
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The impact of side-chain fluorination on proton-bound phenylalanine dimers: a cryogenic infrared spectroscopic study†
Marc
Safferthal‡
 ab,
Kim
Greis‡
abc,
Rayoon
Chang‡
ab,
Chun-Wei
Chang
ab,
Waldemar
Hoffmann
ab,
Gerard
Meijer
b,
Gert
von Helden
b and
Kevin
Pagel
*ab
ab,
Kim
Greis‡
abc,
Rayoon
Chang‡
ab,
Chun-Wei
Chang
ab,
Waldemar
Hoffmann
ab,
Gerard
Meijer
b,
Gert
von Helden
b and
Kevin
Pagel
*ab
aDepartment of Biology, Chemistry, Pharmacy, Freie Universität Berlin, Altensteinstraße 23a, 14195 Berlin, Germany. E-mail: kevin.pagel@fu-berlin.de
bFritz Haber Institute of the Max Planck Society, Faradayweg 4-6, 14195 Berlin, Germany
cDepartment of Chemistry and Applied Biosciences, ETH Zürich, Vladimir-Prelog-Weg 10, 8093, Zürich, Switzerland
First published on 5th November 2024
Abstract
The incorporation of fluorine into amino acids is an important strategy to produce tailored building blocks with unique properties for peptide-based materials. Phenylalanine is frequently modified due to its role in cation–π interactions and the formation of amyloid fibres. Previous studies have utilized gas-phase vibrational spectroscopy to study interactions between canonical amino acids. In this study, we employ a combination of cryogenic gas-phase infrared spectroscopy and density functional theory to study the interactions in proton-bound dimers of side-chain fluorinated phenylalanines. Our findings reveal how the position and number of fluorine atoms affect the interactions and structures of the dimers. Monofluorinated phenylalanines adopt charge-solvated structures in which the two amino acids interact via their ammonium and amine functions (NH3+⋯NH2). The dimer with the perfluorinated side chain forms multiple charge-solvated and salt-bridged structures with varying interaction types. These structural changes are attributed to the significant reduction of electron density in the aromatic systems.
Introduction
The introduction of fluorine into biomolecules such as carbohydrates,1 nucleic acids,2 lipids,3 and amino acids4 has become a powerful strategy for tuning and improving their physical properties. Fluorinated amino acids demonstrate altered hydrophobicities and helix propensities5 compared to their natural analogs. The incorporation of fluorinated amino acids into peptides and proteins can influence protein–protein interactions4 and their stability towards proteases.6 Fluorinated analogs of phenylalanine are particularly interesting as phenylalanine is known to be one of the main driving forces in the formation of amyloids7 and involved in cation–π interactions in peptides and proteins.8Past studies have explored proton-bound amino acid dimers in the gas phase using a combination of infrared (IR) action spectroscopy and first-principle theoretical calculations in order to gain information on the structures and interaction characteristics of specific amino acids.9–24 These models are generally chosen because they contain all the relevant functional groups involved in the interactions while being relatively small in size. Homodimers, for instance, show that the interaction type is strongly correlated with the proton affinity (PA) of the amino acid.19 Amino acids with lower PAs tend to bind via salt-bridge interactions, whereas amino acids with higher PAs are more likely to adopt charge-solvated structures. In 2017, we elucidated the structure of the proton-bound phenylalanine dimer (Phe2H+) using infrared multiple photon dissociation (IRMPD) spectroscopy and density functional theory (DFT).23 The experiments revealed that Phe2H+ adopts a “sandwich-like” structure where the phenyl rings align antiparallel, and the amino acids are bound via the ammonium and amine groups of the two amino acids (NH3+⋯NH2). Cryogenic gas-phase IR spectroscopy yields generally more resolved spectra that can be used to deduce the structures of biopolymers like RNA,25 glycans,26,27 and glycolipids.28 Using cryogenic gas-phase IR spectroscopy in helium nanodroplets, we recently showed that side-chain fluorination in protonated phenylalanines can lead to the formation of short NH+⋯F hydrogen bonds, significantly impacting their structure.29
This study aims to systematically explore proton-bound dimers of side-chain fluorinated phenylalanines using cryogenic IR spectroscopy in helium droplets and DFT. It focuses on how the position and number of fluorine atoms influence the structure and interaction type of these dimers.
Experimental
Materials
2-Fluoro-L-phenylalanine, 3-fluoro-L-phenylalanine, 4-fluoro-L-phenylalanine and pentafluoro-L-phenylalanine were obtained from Tokyo Chemical Industry (purity > 95%). All samples were dissolved in a mixture of acetonitrile and water (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to yield 100 μM solutions for cryogenic infrared spectroscopy experiments and in water to yield 5 mM solutions for the IRMPD experiments. The samples were ionized by nanoelectrospray ionization (nESI) using Pd/Pt coated glass capillaries (Sputter Coater HR 208, Cressington) that were pulled to a tip with an inner diameter of 1–2 μm using a micropipette puller (Model P-1000, Sutter Instrument). Needle voltages ranging from 0.8 to 1.1 kV were applied in the positive ion mode.
:1) to yield 100 μM solutions for cryogenic infrared spectroscopy experiments and in water to yield 5 mM solutions for the IRMPD experiments. The samples were ionized by nanoelectrospray ionization (nESI) using Pd/Pt coated glass capillaries (Sputter Coater HR 208, Cressington) that were pulled to a tip with an inner diameter of 1–2 μm using a micropipette puller (Model P-1000, Sutter Instrument). Needle voltages ranging from 0.8 to 1.1 kV were applied in the positive ion mode.
Instrumental setups
Cryogenic gas-phase IR spectroscopy in superfluid helium droplets was performed using a custom-built instrument, which has been described in detail previously.30 Here, the analytes are ionized using nESI and selected according to their mass-to-charge ratio (m/z) by a quadrupole mass filter. The ions of interest are accumulated and stored in a cooled (ca. 90 K) hexapole ion trap. The analytes are picked up and cooled to 0.4 K by superfluid helium droplets generated using an Even–Lavie valve.31 The helium droplets guide the ions to the detection region, where they interact with incoming IR light that is generated by the free-electron laser of the Fritz Haber Institute (FHI FEL).32 The sequential absorption of resonant photons causes helium atoms to evaporate from the droplet, eventually leading to the bare ion, which is subsequently detected in a time-of-flight mass analyzer. Plotting the ion signal against the tunable wavenumber of the laser yields well-resolved IR spectra. The IR spectra were recorded in the mid-IR region between 1050 and 1800 cm−1.IRMPD spectroscopy was conducted on a custom-built instrument, previously described in detail.33 The ions of interest are generated using a nESI source and accumulated in an entrance funnel. The analytes are pulsed into a linear drift tube, where they collide with helium buffer gas and are separated according to their mass, charge, size and shape. Electrostatic gating with an einzel lens is used to select ions within a certain drift time window. The ions are radially confined and guided to a quadrupole mass filter that allows the selection of ions by their mass-to-charge ratio. In the following, the drift time- and m/z-selected ions are irradiated by IR photons generated by the FHI FEL. Fragment ions are detected by a time-of-flight mass analyzer. Plotting the photofragmentation yield against the tunable wavenumber of the laser yields IRMPD spectra.
Computational methods
The conformational space of the fluorinated phenylalanine dimers was explored using CREST (version 2.11.1)34 with the semiempirical method GFN2-xTB35 as implemented in the xtb package (version 6.4.0) and default settings. Geometry optimization and harmonic frequency calculations of the 20 most stable structures according to GFN2-xTB were performed at the PBE0 + D3BJ/6-311+G(d,p)36–38 level of theory in Gaussian 16 (Revision A.03)39 (see ESI†). This level of theory previously provided reliable results for fluorinated phenylalanine monomers.29 Including dispersion interactions is generally recommended and vital for accurately predicting geometries and energetics of systems with non-covalent interactions.40 All frequencies were scaled by an empirical factor of 0.966 and convoluted using Gaussian-shaped functions with a width of 0.4% of the wavenumber.29 Relative free energies (ΔF) of the conformers were calculated for the ion trap temperature of 90 K and the indicated energies are relative to the lowest-energy conformer. Relative zero-point corrected total energies (ΔE) can be found in the ESI.† Reduced density gradient (RDG) isosurface maps and scatter plots for non-covalent interaction (NCI) analysis were generated and visualized using Multiwfn (version 3.8)41,42 and VMD (version 1.9.3).43Results and discussion
The three monofluorinated positional isomers ortho-, meta-, and para-fluorophenylalanine (oF-Phe, mF-Phe, and pF-Phe), as well as pentafluorophenylalanine (F5-Phe), were selected for this study (Fig. 1). These model systems were selected to compare different degrees of fluorination and varying positions of fluorine atoms on the structures of proton-bound amino acid homodimers. The proton-bound dimers were generated, and their IR signatures were recorded using room-temperature IRMPD spectroscopy and cryogenic (cryo) IR spectroscopy in superfluid helium droplets for comparison. Fig. 2 shows the IRMPD and cryo IR spectra of the proton-bound ortho-fluorophenylalanine homodimer (oF-Phe)2H+. The IRMPD spectrum consists of seven broad features between 1050 and 1800 cm−1. In the cryo IR spectrum, it is apparent that the broad features are composed of multiple absorption bands. The peak broadening and shifting observed in the IRMPD spectrum result from the combined effects of multiple photon excitation and dissociation dynamics.44 Especially the functional group region (1400–1800 cm−1), crucial for structural assignment, is well-resolved and less congested in the cryo IR spectrum. This clearly underscores the higher resolution of cryo IR spectroscopy, which simplifies the assignment of the investigated structures. All further discussions are therefore based on cryo IR spectra, with the corresponding IRMPD spectra provided in the ESI.† | ||
| Fig. 1 Structures of the selected fluorinated phenylalanines. Fluorine atoms are highlighted in green. | ||
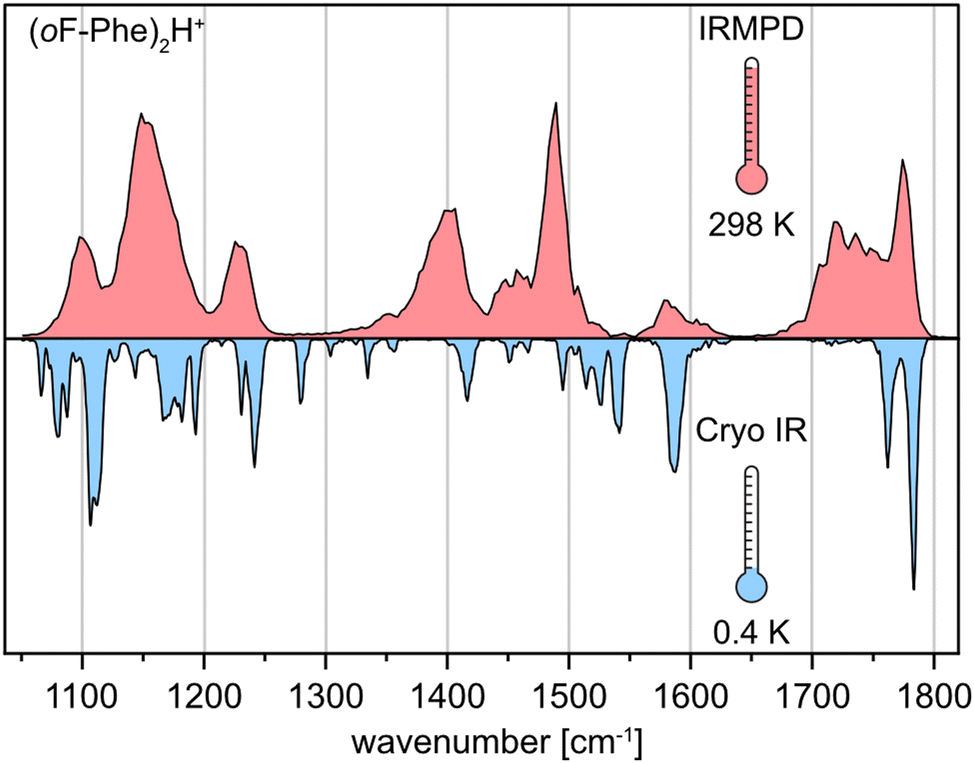 | ||
| Fig. 2 Comparison of experimental IR spectra of the protonated ortho-fluorophenylalanine dimer (oF-Phe)2H+ generated by room-temperature IRMPD spectroscopy (red) and cryogenic IR spectroscopy in helium droplets (0.4 K, blue). | ||
The IR signatures of all three monofluorinated phenylalanine dimers along with computed spectra of selected structures are shown in Fig. 3a. In amino acid dimers, three types of interaction are generally conceivable: type A and type B, in which one residue is neutral and the other one singly protonated as well as type Z in which a protonated and a zwitterionic amino acid are involved (Fig. 3b).23 For each interaction type and amino acid, computed spectra of the lowest-energy conformer were calculated (Fig. 3a). Type A dimers are bound via the terminal ammonium and amine functions of the amino acids (NH3+⋯NH2). Characteristic in their vibrational spectra are two adjacent carbonyl stretching bands between 1750 and 1800 cm−1. In type B, the ammonium function of the first amino acid interacts with the carbonyl moiety of the second amino acid (NH3+⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C). Type B interactions can therefore be assigned in the IR spectrum by a carbonyl stretching band between 1750–1800 cm−1 and a redshifted carbonyl stretching band between 1700 and 1750 cm−1. In structures of the type Z interaction, one zwitterionic amino acid, which carries ammonium and carboxylate functional groups, is bound to the second protonated amino acid. The IR spectra of type Z structures show one carbonyl stretching band between 1750–1800 cm−1 and one redshifted feature between 1650–1700 cm−1 for the antisymmetric carboxylate stretching mode.
C). Type B interactions can therefore be assigned in the IR spectrum by a carbonyl stretching band between 1750–1800 cm−1 and a redshifted carbonyl stretching band between 1700 and 1750 cm−1. In structures of the type Z interaction, one zwitterionic amino acid, which carries ammonium and carboxylate functional groups, is bound to the second protonated amino acid. The IR spectra of type Z structures show one carbonyl stretching band between 1750–1800 cm−1 and one redshifted feature between 1650–1700 cm−1 for the antisymmetric carboxylate stretching mode.
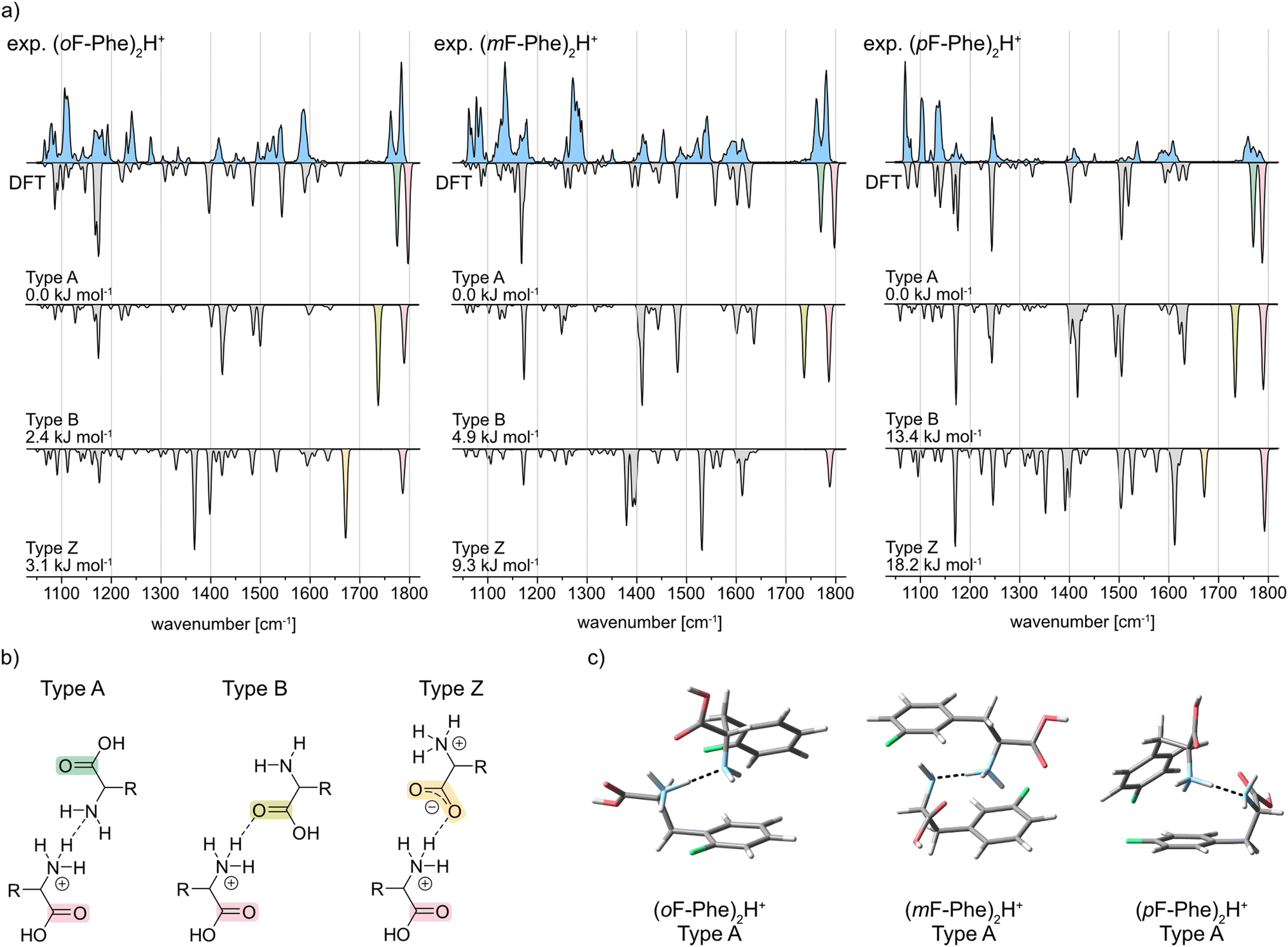 | ||
| Fig. 3 (a) Infrared spectra of the proton-bound homodimers of ortho-, meta-, and para-fluorophenylalanine. Experimental IR spectra are depicted as light blue traces. Computed spectra of the low-energy structures are shown as gray inverted traces with distinct absorption bands highlighted according to the vibration they originate from. Relative free energies at 90 K are indicated. (b) Three common interaction types illustrated as structural formulas. Important carbonyl functional groups are highlighted. (c) Global minimum structures of the proton-bound monofluorinated phenylalanine dimers. | ||
The IR spectrum of (oF-Phe)2H+ clearly reveals two adjacent bands at 1783 and 1762 cm−1 in the carbonyl stretching region. Comparison with the computed spectra of the A, B, and Z type structures between 1400–1800 cm−1 shows that the type A spectrum provides the best match with the experiment. These observations indicate the presence of conformers with type A interactions for (oF-Phe)2H+. The computed spectrum of the lowest-energy type A conformer also matches well with the experimental spectrum in the lower-wavenumber region (1050–1400 cm−1), even though the bands at 1192 and 1280 cm−1 are not reproduced in the computed spectrum. Additional computed structures and spectra of type A (oF-Phe)2H+ structures can be found in the ESI.† The low absorption at 1715 cm−1 indicates a minor presence of type B structures. The type A and B (oF-Phe)2H+ structures are close in free energy, which would also suggest the coexistence of both interaction types. Nevertheless, based on the intensity and position of the carbonyl stretching bands, the experimental spectrum shows that type A is the prevailing interaction in (oF-Phe)2H+.
The experimental IR spectrum of the meta-fluorophenylalanine dimer (mF-Phe)2H+ resembles its ortho-fluorinated counterpart. Two adjacent bands representing the two carbonyl stretching vibrations are observed at 1780 and 1761 cm−1. The computed spectrum of the type A conformer provides the best match with the experimental IR signature between 1400 and 1800 cm−1. The features between 1476 and 1529 cm−1 are not perfectly reproduced in the computed spectrum of type A, which may be due to anharmonic effects.25 Compared to the computed spectrum of the type A conformer, the fingerprint region between 1050 and 1400 cm−1 is slightly more congested in the experiment. Additional computed structures and spectra of type A (mF-Phe)2H+ structures can be found in the ESI.† However, due to the good match of the carbonyl stretching bands, it can be concluded that the type A interaction is dominating in (mF-Phe)2H+.
For the proton-bound para-fluorophenylalanine dimer (pF-Phe)2H+, the experimental IR spectrum is surprisingly different in the carbonyl stretching region. Instead of two adjacent bands, a low intensity feature with three maxima at 1759, 1770, and 1783 cm−1 is observed. This feature cannot be found in any of the computed spectra. However, comparison of the overall IR spectrum with the three computed spectra shows that the type A structure still provides the best match based on the position and intensity of the bands. Additional computed structures and spectra of type A (pF-Phe)2H+ structures can be found in the ESI.† Due to the location of the carbonyl stretching bands above 1750 cm−1 and the good match of experimental and computed spectrum in the fingerprint and functional group region, the type A interaction is the most likely in (pF-Phe)2H+.
Fig. 3c shows the computed lowest-energy type A structures of all three monofluorinated phenylalanine dimers. For the lowest-energy structure of (mF-Phe)2H+, a “sandwich-like” structure similar to that of the proton-bound phenylalanine dimer23 is observed. In contrast, the computed lowest-energy structures of (oF-Phe)2H+ and (pF-Phe)2H+ show geometries in which the phenyl rings are tilted towards each other and not aligned in an antiparallel fashion. NCI analysis (see ESI†) shows that the tilting of the phenyl rings in (oF-Phe)2H+ is stabilized by additional weak C–F⋯H–N and a C–F⋯H–C interactions. Tilting of the phenyl rings in (pF-Phe)2H+ appears to be introduced by additional weak interactions between the phenyl rings.
In contrast, the IR spectrum of the pentafluorophenylalanine dimer (F5-Phe)2H+, is considerably different and presents much more congested features extending from 1400 to 1800 cm−1. The experimental IR signature of (F5-Phe)2H+ and the computed spectra of its A, B, and Z type structures are shown in Fig. 4a. The broad and complex IR feature representing the CO stretching vibrations extends from 1675 to 1795 cm−1. Local maxima within this feature are observed at 1681 cm−1, 1729 cm−1, 1757 cm−1, and 1782 cm−1. The vibrational band at 1782 cm−1 is reproduced in all computed spectra and is therefore not diagnostic for a specific interaction type. However, the local maxima at 1681 cm−1, 1729 cm−1, and 1757 cm−1 are strong indicators for the coexistence of conformers with A, B, and Z type interactions. Next to the local maxima, multiple overlapping bands with low intensity are found, which indicates the presence of multiple conformers with similar structure. In addition, several overlapping bands are found in the region between 1050–1205 cm−1, which cannot be explained by the IR spectrum of a single computed structure. Also geometrically, the low-energy structures of (F5-Phe)2H+ are different compared to the monofluorinated variants (Fig. 4b). Especially the phenyl ring appears less ordered and, when in close proximity, forms a weak interaction between the electron-deficient ring and the electron rich fluorine of the second side chain. This weak interaction is further supported by NCI analysis in the ESI,† which visualizes the weak interactions between the fluorine atoms and the rings as green surfaces.
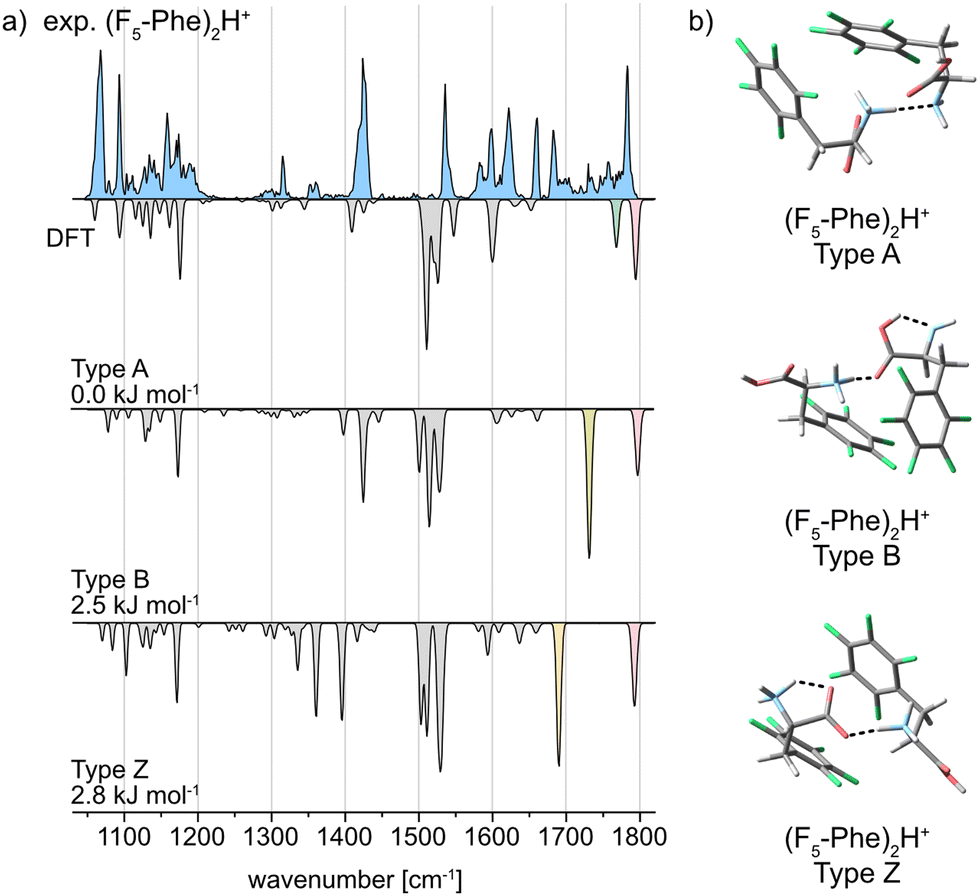 | ||
| Fig. 4 (a) Infrared spectra of the proton-bound homodimer of pentafluorophenylalanine. The experimental IR spectrum is depicted as a light blue trace. Computed spectra of the low-energy structures are shown as gray inverted traces with distinct absorption bands highlighted according to the vibration they originate from. Relative free energies at 90 K are indicated. (b) Global minimum structures of the A, B, and Z type interactions for (F5-Phe)2H+. | ||
A comparison of the interaction types in the monofluorinated phenylalanine dimers with the pentafluorinated variant highlights the strong impact of fluorine atoms on the electron distribution in the underlying amino acids. A drastically altered electron density as in F5-Phe leads to a destabilization of the preferred charge-solvated type A interaction and allows the formation of structures with NH3+⋯CO (type B) and salt-bridge (type Z) interactions.
Conclusions
This study investigated the structures of proton-bound side-chain fluorinated phenylalanine homodimers using IRMPD spectroscopy, cryogenic IR spectroscopy and DFT calculations. The results demonstrate that the position and number of fluorine atoms strongly influence the gas phase structures of the dimers. Monofluorinated phenylalanine dimers predominantly adopt gas-phase structures involving NH3+⋯NH2 interactions (type A), while pentafluorinated variants exhibit a combination of structures with NH3+⋯C = O (type B) and salt bridge interactions (type Z), highlighting the role of fluorine in modulating electron distribution and interaction stability.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. S., R. C., and K. P. thank the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) for support under project number 387284271-SFB 1349. K. G. thanks the Fonds National de la Recherche (FNR) Luxembourg, for funding the project GlycoCat (13549747). Funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – 434130070.References
- T. Oberbillig, C. Mersch, S. Wagner and A. Hoffmann-Röder, Chem. Commun., 2012, 48, 1487–1489 RSC.
- M. Egli, Acc. Chem. Res., 2012, 45, 1237–1246 CrossRef CAS.
- M.-C. Gagnon, M. Auger and J.-F. Paquin, Org. Biomol. Chem., 2018, 16, 4925–4941 RSC.
- M. Salwiczek, E. K. Nyakatura, U. I. M. Gerling, S. Ye and B. Koksch, Chem. Soc. Rev., 2012, 41, 2135–2171 RSC.
- U. I. M. Gerling, M. Salwiczek, C. D. Cadicamo, H. Erdbrink, C. Czekelius, S. L. Grage, P. Wadhwani, A. S. Ulrich, M. Behrends, G. Haufe and B. Koksch, Chem. Sci., 2014, 5, 819–830 RSC.
- S. Huhmann and B. Koksch, Eur. J. Org. Chem., 2018, 3667–3679 CrossRef CAS.
- S. Chowdhary, J. Moschner, D. J. Mikolajczak, M. Becker, A. F. Thünemann, C. Kästner, D. Klemczak, A.-K. Stegemann, C. Böttcher, P. Metrangolo, R. R. Netz and B. Koksch, ChemBioChem, 2020, 21, 3544–3554 CrossRef CAS PubMed.
- S. Tantry, F.-X. Ding, M. Dumont, J. M. Becker and F. Naider, Biochemistry, 2010, 49, 5007–5015 CrossRef CAS.
- H.-B. Oh, C. Lin, H. Y. Hwang, H. Zhai, K. Breuker, V. Zabrouskov, B. K. Carpenter and F. W. McLafferty, J. Am. Chem. Soc., 2005, 127, 4076–4083 CrossRef CAS.
- X. Kong, I.-A. Tsai, S. Sabu, C.-C. Han, Y. T. Lee, H.-C. Chang, S.-Y. Tu, A. H. Kung and C.-C. Wu, Angew. Chem., Int. Ed., 2006, 45, 4130–4134 CrossRef CAS.
- R. Wu and T. B. McMahon, J. Am. Chem. Soc., 2007, 129, 4864–4865 CrossRef PubMed.
- C. G. Atkins, K. Rajabi, E. A. L. Gillis and T. D. Fridgen, J. Phys. Chem. A, 2008, 112, 10220–10225 CrossRef PubMed.
- K. Rajabi and T. D. Fridgen, J. Phys. Chem. A, 2008, 112, 23–30 CrossRef PubMed.
- R. Wu, R. A. Marta, J. K. Martens, K. R. Eldridge and T. B. McMahon, J. Am. Soc. Mass Spectrom., 2011, 22, 1651–1659 Search PubMed.
- U. J. Lorenz and T. R. Rizzo, J. Am. Chem. Soc., 2012, 134, 11053–11055 CrossRef PubMed.
- X. Kong, J. Am. Soc. Mass Spectrom., 2014, 25, 422–426 CrossRef PubMed.
- U. J. Lorenz and T. R. Rizzo, J. Am. Chem. Soc., 2014, 136, 14974–14980 CrossRef PubMed.
- Y. J. Alahmadi, A. Gholami and T. D. Fridgen, Phys. Chem. Chem. Phys., 2014, 16, 26855–26863 RSC.
- H. Yin and X. Kong, J. Am. Soc. Mass Spectrom., 2015, 26, 1455–1461 CrossRef CAS PubMed.
- R. Feng, H. Yin and X. Kong, Rapid Commun. Mass Spectrom., 2016, 30, 24–28 CrossRef CAS PubMed.
- W. Fu, J. Xiong, M. J. Lecours, P. J. J. Carr, R. A. Marta, E. Fillion, T. McMahon, V. Steinmetz and W. S. Hopkins, J. Mol. Spectrosc., 2016, 330, 194–199 CrossRef CAS.
- C. Ieritano, P. J. J. Carr, M. Hasan, M. Burt, R. A. Marta, V. Steinmetz, E. Fillion, T. B. McMahon and W. Scott Hopkins, Phys. Chem. Chem. Phys., 2016, 18, 4704–4710 RSC.
- J. Seo, W. Hoffmann, S. Malerz, S. Warnke, M. T. Bowers, K. Pagel and G. von Helden, Int. J. Mass Spectrom., 2018, 429, 115–120 CrossRef CAS.
- Å. Andersson, M. Poline, M. Kodambattil, O. Rebrov, E. Loire, P. Maître and V. Zhaunerchyk, J. Phys. Chem. A, 2020, 124, 2408–2415 CrossRef.
- K. Greis, C. Kirschbaum, M. I. Taccone, M. Götze, S. Gewinner, W. Schöllkopf, G. Meijer, G. von Helden and K. Pagel, Angew. Chem., Int. Ed., 2022, 61, e202115481 CrossRef CAS PubMed.
- E. Mucha, A. I. González Flórez, M. Marianski, D. A. Thomas, W. Hoffmann, W. B. Struwe, H. S. Hahm, S. Gewinner, W. Schöllkopf, P. H. Seeberger, G. von Helden and K. Pagel, Angew. Chem., Int. Ed., 2017, 56, 11248–11251 CrossRef CAS PubMed.
- M. Grabarics, M. Lettow, C. Kirschbaum, K. Greis, C. Manz and K. Pagel, Chem. Rev., 2022, 122, 7840–7908 CrossRef CAS PubMed.
- C. Kirschbaum, K. Greis, E. Mucha, L. Kain, S. Deng, A. Zappe, S. Gewinner, W. Schöllkopf, G. von Helden, G. Meijer, P. B. Savage, M. Marianski, L. Teyton and K. Pagel, Nat. Commun., 2021, 12, 1201 CrossRef CAS.
- M. Safferthal, K. Greis, R. Chang, C. Kirschbaum, W. Hoffmann, G. Meijer, G. von Helden and K. Pagel, Phys. Chem. Chem. Phys., 2023, 25, 24783–24788 RSC.
- D. A. Thomas, E. Mucha, M. Lettow, G. Meijer, M. Rossi and G. von Helden, J. Am. Chem. Soc., 2019, 141, 5815–5823 CrossRef PubMed.
- U. Even, EPJ Tech. Instrum., 2015, 2, 17 CrossRef.
- W. Schöllkopf, S. Gewinner, H. Junkes, A. Paarmann, G. von Helden, H. Bluem and A. M. Todd, Proc. SPIE-Int. Soc. Opt. Eng., 2015, 9512, 95121L CrossRef.
- S. Warnke, J. Seo, J. Boschmans, F. Sobott, J. H. Scrivens, C. Bleiholder, M. T. Bowers, S. Gewinner, W. Schöllkopf, K. Pagel and G. von Helden, J. Am. Chem. Soc., 2015, 137, 4236–4242 CrossRef PubMed.
- P. Pracht, F. Bohle and S. Grimme, Phys. Chem. Chem. Phys., 2020, 22, 7169–7192 RSC.
- C. Bannwarth, S. Ehlert and S. Grimme, J. Chem. Theory Comput., 2019, 15, 1652–1671 CrossRef.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef PubMed.
- M. J. Frisch, et al., Gaussian 16, Wallingford CT, 2016 Search PubMed.
- M. Bursch, J.-M. Mewes, A. Hansen and S. Grimme, Angew. Chem., Int. Ed., 2022, 61, e202205735 CrossRef PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef PubMed.
- T. Lu, J. Chem. Phys., 2024, 161, 082503 CrossRef PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef PubMed.
- J. Oomens, B. G. Sartakov, G. Meijer and G. von Helden, Int. J. Mass Spectrom., 2006, 254, 1–19 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Mass spectra and IRMPD spectra of all proton-bound dimers; cartesian coordinates of selected geometry-optimized structures; additional computed structures and spectra of type A structures. See DOI: https://doi.org/10.1039/d4cp03823a |
| ‡ These authors contributed equally to this work. |
| This journal is © the Owner Societies 2024 |