Solid-state 1H and 13C NMR studies of new ionic plastic-crystals with branched structures: [NEtxMe(3−x)(i-Pr)][BEt(4−y)Mey] (x = 1–3, y = 0, 1)†
Received
22nd October 2024
, Accepted 15th November 2024
First published on 18th November 2024
Introduction
Many compounds (such as H2O) directly transform from solid to liquid states upon heating, whereas a few chemicals have mesophases between them. The plastic crystal phase is a mesophase state (the liquid crystal phase is also classified as a mesophase). In the plastic crystal phase, the orientation of the constituents is disordered in three dimensions (the molecules undergo isotropic reorientation motion), and the gravity point is ordered; therefore, plastic crystals are often referred to as crystals with liquid characteristics. The rotator phase is called so if the constituents undergo uniaxial rotation in the crystal (orientational disorder in two dimensions). Based on their similarities, some researchers have frequently reclassified rotator crystals as part of the plastic crystal phase. The correct definition of ‘plastic crystal’ is employed in this literature. In the liquid crystal phase, the opposite relationship of the plastic crystal is defined: the orientation is ordered in one or two dimensions, and the gravity point is disordered. In plastic crystals, the constituents can jump to lattice defects with low activation energies, in addition to undergoing isotropic reorientation. This translational motion imparts plasticity to the solid material. Based on the soft property, many researchers have actively developed ion conductors using ionic plastic crystals (e.g. hexamethylguanidinium bis(fluorosulfonyl)imide ([HMG)][FSI])1 and N,N′-ethylmethylpyrrolidinium bis(trifluoromethanesulfonyl)-amide ([C2 mpyr][TFSA]) salts2 show electrical conductivity (σ) of ca. 10−5 S cm−1; 1-methyl-imidazolium triflate,3 [C2 mpyr][N{(SO2)C2F5}2],4 15-crown-5-ether – Li-TFSA complex ([Li 15C5][TFSA]),5 and [NEt4][BEt3Me] compounds have ca. 10−4 S cm−1; [NEt3R][BEt3Me] (R = Me, Pr),6 [SEt2R][FSI] (R = Me, Et),7 and [C3 mpyr][BF4]8 salts indicate ca. 10−3 S cm−1; [N,N-diethyl-3-methylpyrazolium][TFSA] is 2.6 × 10−3 S cm−1;9 the values of ca.10−2 S cm−1 are reported in [C2 mpyr][C(CN)3],4 [NEt4][N(CN)2],10 1-alkyl-2-methyl pyrrolinium ([(C4H6N)MeR]) (R = Et, Pr) with [TFSA] anion,11 and CsHCO212 crystals; [PEt2Me(i-Bu)][C(CN)2NO] shows ca. 6 × 10−2 S cm−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 13 the σ values of 1-alkyl-4-aza-1-azoniabicyclo-[2,2,2]octane (N–R DABCO, R = Me, Et) with [BEt3Me] anion,14 [Cn mpyr][C(CN)2NO] (n = 2–4),13 and [C1 mpyr][SCN]15 compounds reach ca. 10−1 S cm−1, etc. However, the mechanisms underlying the formation of these plastic crystals are poorly understood.
13 the σ values of 1-alkyl-4-aza-1-azoniabicyclo-[2,2,2]octane (N–R DABCO, R = Me, Et) with [BEt3Me] anion,14 [Cn mpyr][C(CN)2NO] (n = 2–4),13 and [C1 mpyr][SCN]15 compounds reach ca. 10−1 S cm−1, etc. However, the mechanisms underlying the formation of these plastic crystals are poorly understood.
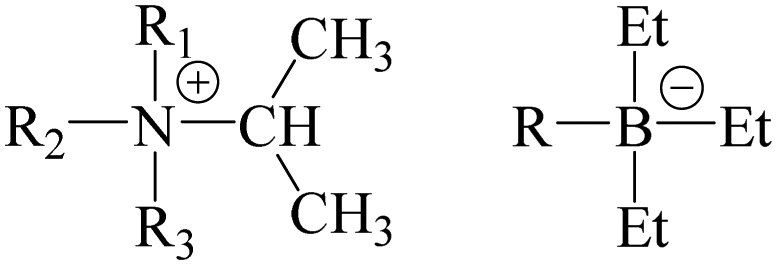
Some globular molecules (fullerene, adamantane, tetrachloromethane, etc).16–23 have plastic crystalline phases. Based on this fact, it can be concluded that both globular shapes and weak intermolecular attractions are key factors in forming molecular plastic crystals. In contrast, a globular ion and a non-globular counter ion (e.g. MNO2 (M = K, Rb, Cs, Tl, and NH4).24–32 [Cn mpyr]X (X = SCN, FSI, TFSA, N{(SO2)C2F5}2, etc.).2,4,15 NEt4X (X = N3, N(CN)2, etc.),33 piperidinium X (X = ClO4, PF6, NO3),34,35 [Fe(C5R4H)2][CH2BrCF3] (R = H, Me),36 [Li 15C5][TFSA])5 form ionic plastic crystals. Thus, planar or rod-like shapes are vital in forming ionic plastic crystals. In these compounds, a revolving door model28,31 can explain the ionic translation in the plastic crystal: the isotropic reorientation velocities of the non-globular ions (the revolving door) are sufficiently small for the counter ions (the human) to translate to lattice defects. However, globular shape ions (e.g. [NR1R2R3R4] (R1, R2, R3, R4 = Me, Et, Pr, etc.)6,37–41 and [N–R DABCO] (R = Me, Et) cations14 with [BEt3Me]− and [BEt4]− anions) also form ionic plastic crystalline phases. From these results, it can be considered that there is a relationship between the shapes of the constituents and the strength of the interactions in the plastic crystals. For the [NR4][BEt4]-type plastic crystals, it can be considered that the Coulomb force acting among the ions is sufficiently weak, similar to a molecular plastic crystal (the alkyl groups of the cation and anion act as spacers between the N and B atoms (centers of charge)). If the Coulomb force is a key factor, it can be regarded that charge shielding by alkyl groups also contributes to physical properties of ionic plastic crystals. To reveal the shielding effect of the alkyl group, compounds substituted from the ethyl group to the isopropyl group were investigated in this study (Fig. 1). Solid-state nuclear magnetic resonance (NMR) spectra provide information on these interactions and their molecular dynamics. Because the line widths of 1H NMR signals are mainly determined by dipole–dipole interactions, information about the interactions among the constituents can be estimated. 1H NMR spin–lattice relaxation time (T1) measurements show the activation energy of molecular motion (e.g., isotropic reorientation). In the case of 13C NMR spectra, the line shapes of the powdered samples are determined by chemical shift anisotropy (CSA); therefore, Lorentz-type lines (three components of the chemical shift (CS) tensor averaged by isotropic reorientation motion) can generally be recorded in plastic crystals. In addition to the NMR observations, differential scanning calorimetry (DSC) and X-ray diffraction (XRD) measurements were performed. In plastic crystalline phases, the isotropic reorientation motion of the constituents results in large and small entropy changes at the phase transition temperature (ΔtrS) from the ordinal crystalline phases to the plastic phase and at the melting point (ΔmpS), respectively. Timmermans reports the relationship ΔmpS < 21 J K−1 mol−1 for plastic crystals.42 XRD measurements provide beneficial information on plastic crystals, which normally have NaCl- or CsCl-type cubic structures,5,6,14,37–41 based on the isotropic reorientation of cations and anions. In addition, electrical conductivity measurements were carried out to estimate the activation energies (Eadiff) of ion diffusion.
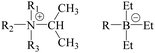 |
| | Fig. 1 Chemical structures treated in this study (R1, R2, R3, R = Me, Et). | |
In this literature, the symbols Ttr1 and Ttr2 indicate the phase-transition temperatures within solid phases upon moving from higher to lower temperatures, and the symbols Δtr1S and Δtr2S are the entropy changes at each transition temperature between the solid phases. The characters of phase I, phase II, etc., indicate the solid phases starting from a high temperature, while phase I lies from Ttr1 to each decomposition temperature.
Experimental
Sample preparation
The cation component of [NEtxMe(3−x)(i-Pr)]I (x = 0–3) was prepared by modifying the previous reported procedure:43 after adding NEtxMe(3−x) and (i-Pr)I in methanol, the solution was refluxed at 70 °C for 2 days under N2 atmosphere. The obtained samples were then dried by evaporation. The same procedures used in previous reports6 were employed for the anionic components of Li[BEt3Me] and Li[BEt4]. The same procedures in the previous reports6 were employed. By adding Li[BEt3Me] or Li[BEt4] to each cation component in an aqueous solution, powder samples of [NEtxMe(4−x)(i-Pr)][BEt(4−y)Mey] (x = 0–3, y = 1, 2) were obtained. The obtained samples were recrystallized from water and dried under vacuum. The product was successfully synthesized, as confirmed by 1H NMR spectroscopy. The samples used in this study were obtained from Kanto Kagaku Co. without LiEt dissolved in diethyl ether, NEt2(i-Pr) (Sigma Aldrich Co.), NEtMe2 and NEt2Me (Tokyo Kasei Co.).
Measurements
Because plastic crystals frequently have metastable phases, applying scan cycles to the DSC measurements is necessary. Virgin crystals are generally heated immediately below each melting point, and DSC measurements are performed. Unfortunately, all the samples treated in this study decomposed below the melting temperature. Therefore, thermogravimetric (TG) analysis was performed using a Hitachi Hightech STA7300 calorimeter before DSC measurements. DSC measurements were performed using a Hitachi Hightech DSC-7020 calorimeter. All samples were placed in aluminum pans (h = 5 mm, ϕ = 5.2 mm) for the TG and DSC measurements. For the DSC measurements, the samples were first cooled from room temperature to approximately 200 K and subsequently heated to 370 K at a rate of 5 K min−1. A second cooling and heating cycle was performed. The Ttr and ΔtrS values were recorded during the second cycle.
XRD patterns were recorded using a Bruker D8 ADVANCE diffractometer with a Cu anticathode. The TTK450 probe was used at various temperatures. The powder patterns were plotted in the scan range of 5°–40° with an angular step size of 0.02°.
Electrical conductivity (σ) measurements were performed at 1 kHz at temperatures ranging from 298 K to below each decomposition point using a two-terminal method employing an Agilent Technologies E4980A Precision LCR meter equipped with an Al sheet. The powdered sample was pressed into a disc with a diameter of 1 cm and thickness of approximately 1 mm at 20 kPa.
Solid-state 1H and 13C NMR spectra were recorded at the Larmor frequency of 600.13 MHz and 150.92 MHz, respectively, using a Bruker Avance 600 spectrometer (14.10 T). The samples of [NEtxMe(3−x)(i-Pr)][BEt(4−y)Mey] (x = 0–3, y = 0, 1) were sealed in handmade glass tubes with an outer diameter of approximately 4 mm. The glass tube was loaded into the solid-state probe, and measurements were performed as a function of temperature starting from room temperature. After waiting for 10 min, the 1H and 13C NMR measurements were performed at each temperature. The spectra were obtained using the Fourier transform (FT) of the free induction decay (FID) signals recorded after a π/2 pulse. Recycling times of 5 s and 20 s were employed for the 1H and 13C measurements, respectively. The 1H and 13C NMR spectra were recorded relative to an external water (δ = 4.80 ppm) and external glycine (δ = 43.16 ppm) reference, respectively. The 13C signals of glycine were recorded at a magic angle spinning (MAS) rate of 5 kHz using the cross-polarization (CP) method. By exchanging the probe, the 1H NMR spectra of the prepared samples dissolved in DMSO-d6 were recorded relative to the solvent signals (δ = 2.50 ppm).
Results and discussion
DSC
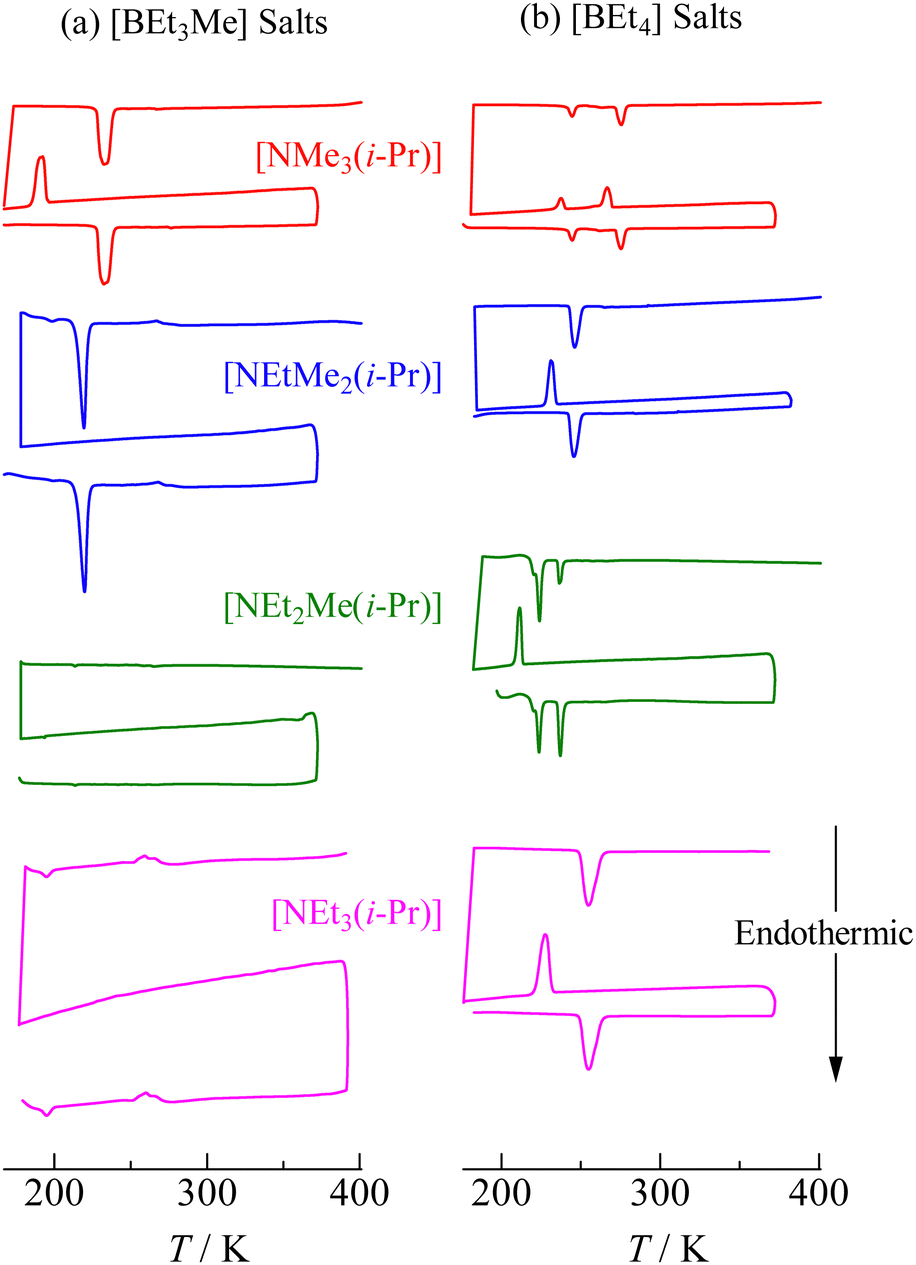
The DSC thermograms of [NEtxMe(3−x)(i-Pr)][BEt(4−y)Mey] (x = 0–3, y = 1, 2) crystals are shown in Fig. 2. The sample was heated in each chart from 180 K to 370 K (first heating run). TG measurements predetermined the latter temperature before the DSC measurements because all the samples treated in this study decomposed below each melting temperature (using test-tube observations, we checked that all compounds decomposed before fusion). The sample temperature in the DSC measurements successively decreased until ca. 180 K from 370 K and then increased again (the second heating run). The same curves were detected in the second heating scan as those in the first run (Fig. 2); therefore, the phase transition temperatures (Ttr) and entropy changes (ΔtrS) listed in Table 1 were determined from the data in the second heating scan. The obtained Δtr1S (+Δtr2S) values, except for [NEt2Me(i-Pr)][BEt3Me] and [NEt3(i-Pr)][BEt3Me], were similar to those of the plastic crystals of [NEtxMe(4−x)][BEt(4−y)Mey] (x = 0–4, y = 0, 1).6,37,38,41 This suggests that motional modes with large degrees of freedom occur in phase I of these crystals. The obtained Ttr1 values were similar to those reported for nonbranched crystals of [NEtxMe(3−x)][BEt(4−y)Mey].6,37–41 (Table S1 and Fig. S1, ESI†).
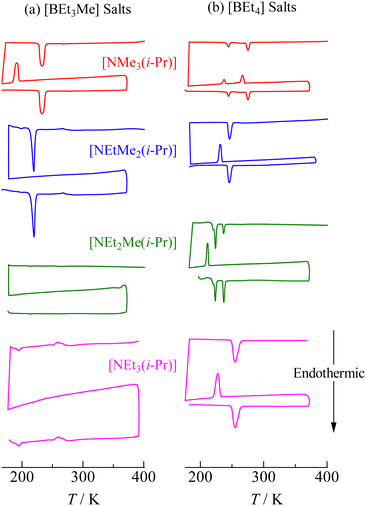 |
| | Fig. 2 DSC thermal traces in the first and second heating run of each [NEtxMe(3−x)(i-Pr)][BEt(4−y)Mey] (x = 0–3, y = 0, 1) compound. The thermograms were recorded from left bottom in each sample. | |
Table 1 Entropy changes (J mol−1 K−1) at each phase transition temperature of [NEtxMe(3−x)(i-Pr)][BEt(4−y)Mey] (x = 0–3, y = 0, 1). The temperatures are shown in parenthesis, and the unit is in K
|
|
Δtr2S/J mol−1 K−1 |
Δtr1S/J mol−1 K−1 |
| [NMe3(i-Pr)][BEt3Me] |
|
64 ± 1 (227 ± 1) |
| [NEtMe2(i-Pr)][BEt3Me] |
|
29 ± 1 (197 ± 1) |
| [NEt2Me(i-Pr)][BEt3Me] |
|
|
| [NEt3(i-Pr)][BEt3Me] |
3 ± 1 (184 ± 1) |
Value was not determined correctly (251 ± 1) |
| [NMe3(i-Pr)][BEt4] |
15 ± 1 (241 ± 1) |
24 ± 1 (271 ± 1) |
| [NEtMe2(i-Pr)][BEt4] |
|
50 ± 1 (242 ± 1) |
| [NEt2Me(i-Pr)][BEt4] |
38 ± 4 (217 ± 1) |
19 ± 5 (235 ± 1) |
| [NEt3(i-Pr)][BEt4] |
|
78 ± 1 (245 ± 1) |
XRD
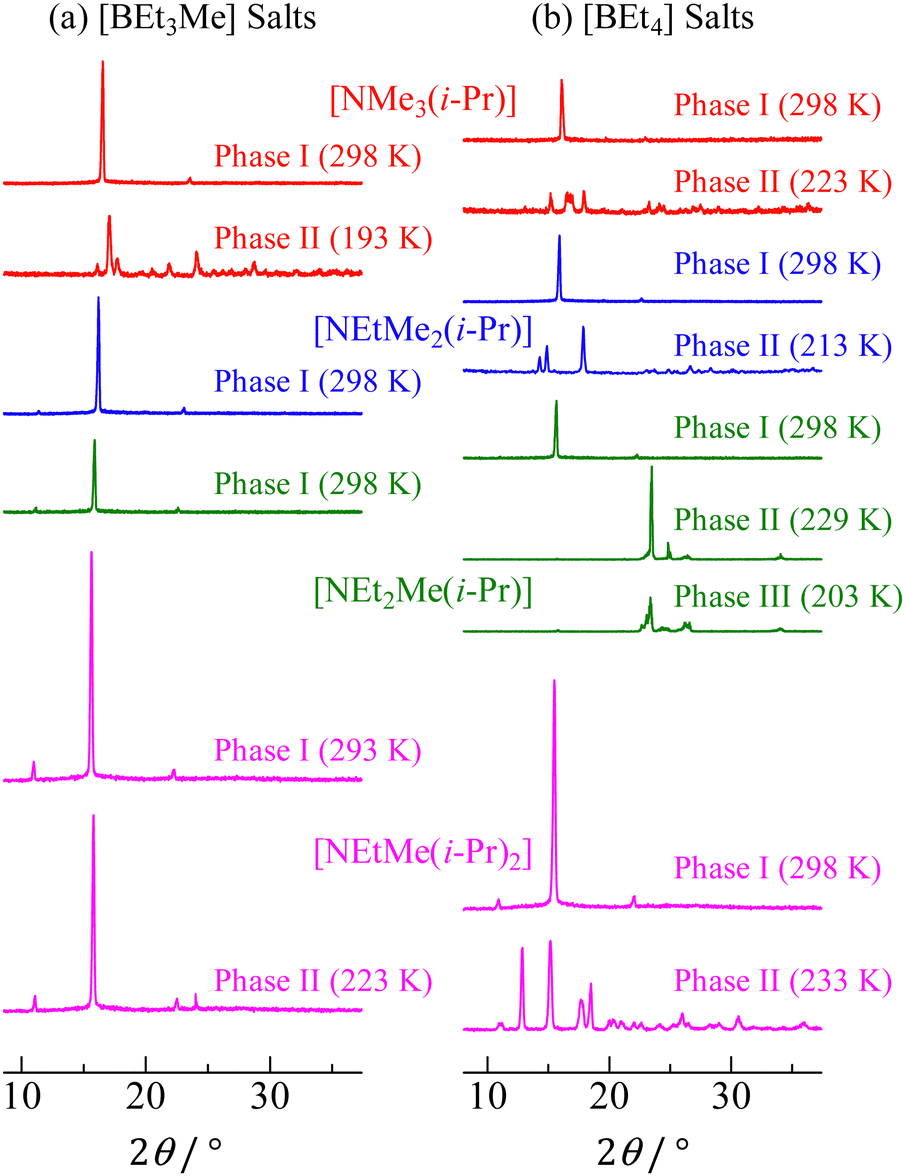
The powder XRD patterns of [NEtxMe(3−x)(i-Pr)][BEt(4−y)Mey] (x = 0–3, y = 0, 1) compounds recorded at various temperatures are shown in Fig. 3. The reflection patterns detected in phase I for each salt could be indexed to the CsCl-type cubic structure. This result suggests that the constituents underwent overall molecular motion with large amplitudes in phase I. In contrast, complex patterns are observed in the lower-temperature phases. Unfortunately, the patterns recorded in phases II and III of each salt could not be fitted to a unique space group. Based on the large Δtr1S (+Δtr2S) values, it can be considered that the ions had a small degree of freedom of motion in phases II and III. Thus, the complex powder patterns were consistent with the DSC results. The estimated lattice constants (a) of the cubic crystal structure phase are summarized in Table 2. Some reported values are also listed in the table for comparison with the lattice constants of other nonbranched plastic crystals of [NEtxMe(4−x)][BEt(4−y)Mey] (x = 0–4, y = 0, 1).It can be assumed that the distance from the N atom to the terminal CH3 of the isopropyl group in the cations is similar to that in the ethyl group: Similar a values would be obtained in [NEtxMe(3−x)(i-Pr)]X and [NEtxMe(3−x)Et]X (x = 0–3, X = [BEt3Me] and [BEt4]). However, the relationship [NEtxMe(3−x)(i-Pr)]X > [NEtxMe(3−x)Et]X was obtained. Thus, the apparent cation radii (rcation) of [NEtxMe(3−x)(i-Pr)]+ and [NEtxMe(3−x)Et]+ were estimated using the [BEt3Me]− radius of 401 pm, which was simulated using the B3LYP/6-311+G** function. The rcation values are summarized in Table 2. The rcation values of [NEtxMe(3−x)(i-Pr)]+ were 19–29 pm higher than those of the corresponding [NEtxMe(3−x)Et]+ cations.
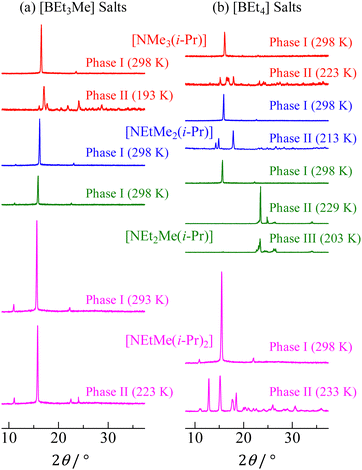 |
| | Fig. 3 XRD powder patterns of [NEtxMe(3−x)(i-Pr)][BEt(4−y)Mey] (x = 0–3, y = 0, 1) recorded at each solid phase. | |
Table 2 Lattice constants (a) of the cubic structure (phase I) of [NEtxMe(3−x)(i-Pr)][BEt(4−y)Mey] (x = 0–3, y = 0, 1), and apparent ionic radius (rcation) of the cations which is estimated by each a value and [BEt3Me]− of 401 pm. In this table, a values reported in some plastic crystals are also summarized
|
|
a/pm |
r
cation/pm |
| [BEt3Me] |
[BEt4] |
|
Ref. 6.
Ref. 41.
|
| [NMe4] |
722 ± 1a |
736 ± 3a |
224 ± 3 |
| [NEtMe3] |
734 ± 3a |
749 ± 3a |
225 ± 3 |
| [NEt2Me2] |
750 ± 1a |
766 ± 3a |
249 ± 3 |
|
[NMe
3
(i-Pr)]
|
755 ± 2
|
772 ± 2
|
254 ± 2 |
| [NEt3Me] |
755 ± 3a |
779 ± 3a |
257 ± 4 |
| [NEtMe2Pr] |
760 ± 2b |
— |
257 ± 2 |
| [NEt4] |
769 ± 2a |
792 ± 3a |
268 ± 3 |
| [NEt2MePr] |
772 ± 2b |
— |
268 ± 2 |
|
[NEtMe
2
(i-Pr)]
|
773 ± 2
|
787 ± 2
|
268 ± 2 |
|
[NEt
2
Me(i-Pr)]
|
788 ± 2 |
800 ± 2 |
280 ± 2 |
|
[NEt
3
(i-Pr)]
|
800 ± 2 |
808 ± 2 |
289 ± 3 |
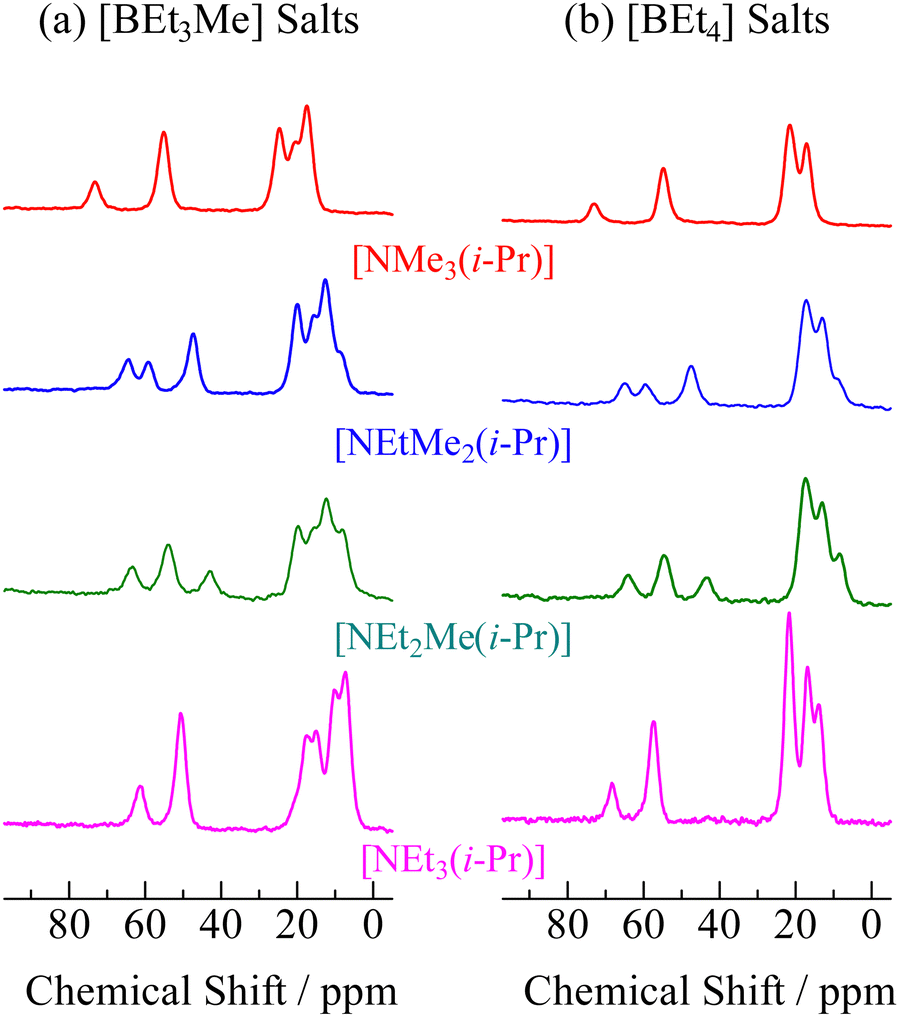
13C NMR
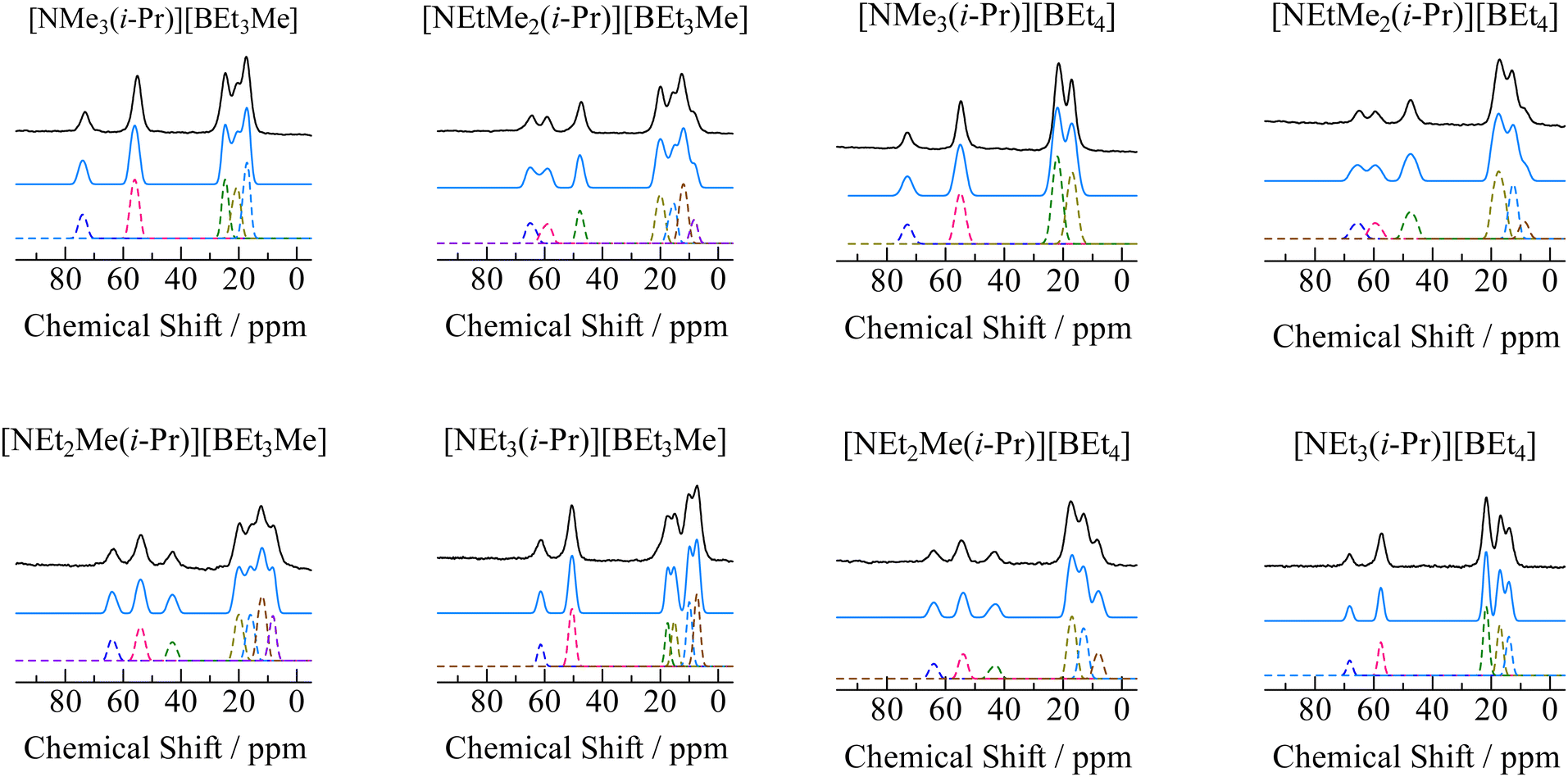
In order to assign ionic motions that occurred in each crystal, 13C NMR measurements of the powdered samples were performed without the MAS method. The 13C NMR spectra recorded in phase I for all crystals are shown in Fig. 4. Narrow line shapes were observed in the spectra. Because decouple pulses were applied to 1H nuclei in the measurement, it can be regarded that the line-breadths and-shapes of the 13C NMR spectra are caused by CSA rather than dipole–dipole interactions between 13C nuclei (ca. 1% 13C nuclei are distributed over the sample). In the case of [NEt4]Br, which forms an ordinal crystal, the 13C NMR signal of α-carbon shows a CSA powder pattern with a linewidth of 60–80 ppm.6 Based on this fact, estimating the line-breadths and -shapes of α-carbon in both ions benefits the discussion of the motional modes. The signals attributed to the cations and anions were fitted using Lorentz functions, as shown in Fig. 5 (the obtained CS values and assignments are listed in Table 3). The 13C NMR results suggest that both the cation and anion undergo isotropic reorientation in phase I, and the compounds treated in this study can be classified as ionic plastic crystals.
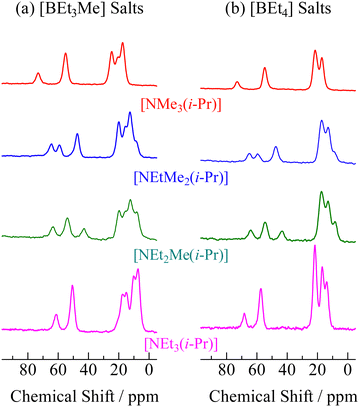 |
| | Fig. 4
13C NMR spectra recorded at 298 K (phase I) of [NEtxMe(3−x)(i-Pr)][BEt(4−y)Mey] (x = 0–3, y = 0, 1). | |
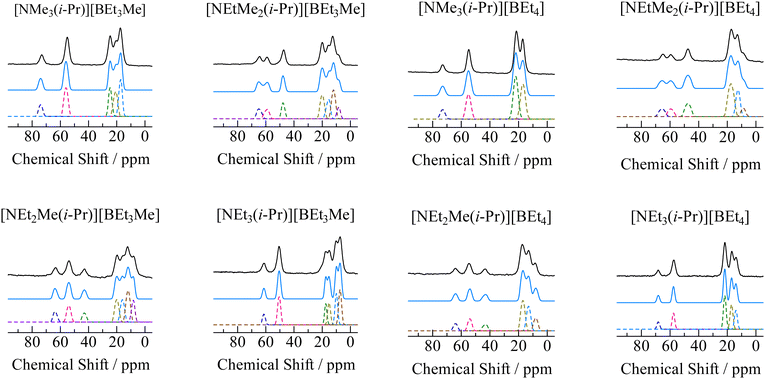 |
| | Fig. 5 Spectral analysis of 13C NMR spectra of [NEtxMe(3−x)(i-Pr)][BEt(4−y)Mey] (x = 0–3, y = 0, 1.). The observed signals are shown as solid black envelopes. The spectra are plotted as broken lines, and the sum of the lines is shown as a blue curve. | |
Table 3 Obtained chemical shift values (δ) in ppm and assignment of 13C NMR lines of [NEtxMe(3−x)(i-Pr)][BEt(4−y)Mey] (x = 0–3, y = 0, 1). Since linewidth was 4–6 ppm, accuracy of δ value is estimated within 2–3 ppm
|
|
N–![[C with combining low line]](https://www.rsc.org/images/entities/b_char_0043_0332.gif) H3 H3 |
N–H2–CH3 |
N–CH2–H3 |
N–H–(CH3)2 |
N–CH–(H3)2 |
B–H3 |
B–H2–CH3 |
B–CH2–H3 |
| [NMe3(i-Pr)][BEt3Me] |
56 |
— |
— |
74 |
21 |
21 |
25 |
17 |
| [NEtMe2(i-Pr)][BEt3Me] |
48 |
59 |
8 |
65 |
21 |
16 |
20 |
12 |
| [NEt2Me(i-Pr)][BEt3Me] |
43 |
54 |
8 |
64 |
21 |
16 |
20 |
12 |
| [NEt3(i-Pr)][BEt3Me] |
— |
51 |
7 |
61 |
18 |
15 |
18 |
10 |
| [NMe3(i-Pr)][BEt4] |
55 |
— |
— |
73 |
22 |
— |
22 |
17 |
| [NEtMe2(i-Pr)][BEt4] |
48 |
59 |
9 |
66 |
18 |
— |
18 |
13 |
| [NEt2Me(i-Pr)][BEt4] |
43 |
54 |
8 |
64 |
17 |
— |
17 |
13 |
| [NEt3(i-Pr)][BEt4] |
– |
57 |
14 |
68 |
22 |
— |
22 |
17 |
1H NMR spectra and relaxation time, T1
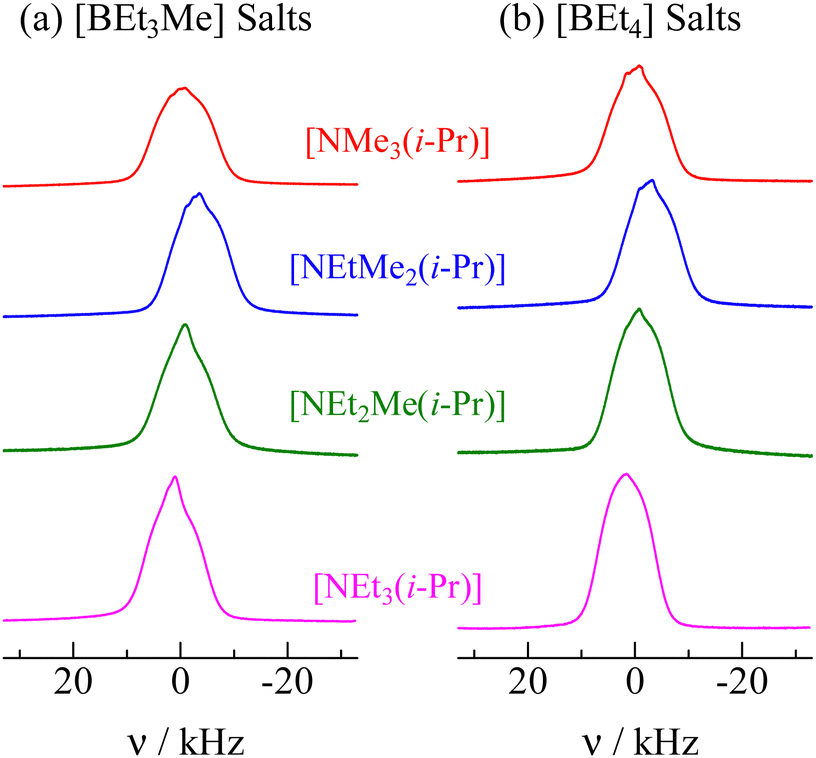
To obtain information about the inter- and intramolecular interactions, 1H NMR measurements of the powdered samples were carried out without the MAS method. The 1H NMR spectra recorded in phase I for all crystals treated in this study are shown in Fig. 6. Because narrow line shapes were recorded, the 1H NMR data also supported the conclusion that the constituents underwent isotropic reorientation in phase I.
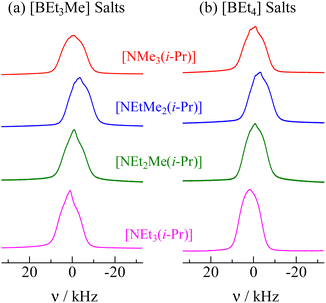 |
| | Fig. 6
1H NMR spectra recorded in phase I of [NEtxMe(3−x)(i-Pr)][BEt(4−y)Mey] (x = 0–3. y = 0, 1.). The whole lines were recorded at 298 K and 600.13 MHz. | |
To obtain information about ionic motion, 1H NMR T1 measurements were also performed. An inversion recovery method (pulse sequence: π – τ – π/2) was applied to estimate T1 values (τ: interval time between two pulses). Using this method, the magnetic recovery curves were fitted to the following single exponential function:44
| | 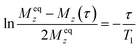 | (2) |
Here,
Meqz and
Mz(
τ) are the magnitudes of the magnetizations along the static magnetic field (
z-direction) at thermal equilibrium and at an interval time (
τ), respectively. Applying this equation to the recovery curve obtained, the
T1 value was determined for each sample at each temperature. This result indicates that one type of motion mainly contributes to the relaxation of the
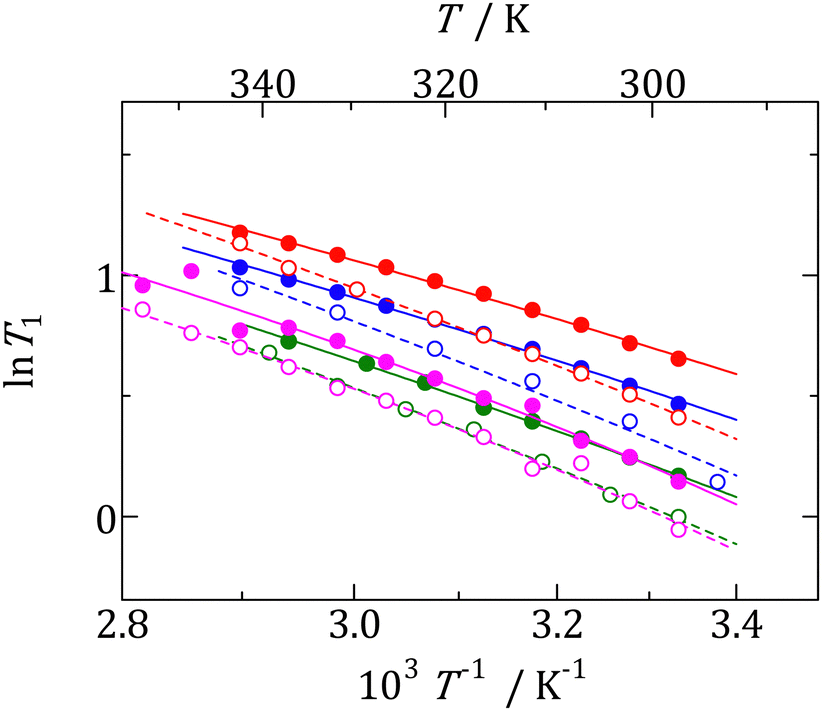
1H nuclear spins in phase I. The temperature dependence of
1H NMR
T1 values obtained for each compound are shown in
Fig. 7. The
T1 values increase with increasing temperature. This tendency is similar to that reported for plastic crystals with nonbranched alkyl groups, [NEt
xMe
(4−x)][BEt
(4−y)Me
y] (
x = 0–4,
y = 0, 1).
6,41 This monotonic increase indicates that the motion is sufficiently fast compared with the observed frequency (600.13 MHz). In this case, the following relation (high-temperature approximation) can be applied to
T1 analysis:
44| | 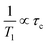 | (3) |
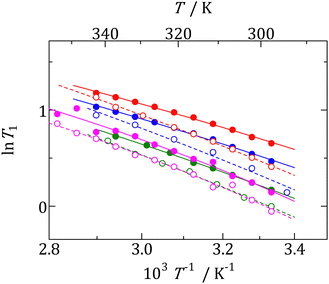 |
| | Fig. 7 Temperature dependences of 1H NMR T1 obtained in phase I of each compound. [NMe3(i-Pr)][BEt3Me] ( ), [NEtMe2(i-Pr)][BEt3Me] ( ), [NEtMe2(i-Pr)][BEt3Me] ( ), [NEt2Me(i-Pr)][BEt3Me] ( ), [NEt2Me(i-Pr)][BEt3Me] ( ), [NEt3(i-Pr)][BEt3Me] ( ), [NEt3(i-Pr)][BEt3Me] ( ), [NMe3(i-Pr)][BEt4] ( ), [NMe3(i-Pr)][BEt4] ( ), [NEtMe2(i-Pr)][BEt4] ( ), [NEtMe2(i-Pr)][BEt4] ( ), [NEt2Me(i-Pr)][BEt4] ( ), [NEt2Me(i-Pr)][BEt4] ( ), [NEt3(i-Pr)][BEt4] ( ), [NEt3(i-Pr)][BEt4] ( ). ). | |
In general, the correlation time (τc) can be linked to the Arrhenius relation:
| | 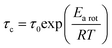 | (4) |
Here,
τ0,
Earot, and
R are the correlation time at
T = ∞, the activation energy of molecular motion, and the gas constant, respectively. Using
eqn (3) and (4), the straight line displayed in
Fig. 7 and the
Earot values listed in
Table 4 were obtained. Based on the result of the
13C NMR spectra, it can be concluded that the isotropic reorientation motion contributes to
T1 relaxation. The obtained
Ea values were slightly smaller than those of the non-branched plastic crystals of [NEt
xMe
(4−x)][BEt
(4−y)Me
y] (
x = 0–4,
y = 0, 1).
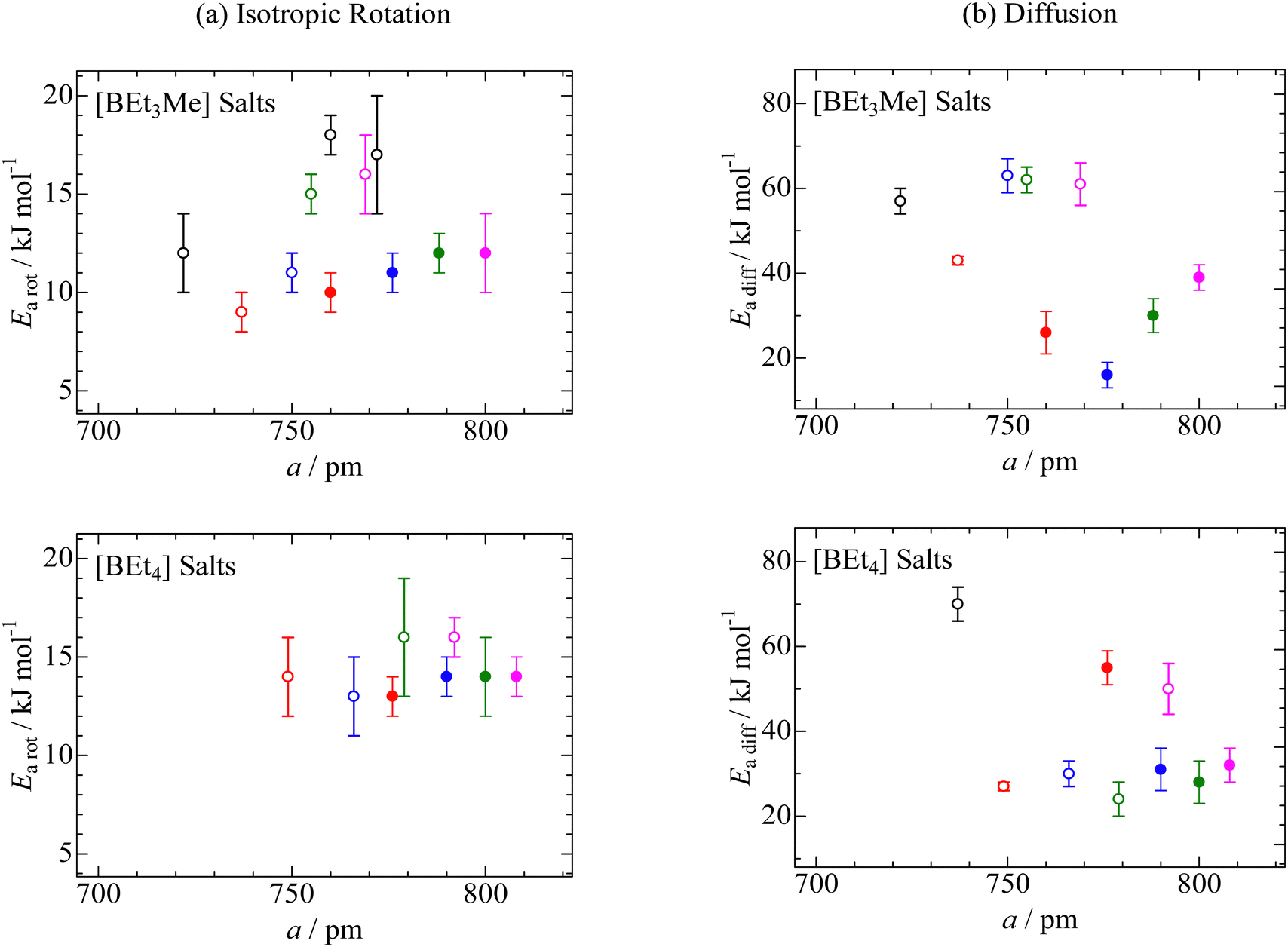
6,41 To reveal the substituent effects on isotropic reorientation, we plotted the
Earot values as a function of
a, estimated from the XRD data (
Fig. 8a). The figure shows that the
Earot values of the compounds containing i-Pr group are related to the
a values. In addition, the
Earot values were lower than those of the corresponding salts without branched structures in the cation. As described in the XRD section, compounds with i-Pr groups have larger lattice constants than salts without a branched cation structure. Based on the NMR and XRD results, it can be concluded that the i-Pr group reduced the Coulombic interaction between the cation and anion, and the shielding effect of the CH
3 group resulted in large apparent cation sizes and low
Earot values for isotropic rotation.
Table 4 Activation energies Earot in kJ mol−1 of isotropic reorientation motion of each crystal
|
|
[BEt3Me] |
[BEt4] |
|
[BEt3Me] |
[BEt4] |
|
Ref. 6.
Ref. 41.
|
| [NMe3(i-Pr)] |
10 ± 1 |
13 ± 1 |
[NEtMe3] |
9 ± 1a |
14 ± 1b |
| [NEtMe2(i-Pr)] |
11 ± 1 |
14 ± 1 |
[NEt2Me2] |
11 ± 1a |
13 ± 1b |
| [NEt2Me(i-Pr)] |
12 ± 1 |
14 ± 1 |
[NEt3Me] |
15 ± 1a |
16 ± 1b |
| [NEt3(i-Pr)] |
12 ± 1 |
14 ± 1 |
[NEt4] |
16 ± 2a |
16 ± 1b |
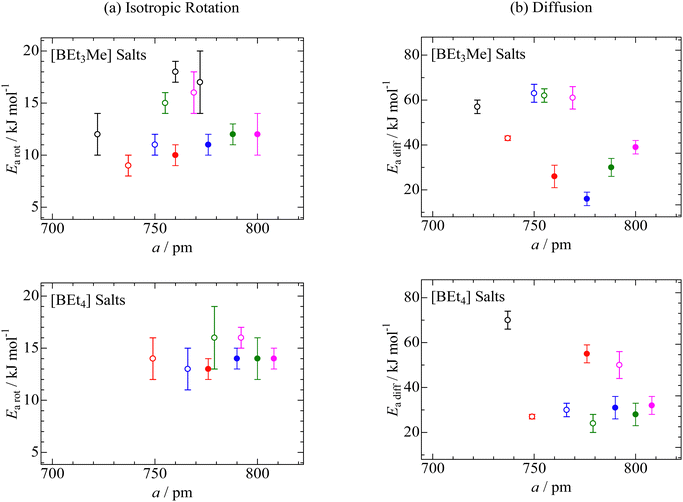 |
| | Fig. 8 Activation energies of (a) isotropic reorientation (Earot) and (b) ion diffusion (Eadiff) as a function of lattice constant (a) in CsCl-type cubic crystal structures of [BEt3Me] and [BEt4] compounds with [NMe3(i-Pr)] ( ), [NEtMe2(i-Pr)] ( ), [NEtMe2(i-Pr)] ( ), [NEt2Me(i-Pr)] ( ), [NEt2Me(i-Pr)] ( ), and [NEt3(i-Pr)] ( ), and [NEt3(i-Pr)] ( ). In the figures, the reported data of [BEt3Me] and [BEt4] salts with [NMe4] ( ). In the figures, the reported data of [BEt3Me] and [BEt4] salts with [NMe4] ( ), [NMe3Et] ( ), [NMe3Et] ( ), [NEt2Me2] ( ), [NEt2Me2] ( ), [NEt3Me] ( ), [NEt3Me] ( ), and [NEt4] ( ), and [NEt4] ( ) and the others ( ) and the others ( ),6,38,41 are also plotted. ),6,38,41 are also plotted. | |
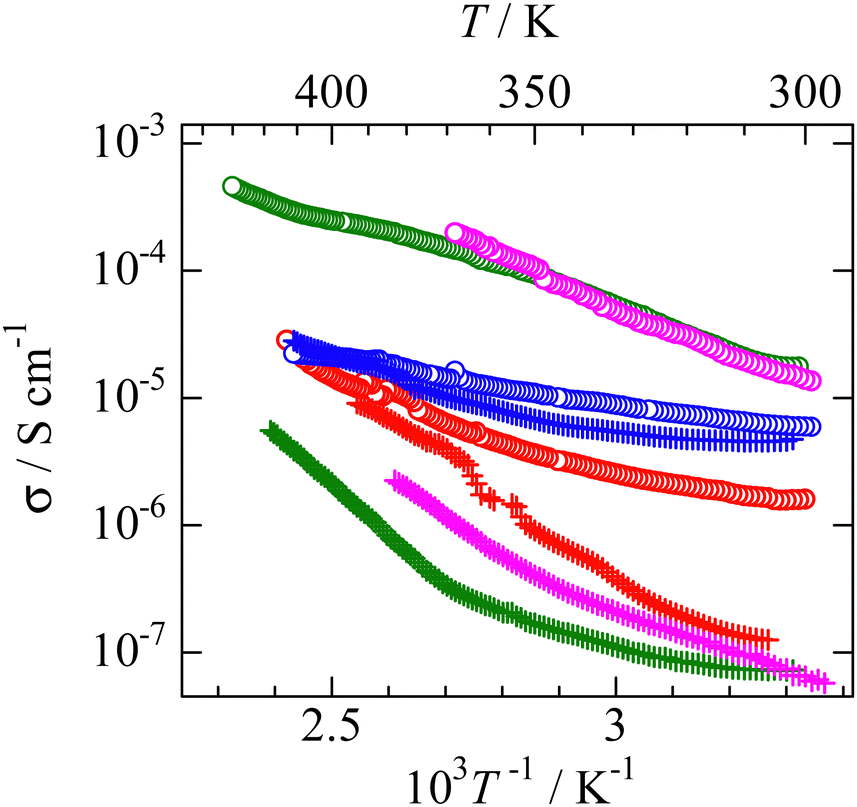
Electrical conductivity
Since it is well known that the constituents in the plastic crystal can jump to the neighbor crystal defect with small activation energies,6,37–41 electrical conductivity (σ) measurements were performed as a function of temperature. The results are presented in Fig. 9. The obtained σ values were similar to those reported for nonbranched plastic crystals of [NEtxMe(4−x)][BEtyMe(y−3)] (x = 0–4, y = 0, 1).6,41 Here, the data fluctuation was recorded at 360 K in the [NMe3(i-Pr)][NEt4] crystal. Because the DSC (Fig. 2) charts do not show any change around the temperature, a measurement error is considered in the σ observation. The activation energy of ion diffusion (Eadiff) was estimated using the following equation:| | 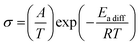 | (5) |
Here, A is a constant that depends on the number of vacancies, ion jump distance, and crystal structure. Plotting ln (σT) as a function of T−1 (Fig. S2, ESI†), the Eadiff values listed in Table 5 were obtained. Some compounds showed a change in the slope (Fig. S2, ESI†). Such phenomena are frequently recorded when the mobilities of cations and anions differ in the plastic phase6,14,25–29,34–41 The similar Eadiff values are obtained in the [NMe3(i-Pr)][BEtyMe(y−3)] crystals to those of [NEtxMe(4−x)][BEtyMe(y−3)], as shown in Table 5. The Eadfft values were plotted as a function of the lattice constant, similar to those for isotropic rotation, to reveal the substituent effects on ion diffusion. The results are presented in Fig. 8b. A branching effect on the Eadfft values was observed for the [BEt3Me] compounds; conversely, little contribution was recorded for the [BEt4] salts. The difference between [BEt3Me] and [BEt4] can also be explained by the shielding effect of the alkyl groups; because the [BEt4]− anion has one more CH2 group, it can be considered that the shielding effect is sufficient. Therefore, the shielding effect of the i-Pr group on the cations decreased in the compounds.
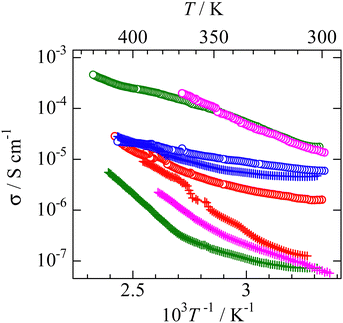 |
| | Fig. 9 Temperature dependences of electrical conductivity (s) in phase I of each salt. [NMe3(i-Pr)][BEt3Me] ( ), [NEtMe2(i-Pr)][BEt3Me] ( ), [NEtMe2(i-Pr)][BEt3Me] ( ), [NEt2Me(i-Pr)][BEt3Me] ( ), [NEt2Me(i-Pr)][BEt3Me] ( ), [NEt3(i-Pr)][BEt3Me] ( ), [NEt3(i-Pr)][BEt3Me] ( ), [NMe3(i-Pr)][BEt4] ( ), [NMe3(i-Pr)][BEt4] ( ), [NEtMe2(i-Pr)][BEt4] ( ), [NEtMe2(i-Pr)][BEt4] ( ), [NEt2Me(i-Pr)][BEt4] ( ), [NEt2Me(i-Pr)][BEt4] ( ), [NEt3(i-Pr)][BEt4] ( ), [NEt3(i-Pr)][BEt4] ( ). ). | |
Table 5 Activation energies Eadiff (kJ mol−1) obtained by electrical conductivity measurements. (Eadiff values in parenthesis are estimated in each high-temperature region)
|
|
[BEt3Me] |
[BEt4] |
|
[BEt3Me] |
[BEt4] |
|
Ref. 6.
Ref. 41.
|
| [NMe3(i-Pr)] |
26 ± 5 |
55 ± 4 (49 ± 5) |
[NEtMe3] |
43 ± 1a |
27 ± 1b |
| [NEtMe2(i-Pr)] |
16 ± 3 |
31 ± 5 |
[NEt2Me2] |
63 ± 4a |
30 ± 3b |
| [NEt2Me(i-Pr)] |
30 ± 4 |
28 ± 5 (80 ± 4) |
[NEt3Me] |
62 ± 3a |
24 ± 4b |
| [NEt3(i-Pr)] |
39 ± 3 |
32 ± 4 (65 ± 4) |
[NEt4] |
61 ± 5a |
50 ± 6b |
Conclusion
In this study, eight new ionic plastic crystals [NEtxMe(3−x)(i-Pr)][BEt(4−y)Mey] (x = 1–3, y = 0, 1) was observed, revealing that the shielding effect of the alkyl group contributed to the formation of an ionic plastic crystalline phase. DSC measurements showed large entropy changes at the phase transition temperature to the plastic phase (phase I) (except for [NEt2Me(i-Pr)][BEt3Me] and [NEt3(i-Pr)][BEt3Me]). The powder XRD patterns were indexed to a CsCl-type cubic structure in each plastic phase. The line shapes of the solid-state 13C NMR spectra revealed that both cations and anions underwent isotropic reorientation in the plastic crystalline phases. The solid-state 1H NMR spectra also confirmed this motion. The Earot values were determined by 1H NMR T1 measurements. The temperature dependence of the electrical conductivity was estimated using the Eadiff values. Analysis of the XRD data reveals that the apparent size (rcation) of the [NEtxMe(3−x)(i-Pr)] cation is slightly larger than that of the corresponding [NEtxMe(3−x)Et] cation. In addition, the Earot and Eadiff values of the [BEt3Me] compounds with branched cations were lower than those of the non-branched salts. In contrast, the shielding effect was reduced to the Eadiff values in the [BEt3Me] salts. Although preparing and comparing more compounds is necessary to investigate the shielding effect quantitatively, qualitative validation was achieved in this study.
Author contributions
K. N.: investigation, formal analysis, methodology; Y. O.: investigation, formal analysis, methodology; H. H.: conceptualization, supervision, project administration, writing – original draft.
Data availability
The NMR, DSC, XRD, and conductivity data discussed in this manuscript can be found in the ESI,† and at the following site: DOI: https://doi.org/10.57760/sciencedb.12344.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was partly supported by JSPS KAKENHI Grant Number JP23K04693.
References
- K. Biernacka, D. Al-Masri, R. Yunis, H. Zhu, A. F. Hollenkamp and J. M. Pringle, Electrochim. Acta, 2020, 357, 136863 CrossRef CAS.
- Y. Shekibi, A. Gray-Weale, D. R. MacFarlane, A. J. Hill and M. Forsyth, J. Phys. Chem. C, 2007, 111, 11463 CrossRef CAS.
- H. Zhu, X. Wang, R. Vijayaraghava, Y. Zhou, D. R. MacFarlane and M. Forsyth, J. Phys. Chem. Lett., 2018, 9, 3904 CrossRef CAS PubMed.
- Y. Shun, Y. Hiromasa, T. Yuko, R. Masahiro and Y.-F. Masahiro, New J. Chem., 2019, 43, 4008 RSC.
- K. Akira, Y. Jun, R. Subham, T. Satoshi and H. Hisashi, Phys. Chem. Chem. Phys., 2023, 25, 2783 Search PubMed.
- H. Tomoyuki, H. Satoru and H. Hisashi, Bull. Chem. Soc. Jpn., 2013, 86, 993 CrossRef.
- H. B. Han, J. Nie, K. Liu, W. K. Li, W. F. Feng, M. Armand, H. Matsumoto and Z. B. Zhou, Electrochim. Acta, 2009, 55, 1221 CrossRef.
- J. Efthimiadis, S. J. Pas, M. Forsyth and D. R. MacFarlane, Solid State Ionics, 2002, 154–155, 279 CrossRef CAS.
- Y. Abu-Lebdeh, A. Abouimrane, P.-J. Alarco and M. Armand, J. Power Sources, 2006, 154, 255 CrossRef CAS.
- G. Annat, J. Adebahr, I. R. McKinnon, D. R. MacFarlane and M. Forsyth, Solid State Ionics, 2007, 178, 1065 CrossRef CAS.
- J. Sun, D. R. MacFarlane and M. Forsyth, Solid State Ionics, 2002, 148, 145 CrossRef CAS.
- R. Ikeda, S. Ishimaru, T. Tanabe and D. Nakamura, J. Mol. Struct., 1995, 345, 151 CrossRef CAS.
- J. Janikowski, M. R. Razali, S. R. Batten, D. R. MacFarlane and J. M. Pringle, ChemPlusChem, 2012, 77, 1039 CrossRef CAS.
- H. Satoru, K. Yuka, H. Tomoyuki and H. Hisashi, Bull. Chem. Soc. Jpn., 2015, 88, 1735 CrossRef.
- J. Adebahr, M. Grimsley, N. M. Rocher, D. R. MacFarlane and M. Forsyth, Solid State Ionics, 2008, 178, 1798 CrossRef CAS.
- Y. M. Solonin, V. F. Gorban and E. A. Graivoronskaya, Dopov. Nats. Akad. Nauk Ukr, 2008, 4, 114 Search PubMed.
- W. I. F. David, Appl. Radiat. Isot., 1995, 46, 519 CrossRef CAS.
- Y. Jin, A. Xenopoulos, J. Cheng, W. Chen, B. Wunderlich, M. Diack, C. Jin, R. L. Hettich, R. N. Compton and G. Guiochon, Mol. Cryst. Liq. Cryst. Sci. Technol., Sect. A, 1994, 257, 235 CrossRef CAS.
- S. Takeda and T. Atake, Kotai Butsuri, 1993, 28, 183 CAS.
- G. Burns, F. H. Dacol and B. Welber, Solid State Commun., 1979, 32, 151 CrossRef CAS.
- M. Debeau and P. Depondt, J. Chim. Phys. Phys. Chim. Biol., 1985, 82, 233 CrossRef CAS.
- S. Ganguly, J. R. Fernandes and C. N. R. Rao, Adv. Mol. Relax. Interact. Proc., 1981, 20, 149 CrossRef CAS.
- K. Kobashi and R. D. Etters, Mol. Phys., 1982, 46, 1077 CrossRef CAS.
- Y. Furukawa, H. Nagase, R. Ikeda and D. Nakamura, Bull. Chem. Soc. Jpn., 1991, 64, 3105 CrossRef CAS.
- M. Kenmotsu, H. Honda, H. Ohki, R. Ikeda, T. Erata, A. Tasaki and Y. Furukawa, Z. Naturforsch., A: Phys. Sci., 1994, 49, 247 CrossRef CAS.
- H. Honda, M. Kenmotsu, H. Ohki, R. Ikeda and Y. Furukawa, Ber. Bunsenges. Phys. Chem., 1995, 99, 1009 CrossRef CAS.
- H. Honda, S. Ishimaru, N. Onoda-Yamamuro and R. Ikeda, Z. Naturforsch., A: Phys. Sci., 1995, 50, 871 CrossRef CAS.
- H. Honda, M. Kenmotsu, N. Onoda-Yamamuro, H. Ohki, S. Ishimaru, R. Ikeda and Y. Furukawa, Z. Naturforsch., A: Phys. Sci., 1996, 51, 761 CrossRef CAS.
- H. Honda, N. Onoda-Yamamuro, S. Ishimaru, R. Ikeda, O. Yamamuro and T. Matsuo, Ber. Bunsen-Ges. Phys. Chem., 1998, 102, 148 CrossRef CAS.
- N. Onoda-Yamamuro, H. Honda, R. Ikeda, O. Yamamuro, T. Matsuo, K. Oikawa, T. Kamiyama and F. Izumi, J. Phys.: Condens. Matter, 1998, 10, 3341 CrossRef CAS.
- H. Honda, Z. Naturforsch., A: Phys. Sci., 2007, 62, 633 CrossRef CAS.
- R. Ikeda, Recent Res. Dev. Chem. Phys., 2004, 5, 257 CAS.
- A. J. Seeber, M. Forsyth, C. M. Forsyth, S. A. Forsyth, G. Annat and D. R. MacFarlane, Phys. Chem. Chem. Phys., 2003, 5, 2692 RSC.
- H. Ono, S. Ishimaru and R. Ikeda, Ber. Bunsenges. Phys. Chem., 1998, 102, 650 CrossRef CAS.
- H. Ono, S. Ishimaru, R. Ikeda and H. Ishida, Bull. Chem. Soc. Jpn., 1999, 72, 2049 CrossRef CAS.
- M. Tomoyuki, J. Org. Chem., 2019, 895, 23 CrossRef.
- H. Tomoyuki, H. Satoru and H. Hisashi, Z. Naturforsch., A: Phys. Sci., 2014, 69, 433 CrossRef.
- K. Yuka and H. Hisashi, Bull. Chem. Soc. Jpn., 2019, 92, 768 CrossRef.
- Y. Yamada, E. Kashimoto and H. Honda, Bull. Chem. Soc. Jpn., 2019, 92, 1289 CrossRef CAS.
- K. Nagai and H. Honda, Z. Naturforsch., A: Phys. Sci., 2022, 77, 899 CrossRef CAS.
- S. Hirakawa and H. Honda, Z. Naturforsch., A: Phys. Sci., 2015, 70, 521 CrossRef CAS.
- J. Timmermans, J. Phys. Chem. Solids, 1961, 18, 1 CrossRef CAS.
- A. T. Liu, M. Nag, W. R. Caroll and J. D. Roberts, Magn. Reson. Chem., 2013, 51, 701 CrossRef CAS PubMed.
-
Nuclear Magnetic Resonance and Relaxation, ed. B. Cowan, Cambridge University Press, Cambridge, 1997 Search PubMed.
|
| This journal is © the Owner Societies 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*
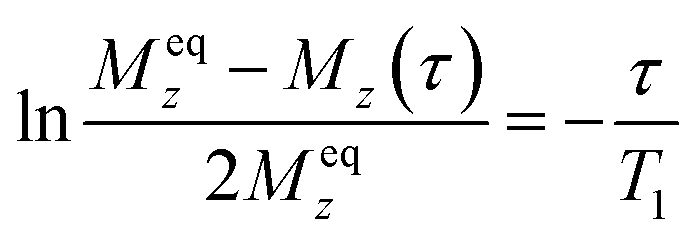
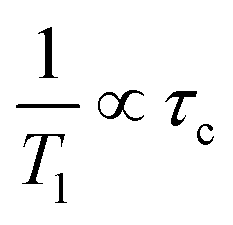
 ), [NEtMe2(i-Pr)][BEt3Me] (
), [NEtMe2(i-Pr)][BEt3Me] ( ), [NEt2Me(i-Pr)][BEt3Me] (
), [NEt2Me(i-Pr)][BEt3Me] ( ), [NEt3(i-Pr)][BEt3Me] (
), [NEt3(i-Pr)][BEt3Me] ( ), [NMe3(i-Pr)][BEt4] (
), [NMe3(i-Pr)][BEt4] ( ), [NEtMe2(i-Pr)][BEt4] (
), [NEtMe2(i-Pr)][BEt4] ( ), [NEt2Me(i-Pr)][BEt4] (
), [NEt2Me(i-Pr)][BEt4] ( ), [NEt3(i-Pr)][BEt4] (
), [NEt3(i-Pr)][BEt4] ( ).
).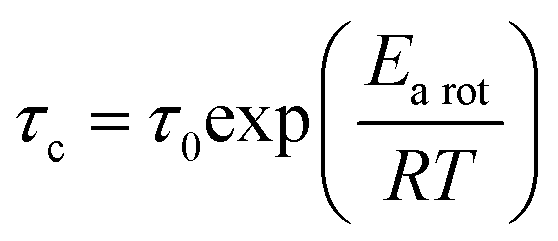
 ), [NEtMe2(i-Pr)] (
), [NEtMe2(i-Pr)] ( ), [NEt2Me(i-Pr)] (
), [NEt2Me(i-Pr)] ( ), and [NEt3(i-Pr)] (
), and [NEt3(i-Pr)] ( ). In the figures, the reported data of [BEt3Me] and [BEt4] salts with [NMe4] (
). In the figures, the reported data of [BEt3Me] and [BEt4] salts with [NMe4] ( ), [NMe3Et] (
), [NMe3Et] ( ), [NEt2Me2] (
), [NEt2Me2] ( ), [NEt3Me] (
), [NEt3Me] ( ), and [NEt4] (
), and [NEt4] ( ) and the others (
) and the others ( ),6,38,41 are also plotted.
),6,38,41 are also plotted.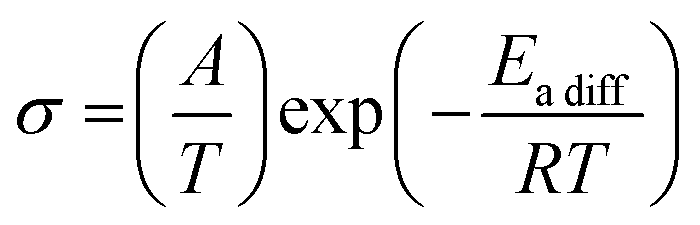
 ), [NEtMe2(i-Pr)][BEt3Me] (
), [NEtMe2(i-Pr)][BEt3Me] ( ), [NEt2Me(i-Pr)][BEt3Me] (
), [NEt2Me(i-Pr)][BEt3Me] ( ), [NEt3(i-Pr)][BEt3Me] (
), [NEt3(i-Pr)][BEt3Me] ( ), [NMe3(i-Pr)][BEt4] (
), [NMe3(i-Pr)][BEt4] ( ), [NEtMe2(i-Pr)][BEt4] (
), [NEtMe2(i-Pr)][BEt4] ( ), [NEt2Me(i-Pr)][BEt4] (
), [NEt2Me(i-Pr)][BEt4] ( ), [NEt3(i-Pr)][BEt4] (
), [NEt3(i-Pr)][BEt4] ( ).
).