
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Light-induced selectivity in an exemplary photodimerization reaction of varied azaanthracenes†
Adam
Mames
 a,
Aleksander
Gorski
a,
Joanna
Jankowska
b,
Tomasz
Ratajczyk
*a and
Mariusz
Pietrzak
*a
a,
Aleksander
Gorski
a,
Joanna
Jankowska
b,
Tomasz
Ratajczyk
*a and
Mariusz
Pietrzak
*a
aInstitute of Physical Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01- 224 Warsaw, Poland. E-mail: tratajczyk@ichf.edu.pl; mpietrzak@ichf.edu.pl
bFaculty of Chemistry, University of Warsaw, Pasteura 1, 02-093 Warsaw, Poland
First published on 29th October 2024
Abstract
Currently, there is intense interest in light-driven chemical reactions, including photocatalytic processes, photopolymerization and photodimerization. The need for regiocontrol in such reactions is obvious, especially in cases where many products can potentially be formed. Here, the photodimerization involving various azaanthracenes is presented for the first time. Specifically, 2-azaanthracene (A) and N-methyl-2-azaanthracene (M) are considered. Photoreactions of A, M and the A + M mixture under two irradiation wavelengths (365 and 420 nm) and in two solvents (methanol, dichloromethane) were carried out. In the case of A, four regiomers were obtained, in contrast to the available literature data, where only two products were reported. The relative ratio of these products is a function of the irradiation wavelength, the solvent used, and the irradiation time. In the case of M, we have identified two main products and a small amount of a third one, again contradicting the literature data. Irradiation of an equimolar A and M mixture at 365 nm led to a mixture of several products, where the yield of the AM dimers was about 40%. Importantly, the change of the irradiation wavelength to 420 nm significantly increased the AM yield (to about 80%). We demonstrated that only two AM dimers were formed (out of a possible four). The products were comprehensively characterized by NMR spectroscopy. We have determined the photophysical parameters of A and M and measured the quantum yield of photodimerization using UV-vis spectroscopy. The quantum-chemical calculations in the excited state allowed us to propose a plausible explanation for why only two AM dimers are formed upon irradiation. The presented results indicated that photodimerization among various molecules can have advantages and, in particular, does not need to give a complex mixture of multiple products. Importantly, it has been observed that the wavelength shift can significantly improve the photoreaction selectivity.
Introduction
Light-induced processes are currently of significant interest in the scientific community, and this interest is growing rapidly.1,2 A few of the most important reasons for this are the photosynthesis of added-value chemical compounds, the utilization of photoreactions in cancer therapy, and the synthesis of photoactive materials.3–7 Many other potential applications of photoreactions can be easily found in the scientific literature. The advantages of photoreactions make them attractive in the field of materials chemistry, where these reactions can be harnessed for the photofabrication of various functional materials.8–10 In many cases, the photofabricated materials are photoactive, which facilitates the production of light-responsive materials. Of course, an integral aspect of photochemistry is molecules that exhibit appropriate photochemical properties which can be utilized for specific purposes; for example, in medicine, material sciences and other applied chemistry areas.A lot of photochemical reactions are referred to as “photo-click” chemistry.11 This concept is used widely in the chemical and biological sciences. The most important advantages of the combination of “click” chemistry with light are its relatively easy applications, typically fast reaction timescales, high selectivity and excellent product yield.12 However, each “photo-click” reaction has its own advantages and disadvantages.
One class of “photo-click” reactions is photoinduced dimerization, which plays a role in industrial and biochemical processes. For example, photodimerization of cytosine and thymine in DNA seems to be linked to skin cancer development.13,14 There are many types of photodimerization reactions.15,16 Regardless of their classifications, these photoreactions have an eye-catching feature due to the fact that they enable, figuratively speaking, a transition from 2D to 3D molecular complexities, and this makes them of central importance for the construction of complex rigid systems.17,18 A good example is the photodimerization of anthracenes, where two “flat” molecules dimerize, and a rigid 3D dimer is yielded. Apart from that, photodimerization also allows for the construction of functional polymeric molecular systems by the simple photoinduced clipping of two or more units.
While photodimerization offers a straightforward vehicle for the construction of a more complex and functional system, there is also a critical element that, in many cases, makes the application mentioned above not straightforward. This includes situations where many isomers can be produced during photoreactions. Depending on the molecule and its symmetry, a variety of isomers are often yielded.
The method of controlling the photoreaction, especially with regard to regioselectivity, is a key aspect.19–22
Complex reactions with many possible products are common but problematic because of smaller yields and problems with purification from unwanted products with very similar structures and properties. In this work, we focus on a method to control the selectivity of photoreactions with light and to characterize and explain the natural regioselectivity observed in some photocycloaddition reactions.
There are many examples of photoactive molecules and materials that can be constructed via their photoactivity. Examples of photoactive molecules in this field include acenes, and among them, anthracenes and azaanthracenes.23–25 Anthracenes display many photoreactive facets, which are being utilized in materials science, organic synthesis and medicine. In particular, anthracenes are some of the most convenient building blocks that can be used for the photofabrication of advanced materials.26,27 This is because anthracenes have [4 + 4] photodimerization capabilities, which allow photopolymerization to occur in the case of two anthracene molecules linked together.28,29 Numerous papers have been published about anthracene-based polymers and their applications in materials sciences.26,30,31 Notably, the interest in anthracene applications is still growing, but in this publication, we would like to concentrate on azaanthracenes – a different class of photoactive molecules that are analogs of anthracenes. We think that these molecules can have an even greater potential in the field of materials sciences, or that they can at least be complementary to anthracenes and other photoactive molecular-based systems. Azaanthracenes have one (or more) nitrogen atoms in the molecule.32–35 These nitrogen atoms provide a center for further derivatization and for the tuning of the physicochemical properties of more complex azaanthracene-based systems. Moreover, the azanthracenes are much more resistant to oxidation compared to anthracenes.28,32
The photophysics of many azaantracenes have been known since the 80's. The series of articles by Kreysig et al. covers the lifetimes of electronic excited states, photodimerization mechanisms, quantum yields of photoreactions as a function of solvents and concentrations, and many other physico-chemical factors.36,37 However, this excellent dataset does not address the fact that the photodimerization under study is not well defined, as it may result in different isomers. For example, one of the first investigations of the mechanistic aspects of azaanthracene photodimerization was carried out for 1-azaanthracene. The authors assumed that only one regiomer (anti-HT) is formed, which is questionable.36
In the case of non-symmetric azaanthracenes, four different regiomers can potentially be formed during photodimerization. In a paper by Yamada and Kawamura, the authors showed that three photodimers of 1-azaanthracene and two photodimers of 2-azaanthracene were formed in solution under strong and prolonged illumination.38 In the case of the photodimerization of N-methyl-azaanthracenes, 2 regiomers have also been reported by Li et al.39 In both cases, the explanation as why a particular isomer is or is not formed was based on the energies of the end products calculated by DFT methods, which seems to be an oversimplification.
Several studies have been devoted to the problem of how to control the regioselectivity in photodimerization. The parameter that can modify the regioselectivity is the pH, which leads to protonation/deprotonation of the nitrogen atom. It has been recently demonstrated that the host–guest interaction can also be harnessed to control regioselectivity. More specifically, the presence of curcubit[10]uril (CB) led to the highest yield of one regiomer of an azaanthracene dimer.39 Another example of regiocontrol is the photochemical [4 + 4] cycloaddition of 2,6-disubstituted anthracenes. This example explores the regioselectivity over supramolecular moieties containing silver cations.40 In addition, regioselectivity reversal during photodimerization of 2-anthracenecarboxylic acid in a water-soluble organic cavitand has been achieved and reported in the literature.41
Another case is when two different photoactive molecules react with each other. Competitive cross- and homo-photoreactions can occur in solution, leading to a mixture of products. Various strategies are presented to control the selectivity of reactions. The use of a chiral hydrogen-bonding template has been demonstrated for anthracene and 2-anthracenecarboxylic acid.42 A similar example that used host/guest interaction with cucurbit[10]uril for (4a-azonia)-anthracenes led to highly selective cross-photodimerization.43 Another method that is utilized is cross-photodimerization in the solid state. In this case, the key step is to obtain a suitable co-crystal in which two different molecules can react with each other under illumination. Such examples have been presented for the solid-state [4 + 2] cycloaddition of anthracene with bis(N-ethylimino)-1,4-dithiin44 and for the [2 + 2] cross-photoreaction of asymmetric olefins to form a cyclobutane ring with different substituents.45,46 Another strategy was to mix two different anthracene molecules and then excite only one of them. This has led to selective cross-photodimerization via a [4 + 4]-cycloaddition reaction.47
Notably, no reaction between two different azaanthracene molecules has been reported until now. In the case of molecules with lower symmetry, the selectivity control will be more complex, because the number of possible products is multiplied. A proper choice of the illumination wavelength, in such a way that only the less reactive substrate is efficiently excited, may favor the cross-photodimerization. Here, we would like to demonstrate photodimerization between varied azaanthracenes, which can be controlled to some extent. In addition, a non-obvious explanation will be presented as to why some regiomers are formed and others are not.
Results
The following azaaanthracenes were chosen for the purpose of our study: 2-azaanthracene (A) and N-methyl-2-azaanthracene (M) iodide. Both molecules have very similar structures that differ by a methyl substituent on the nitrogen atom. However, they exhibit very different electronic properties (UV-vis absorption spectra), which allows the photocyclization reaction to be controlled by light.Both compounds are known, and there have been several reports on their physicochemical properties, including efficient fluorescence. They are also used as fluorescence dyes, as their luminescence wavelength can be easily adjusted by adding substituents. Moreover, there is also some literature about the photodimerization reaction of both, azaanthracene and N-methylazaanthracene.
Therefore, we focus on the investigation of photodimerization in a mixture containing two different substrates. As the absorption spectra of these compounds differ, there is the important question of the interplay between the illumination wavelength and the reaction fate, yield, and other parameters. This may enable manipulation of the illumination conditions toward a specific product or several products.
Azaanthracenes were prepared and purified according to the literature data (see ESI† Fig. S1).48 The structure and purity of the obtained compounds were confirmed via standard NMR (see Fig. S3–S6, ESI†) and mass spectrometry methods (see Fig. S30–S31, ESI†).
The photo-optical properties of both azaanthracenes, A and M, were described before.49,50,51 Here, we focus on UV absorption and fluorescence spectra recorded for A and M in two solvents: methanol (Fig. 1) and dichloromethane.
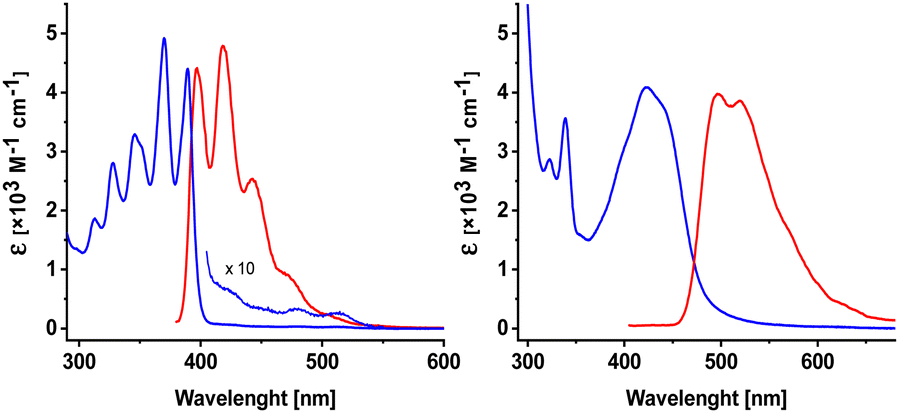 | ||
| Fig. 1 Electronic absorption (blue) and fluorescence (red) spectra of 2-azaanthracene (A, left) and N-methyl-2-azaanthracene (M, right) in methanol. | ||
The absorption band for azaanthracene (A) exhibits a subtle structure with a maximum at 390 nm. The maximum of the fluorescence band is slightly (452 cm−1) Stokes-shifted and located at 397 nm. A very weak, structured absorption band was observed at longer wavelengths above 400 nm. The origin of this band is unclear. One possible explanation is the formation of a complex between A and solvent's impurities. In the case of N-methylanthracene (M), the first absorption band is broad and unstructured. This band is red-shifted compared to A, with a maximum at 420 nm, but has nearly the same intensity. The fluorescence band of M is significantly (3690 cm−1) Stokes-shifted with a maximum at 497 nm. The larger Stokes shift of M suggests the greater stabilization of M by a polar solvent. The difference between the absorption and fluorescence spectra of A and M is because of the N-methylation, i.e., the involvement of the lone electron pair from the nitrogen atom. This effect is consistent with the literature data.37
For the purpose of photodimerization, we decided to use two irradiation lengths: 365 nm, where A is excited to more extent than M, and 420 nm where M is mainly excited. For the chosen wavelengths, the illumination was conducted for the solution with pure A, pure M, and the mixture of A and M (∼1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar). The repetition of experiments with pure A and pure M was a starting point for the mixed dimerization. These experiments were carried out in two solvents, methanol and dichloromethane.
:1 molar). The repetition of experiments with pure A and pure M was a starting point for the mixed dimerization. These experiments were carried out in two solvents, methanol and dichloromethane.
2-Azaanthracene (A)
In the case of 2-azaanthracene, four photocycloaddition products are possible: anti-HT, anti-HH, syn-HT, and syn-HH (anti, syn refer to the location of the N atom versus the long axis of the azaantracene unit, HT and HH means head-to-tail and head-to-head and refer to the location of the N atom versus the short axis, Scheme 1). The illumination of A in methanol at the maximum of the absorption band at 365 nm revealed that all four possible homodimers were formed. This was a bit surprising, as Yamada et al. reported only two products after 48 hours of irradiation in methanol.38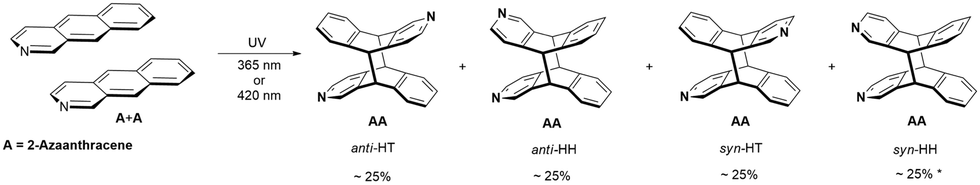 | ||
| Scheme 1 Products of the photocycloaddition of 2-Azaanthracene (A) * – yield values vary (see text). | ||
The analysis of relatively well resolved NMR lines above 8 ppm stemming from protons H1 and H3 (neighboring the nitrogen atom) showed that the relative amount of all four regiomers b:a:d:c is: 24:24:26:26, where b, d are anti, and a, c are syn isomers (Fig. 2, the ratios are given according to the order of H3 signals, where four doublets from four regiomers can be easily observed).
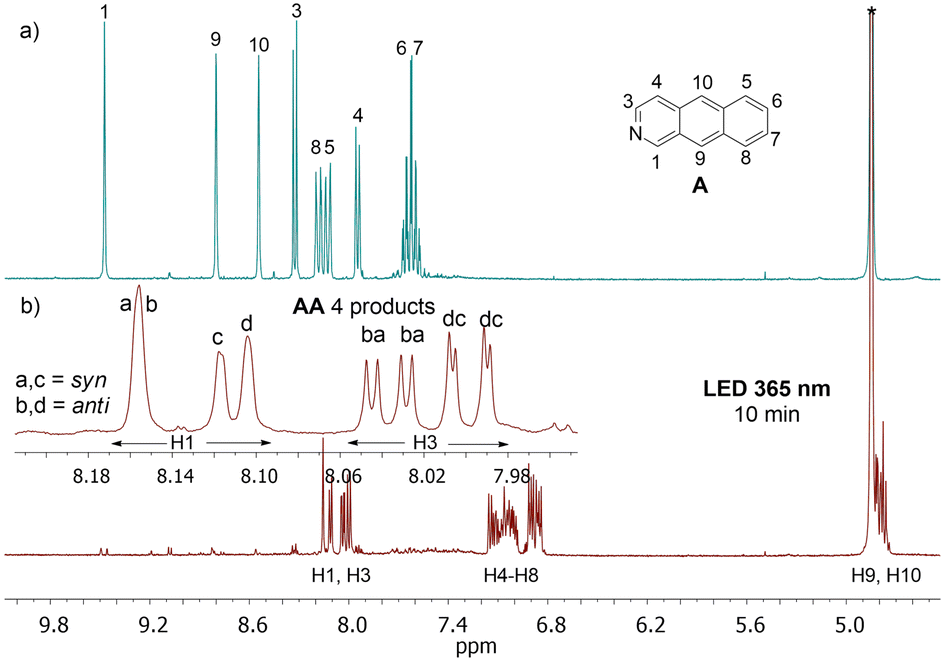 | ||
| Fig. 2 1H NMR spectrum of 2-azanthracene (A) (a) and the photoproduct after 10 min of irradiation at 365 nm (mixture of AA isomers) (b) measured in methanol-d4 at 298 K at 300 MHz. The insert shows the region of the spectrum above 7.95 ppm with signals of protons H1 (singlets) and H3 (doublets) -near the nitrogen atom. The initial concentration of A was 4.7 × 10−3 M. | ||
Further experiments revealed that this ratio may vary depending on the duration of irradiation and possibly the intensity of the irradiation. After 20 hours of irradiation, the relative concentration in the solution changes to b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) :a:d:c = 35:21:31:13 (see Fig. S16, ESI†). Generally, the relative amount of syn isomers decreased in comparison to anti isomers. The opposite effect was achieved just by storing the sample, irradiated for 10 min, at 273 K overnight (b:a:d:c = 22:33:15:30) (see Fig. S12, ESI†). The mechanism behind this is not clear to us, but it could involve a reverse reaction, decomposition of products, or precipitation of some crystals from the solution at lower temperatures. Each of these processes may proceed in a slightly different way for different isomers, leading to a change in their relative concentration.
:a:d:c = 35:21:31:13 (see Fig. S16, ESI†). Generally, the relative amount of syn isomers decreased in comparison to anti isomers. The opposite effect was achieved just by storing the sample, irradiated for 10 min, at 273 K overnight (b:a:d:c = 22:33:15:30) (see Fig. S12, ESI†). The mechanism behind this is not clear to us, but it could involve a reverse reaction, decomposition of products, or precipitation of some crystals from the solution at lower temperatures. Each of these processes may proceed in a slightly different way for different isomers, leading to a change in their relative concentration.
Irradiation at 420 nm, at the tail of an absorption band, resulted in four regiomers, as in the case of irradiation at 365 nm. Interestingly, the ratio b:a:d:c between them from the very beginning of irradiation led to an excess of anti-isomers (see Fig. S8, ESI†), and after 30 min of illumination amounted to 35:13:37:15 (see Fig. S15, ESI†). In this way, some tuning of the ratio of end products is possible. The illumination with the 365 nm wavelength causes a much faster photoreaction than irradiation at 420 nm, which is caused by the differences in absorption coefficients at both wavelengths.
In the case of dichloromethane (DCM), the photodimerization is also faster at 365 nm than at 420 nm, and the ratio of isomers depends on the irradiation wavelength. However, an exact analysis of the resulting regiomers was uncertain, due to broad H1 and H3 signals and overlapping of the H9 and H10 lines. Irradiation at 365 nm leads to four regiomers where anti isomers slightly dominate, but the irradiation at 420 nm leads to a large excess of anti-forms (see Fig. S13 and S14, ESI†).
All experiments described above were performed with degassed samples. When the sample contained some oxygen dissolved in the solution, the photoreaction occurred in a similar way to the reaction without oxygen and yielded the same products. However, some additional resonances appeared, coming from the oxidation processes of substrates and/or products (see Fig. S11, ESI†).
N-Methyl-2-azaanthracene (M) iodide
The case of N-methyl-2-azaanthracene (M) is different compared to 2-azaanthracene, as this molecule is a cation with a positive charge at the nitrogen atom. The photocycloaddition reaction leads to the formation of a bication. The irradiation at 420 nm or 365 nm in methanol produces two main products (with the 3:2 ratio) and a third in a lower amount (Scheme 2).
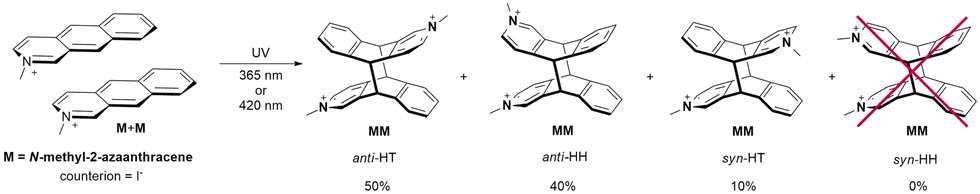 | ||
| Scheme 2 Products of photocycloaddition of N-methyl-2-azaanthracene (M) iodide. | ||
The proton spectrum measured at 300 MHz shows two lines in the methyl region at 4.22 and 4.28 ppm and a multiplet in the region of 5.2–5.4 ppm from protons connected to the bridging carbon atoms (H9 and H10) (Fig. 3, see Fig. S17, ESI†).
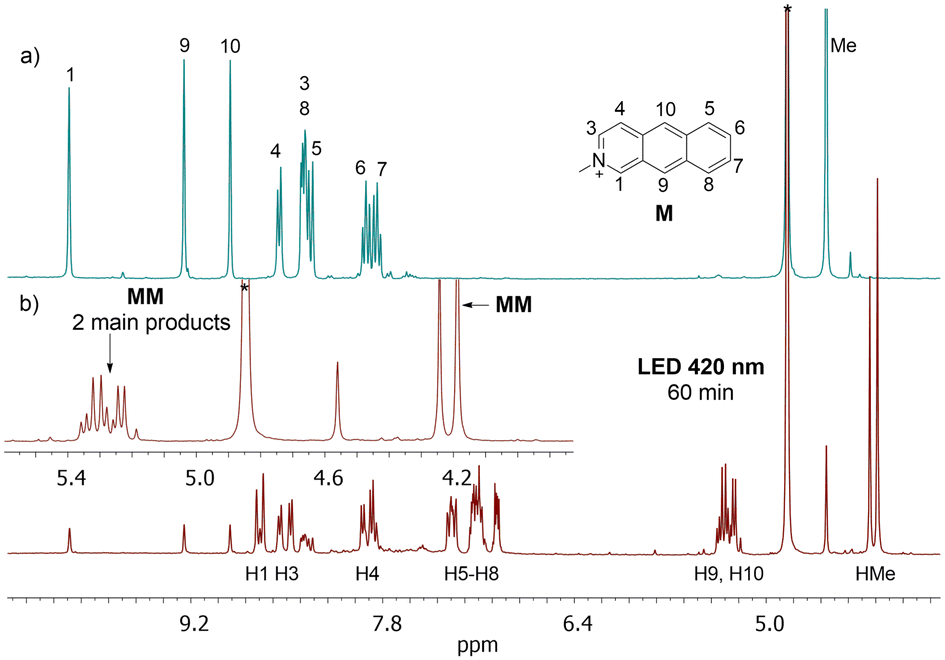 | ||
| Fig. 3 1H NMR spectrum of N-methyl-2-azaanthracene (M) iodide (a) and the photoproducts after irradiation at 420 nm (mixture of MM isomers) (b) measured in methanol-d4 at 298 K at 300 MHz. The initial concentration of M was 4.7 × 10−3 M. | ||
The proton spectrum measured at 500 MHz shows two strongly coupled doublet of doublets from H9 and H10 protons however, two small singlets are visible as well (see Fig. S18, ESI†) also in HSQC spectrum (see Fig. S20, ESI†). This suggests that the main products are anti isomers: anti-HT and anti-HH, because in these isomers neighboring H9/H10 protons from two azaanthracene units are not chemically equivalent and, therefore, coupled with each other. The additional line with low intensity in the methyl region, as well as two low singlets at 5.33 and 5.39 ppm, were ascribed to syn-HT isomer. The analysis of the HSQC spectra confirmed this assignment. The NOESY spectrum of the products shows weak contacts between protons H1 and protons H4′ or H5′ from another azaanthracene unit, allowing for differentiation of the anti-HT and anti-HH isomers. (see Fig. S22, ESI†)
It is worth mentioning that Li et al. reported the photocycloaddition of N-methyl-2-azaanthracene (M) bromide irradiated at 365 nm in D2O.39 However, they identified two products that were ascribed to anti-HT and syn-HT isomers. This discrepancy could be caused by the influence of the solvent on the course of the reaction (D2O vs. methanol), the counterion interaction (Br−vs. I−), or the fact that their assignment was questionable. The authors based their assignment on the calculated energies of products in the ground state where anti-HT and syn-HT isomers had slightly lower energy than the other two regiomers, but the careful analysis of the obtained 1H NMR spectra was not considered. In turn, Wang et al. reported photodimerization of N-methyl-1,8-diazaanthracene,52 which is a close analog to M. In this case, two products were also obtained, which were assigned to anti-HT and syn-HT isomers based on DFT calculations of the relative energies of photodimers in the ground state. As shown above, such reasoning may sometimes not be sufficient.
The photoreaction of M is faster when irradiated at 420 nm compared to 365 nm. This is not surprising as the 420 nm wavelength is closer to the maximum absorption band and the absorption coefficient is approximately twice as high as at 365 nm.
Interestingly, in DCM, the photocycloaddition reaction did not take place. This observation demonstrates the importance of solvation of the substrate molecules for the reaction progress.
Mixture of 2-azaanthracene (A) and N-methyl-2-azaanthracene (M)
When A and M are present in the solution, several homo- and heterodimers can be created. In theory, 12 different products of photodimerization can be formed: 4 AA, 4 MM, and 4 AM regiomers. This raises the question of whether we will be able to control this process to get only mixed dimers (AM), which have not been reported so far.When the nearly equimolar mixture of A and M was illuminated at 365 nm in methanol-d4, AM, AA and MM dimers were formed. After 30 min of irradiation, the ratio of products was 40:52:8, respectively. Thus, heterodimers were obtained, but the contribution of the AA homodimer was even higher, and it would be an asset to be able to limit the production of AA. Having the results for the solution with A, we know that the reaction between A and A can be slowed down by the irradiation at 420 nm, i.e., at the tail of the absorption band. At this wavelength the absorption coefficient for M is several magnitudes higher than for A. Indeed, the illumination at 420 nm leads to the efficient excitation of M while the excitation of A is less effective. As a consequence, the production of dimers containing M should be facilitated. Indeed, under irradiation at 420 nm, the dimers AM, AA and MM were produced in the ratio 80:12:8 (Fig. 4).
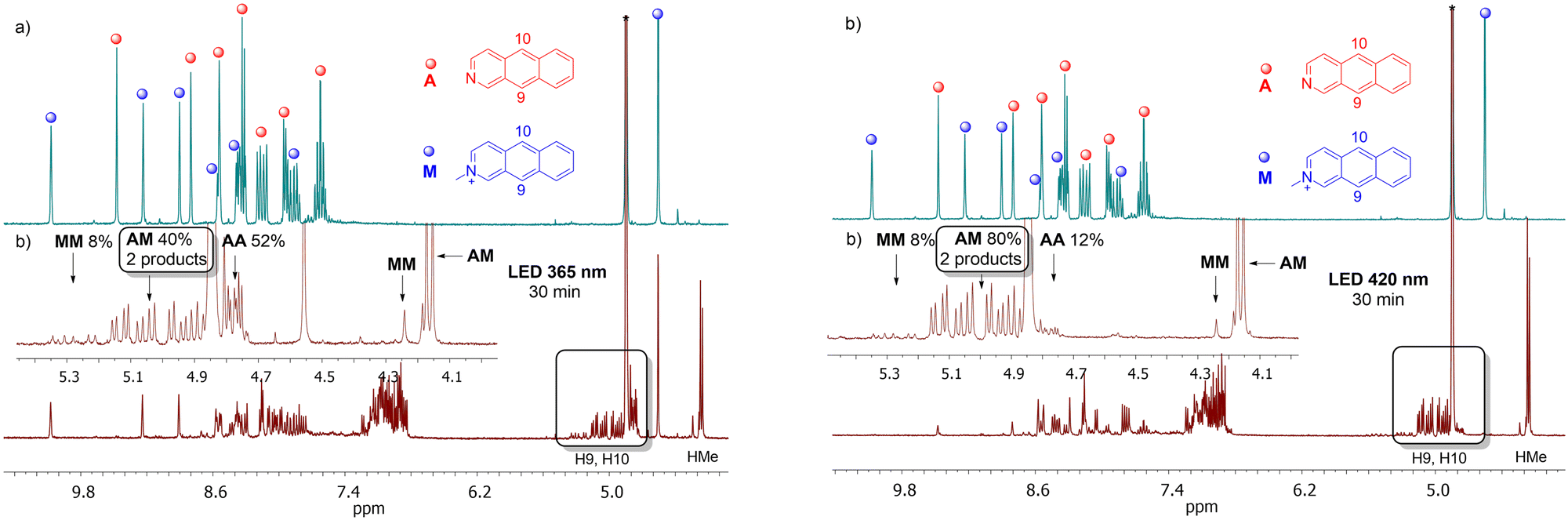 | ||
| Fig. 4 1H NMR spectrum of the mixture of N-methyl-2-azaanthracene (M) iodide and azaanthracene (A) before (a) and after (b) 30 min of irradiation at 365 nm (left) and 420 nm (right) measured in methanol-d4 at 298 K at 300 MHz. The inserts show the aliphatic regions of the spectra. Region 5.4–4.7 ppm is characteristic for H9 and H10 protons of dimers: 5.4–5.2 ppm for MM, 5.2–4.9 ppm for AM, 4.9–4.7 ppm for A. | ||
Next, we analyzed which isomers were produced. Two intense methyl lines, and well-separated resonances in the region 5.2–4.9 ppm from H9 and H10 (four doublet of doublets) suggest that two isomers were created in a ratio close to 1:1 (Fig. 4). In order to assign the 1H lines to the certain regiomers, the NOESY spectrum was collected (Fig. 5).
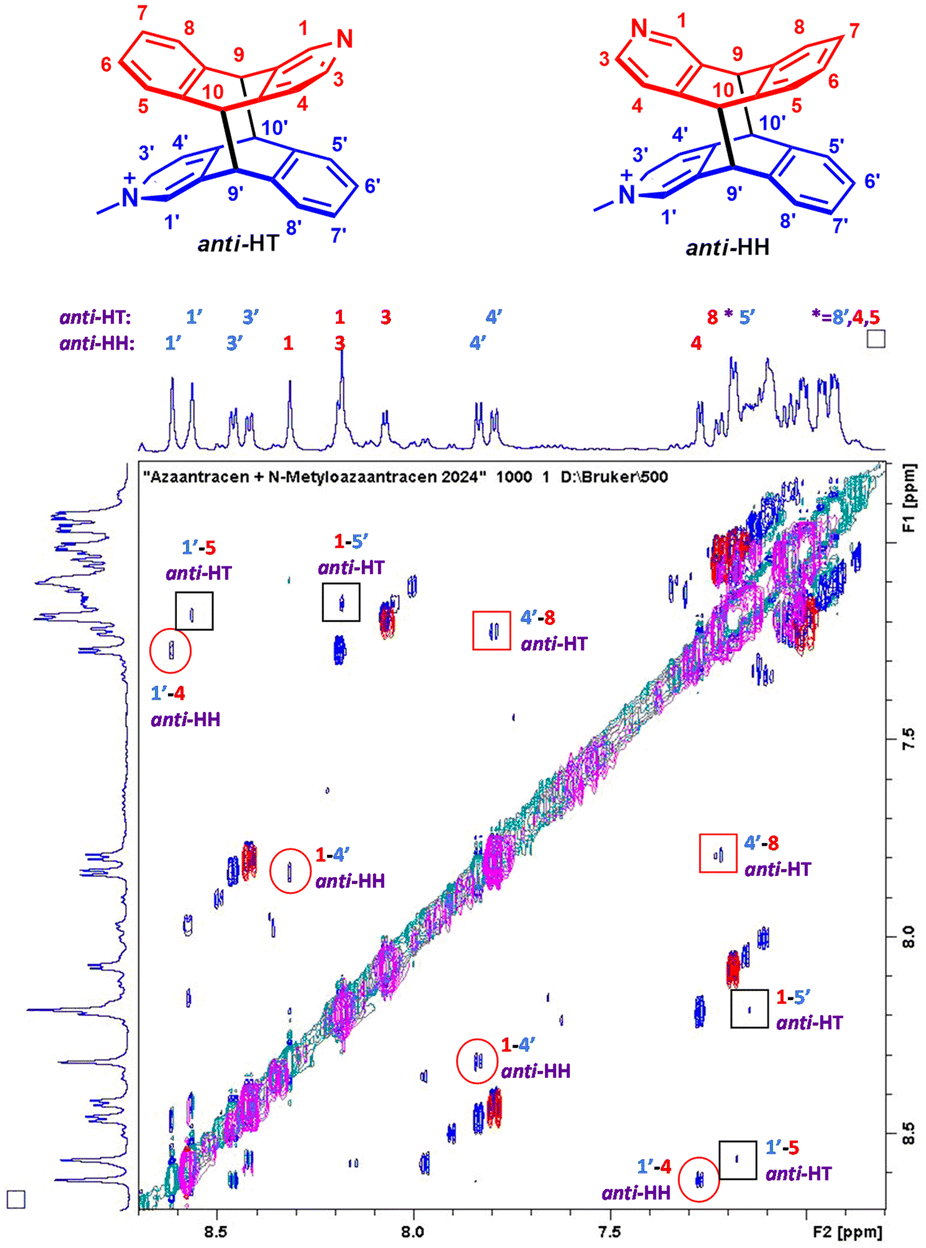 | ||
| Fig. 5 1H–1H NOESY spectrum of an equimolar mixture of A and M in methanol-d4 irradiated for 2 hours at 420 nm at 298 K at 500 MHz, mixing time 1.2 s (blue crosspeaks) and 1H–1H NOESY spectrum of the purified anti-HT isomer at 298 K at 300 MHz, mixing time 1 s (red crosspeaks). Red circles and red squares indicate essential contacts between A and M units for anti-HH and anti-HT signals, respectively. Black squares label non-diagnostic crosspeaks for anti-HT (due to signal overlapping). Red and blue numbers correspond to the proton signals from the A and M units, respectively. | ||
The crosspeaks between aromatic protons from the A and M units were observed. H1 to H4′ and H1′ to H4 contacts are characteristic for anti-HH isomer, whereas H8 to H4′ crosspeak proves formation of anti-HT isomer (numbers with primes refer to the M unit; contacts H1 to H5′ and H5 to H1′ were not diagnostic due to the line overlapping) (Fig. 5). Therefore, the ratio anti-HH:anti-HT is 55:45. No traces of syn isomers have been observed (Scheme 3).
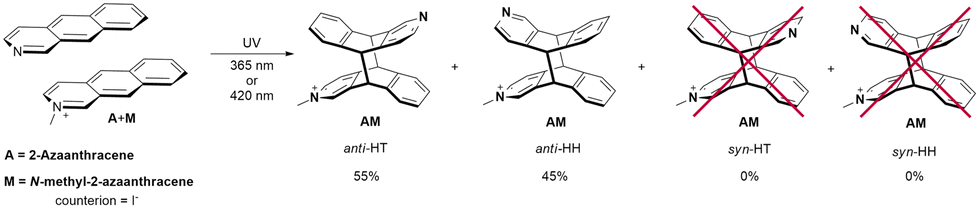 | ||
| Scheme 3 Mixed products of the photocycloaddition of N-methyl-2-azaanthracene (M) iodide and 2-azaanthracene (A). | ||
Our attempt to separate both main products was partially successful. Column chromatography delivered one pure isomer, whereas the second product was lost during purification. The 1H, COSY and NOESY NMR spectra of this pure regiomer anti-HT are shown in the ESI† (see Fig. S26–S29).
The question, as to whether only anti isomers of AM were produced during irradiation, is addressed in the quantum-chemical calculations section below.
The mass spectrum of the A and M mixture after irradiation reveals molecular peaks corresponding to AA, AM and MM (see Fig. S32–S34, ESI†).
Finally, the mixture of A and M in DCM produced only the AA isomers upon irradiation at both 365 nm and 420 nm.
UV-vis measurements
In order to better characterize the photocycloaddition reaction between A and M, UV-vis absorption spectra were recorded during irradiation of the samples A, M and the A/M mixture (Fig. 6). The absorption spectra of substrates reveal two bands in the region of 300–500 nm. However, the absorption of AA, MM and AM dimers in this region is very weak. Therefore, UV-vis absorption spectra can only track substrates loss and cannot monitor photoreaction products.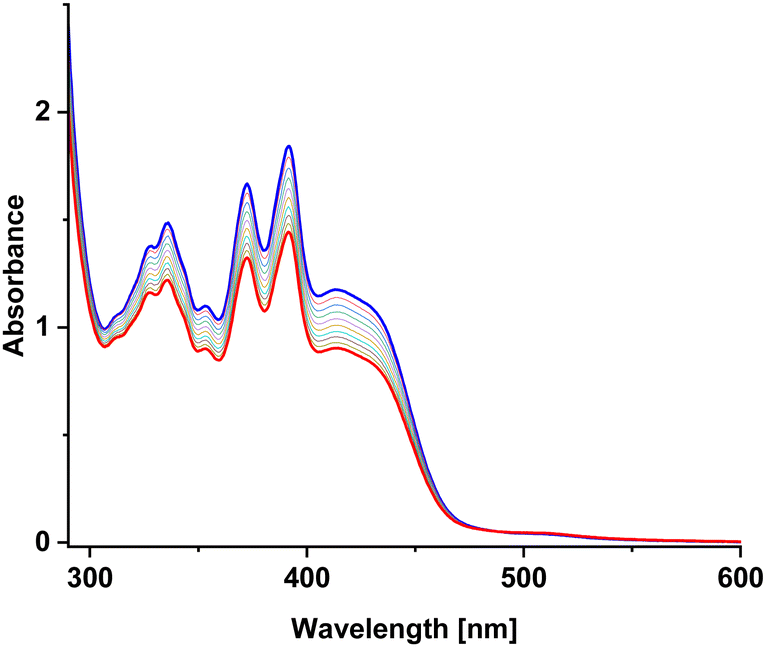 | ||
| Fig. 6 Changes in the absorption spectrum of the A:M (molar 1:1) mixture in MeOH under light irradiation at 420 nm by 170 mW LED. Spectra acquired after every 10 seconds of irradiation. Blue: the absorption spectrum before irradiation. Red: the absorption spectrum after 90 seconds of irradiation. | ||
The obtained quantum yields of the photocycloaddition reaction (Table 1) can lead to the formulation of several observations. First, the A + A photodimerization exhibits an order of magnitude higher quantum yield than M + M. This effect can be justified by a positive charge and a methyl substituent on the nitrogen atom for M molecule. Both factors make it difficult to bring the reacting molecules closer to each other. Second, photoreaction for the A + M mixture is two times faster than for M + M, again probably due to a higher steric hindrance in the case of M + M. Third, the presence of oxygen does not significantly change the quantum yield of the decay of the substrates. This is because excited azaanthracene reacts with oxygen slowly in contrast to anthracenes which are very sensitive to air.53 Whereas, N-methyl-anthracene seems to be even more resistant to oxidation.
| λ (nm) | A | M |
A + M, 1:1 |
|||
|---|---|---|---|---|---|---|
| Norm. | Air free | Norm. | Air free | Norm. | Air free | |
| 372 | 18.5 | 25.4 | — | — | 4.5 | 4.6 |
| 392 | 18.7 | 26.1 | — | — | 4.7 | 4.9 |
| 415 | — | — | 2.6 | 2.0 | 5.0 | 5.2 |
| 450 | — | — | 2.7 | 2.1 | 4.6 | 5.1 |
| Averaged | 18.6 | 25.7 | 2.7 | 2.1 | 4.7 | 5.0 |
Quantum-chemical calculations
To evaluate the possible mechanism of the formation of AM dimer, first, the ground-state geometries of all four AM regiomers have been optimized with use of a fast ODM2/MRCI-SD semiempirical method allowing for a multireference electronic structure treatment, and including inherently the dispersion interactions, (for details regarding this approach, please refer to the ‘Methods’ section). The obtained regiomers’ energies, presented in Table 2 along with the corresponding optimized structures, turned out to be comparable, differing by less than 1.00 kcal mol−1. Therefore, the thermodynamic stability of the products at room temperature did not seem to be the parameter controlling the reaction.|
|
|
|
|
|---|---|---|---|
| syn-HH | anti-HH | syn-HT | anti-HT |
| 0.031 eV | 0.039 eV | 0.000 eV | 0.032 eV |
| 0.72 kcal mol−1 | 0.90 kcal mol−1 | 0.00 kcal mol−1 | 0.74 kcal mol−1 |

A subsequent attempt to optimize four AM pairs in the ground electronic state (S0) led to all complexes adopting a slack geometry of the J-aggregate type, with the most stable one (syn-hh) characterized by an observable mutual rotation of the A and M+ symmetry axes (Fig. 7a). This rotation has been observed to induce a stabilizing effect also in complementary density functional theory (DFT) calculations performed with implicit inclusion of the methanol environment, the results of which can be found in Table S2 in the ESI† (for details regarding the applied DFT methodology, please refer as well to the ‘Methods’ section). Together with relatively large obtained interatomic distances, this dimer arrangement suggests that the S0-state complexation is unlikely to provide favorable starting conditions for the eventual photoproducts’ formation, in which both molecules are fully aligned. Hence, a different mechanism explaining the regioselectivity of the observed photodimerization can be anticipated.
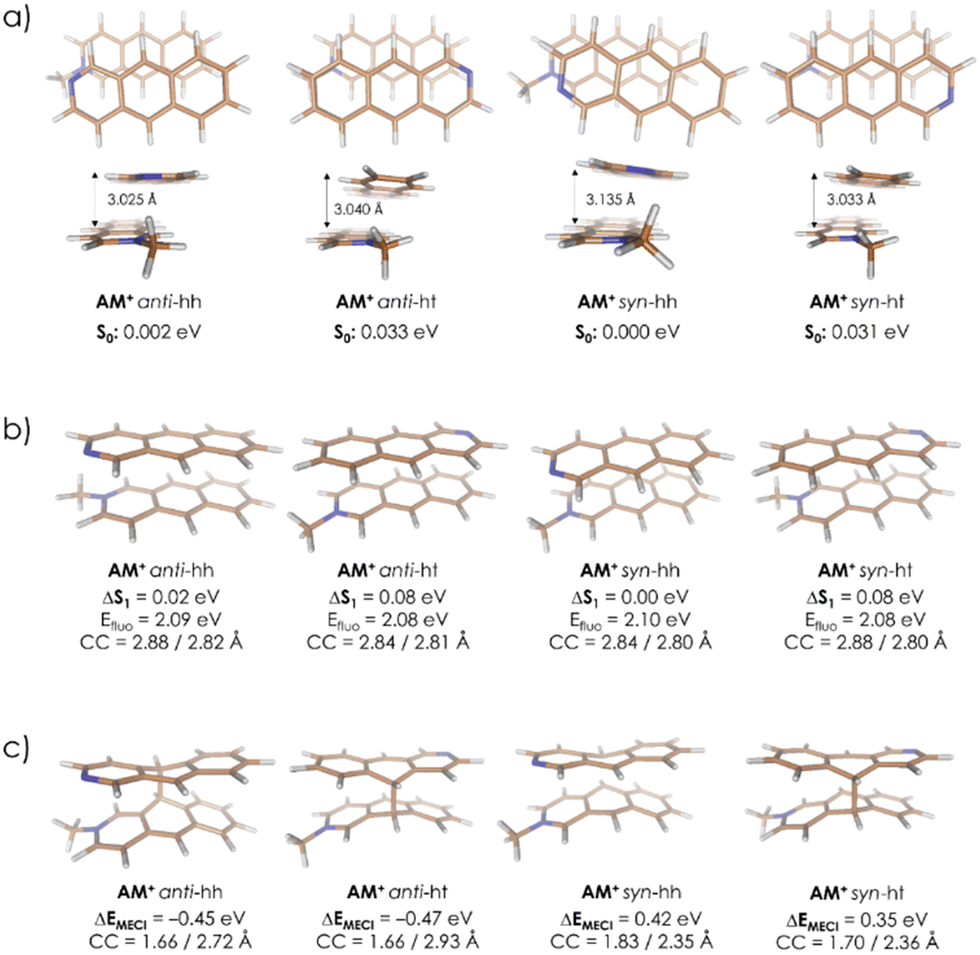 | ||
| Fig. 7 Geometrical structures and characteristic energies of four AM regiomers optimized in the S0 state (a), S1 state (b), and at MECI points (c) at the ODM2/MRCI-SD level of theory. The relative energies in the ground (S0) and in the first excited state (ΔS1) are calculated against corresponding values for the syn-hh form. The indicated CC values show distances between C9/C10 carbon atom pairs involved in the formation of the A–M bonds. The MECI energies (ΔEMECI) are calculated against the energies of the respective S1-minima. | ||
Eventually, in the next step, the dimer pairs were reoptimized in the first excited singlet state, S1. In all regiomers, this led to a similar reorganization of A and M, which adopted a parallel orientation with decreased intermolecular distance. Fig. 7b shows geometries and relative energies. A similar trend has also been observed at the Tamm–Dancoff time-dependent DFT (TDA-TDDFT) level of theory, confirming the S1 state favorable dimerization properties, as can be seen from Table S3 in the ESI.† Finally, the S1/S0 minimum-energy conical intersection (MECI) points were optimized. In this case, a clear distinction between anti and syn forms was observed. While all obtained structures show asymmetric formation of a C–C bond between A and M, the asymmetry in the resulting C–C distances at two centers which are connected in the final product in the anti-structures is much stronger than in the syn pair. This also comes with a difference in MECI energies calculated against corresponding S1-minima: the anti regiomers are expected to undergo an exoergic reaction to form MECI, while for syn regiomers the reaction is endoergic (Fig. 7c). Eventually, at the employed semi-empirical level of theory, the two anti-dimers are expected to prevail as the AM photoreaction products. This result has been confirmed also at an ab initio complete active space self-consistent field (CASSCF) level of theory (for details, please refer to the ‘Methods’ section and to Table S4 in the ESI†). On these grounds, the excited-state dimer relaxation pathway through a regiomer-specific conical intersection is proposed to be the key to explaining the observed anti alignment preference.
A similar analysis has been also performed for AA and MM dimers. In the MM case, the results resemble quite closely those obtained for AM (Table S6 in the ESI†), indicating asymmetric MECI structures and lower relative energies of the anti-forms which at this time, however, do not differ so strongly from the syn values. At the same time, while the latter observation is generally correct also for AA, in this system optimized MECI structures are almost fully symmetric, i.e., they characterize with almost identical C–C distances in both newly-forming bonds (Table S5 in the ESI†). Hence, while still the anti-dimers are expected to dominate over other possible products in AA and MM, it appears that presence of the methyl group attached to one or both N centers promotes a different (asymmetric) photoreaction mechanism, as compared to the parent AA system which is expected to undergo a symmetric cyclization reaction.
Photodimerization mechanism
The photodimerization of anthracenes has been studied many times. The mechanism of the reaction covers two pathways: directly through a singlet excimer 1(AA)* or through a triplet–triplet annihilation (3A*3A*).54 In the case of 1-azaanthracene, the reaction pathway involved solely excimers has been proposed.36In our case, the quantum yield of the photodimerization (more precisely the loss of substrates) does not depend much on the presence of oxygen. This strongly suggests that the triplet–triplet pathway does not play a role for 2-azaanthracenes, and we can focus on the pathway through a singlet excimer. Considering that the excited singlet state lives on the scale of tens of nanoseconds. (we have measured 13 ns for A and 27 ns for M in MeOH (see Table S1 in ESI†), whereas it is 19 ns for A and 37 ns for M in glycerol at 298 K37), one could suspect that the formation of molecule pairs via π–π-stacking interactions may already be taking place at the ground state. However, quantum chemical calculations do not confirm such a possibility. According to the calculated data, the pair of molecules is formed rather after excitation leading to an excimer.
The relative MECI energies in the S1 state are negative for anti-isomers of AM (−0.45/−0.47 eV) and positive for syn-isomers of AM (+0.42/+0.35 eV) (Fig. 7b) and, during irradiation, only anti-isomers are produced. In the case of MM, the MECI energies are negative for anti-HT, anti-HH and syn-HT (−0.13/−0.31/−0.05 eV) and positive for syn-HH (+0.19 eV) (see Table S7b in ESI†) and during irradiation anti-isomers are formed with a small amount of syn-HT. This coincidence may suggest that the controlling step of the photocyclization of A + M and M + M is the formation of the first C–C bond between azaanthracene units in the excimer. Such an interpretation leads to the conclusion, that this reaction is stepwise. In general, the cycloaddition reactions are considered to be a concerted process. However, there are more and more exceptions to this statement in the literature.55
In the case of photodimerization of A, the calculated MECI energies all show small positive values, and all four AA isomers are produced upon irradiation. Here, the reaction mechanism may be different, however, especially that no cations take part in the reaction in contrast to M.
The role of the solvent, in which photocyclization takes place, is important. It can influence the regioselectivity, as was shown on the example of the A + A photodimerization. In general, DCM favors anti-dimers over syn-dimers in comparison to methanol. In the case of A + M and M + M, the reaction in DCM does not run at all. This lack of reactivity may be twofold. First, the solvation of the excimers in methanol may push both molecules together enabling the reaction. The second reason is connected with the photophysics of M in DCM. In this case, the quantum yield of fluorescence is small (0.1) in comparison to M in methanol (0.61). Also, the quantum yield of singlet oxygen generation is small in DCM (0.02) in comparison to methanol (0.31) (see Table S1 in ESI†). This observation shows that in DCM nonradiative relaxation path is important, and the S1 state is short-lived, which may prevent the reaction in the excited state.
Eventually, the role of the irradiation wavelength is important. It allows some controlling of photoreaction in the solution with a mixture of substrates. This influences the reaction selectivity. The effect of the different proportions of regiomers produced after irradiation at different wavelengths is puzzling and difficult to explain.
Methods
NMR samples and measurements
Deuterated CD3OD and CD2Cl2 in 0.75 ml ampules were obtained from Deutero GmbH (99.6% D) and were used without further purification. Sample concentrations were about 1–2 mM. Samples were prepared with contact with atmospheric air, and therefore they contained dissolved air. Degassed samples were obtained by cool-pump-thaw procedure repeated 3 times in tight NMR tubes sealed with septum.NMR spectra were acquired on two spectrometers: a Bruker AVANCE II 300 MHz with a WB magnet equipped with a 5 mm BBI Z-gradient probe head and a Bruker AVANCE II 500 MHz spectrometer with a WB magnet equipped with a 5 mm BBO Z-gradient probe. The temperature was controlled by a BVT-3000 unit. The spectrometers were operated via the TOPSPIN program (version 2.0 at 500 MHz and 3.2 at 300 MHz). The NOESY (mixing time 1.2 s), COSY and HSQC experiments were performed with standard Bruker pulse programs (noesygpph, cosygpmfph, hsqcgpph). The reference for 1H NMR was the signal of CHD2OD and CHDCl2 set to 3.32 and 5.32 ppm, respectively.
Simulation of multiplets was performed with DAISY Spectrum Simulation Module (version 3.5.2) built in Topsin 4.0.7.
Irradiation
The irradiation was realized with a UV-diode emitting at 365 nm (Thorlabs M365LP1-C4) and at 420 nm (Thorlabs M420L3). The irradiation outside the magnet was done directly in an NMR tube through the glass (it was checked that the transmittance of light at 365 nm was above 95%, roughly estimated light beam power delivered to the sample was about 20 mW).Electronic absorption
The electronic absorption measurements were carried out on Shimadzu 2700 absorption spectrophotometer. Spectral grade methanol and dichloromethane (for spectroscopy Uvasol, Merck) were used to prepare all the solutions.Determination of photodegradation quantum yield
The azaanthracene derivatives dissolved in methanol or dichloromethane, placed in sealed quartz cuvettes, were irradiated with light emitted by LED diodes (λ = 365 or 420 nm). High power LEDs (Thorlabs M420L3 and M365L) were used for irradiation. The light intensity was determined by power meter as 102 and 170 mW for 365 nm and 420 nm LEDs, respectively. The sample solutions were stirred during the irradiation. The photodegradation process was monitored by UV-vis absorption measurements (Shimadzu UV2700). The photodegradation quantum yield was determined by measuring the sample absorbance before irradiation (A0) and at time t after the beginning of irradiation (A(t)). Next, the A0/A(t) ratio was plotted as a function of F(t), defined as Ntot(t)/A(t), where Ntot(t) denotes the total number of photons absorbed after irradiating the sample for time t. The photodegradation quantum yield was calculated based on the following equation: Φpho = (b × NAv × V)/(1000 × ε × l); where b is the slope in the equation: A0/A(t) = 1 + bF(t), NAv is the Avogadro number, V is the sample volume (in mL), ε is the molar absorption coefficient at the wavelength selected to monitor absorbance decrease, and l is the optical path length (in cm). The detailed procedure of the determination and calculation of the quantum yield of photodegradation is described elsewhere.56Quantum-chemical calculations.
All semi-empirical calculations have been performed with the MNDO99 code (v7 with upgrades),57 at the ODM2/MRCI-SD level of theory58–61 in which the ODM2 electronic structure model has been combined with the multi-reference configuration interaction method with single and double excitations. In these calculations, the half-electron restricted open-shell Hartree–Fock (HF) formalism62 was applied in the self-consistent field (SCF) treatment.The active space in the MRCI calculations included 12 electrons in 12 orbitals for AA and AM systems, and 10 electrons in 10 orbitals for the MM system (for the orbitals plots, please check Table S1 in the ESI†). Three configuration state functions were chosen as references for the MRCI treatment: the SCF configuration and the two closed-shell configurations derived therefrom. The MRCI wave function was built by allowing all single and double excitations from these three references within the defined active space.
The DFT calculations have been performed with the Becke 3-parameter-Lee–Yang–Parr (B3LYP) hybrid functional63,64 with the Grimme empirical dispersion correction65,66 modified by the Becke–Johnson damping function,67,68 and with use of the def2-SVP basis set.69 The solvent effects have been accounted for with the Polarizable Continuum Model (PCM) in its integral equation formalism variant (IEFPCM).70–72 For the excited electronic states, time-dependent density functional theory (TD-DFT) calculations have been performed with the Tamm–Dancoff approximation (TDA).73 All DFT calculations have been performed with the Gaussian 16 (rev. C.01) suite of programs74
In the ab initio minimum energy conical intersection (MECI) benchmark optimization the complete active space self-consisted field (CASSCF)75,76 method was employed, with the dunning correlation-consistent double-zeta basis set (cc-pVDZ).77
The calculations were state-averaged (SA) with equal weights between the S0 and S1 states. The employed reduced active space, consisting of 4 electrons in 4 orbitals, can be found in Table S4 in the ESI.† In this part, we used the BAGEL software.78,79
Conclusions
The photochemistry in systems containing different photoactive molecules, which can react with each other, can be complex. Here, such complexity was demonstrated for photodimerization in a mixture of two varied azaanthracenes. It has been demonstrated that the complexity can be substantially ameliorated by tuning the illumination wavelength.We have demonstrated an example where we were able to optimize the reaction in such a way that it primarily yields two AM heterodimers from twelve theoretically possible structures. Excitation with the proper wavelength boosts the AM amount from 40% (at 365 nm) to 80% (at 420 nm). We were able to identify the obtained isomers by the means of NMR methods. The obtained structures were anti isomers (anti-HH and anti-HT in both cases, A + M and M + M). For M + M, a small amount of syn-HT was also detected.
The calculations in the excited state allowed us to propose an explanation as to why, in the case of AM, anti regiomers are favored. Namely, the formation of the first bond between A and M in the excited state is energetically favored for antiAM isomers (−0.45/−0.47 eV) and unfavored for synAM isomers (+0.42/+0.35 eV). This suggests that the controlling step of the reaction process is the formation of the first intermolecular C–C bond in the electronic excited state. Therefore, the reaction mechanism of this photodimerization seems to be stepwise. This is surprising, but such mechanisms have been already suggested in the literature for some photodimerization processes.
We have illustrated the role of solvents in this photoreaction. Protic methanol enables the photocycloaddition of A + M and M + M, whereas DCM does not. The photophysics reveals that the singlet lifetime in DCM is much shorter for some reasons, which may stop the reaction.
Last but not least, we have obtained heterodimers of azaanthracenes for the first time.
Notably, the presented results indicate that when two photoactive compounds react with each other, the yield and selectivity of the reaction can be manipulated by an appropriate irradiation wavelength. The NMR data collected during the investigation of the photodimerization of azaanthracenes can significantly facilitate NMR data interpretation in the future for similar systems.
Author contributions
MP – research concept and methodology; AM – synthesis; MP – NMR measurements, NMR data analysis and simulations; AG and MP – optical spectroscopy measurements; AG – optical spectroscopy data analysis and description; JJ – quantum chemical calculations, data analysis and description; MP – supervision; MP and TR – writing of the main part of the manuscript; AM, MP, AG and JJ – preparing of figures.Conflicts of interest
There are no conflicts to declare.Availability statement
The data supporting this article have been included as part of the ESI.† Data for this article are available at RepOD at https://doi.org/10.18150/GUJCHR.Acknowledgements
JJ gratefully acknowledges Polish high-performance computing infrastructure PLGrid (HPC Center: ACK Cyfronet AGH) for providing computer facilities and support within computational grant no. PLG/2023/016751. TR acknowledges financial support from the National Science Centre, Poland, grant 2018/30/E/ST4/00004. MP acknowledges financial support from the National Science Centre, Poland in the form of grant SONATA (project no. 2022/47/D/ST4/00846).Notes and references
- P. C. Ford, Chem. Sci., 2016, 7, 2964–2986 RSC.
- H. E. Bonfield, T. Knauber, F. Lévesque, E. G. Moschetta, F. Susanne and L. J. Edwards, Nat. Commun., 2020, 11, 804 CrossRef CAS.
- D. Cambié, C. Bottecchia, N. J. W. Straathof, V. Hessel and T. Noël, Chem. Rev., 2016, 116, 10276–10341 CrossRef PubMed.
- E. G. Moschetta, G. C. Cook, L. J. Edwards, M. A. Ischay, Z. Lei, F. Buono, F. Lévesque, J. A. O. Garber, M. MacTaggart, M. Sezen-Edmonds, K. P. Cole, M. G. Beaver, J. Doerfler, S. M. Opalka, W. Liang, P. D. Morse and N. Miyake, Org. Process Res. Dev., 2024, 28, 831–846 CrossRef CAS.
- M. Ogawa and H. Takakura, Bioorg. Med. Chem., 2021, 43, 116274 CrossRef CAS PubMed.
- Y.-M. Tian, W. Silva, R. M. Gschwind and B. König, Science, 2024, 383, 750–756 CrossRef CAS.
- S. Chatani, C. J. Kloxin and C. N. Bowman, Polym. Chem., 2014, 5, 2187–2201 RSC.
- L.-M. Zhao, J. Mater. Chem. C, 2023, 11, 2826–2830 RSC.
- T. Noguchi, N. Akioka, Y. Kojima, A. Kawamura and T. Miyata, Adv. Mater. Interfaces, 2022, 9, 2101965 CrossRef CAS.
- S. He, X. Shu, Z. Wang, X.-W. Gao, K. Feng, S. Yang, J. Shao and N. Xie, Mater. Adv., 2024, 5, 2888–2897 RSC.
- B. D. Fairbanks, L. J. Macdougall, S. Mavila, J. Sinha, B. E. Kirkpatrick, K. S. Anseth and C. N. Bowman, Chem. Rev., 2021, 121, 6915–6990 CrossRef CAS PubMed.
- M. A. Tasdelen and Y. Yagci, Angew. Chem., Int. Ed., 2013, 52, 5930–5938 CrossRef CAS PubMed.
- W. J. Schreier, T. E. Schrader, F. O. Koller, P. Gilch, C. E. Crespo-Hernández, V. N. Swaminathan, T. Carell, W. Zinth and B. Kohler, Science, 2007, 315, 625–629 CrossRef PubMed.
- H. G. Khorana, W. E. Razzell, P. T. Gilham, G. M. Tener and E. H. Pol, J. Am. Chem. Soc., 1957, 79, 1002–1003 CrossRef.
- I. Tomatsu, K. Peng and A. Kros, Adv. Drug Delivery Rev., 2011, 63, 1257–1266 CrossRef.
- Q.-Q. Zhou, Y.-Q. Zou, L.-Q. Lu and W.-J. Xiao, Angew. Chem., Int. Ed., 2019, 58, 1586–1604 CrossRef PubMed.
- M. Fukumitsu, T. Fukui, Y. Shoji, T. Kajitani, R. Khan, N. V. Tkachenko, H. Sakai, T. Hasobe and T. Fukushima, Sci. Adv., 2024, 10, eadn7763 CrossRef PubMed.
- M. Woźny, A. Mames and T. Ratajczyk, Molecules, 2022, 27, 250 CrossRef.
- H. Ihmels, C. J. Mohrschladt, A. Schmitt, M. Bressanini, D. Leusser and D. Stalke, Eur. J. Inorg. Chem., 2002, 2624–2632 CrossRef.
- R. D. Nikolova, G. N. Vayssilov, N. Rodios and A. Bojilova, Molecules, 2002, 7, 420–432 CrossRef.
- T.-G. Hsu, H.-C. Chou, M.-J. Liang, Y.-Y. Lai and Y.-J. Cheng, Chem. Commun., 2019, 55, 381–384 RSC.
- X. Yu, D. Scheller, O. Rademacher and T. Wolff, J. Org. Chem., 2003, 68, 7386–7399 CrossRef CAS.
- S. Dong, A. Ong and C. Chi, J. Photochem. Photobiol., C, 2019, 38, 27–46 CrossRef CAS.
- C. Tönshoff and H. F. Bettinger, Chem. – Eur. J., 2021, 27, 3193–3212 CrossRef PubMed.
- K. Kristinaityte, M. Urbańczyk, A. Mames, M. Pietrzak and T. Ratajczyk, Molecules, 2021, 26, 6695 CrossRef CAS PubMed.
- F. Dumur, Eur. Polym. J., 2022, 169, 111139 CrossRef CAS.
- K. Kristinaityte, A. Mames, M. Pietrzak, F. F. Westermair, W. Silva, R. M. Gschwind, T. Ratajczyk and M. Urbańczyk, J. Am. Chem. Soc., 2022, 144, 13938–13945 CrossRef CAS.
- H. Bouas-Laurent, A. Castellan, J.-P. Desvergne and R. Lapouyade, Chem. Soc. Rev., 2000, 29, 43–55 RSC.
- J.-F. Xu, Y.-Z. Chen, L.-Z. Wu, C.-H. Tung and Q.-Z. Yang, Org. Lett., 2013, 15, 6148–6151 CrossRef.
- J. Van Damme and F. Du Prez, Prog. Polym. Sci., 2018, 82, 92–119 CrossRef.
- R. Bhola, P. Payamyar, D. J. Murray, B. Kumar, A. J. Teator, M. U. Schmidt, S. M. Hammer, A. Saha, J. Sakamoto, A. D. Schlüter and B. T. King, J. Am. Chem. Soc., 2013, 135, 14134–14141 CrossRef PubMed.
- O. S. Taniya, D. S. Kopchuk, A. F. Khasanov, I. S. Kovalev, G. V. Zyryanov, V. N. Charushin and O. N. Chupakhin, Chem. Heterocycl. Compd., 2019, 55, 505–507 CrossRef.
- O. S. Taniya, D. S. Kopchuk, A. F. Khasanov, I. S. Kovalev, S. Santra, M. Rahman, G. V. Zyryanov, A. Majee, V. N. Charushin and O. N. Chupakhin, New J. Chem., 2019, 43, 11382–11390 RSC.
- U. H. F. Bunz, J. U. Engelhart, B. D. Lindner and M. Schaffroth, Angew. Chem., Int. Ed., 2013, 52, 3810–3821 CrossRef PubMed.
- Y. Wang, Y. Xu, M. K. Ravva, Y. Yu, M. Xiao, X. Xue, X. Yang, Y. Chen, Z. Li and W. Yue, Org. Chem. Front., 2019, 6, 2974–2980 RSC.
- J. Bendig, J. Fischer and D. Kreysig, Tetrahedron, 1981, 37, 1397–1401 CrossRef.
- J. Bendig, D. Kreysig and A. Kawski, Z. Naturforsch., 1981, 36a, 30–33 CrossRef.
- S. Yamada and C. Kawamura, Org. Lett., 2012, 14, 1572–1575 CrossRef PubMed.
- H. Li, X. Hu, F. Liu, D. Sun, Y. Wu and S. Liu, Chin. Chem. Lett., 2022, 33, 5124–5127 CrossRef.
- S. Bai, L.-L. Ma, T. Yang, F. Wang, L.-F. Wang, F. E. Hahn, Y.-Y. Wang and Y.-F. Han, Chem. Sci., 2021, 12, 2165–2171 RSC.
- X. Wei, A. M. Raj, J. Ji, W. Wu, G. B. Veerakanellore, C. Yang and V. Ramamurthy, Org. Lett., 2019, 21, 7868–7872 CrossRef.
- Y. Kawanami, H. Umehara, J. Mizoguchi, M. Nishijima, G. Fukuhara, C. Yang, T. Mori and Y. Inoue, J. Org. Chem., 2013, 78, 3073–3085 CrossRef PubMed.
- X. Hu, J. Yang, F. Gao, Z. Zhao and S. Liu, Chin. Chem. Lett., 2024, 109967 CrossRef.
- J. H. Kim, S. M. Hubig, S. V. Lindeman and J. K. Kochi, J. Am. Chem. Soc., 2001, 123, 87–95 CrossRef PubMed.
- G. W. Coates, A. R. Dunn, L. M. Henling, J. W. Ziller, E. B. Lobkovsky and R. H. Grubbs, J. Am. Chem. Soc., 1998, 120, 3641–3649 CrossRef.
- M. A. Sinnwell, R. H. Groeneman, B. J. Ingenthron, C. Li and L. R. MacGillivray, Commun. Chem., 2021, 4, 60 CrossRef.
- D. Bailey and V. E. Williams, J. Org. Chem., 2006, 71, 5778–5780 CrossRef PubMed.
- A. P. Krapcho and T. P. Gilmor, J. Heterocycl. Chem., 1998, 35, 669–674 CrossRef.
- N. Nijegorodov and R. Mabbs, Spectrochim. Acta, Part A, 2002, 58, 349–361 CrossRef PubMed.
- N. Nijegorodov, R. Mabbs and D. P. Winkoun, Spectrochim. Acta, Part A, 2003, 59, 595–606 CrossRef PubMed.
- V. P. Zvolinskii, N. I. Nizhegorodov, V. V. Nikiforov, V. V. Dorogov and A. N. Gusarov, Sov. Phys. J., 1977, 20, 701–706 CrossRef.
- R. Wang, H. Liu, J. Li, J. Tian, Z. Li and Y. Zhao, Asian J. Org. Chem., 2018, 7, 906–909 CrossRef CAS.
- D. Sasikumar, R. Kohara, Y. Takano, K. Yuyama and V. Biju, J. Chem. Sci., 2019, 131, 5 CrossRef.
- J. L. Charlton, R. Dabestani and J. Saltiel, J. Am. Chem. Soc., 1983, 105, 3473–3476 CrossRef.
- R. Jasiński, Symmetry, 2021, 13, 1911 CrossRef.
- J. Ostapko, A. Gorski, J. Buczyńska, B. Golec, K. Nawara, A. Kharchenko, A. Listkowski, M. Ceborska, M. Pietrzak and J. Waluk, Chem. – Eur. J., 2020, 26, 16666–16675 CrossRef.
- W. Thiel, MNDO99 v7.0 with upgrades, Max-Planck-Institut für Kohlenforschung, 2017.
- P. O. Dral, X. Wu and W. Thiel, J. Chem. Theory Comput., 2019, 15, 1743–1760 CrossRef.
- W. Weber and W. Thiel, Theor. Chem. Acc., 2000, 103, 495–506 Search PubMed.
- A. Koslowski, M. E. Beck and W. Thiel, J. Comput. Chem., 2003, 24, 714–726 CrossRef.
- P. O. Dral, X. Wu, L. Spörkel, A. Koslowski, W. Weber, R. Steiger, M. Scholten and W. Thiel, J. Chem. Theory Comput., 2016, 12, 1082–1096 CrossRef PubMed.
- M. J. S. Dewar, J. A. Hashmall and C. G. Venier, J. Am. Chem. Soc., 1968, 90, 1953–1957 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- S. Grimme, J. Comput. Chem., 2004, 25, 1463–1473 CrossRef CAS.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- R. Improta, V. Barone, G. Scalmani and M. J. Frisch, J. Chem. Phys., 2006, 125, 54103 CrossRef.
- R. Improta, G. Scalmani, M. J. Frisch and V. Barone, J. Chem. Phys., 2007, 127, 74504 CrossRef PubMed.
- G. Scalmani, M. J. Frisch, B. Mennucci, J. Tomasi, R. Cammi and V. Barone, J. Chem. Phys., 2006, 124, 94107 CrossRef PubMed.
- S. Hirata and M. Head-Gordon, Chem. Phys. Lett., 1999, 314, 291–299 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2019 Search PubMed.
- B. O. Roos, Int. J. Quantum Chem., 1980, 18, 175–189 CrossRef.
- B. O. Roos, P. R. Taylor and P. E. M. Sigbahn, Chem. Phys., 1980, 48, 157–173 CrossRef.
- T. H. Dunning Jr., J. Chem. Phys., 1989, 90, 1007–1023 CrossRef.
- BAGEL, Brilliantly Advanced General Electronic-structure Library. https://www.nubakery.org under the GNU General Public License.
- T. Shiozaki, WIREs Comput. Mol. Sci., 2018, 8, e1331–e1331 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp03899a. |
| This journal is © the Owner Societies 2024 |