
Biocatalytic reductive aminations with NAD(P)H-dependent enzymes: enzyme discovery, engineering and synthetic applications†
Bo
Yuan
 *ab,
Dameng
Yang
a,
Ge
Qu
ab,
Nicholas J.
Turner
*c and
Zhoutong
Sun
*ab
*ab,
Dameng
Yang
a,
Ge
Qu
ab,
Nicholas J.
Turner
*c and
Zhoutong
Sun
*ab
aTianjin Institute of Industrial Biotechnology, Chinese Academy of Sciences, Tianjin 300308, China. E-mail: sunzht@tib.cas.cn; yuanb@tib.cas.cn
bKey Laboratory of Engineering Biology for Low-Carbon Manufacturing, Tianjin Institute of Industrial Biotechnology, Chinese Academy of Sciences, 32 West 7th Avenue, Tianjin Airport Economic Area, Tianjin 300308, China
cDepartment of Chemistry, Manchester Institute of Biotechnology, University of Manchester, Manchester M1 7DN, UK. E-mail: nicholas.turner@manchester.ac.uk
First published on 7th December 2023
Abstract
Chiral amines are pivotal building blocks for the pharmaceutical industry. Asymmetric reductive amination is one of the most efficient and atom economic methodologies for the synthesis of optically active amines. Among the various strategies available, NAD(P)H-dependent amine dehydrogenases (AmDHs) and imine reductases (IREDs) are robust enzymes that are available from various sources and capable of utilizing a broad range of substrates with high activities and stereoselectivities. AmDHs and IREDs operate via similar mechanisms, both involving a carbinolamine intermediate followed by hydride transfer from the co-factor. In addition, both groups catalyze the formation of primary and secondary amines utilizing both organic and inorganic amine donors. In this review, we discuss advances in developing AmDHs and IREDs as biocatalysts and focus on evolutionary history, substrate scope and applications of the enzymes to provide an outlook on emerging industrial biotechnologies of chiral amine production.
 Bo Yuan | Dr Bo Yuan obtained her doctoral degree in Biochemistry from the Turner Group at University of Manchester in 2011, and then joined Croda Europe Ltd. as a Research Scientist. In 2016 she moved to Xi’an Jiaotong University as a Lecturer. She joined Tianjin Institute of Industrial Biotechnology, Chinese Academy of Sciences as an Associate Professor in 2021. Her research interests are in protein engineering, asymmetric synthesis and chemoenzymatic cascade reactions. |
 Dameng Yang | Dr Dameng Yang received his PhD degree in Biochemistry from Tianjin Institute of Industrial Biotechnology, Chinese Academy of Sciences in 2022 under the guidance of Prof. Zhoutong Sun, and then joined Meihua Holdings Group Co., Ltd as a project leader. His research interests are in synthetic biology, enzyme engineering and metabolic engineering. |
 Ge Qu | Dr Ge Qu received his doctoral degree in bioinformatics from Adam Mickiewicz University in Poland in 2015. After graduation, he joined the group of Professor Zhoutong Sun at Tianjin Institute of Industrial Biotechnology (TIB), Chinese Academy of Sciences. Currently he is an Associate Professor at TIB and working in the fields of rational enzyme design and biosynthesis. |
 Nicholas J. Turner | Prof. Nicholas Turner obtained his DPhil in 1985 with Professor Sir Jack Baldwin and from 1985–1987 was a Royal Society Junior Research Fellow at Harvard University with Prof. George Whitesides. He was appointed lecturer in 1987 at Exeter University and moved to Edinburgh in 1995, initially as a Reader and subsequently Professor in 1998. In October 2004 he joined Manchester University as Professor of Chemical Biology. He is Director of the Centre of Excellence in Biocatalysis (CoEBio3) and a Co-Director of SYNBIOCHEM, the BBSRC Synthetic Biology Research Centre. His research interests are in the following fields: use of enzymes as catalysts for organic synthesis; directed evolution of enzymes; protein engineering; biocatalytic retrosynthesis using Computer Assisted Synthesis Planning (CASP) tools; biocatalytic cascade reactions; enzymatic synthesis of therapeutic oligonucleotides. |
 Zhoutong Sun | Prof. Zhoutong Sun obtained his doctoral degree in microbiology with Prof. Sheng Yang at Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences in 2012, he then moved to Nanyang Technological University in Singapore as a research fellow in metabolic engineering. One year later, he joined the Reetz group as a postdoc at MPI für Kohlenforschung and Marburg University in directed evolution and biocatalysis. In 2016, he became a full professor at Tianjin Institute of Industrial Biotechnology, Chinese Academy of Sciences. His research interests are in enzyme engineering, metabolic engineering and synthetic biology. |
1. Introduction
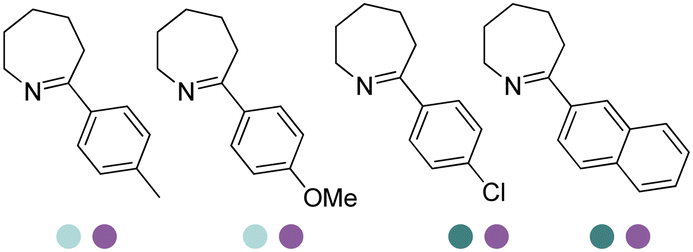
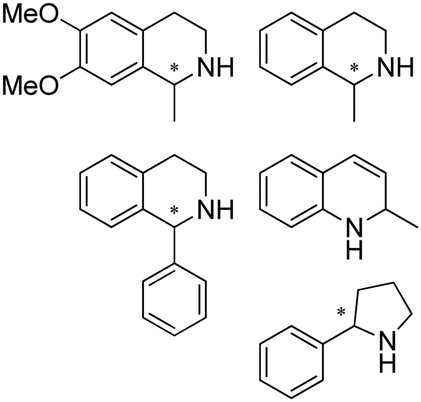
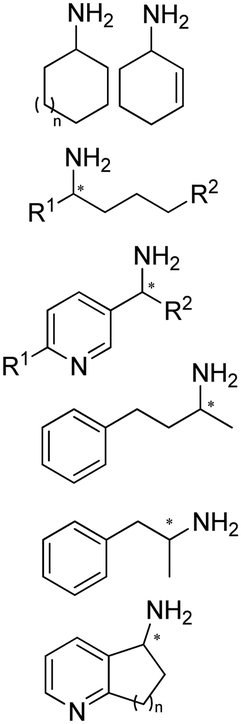
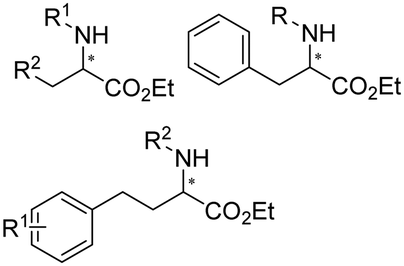
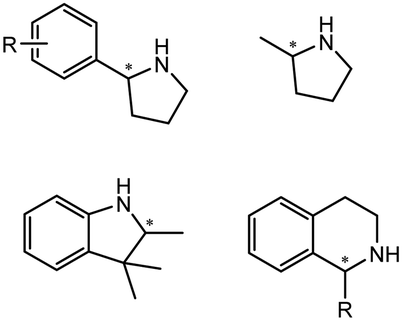
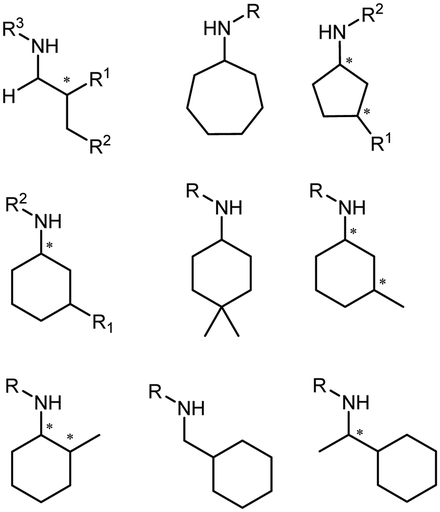
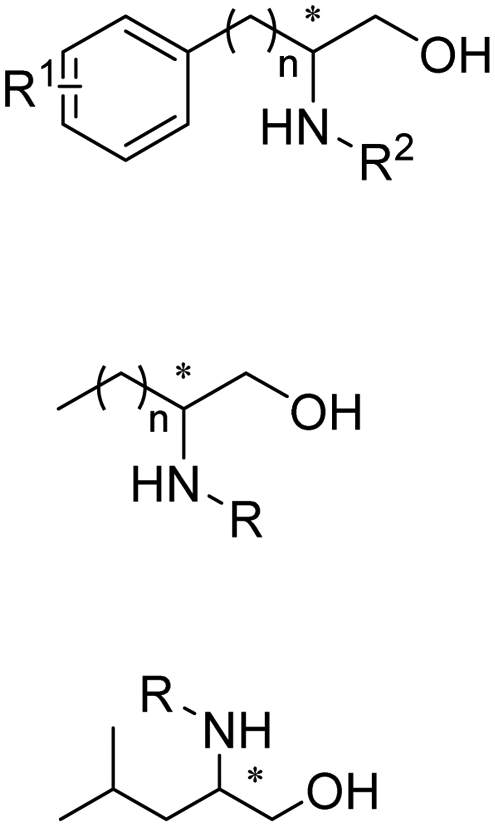
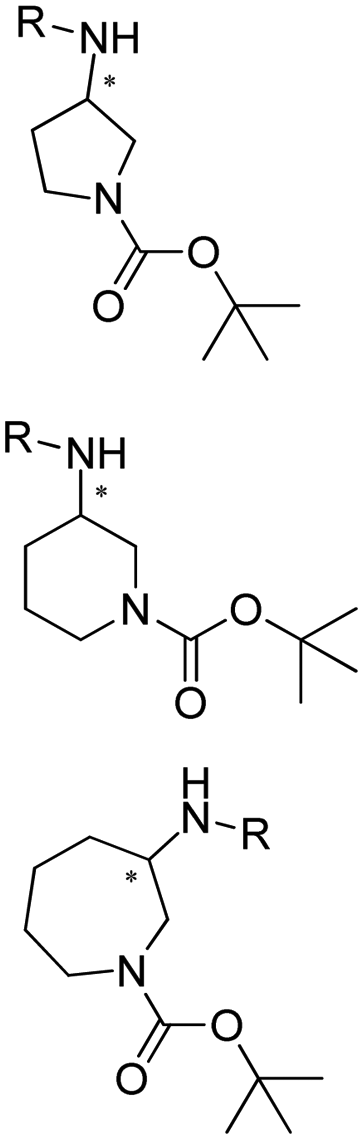
Enantiomerically pure amines are present in numerous pharmaceutical products, agrochemicals, natural products, and functional materials.1–4 Many asymmetric synthetic methods have been developed to synthesize this class of compounds. Among them, asymmetric reductive amination (ARA) plays a pivotal role in the construction of C–N bonds in pharmaceutical chemistry.5 For example, suvorexant (hypnotic),6 sitagliptin (antidiabetic),7 sertraline (antidepressant),8 escitalopram (antidepressant),9 levobupivacaine (anesthetic),10 rivastigmine (dementia treatment)11 (Fig. 1) are highly valuable drugs that have been synthesized employing at least one reductive amination step. Although transition-metal catalyzed ARA has been showcased by a number of successful strategies,12 it is less developed than imine reduction.13,14 It is difficult partially due to the inhibition caused by coordination of the amine to the metal catalyst or the incompatibility of transition metal hydrides with ketones.15 On the other hand, enzymatic ARA can be achieved with benefits such as generally higher stereoselectivities, superior chemo- and regioselectivities, higher turnover rates, and mild reaction conditions.16,17 Among the biocatalysts that transform various substrates into amines, several classes of enzymes are prominent due to wide applicability, including flavin-dependent monoamine oxidases (MAOs),18 pyridoxal 5-phosphate (PLP)-dependent ω-transaminases (ATAs),19 phenylalanine ammonia lyases (PALs)20 and P450 monooxgenases (P450s). Alternatively, NAD(P)H-dependent amine dehydrogenases (AmDHs),21–25 imine reductases (IREDs)26 and reductive aminases (RedAms)27 are classes of enzymes that utilizes both organic and inorganic ammonia sources27,28 and a broad range of substrates such as aldehydes, ketones, keto acids or keto esters to form amine products. The catalytic mechanisms of these classes of enzyme are similar, involving the formation of a carbinolamine intermediate with subsequent hydride transfer from the nicotinamide cofactor.29,30 Although AmDHs traditionally use free ammonia as the amine donor, generating primary amines as the products, it has also been shown that AmDHs can accept organic amine donors and produce secondary amines.31,32 Similarly, IREDs and RedAms can also use free ammonia as amine donors.33 In this review, we describe the discovery, evolutionary history, substrate scope and synthetic applications of AmDHs, IREDs and RedAms, in order to provide a comprehensive overview of these classes of enzymes and their applications in synthesis.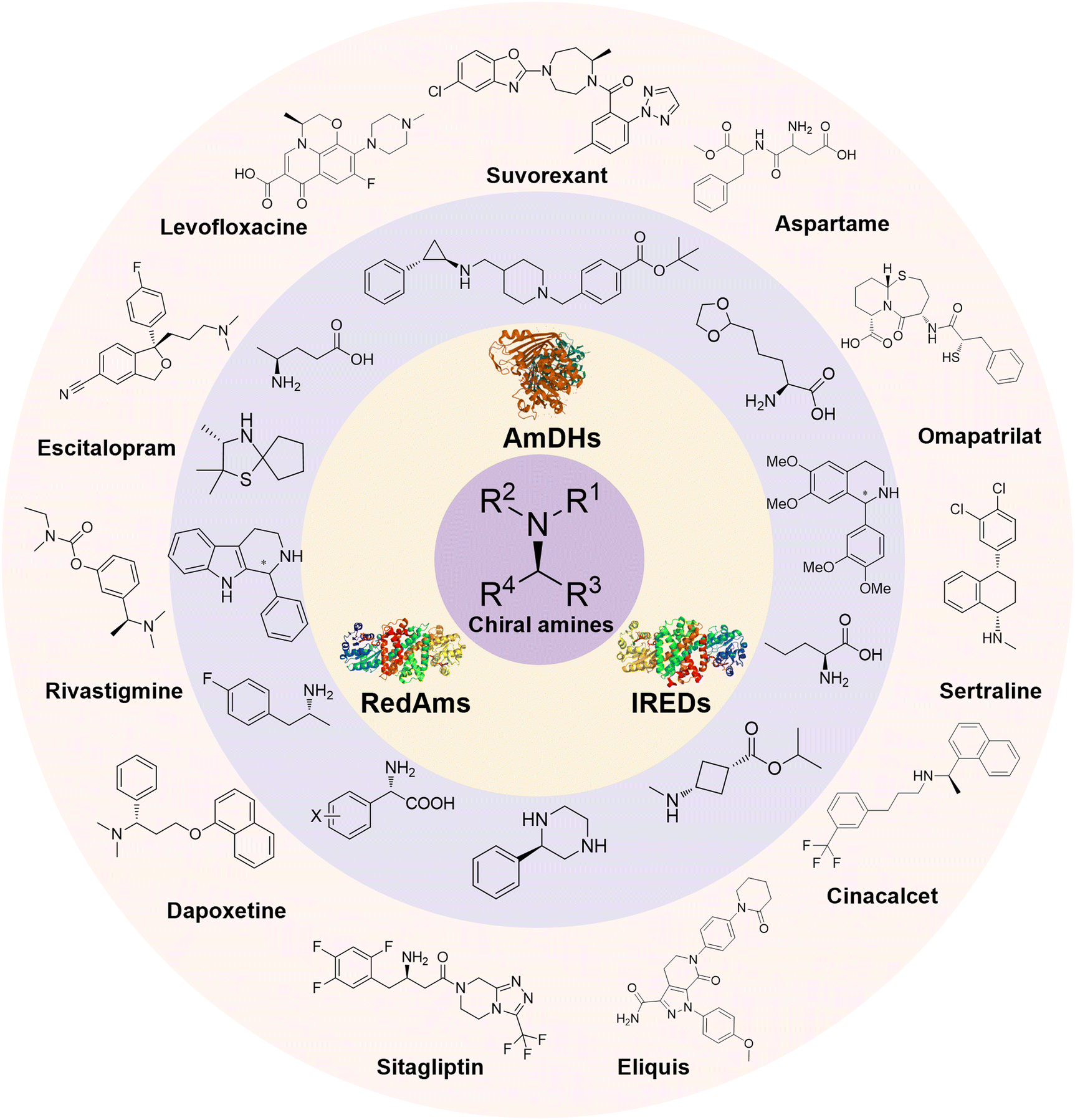 | ||
| Fig. 1 Chiral amine building blocks and drug intermediates synthesized by reductive amination. | ||
2. Amine dehydrogenases (AmDHs)
In 2012, LeuDH was engineered to accept ketones as substrates as reported firstly by Abrahamson et al.34 Following this breakthrough, many efforts have been invested in the identification, engineering, and generation of AmDHs from various sources. In addition, native AmDHs have also been discovered,29 which do not share sequence homology with the engineered AmDHs. In the following sections, we review the discovery of AmDHs from the above-mentioned pathways, and summarize the structural, mechanistic, engineering and synthetic application of AmDHs.2.1. Structural and mechanistic insights
A variety of AmDHs have been characterized and their crystal structures determined in apo form or in complex with cofactors or substrates. Based on the structural analysis, the catalytic mechanisms of AmDHs have been investigated in-depth. Table 1 lists the crystal structures of experimentally validated LeuDHs, PheDHs, and AmDHs.| Names | Origin | PDB ID | In complex with | Ref. |
|---|---|---|---|---|
| LeuDHs | ||||
| WT | Lysinibacillus sphaericus | 1LEH | — | 35 |
| WT | Sporosarcina psychrophila | 3VPX | — | 36 |
| WT | Geobacillus stearothermophilus 10 | 6ACF | — | 37 |
| WT | Geobacillus stearothermophilus 10 | 6ACH | NAD+ | 37 |
| WT | Exiguobacterium sibiricum | 8GXD | — | 38 |
| PheDHs | ||||
| WT | Rhodococcus sp. | 1BXG | NAD+ and β-phenylpropionate | 39 |
| WT | Rhodococcu sp. | 1BW9 | NAD+ and phenylpyruvate | 39 |
| WT | Rhodococcus sp. | 1C1X | NAD+ and L-3-phenyllactate | 40 |
| WT | Rhodococcus sp. | 1C1D | NADH and L-phenylalanine | 40 |
| AmDHs | ||||
| WT | Petrotoga mobilis SJ95 | 6G1H | NAD+ | 29 |
| WT | Petrotoga mobilis SJ95 | 6G1M | NAD+ | 29 |
| WT | Cystobacter fuscus | 6IAU | NADP+ and cyclohexylamine | 29 |
| W145A | Cystobacter fuscus | 7QZN | NAD+ | 41 |
| W145A | Cystobacter fuscus | 7QZL | NADP+ and pentylamine | 41 |
| WT | Mycolicibacterium smegmatis | 6IAQ | NADP+ | 29 |
| WT | “MATOUAmDH2” from an eukaryotic organism | 7R09 | NADP+ and cyclohexylamine | 42 |
As an example, the crystal structure of the native AmDH4 is shown in Fig. 2A. The two dimers are in ‘open’ and ‘closed’ forms. Each of the monomer consists of a Rossmann fold domain at the N-terminal, and a six-strand β-sheet domain at the C-terminal. The structures resemble that of the dihydropicolinate reductase (DHDPRs, PDB ID: 5KT0) and some natural amino acid dehydrogenases such as meso-diaminopimelate dehydrogenases (DAPDHs, PDB ID: 1F06 and 3WBF), but the sequences do not share significant homology. The rossmann fold is well-conserved among the native AmDHs, DHDPRs, and DAPDHs, but the β-sheet domain bears significant differences. The key residues that shape the active site pocket are E102, H197, I198h, H264, Q266, G299, and T303. The structure of the active site of AmDH4 in complex with the substrate (2R,4S)-2,4-diaminopentanoate (2,4-DAP) is shown in Fig. 2B. The key residues that interact with the substrate are R161, H264, H197, E102. The residue E102 is conserved between DAPDHs and AmDH4, which is considered to be involved in the activation of ammonia. Q266 at the bottom of the active site and F168 situated at the flexible lid of the active site are considered to involve in the catalytic events.
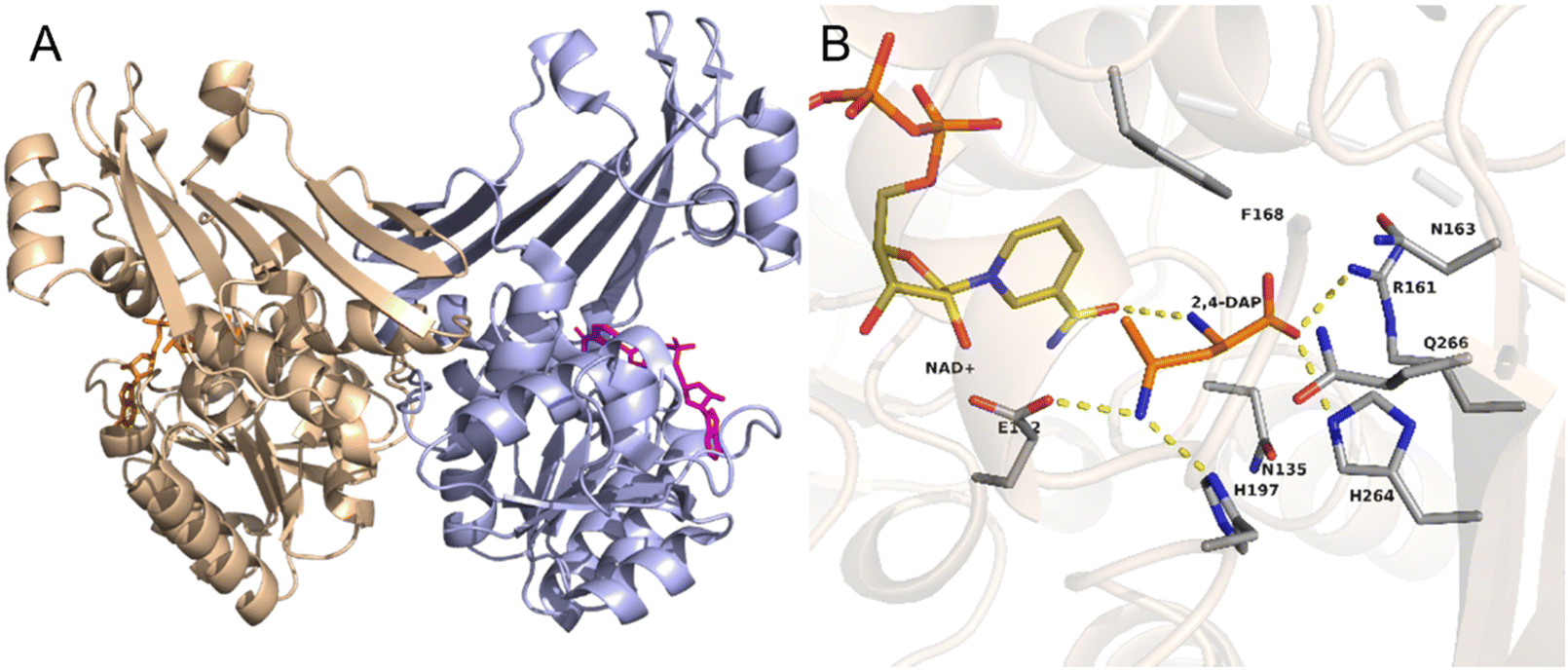 | ||
| Fig. 2 Structures of (A) AmDH4 dimers in complex with NAD+ and (B) the active site of AmDH4 in complex with the substrate 2,4-DAP (PDB ID: 6G1M). | ||
The proposed mechanism of the native AmDH catalyzed reductive amination involves several key steps (Fig. 3).29,31,43 Firstly, ammonia is activated by glutamate (E108 in the case of CfusAmDH) (II), then electrophilic attack takes place on the carbon from the carbonyl moiety, which is stabilized via hydrogen bonds with tyrosine (Y168 for CfusAmDH) at the active site (III). The carbinolamine intermediate is then formed, subsequently, reduction of the carbinolamine by the hydride of the nicotinamide cofactor occurs and then this is dehydrated to form the amine product (IV and V). This mechanism resembles the reductive amination catalyzed by RedAms,30 where both mechanisms involve a tyrosine proton donor for substrate stabilization, and a carbinolamine intermediate followed by hydride transfer from the co-factor NAD(P)H.
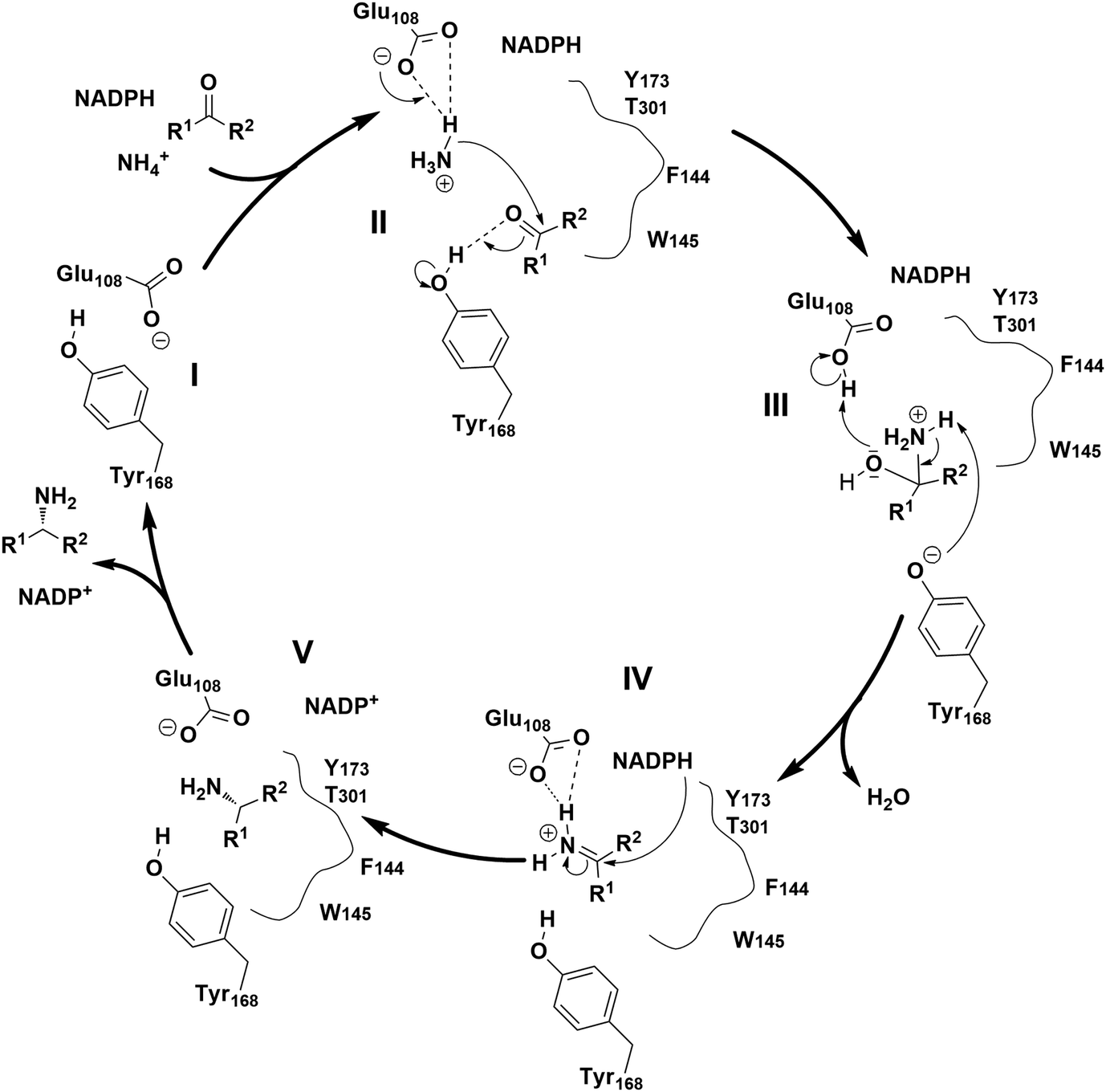 | ||
| Fig. 3 Proposed catalytic mechanisms of CfusAmDHs.29 | ||
2.2. Discovery of AmDHs
PheDH and LeuDH have been the first targets as the sources for conversions to AmDHs. Two key mutations with interactions of the substrate side chains have been identified from various PheDHs and LeuDHs. For example, Jiang et al. engineered a PheDH from Bacillus halodurans to utilize a range of benzylic and aliphatic ketone substrates with double mutations (E113D/N276L).48 Ye et al. engineered a PheDH from Rhodococcus sp. M4 to an AmDH with a triple mutation K66Q/S149G/N262C.49 Furthermore, Liu et al. engineered a thermostable PheDH from Geobacillus kaustophilus to develop a GkAmDH32 with the key double mutations (K78S/N276L). The Tm of GkAmDH is up to 71.5 °C with high activity towards a range of benzylic ketones using inorganic and organic ammonia sources to afford both primary and secondary amine or amino alcohol products. Pushpanath et al. developed a CalAmDH from PheDH from Caldalkalibacillus thermarum, bearing the 2 key mutations (K68S/N266L), with a high Tm up to 83.5 °C.50
Although a majority of AmDHs have been converted from LeuDH and PheDH, efforts have also been invested in engineering other classes of amino acid dehydrogenases to generate AmDHs, as exemplified by L-lysine dehydrogenase from Geobacillus stearothermophilus (LysEDH) by Tseliou et al. Benefitting from the thermophile origin of LysEDH, the AmDH variant (LE-AmDH-v1) showed high stability with Tm up to 69 °C, which also retained activity at room temperature, as well as 80% of the activity at 50 °C. A range of aliphatic and aryl ketones were reductively aminated to the corresponding amines in up to 99% ee and conversions (Fig. 4). The F173A mutation was proposed to enlarge the substrate binding pocket that enabled the acceptance of aromatic substrates. For the aliphatic ω-amino acid, the variant afforded (S)-configured product, whereas for all of the other aliphatic and aromatic substrates tested, (R)-configuration was observed.51 Later, the same variant has been coupled with a NADH oxidase (NOX) in the kinetic resolution of α-chiral amines.52 Harnessing the promiscuity of LysEDH, Tseliou et al. performed further engineering on the enzyme to generate dual alcohol dehydrogenase and amine dehydrogenase activity,53 thereby completing a one-enzyme “hydrogen-borrowing” cascade, albeit with low yields (up to 34%). While F173A variant favored the AmDH activity as described, the authors found that the introduction of F173S mutation enhanced the alcohol dehydrogenase activity.
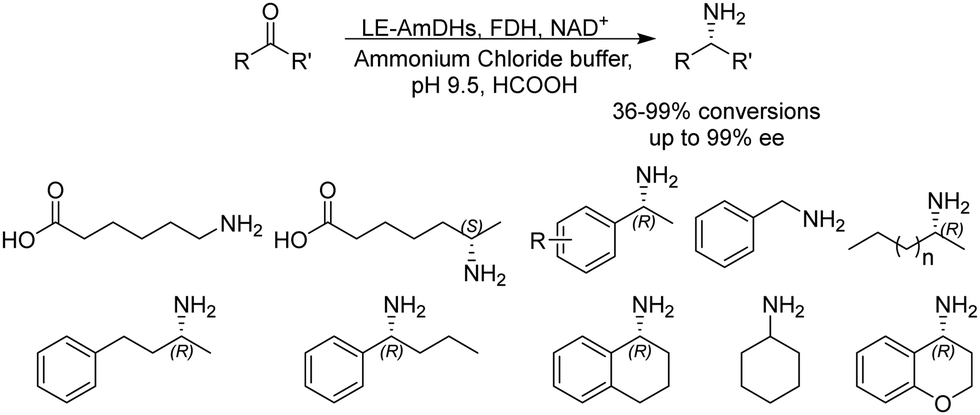 | ||
| Fig. 4 Biocatalytic reductive amination performed by LE-AmDH variants. | ||
2.3. The history of evolution for AmDHs
The pioneering work for the generation of AmDHs has sparked research interests in the protein engineering of AmDHs. Although these enzymes are important as they allow the reductive amination to utilize ketone substrates with many merits including generating only water as the by-product and using cheap inorganic amine donors, they suffer from narrow substrate scope and relatively low stability. Therefore, protein engineering of AmDHs has been carried out on the purpose of expanding the substrate scope and improving stability while maintaining or increasing activity of the AmDH.The AmDH structures contain an independent substrate-binding domain and cofactor-binding domains. Domain shuffling has been firstly applied in the engineering of AmDHs generated from LeuDH and PheDH by the Bommarius group in 2014. The final variant cFL1-AmDH consisted of the ketone/amine binding domain of F-AmDH from Rhodococcus sp. M4 and cofactor-binding domain from L-AmDH from Bacillus sphaericus. The variant exhibited expanded substrate scope to benzylic carbonyls and higher thermostability.58 The same group also performed the reactions in a biphasic system using heptane and ammonium formate buffer, enabling the hydrophobic substrates to be accepted with improved enzymatic activity.59 Later in 2018, this variant was used by Löwe et al. as the model enzyme to investigate the substrate scope, synthetic processes and solvent tolerance in detail.60 Recently, Li et al. continued with domain engineering on AmDH engineered from a PheDH.61 The resulting variant cFLF-AmDH combined the coenzyme-binding domain of L-AmDH from Bacillus stereothermophilus and the core structure of F-AmDH from Bacillus stereothermophilus with lower coenzyme affinity than L-AmDH. Therefore, cFLF exhibited 2-fold increase in NADH binding affinity and 4.4-fold increase in kcat/Km comparing with F-AmDH.
Co-factors and the recycling systems drastically affect the economic viability for the applications of AmDHs. To achieve high atom efficiency and efficient use of cofactors, the Turner group has developed a dual-enzyme hydrogen-borrowing process, which has utilized a range of primary and secondary alcohols for the synthesis of corresponding amines (Fig. 5).62 AmDH was applied with alcohol dehydrogenases (ADHs from Aromatoleum sp., Lactobacillus sp., or Bacillus sp.) to achieve a redox-neutral, self-sufficient process. The amine products were synthesized with up to 99% ee and 96% conversions in (R)-configuration starting from either (R)-, (S)- or racemic alcohols. This concept has also been reported by Chen et al, who introduced 2 knowledge-based key residues (K77S/N270L) to EsLeuDH from Exiguobactertium sibiricum and considerably improved activities for a variety of ketones. The EsAmDH was then coupled with ADHs to achieve the self-sufficient process. In preparative scale, 2-pentanamine was obtained in >90% conversions starting from 2-pentanol.63 The similar approach has been applied to a variety of cascade reactions involving several different classes of enzymes (with ADHs) such as transaminases,64 reductive aminase,65 and ene-reductases.66 Other than the redox neutral cascades, two reductive enzymatic processes can also be performed in one-pot employing ene-reductases (old yellow enzyme, OYE) and AmDHs to convert α,β-unsaturated enones to amines.67
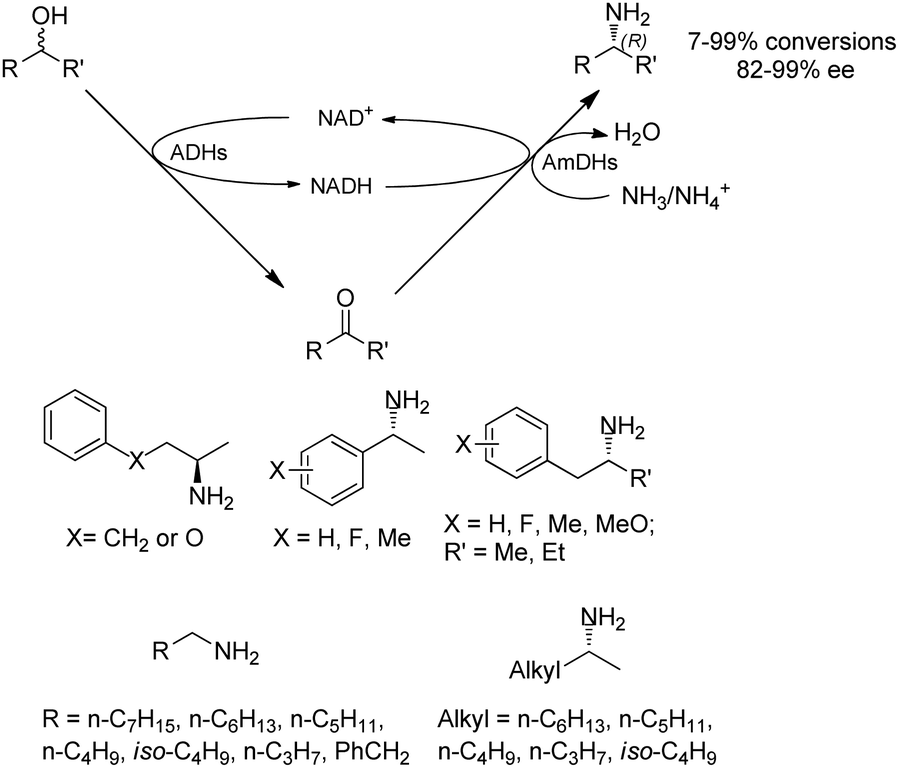 | ||
| Fig. 5 Hydrogen-borrowing self-sufficient cascades. | ||
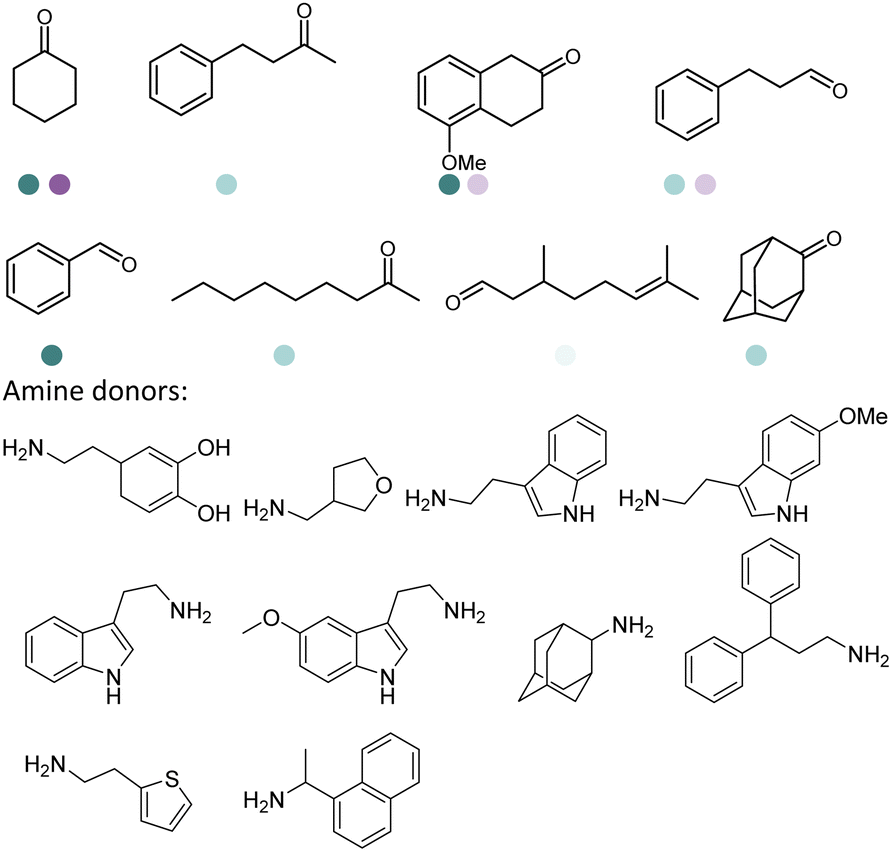
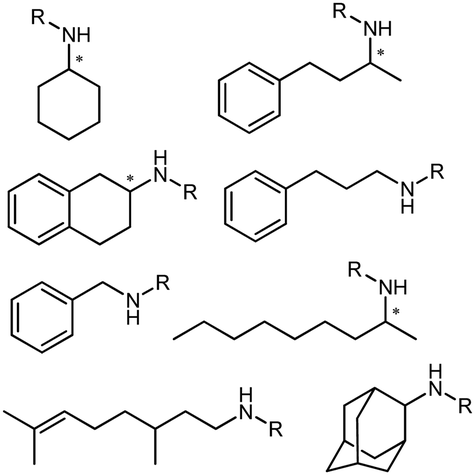
Early examples of AmDHs exhibited narrow substrate scope, and efforts were invested to expand the scope for bulky, aliphatic and aromatic substrates. In 2018, Chen et al. synthesized bulky aliphatic amines via an engineered LfAmDH from Lysinibacillus fusiformis from LeuDH with up to 400 mM substrate loading. The same key mutations (K68S/N261L) that were responsible for altering the substrate specificities were introduced to three amine dehydrogenases (EsAmDH from Exiguobacterium sibiricum, LfAmDH from Lysinibacillus fusiformis, and BspAmDH from Bacillus sphaericus) and the best one was identified as the LfAmDH. They identified key mutations A113G and T134G that enlarged the volume of the substrate-binding cavity, which accommodated the large aliphatic substrates well, giving excellent enantio-selectivity (>99% ee, (R)-selectivity) with >99% conversions.68 This variant has greatly expanded the substrate scope of AmDHs. Later the same group further engineered the variant to expand the substrate scope to include α-hydroxy ketones. Two variants (LfAmDH-M0 and LfAmDH-M3) carrying further mutations (K68T/N261L and K68T/N261L/A113G/T134G, respectively) afforded the (S)-α-hydroxyl amines with excellent ee (>99%%) and conversions (>70%).69 Therefore, both (R)- and (S)-products can be obtained with the two enantio-complementary variants provided by above studies (Fig. 6).
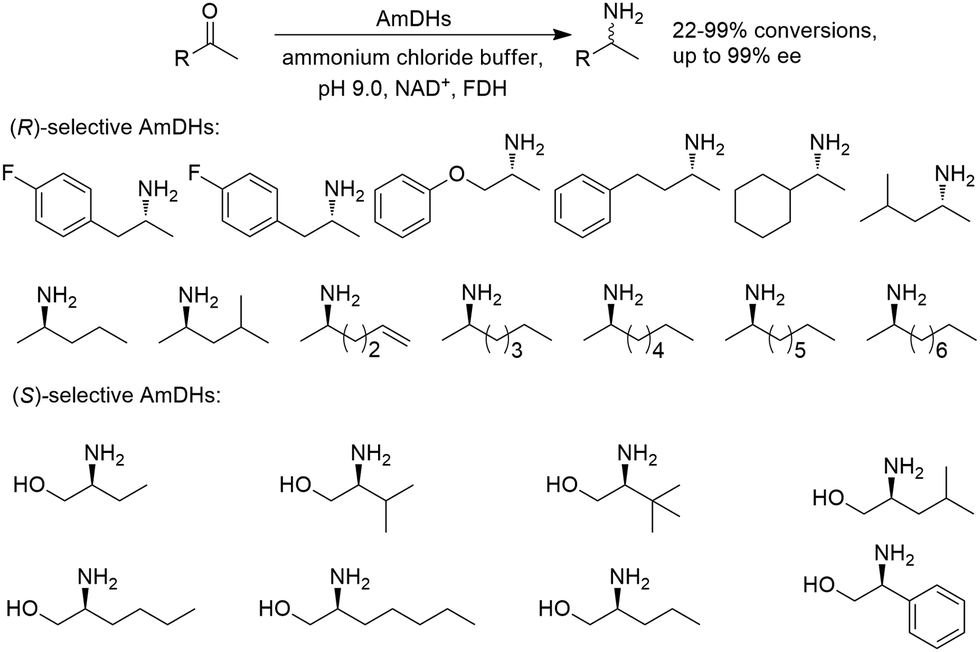 | ||
| Fig. 6 Reductive amination by AmDHs affording both (R)- and (S)-configured amines. | ||
There have been growing interests in the protein engineering of AmDHs to expand the substrate scope to aliphatic ketones68 or aromatic ketones,70 and improving the thermostability.71 Later in 2021, Wang et al. reported a further engineered variant of the GkAmDH from Geobacillus kaustophilus.72 By combining a semi-rational design strategy and directed evolution (error-prone PCR and DNA shuffling), the authors identified further key residues that rendered up to 110-fold increase in activity comparing to the starting variant for the synthesis of bulky aromatic ketone substrates. The substrate scope has also been extended to methyl ketones with alkyl chains, remote functional groups, or heterocycles, as well as ethyl and propyl ketones with alkyl chains. Furthermore, the authors employed the variant M3 and M8 with high activity in preparative synthesis of several pharmaceutically useful building blocks (dilevalol and medroxalol), both afforded good to high yields and excellent enantio-selectivity (Fig. 7).
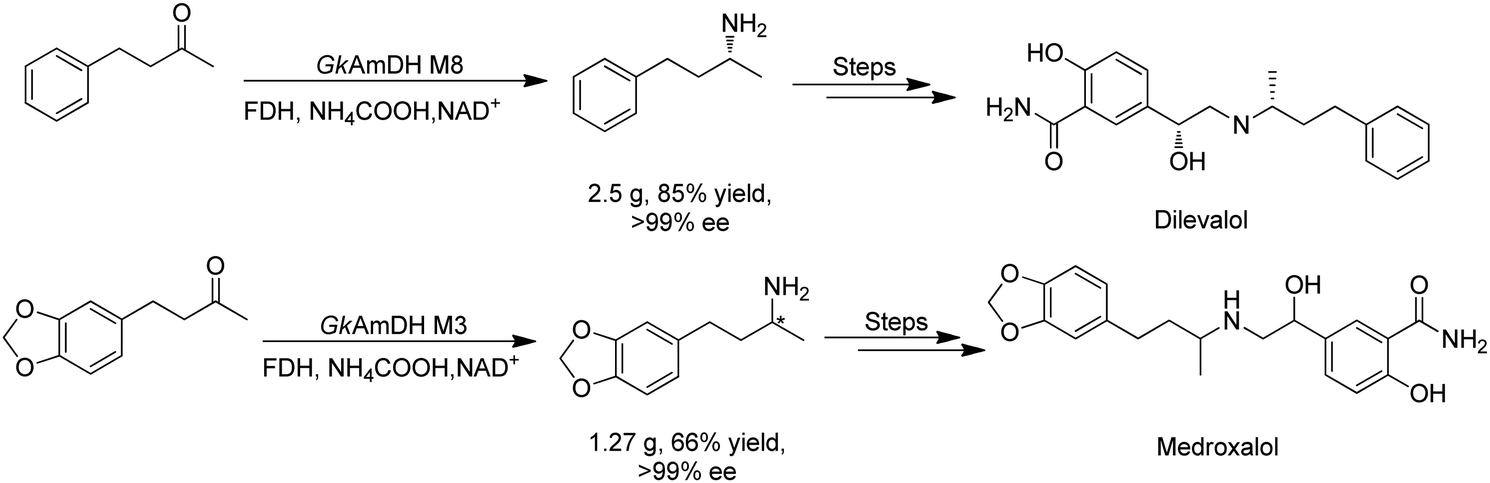 | ||
| Fig. 7 Engineering GkAmDH to catalyze the synthesis of intermediates of dilevalol and medroxalol. | ||
Knaus et al. investigated and expanded the substrate scope of AmDHs by testing 3 previously reported variants, namely Bb-PhAmDH, Rs-PhAmDH, and Chi-AmDH.73 Upon optimization, (R)-configured bulky-bulky ketones were tested with high activity and selectivity (up to >99% conversions and ee) when catalyzed by Chi-AmDH and Rs-PhAmDH. Formate dehydrogenase (FDH) was used to improve the atom economy and carbonate was produced as the sole by-product.
Recently Kong et al. engineered an AmDH from Jeotgalicoccus aerolatus (JaAmDH) to achieve enhanced activities for aryl ketones and heteroaryl ketones.74 The authors identified three AmDHs with the best one being JaAmDH. They introduced the preserved K68 and N261 amino acids for converting the amino acid dehydrogenase (AADH) activity to AmDH activity. Further engineering of JaAmDH has significantly improved specific activity towards ketones bearing heterocycles moiety, which were previously found difficult to be utilized by AmDHs. Key mutations including K67S/N260L/E113V/V291G were introduced to create the variants M33, with kcat/Km improving up to 46 folds for the model substrate 1-(6-methylpyridin-2-yl)ethan-1-one. Up to 18 heteroaryl ketones and 12 aryl ketones were subjected to reductive amination and ee were all above 99% with conversions ranging from 74% to 99% (Fig. 8). Preparative scale reactions on 200–1000 mM substrate concentration afforded conversions and ee both up to 99%.
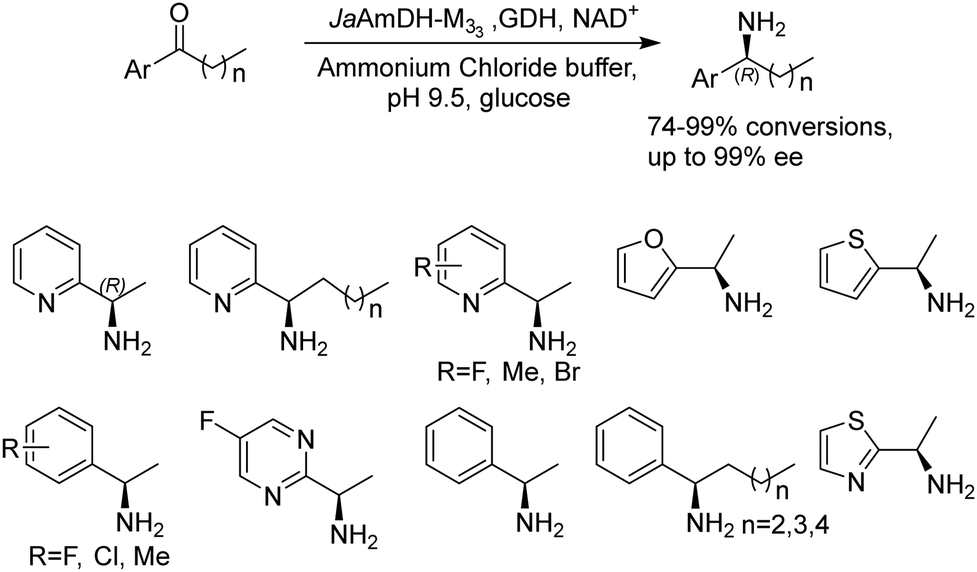 | ||
| Fig. 8 Reductive amination of aryl and heteroaryl ketones by AmDHs. | ||
In addition to the AmDHs converted from LeuDHs and PheDHs, native AmDHs have also been targeted for protein engineering, which has enabled the production of industrial relevant compounds in high substrate loadings. Cai et al. engineered PmAmDH (or AmDH4) by directed evolution using techniques including error-prone PCR and DNA shuffling, and the kcat/Km was elevated by 18 folds compared with the wild type AmDH with key mutations I80T/P224S/E296G, while the Tm of the variant reached 60 °C. Reductive amination was performed on levulinic acid in preparative scales at a 0.5 M (58 g L−1) substrate concentration, and 97% conversions was achieved in 24 h with >99% ee.75
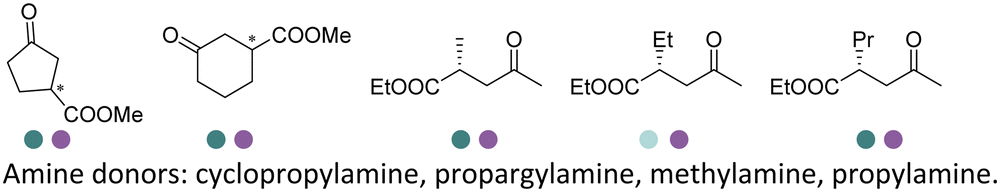
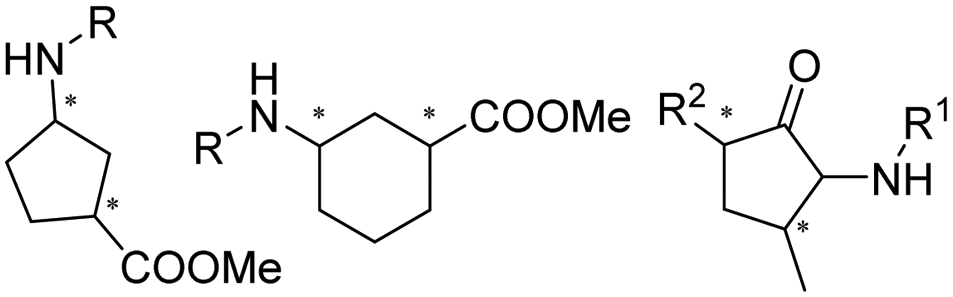
Desymmetrization strategies have been firstly applied on 1,3-cyclodiketones using AmDHs very recently. The reductive amination was carried out to stereoselectively aminate one of the ketone groups to produce 2,2-disubstituted β-amino ketones containing a quaternary carbon center. A library of 51 AmDHs were screened, and LsPheDH from Lysinibacillus sphaericus bearing the key mutations K79S/N277L was selected. Saturation libraries for residues within 5 Å of the substrate and NADH were constructed and screened, and subsequently the combinatorial libraries were constructed based on the positive hits. Finally, 5 variants were employed in the biotransformation of a range of substrates with expanded scope, namely W0 (LsPheDH K79S/N277L), W2 (T125A), W3 (N145L), W5 (T125A/N145L), and W8 (L51A/T125A/N145L/S157P). Using these variants, up to 97% conversion, 99% de and 99% ee have been achieved. Preparative-scale reactions were also performed with good yields (60–85%), and by conducting a two-step reaction followed by Buchwald–Hartwig cross coupling, a cyclopenta[b]hydroquinoline derivative was obtained in up to 55% yield (Fig. 9).76
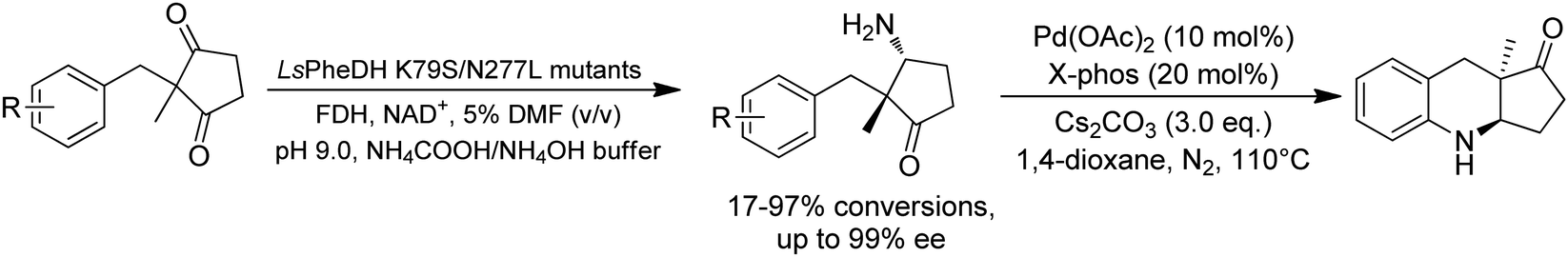 | ||
| Fig. 9 Desymmetric reductive amination of 2,2-disubstituted-1,3-ketones by AmDHs. | ||
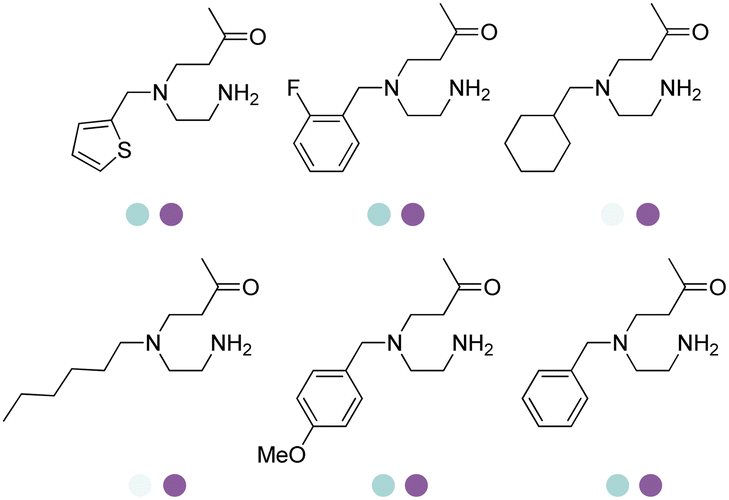
The Sun group conducted data mining, discovery and protein engineering of AmDHs on purpose of identifying new enzymes and improving enzymatic activity towards hydroxyl ketone substrates.77,78 They developed a high throughput assay with fluorescent probe to quantitatively determine the ketones for AmDH library screenings.79 Five AmDHs have been discovered, namely GsAmDH from Geobacillus stearothermophilus, BsAmDH from Bacillus stearothermophilus, LsAmDH from Lysinibacillus sphaericus CBAM5, SpAmDH from Sporosarcina psychrophile, and TiAmDH from Thermoactinomyces intermedius, which exhibited significant activity towards 1-hydroxybutan-2-one after the introduction of the two key residues responsible of converting AADH to AmDH. 99% conversion and 99% ee were obtained by GsAmDH on the model substrate. In addition, good to excellent conversions and ee were obtained for a range of aliphatic hydroxyl ketone and benzylic hydroxyl ketone substrates. Later, site-directed saturation mutagenesis (SSM), CAST/ISM strategies were employed to further engineering the SpAmDH and GsAmDH for several rounds to achieve up to 4.2 fold improved activity towards hydroxyl ketones, with >99% ee and 99% conversions (Fig. 10).78,80
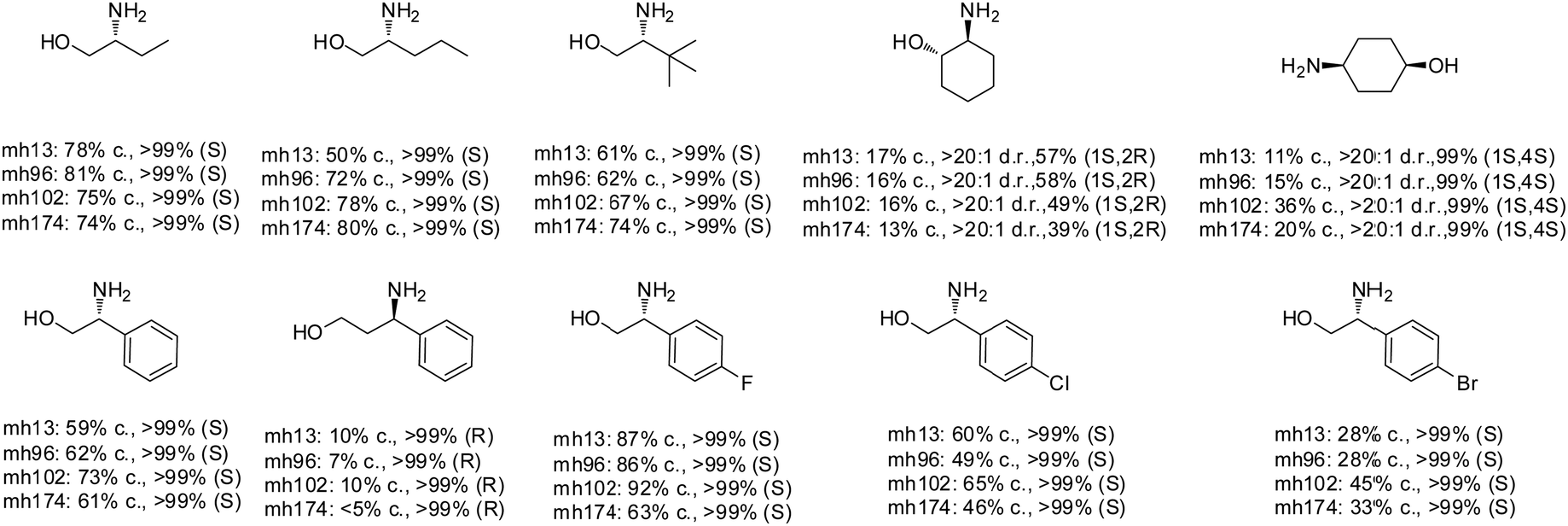 | ||
| Fig. 10 Expanded substrate scope by engineered AmDHs. | ||
2.4. Applications of AmDHs
Immobilization of enzymes represents a practical technique which possesses several advantages comparing to free enzymes such as improved thermostability and higher organic solvent tolerance.81,82 Immobilization often enables the biocatalysts to be reused for a number of batches and with continuous flow processes, the overall economy could be considerably improved. AmDHs have been employed in several studies involving enzyme immobilization on various supports and flow processes. For example, in 2017, Liu et al. immobilized an AmDH (crude enzyme) engineered from PheDH from Rhodococcus sp. M4 on magnetic nanoparticles (MNP) (Fig. 11).83 Additionally, they also immobilized a glucose dehydrogenase (GDH) used for cofactor recycling systems, which reduced the cost of the overall reaction while retaining the productivity. Ren et al. coated the AmDH by polyethylenimine (PEI), and then the AmDH-PEI hybrid was entrapped by titania to form the AmDH-PEI-Ti nanoparticles. The immobilized enzyme displayed improved thermostability, and can be used for 6 cycles while retaining 50% of the initial activity.84 In 2018, Caparco et al. investigated the effect of peptide linkers of leucine zipper-AmDH/FDH fusion proteins on the catalytic efficiency of the immobilized enzymes on inorganic supraparticles.85 Later in 2020, the authors immobilized an AmDH and a formate dehydrogenase with fluorescent model proteins and a leucine zipper binding domain onto the calcium-phosphate–protein supraparticles, and successfully converted ketone substrates to amines.86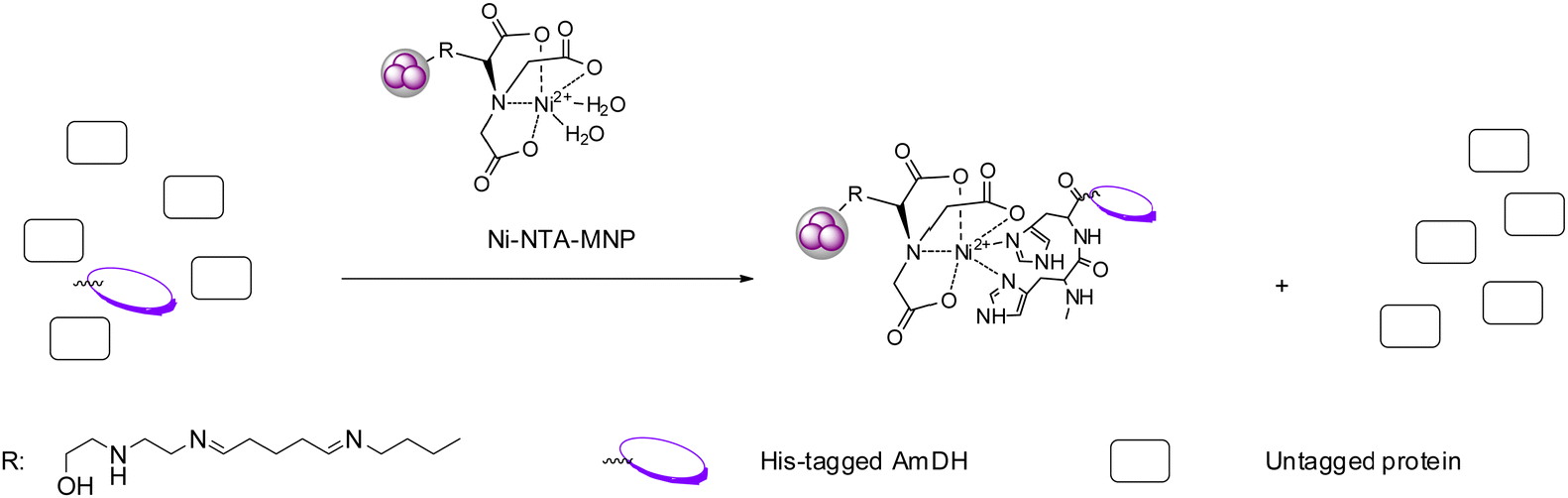 | ||
| Fig. 11 Highly specific affinity attachment-enabled immobilization of his-tagged AmDH from cell-free extract on Ni-NTA-MNP. | ||
Recently, continuous flow was attempted using a fixed-bed reactor packed with immobilized Ch1-AmDH and FDH. The Nuvia® IMAC resin from Bio-Rad was used as the carrier, and amination of 5-methyl-2-hexanone was performed with up to 443 g L−1 day−1 productivity (Fig. 12).87
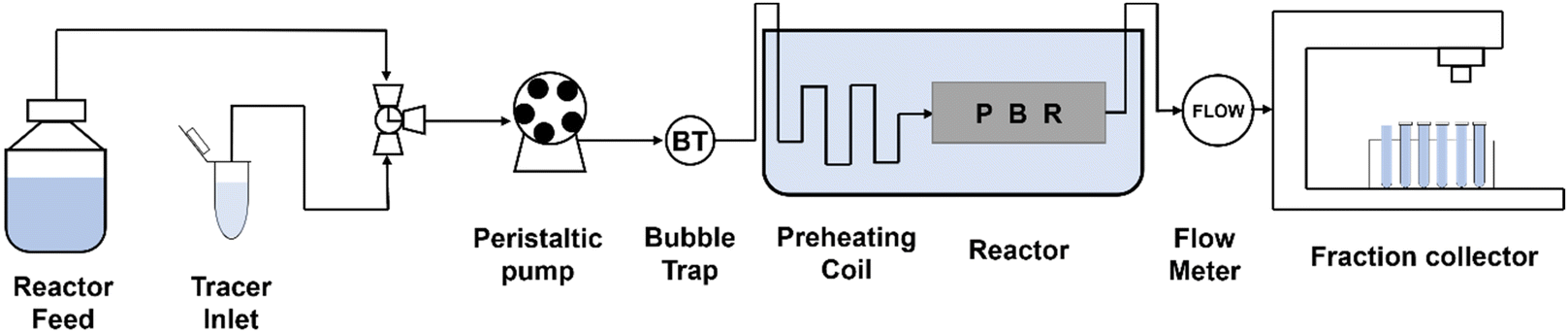 | ||
| Fig. 12 Block flow diagram of the continuous flow packed bed reactor system. | ||
Synthetic biology utilizes enzymes in metabolic pathways to construct whole-cell systems with several enzymatic routes to targeted products. In 2018, Yu et al. constructed a C–H amination cascade in vivo, cascading P450 BM3 (19A12) catalyzed C–H hydroxylation, alcohol dehydrogenase (ADH) ScCR catalyzed oxidation of the alcohol, and EsAmDH catalyzed amination in one-pot (Fig. 13).88 Non-interfering cofactor recycling systems have been employed and eventually a one-pot two-step cascade reaction has been established. The atom economy reached 100% while the reaction could be performed at scale by whole-cell. Houwman et al. has constructed an ADH with AmDH in resting E. coli cells implementing the “hydrogen-borrowing” concept in vivo.89 Liu et al. coupled whole-cell with cell-free systems, and the cofactor recycling system was enhanced by separation.90 A NADH oxidase (NOX) was coupled with an ADH to obtain full conversion from racemic alcohols to ketones, and a GDH with AmDH were used for the reductive amination of the ketones, as a result, a TTN for NADH recycling of 1410 was achieved, which was reported as the highest TTN thus far.
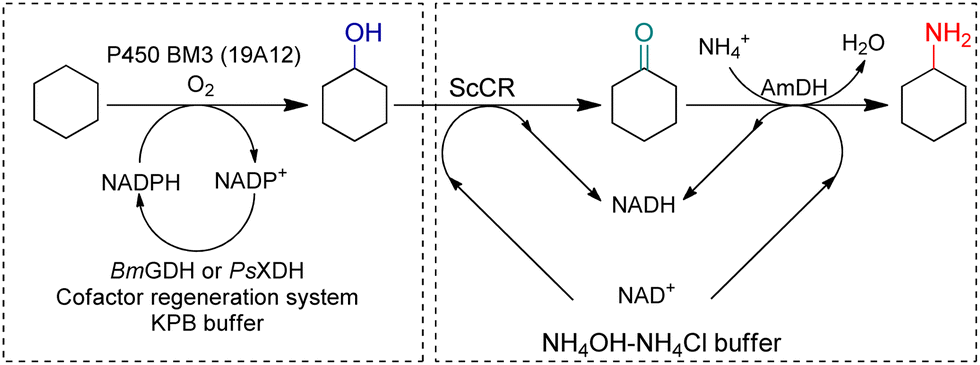 | ||
| Fig. 13 One-pot two-step strategy to produce cyclohexylamine. Adapted with permission from ref. 88 with modifications. Copyright 2018 Elsevier Inc. | ||
Cascading AmDH with organic/metal catalysts represents major opportunities in organic synthesis for the production of important drug intermediates. However, it is difficult to identify suitable catalysts and conditions for the two previously incompatible systems to work in synergy in aqueous phase. The Turner group reported a one-pot reaction with a Pd catalyst and AmDH to produce chiral N-arylamines.91 A Buchwald–Hartwig cross-coupling (BHA) reaction was performed with the addition of a surfactant DL-a-tocopherol methoxypolyethylene glycol succinate (TPGS-750-M) to improve the water miscibility of the catalyst and accelerate the rates of the coupling reaction in enzymatic compatible conditions (Fig. 14). In the end, up to 90% conversions of the chiral N-arylamines were obtained. Additionally, multi-enzyme cascades have also been alternative methodologies, for example, Chen et al. reported a trienzymatic cascade involving a ω-transaminase, an AmDH and a FDH.92
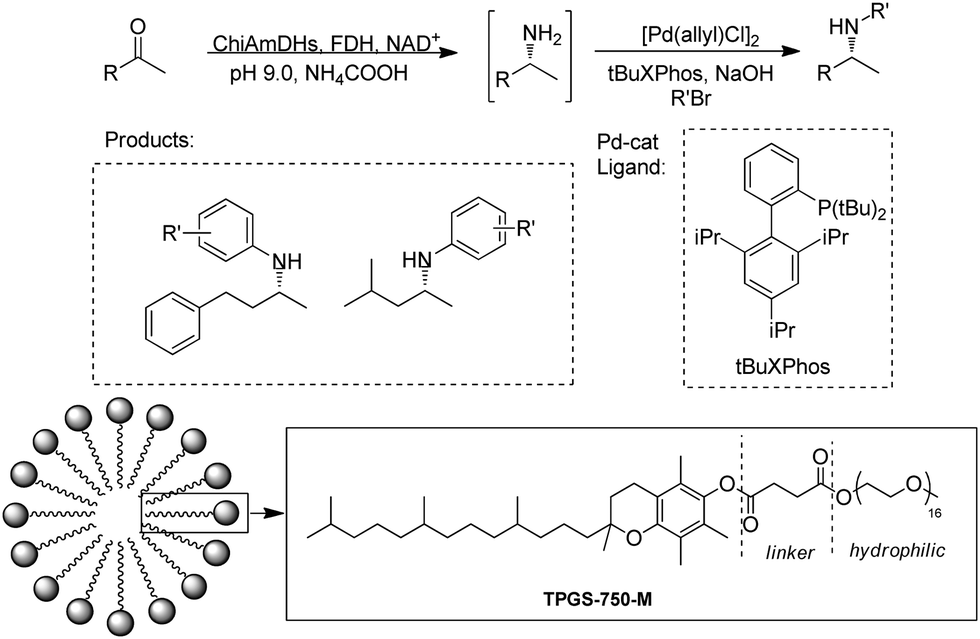 | ||
| Fig. 14 Cascade of AmDHs catalyzed RA and Buchwald–Hartwig cross-coupling (BHA). | ||
In summary, AmDHs have exhibited enormous potential in the synthetic applications of chiral amines. The key advantage of AmDHs is the ability to utilize inorganic ammonia donors producing only water as the side products, which brings environmental and economic benefits in the industrial applications. In addition, investigations have also been carried out to explore the synthesis of secondary amines catalyzed by AmDHs, although the scope is very limited and requires further efforts to enhance the ability to produce chiral secondary amines. On the other hand, in the following section, we introduce imine reductases (IREDs) which advances in the synthesis of secondary and tertiary chiral amines.
3. Imine reductases and reductive aminases (IREDs and RedAms)
Imine reductases (IREDs) have been firstly discovered by Mitsukura et al. in 2010.93 Both (R)- and (S)-selective IREDs were identified by screening against a large number of bacteria, actinomycetes, and fungi. In the following decade, IREDs have sparked significant research interests and enormous efforts have been invested in the engineering of IREDs to find applications across the industrial sectors. IREDs are NAD(P)H-dependent oxidoreductases that are capable of catalyzing reductive amination not only of cyclic imines, but also between ketone substrates and amine donors to produce primary, secondary and tertiary amines.25,26,94–100 Hereafter, we summarize the structures, mechanisms, evolutionary history and applications of IREDs.3.1. Structures and mechanistic insights into IREDs and RedAms
The first X-ray crystal structure of IRED from Streptomyces kanamyceticus was reported by Rodriguez-Mata et al.101 Subsequently, many crystal structures of IREDs from various origins were resolved as presented in Table 2.102–105| Names | Origin | PDB ID | In complex with | Ref. |
|---|---|---|---|---|
| SkIRED | Streptomyces kanamyceticus | 3ZHB | NADP+ | 101 |
| SkIRED | Streptomyces sp. GF3546 | 4OQY | NADPH | 103 |
| SaIRED | Streptomyces aurantiacus | 4OQZ | — | 103 |
| NhIRED | Nocardiopsis halophila | 4D3S | n-Octyl β-D-glucoside (OBDG) | 104 |
| BcIRED | Bacillus cereus BAG3X2 | 4D3F | NADP+ | |
| AoIRED | Amycolatopsis orientalis | 5A9T/5FWN | NADP+/ | 102 |
| (R)-Methyltetrahydroisoquinoline | ||||
| AtIRED | Aspergillus terreus | 5OJL | NADPH4 and dibenz[c,e]azepine | |
| AtIRED | Aspergillus terreus | 6EOD | NADP(H) | 106 |
| AtIRED | Aspergillus terreus | 6EOH | NADPH and ethyl levulinate | 30 |
| AtIRED | Aspergillus terreus | 6EOI | NADPH and ethyl-5-oxohexanoate | |
| AtIRED | Aspergillus terreus | 6H7P | NADPH4, cyclohexanone and allylamine | |
| AspRedAm | Aspergillus oryzae | 5G6R/5G6S | NADPH and (R)-rasagiline | |
| SrIRED | Streptosporangium roseum | 5OCM | NADP+ and 2,2,2-trifluoroacetophenone hydrate | 28 |
| MsIRED | Myxococcus stipitatus | 6TO4 | NADP+ | 107 |
| MsIRED | Myxococcus stipitatus | 6TOE | NAD+ | 108 |
| IR-G36-M5 | Actinoalloteichus hymeniacidonis | 7WNW | NADP+ | 109 |
| EneIRED | Pseudomonas putida | 7A3W | NADPH | 110 |
| EneIRED-07 | Micromonospora sp. Rc5 | 7OSN | NADP+ | 111 |
| A208N variant | Ensifer adhaerens | 8A5Z | NADP+ | 112 |
As an example, the crystal structure of AtRedAm in complex with NADPH is shown in Fig. 15A.30 The structure consists of two monomers with a typical domain-swapping feature that exists in most of the IREDs and RedAms. Each monomer is constructed with a Rossmann domain at the N-terminal and a helical bundle at the C-terminal. The active site is shaped by the N-terminal domain of one monomer and C-terminal domain of the other monomer, which exists in ‘open’ (apo-) and ‘closed’ (bound with NADPH) form. The proposed catalytic residues are Y183, D175, and N98. A hydrogen bond is formed between the ketone carbonyl group of the substrate ethyl levulinate and the hydroxyl group from Y183, and the carbon atom of the carbonyl group is placed within 4.5 Å from the C4 atom of NADPH (Fig. 15B). This distance is not sufficient for efficient hydride transfer. The quaternary complex of cyclohexanone, allylamine, the redox-inactive NADPH4 were resolved, and the crystal structure is shown in Fig. 15C. Y183 is coordinated with a water molecule that interact with carbonyl group of cyclohexanone. The allylamine is observed near D175 and N98, with interactions between the amine group and the side chains of the residues. This structure of the quaternary complex provides evidence for both ketone and amine binding, which leads to the imine formation and reduction.
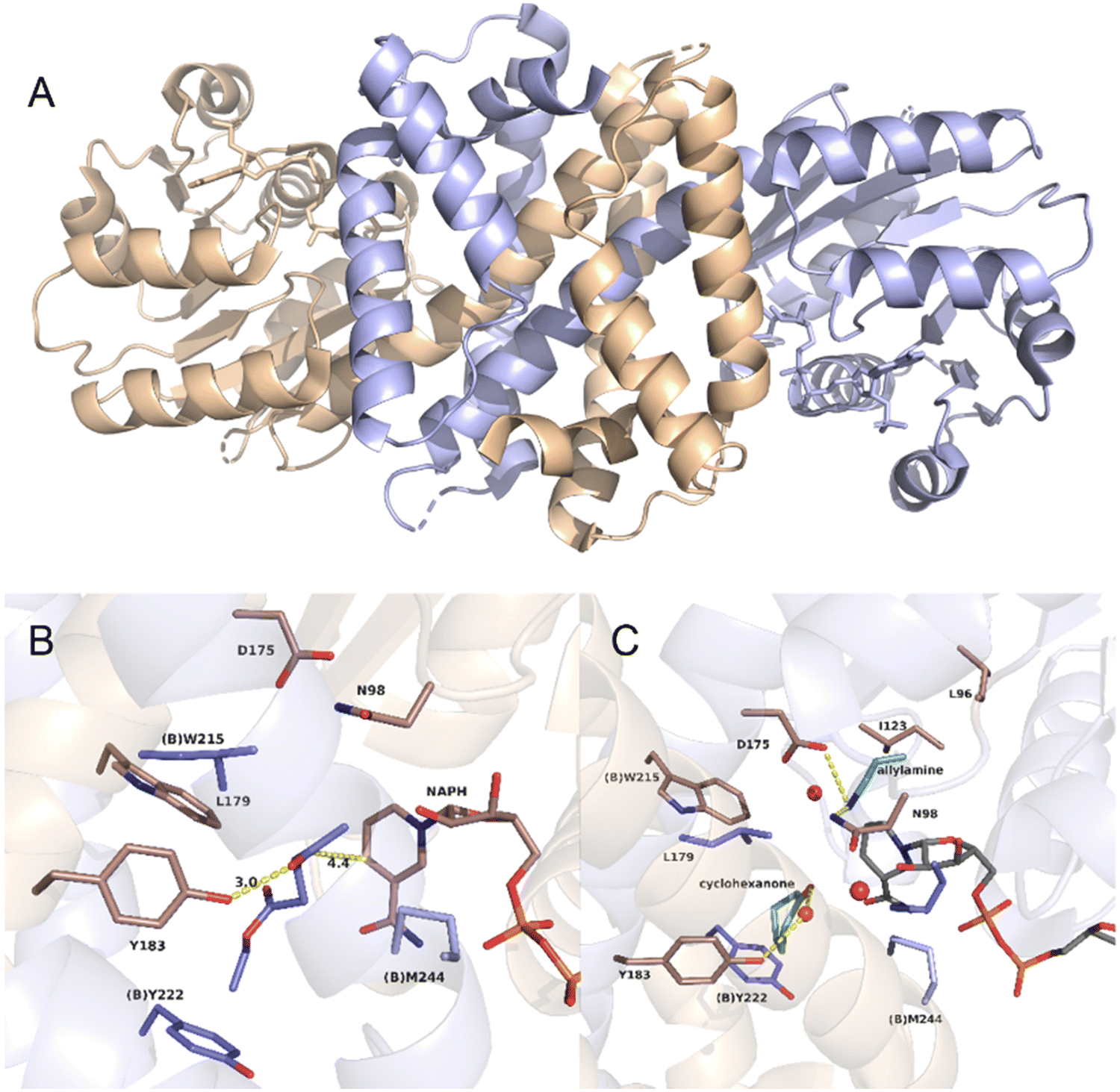 | ||
| Fig. 15 Crystal structures of AtIRED. (A) AtIRED in complex with NADPH; (B) the active site of AtIRED with the substrate ethyl levulinate; (C) the quaternary complex of cyclohexanone and allylamine. (PDB ID: 6H7P). | ||
The catalytic mechanisms have been determined to start with ketone binding to Y177, and deprotonation of the amine by D169 (II). Then the carbonyl group of the ketone is attached by the deprotonated amine again and forms a carbinolamine intermediate (III). Subsequently, the hydroxyl group of the intermediate is protonated, and water as a leaving group is eliminated while Y177 deprotonates the amine. Finally the prochiral iminium ion intermediate is obtained (IV), which is then reduced by the NADPH hydride to yield the final amine product (V) (Fig. 16).30
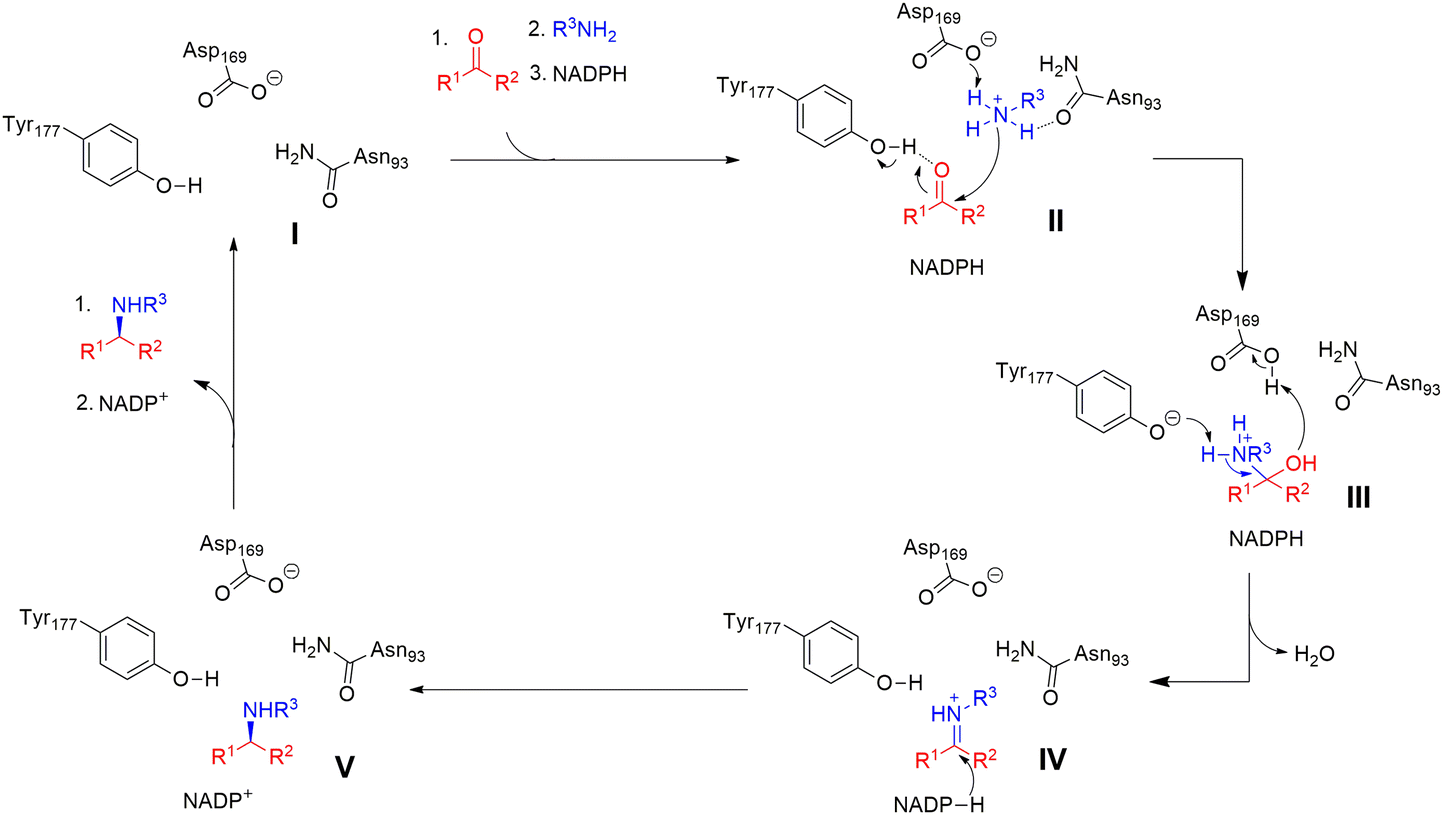 | ||
| Fig. 16 The hypothetical mechanisms for RedAms.30 | ||
3.2. Discovery, history of evolution and substrate scope of IREDs
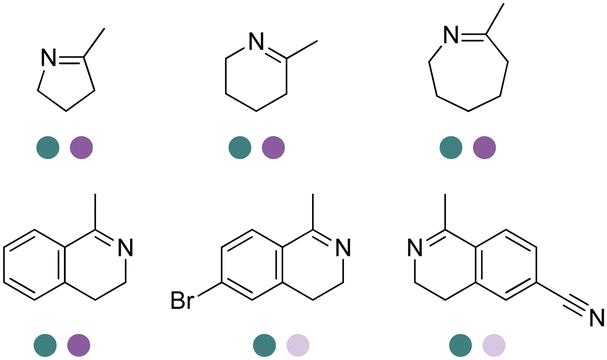
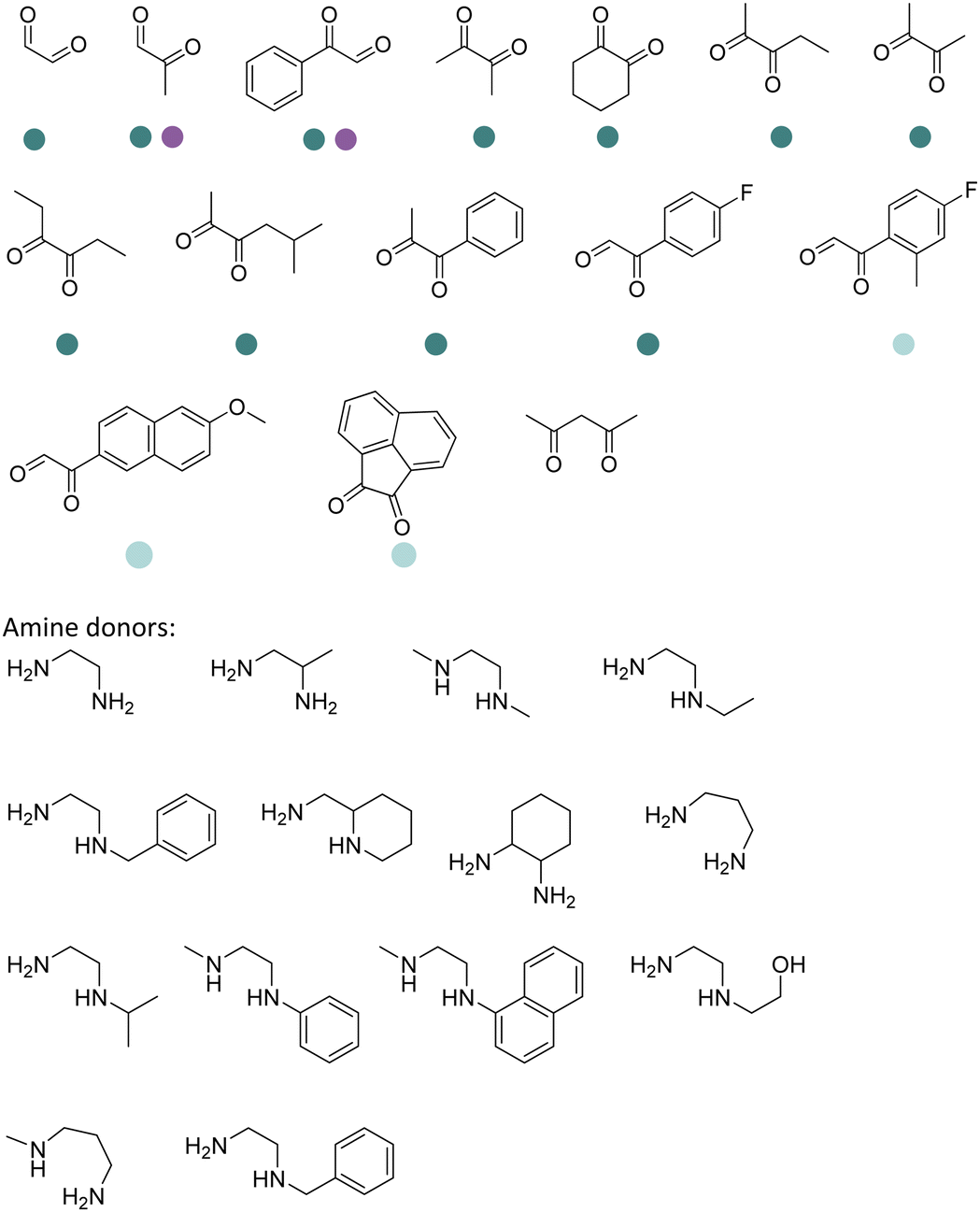
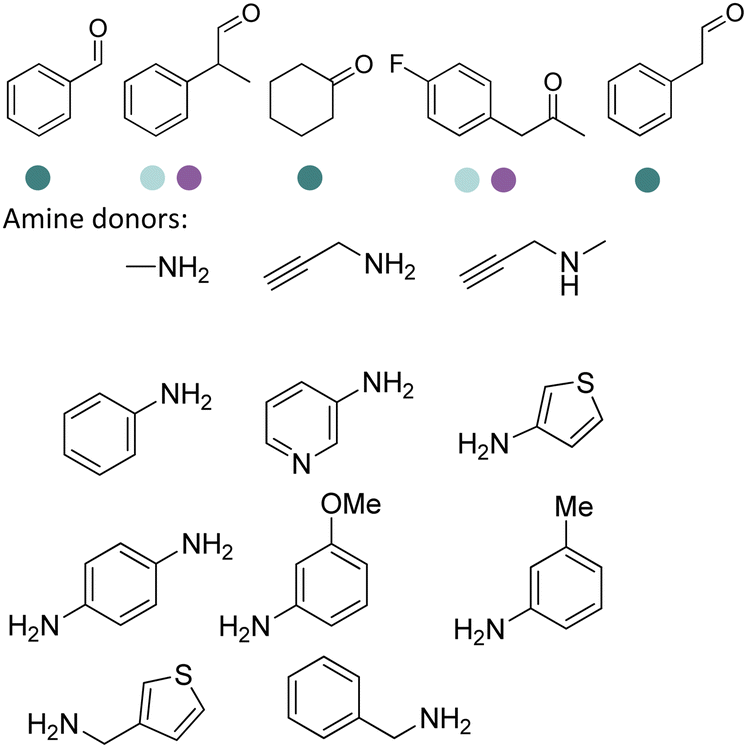
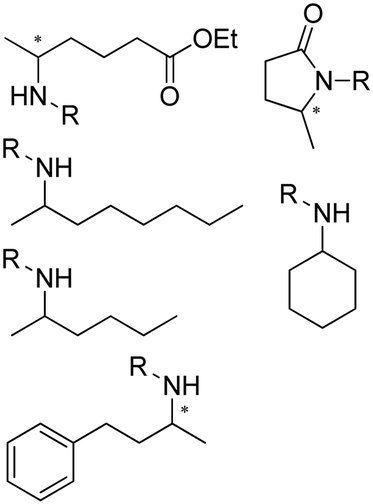
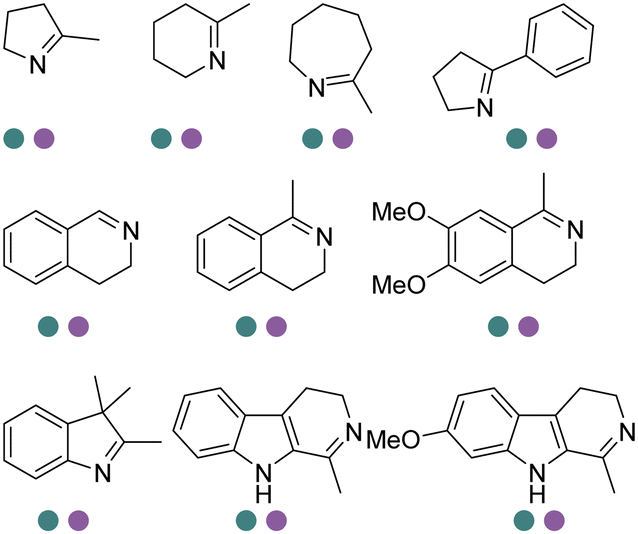
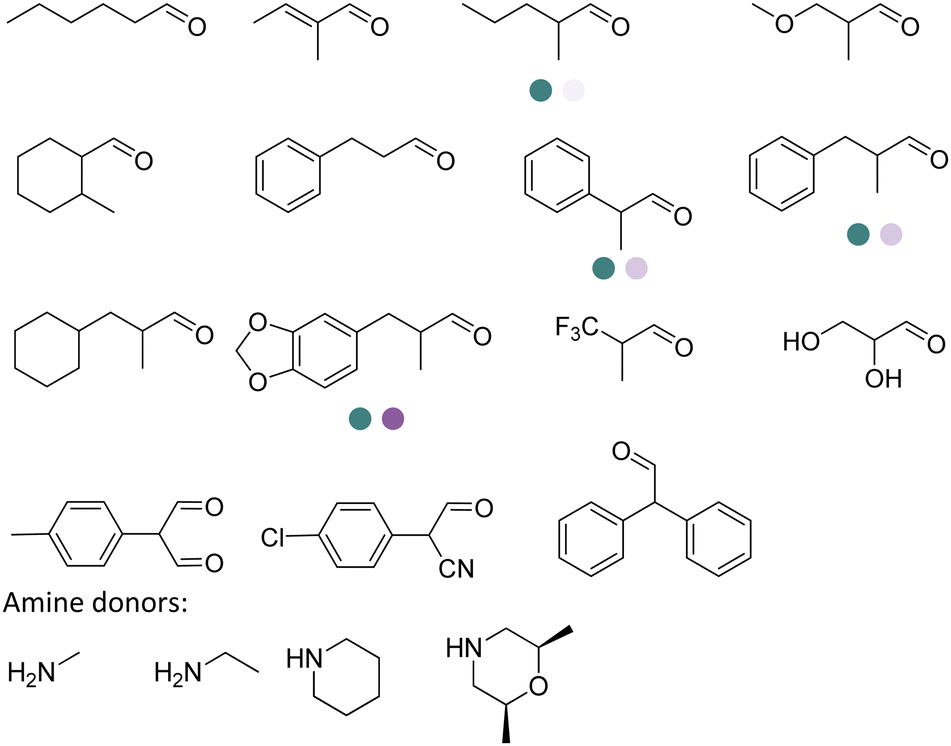
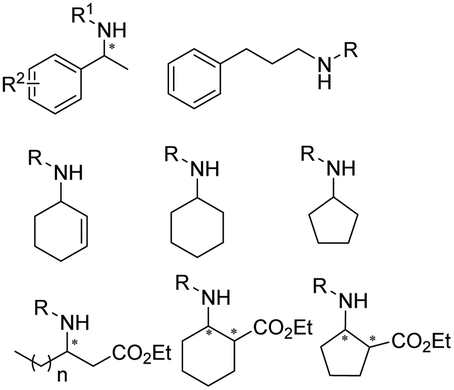
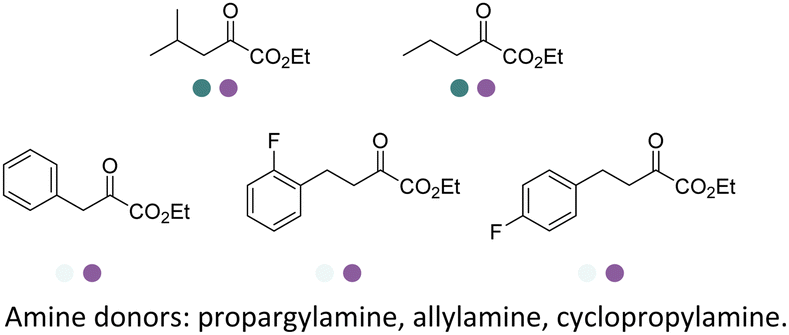
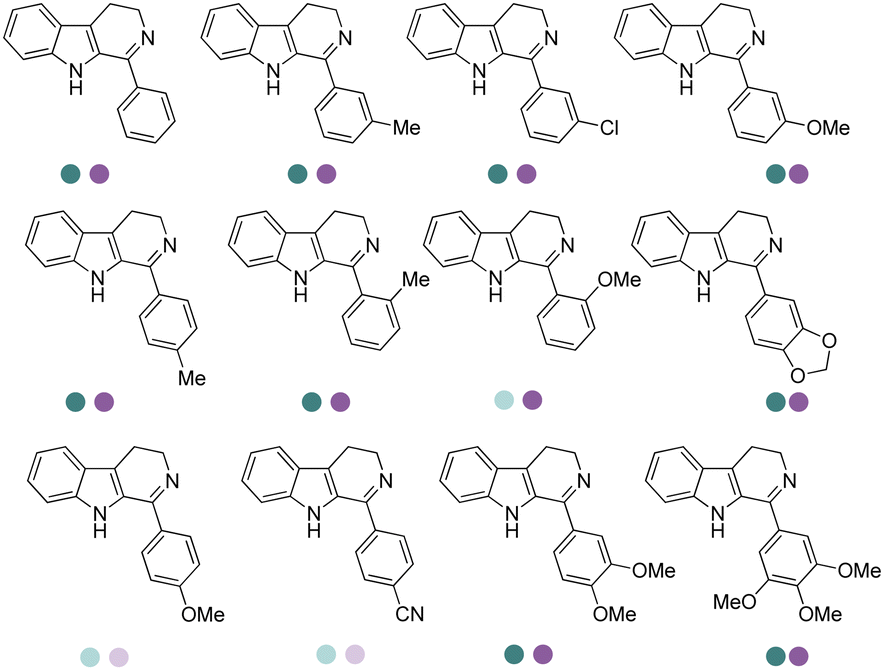
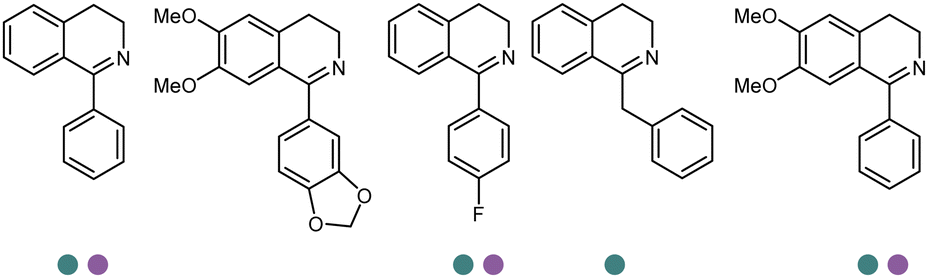
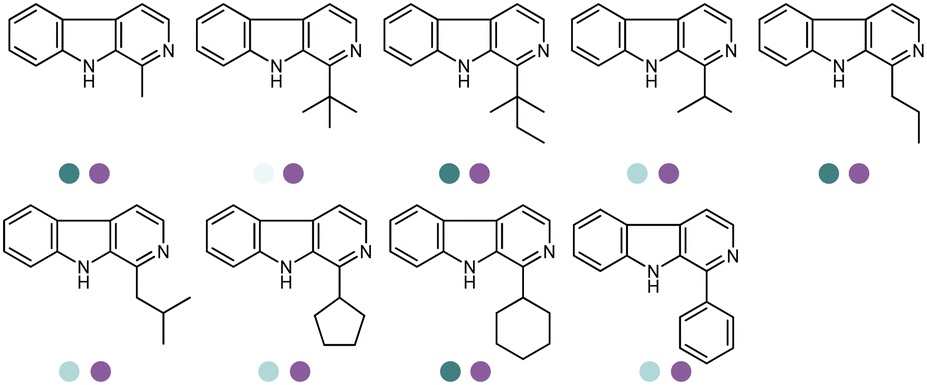
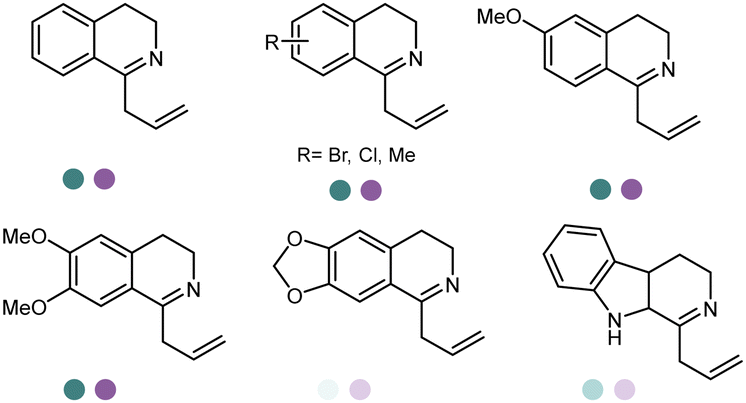
The discovery of IREDs was by Nagasawa et al. in 2010, where 2-methyl-1-pyrroline, a cyclic imine, was reduced by the whole-cell strains Streptomyces sp. GF3587 and 3546. The genes encoding them were subjected to heterologous expression in E. coli and employed in the biotransformations to afford both (R)- and (S)-configured amine products.93,113–115 The synthetic potential of Streptomyces sp. GF3546 has been explored and the substrate scope has been expanded to a variety of cyclic imines.116 Scheller et al. identified three new IREDs ((R)-SrIRED from Streptosporangium roseum DSM 43021, (R)-StIRED from Streptomyces turgidiscabies and (S)-PeIRED from Paenibacillus elgii),117 the substrate scope was expanded for the (R)-configured IRED from Streptomyces sp. GF3587 with further mutagenesis performed and variants D172A and D172L generated with moderate improvements (Table 3, entry 1).118 Following this line of work, IRED toolbox has been expanded further by screening IRED candidates and characterized a library of 20 IREDs (Table 3, entry 2).104,119,120| Entry | Enzymes | Sources | Substratesa | Products | Product informationb | pH | T/°C | T m/°C | Ref. |
|---|---|---|---|---|---|---|---|---|---|
a
 : conversions ≥ 80%; : conversions ≥ 80%;  : 40% < conversions < 80%; : 40% < conversions < 80%;  : conversions ≤ 40%; : conversions ≤ 40%;  : ee ≥ 80%; : ee ≥ 80%;  : 40% < ee < 80%; : 40% < ee < 80%;  : ee ≤ 40%.
b
R/S: the absolute configuration of the products; conv.: conversions. : ee ≤ 40%.
b
R/S: the absolute configuration of the products; conv.: conversions.
|
|||||||||
| 1 | IREDs | Streptomyces sp. |
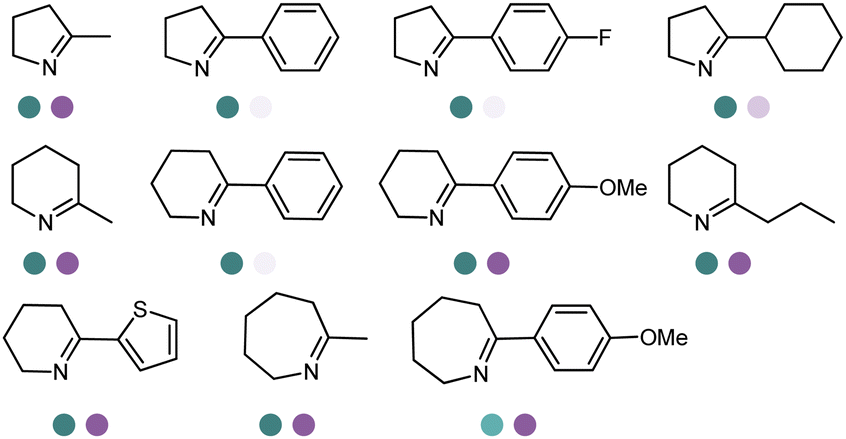
|
|
R or S, 4–99% conv., 8–98% ee | 7.0 | 30 | 118 | |
| 2 | IR-20/IR-23 | Streptomyces tsukubaensis/Streptomyces vidiochromogenes |
|
|
R or S, up to 99% conv., up to 99% ee | 7.0 | 30 | 119 | |
| 3 | R-IRED-Sr/R-IRED_Ms/S-IRED_Pe | Streptosporangium roseum/Myxococcus stipitatus/Paenibacillus elgii |
|
|
R or S, 39–99% conv., up to 87% isolated yields up to 94% de, 90–99% ee | 7.0 | 25 | 130 | |
| 4 | IREDs | Various sources |
|
|
R or S, up to 99% conv., up to 99% ee | 7.0 | 30 | 129 | |
| 5 | AspRedAm | Aspergillus oryzae |
|
|
R or S, up to 99% conv., up to 97% ee | 7.0 | 30 | 65 | |
| 6 | IRED14/IR-sip | Nocardia cyriacigeorgica GUH-2/Streptomyces ipomoeae 91-03 |
|
|
R or S, 71–94% conv., 52–90% ee | 9.5 | 30 | 128 | |
| 7 | IRED5/8/24 | Mycobacterium smegmatis/Glycomyces tenuis/Cupriavidus sp. HPC(L) |
|
|
S, 82–98% conv., 96–99% ee | 7.0 | 30 | 122 | |
| 8 | CfIRED | Cystobacter ferrugineus |
|

|
R or S, up to 92% conv., up to 99% ee | 8.0 | 30 | 146 | |
| 9 | AtRedAm/AdRedAm | Aspergillus terreus/Ajellomyces dermatitidis |
|
|
R or S, up to 96% conv., up to 96% ee | 9.0 | 30 | 30 | |
| 10 | IRED40/IRED30 | Sandaracinus amylolyticus/Sciscionella marina |
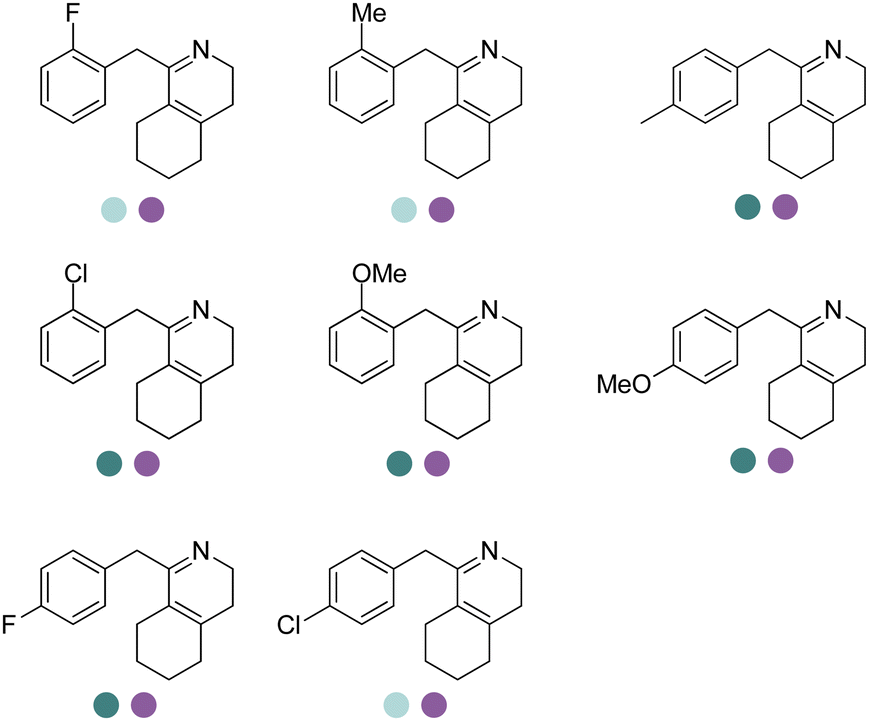
|
|
R or S, 47–95% isolated yields, 94–99% ee | 9.0 | 30 | 147 | |
| 11 | IREDs | Various sources |
|
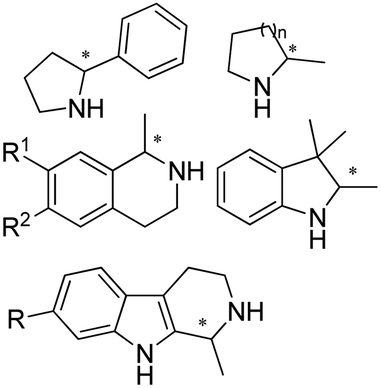
|
R or S, up to 99% conv., up to 99% ee | 7.5/6.0 | 30 | 148 | |
| 12 | MsIRED | Mycobacterium smegmatis |
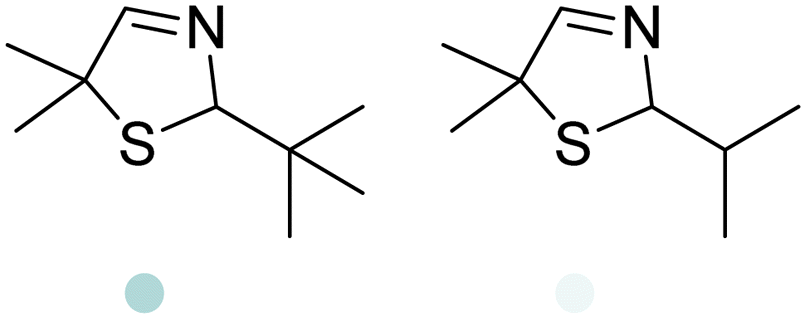
|
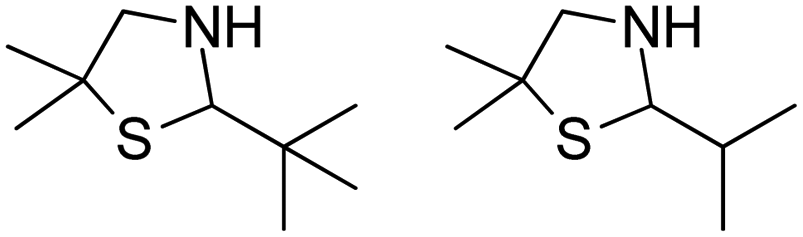
|
21–77% conv., 0% ee | 7.0 | 30 | 124 | |
| 13 | IRED12/IRED5 | Nocardia cyriacigeorgica GUH-2 /Cupriavidus sp. HPC(L) |
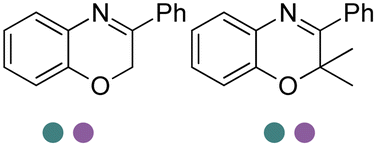
|
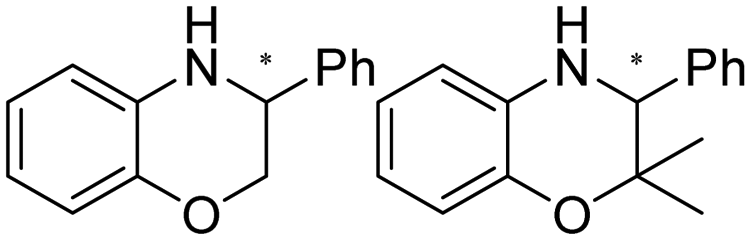
|
R or S, 9–99% conv., 26–99% ee | 7.4 | 30 | 125 | |
| 14 | IRED-E/IRED-D | Nocardiopsis alba/Mesorhizobium sp. L2C089B000 |
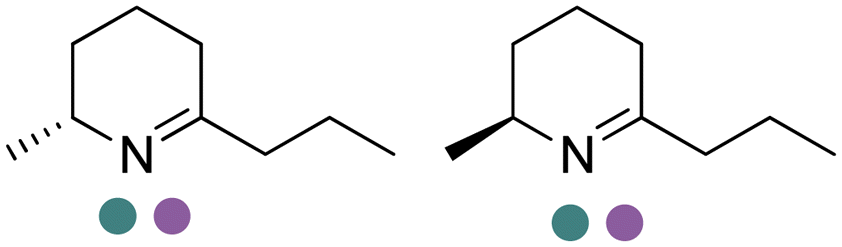
|
|
R or S, 2–99% conv., 94–99% de | 8.0 | 30 | 149 | |
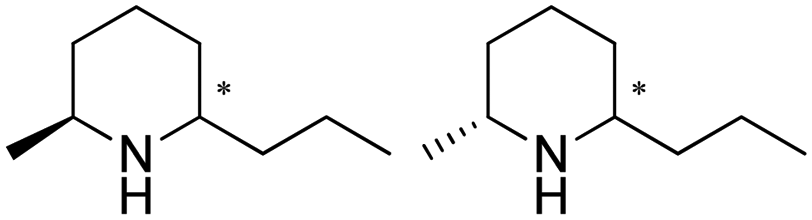
| 15 | IR46-M3 | Saccharothrix espanaensis |
|
|
1R, 2S, 95.6% conv., 99.9% ee, 84.4% isolated yields, | 4.6 | 30 | 70.0 | 145 |
| 16 | IR-Sip | Streptomyces ipomoeae 91-03 |
|
|
R or S, 86–95% conv., 28–95% ee, 3–81% isolate yields | 9.5 | 30 | 28 | |
| 17 | Artificial IREDs |
|
|
R or S, up to 100% conv., up to 96% ee | 150 | ||||
| 18 | SpRedAm | Streptomyces purpureus |
|
|
S, 96–96.5% conv., up to 99.5% dr, up to 80% isolated yields | 7.0 | 25–30 | 151 | |
| 19 | NfRedAm/NfisRedAm | Neosartorya fumigata/Neosartorya fischeri |
|
|
R or S, up to 97% conv., up to 97% ee | 9.0 | 30 | 50–54 | 33 |
| 20 | AdRedAm/NfRedAm/NfisRedAm | Ajellomyces dermatitidis/Neosartorya fumigate/Neosartorya fischeri |
|
|
S, 34–84% conv., 85–99% ee | 9.0 | 30 | 127 | |
| 21 | pIRs | Various sources |
|
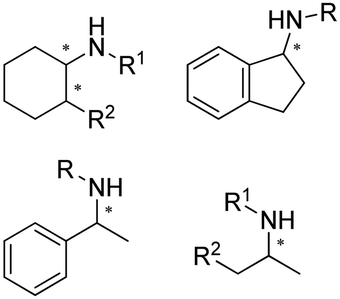
|
20–99% conv. | 8.0 | 25 | 132 | |
| 22 | IR1-Y194F/D232H and IR25 | Leishmania major/Micromonospora echinaurantiaca |
|
|
R or S, up to 99% conv., 93–99% ee, 31–86% isolated yields | 7.5 | 30 | 59 | 136 |
| 23 | IR45 and variants | Streptomyces aurantiacus |
|
|
S, 75–100% conv., 99% ee | 7.0 | 30 | 152 | |
| 24 | ScIR/SvIR | Streptomyces clavuligerus/Streptomyces viridochromogenes |
|
|
R or S, >99% conv., > 99% ee, 60–80% isolated yields | 7.0 | 30 | 137 | |
| 25 | pIRs | Various sources |
|
|
R or S, up to 99% conv., up to 99% ee, 31–91% isolated yields | 7.0–8.0 | 30–40 | 53.5 | 133 |
| 26 | pIR-88/pIR-202 and others | Various sources |
|
|
R or S, 18–99% conv., 36–99% ee, 21–91% isolated yields | 7.0 | 30 | 134 | |
| 27 | pIRs | Various sources |
|
|
R or S, up to 99% conv., up to 99% ee, 27–80% isolated yields | 7.5 | 30 | 153 | |
| 28 | IRED45 | Streptomyces aurantiacus |
|
|
S, 58–99% conv., 77–99% ee | 7.0 | 30 | 154 | |
| 29 | ScIRED-R3-V4 | Streptomyces clavuligerus |
|
|
R, up to 99% conv., up to 99% ee, 82.5% isolated yield | 6.0 | 30 | 49.8 | 138 |
| 30 | IREDs | Various sources |
|
|
R or S, 50–99% conv., 45–99% ee, 21–77% isolated yields | 7.0 | 30 | 155 | |
| 31 | EneIRED | Pseudomonas sp. |
|
|
R or S, up to 99% conv., up to 99% ee, up to 99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr :1 dr |
9.0 | 25 | 110 | |
| 32 | IREDs | Various sources |
|
|
R or S, 24–99% conv., up to 99:1 dr, 60–72% isolated yields |
7.5 | 25 | 156 | |
| 33 | IR-36 and others | Mesorhizobium sp. 1M-11 |
|
|
R or S, 4–99% conv., 23–99% ee, 41–84% isolated yields | 7.5–8.0 | 25 | 135 | |
| 34 | IR-G36-M5 | Actinoalloteichus hymeniacidonis |

|
|
R, up to 99% conv., up to 99% ee, 73–84% isolated yields | 7.0 | 30 | 63.5 | 109 |
| 35 | IR88/IR86/IR51/IR64 | Various sources |
|
|
R or S, 57–99% yields, 98–99% ee | 7.5 | 25 | 141 | |
| 36 | IR-G02/IR-G21/IR-G35/IRED2 | Various sources |
|
|
R or S, up to 99% conv., up to 99% ee, 39% isolated yield | 7.0 | 30 | 52.0 | 139 |
| 37 | PcIRED M3 | Penicillium camemberti |
|
|
R or S, 4–99% conv., up to 99% ee, 84.7% isolated yield | 7.0 | 30 | 51.5 | 142 |
| 38 | IREDs | Various sources |
|
|
R or S, up to 98% conv., up to 99% ee | 7.0 | 30 | 157 | |
| 39 | AtIRED | Amycolatopsis thermoflava |
|
|
S, 24–99% conv., 97–99% ee, 30–87% isolated yields | 7.5 | 30 | 158 | |
| 40 | R-IRED | Streptomyces sp. GF3587 |
|
|
R, 11–99% conv., 20–99% ee | 7.8 | 30 | 159 | |
Although the majority of IREDs are (R)-selective, the Grogan and Turner groups reported the structure of an (S)-IRED from Amycolatopsis orientalis (AoIRED).102AoIRED exhibited (S)-selectivity towards a range of cyclic imines with high activity, although interestingly (R)-configured products were observed upon prolonged storage of the enzyme or minor changes in the structure of the substrate. Based on crystal structures in complex with the cofactor NADPH or the substrate (R)-1-methyl-1,2,3,4-tetrahydroisoquinoline ((R)-MTQ), site-directed mutagenesis on the residues Y179, N241, and N171 was performed, and kinetic properties were investigated. Recently, Prejanò et al. conducted density functional theory (DFT) calculations to further investigate the mechanisms involved in the inversion of stereoselectivity of AoIRED.121 The transition state models were constructed with the substrate 1-methyl-3,4-dihydroisoquinoline, and revealed that the energy barrier for the (S)-selective path is lower than the (R)-selective path by 3.2 kcal mol−1, which correlated well with the experimental data where the product was obtained in 81% ee. In particular, the variants Y179A and N171A were introduced to the models, and the energies for (R)- pathways were both lower than (S)-pathways in the variants, which correlated well with the enantio-selectivity switch of the experimental data.
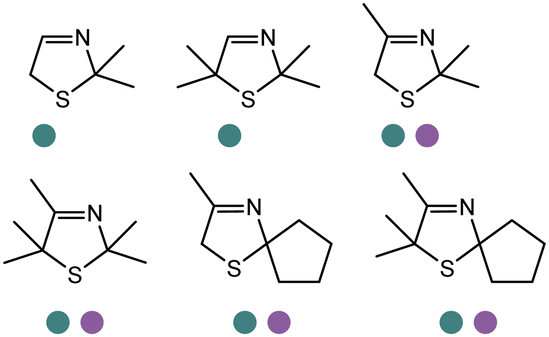
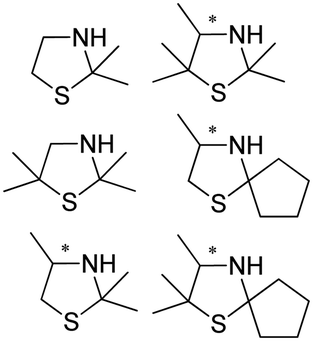
Also targeting cyclic imines, Zumbrägel et al. expanded the substrate scope of IREDs to 3-thiazolidine derivatives with sulfur-containing heterocycles. A pH-indicator assay was developed where color changes were correlated to the pH changes from the 3-thiazolines to the products. High enantioselectivity (>99% ee) and conversions (>96%) have been obtained for reduction of 3-thiazolines and 2H-1,4-benzothiazines, especially sulfur-containing spiro-heterocyclic amines were synthesized in high stereoselectivity (Table 3, entries 7, 12 and 13). Co-expression of IRED and GDH in E. coli enabled in situ cofactor recycling and preparative synthesis of a (S)-3-thiazolidine derivative at a 18 g L−1 substrate loading, 78% isolated yields and 99% ee.122–124 In addition to the sulfur-containing cyclic imines, the same group has also investigated N and O containing heterocycles such as 2H-1,4-benzoxazines.125
The discovery and expansion of the IREDs toolbox have provided important tools to the construction of chiral amine products. However, the formation of the imine intermediate has not been described by then. Therefore, initially IREDs were believed to primarily catalyze the reduction of cyclic imines. Nevertheless, it was soon to be found that intermolecular reductive amination could also be achieved and IREDs have been capable of catalyzing the coupling of the ketones with amine nucleophiles. Huber et al. firstly described an intermolecular reductive amination on ketones and in methylammonium buffer with IREDs from Streptomyces sp. GF3546 and S. aurantiacus with low conversions.103 Going forward, Scheller et al. employed a previously reported (R)-SrIRED in the amination of a variety of aldehydes and ketones.126 Primary and secondary amine products have been obtained using ammonia, methylamine or aniline as amine donors, albeit moderate conversions.
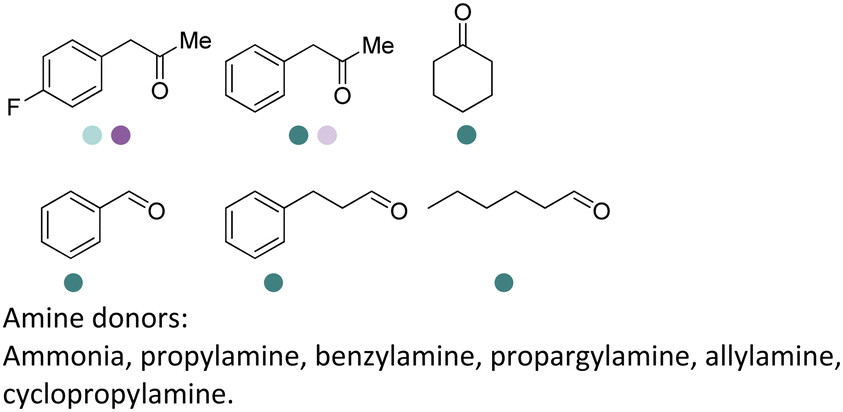
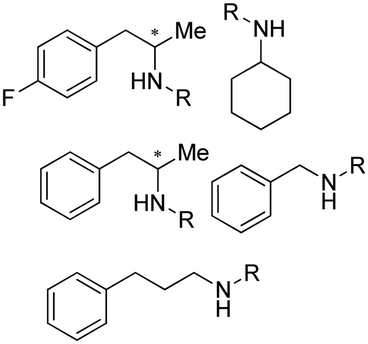
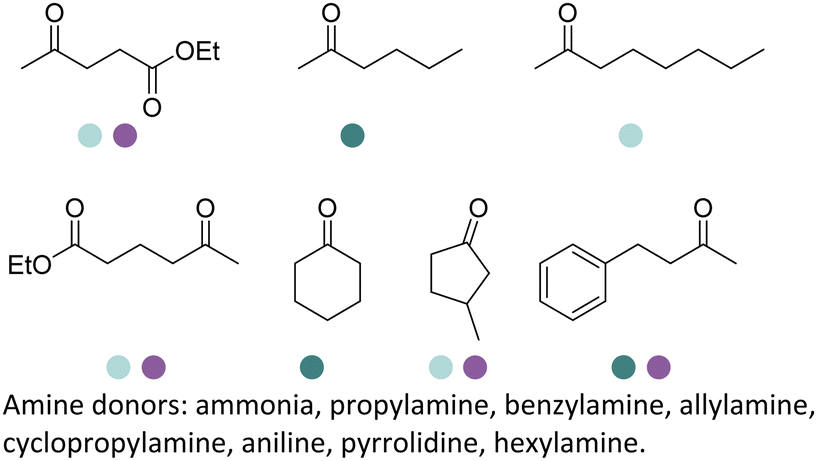
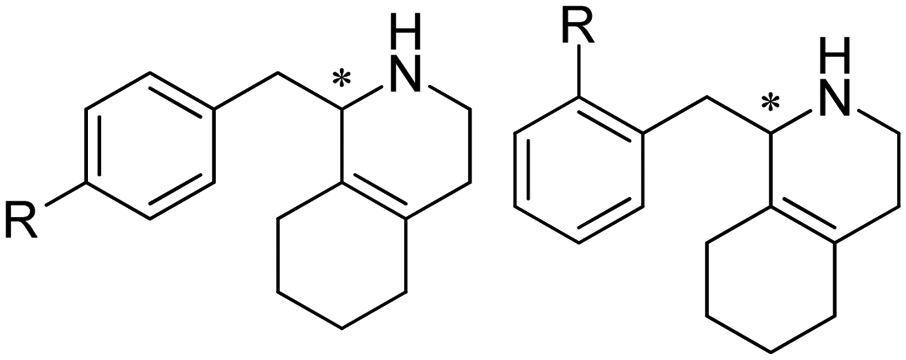
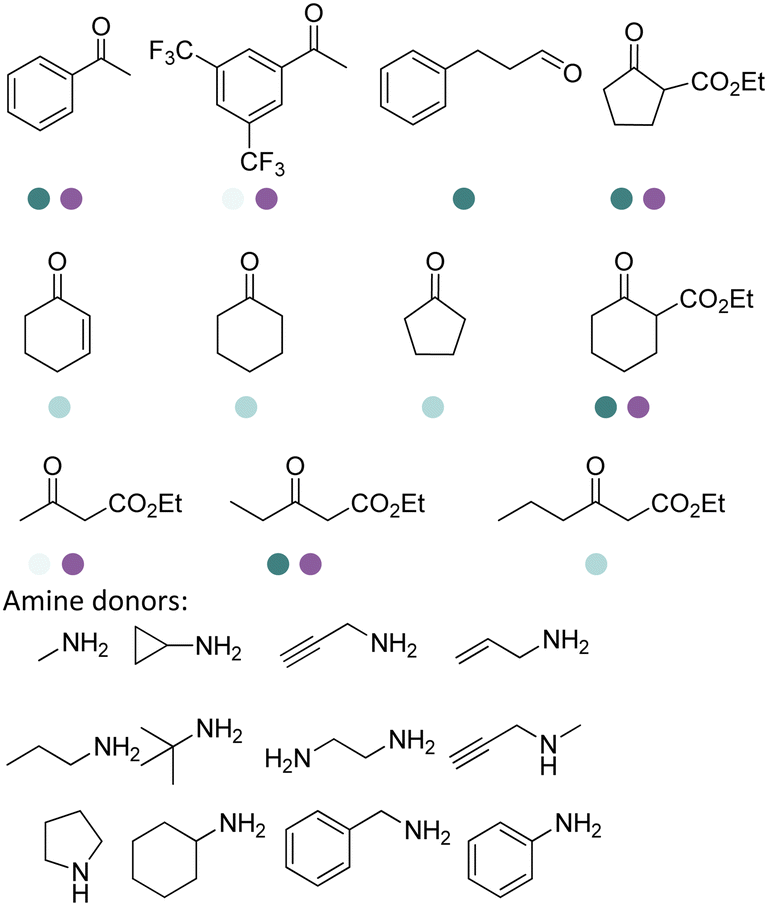
Aleku et al. reported a reductive aminase from Aspergillus oryzae (AspRedAm) that can catalyze both the formation of the imines and the imine reduction to produce chiral amines.28 A broad range of 32 ketone acceptors with 19 different amine donors have been tested for activity of the AspRedAm. From 1 eq. to 50 eq. of amine donors were used, and high conversions and ee have been achieved. With the crystal structure of RedAm, 6 residues including N93, D169, Y177, W210, M239 and Q240 were pointed out to be the guidance for distinctive RedAm activities. Two more RedAms were identified from Aspergillus terreus (AtRedAm) and Ajellomyces dermatitidis (AdRedAm) which contained the equivalent six residues. The studies indicate that the coupling of the ketone substrate with the amine donor follows an ordered sequential Ter Bi mechanism. For the first time, further evidence demonstrated that AspRedAm can catalyze imine formation. Later their crystal structures have been established in complex with cofactors and substrates and the catalytic mechanism by AtRedAm was proposed.30,106 The substrate scope of this enzyme has been expanded to fluorine-containing arylkontones.127
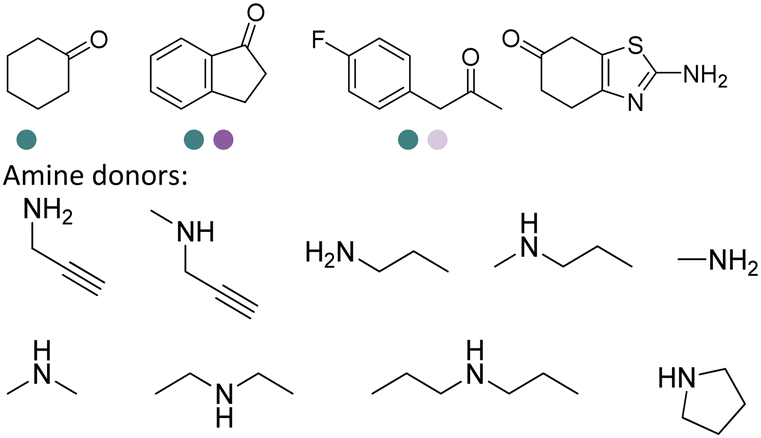
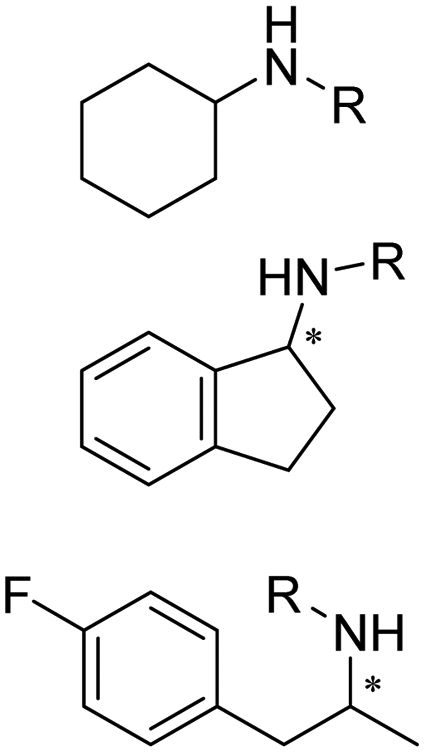
IREDs can be used in the synthesis of a number of pharmaceuticals and their amine-containing intermediates. One of the early examples was reported by Matzel et al, who screened a panel of 39 IREDs targeting the pharmaceutically relevant (R)-rasagiline in preparative scale with high yields (up to 81%) and ee (up to 90%) (Table 3, entry 6). Tertiary amines have also been synthesized in preparative scale using N-methyl propargylamine as the amine donor, albeit long reaction time (7 days) and excessive amine utilization (40–100 eq.).128
Although the reductive amination has been shown to proceed with high efficiency, the previously used large excess of amine donors used (up to 100 eq.) prohibits the applications in industry. Therefore, identifying more powerful biocatalysts, reducing the excess use of amine and improving the activity and stereoselectivity have become a focus in the following investigations of IREDs. Indeed, Roiban et al. expanded the application of the reaction to the synthesis of a range of secondary and tertiary amines including anilines and heteroaromatic amines with only 1.0–1.1 eq. of amine donors (Table 3, entry 4).129 Using 6.25 mM (1.25 eq.) 1,2-dicarbonyl substrates and 5 mM 1,2-diamines (Table 3, entry 3), Borlinghaus et al. reported a double reductive amination, leading to the formation of piperazine derivatives, which were key intermediates for pharmaceuticals such as Indinavir or Mirtazapin. Both (R)-IREDs and (S)-IREDs were tested, and MsIRED from Myxococcus stipitatus was identified with good reactivity.130 Later, key residues have been recognized to be responsible for changing the stereo-preference of MsIRED from Myxococcus stipitatus. The same group constructed SSM libraries at the regions 240–246. Beneficial mutations were recombined, and the final variant containing five mutations (A241V/H242Y/N243D/V244Y/A245L) exhibited a switch of enantio-selectivity from (R)-configuration (99% ee) to (S)-configuration with 91% ee.108
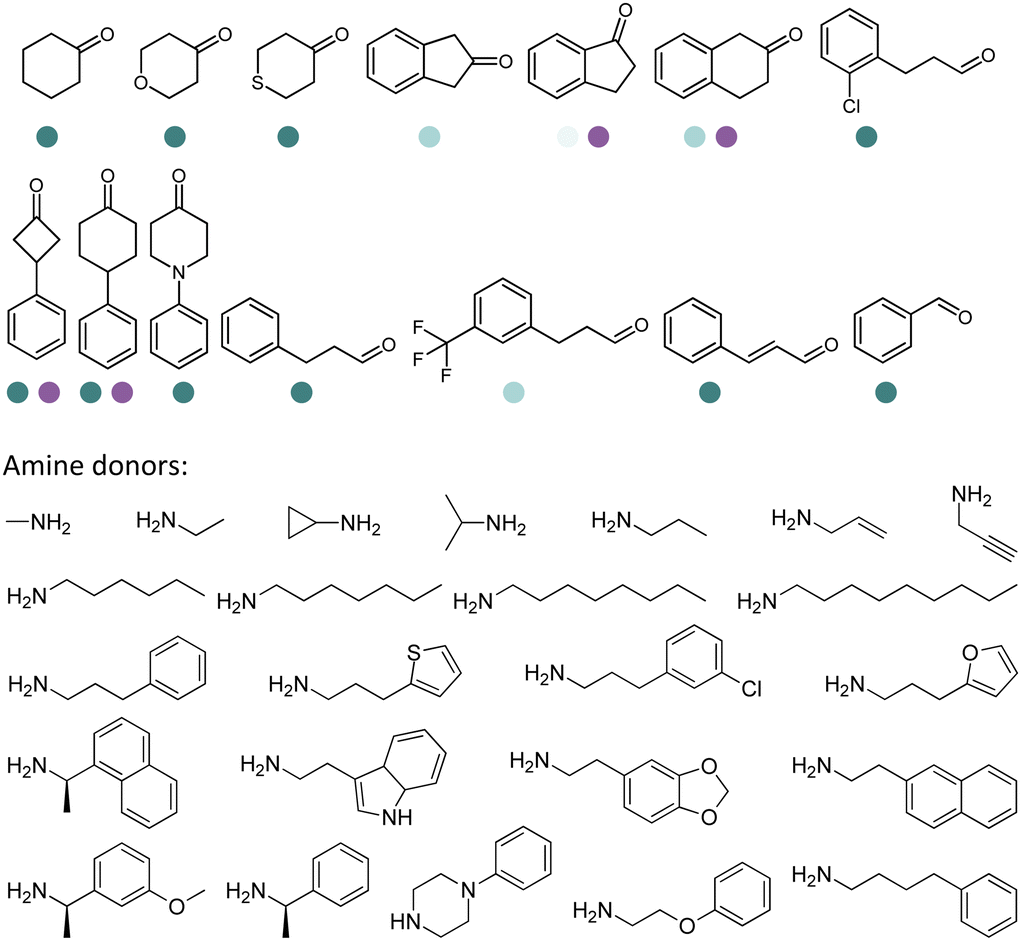
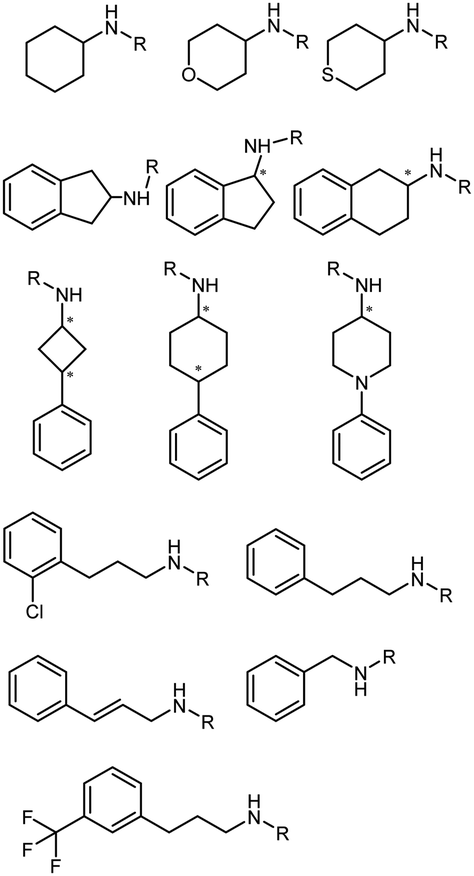
The IREDs toolbox was further expanded by mining and characterizing novel IREDs using tools such as BLAST. Several panels of bacterial IREDs have been identified to catalyze the formation of primary and secondary amines.131 The discovery and characterization of 80 putative IREDs based on search with the sequence identity >30% with AspRedAm were reported by Montgomery et al.132 The sequence-structure analysis was performed using a SmartScaffold approach. It was revealed that (R)-selective IREDs have the AspRedAm-equivalent P121, F176, M/I210, and S243 residues, and AspRedAm equivalent T121, M/L176, F/W210, and A243 for (S)-selective IREDs. They concluded that the IRED sequence–activity relationships are dependent heavily on the substrates, and IREDs known to catalyze the reductive amination of cyclic imines are also capable of catalyzing the intermolecular reductive aminations (Table 3, entry 21).
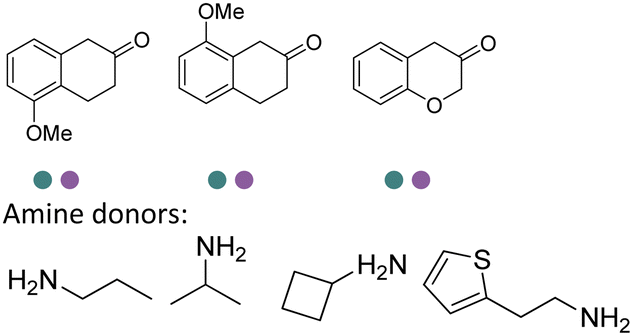
The IREDs catalyzed synthesis of chiral amine intermediates have been significantly advanced by screening of a metagenomic panel of 384 enzymes including the use of a colorimetric high throughput (HTS) method.133 The substrate scope has been greatly expanded to include the synthesis of β-amino esters from β-keto esters via a dynamic kinetic resolution (DKR) process at preparative scales, affording high yields and ee (Table 3, entry 25). A wide range of acetophenone derivatives, dimethylamine derivatives, β-keto esters derivatives were demonstrated to be synthesized by the IREDs catalyzed reductive amination. Later, the same metagenomic panel were screened towards the synthesis of intermediates for the drug Rotigotine used for Parkinsons disease therapy (Table 3, entry 26), which reduced the synthetic steps from 5 to 3.134 Further screening has identified IREDs from the same panel to synthesize N-alkyl-α-amino esters from α-ketoesters (Table 3, entry 33).135 The above work did not require engineering efforts but afford products in near-perfect enantio-selectivity and high conversions, which demonstrated the powerful potential of the metagenomic enzyme discovery and high-throughput screening.
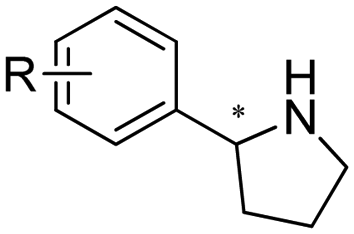
With the rapid expansion of the IRED toolbox, applications in the synthesis of valuable pharmaceuticals have been developed and exhibited promising potential in upscaled productions of important drugs. For example, Xu et al. attempted the IREDs catalyzed synthesis of 1,4-diazepanes via intramolecular reductive amination. (R)-selective LmIRED from Leishmania major and (S)-selective MeIRED from Micromonospora echinaurantiaca have been identified from screening a panel of 48 IREDs. The Y194F/D232H variant from LmIRED exhibited synergistic effect with the highest catalytic efficiency and highest selectivity for a range of substrates (Table 3, entry 22). Preparative scale biotransformations at 100 mM substrate concentration were performed affording excellent ee (99%) for the synthesis of a Suvorexant intermediate.136
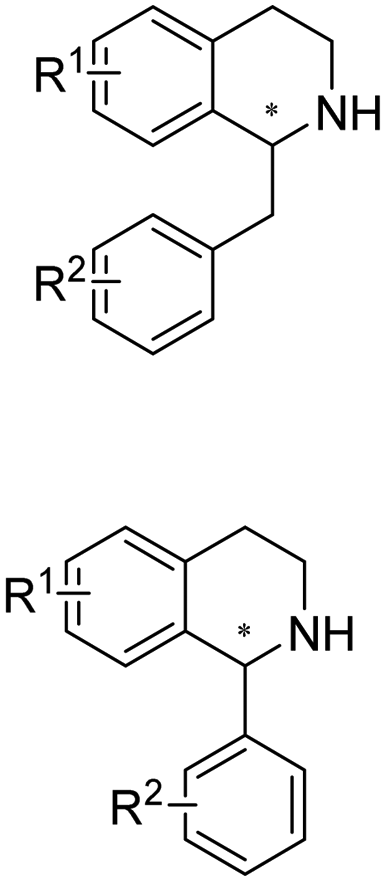
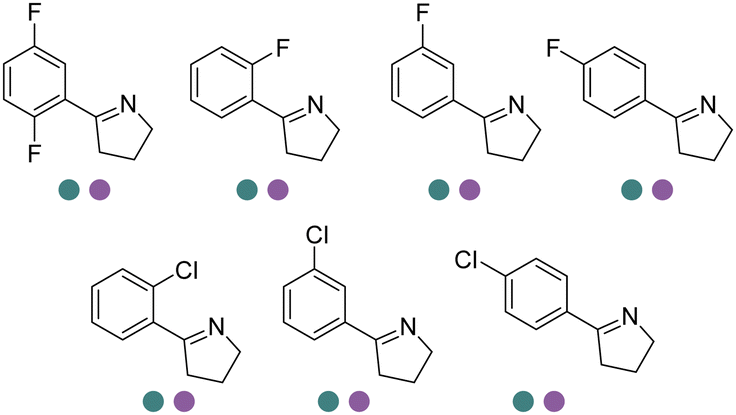
Chiral 2-aryl-substituted pyrrolidines are important building blocks for a number of potential pharmaceutical candidates including CDK8 inhibitor MSC2530818, tropomyosin receptor kinase inhibitor Larotrectinib (LOXO-101), etc. The Zheng group screened a panel of IREDs towards 2-aryl-substituted pyrrolines and obtained two best performing IREDs: (R)-selective ScIR from Streptomyces clavuligerus and (S)-selective SvIR from Streptomyces viridochromogenes. They developed preparative scale (100 mL) processes for both (R)- and (S)-IREDs to afford conversions >99% with up to 79.7% isolated yields and >99% ee at up to 40 mM substrate loadings (Table 3, entry 24).137 Recently, the same group further engineered ScIR for 3 rounds, and with the support of crystal structure of WT ScIRED, they have been able to select 10 residues in proximity with the substrate and constructed SSM libraries.138 The variant ScIRED-R1-V1 was generated, resulting in a 10-fold increase in activity. For the second round, all 27 sites were selected from 4–8 Å in the second shell. The resulting variants ScIRED-R2-V2 displayed over 70-fold improvement in activity over the WT enzyme, and ScIRED-R2-V3 displayed higher thermostability. Finally, based on B-factor analysis, the best variant ScIRED-R2-V4 was obtained which was able to transform 80 g L−1 substrate, affording >99.5% ee and >99% conversion (Table 3, entry 29). In the upscaled synthesis of Larotrectinib intermediate (R)-2-(2,5-difluorophenyl) pyrrolidine on 20 L scale, 1.32 kg of the product was obtained in 4.5 h with 82.5% isolated yield, >98.5% purity and >99% ee, which resulted in a 352 g L−1 day−1 space-time-yield (STY).
Similarly, targeting N-containing heterocycles, Zhang et al., screened a panel of 86 enzymes against N-Boc-3-piperidinone with benzyl amine as the amine donor. IR-G36 was identified with the highest (R)-selective product (78% ee), albeit only 8% conversion. Upon examining the crystal structure and docking structures, we used site-saturation mutagenesis (SSM) and CAST to perform several rounds of engineering on IR-G36. The final variant IR-G36-M5 bearing mutations at various positions (N121T/S260F/F207I/M238A/M264H/A267H/L197I/N271K/H189A/G145K) showed 5.4 °C improvement in Tm, and 4193-fold in kcat/Km. The crystal structure of the variant IR-G36-M5 in complex with NADP+ was obtained, and additional hydrogen bonds and salt bridges were identified, which provided insights to the stability of the variant. Further preparative scale biotransformations afforded N-boc protected azacycloalkylamines in ketone loadings up to 47 g L−1 and up to 99% conversions and ee (Table 3, entry 34).109 Later, bulky amines as donors have been included into the scope (Table 3, entry 36).139
Large panel screenings and further engineering efforts have enabled a number of target pharmaceuticals to be synthesized by IREDs. Zhan et al. developed the IRED catalyzed synthesis of a key intermediate to the tyrosine kinase inhibitor Tofacitinib, which has been a successful drug commercialized in more than 50 countries.140 A panel of 175 IREDs were screened against the racemic ketone substrate rac-1-benzyl-4-methylpiperidin-3-one to obtain (3R,4R)-1-benzyl-N,4-dimethylpiperidin-3-amine bearing two consecutive stereocenters. 43 active enzymes were identified. Further exploration and optimization of amine donors, substrate concentrations led to the identification of two IREDs with the highest activity and enantio-selectivity. Preparative reactions were carried out using 100 mM rac-1-benzyl-4-methylpiperidin-3-one at 25 mL scale afforded both (3R,4R)-1-benzyl-N,4-dimethylpiperidin-3-amine or (3S,4S)-1-benzyl-N,4-dimethylpiperidin-3-amine in high isolated yields (83% and 91%), excellent stereoselectivity (97% and >99% ee values) and diastereoselectivity (>99:1 dr). The same group reported later another application of IREDs in the synthesis of enantiocomplementary fused-ring tetrahydroisoquinoline (THIQs) and tetrahydro β-carboline (THβCs). A panel of 104 IREDs were screened and high stereoselectivity (up to >99% ee) were obtained. A number of fused-ring THIQ and THβCs were synthesized at preparative scales including natural alkaloids with stereo-complementary IREDs in high yields (up to 85%) (Table 3, entry 35).141
Very recently, the Zheng group discovered an IRED from Penicillium camemberti (PcIRED). Targeting the calcimimetic drug Cinacalcet, ISM was performed on this enzyme. With three rounds of engineering combining site-directed saturation mutagenesis and combination of beneficial mutations, a variant M3 was obtained with 488-fold increase in activity and 11 °C improvement in melting temperature over the wild-type enzyme. 100 mL scale synthesis of cinacalcet was performed with 500 mM (R)-1-(1-naphthyl)ethylamine and 1.2 eq. of the ketone substrate 3-[3-(trifluoromethyl)phenyl]propanal, and the final isolated yield reached 84.7% with >99% ee. In particular, broad scope for the bulky ketone and amine nucleophiles were well tolerated by the variants, and high cis/trans selectivity was obtained towards a range of 3-phenyl-cyclobutanone or 4-phenyl-cyclohexanone derivatives (Table 3, entry 37).142
It has been established that IREDs catalyze both the imine formation and asymmetric reduction of the imines. However, there is more evidence towards the latter than the former. Gilio et al. characterized the imine formation as well as the reductive amination by IREDs and showed that larger amine donors may undergo spontaneous formation of imines which were then recruited from solution and reductively aminated by IREDs. Site saturation mutagenesis library of a knowledge-based site A208 has led to an A208N variant with superior activity towards bulky amines such as isoindoline or octahydrocyclopenta(c)pyrrole coupled with cyclohexanone. Preparative scale biotransformations with 1.2 molar eq. of amines afforded up to 93% isolated yields.112
With the use of large data sets, machine learning models have been trained and combined with directed evolution to explore the sequence space of the enzymes more efficiently. Ma et al. conducted deep mutational scanning (DMS), error-prone PCR, structure-guided machine-directed evolution and low N engineering to IRED-88 that was identified from genome mining. The authors demonstrated that machine-learning combined with directed evolution allowed the screening of a smaller library to obtain comparable activities and stereoselectivities to the traditional strategies. Machine-learning models can be helpful when the throughput of the screening is limited due to cost, scale, or time and a combination use of the methodologies might be necessary depending on the available resources.143
Most recently, a team at Manchester established the IREDFisher workflow by designing an in silico streamline with sequence analysis, template-based homology modelling, and molecular docking. The final scoring algorithm consist of Autodock Vina scoring, key catalytic hydride transfer distance and iminium intermediate-stabilizing residues. Experimental validation results from IREDFisher-selected panels outperformed random selection in hits with conversions over 50%. In addition, user-friendly web interface (https://enzymeevolver.com/IREDFisher) was provided which enabled users with no computational experiences to exploit the tools with convenience.144
Since the discovery of IREDs and the advancement of high throughput screening, directed evolution and rational design methodologies, extensive research efforts have been invested in expanding the substrate scope of IREDs. Herein a selection of substrate structures from various literature precedence are listed in Table 3. IREDs from various sources have been identified, and both discovery and engineering of IREDs have enabled the biotransformation of a large number of substrates carrying various moieties. In particular, the thermostability of IREDs has been improved dramatically. For example, in the work reported by Schober et al, the melting point (Tm) of the IRED was improved from 41.3 °C to 70.0 °C through 3 rounds of mutagenesis.145 Therefore, Tm of the various wild-type IREDs or their variants are included in Table 3. The pH and temperature are also critical when designing the enzymatic reactions, and generally IREDs prefer slightly basic conditions (pH 7.0–9.0) and room temperature (30 °C). These conditions are also summarized in Table 3.
3.3. Towards the applications of IREDs
3.3.1.1. Multi-enzymatic cascades. One-pot multi-enzymatic approaches as well as whole-cell biocatalysis have received increasing research attention as they fulfil the industrial needs for the synthesis of structurally diverse molecules. In particular, chemoenzymatic methodologies are important since they harness the benefits of selectivity of biocatalysis and highly diverse reactivity of organic/metal catalysis. Hereafter we discuss the applications of IREDs in cascades with either multi-enzymatic biocatalysis or chemo-catalysis for the synthesis of industrially relevant building blocks.
Piperidines and pyrrolidines are core structures for a variety of biologically active molecules. Using multi-enzymatic cascades, the synthesis of chiral piperidines or pyrrolidines could be achieved using carboxylic acid reductase (CAR), ω-transaminase (ω-TA), and imine reductase (IRED) in one-pot.161 On the other hand, whole cells are often important sources of the enzymes for biotransformations. Targeting the synthesis of chiral substituted piperidines, the Flitsch group designed a four-enzyme, four-step cascade in whole cells starting from keto acids using a bioretrosynthetic approach. Carboxylic acid reductase, ω-transaminase and IREDs have been constructed in the whole-cell system. Under optimized reaction conditions, the 4-methyl-2-phenylpiperidine was obtained in >98% de and 93% conversions, with isolated yield of 59% on preparative scale (Fig. 17).160
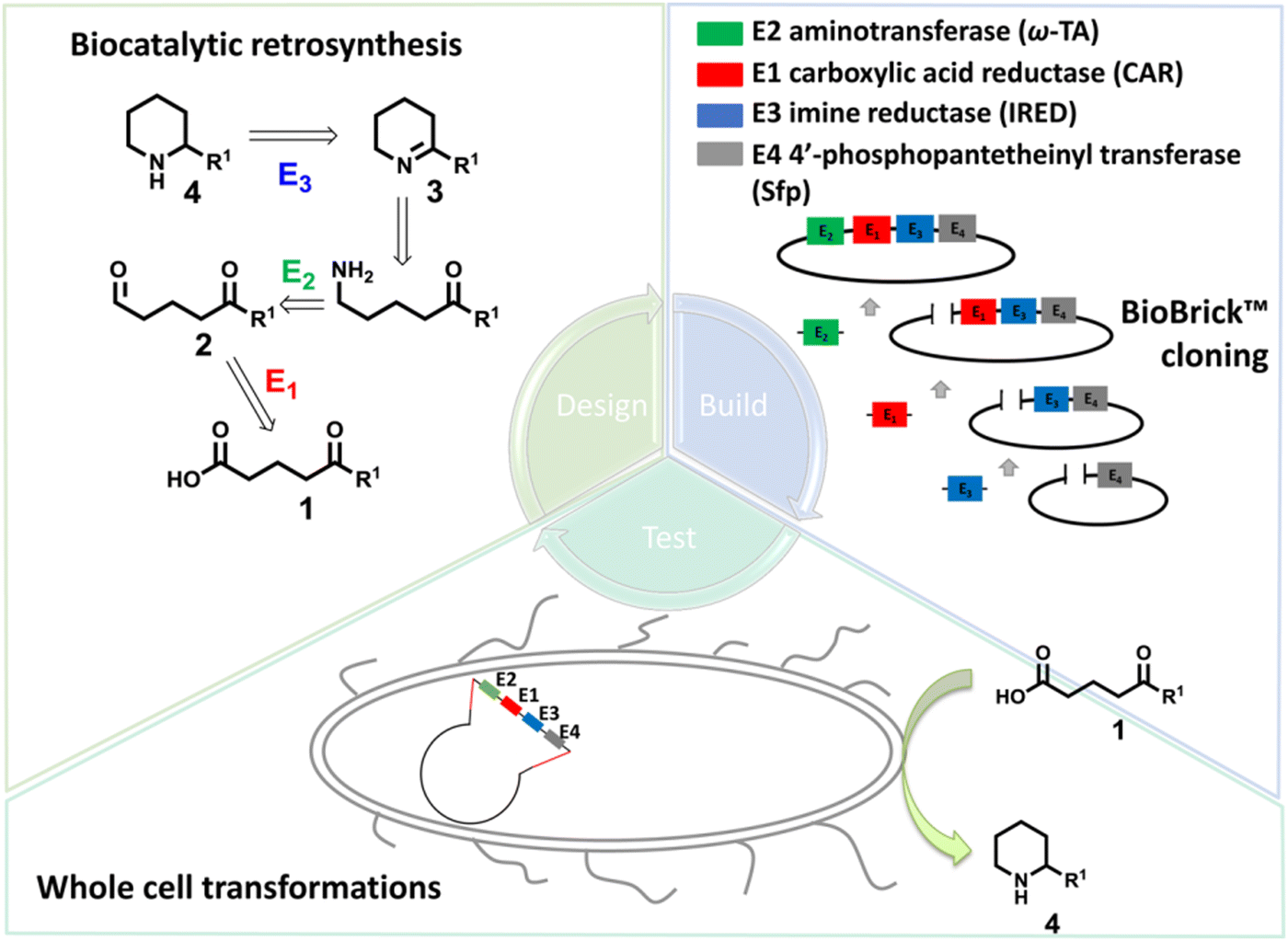 | ||
| Fig. 17 The whole-cell enzymatic cascades achieved by the design–build–test workflow. Adapted with permission from ref.160. Copyright 2017 American Chemical Society. | ||
IREDs have also been employed in a cascade with P450 monooxygenases to produce secondary amines from cyclo-alkanes. Formate dehydrogenase from Candida boidinii (CboFDH) was used for the cofactor regeneration of P450, and a variant of ADH from Thermoanaerobacter ethanolicus (TeSADH W110A) was used to construct a hydrogen-borrowing cascade for IREDs.162 Further cascades of IREDs have been built with ene-reductases (EREDs) for the synthesis the 2-substituted saturated amine heterocycles with 2 stereocenters,163 and galactose oxidase (GOase) to afford L-3-N-aminopiperidines and L-3-N-aminoacepanes using GOase variants M3–5 and F2164,165 to oxidize the aminoalcohol, followed by spontaneous cyclization to form an imine intermediate, which was subsequently reduced by AdRedAm.166
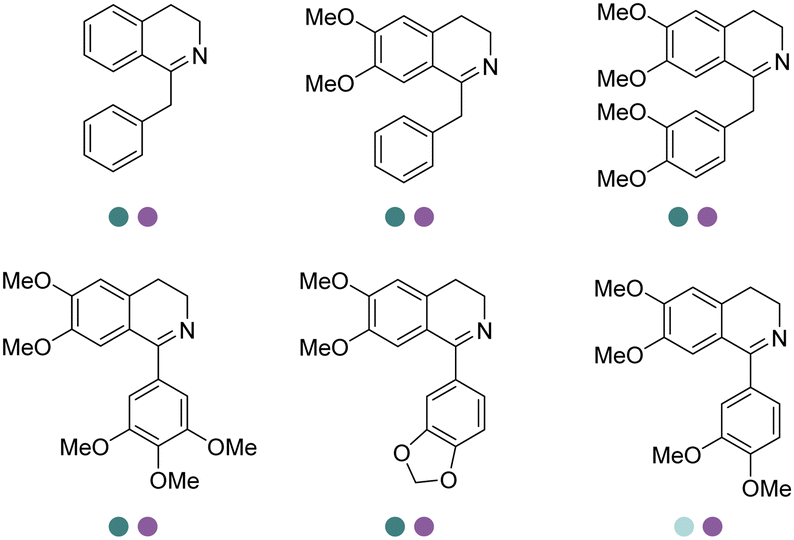
Yang et al. screened and engineered IREDs for the production of a series of tetrahydroisoquinoline alkaloids (THIQAs) from dihydroisoquinoline (DHIQ) precursors (Table 3, entry 23). The IRED variant with improved enzymatic activity was then combined with a coclaurine N-methyltransferase (CNMT) from Coptis japonica, and a glucose dehydrogenase (GDH) either in a single cell (pACYCDuet and pET28a) or one-pot cascade where IRED and GDH were co-expressed and then added to the CNMT culture. The biosynthetic pathway led to full conversions in 1–3 days and isolated yields of the final THIQAs reached 93% and 96%.152 Later, the same group reported the applications of IREDs and CNMT cascades in the synthesis of chiral N-methylated 1-aryl-tetrahydro-β-carboline (THβC), which is a key intermediate for drugs such as tadalafil that are used for the treatment of erectile dysfunction (Table 3, entry 28).154 Key active-site residues were identified and mutated by analyzing the interactions of the bulky substrate 1-phenyl-DHβC with the IRED, and further combinations were constructed with the beneficial mutations. The final triple variant IR45-L228′A/M250′L/E251′M was able to reduce most of the substituted 1-phenyl-DHβCs in >99% ee and conversions, and preparative scale biotransformations were also performed in 1 L scale to achieve full conversions. Targeting THβCs and THIQs, further discovery and engineering work has revealed the powerful potential of IREDs to catalyze this class of stereoselective synthesis of the sterically demanding molecules (Table 3, entries 38–40).157–159
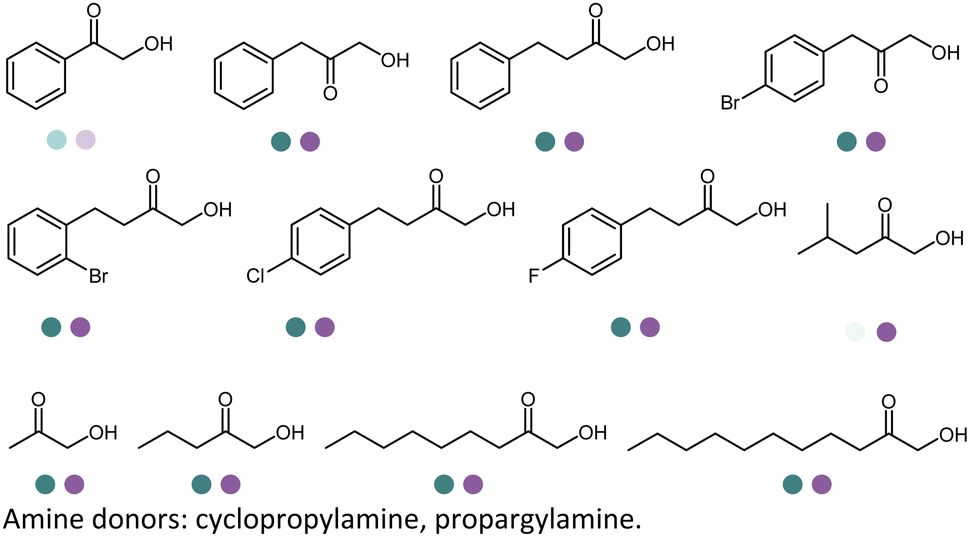
In 2022, Li et al. reported the two-step sequence employing the EREDs and IREDs for the production of N-substituted γ-amino esters and γ-lactams using cyclopropylamine as the amine partner (Table 3, entry 31). Screening efforts were invested in identification of the most active ERED and IRED towards α,β-unsaturated γ-ketoesters substrates such as methyl 3-oxocyclohex-1-ene-1-carboxylate and ethyl (E)-4-oxo-2-propylpent-2-enoate. The carbonyl reduction was the first step and then upon completion the IRED catalyzed reductive amination was initiated as the second step. All of the four diastereomers of the γ-amino esters and two enantiomers of the γ-lactams were obtained using various combinations of (R)- and (S)-selective EREDs and IREDs in 60–72% yields and >99% ee or dr on preparative scales.156
3.3.1.2. Chemo-enzymatic cascades. Chemoenzymatic approaches have proved to be potentially valuable methods for the synthesis of complex target molecules. Azepanes and benzazepines are core motifs of both medicinal and pesticidal active molecules. Zawodny et al. utilized IREDs in the reduction of heterocyclic 7-membered rings containing an imine moiety, followed by treatment of the amine reduction products with base (LDA in a 4
:1 mixture of Et2O and DMPU), enabling aryl migration to take place, thereby generating enantiopure α-functionalized azepine-derived ureas in good yields. This is an excellent example of chemoenzymatic synthesis of heterocyclic tertiary amines (Table 3, entry 8), taking advantage of the enantioselectivity of enzymes to prepare steric challenging amine products (Fig. 18).146
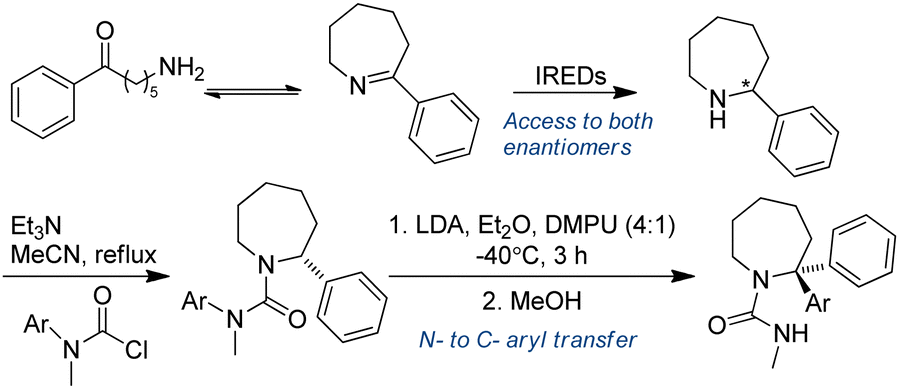 | ||
| Fig. 18 IREDs-catalyzed reductive amination followed by organolithium mediated α-functionalization of azepine-derived ureas. | ||
Further chemoenzymatic approaches were developed with IREDs with ammonia borane for the deracemization of racemic amines. The oxidation of amines in presence of NADP+ was exploited in combination with imine reduction by ammonia borane and cofactor regeneration by NADPH oxidase (NOX) using oxygen. This system was applied to a broad range of cyclic and acyclic substrates and represents a useful addition to the synthetic toolbox of chiral amines starting from racemic amines.167
Electrical energy has been extensively used in the cofactor regeneration of oxidoreductases to take advantage of sustainable and clean energy sources. Al-Shameri et al. used molecular H2 for NAD+-reducing hydrogenase (SH) coupled with putrescine oxidase catalyzed oxidation and IRED catalyzed reductive amination for the synthesis of piperidine derivatives from diamine substrates. A gas-selective permeable membrane was used to facilitate the transfer of gases and enzymes were immobilized on commercially available carriers. The system was easily scaled up to 300 mL and the immobilized enzymes can be reused up to six times.168
Dihydropinidines are a class of piperidines with antifeedant activity. The Kroutil group reported the synthesis of dihydropinidines using both chemo- and bio-cascades involving IREDs on multigram scale. The steps are: (1) Lewis acid catalyzed Michael addition; (2) pig liver esterase (PLE) catalyzed hydrolysis followed by acid-promoted decarboxylation to afford diketone nonane-2,6-dione; (3) two stereo-complementary transaminases from Arthrobacter (ArR-TA and ArS-TA) catalyzed transamination of the diketone nonane-2,6-dione followed by spontaneous cyclization to the imine intermediate; (4) imine reduction of 4 by screening a panel of 14 IREDs and identification of highly active (R)- and (S)-selective IREDs. Finally, implementation of the TA and IRED catalyzed reactions enabled a one-pot concurrent cascade reaction on preparative scale, affording >99% ee and 57% overall isolated yields for the final product (2R,6S)-dihydropinidine (Fig. 19).149 Similarly, using the TA-IRED cascade or Pt/C catalyzed hydrogenation in a continuous flow reactor, tri-substituted piperidines with three chiral centres have been obtained in high diastereomeric ratio (dr) (>99:1) and good yield (up to 73%) on preparative scale.169
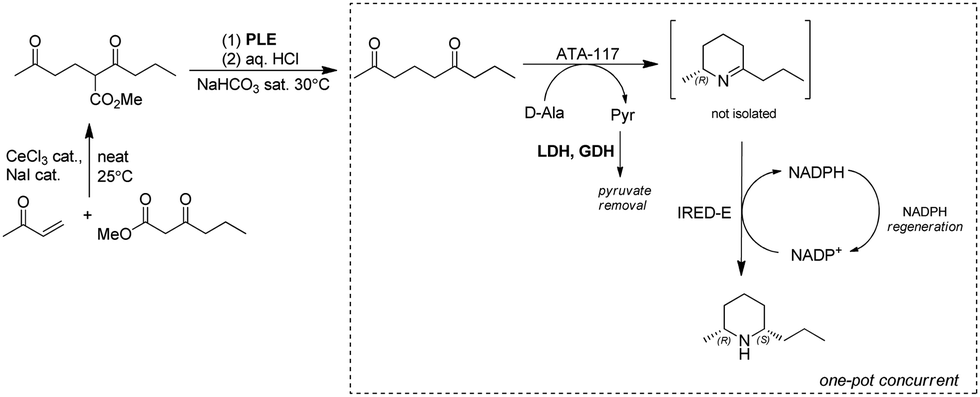 | ||
| Fig. 19 Chemo-enzymatic approaches to the synthesis of dihydropinidine. | ||
Very recently, the deracemization methodology to combine MAO-N, IREDs and metal catalyzed allylation has been achieved in one-pot to produce a range of C(1)-substituted THIQs. The allylBPin intermediate was generated by Yb(OTf)3 catalyzed allylboration to form racemic THIQs, subsequently biocatalytic deracemization afforded the C(1)-substituted THIQs in high yields and good to high ee. The system could also be scaled up with up to 64% isolated yield and 98% ee (Table 3, entry 40).159
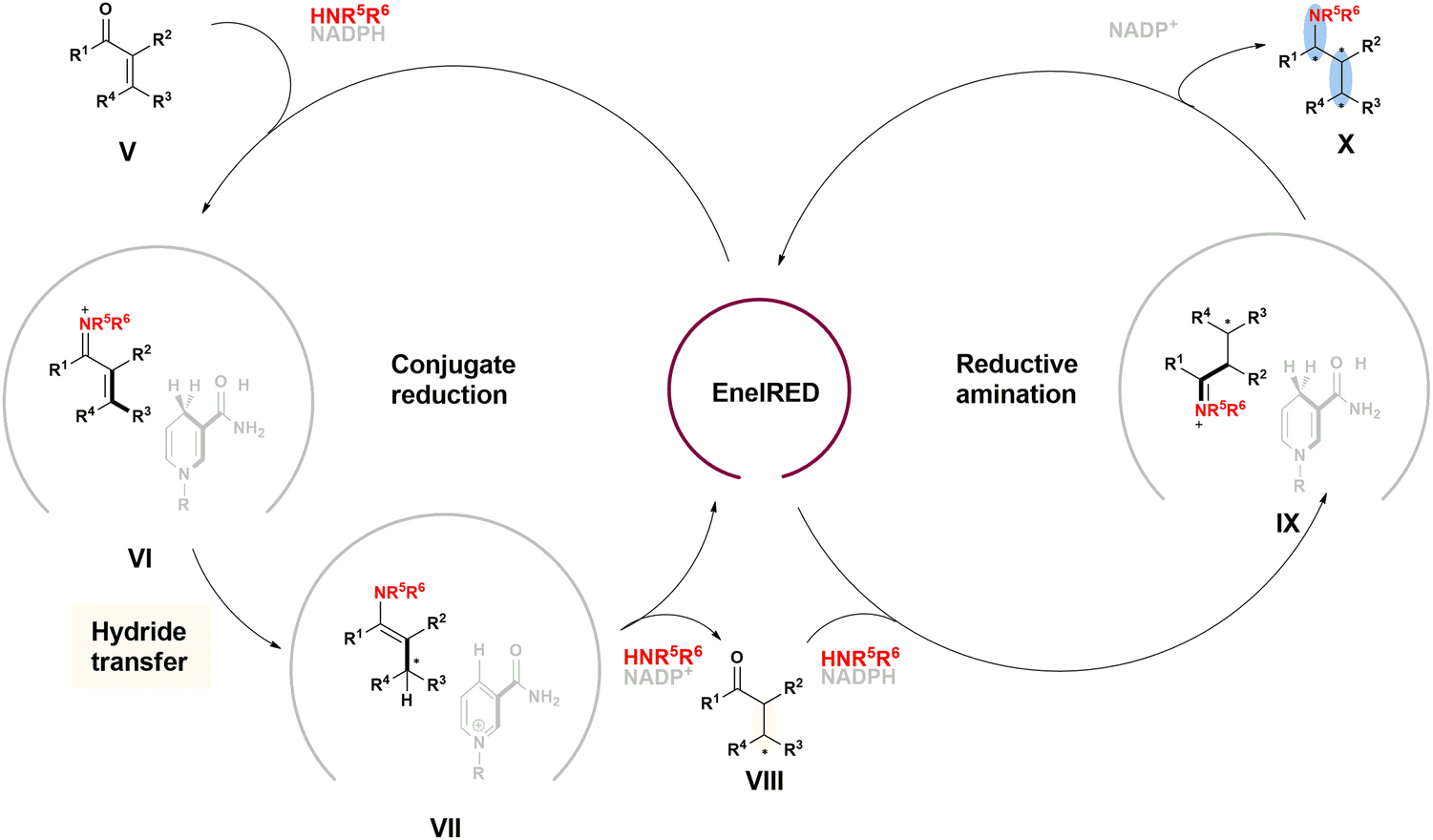 | ||
| Fig. 20 Proposed catalytic cycle of the EneIRED catalyzed carbonyl reduction and reductive amination. | ||
As discussed in previous sections, piperidines are often target products for applications of IREDs. However, most of the approaches employ multi-enzymatic cascades starting from ketoacids,160,161 diamines171 or amino alcohols.166 Alternatively, pyridines could be obtained by mild and naturally occurring pathways and serve as a source through dearomatization by quaternization-activation of the pyridine nitrogen. Very recently, the Turner group reported a chemoenzymatic cascade whereby pyridines were subjected to chemical reduction to produce tetrahydropyridines (THP), followed by biocatalytic amine oxidation by a 6-hydroxy-D-nicotine oxidase (6-HDNO) variant and conjugate reduction of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CN bonds with the multifunctional EneIRED (Fig. 21). The chemo-enzymatic cascade could be applied to the synthesis of both (R)- and (S)-configured products on preparative scale by using EneIREDs with complementary stereoselectivity. For example, the antipsychotic drug Preclamol was obtained in both (R)-(+)- and (S)-(−)-conformation with 96% ee and >50% yields. This system was also applied to the synthesis of other APIs such as OSU6162 and Niraparib in high ee.111
C and CN bonds with the multifunctional EneIRED (Fig. 21). The chemo-enzymatic cascade could be applied to the synthesis of both (R)- and (S)-configured products on preparative scale by using EneIREDs with complementary stereoselectivity. For example, the antipsychotic drug Preclamol was obtained in both (R)-(+)- and (S)-(−)-conformation with 96% ee and >50% yields. This system was also applied to the synthesis of other APIs such as OSU6162 and Niraparib in high ee.111
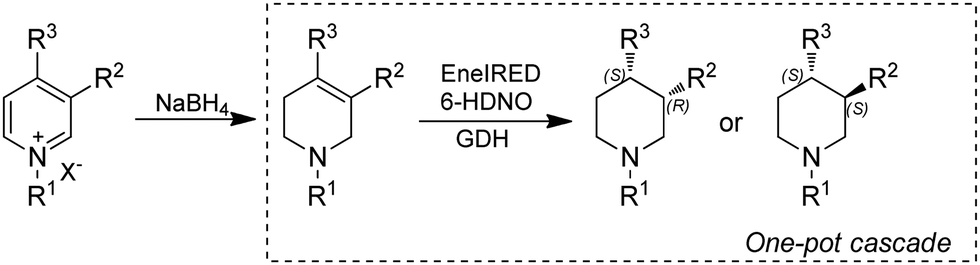 | ||
| Fig. 21 Chemo-enzymatic dearomatization of pyridines for the synthesis of chiral 3- and 3,4-substituted piperidines. | ||
In addition to multifunctional IREDs, short-chain dehydrogenases (SDR), whose native activity is carbonyl reduction, were also reported to catalyze reductive amination reactions. For example, SDRs from Narcissus pseudonarcissus, Zephyranthes treatiae, and Amaryllidacea Zephyranthes treatiae were all reported to reduce imines.172,173 Furthermore, the β-hydroxyacid dehydrogenases from Thermocrinus albus (TaβHADs) were also discovered to reduce cyclic imines to the corresponding piperidine products.107 Structural investigations revealed both common and different features for the βHADs and IREDs.174
000 fold increased compared to wild type enzyme. This work not only demonstrated the potential of IREDs in commercial productions at low amine excess, high substrate loadings and large scales, but also showed the robustness of the enzyme that could withstand a buffer pH at 4.2, and preincubation at 40 °C.
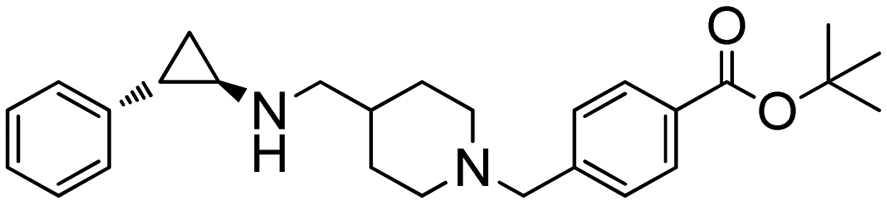
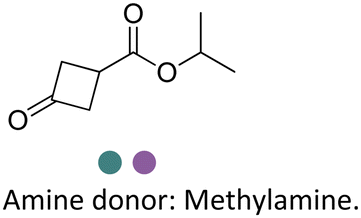
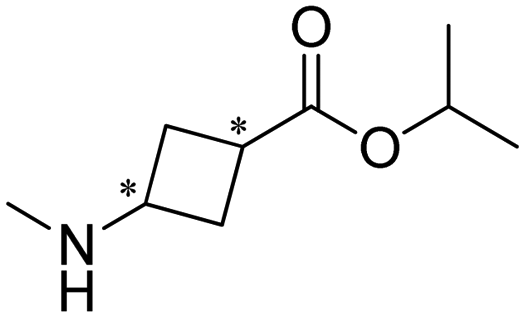
Researchers at Pfizer employed IREDs to perform the reductive amination with methylamine of a key ketone intermediate for the drug abrocitinib to directly obtain the intermediate isopropyl (1S,3S)-3-(methylamino)cyclobutane-1-carboxylate on a 230 kg commercial plant scale production (Table 3, entry 18).151 The drug abrocitinib is a Janus kinase 1 (JAK1) inhibitor and is in development to treat atopic dermatitis. Screening a panel of wild-type IRED enzymes and 3 rounds of further engineering with single site saturation mutagenesis (SSM) and recombination of beneficial mutations yielded a final variant IR021-R3-V6, bearing N131H/A170C/F180M/G217D mutations that locate mostly at the substrate or cofactor binding sites. Pilot plant and plant productions were carried out on 65 kg and 230 kg scales, and the conversions and ee were up to 92.5% and >99.5%. Results in various scales were shown to be perfectly in accordance without the decline that normally would be observed for scale-up reactions.
Another example was also demonstrated by researchers at Pfizer where they showcased a multi-step, chemobiocatalytic synthesis of a CDK 2/4/6 Inhibitor, which was developed as an oncology candidate. A key step to the intermediate is the reductive amination of the ketone 2-hydroxy-2-methylcyclopentanone to the corresponding amine (1R)-2-(benzylamino)-1-methylcyclopentanol.175 A panel of 88 IREDs were screened, and 98 further SSM libraries were constructed and screened. Finally, a variant IR007-143 achieved 43% conversion at a 50 g L−1 substrate concentration and 2.5 wt% enzyme loading.
Process aspects of using IREDs in downstream processing (DSP) have been further investigated to showcase the applicational potentials.176 Employing IREDs in continuous flow process would enable the combinations of incompatible processes in one streamline. For example, the Turner group used a multi-point injection reactor for inline mixing and packed bed systems for various enzymes including transaminases, galactose oxidases, and IREDs to perform the cascade reactions in a single continuous flow system.177
Initially, the cofactors for IREDs had been strictly limited to NADPH, which limited the possibilities of using a variety of cofactor recycling systems, since the availability of NADPH regenerating systems is more rare and the cost of NADH is lower.178 Borlinghaus et al. reported a CSR-SALAD (namely “cofactor specificity reversal-structural analysis and library design”) strategy to semi-rationally construct mutant libraries with the guidance of crystal structures with the NADPH binding pocket and coordinating residues to the phosphate moiety at the NADPH molecule. The final variant V10 gave a TON towards NADH of 38 min−1, comparing to a lower NADPH TON of 0.6 min−1, demonstrating that the cofactor preference has been successfully switched. Other research regarding the recycling of NADPH has also been reported, including H2-driven cofactor recycling.179 Vidal et al. reported an approach using CSR-SALAD to predict residues crucial for cofactor preference, and then artificial selection platform was built to identify highly active variants with switched cofactor preference.180 They manipulated the E. coli genes to restrict the growth under anaerobic conditions, therefore, the restoration of growth can be linked to the presence of a variant that is active for the un-nature substrate. MsIRED was used as one of the model enzymes, and a higher activity was obtained with NAD+ preference instead of NADP+ with just a single round of mutagenesis.
IREDs are mostly NADPH dependent enzymes, and traditional cofactor recycling systems using glucose and GDH have been coupled with IREDs to enable efficient biocatalytic reactions. However, glucose as a co-substrate affords gluconic acid as the product with very poor atom economy. Alternatively, cyanobacteria have been shown to utilize water splitting for electron supply for reductive reaction, offering good atom economy and ease for product extraction. Büchsenschütz et al. integrated the genes encoding IREDs into the cyanobacterium Synechocystis sp. PCC 6803 under the control of the light-inducible promotor PpsbA2.181 Initially low conversions were obtained (<36%), however, upon promotor optimization, the substrate 6-methyl-2,3,4,5-tetrahydropyridine can be reduced in 83% conversion to form the corresponding amine, and up to 99% ee were obtained for the substrates.
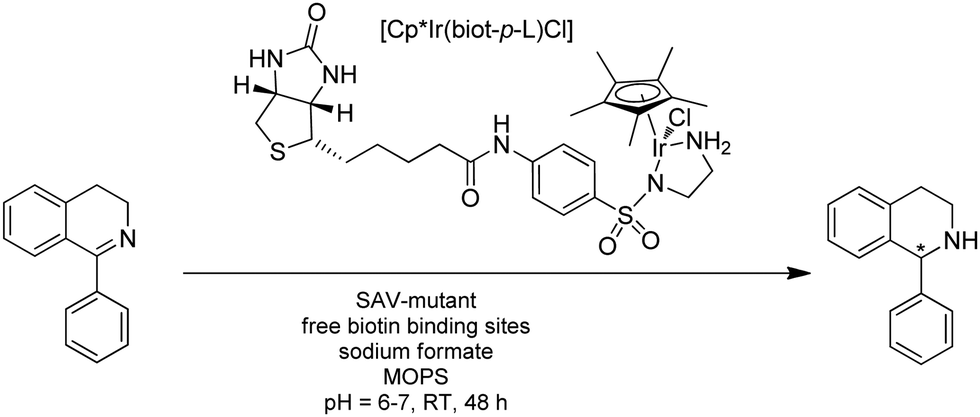 | ||
| Fig. 22 Reduction of a cyclic imine using ATHase based on biotin–streptavidin technology. | ||
4. Future perspectives
In this review, we have focused on NAD(P)H-dependent enzymes, namely AmDHs and IREDs (or RedAms) that are capable of catalyzing the reductive amination of ketones utilizing both inorganic and organic amines to produce primary, secondary and tertiary chiral amines. The phylogenetic tree has been constructed against a variety of AmDHs, IREDs and RedAms (Fig. S1, ESI†), which shows that the sequence homology between IREDs and RedAms are more prominent than that with AmDHs. The importance of enzymatic reductive amination has been clearly established for the synthetic applications of chiral amines. In terms of the discovery of novel enzymes, developing advanced enzyme toolbox, novel design of synthetic routes and large-scale industrial applications, both AmDHs and IREDs are among the most robust enzymes which have attracted considerable attention in recent years. Going forward, there remain considerable opportunities and challenges for the industrialization of the enzymes. Some priorities for the future development of AmDHs and IREDs are: (1) synthetic biology enables the manufacturing of high-value products from sustainable feedstock, and applications of engineered enzymes in construction of non-nature pathways could be valuable for industrialization of the enzymes. (2) Artificial intelligence (AI) is drawing attention across the globe, including within the biocatalysis community.185 There are already reports of using machine learning methods in the directed evolution of IREDs,143 and Alphafold2 has now been used routinely to predict protein structures. In the future, more in-depth applications of AI-assisted protein engineering could be expected. (3) High throughput screening has been a bottleneck for engineering of many enzymes. Currently the HTS method reported for IREDs133 utilizes the reverse reaction of IREDs (oxidation of amines) which limits its applications. (4) Continuous flow systems enable the reuse and recycling of biocatalysts and dramatically reduce the cost of the enzymes in industrial application, thereby implying the vital roles for enzyme immobilization techniques. Especially for enzymes require co-factors and regeneration systems, co-immobilization of enzymes require further in-depth investigations. (5) For AmDHs, the substrate scope of ketone substrates and amine nucleophiles are still relatively narrow, with only a few reported examples showing that AmDHs can accept secondary amine donors. Future engineering work could focus on the development of AmDHs for the synthesis of more structurally diverse products.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Research and Development Program of China (No. 2019YFA0905100 and 2021YFA0910400), the National Natural Science Foundation of China (No. 32171462 and 32171268), Tianjin Synthetic Biotechnology Innovation Capacity Improvement Project (No. TSBICIP-CXRC-040), the Natural Science Foundation of Tianjin (No. 21JCJQJC00110).References
- D. Ghislieri and N. J. Turner, Top. Catal., 2014, 57, 284–300 CrossRef CAS.
- C. E. Paul, M. Rodríguez-Mata, E. Busto, I. Lavandera, V. Gotor-Fernández, V. Gotor, S. García-Cerrada, J. Mendiola, Ó. de Frutos and I. Collado, Org. Process Res. Dev., 2014, 18, 788–792 CrossRef CAS.
- D. Wu, P. Cai, X. Zhao and Y. Pan, Org. Lett., 2017, 19, 5018–5021 CrossRef CAS PubMed.
- M. Höhne and U. T. Bornscheuer, ChemCatChem, 2009, 1, 42–51 CrossRef.
- O. I. Afanasyev, E. Kuchuk, D. L. Usanov and D. Chusov, Chem. Rev., 2019, 119, 11857–11911 CrossRef CAS PubMed.
- N. A. Strotman, C. A. Baxter, K. M. J. Brands, E. Cleator, S. W. Krska, R. A. Reamer, D. J. Wallace and T. J. Wright, J. Am. Chem. Soc., 2011, 133, 8362–8371 CrossRef CAS PubMed.
- D. Q. Pham, A. Nogid and R. Plakogiannis, Am. J. Health-Syst. Pharm., 2008, 65, 521–531 CrossRef CAS PubMed.
- B. L. S. H. Berman and C. Salzman, Ment. Hosp., 1992, 43, 671–672 Search PubMed.
- W. J. Burke, Expert Opin. Invest. Drugs, 2002, 11, 1477–1486 CrossRef CAS PubMed.
- M. S. A. G. M. Keating, Drugs, 2010, 70, 761–791 CrossRef PubMed.
- G. G. Abhilash Desai, Expert Opin. Pharmacother., 2001, 2, 653–666 CrossRef PubMed.
- Q. Yin, Y. Shi, J. Wang and X. Zhang, Chem. Soc. Rev., 2020, 49, 6141 RSC.
- Y. Tian, L. a Hu, Y.-Z. Wang, X. Zhang and Q. Yin, Org. Chem. Front., 2021, 8, 2328–2342 RSC.
- T. Irrgang and R. Kempe, Chem. Rev., 2020, 120, 9583–9674 CrossRef CAS PubMed.
- X. Tan, S. Gao, W. Zeng, S. Xin, Q. Yin and X. Zhang, J. Am. Chem. Soc., 2018, 140, 2024–2027 CrossRef CAS PubMed.
- S. Simic, E. Zukic, L. Schmermund, K. Faber, C. K. Winkler and W. Kroutil, Chem. Rev., 2022, 122, 1052–1126 CrossRef CAS PubMed.
- H. Kohls, F. Steffen-Munsberg and M. Hohne, Curr. Opin. Chem. Biol., 2014, 19, 180–192 CrossRef CAS PubMed.
- V. F. Batista, J. L. Galman, D. C. G. A. Pinto, A. M. S. Silva and N. J. Turner, ACS Catal., 2018, 8, 11889–11907 CrossRef CAS.
- D. Koszelewski, K. Tauber, K. Faber and W. Kroutil, Trends Biotechnol., 2010, 28, 324–332 CrossRef CAS PubMed.
- F. Parmeggiani, N. J. Weise, S. T. Ahmed and N. J. Turner, Chem. Rev., 2018, 118, 73–118 CrossRef CAS PubMed.
- J. Liu, W. Kong, J. Bai, Y. Li, L. Dong, L. Zhou, Y. Liu, J. Gao, R. T. Bradshaw Allen, N. J. Turner and Y. Jiang, Chem. Catal., 2022, 2, 1–27 CrossRef.
- M. Sharma, J. Mangas-Sanchez, N. J. Turner and G. Grogan, Adv. Synth. Catal., 2017, 359, 2011–2025 CrossRef CAS PubMed.
- M. D. Patil, G. Grogan, A. Bommarius and H. Yun, ACS Catal., 2018, 8, 10985–11015 CrossRef CAS.
- J. J. Sangster, J. R. Marshall, N. J. Turner and J. Mangas-Sanchez, ChemBioChem, 2022, 23, e202100464 CrossRef CAS PubMed.
- G. Grogan, Curr. Opin. Chem. Biol., 2018, 43, 15–22 CrossRef CAS PubMed.
- J. Mangas-Sanchez, S. P. France, S. L. Montgomery, G. A. Aleku, H. Man, M. Sharma, J. I. Ramsden, G. Grogan and N. J. Turner, Curr. Opin. Chem. Biol., 2017, 37, 19–25 CrossRef CAS PubMed.
- G. A. Aleku, S. P. France, H. Man, J. Mangas-Sanchez, S. L. Montgomery, M. Sharma, F. Leipold, S. Hussain, G. Grogan and N. J. Turner, Nat. Chem., 2017, 9, 961–969 CrossRef CAS PubMed.
- P. Matzel, S. Wenske, S. Merdivan, S. Guenther and M. Hoehne, ChemCatChem, 2019, 11, 4281–4285 CrossRef CAS.
- O. Mayol, K. Bastard, L. Beloti, A. Frese, J. P. Turkenburg, J.-L. Petit, A. Mariage, A. Debard, V. Pellouin, A. Perret, V. de Berardinis, A. Zaparucha, G. Grogan and C. Vergne-Vaxelaire, Nat. Catal., 2019, 2, 324–333 CrossRef CAS.
- M. Sharma, J. Mangas-Sanchez, S. P. France, G. A. Aleku, S. L. Montgomery, J. I. Ramsden, N. J. Turner and G. Grogan, ACS Catal., 2018, 8, 11534–11541 CrossRef CAS.
- V. Tseliou, M. F. Masman, W. Bohmer, T. Knaus and F. G. Mutti, ChemBioChem, 2019, 20, 800–812 CrossRef CAS PubMed.
- L. Liu, D.-H. Wang, F.-F. Chen, Z.-J. Zhang, Q. Chen, J.-H. Xu, Z.-L. Wang and G.-W. Zheng, Catal. Sci. Technol., 2020, 10, 2353–2358 RSC.
- J. Mangas-Sanchez, M. Sharma, S. C. Cosgrove, J. I. Ramsden, J. R. Marshall, T. W. Thorpe, R. B. Palmer, G. Grogan and N. J. Turner, Chem. Sci., 2020, 11, 5052–5057 RSC.
- M. J. Abrahamson, E. Vázquez-Figueroa, N. B. Woodall, J. C. Moore and A. S. Bommarius, Angew. Chem., Int. Ed., 2012, 51, 3969 CrossRef CAS PubMed.
- P. J. Baker, A. P. Turnbull, S. E. Sedelnikova, T. J. Stillman and D. W. Rice, Structure, 1995, 3, 693–705 CrossRef CAS PubMed.
- Y. Zhao, T. Wakamatsu, K. Doi, H. Sakuraba and T. Ohshima, J. Mol. Catal. B: Enzym., 2012, 83, 65–72 CrossRef CAS.
- H. Yamaguchi, A. Kamegawa, K. Nakata, T. Kashiwagi, T. Mizukoshi, Y. Fujiyoshi and K. Tani, J. Struct. Biol., 2019, 205, 11–21 CrossRef CAS PubMed.
- T. Wu, Y. Wang, N. Zhang, D. Yin, Y. Xu, Y. Nie and X. Mu, ACS Catal., 2023, 13, 158–168 CrossRef CAS.
- J. L. Vanhooke, J. B. Thoden, N. M. W. Brunhuber, J. S. Blanchard and H. M. Holden, Biochemistry, 1999, 38, 2326–2339 CrossRef CAS PubMed.
- N. M. W. Brunhuber, J. B. Thoden, J. S. Blanchard and J. L. Vanhooke, Biochemistry, 2000, 39, 9174–9187 CrossRef CAS PubMed.
- L. Ducrot, M. Bennett, G. André-Leroux, E. Elisée, S. Marynberg, A. Fossey-Jouenne, A. Zaparucha, G. Grogan and C. Vergne-Vaxelaire, ChemCatChem, 2022, 14, e202200880 CrossRef CAS.
- M. Bennett, L. Ducrot, C. Vergne-Vaxelaire and G. Grogan, ChemBioChem, 2022, 23, e202200136 CrossRef CAS PubMed.
- R. D. Franklin, J. A. Whitley, J. M. Robbins and A. S. Bommarius, Chem. Eng. J., 2019, 369, 634–640 CrossRef CAS.
- M. J. Abrahamson, J. W. Wong and A. S. Bommarius, Adv. Synth. Catal., 2013, 355, 1780–1786 CrossRef CAS.
- M. T. Reetz, Angew. Chem., Int. Ed., 2011, 50, 138–174 CrossRef CAS PubMed.
- M. T. Reetz and J. D. Carballeira, Nat. Protoc., 2007, 2, 891–903 CrossRef CAS PubMed.
- G. Qu, A. Li, C. G. Acevedo-Rocha, Z. Sun and M. T. Reetz, Angew. Chem., Int. Ed., 2020, 59, 13204–13231 CrossRef CAS PubMed.
- W. Jiang and Y. Wang, J. Microbiol. Biotechnol., 2020, 30, 146–154 CrossRef CAS PubMed.
- L. J. Ye, H. H. Toh, Y. Yang, J. P. Adams, R. Snajdrova and Z. Li, ACS Catal., 2015, 5, 1119–1122 CrossRef CAS.
- A. Pushpanath, E. Siirola, A. Bornadel, D. Woodlock and U. Schell, ACS Catal., 2017, 7, 3204–3209 CrossRef CAS.
- V. Tseliou, T. Knaus, M. F. Masman, M. L. Corrado and F. G. Mutti, Nat. Commun., 2019, 10, 3717 CrossRef PubMed.
- V. Tseliou, T. Knaus, J. Vilim, M. F. Masman and F. G. Mutti, ChemCatChem, 2020, 12, 2184–2188 CrossRef CAS PubMed.
- V. Tseliou, D. Schilder, M. F. Masman, T. Knaus and F. G. Mutti, Chem. – Eur. J., 2021, 27, 3315–3325 CrossRef CAS PubMed.
- N. Itoh, C. Yachi and T. Kudome, J. Mol. Catal. B: Enzym., 2000, 10, 281–290 CrossRef CAS.
- O. Mayol, S. David, E. Darii, A. Debard, A. Mariage, V. Pellouin, J.-L. Petit, M. Salanoubat, V. de Berardinis, A. Zaparucha and C. Vergne-Vaxelaire, Catal. Sci. Technol., 2016, 6, 7421–7428 RSC.
- K. Bastard, A. A. T. Smith, C. Vergne-Vaxelaire, A. Perret, A. Zaparucha, R. De Melo-Minardi, A. Mariage, M. Boutard, A. Debard, C. Lechaplais, C. Pelle, V. Pellouin, N. Perchat, J.-L. Petit, A. Kreimeyer, C. Medigue, J. Weissenbach, F. Artiguenave, V. De Berardinis, D. Vallenet and M. Salanoubat, Nat. Chem. Biol., 2014, 10, 42–49 CrossRef CAS PubMed.
- A. A. Caparco, E. Pelletier, J. L. Petit, A. Jouenne, B. R. Bommarius, V. de Berardinis, A. Zaparucha, J. A. Champion, A. S. Bommarius and C. Vergne-Vaxelaire, Adv. Synth. Catal., 2020, 362, 2427–2436 CrossRef CAS.
- B. R. Bommarius, M. Schurmann and A. S. Bommarius, Chem. Commun., 2014, 50, 14953–14955 RSC.
- S. K. Au, B. R. Bommarius and A. S. Bommarius, ACS Catal., 2014, 4, 4021–4026 CrossRef CAS.
- J. Loewe, A. A. Ingram and H. Groeger, Bioorg. Med. Chem., 2018, 26, 1387–1392 CrossRef CAS PubMed.
- J. Li, X. Mu, T. Wu and Y. Xu, Bioresour. Bioprocess., 2022, 9, 33 CrossRef.
- F. G. Mutti, T. Knaus, N. S. Scrutton, M. Breuer and N. J. Turner, Science, 2015, 349, 1525–1529 CrossRef CAS PubMed.
- F.-F. Chen, Y.-Y. Liu, G.-W. Zheng and J.-H. Xu, ChemCatChem, 2015, 7, 3838–3841 CrossRef CAS.
- J. H. Sattler, M. Fuchs, K. Tauber, F. G. Mutti, K. Faber, J. Pfeffer, T. Haas and W. Kroutil, Angew. Chem., Int. Ed., 2012, 51, 9156–9159 CrossRef CAS PubMed.
- S. L. Montgomery, J. Mangas-Sanchez, M. P. Thompson, G. A. Aleku, B. Dominguez and N. J. Turner, Angew. Chem., Int. Ed., 2017, 56, 10491–10494 CrossRef CAS PubMed.
- T. Knaus, F. G. Mutti, L. D. Humphreys, N. J. Turner and N. S. Scrutton, Org. Biomol. Chem., 2015, 13, 223 RSC.
- E. P. J. Jongkind, A. Fossey-Jouenne, O. Mayol, A. Zaparucha, C. Vergne-Vaxelaire and C. E. Paul, ChemCatChem, 2021, 14, e202101576 CrossRef.
- F.-F. Chen, G.-W. Zheng, L. Liu, H. Li, Q. Chen, F.-L. Li, C.-X. Li and J.-H. Xu, ACS Catal., 2018, 8, 2622–2628 CrossRef CAS.
- F.-F. Chen, S. C. Cosgrove, W. R. Birmingham, J. Mangas-Sanchez, J. Citoler, M. P. Thompson, G.-W. Zheng, J.-H. Xu and N. J. Turner, ACS Catal., 2019, 9, 11813–11818 CrossRef CAS.
- X. Mu, T. Wu, Y. Mao, Y. Zhao, Y. Xu and Y. Nie, ChemCatChem, 2021, 13, 5243–5253 CrossRef CAS.
- R. D. Franklin, C. J. Mount, B. R. Bommarius and A. S. Bommarius, ChemCatChem, 2020, 12, 2436–2439 CrossRef CAS.
- D.-H. Wang, Q. Chen, S.-N. Yin, X.-W. Ding, Y.-C. Zheng, Z. Zhang, Y.-H. Zhang, F.-F. Chen, J.-H. Xu and G.-W. Zheng, ACS Catal., 2021, 11, 14274–14283 CrossRef CAS.
- T. Knaus, W. Bohmer and F. G. Mutti, Green Chem., 2017, 19, 453–463 RSC.
- W. Kong, Y. Liu, C. Huang, L. Zhou, J. Gao, N. J. Turner and Y. Jiang, Angew. Chem., Int. Ed., 2022, 61, e202202264 CrossRef CAS PubMed.
- R.-F. Cai, L. Liu, F.-F. Chen, A. Li, J.-H. Xu and G.-W. Zheng, ACS Sustainable Chem. Eng., 2020, 8, 17054–17061 CrossRef CAS.
- T. Lv, J. Feng, X. Chen, Y. Luo, Q. Wu, D. Zhu and Y. Ma, ACS Catal., 2023, 13, 5053–5061 CrossRef CAS.
- H. Wang, G. Qu, J.-K. Li, J.-A. Ma, J. Guo, Y. Miao and Z. Sun, Catal. Sci. Technol., 2020, 10, 5945–5952 RSC.
- F. Tong, Z. Qin, H. Wang, Y. Jiang, J. Li, H. Ming, G. Qu, Y. Xiao and Z. Sun, Front. Bioeng. Biotechnol., 2021, 9, 778584 CrossRef PubMed.
- Z. Mei, K. Zhang, G. Qu, J.-K. Li, B. Liu, J.-A. Ma, R. Tu and Z. Sun, ACS Omega, 2020, 5, 13588–13594 CrossRef CAS PubMed.
- H. Ming, B. Yuan, G. Qu and Z. Sun, Catal. Sci. Technol., 2022, 12, 5952–5960 RSC.
- M. P. Thompson, I. Penafiel, S. C. Cosgrove and N. J. Turner, Org. Process Res. Dev., 2019, 23, 9–18 CrossRef CAS.
- J. Coloma, Y. Guiavarc’h, P.-L. Hagedoorn and U. Hanefeld, Chem. Commun., 2021, 57, 11416–11428 RSC.
- J. Liu, B. Q. W. Pang, J. P. Adams, R. Snajdrova and Z. Li, ChemCatChem, 2017, 9, 425–431 CrossRef CAS.
- H. Ren, Y. Zhang, J. Su, P. Lin, B. Wang, B. Fang and S. Wang, J. Biotechnol., 2017, 241, 33–41 CrossRef CAS PubMed.
- A. A. Caparco, A. S. Bommarius and J. A. Champion, AIChE J., 2018, 64, 2934–2946 CrossRef CAS.
- A. A. Caparco, B. R. Bommarius, A. S. Bommarius and J. A. Champion, Biotechnol. Bioeng., 2020, 117, 1979–1989 CrossRef CAS PubMed.
- R. D. Franklin, J. A. Whitley, A. A. Caparco, B. R. Bommarius, J. A. Champion and A. S. Bommarius, Chem. Eng. J., 2021, 407, 127065 CrossRef CAS.
- H. L. Yu, T. Li, F. F. Chen, X. J. Luo, A. T. Li, C. Yang, G.-W. Zheng and J. H. Xu, Metab. Eng., 2018, 47, 184–189 CrossRef CAS PubMed.
- J. A. Houwman, T. Knaus, M. Costa and F. G. Mutti, Green Chem., 2019, 21, 3846–3857 RSC.
- J. Liu and Z. Li, Biotechnol. Bioeng., 2019, 116, 536–542 CrossRef CAS PubMed.
- S. C. Cosgrove, M. P. Thompson, S. T. Ahmed, F. Parmeggiani and N. J. Turner, Angew. Chem., Int. Ed., 2020, 59, 18156–18160 CrossRef CAS PubMed.
- F.-F. Chen, Y.-H. Zhang, Z.-J. Zhang, L. Liu, J.-P. Wu, J.-H. Xu and G.-W. Zheng, J. Org. Chem., 2019, 84, 14987–14993 CrossRef CAS PubMed.
- K. Mitsukura, M. Suzuki, K. Tada, T. Yoshida and T. Nagasawa, Org. Biomol. Chem., 2010, 8, 4533–4535 RSC.
- A. K. Gilio, T. W. Thorpe, N. Turner and G. Grogan, Chem. Sci., 2022, 13, 4697–4713 RSC.
- M. Lenz, N. Borlinghaus, L. Weinmann and B. M. Nestl, World J. Microbiol. Biotechnol., 2017, 33, 199 CrossRef PubMed.
- J. H. Schrittwieser, S. Velikogne and W. Kroutil, Adv. Synth. Catal., 2015, 357, 1655–1685 CrossRef CAS.
- S. C. Cosgrove, A. Brzezniak, S. P. France, J. I. Ramsden, J. Mangas-Sanchez, S. L. Montgomery, R. S. Heath and N. J. Turner, Methods Enzymol., ed. N. Scrutton, 2018, 608, 131–149 Search PubMed.
- G. Grogan and N. J. Turner, Chem. – Eur. J., 2016, 22, 1900–1907 CrossRef CAS PubMed.
- L. Ducrot, M. Bennett, G. Grogan and C. Vergne-Vaxelaire, Adv. Synth. Catal., 2020, 363, 328–351 CrossRef.
- K. Wu, J. Huang and L. Shao, ChemCatChem, 2022, 14, e202200921 CrossRef CAS.
- M. Rodriguez-Mata, A. Frank, E. Wells, F. Leipold, N. J. Turner, S. Hart, J. P. Turkenburg and G. Grogan, ChemBioChem, 2013, 14, 1372–1379 CrossRef CAS PubMed.
- G. A. Aleku, H. Man, S. P. France, F. Leipold, S. Hussain, L. Toca-Gonzalez, R. Marchington, S. Hart, J. P. Turkenburg, G. Grogan and N. J. Turner, ACS Catal., 2016, 6, 3880–3889 CrossRef CAS.
- T. Huber, L. Schneider, A. Präg, S. Gerhardt, O. Einsle and M. Müller, ChemCatChem, 2014, 6, 2248–2252 CrossRef CAS.
- H. Man, E. Wells, S. Hussain, F. Leipold, S. Hart, J. P. Turkenburg, N. J. Turner and G. Grogan, ChemBioChem, 2015, 16, 1052–1059 CrossRef CAS PubMed.
- T. Meyer, N. Zumbraegel, C. Geerds, H. Groeger and H. H. Niemann, Biomolecules, 2020, 10(8), 1130 CrossRef CAS PubMed.
- S. P. France, G. A. Aleku, M. Sharma, J. Mangas-Sanchez, R. M. Howard, J. Steflik, R. Kumar, R. W. Adams, I. Slabu, R. Crook, G. Grogan, T. W. Wallace and N. J. Turner, Angew. Chem., Int. Ed., 2017, 56, 15589–15593 CrossRef CAS PubMed.
- M. Lenz, S. Fademrecht, M. Sharma, J. Pleiss, G. Grogan and B. M. Nestl, Protein Eng., Des. Sel., 2018, 31, 109–120 CrossRef CAS PubMed.
- P. Stockinger, N. Borlinghaus, M. Sharma, B. Aberle, G. Grogan, J. Pleiss and B. M. Nestl, ChemCatChem, 2021, 13, 5210–5215 CrossRef CAS PubMed.
- J. Zhang, D. Liao, R. Chen, F. Zhu, Y. Ma, L. Gao, G. Qu, C. Cui, Z. Sun, X. Lei and S. S. Gao, Angew. Chem., Int. Ed., 2022, 61, e202201908 CrossRef CAS PubMed.
- T. W. Thorpe, J. R. Marshall, V. Harawa, R. E. Ruscoe, A. Cuetos, J. D. Finnigan, A. Angelastro, R. S. Heath, F. Parmeggiani, S. J. Charnock, R. M. Howard, R. Kumar, D. S. B. Daniels, G. Grogan and N. J. Turner, Nature, 2022, 604, 86–91 CrossRef CAS PubMed.
- V. Harawa, T. W. Thorpe, J. R. Marshall, J. J. Sangster, A. K. Gilio, L. Pirvu, R. S. Heath, A. Angelastro, J. D. Finnigan, S. J. Charnock, J. W. Nafie, G. Grogan, R. C. Whitehead and N. J. Turner, J. Am. Chem. Soc., 2022, 144, 21088–21095 CrossRef CAS PubMed.
- A. K. Gilio, T. W. Thorpe, A. Heyam, M. R. Petchey, B. Pogranyi, S. P. France, R. M. Howard, M. J. Karmilowicz, R. Lewis, N. Turner and G. Grogan, ACS Catal., 2023, 13, 1669–1677 CrossRef CAS PubMed.
- T. Nagasawa, T. Yoshida, K. Ishida, H. Yamamoto and N. Kimoto, US Pat., WO2010024445A1, 2009 Search PubMed.
- K. Mitsukura, T. Kuramoto, T. Yoshida, N. Kimoto, H. Yamamoto and T. Nagasawa, Appl. Microbiol. Biotechnol., 2013, 97, 8079–8086 CrossRef CAS PubMed.
- K. Mitsukura, M. Suzuki, S. Shinoda, T. Kuramoto, T. Yoshida and T. Nagasawa, Biosci., Biotechnol., Biochem., 2011, 75, 1778–1782 CrossRef CAS PubMed.
- F. Leipold, S. Hussain, D. Ghislieri and N. J. Turner, ChemCatChem, 2013, 5, 3505–3508 CrossRef CAS.
- P. N. Scheller, S. Fademrecht, S. Hofelzer, J. Pleiss, F. Leipold, N. J. Turner, B. M. Nestl and B. Hauer, ChemBioChem, 2014, 15, 2201–2204 CrossRef CAS PubMed.
- S. Hussain, F. Leipold, H. Man, E. Wells, S. P. France, K. R. Mulholland, G. Grogan and N. J. Turner, ChemCatChem, 2015, 7, 579–583 CrossRef CAS PubMed.
- D. Wetzl, M. Berrera, N. Sandon, D. Fishlock, M. Ebeling, M. Muller, S. Hanlon, B. Wirz and H. Iding, ChemBioChem, 2015, 16, 1749–1756 CrossRef CAS PubMed.
- D. Wetzl, M. Gand, A. Ross, H. Müller, P. Matzel, S. P. Hanlon, M. Müller, B. Wirz, M. Höhne and H. Iding, ChemCatChem, 2016, 8, 2023–2026 CrossRef CAS.
- M. Prejano, X. Sheng and F. Himo, ChemistryOpen, 2022, 11, e202100250 CrossRef CAS PubMed.
- N. Zumbraegel, C. Merten, S. M. Huber and H. Groeger, Nat. Commun., 2018, 9, 1949 CrossRef.
- N. Zumbraegel and H. Groeger, J. Biotechnol., 2019, 291, 35–40 CrossRef CAS PubMed.
- N. Zumbraegel, K. Wagner, N. Weissing and H. Groeger, J. Heterocycl. Chem., 2019, 56, 788–794 CrossRef CAS.
- N. Zumbraegel, P. Machui, J. Nonnhoff and H. Groeger, J. Org. Chem., 2019, 84, 1440–1447 CrossRef CAS PubMed.
- P. N. Scheller, M. Lenz, S. C. Hammer, B. Hauer and B. M. Nestl, ChemCatChem, 2015, 7, 3239–3242 CrossRef CAS.
- D. González-Martínez, A. Cuetos, M. Sharma, M. García-Ramos, I. Lavandera, V. Gotor-Fernández and G. Grogan, ChemCatChem, 2020, 12, 2421–2425 CrossRef.
- P. Matzel, M. Gand and M. Höhne, Green Chem., 2017, 19, 385–389 RSC.
- G.-D. Roiban, M. Kern, Z. Liu, J. Hyslop, P. L. Tey, M. S. Levine, L. S. Jordan, K. K. Brown, T. Hadi, L. A. F. Ihnken and M. J. B. Brown, ChemCatChem, 2017, 9, 4475–4479 CrossRef CAS.
- N. Borlinghaus, S. Gergel and B. M. Nestl, ACS Catal., 2018, 8, 3727–3732 CrossRef CAS.
- S. P. France, R. M. Howard, J. Steflik, N. J. Weise, J. Mangas-Sanchez, S. L. Montgomery, R. Crook, R. Kumar and N. J. Turner, ChemCatChem, 2018, 10, 510–514 CrossRef CAS.
- S. L. Montgomery, A. Pushpanath, R. S. Heath, J. R. Marshall, U. Klemstein, J. L. Galman, D. Woodlock, S. Bisagni, C. J. Taylor, J. Mangas-Sanchez, J. I. Ramsden, B. Dominguez and N. J. Turner, Sci. Adv., 2020, 6, eaay9320 CrossRef CAS PubMed.
- J. R. Marshall, P. Yao, S. L. Montgomery, J. D. Finnigan, T. W. Thorpe, R. B. Palmer, J. Mangas-Sanchez, R. A. M. Duncan, R. S. Heath, K. M. Graham, D. J. Cook, S. J. Charnock and N. J. Turner, Nat. Chem., 2021, 13, 140–148 CrossRef CAS PubMed.
- J. Citoler, V. Harawa, J. R. Marshall, H. Bevinakatti, J. D. Finnigan, S. J. Charnock and N. J. Turner, Angew. Chem., Int. Ed., 2021, 60, 24456–24460 CrossRef CAS PubMed.
- Y. Li, N. Hu, Z. Xu, Y. Cui, J. Feng, P. Yao, Q. Wu, D. Zhu and Y. Ma, Angew. Chem., Int. Ed., 2022, 61, e202116344 CrossRef CAS PubMed.
- Z. Xu, P. Yao, X. Sheng, J. Li, J. Li, S. Yu, J. Feng, Q. Wu and D. Zhu, ACS Catal., 2020, 10, 8780–8787 CrossRef CAS.
- Y.-H. Zhang, F.-F. Chen, B.-B. Li, X.-Y. Zhou, Q. Chen, J.-H. Xu and G.-W. Zheng, Org. Lett., 2020, 22, 3367–3372 CrossRef CAS PubMed.
- Q. Chen, B.-B. Li, L. Zhang, X.-R. Chen, X.-X. Zhu, F.-F. Chen, M. Shi, C.-C. Chen, Y. Yang, R.-T. Guo, W. Liu, J.-H. Xu and G.-W. Zheng, ACS Catal., 2022, 12, 14795–14803 CrossRef CAS.
- J. Zhang, X. Li, R. Chen, X. Tan, X. Liu, Y. Ma, F. Zhu, C. An, G. Wei, Y. Yao, L. Yang, P. Zhang, Q. Wu, Z. Sun, B. G. Wang, S. S. Gao and C. Cui, Commun. Chem., 2022, 5, 123 CrossRef CAS PubMed.
- Z. Zhan, Z. Xu, S. Yu, J. Feng, F. Liu, P. Yao, Q. Wu and D. Zhu, Adv. Synth. Catal., 2022, 364, 2380–2386 CrossRef CAS.
- L. Yang, J. Li, Z. Xu, P. Yao, Q. Wu, D. Zhu and Y. Ma, Org. Lett., 2022, 24, 6531–6536 CrossRef CAS PubMed.
- F. F. Chen, X. F. He, X. X. Zhu, Z. Zhang, X. Y. Shen, Q. Chen, J. H. Xu, N. J. Turner and G.-W. Zheng, J. Am. Chem. Soc., 2023, 145, 4015–4025 CrossRef CAS PubMed.
- E. J. Ma, E. Siirola, C. Moore, A. Kummer, M. Stoeckli, M. Faller, C. Bouquet, F. Eggimann, M. Ligibel, D. Huynh, G. Cutler, L. Siegrist, R. A. Lewis, A.-C. Acker, E. Freund, E. Koch, M. Vogel, H. Schlingensiepen, E. J. Oakeley and R. Snajdrova, ACS Catal., 2021, 11, 12433–12445 CrossRef CAS.
- Y. Yu, A. Rué Casamajo, W. Finnigan, C. Schnepel, R. Barker, C. Morrill, R. S. Heath, L. De Maria, N. J. Turner and N. S. Scrutton, ACS Catal., 2023, 13, 12310–12321 CrossRef CAS PubMed.
- M. Schober, C. MacDermaid, A. A. Ollis, S. Chang, D. Khan, J. Hosford, J. Latham, L. A. F. Ihnken, M. J. B. Brown, D. Fuerst, M. J. Sanganee and G.-D. Roiban, Nat. Catal., 2019, 2, 909–915 CrossRef CAS.
- W. Zawodny, S. L. Montgomery, J. R. Marshall, J. D. Finnigan, N. J. Turner and J. Clayden, J. Am. Chem. Soc., 2018, 140, 17872–17877 CrossRef CAS PubMed.
- P. Yao, Z. Xu, S. Yu, Q. Wu and D. Zhu, Adv. Synth. Catal., 2019, 361, 556–561 CrossRef CAS.
- S. Velikogne, V. Resch, C. Dertnig, J. H. Schrittwieser and W. Kroutil, ChemCatChem, 2018, 10, 3236–3246 CrossRef CAS PubMed.
- N. Alvarenga, S. E. Payer, P. Petermeier, C. Kohlfuerst, A. L. Meleiro Porto, J. H. Schrittwieser and W. Kroutil, ACS Catal., 2020, 10, 1607–1620 CrossRef CAS.
- R. L. Booth, G. Grogan, K. S. Wilson and A.-K. Duhme-Klair, RSC Chem. Biol., 2020, 1, 369–378 RSC.
- R. Kumar, M. J. Karmilowicz, D. Burke, M. Burns, L. A. Clark, C. G. Connor, E. Cordi, N. M. Do, K. M. Doyle, S. Hoagland, C. A. Lewis, D. Mangan, C. A. Martinez, E. L. McInturff, K. Meldrum, R. Pearson, J. Steflik, A. Rane and J. Weaver, Nat. Catal., 2021, 4, 775–782 CrossRef CAS.
- L. Yang, J. Zhu, C. Sun, Z. Deng and X. Qu, Chem. Sci., 2020, 11, 364–371 RSC.
- P. Yao, J. R. Marshall, Z. Xu, J. Lim, S. J. Charnock, D. Zhu and N. J. Turner, Angew. Chem., Int. Ed., 2021, 60, 8717–8721 CrossRef CAS PubMed.
- J. Zhu, L. Yang, J. Wu, Z. Deng and X. Qu, ACS Catal., 2022, 12, 9823–9830 CrossRef CAS.
- J. Nonnhoff, H. G. Stammler and H. Groger, J. Org. Chem., 2022, 87, 11369–11378 CrossRef CAS PubMed.
- M. Li, Y. Cui, Z. Xu, X. Chen, J. Feng, M. Wang, P. Yao, Q. Wu and D. Zhu, Adv. Synth. Catal., 2022, 364, 372–379 CrossRef CAS.
- M. Cárdenas-Fernández, R. Roddan, E. M. Carter, H. C. Hailes and J. M. Ward, ChemCatChem, 2023, 15, e202201126 CrossRef PubMed.
- Y. Li, X. Yue, Z. Li, Z. Huang and F. Chen, Org. Lett., 2023, 25, 1285–1289 CrossRef CAS PubMed.
- J. J. Sangster, R. E. Ruscoe, S. C. Cosgrove, J. Mangas-Sanchez and N. J. Turner, J. Am. Chem. Soc., 2023, 145(8), 4431–4437 CrossRef CAS PubMed.
- L. J. Hepworth, S. P. France, S. Hussain, P. Both, N. J. Turner and S. L. Flitsch, ACS Catal., 2017, 7, 2920–2925 CrossRef CAS.
- S. P. France, S. Hussain, A. M. Hill, L. J. Hepworth, R. M. Howard, K. R. Mulholland, S. L. Flitsch and N. J. Turner, ACS Catal., 2016, 6, 3753–3759 CrossRef CAS.
- M. Tavanti, J. Mangas-Sanchez, S. L. Montgomery, M. P. Thompson and N. J. Turner, Org. Biomol. Chem., 2017, 15, 9790–9793 RSC.
- T. W. Thorpe, S. P. France, S. Hussain, J. R. Marshall, W. Zawodny, J. Mangas-Sanchez, S. L. Montgomery, R. M. Howard, D. S. B. Daniels, R. Kumar, F. Parmeggiani and N. J. Turner, J. Am. Chem. Soc., 2019, 141, 19208–19213 CrossRef CAS PubMed.
- F. Escalettes and N. J. Turner, ChemBioChem, 2008, 9, 857–860 CrossRef CAS PubMed.
- J. B. Rannes, A. Ioannou, S. C. Willies, G. Grogan, C. Behrens, S. L. Flitsch and N. J. Turner, J. Am. Chem. Soc., 2011, 133, 8436–8439 CrossRef CAS PubMed.
- G. J. Ford, N. Kress, A. P. Mattey, L. J. Hepworth, C. R. Baldwin, J. R. Marshall, L. S. Seibt, M. Huang, W. R. Birmingham, N. J. Turner and S. L. Flitsch, Chem. Commun., 2020, 56, 7949–7952 RSC.
- G. A. Aleku, J. Mangas-Sanchez, J. Citoler, S. P. France, S. L. Montgomery, R. S. Heath, M. P. Thompson and N. J. Turner, ChemCatChem, 2018, 10, 515–519 CrossRef CAS.
- A. Al-Shameri, M.-C. Petrich, K. Junge Puring, U.-P. Apfel, B. M. Nestl and L. Lauterbach, Angew. Chem., Int. Ed., 2020, 59, 10929–10933 CrossRef CAS PubMed.
- P. Petermeier, C. Kohlfuerst, A. Torvisco, R. Fischer, A. Mata, D. Dallinger, C. O. Kappe, J. Schrittwieser and W. Kroutil, Adv. Synth. Catal., 2023, 365, 2057–2278 CrossRef.
- M. Lenz, J. Meisner, L. Quertinmont, S. Lutz, J. Kastner and B. M. Nestl, ChemBioChem, 2017, 18, 253–256 CrossRef CAS PubMed.
- N. Borlinghaus, L. Weinmann, F. Krimpzer, P. N. Scheller, A. Al-Shameri, L. Lauterbach, A.-S. Coquel, C. Lattemann, B. Hauer and B. M. Nestl, ChemCatChem, 2019, 11, 5738–5742 CrossRef CAS.
- S. Roth, M. B. Kilgore, T. M. Kutchan and M. Mueller, ChemBioChem, 2018, 19, 1849–1852 CrossRef CAS PubMed.
- S. Roth, P. Stockinger, J. Steff, S. Steimle, V. Sautner, K. Tittmann, J. Pleiss and M. Mueller, ChemBioChem, 2020, 21, 2615–2619 CrossRef CAS PubMed.
- P. Stockinger, S. Roth, M. Mueller and J. Pleiss, ChemBioChem, 2020, 21, 2689–2695 CrossRef CAS PubMed.
- S. Duan, D. W. Widlicka, M. P. Burns, R. Kumar, I. Hotham, J.-N. Desrosiers, P. Bowles, K. N. Jones, L. D. Nicholson, M. T. Buetti-Weekly, L. Han, J. Steflik, E. Hansen, C. M. Hayward, H. Strohmeyer, S. Monfette, S. C. Sutton and C. Morris, Org. Process Res. Dev., 2021, 26, 879–890 CrossRef.
- L.-E. Meyer, H. Brundiek and J. von Langermann, Biotechnol. Prog., 2020, 36, e3024 CrossRef CAS PubMed.
- A. P. Mattey, G. J. Ford, J. Citoler, C. Baldwin, J. R. Marshall, R. B. Palmer, M. Thompson, N. J. Turner, S. C. Cosgrove and S. L. Flitsch, Angew. Chem., Int. Ed., 2021, 60, 18660–18665 CrossRef CAS PubMed.
- N. Borlinghaus and B. M. Nestl, ChemCatChem, 2018, 10, 183–187 CrossRef CAS.
- J. Preissler, H. A. Reeve, T. Zhu, J. Nicholson, K. Urata, L. Lauterbach, L. L. Wong, K. A. Vincent and O. Lenz, ChemCatChem, 2020, 12, 4853–4861 CrossRef CAS.
- L. S. Vidal, J. W. Murray and J. T. Heap, Nat. Commun., 2021, 12, 6859 CrossRef PubMed.
- H. C. Buechsenschuetz, V. Vidimce-Risteski, B. Eggbauer, S. Schmidt, C. K. Winkler, J. H. Schrittwieser, W. Kroutil and R. Kourist, ChemCatChem, 2020, 12, 726–730 CrossRef CAS.
- F. Schwizer, Y. Okamoto, T. Heinisch, Y. Gu, M. M. Pellizzoni, V. Lebrun, R. Reuter, V. Kohler, J. C. Lewis and T. R. Ward, Chem. Rev., 2018, 118, 142–231 CrossRef CAS PubMed.
- M. Hestericova, T. Heinisch, L. Alonso-Cotchico, J.-D. Marechal, P. Vidossich and T. R. Ward, Angew. Chem., Int. Ed., 2018, 57, 1863–1868 CrossRef CAS PubMed.
- M. Hestericova, T. Heinisch, M. Lenz and T. R. Ward, Dalton Trans., 2018, 47, 10837–10841 RSC.
- T. Yu, A. G. Boob, M. J. Volk, X. Liu, H. Cui and H. Zhao, Nat. Catal., 2023, 6, 137–151 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cs00391d |
| This journal is © The Royal Society of Chemistry 2024 |