the 2.6 ns degradation of TODGA˙+ is bond scission in the backbone of the radical cation. While we were unable to directly observe products, calculations were again helpful to identify favourable degradation pathways. Inspired by previous reports16,17 in the absence of an acidic phase, a degradation scheme of the mostly likely bonds to be broken was prepared, seen in Fig. S11 (ESI†). Computed free energies of these reactions (Table S6, ESI†), suggests only one favourable degradation pathway (ΔG = −0.31 eV), where the C–C bond between one of the amide groups and the central part of TODGA˙+ breaks, giving the N,N-dioctylcarboxamide radical and 2-methoxy-N,N-dioctylacetamide cation (DPV˙ and DPVIII+ in Fig. S11, ESI†). These products are predicted by TDDFT calculations to absorb at 240 and 229 nm respectively, so were unobservable in our experiments due to a combination of small oscillator strength and overlap with other absorbing species, such as alkyl radicals. Such products are expected to be highly reactive species and thus short lived, contributing to the difficulty in observing them.
3.4. Magnitude of TODGA damage
As noted in the introduction, many reports have inferred various damage mechanisms following radiolysis for organic phase extractants like TODGA. In this work we have shown for the first time that TODGA˙+ is damaged rapidly, with a short < 2.6 ns lifetime (at 200 mM TODGA). Here we ask a simple question: how does the magnitude of damage found here compare to previous works,15 and what does it imply about when most damage occurs?While Section 2 reports on the magnitude of damage to homogenous TODGA˙+, we need an estimate of the total damage to all TODGA˙+ formed, including the geminate ions. The data in Fig. 3 shows that at 200 mM TODGA, more than half of the initial DD˙+ are captured by TODGA in <10 ps, and the remainder, including the small fraction of homogeneous ions, in well under 0.5 ns. Most of these ions are geminate and are recombining with anions rapidly. In neat DD˙+ much of the recombination is occurring with a ∼5 ns lifetime (Fig. 3); we assume that geminate TODGA˙+ will recombine with a similar lifetime. TODGA˙+ recombination is in competition with the 2.6 ns degradation, suggesting that about 2/3's of TODGA˙+ decompose. It is not known if the remaining 1/3 of ions that recombine may also result in damage to TODGA, so are neglected here. Reports give the initial G value for ionization of alkanes is in the rage of 0.31–0.41 μM J−1.82–84 Using an average of 0.36 μM J−1, thus gives a G value for TODGA degradation of 0.24 μM J−1. Previous gamma radiolysis work15 in a similar DD solution, albeit with only 50 mM TODGA, was described using an exponential degradation dose constant of 5.8 × 10−3 kGy−1. This data can also be fit to give an initial G value for degradation of 0.24 μM J−1, identical to the estimate for damage in the current work. We note that the gamma radiolysis study may underestimate the G value due to the lower TODGA concentration. Despite this, the results in this paper suggest that the majority of radiolytic damage to TODGA occurs within a few nanoseconds, and not at long times as the result of subsequent radical reactions. This conclusion has important ramifications for ways to improve extraction systems for UNF. Separation efficiency may be improved by avoiding this rapid damage. This might take the form of avoiding oxidation of TODGA in the first place, but also by blocking damage mechanisms such as PT described above.
4. Conclusions
This paper presents a mechanistic study of TODGA degradation at early-stages (pico- to nanoseconds) after exposure to ionizing radiation. By comparison to the similar IP solute p-xylene, the primary mechanism of the reaction between DD˙+ and TODGA was found to be predominantly HT. The HT process was characterized by a sub-10 ps reaction of pre-solvated holes with a C37 = 300 mM, followed by the relatively slower capture of solvated holes with a rate of k = (2.38 ± 0.15) × 1010 M−1 s−1. A finding of this work is that while holes can be transferred from p-xylene to lower IP hole indicators such as TTA and MePTZ with essentially unit quantum yield, the same is not true for TODGA, which only transferred ∼10% of holes in samples with 200 mM TODGA and 2 mM indicators. This is likely due to the rapid cleavage of bonds within TODGA˙+, resulting in the formation of degradation/fragmentation products that can no longer undergo HT. Such radiolytically caused damage to extractants like TODGA is very important to their use, and a commonly explored theme in the literature. We further found that with 200 mM TODGA, much of the initial radiation-induced damage occurs in 2.6 ns. Although the damage mechanism is not clear, calculations suggest either PT from the weakened C–H bonds next to the central ether in TODGA˙+ (though also possible from less energetically favourable alkyl C–H bonds) or rupture of the C–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond of TODGA˙+ are likely sites for initial damage. While both pathways are calculated to be energetically favourable, the first path is supported by kinetics measurements and may produce products that explain the 600 nm band seen in the absorption spectrum of TODGA˙+. We note that if the PT reaction is bimolecular, the lifetime of TODGA˙+ under typical separation applications will be even shorter, as concentrations of TODGA will exceed the 200 mM used in this study.
O bond of TODGA˙+ are likely sites for initial damage. While both pathways are calculated to be energetically favourable, the first path is supported by kinetics measurements and may produce products that explain the 600 nm band seen in the absorption spectrum of TODGA˙+. We note that if the PT reaction is bimolecular, the lifetime of TODGA˙+ under typical separation applications will be even shorter, as concentrations of TODGA will exceed the 200 mM used in this study.
Overall, this study provides a deeper fundamental understanding of TODGA damage at early stages. Results suggest that decreasing TODGA oxidation or blocking weak C–H bonds in the radical cation might be viable routes to avoid much damage. To test these ideas, future work will explore the impact of other high concentration species typically present in the organic phase of separation systems, such as phase modifiers, that may reduce the number of holes transferred to TODGA and thus inhibit degradation. Work will also seek to further explore the impact of the suggested PT damage mechanism, also found to be important by the work of Galán et al.,25 who reported reduced damage when PT was blocked by both protons on one carbon next to the central ether by methyls. Might such substitution of TODGA molecule at the etheric carbons increase the lifetime of TODGA˙+ allowing recombination without damage, and thus ultimately increase the extraction efficiency of UNF?
Author contributions
Rupali G. Deokar: experiments, data analysis, theoretical calculations, writing. Andrew R. Cook: conceptualization, data analysis, writing, supervision.Data availability
Data from this publication will be openly available, within 30 days of publication, in machine-readable format at https://doi.org/10.5281/zenodo.13776907.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work and use of the Laser Electron Accelerator Facility of the Accelerator Center for Energy Research at BNL, was supported by the U.S. Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences, Division of Chemical Sciences, Geosciences and Biosciences under contract DE-SC0012704. The authors would like to thank Dr John R. Miller, Dr Stephen P. Mezyk and Dr Gregory P. Holmbeck for valuable discussions and comments on this work.Notes and references
- Y. Sasaki, Y. Tsubata, Y. Kitatsuji, Y. Sugo, N. Shirasu and Y. Morita, Solvent Extr. Ion Exch., 2014, 32, 179–188 CrossRef CAS.
- A. Kimberlin, G. Saint-Louis, D. Guillaumont, B. Camès, P. Guilbaud and L. Berthon, Phys. Chem. Chem. Phys., 2022, 24, 9213–9228 RSC.
- A. Wilden, G. Modolo, P. Kaufholz, F. Sadowski, S. Lange, M. Sypula, D. Magnusson, U. Müllich, A. Geist and D. Bosbach, Solvent Extr. Ion Exch., 2015, 33, 91–108 CrossRef CAS.
- S. Tachimori, Y. Sasaki and S.-I. Suzuki, Solvent Extr. Ion Exch., 2002, 20, 687–699 CrossRef CAS.
- S. Suzuki, Y. Sasaki, T. Yaita and T. Kimura, Study on selective separation of uranium by N,N-dialkyl-amide in ARTIST process, France, 2004.
- Y. Sasaki, Y. Sugo, S. Suzuki and S. Tachimori, Solvent Extr. Ion Exch., 2001, 19, 91–103 CrossRef CAS.
- G. Modolo, A. Wilden, P. Kaufholz, D. Bosbach and A. Geist, Prog. Nucl. Energy, 2014, 72, 107–114 CrossRef CAS.
- M. Carrott, A. Geist, X. Hères, S. Lange, R. Malmbeck, M. Miguirditchian, G. Modolo, A. Wilden and R. Taylor, Hydrometallurgy, 2015, 152, 139–148 CrossRef CAS.
- J. Brown, F. McLachlan, M. Sarsfield, R. Taylor, G. Modolo and A. Wilden, Solvent Extr. Ion Exch., 2012, 30, 127–141 CrossRef CAS.
- A. V. Gelis and G. J. Lumetta, Ind. Eng. Chem. Res., 2014, 53, 1624–1631 CrossRef CAS.
- Y. Sugo, Y. Sasaki and S. Tachimori, Radiochim. Acta, 2002, 90, 161–165 CrossRef CAS.
- Y. Sugo, Y. Izumi, Y. Yoshida, S. Nishijima, Y. Sasaki, T. Kimura, T. Sekine and H. Kudo, Radiat. Phys. Chem., 2007, 76, 794–800 CrossRef CAS.
- S. P. Mezyk, G. P. Horne, B. J. Mincher, P. R. Zalupski, A. R. Cook and J. F. Wishart, Procedia Chem., 2016, 21, 61–65 CrossRef.
- R. Malmbeck and N. L. Banik, J. Radioanal. Nucl. Chem., 2020, 326, 1609–1615 CrossRef CAS.
- G. P. Horne, C. A. Zarzana, C. Rae, A. R. Cook, S. P. Mezyk, P. R. Zalupski, A. Wilden and B. J. Mincher, Phys. Chem. Chem. Phys., 2020, 22, 24978–24985 Search PubMed.
- C. A. Zarzana, G. S. Groenewold, B. J. Mincher, S. P. Mezyk, A. Wilden, H. Schmidt, G. Modolo, J. F. Wishart and A. R. Cook, Solvent Extr. Ion Exch., 2015, 33, 431–447 CrossRef CAS.
- A. Kimberlin, D. Guillaumont, S. Arpigny, B. Camès, P. Guilbaud, G. Saint-Louis, H. Galán and L. Berthon, New J. Chem., 2021, 45, 12479–12493 RSC.
- Y. V. Serenko, A. V. Ponomarev, E. V. Belova and N. V. Yudin, J. Radioanal. Nucl. Chem., 2021, 328, 1319–1328 CrossRef CAS.
- Y. V. Nikitina, N. V. Yudin, E. V. Belova and A. V. Ponomarev, J. Radioanal. Nucl. Chem., 2020, 326, 1185–1192 CrossRef CAS.
- H. Zhang, Y.-Y. Ao, Y. Wang, S.-J. Zhao, J.-Y. Sun, M.-L. Zhai, J.-Q. Li, J. Peng and H.-B. Li, Nucl. Sci. Tech., 2023, 34, 48 CrossRef CAS.
- H. Galán, A. Núñez, A. G. Espartero, R. Sedano, A. Durana and J. de Mendoza, Procedia Chem., 2012, 7, 195–201 CrossRef.
- I. Sánchez-García, H. Galán, J. M. Perlado and J. Cobos, EPJ Nuclear Sci. Technol., 2019, 5, 1–7 CrossRef.
- I. Sánchez-García, R. J. M. Egberink, W. Verboom and H. Galán, New J. Chem., 2024, 48, 2087–2096 Search PubMed.
- V. Hubscher-Bruder, V. Mogilireddy, S. Michel, A. Leoncini, J. Huskens, W. Verboom, H. Galán, A. Núñez, J. Cobos, G. Modolo, A. Wilden, H. Schmidt, M. C. Charbonnel, P. Guilbaud and N. Boubals, New J. Chem., 2017, 41, 13700–13711 RSC.
- H. Galán, C. A. Zarzana, A. Wilden, A. Núñez, H. Schmidt, R. J. M. Egberink, A. Leoncini, J. Cobos, W. Verboom, G. Modolo, G. S. Groenewold and B. J. Mincher, Dalton Trans., 2015, 44, 18049–18056 RSC.
- Y. Sugo, Y. Sasaki, T. Kimura, T. Sekine and H. Kudo, Proceeding of the international conference on advanced nuclear fuel cycles and systems, 2005.
- S. P. Mezyk, B. J. Mincher, S. B. Dhiman, B. Layne and J. F. Wishart, J. Radioanal. Nucl. Chem., 2016, 307, 2445–2449 CrossRef CAS.
- G. P. Horne, C. Celis-Barros, J. K. Conrad, T. S. Grimes, J. R. McLachlan, B. M. Rotermund, A. R. Cook and S. P. Mezyk, Phys. Chem. Chem. Phys., 2023, 25, 16404–16413 RSC.
- G. P. Horne, C. Celis-Barros, J. K. Conrad, T. S. Grimes, J. R. McLachlan, B. M. Rotermund, A. R. Cook and S. P. Mezyk, Phys. Chem. Chem. Phys., 2024 10.1039/D4CP90191F.
- B. J. Mincher, C. A. Zarzana and S. P. Mezyk, Radical Cations and Acid Protection during Radiolysis, https://www.osti.gov/biblio/1389193, United States, 2016.
- S. Tagawa, N. Hayashi, Y. Yoshida, M. Washio and Y. Tabata, Int. J. Radiat. Appl. Instrum., Part C, 1989, 34, 503–511 Search PubMed.
- F. Wang, U. Schmidhammer, A. de La Lande and M. Mostafavi, Phys. Chem. Chem. Phys., 2017, 19, 2894–2899 RSC.
- M. J. Bird, A. R. Cook, M. Zamadar, S. Asaoka and J. R. Miller, Phys. Chem. Chem. Phys., 2020, 22, 14660–14670 RSC.
- A. R. Cook, J. Phys. Chem. A, 2021, 125, 10189–10197 CrossRef CAS PubMed.
- A. de la Lande, S. Denisov and M. Mostafavi, Phys. Chem. Chem. Phys., 2021, 23, 21148–21162 RSC.
- J. F. Wishart, A. R. Cook and J. R. Miller, Rev. Sci. Instrum., 2004, 75, 4359–4366 CrossRef CAS.
- A. R. Cook and Y. Shen, Rev. Sci. Instrum., 2009, 80, 073106 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg Williams, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian16, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- T. K. Roy Dennington and John Millam, GaussView, Version 6.1.1, Semichem Inc., Shawnee Mission, KS, 2019 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. DeFrees and J. A. Pople, J. Chem. Phys., 1982, 77, 3654–3665 CrossRef CAS.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS PubMed.
- C. P. Kelly, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2007, 111, 408–422 CrossRef CAS.
- C. P. Kelly, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2006, 110, 16066–16081 CrossRef CAS PubMed.
- A. D. Laurent, C. Adamo and D. Jacquemin, Phys. Chem. Chem. Phys., 2014, 16, 14334–14356 RSC.
- C. Adamo and D. Jacquemin, Chem. Soc. Rev., 2013, 42, 845–856 RSC.
- D. W. Werst and A. D. Trifunac, Radiat. Phys. Chem., 1993, 41, 127–133 CrossRef CAS.
- I. M. Salih, T. Söylemez and T. I. Balkaş, Radiat. Res., 1976, 67, 235–243 CrossRef CAS.
- T. I. Balkaş, Int. J. Radiat. Phys. Chem., 1972, 4, 199–208 CrossRef.
- I. A. Shkrob, M. C. Sauer and A. D. Trifunac, in Studies in Physical and Theoretical Chemistry, ed. C. D. Jonah and B. S. M. Rao, Elsevier, 2001, vol. 87, pp. 175–221 Search PubMed.
- J. Ceulemans, Acc. Chem. Res., 2002, 35, 523–531 CrossRef CAS PubMed.
- T. Toigawa, D. R. Peterman, D. S. Meeker, T. S. Grimes, P. R. Zalupski, S. P. Mezyk, A. R. Cook, S. Yamashita, Y. Kumagai, T. Matsumura and G. P. Horne, Phys. Chem. Chem. Phys., 2021, 23, 1343–1351 RSC.
- R. Mehnert, O. Brede and W. Naumann, J. Radioanal. Nucl. Chem., 1986, 101, 307–318 CrossRef CAS.
- A. R. Cook, M. J. Bird, S. Asaoka and J. R. Miller, J. Phys. Chem. A, 2013, 117, 7712–7720 CrossRef CAS PubMed.
- A. R. Cook, P. Sreearunothai, S. Asaoka and J. R. Miller, J. Phys. Chem. A, 2011, 115, 11615–11623 CrossRef CAS.
- F. B. Sviridenko, D. V. Stas and Y. N. Molin, Dokl. Phys. Chem., 2001, 377, 80–82 CrossRef.
- F. B. Sviridenko, D. V. Stass and Y. N. Molin, Mol. Phys., 2003, 101, 1839–1850 CrossRef CAS.
- R. K. Wolff, J. E. Aldrich, T. L. Penner and J. W. Hunt, J. Phys. Chem., 1975, 79, 210–219 CrossRef CAS.
- K. Y. Lam and J. W. Hunt, Int. J. Radiat. Phys. Chem., 1975, 7, 317–338 CrossRef CAS.
- I. R. Gould, D. Ege, J. E. Moser and S. Farid, J. Am. Chem. Soc., 1990, 112, 4290–4301 CrossRef CAS.
- L. Wojnárovits, in Handbook of Nuclear Chemistry, ed. A. Vértes, S. Nagy, Z. Klencsár, R. G. Lovas and F. Rösch, Springer US, Boston, MA, 2011, pp. 1263–1331 DOI:10.1007/978-1-4419-0720-2_23.
- A. O. Allen, Yields of free ions formed in liquids by radiation, National Institute of Standards and Technology, Gaithersburg, MD, 1976.
- A. M. Funston and J. R. Miller, Radiat. Phys. Chem., 2005, 72, 601–611 CrossRef CAS.
- W. Hayduk, Solubility Data Series: Ethene, Oxford University Press, Oxford, UK, 1994 Search PubMed.
- F. S. C. Lee and F. S. Rowland, J. Phys. Chem., 1977, 81, 1235–1239 CrossRef CAS.
- R. E. BÜHler and M. Ebert, Nature, 1967, 214, 1220–1221 CrossRef.
- R. E. Bühler, Helv. Chim. Acta, 1968, 51, 1558–1571 CrossRef.
- M. H. Litt and J. Radovic, J. Phys. Chem., 1974, 78, 1750–1754 CrossRef CAS.
- R. E. Hester and K. P. J. Williams, J. Chem. Soc., Perkin Trans. 2, 1981, 852–859, 10.1039/P29810000852.
- A. Wilden, B. J. Mincher, S. P. Mezyk, L. Twight, K. M. Rosciolo-Johnson, C. A. Zarzana, M. E. Case, M. Hupert, A. Stärk and G. Modolo, Solvent Extr. Ion Exch., 2018, 36, 347–359 CrossRef CAS.
- K. M. Roscioli-Johnson, C. A. Zarzana, G. S. Groenewold, B. J. Mincher, A. Wilden, H. Schmidt, G. Modolo and B. Santiago-Schübel, Solvent Extr. Ion Exch., 2016, 34, 439–453 CrossRef CAS.
- G. P. Horne, A. Wilden, S. P. Mezyk, L. Twight, M. Hupert, A. Stärk, W. Verboom, B. J. Mincher and G. Modolo, Dalton Trans., 2019, 48, 17005–17013 RSC.
- R. B. Gujar, S. A. Ansari, A. Bhattacharyya, A. S. Kanekar, P. N. Pathak, P. K. Mohapatra and V. K. Manchanda, Solvent Extr. Ion Exch., 2012, 30, 278–290 Search PubMed.
- A. Sengupta, S. M. Ali and K. T. Shenoy, Polyhedron, 2016, 117, 612–622 CrossRef CAS.
- S. A. Ansari, P. N. Pathak, M. Husain, A. K. Prasad, V. S. Parmar and V. K. Manchanda, Radiochim. Acta, 2006, 94, 307–312 CrossRef CAS.
- I. S. Sosulin and A. Lisouskaya, Radiat. Phys. Chem., 2024, 222, 111785 CrossRef CAS.
- F. Wang, G. P. Horne, P. Pernot, P. Archirel and M. Mostafavi, J. Phys. Chem. B, 2018, 122, 7134–7142 CrossRef CAS.
- A. Kimberlin and K. L. Nash, Solvent Extr. Ion Exch., 2021, 39, 38–55 CrossRef CAS.
- H. Naganawa and S. Tachimori, Anal. Sci., 1994, 10, 309–314 CrossRef CAS.
- Q. N. Vo, J. L. Unangst, H. D. Nguyen and M. Nilsson, J. Phys. Chem. B, 2016, 120, 6976–6984 CrossRef CAS PubMed.
- P. J. Dyne, Can. J. Chem., 1965, 43, 1080–1092 CrossRef CAS.
- L. Wojnarovits and G. Foldiak, Radiat. Res., 1982, 91, 638–643 CrossRef CAS.
- W. F. Schmidt, Radiat. Res., 1970, 42, 73–78 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional figures, tables, details as referenced in the text, and calculated molecular geometries. See DOI: https://doi.org/10.1039/d4cp03678f |
| This journal is © the Owner Societies 2024 |