
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photoinduced removal of molecular oxygen from solutions
Barbara
Golec
 ab,
Natalia
Dutkiewicz
a,
Jakub
Ostapko
a,
Jacek
Waluk
ab and
Aleksander
Gorski
*a
ab,
Natalia
Dutkiewicz
a,
Jakub
Ostapko
a,
Jacek
Waluk
ab and
Aleksander
Gorski
*a
aInstitute of Physical Chemistry, Polish Academy of Sciences Kasprzaka 44/52, 01-224 Warsaw, Poland. E-mail: agorski@ichf.edu.pl
bFaculty of Mathematics and Science, Cardinal Stefan Wyszyński University Dewajtis 5, 01-815 Warsaw, Poland
First published on 18th November 2024
Abstract
We propose a new, simple and efficient procedure of light-driven deoxygenation of solutions based on hydroperoxides formation upon irradiation. Efficient and fast removal of molecular oxygen is caused by photosensitized generation of singlet oxygen, which then reacts with the solvent (2-methyltetrahydrofuran or tetrahydrofuran). Oxygen depletion makes it possible to observe processes normally undetectable in non-degassed liquid samples at room temperature, such as phosphorescence and triplet–triplet annihilation. The potential of the proposed protocol is demonstrated by recording of previously unknown phosphorescence of palladium complex of octaethylporphycene.
Introduction
Numerous photochemical and photophysical processes occur in triplet excited states of compounds involved in light-driven reactions. Triplet excited states of organic molecules play a crucial role in fluorescence upconversion processes based on triplet–triplet annihilation,1,2 thermally activated delayed fluorescence suitable for OLEDs development,3,4 singlet oxygen generation and developing of new agents for photodynamic therapy (PDT)5 and photodynamic inactivation of bacteria (PDI),6 photocatalysis,7 and photodegradation of organic dyes.8,9In solutions, one of the most efficient triplet state quenchers is molecular oxygen.10 Effective exploitation of the potential of triplet state-related phenomena in applications based on photoinduced processes requires the development of a strategy to protect against quenching by molecular oxygen and the subsequent production of highly reactive singlet oxygen species.
Various methodologies that allow reducing the detrimental effects caused by oxygen have been proposed: (i) passive or physical, based on decreasing oxygen mobility by, e.g., placing chromophores in polymer films11 or their incorporation into supramolecular complexes;12 (ii) active or chemical, by means of oxygen scavenging compounds,13–15 or (iii) combination of scavenging and incorporation strategies.16
One of the useful methods to prevent quenching of the excited state by molecular oxygen is chemical scavenging which involves the use of compounds capable of forming organic peroxides under photoexcitation.17 This phenomenon was initially observed and described in the early 20th century regarding rubrene behavior under light irradiation.18 Subsequently, numerous scientists have described similar reactions with polycyclic aromatic hydrocarbons.17,19
Inspired by these ideas, we propose a straightforward deoxygenation procedure that avoids the need to add a specific oxygen-removing substance to the solution. Instead, the solvent itself is used as a scavenger. It is known that such solvents as 2-methyltetrahydrofuran (2-MTHF) or tetrahydrofuran (THF) readily form hydroperoxides (Scheme 1).20 We show that this reaction is dramatically accelerated when molecular oxygen is excited to its lowest singlet state. Using a photosensitizer, it is possible to remove oxygen from the liquid sample in a few minutes. The efficiency of oxygen depletion is proven by the observation of processes that would not have been observed in the presence of oxygen: room-temperature phosphorescence and triplet–triplet annihilation.
 | ||
| Scheme 1 Oxidation of 2-methyltetrahydrofuran. Two isomers of 2-methyltetrahydrofuran hydroperoxide are formed: 2-methyl-2-peroxytetrahydrofuran (left) and 2-methyl-5-peroxytetrahydrofuran (right). | ||
The deoxygenation procedure is both simple and efficient, with the concentration of molecular oxygen reduced to 10−6 M or less after irradiation, which is comparable to conventional methods, such as degassing by inert gas bubbling or freeze–pump–thaw cycles.21,22 The protocol is easy to use and can be extended to other solvents by adding 2-MTHF, as demonstrated here for 2-MTHF/acetonitrile and 2-MTHF/toluene mixtures.
Experimental
Chemicals
All chemicals and commercially available reagents were used as purchased without further purification. 2,3,7,8,12,13,17,18-Octaethylporphyrin (OEP) was acquired from PorphyChem (purity 98%). Palladium(II) complex of OEP was synthesized and purified following the procedure described in ref. 23 palladium(II) 2,3,6,7,12,13,16,17-octaethylporphycene was synthesized and purified following the procedure described in ref. 24. 2-Methyltetrahydrofuran (2-MTHF), tetrahydrofuran (THF), toluene, and acetonitrile (ACN) from Aldrich (Spectroscopic Grade) were used as solvents. Rubrene, perylene, and 9,10-diphenylanthracene (Light & Co., Ltd., Colnbrook, England) were available in the laboratory. Electronic absorption measurements did not indicate the presence of impurities.Electronic absorption spectra were measured with a Shimadzu 2700 UV spectrophotometer.
Fluorescence, phosphorescence, and fluorescence upconversion measurements were carried out on the home-built setup equipped with a two-channel CCD spectrometer (Avantes AvaSpec-ULS2048-2-USB2). As the excitation light source, an Opotek RADIANT 355 laser was used (200–2500 nm tunable spectral range, 5–7 cm−1 spectral bandwidth, 5 ns pulse duration). The repetition rate was 10 Hz and the energy of 100 μJ per pulse was used. The second setup was an Edinburgh FS 900 CDT spectrofluorometer.
Irradiation of the samples was performed using 1 cm path-length quartz cells. The solution was continuously stirred at 1000 rpm on a magnetic stirrer (ROTH ROTILABO M3) during irradiation using a small magnetic bar placed in the fluorescence cuvette. A Thorlabs M385L continuous light-emitting diode (385 nm wavelength, power of 102 mW) was used for irradiation. The wavelength was selected to ensure good overlap with the Soret absorption band. The diode spectral full width at half maximum was 11.6 nm.
Triplet–triplet absorption decays were measured on a home-built spectrometer equipped with a Hamamatsu R955 photomultiplier and Yokogawa DL9140 oscilloscope. As the excitation light source, an Opotek RADIANT 355 laser was used. The continuous output of a laser-driven Xe lamp (Energetiq EQ-99-Plus-EU) was used as a probe beam.
Singlet oxygen lifetimes were measured with a homemade experimental setup based on a BENTHAM DTMc300 double monochromator, equipped with a thermoelectrically cooled photomultiplier (Hamamatsu H10330C75, 950–1700 nm registration range) and Yokogawa DL9140 fast oscilloscope. The previously described Opotek laser was used as the excitation light source.
Results and discussion
The structures of compounds used as photosensitizers in this work – palladium(II) metallocomplexes of 2,3,7,8,12,13,17,18-octaethylporphyrin (PdOEP) and 2,3,6,7,12,13,16,17-octaethylporphycene (PdOEPc) are depicted in Fig. 1. | ||
| Fig. 1 Palladium(II) 2,3,7,8,12,13,17,18-octaethylporphyrin (a) and palladium(II) 2,3,6,7,12,13,16,17-octaethylporphycene (b). | ||
PdOEP is a derivative of porphyrin with well-known photophysical properties: nearly 100% quantum yields of triplet state formation and singlet oxygen generation and very weak fluorescence (quantum yield of 3 × 10−4).25 In organic solvents under atmospheric conditions and at room temperature PdOEP exhibits extremely weak phosphorescence (10 times weaker than fluorescence) in the red part of the visible spectral region. The second photosensitizer – PdOEPc – is a derivative of porphycene, a constitutional isomer of porphyrin, with photophysical properties significantly different from those of porphyrin derivatives. The electronic absorption spectrum of PdOEPs is red-shifted compared to PdOEP and fluorescence is also very weak. The triplet state energy is unknown so far. The procedure of oxygen depletion presented in this work allows us to determine the triplet state energy of PdOEPc via observation of phosphorescence.
Room temperature phosphorescence
Luminescence of PdOEP excited at 400 nm was measured in 2-MTHF solution at room temperature (Fig. 2). The low-energy band between 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–16000 cm−1 corresponds to phosphorescence, while the high-energy part of the spectrum, 16000–19000 cm−1, represents fluorescence. At the beginning, for a freshly prepared solution, phosphorescence intensity is extremely weak. The behavior of phosphorescence changes with systematic irradiation of the solution by 102 mW LED light at 385 nm. The phosphorescence intensity of PdOEP increases gradually with the consecutive steps of irradiation (30 seconds each), exceeding several orders of magnitude (Fig. 2) at the end of irradiation (150 seconds). The intensity of very weak fluorescence remains unchanged during irradiation.
000–16000 cm−1 corresponds to phosphorescence, while the high-energy part of the spectrum, 16000–19000 cm−1, represents fluorescence. At the beginning, for a freshly prepared solution, phosphorescence intensity is extremely weak. The behavior of phosphorescence changes with systematic irradiation of the solution by 102 mW LED light at 385 nm. The phosphorescence intensity of PdOEP increases gradually with the consecutive steps of irradiation (30 seconds each), exceeding several orders of magnitude (Fig. 2) at the end of irradiation (150 seconds). The intensity of very weak fluorescence remains unchanged during irradiation.
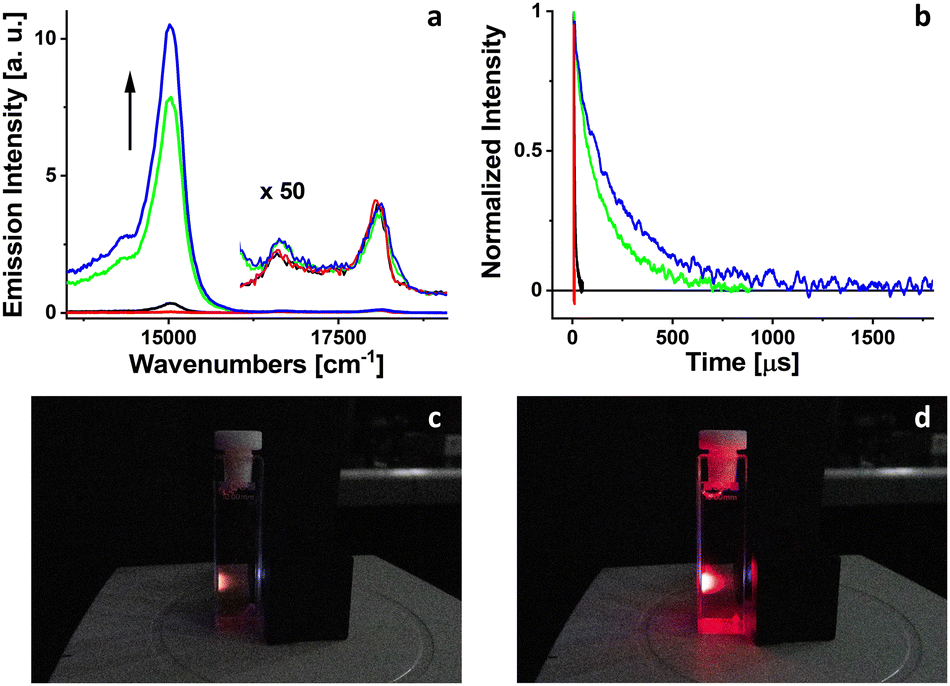 | ||
| Fig. 2 Changes in phosphorescence intensity (a) and triplet lifetimes (b) of PdOEP in 2-MTHF during systematic irradiation at 385 nm: red, black, green, and blue curves correspond to 0, 90, 120, and 150 seconds of irradiation, respectively. The intensity of fluorescence (×50), (a) remains unchanged during irradiation. Snapshots of cuvette with PdOEP solution in 2-MTHF excited at 385 nm before (c) and after (d) 2 minutes of irradiation by 102 mW LED at 385 nm. | ||
The quantum yield of triplet state formation of PdOEP at room temperature approaches 100% due to strong spin–orbit coupling induced by palladium. The phosphorescence of PdOEP in solutions at room temperature is typically quenched by molecular oxygen,26 leading to the production of singlet oxygen. Singlet oxygen, an extremely reactive species, reacts with 2-MTFH molecules resulting in the formation of hydroperoxides.20 Assuming the long-term stability of hydroperoxides at room temperature, the concentration of molecular oxygen in solution decreases during irradiation. The absence of molecular oxygen, the primary phosphorescence quencher, significantly increases the quantum yield and decay time of phosphorescence of PdOEP.
The concentration of molecular oxygen (Table 1) remaining in solution after irradiation can be estimated using the Stern–Volmer equation:
| (τ0T/τT) − 1 = τ0Tkq[O2], |
| Irradiation time, s | τ, μs | [O2], mM |
|---|---|---|
| 0 | 0.23 | 1 |
| 30 | 0.36 | 0.65 |
| 60 | 0.68 | 0.34 |
| 90 | 6 | 0.039 |
| 120 | 141 | 0.00165 |
| 150 | 242 | 0.00096 |
Using the triplet lifetime data obtained for aerated and deoxygenated solutions (Table 1) and assuming 10−3 M concentration of molecular oxygen in 2-MTHF at room temperature (it is 2.11 mM for THF27), leads to kq = 4.3 × 109 s−1 M−1. One can then estimate the upper limit for the concentration of molecular oxygen in 2-MTHF solution after irradiation as 10−6 M or less, at least three orders of magnitude lower than oxygen concentration at atmospheric pressure.
Triplet–triplet absorption
Simultaneously with the stationary luminescence measurements, time profile of the triplet–triplet absorption of PdOEP was measured (Fig. 2) for the same sample. PdOEP was excited at 385 nm by 5 ns, 0.1 mW laser pulses and was probed using a continuous light source at the maximum of triplet–triplet absorption band (420 nm, ref. 25) At the end of irradiation, the triplet lifetime of PdOEP increased more than one thousand times (Table 1). The increase of triplet state lifetime of PdOEP during systematic irradiation proves the decrease of molecular oxygen concentration in the investigated solution.Fluorescence upconversion
The fluorescence upconversion in solutions is a process based on energy transfer from an excited donor chromophore to an acceptor chromophore, followed by triplet–triplet annihilation (TTA) of two excited state acceptors. One acceptor molecule returns to the ground state, while the other one reaches the first excited singlet state and is able to emit a photon of fluorescence. The beauty of this method is the appearance of fluorescence of the acceptor at a higher energy than the excitation energy of the donor. The fluorescence upconversion donor/acceptor pairs as well as limitations of triplet–triplet annihilation based processes and their applications have been widely described in the literature.1 The main limitation of the TTA process is quenching by molecular oxygen, which is common to all triplet-based reactions in solutions.289,10-Diphenylanthracene (DPA) and perylene, depicted in Fig. 3, are well-known energy acceptor compounds, suitable to be used in fluorescence upconversion processes with PdOEP acting as an energy donor.1,2
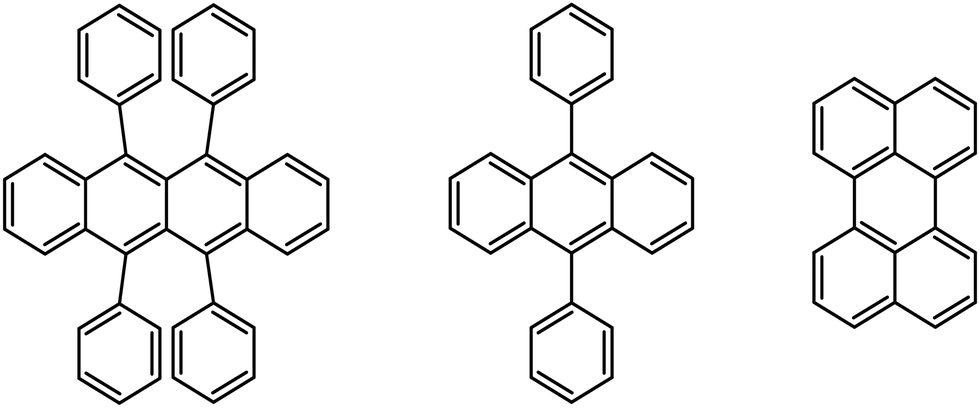 | ||
| Fig. 3 From left to right: rubrene, 9,10-diphenylanthracene, and perylene. | ||
Two fluorescence upconversion donor/acceptor pairs were prepared in 2-MTHF under atmospheric conditions. In both cases PdOEP was used as an energy donor chromophore, with a concentration of 2.4 × 10−5 M. Perylene and DPA were used as energy acceptors with concentrations of 6.4 × 10−4 and 1.9 × 10−4 M, respectively. Donor/acceptor pairs concentrations align with ranges recommended in the literature.1 When the freshly prepared samples were excited into the first absorption band of PdOEP at 545 nm by weak laser light (energy ∼ 0.1 mJ, 10 Hz) used as a probe, no upconverted fluorescence signals were observed (Fig. 4). After 10 minutes of irradiation at 385 nm with 102 mW LED, an upconverted fluorescence appeared in both cases for perylene and DPA acceptors. The emission with the maximum at 21053 cm−1 (475 nm) corresponds to the fluorescence of perylene. The upconverted fluorescence of DPA is located at 23095 cm−1 (433 nm).
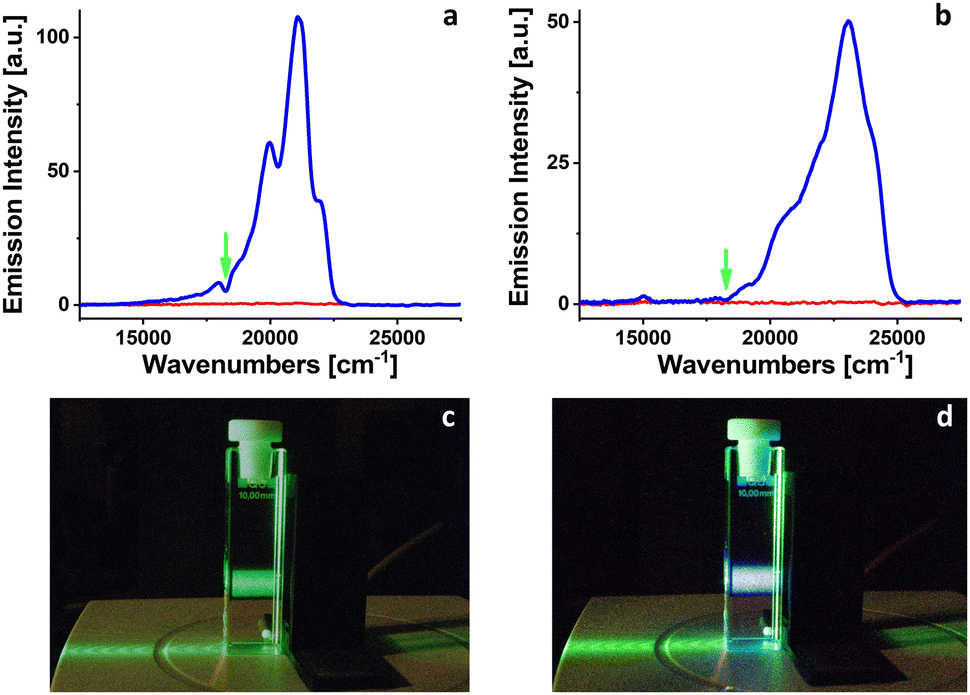 | ||
| Fig. 4 The appearance of fluorescence upconversion from PdOEP/perylene pair (a) and from PdOEP/DPA pair (b) excited at 545 nm (green arrow) in 2-MTHF. Red line: the signal before irradiation. Blue line: the signal after 10 min of irradiation at 385 nm. Snapshots of a cuvette with PdOEP/DPA mixture in 2-MTHF excited at 520 nm before (c) and after (d) irradiation at 385 nm for 2 minutes. | ||
The appearance of fluorescence upconversion signals for PdOEP/perylene or PdOEP/DPA pairs in 2-MTHF after 10 minutes of irradiation at 385 nm confirmed the decrease of molecular oxygen concentration (due to the formation of 2-MTHF hydroperoxides) below the limit that precludes the observation of TTA processes.
Deoxygenation of different solvents
To propose the protocol for light-driven deoxygenation in various organic solvents, 2-MTHF, THF and mixtures of 2-MTHF with ACN and toluene in different ratios were tested. Fresh solutions with PdOEP concentrations of 2.4 × 10−5 M were prepared for each solvent and six mixtures. Each sample was irradiated at the same configuration, using a 102 mW LED emitting at 385 nm with continuous stirring during irradiation. The optical density (OD) of solutions exceeded 3 at the irradiation wavelength, so that 99.9% of photons were absorbed. The intensity of the phosphorescence of PdOEP was monitored throughout irradiation and the experiment was halted upon reaching the maximum (plateau) of phosphorescence of PdOEP. The duration of irradiation varied, ranging from several minutes for 2-MTHF to nearly 6 hours for 2-MTHF: toluene mixture (see Table 2).| t irrad, s | τ, μs | [1O2], mM | Φ | k, s−1 | |
|---|---|---|---|---|---|
| a Assumed value for THF, in two-component solvents the oxygen concentration for ACN27 or toluene29 was used. b Ref. 27. c Ref. 29. t irrad – time of irradiation, τ – singlet oxygen lifetime, Φ – quantum yield of photoreaction, k – rate constant of photoreaction. | |||||
| 2-MTHF | 195 | 15 | 2.1a | 1.0 × 10−1 | 6684 |
| THF | 7590 | 23 | 2.1b | 2.6 × 10−3 | 112 |
| 2-MTHF:ACNb |
|||||
| 1:1 |
660 | 23 | 2.4 | 34 × 10−3 | 1472 |
| 1:2 |
2460 | 31 | 2.4 | 9.1 × 10−3 | 293 |
| 1:4 |
12600 |
48 | 2.4 | 1.8 × 10−3 | 37 |
| 2-MTHF:TOLc |
|||||
| 1:1 |
570 | 25 | 2.5 | 35 × 10−3 | 1633 |
| 1:2 |
2520 | 27 | 2.5 | 8.0 × 10−3 | 342 |
| 1:4 |
21600 |
28 | 2.5 | 1.1 × 10−3 | 38 |
The quantum yield of molecular oxygen consumption was determined assuming 100% absorption of irradiating photons and 100% quantum yield of singlet oxygen formation (the value of 93% has been reported ref. 25). To assess the rate constant of molecular oxygen consumption during irradiation, we measured lifetimes of singlet oxygen for the solutions under investigation. The lifetimes of singlet oxygen measured for pure ACN and toluene, 82 and 31 μs, respectively, agree with the literature data.30 Utilizing literature data on molecular oxygen concentration in different solvents and the obtained lifetimes of singlet oxygen in two solvents and six mixtures, we calculated the rate constant (k = Φ/τ) for the reaction of molecular oxygen consumption in eight cases, as presented in Table 2.
The oxygen consumption in 2-MTHF is much faster than in THF, in agreement with the previous investigations of polycyclic aromatic compounds which reported that oxygen is preferentially attached to the carbon substituted with the methyl group.31 The latter acts as an electron donor. Interestingly, similar deoxygenation rates were observed for mixtures of 2-MTHF with nonpolar toluene and polar acetonitrile. Further studies are required to determine the reaction mechanisms in one and two-component solvents.
Testing the procedure: determination of triplet state energy
To evaluate the proposed protocol, we decided to use a derivative of porphycene – PdOEPc (Fig. 1). The triplet state location in PdOEPc was previously unknown, but considering that the triplet of unsubstituted porphycene is located at 10136 cm−1 (987 nm),32 we expected the triplet state of PdOEPc to lie near 10000 cm−1. To estimate the energy of PdOEPc triplet state, three fluorescence upconversion donor/acceptor pairs were prepared in 2-MTHF. PdOEPc at a concentration close to 3.0 × 10−5 M was used as an energy donor. DPA, perylene, and rubrene (Fig. 3) with triplet states located at 14305 cm−1 (699 nm),33 12620 cm−1 (792 nm)34 and 9195 cm−1 (1088 nm),35 respectively, were used as energy acceptors. The fluorescence upconversion was tested for both fresh solutions and solutions irradiated from 1 to 5 minutes by 102 mW LED light at 385 nm. The appearance of a fluorescence upconversion signal only in the irradiated solution with rubrene (c = 1.5 × 10−4 M) used as the energy acceptor (Fig. 5) suggests that the lowest triplet state of PdOEPc is located in the region between the triplets of perylene and rubrene (12620–9195 cm−1).
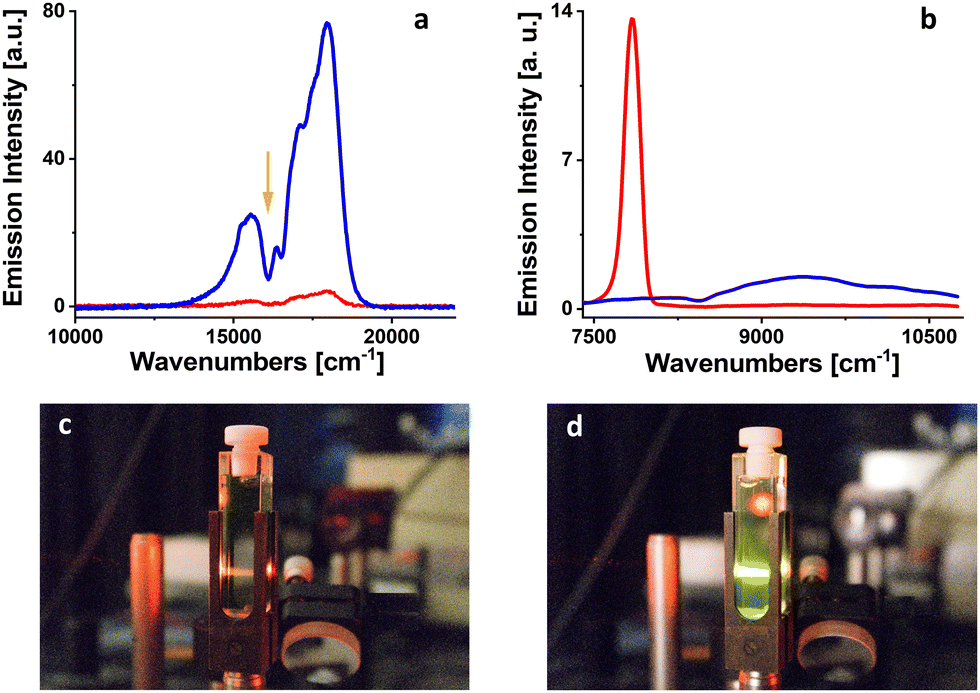 | ||
| Fig. 5 The appearance of fluorescence upconversion from PdOEPc/rubrene pair excited at 620 nm (orange arrow) in 2-MTHF (a). Red line: the signal before irradiation; blue line: the signal after 1 min of irradiation at 385 nm. The appearance of phosphorescence of PdOEPc in 2-MTHF at room temperature (b). Red line: the signal before irradiation; blue line: the signal after 5 minutes of irradiation at 385 nm. Snapshots of a cuvette with PdOEP/rubrene mixture in 2-MTHF excited at 620 nm before (c) and after (d) irradiation at 385 nm for 1 minute. | ||
In the next step, we tried to measure the phosphorescence of PdOEPc in 2-MTHF at room temperature for both, freshly prepared and irradiated solutions in the near-infrared region. For the fresh solution, the signal was dominated by singlet oxygen phosphorescence located at 7849 cm−1 (1274 nm, Fig. 5). A weak but detectable signal of PdOEPc phosphorescence appeared with the maximum at 9242 cm−1 (1082 nm) for the solution irradiated at 385 nm, 102 mW LED, for 5 minutes (Fig. 5). At the same time, phosphorescence of singlet oxygen disappeared, indicating efficient removal of oxygen from the sample. One should add that a successful observation of upconversion in rubrene, which itself is known to readily form peroxides,18 demonstrates that singlet oxygen scavenging by the solvent is more efficient than photooxidation of rubrene.
Comparison with other deoxygenation procedures
The present method represents a chemical tool to remove oxygen from solution. It can be considered an extension of previously applied protocols that used specific oxygen scavengers, such as dimethylthiomethane15 or limonene.16 Avoiding the need of introducing a scavenger into solution is an obvious advantage. However, the disadvantage of all “chemical” approaches is that the oxygenation products may affect the photophysics of studied systems. A “physical” freeze and thaw methodology avoids this problem. It is, however, much more time-consuming and requires specific equipment.The possibility of physical quenching of excited states by peroxides can be excluded due to their absorption at higher energy. To check the chemical activity of peroxides, PdOEP in photodeoxygenated 2-MTHF was stored in the dark for four days. According to the absorption measurements PdOEP decomposed after that period by approximately 5%.
Our methodology requires the use of a sensitizer to generate singlet oxygen. In many cases (as in the present work), this role is played by the molecule under investigation.
An important parameter when comparing the “chemical” and “physical” approaches is the concentration of the oxygen remaining in solution. For the systems described in this work, practically the same triplet lifetimes (of the order of several hundred microseconds) were obtained using both techniques. In addition, we did not observe any significant spectral or photophysical changes while using the solvent as an oxygen scavenger.
We conclude that, in the case when the formation of hydroperoxide is not a problem, the presently proposed approach can be considered a method of choice for oxygen removal.
Conclusions
A novel method for efficient deoxygenation of liquid solutions at room temperature has been proposed. It is based on the photoinduced generation of singlet oxygen, which then reacts with the solvent. 2-MTHF emerges as the best medium for this procedure, but other solvents (THF) can also be used, as well as mixtures of 2-MTHF with both polar and non-polar liquids.The simplicity of the protocol makes it highly attractive for experiments that require the presence of a considerable population of sufficiently long-lived triplet states. If this requirement is fulfilled, one gains access to processes that are normally not observed, due to quenching of the excited state by oxygen. These include room temperature phosphorescence, triplet–triplet annihilation, thermally activated delayed fluorescence, or photodegradation of excited molecules by factors not related to the presence of oxygen. With regard to the latter, a note of caution seems relevant. Lengthening of the triplet lifetime may open up photodegradation channels that are normally not active because of quenching by oxygen. In such cases, the loss of a beneficial role of oxygen as a quencher may lead to much higher photobleaching yields. In fact, our initial studies of PdOEP show that its photostability decreases by two orders of magnitude in deoxygenated solutions. This result indicates that, in order to fully exploit the merits of the deoxygenation protocol, it is important to select an appropriate photosensitizer for each particular kind of study.
Author contributions
Aleksander Gorski did literature research and wrote the original draft. Jacek Waluk reviewed and edited the manuscript. Barbara Golec and Natalia Dutkiewicz provided part of the research data. Jakub Ostapko did synthesis of PdOEP and PdOEPc.Data availability
Data for this article are available at RepOD at https://doi.org/10.18150/QSSZ1K.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the Polish National Science Centre, grant numbers 2020/39/B/ST4/01956 and 2019/35/B/ST4/00297.Notes and references
- J. Zhou, Q. Liu, W. Feng, Y. Sun and F. Y. Li, Chem. Rev., 2015, 115, 395–465 CrossRef CAS PubMed.
- V. Gray, D. Dzebo, A. Lundin, J. Alborzpour, M. Abrahamsson, B. Albinsson and K. Moth-Poulsen, J. Mater. Chem. C, 2015, 3, 11111–11121 RSC.
- Y. C. Liu, C. S. Li, Z. J. Ren, S. K. Yan and M. R. Bryce, Nat. Rev. Mater., 2018, 3, 18020 CrossRef CAS.
- H. Yersin, Top. Curr. Chem., 2004, 241, 1–26 CrossRef CAS.
- B. Habermeyer and R. Guilard, Photochem. Photobiol. Sci., 2018, 17, 1675–1690 CrossRef CAS PubMed.
- N. Masiera, J. Ostapko, A. Gorski, A. Bojarska, I. Gawryszewska, E. Sadowy, W. Hryniewicz and J. Waluk, Eur. J. Med. Chem., 2020, 200, 112472 CrossRef CAS PubMed.
- M. C. DeRosa and R. J. Crutchley, Coord. Chem. Rev., 2002, 233, 351–371 CrossRef.
- B. Golec, A. Gorski, R. P. Thummel, M. Sierakowski and J. Waluk, Photochem. Photobiol. Sci., 2023, 22, 333–344 CrossRef CAS PubMed.
- B. Golec, J. Buczynska, K. Nawara, A. Gorski and J. Waluk, Photochem. Photobiol. Sci., 2023, 22, 2725–2734 CrossRef CAS PubMed.
- O. L. Gijzeman, F. Kaufman and G. Porter, J. Chem. Soc., Faraday Trans. 2, 1973, 69, 708–720 RSC.
- R. R. Islangulov, J. Lott, C. Weder and F. N. Castellano, J. Am. Chem. Soc., 2007, 129, 12652–12653 CrossRef CAS PubMed.
- L. J. C. Love, M. Skrilec and J. G. Habarta, Anal. Chem., 1980, 52, 754–759 CrossRef CAS.
- A. S. Carretero, C. C. Blanco, B. C. Díaz and A. F. Gutiérrez, Anal. Chim. Acta, 1998, 361, 217–222 CrossRef CAS.
- S. H. C. Askes and S. Bonnet, Nat. Rev. Chem., 2018, 2, 437–452 CrossRef.
- D. Dzebo, K. Moth-Poulsen and B. Albinsson, Photochem. Photobiol. Sci., 2017, 16, 1327–1334 CrossRef CAS PubMed.
- S. G. Wan, D. X. Wang, M. Q. Cai, Y. Z. Shi, Y. S. Zhang, S. R. Chen, C. Q. Ye and Y. L. Song, Chem. Commun., 2023, 59, 13895–13898 RSC.
- M. A. Filatov and M. O. Senge, Mol Syst. Des. Eng., 2016, 1, 258–272 RSC.
- C. Dufraisse, C. Moureu and P. M. Dean, C. R. Hebd. Seances Acad. Sci., 1926, 182, 1440 Search PubMed.
- C. S. Foote and S. Wexler, J. Am. Chem. Soc., 1964, 86, 3879–3880 CrossRef CAS.
- G. I. Nikishin, V. G. Glukhovtsev, M. A. Peikova and A. V. Ignatenko, Acad. Sci. USSR, Div. Chem. Sci., 1971, 2323–2325 CAS.
- N. Barboy and J. Feitelson, Anal. Biochem., 1989, 180, 384–386 CrossRef CAS PubMed.
- J. Feitelson and D. Mauzerall, J. Phys. Chem., 1982, 86, 1623–1628 CrossRef CAS.
- A. D. Adler, F. R. Longo, F. Kampas and J. Kim, J. Inorg. Nucl. Chem., 1970, 32, 2443 CrossRef CAS.
- A. Rana and P. K. Panda, Org. Lett., 2014, 16, 78–81 CrossRef CAS PubMed.
- A. Gorski, V. Knyukshto, E. Zenkevich, A. Starukhin, M. Kijak, J. Solarski, A. Semeikin and T. Lyubimova, J. Photochem. Photobiol., A, 2018, 354, 101–111 CrossRef CAS.
- A. Gorski, M. Kijak, E. Zenkevich, V. Knyukshto, A. Starukhin, A. Semeikin, T. Lyubimova, T. Rolinski and J. Waluk, J. Phys. Chem. A, 2020, 124, 8144–8158 CrossRef CAS PubMed.
- M. Quaranta, M. Murkovic and I. Klimant, Analyst, 2013, 138, 6243–6245 RSC.
- Y. Y. Cheng, T. Khoury, R. G. C. R. Clady, M. J. Y. Tayebjee, N. J. Ekins-Daukes, M. J. Crossley and T. W. Schmidt, Phys. Chem. Chem. Phys., 2010, 12, 66–71 RSC.
- A. R. Li, S. W. Tang, P. H. Tan, C. J. Liu and B. Liang, J. Chem. Eng. Data, 2007, 52, 2339–2344 CrossRef CAS.
- M. Bregnhoj, M. Westberg, F. Jensen and P. R. Ogilby, Phys. Chem. Chem. Phys., 2016, 18, 22946–22961 RSC.
- N. J. Turro, M. F. Chow and J. Rigaudy, J. Am. Chem. Soc., 1981, 103, 7218–7224 CrossRef CAS.
- B. Golec, A. Gorski and J. Waluk, Photochem, 2022, 2, 217–224 CrossRef CAS.
- J. S. Brinen and J. G. Koren, Chem. Phys. Lett., 1968, 2, 671–672 CrossRef CAS.
- C. A. Parker, Photoluminescence of Solutions, Elsevier Publishing Co., Amsterdam-London-New York, 1968, p. 544 Search PubMed.
- A. P. Damranyan and V. A. Kuz'min, Dokl. Phys. Chem., 1981, 260, 938–941 Search PubMed.
| This journal is © the Owner Societies 2024 |