DOI:
10.1039/D3CS00837A
(Review Article)
Chem. Soc. Rev., 2024,
53, 1624-1692
Organoboron-based multiple-resonance emitters: synthesis, structure–property correlations, and prospects†
Received
30th September 2023
First published on 3rd January 2024
Abstract
Boron-based multiple-resonance (MR) emitters exhibit the advantages of narrowband emission, high absolute photoluminescence quantum yield, thermally activated delayed fluorescence (TADF), and sufficient stability during the operation of organic light-emitting diodes (OLEDs). Thus, such MR emitters have been widely applied as blue emitters in triplet–triplet-annihilation-driven fluorescent devices used in smartphones and televisions. Moreover, they hold great promise as TADF or terminal emitters in TADF-assisted fluorescence or phosphor-sensitised fluorescent OLEDs. Herein we comprehensively review organoboron-based MR emitters based on their synthetic strategies, clarify structure–photophysical property correlations, and provide design guidelines and future development prospects.

Masashi Mamada
| Masashi Mamada obtained his PhD degree in 2010 from Tokyo Institute of Technology under the supervision of Prof. Yoshiro Yamashita. After working as a postdoctoral researcher in Prof. Pavel Anzenbacher's group at Bowling Green State University, USA, until 2011, he joined Innovation Center for Organic Electronics (INOEL) of Yamagata University as an assistant professor. In 2016, he moved to Prof. Chihaya Adachi's group of Center for Organic Photonics and Electronics Research (OPERA) at Kyushu University as an assistant professor to contribute to the Molecular Exciton Engineering Project. He is currently an associate professor in the Department of Chemistry, Graduate School of Science at Kyoto University. His research is focused on synthesis and photophysics of novel organic materials for optoelectronic applications such as OLEDs, OFETs, and organic lasers. |

Masahiro Hayakawa
| Masahiro Hayakawa received his BSc (2017), MSc (2019), and PhD (2022) degrees in Chemistry from Nagoya University under the supervision of Prof. Shigehiro Yamaguchi and Prof. Aiko Fukazawa. In 2022, he joined the group of Prof. Takuji Hatakeyama and started his academic career as an assistant professor in the Department of Chemistry, Graduate School of Science at Kyoto University. His current research interest is the development of unusual π-conjugated compounds. |

Junki Ochi
| Junki Ochi received his BSc (2018), MSc (2020), and PhD (2023) degrees in Chemistry from Kyoto University under the supervision of Prof. Yoshiki Chujo and Prof. Kazuo Tanaka. In 2023, he joined the group of Prof. Takuji Hatakeyama in the Department of Chemistry, Graduate School of Science at Kyoto University, as a Program-Specific Assistant Professor. His current research interest is developing heteroatom-containing luminescent materials. |

Takuji Hatakeyama
| Takuji Hatakeyama received his PhD degree in 2005 from the University of Tokyo under the supervision of Prof. Eiichi Nakamura. After working as a postdoctoral researcher in Prof. Rustem F. Ismagilov's group at Chicago University, he joined the group of Prof. Masaharu Nakamura at Kyoto University as an assistant professor in 2006. In 2013, he initiated his independent research career as an associate professor at Kwansei Gakuin University, and was promoted to a full professor in 2018. Since 2022, he has been a full professor in the Department of Chemistry, Graduate School of Science at Kyoto University. His research interests include the development of carbon–heteroatom bond formation reactions and synthesis of novel boron-based materials for optoelectronic applications. |
1. Introduction
Given their advantageous optical and electrochemical properties, π-conjugated compounds featuring tricoordinated sp2-hybridised boron (Bsp2) atoms with empty pz orbitals have attracted significant attention in the field of materials chemistry.1–21 The inherent vulnerability of Bsp2–C bonds to moisture and oxygen necessitates the adoption of protective measures such as steric shielding22–24 (e.g., Mes2B group (Mes = mesityl)), intramolecular π-electron donation25–34 (e.g., aza- and oxaborines), and planarity constraints35,36 (e.g., boron-doped polycyclic aromatic hydrocarbons (PAHs)) to achieve sufficient stability for use in, e.g., nonlinear optics, optoelectronics, stimuli-responsive systems, bioimaging, sensing, and catalysis. Although the observed functionalities mostly originate from the π-electron-accepting ability or Lewis acidity of Bsp2, the resonance effect of such centres has not been intentionally exploited in material design (Fig. 1).
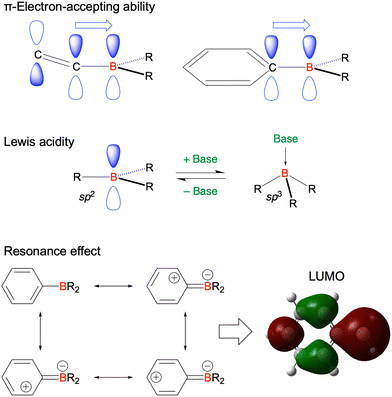 |
| | Fig. 1 Representative features of a tricoordinated sp2-hybridised boron (Bsp2) atom. | |
In 2015 and 2016, a new molecular design utilising the opposite resonance effects of Bsp2 and electron-donating atoms (e.g., oxygen37 and nitrogen38) at relative ortho-positions in a polycyclic aromatic skeleton was proposed by Hatakeyama et al. (Fig. 2). In such structures, the highest occupied molecular orbital (HOMO) is mainly localised on electron-donating atoms and carbons ortho/para relative to them, whereas the lowest unoccupied molecular orbital (LUMO) is mainly localised on the boron atom and carbons ortho/para relative to it in adjacent benzene rings. This alternating localisation of frontier molecular orbitals (FMOs) results in narrowband emission, high absolute photoluminescence (PL) quantum yield (ΦPL), thermally activated delayed fluorescence (TADF),39–41 and sufficient stability during the operation of organic light-emitting diodes (OLEDs).42,43 Consequently, more than 400 organoboron-based multiple-resonance (MR) emitters have been developed44–53 after the pioneering reports on 5,9-dioxa-13b-boranaphtho[3,2,1-de]anthracene (DOBNA)37 and 5,9-diaza-13b-boranaphtho[3,2,1-de]anthracene (DABNA).38
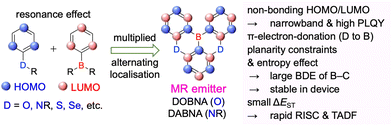 |
| | Fig. 2 Molecular design and physical properties of multiple-resonance (MR) emitters. | |
For organic emitters, the full width at half maximum (FWHM) of the emission peak is generally correlated with the degree of structural relaxation in the first singlet excited state (S1).54,55 The number of vibrational modes (stretching, bending, scissoring, rocking, wagging, twisting, etc.) and vibronic transitions (0–1, 0–2, 0–3, etc.) in each mode that can couple with electronic excitation and, hence, the extent of emission peak broadening, are positively correlated with the extent of structural differences between the ground (S0) and S1 states (Fig. 3a). This correlation is the underlying reason for the broad emission (FWHM > 70 nm) of donor–acceptor (D–A)-type emitters featuring intramolecular charge-transfer (ICT) excited states. For π-conjugated compounds with a rigid framework and no strong donor or acceptor groups (e.g., pyrene and perylene), locally excited (LE) character becomes dominant to result in narrower emission (FWHM < 50 nm). Moreover, LE emitters show small Stokes shifts (10–20 nm) because of their minimal structural relaxation in the S1 state. However, in terms of colour purity, even LE emitters cannot compete with inorganic ones (e.g., GaN-based light-emitting diodes (LEDs)56 and CdS/ZnS quantum dots57,58) because of the unfavourable vibronic peaks with energy intervals of ∼1500 cm−1 observed in the former case (Fig. 3b).42,57,58 This behaviour indicates strong vibronic coupling between the S1–S0 electronic transition and stretching vibrations originating from the bonding/anti-bonding character of FMOs.54,59 In contrast, MR emitters do not show obvious vibronic peaks because of the non-bonding character of their FMOs and feature S1–S0 electronic transition coupling with the scissoring/twisting vibrations at <200 cm−1 and not with the stretching vibrations at ∼1500 cm−1.60 As a result, all vibronic transitions with energy intervals of <200 cm−1 are included in one emission peak with a FWHM of <30 nm (Fig. 3c). Given the minimal vibronic coupling between the electronic transition and stretching vibrations, the non-radiative transition from S1 to S0 is efficiently suppressed, and most MR emitters therefore exhibit high ΦPL (>90%).61–63
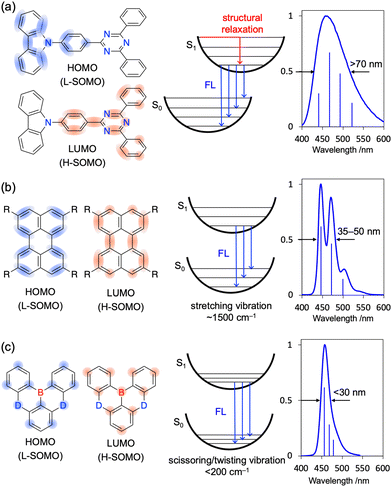 |
| | Fig. 3 Typical vibronic spectra of (a) D–A-type, (b) LE, and (c) MR emitters. | |
The pioneering work of Adachi et al. (2012)41,64,65 inspired numerous studies on organic TADF emitters for optoelectronic devices. Unlike conventional fluorescent emitters, which can harvest only singlet excitons, TADF emitters can harvest both singlet and triplet excitons via efficient reverse intersystem crossing (RISC) from the triplet excited (T1) state to the S1 state, which enables the realisation of OLEDs with internal quantum efficiencies (IQEs) of up to 100% (Fig. 4).41 According to first-order perturbation theory, namely Fermi's golden rule, the rate constant for the RISC between two states (kRISC) is proportional to the spin–orbit coupling (SOC) matrix element (〈Sn|ĤSOC|Tn〉) and inversely proportional to the exciton singlet–triplet energy gap (ΔEST), i.e., kRISC ∝ 〈Sn|ĤSOC|Tn〉 exp(−ΔEST/kBT), where kB is the Boltzmann constant and T is the absolute temperature.66–69 To simultaneously realise a small ΔEST and sufficient transition oscillator strength (f) of conventional TADF emitters, twisted D–A structures with D and A units of variable electron-donating and -accepting abilities can be used to achieve an appropriate overlap of FMO wavefunctions.70–73 The balance between ΔEST and f can be also optimised by introducing π-spacer introduction. However, as mentioned above, D–A-type TADF emitters exhibit broad emission peaks because of their substantial structural relaxation in the S1 state. In contrast, MR emitters feature both TADF properties and narrowband emission because their alternating HOMO–LUMO separation efficiently suppresses the exchange interaction between FMOs and vibronic coupling with stretching vibrations in excited states. Therefore, MR emitters are usually denoted as MR-TADF emitters to clearly distinguish them from D–A-type TADF emitters. From a theoretical viewpoint, MR-TADF emitters are characterised by short-range charge-transfer (CT) character and long-range electronic delocalisation in the excited state, which collectively result in a small ΔEST + large f, which is ideal for TADF emitters.74 These distinctive features enable light amplification for solid-state laser applications despite the difficulty of lasing with most TADF materials.75–77 Moreover, because of their planar structures, MR-TADF emitters prefer a horizontal orientation in vacuum-deposited OLEDs, which results in high outcoupling efficiency and external quantum efficiency (EQE).78–81 One drawback of MR-TADF emitters is their insufficiently fast RISC (kRISC = 104–105 s−1) due to the weak SOC between their S1 and Tn states. Although state-of-the-art D–A-type TADF emitters exhibit strong SOC between their 3LE (Tn) and 1CT (S1) states because of the twisted D–A structures,67–74 the generation of sufficient SOC in the planar and rigid π-conjugated frameworks of MR emitters is challenging.82–85
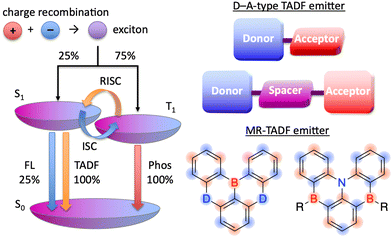 |
| | Fig. 4 HOMO–LUMO separation in D–A-type TADF and MR-TADF emitters (D = O, NR, S, Se, etc.; A = BR, CO, etc.). | |
Over the last three decades, much effort has been dedicated to developing emitters based on Bsp2,1–21 although their industrial applicability is questionable because of their rapid degradation via the cleavage of Bsp2–C bonds during device operation (Fig. 5). This behaviour is due to the critically small bond dissociation energy (BDE)86 of the radical-cation species that decomposes to produce a borinium ion and Csp2-centred radical species; the BDE (ΔH‡) of triphenylborane (Ph3B) in radical-cation and neutral states is 51.1 and 116.9 kcal mol−1, respectively. Although the incorporation of electron-donating atoms in phenoxaborin and phenazaborin increases the BDE (ΔH‡) of the radical-cation state by 35.7 and 39.0 kcal mol−1, respectively, through the stabilisation of this state and destabilisation of borinium ions, the ΔG‡ of bond dissociation is much smaller than the ΔH‡ because of the entropy gain due to the generation of two species from one. For MR emitters, the BDE (ΔH‡) of the radical-cation state is even higher, equalling 105.5 kcal mol−1 for DOBNA and 116.6 kcal mol−1 for DABNA. Notably, the differences between the ΔH‡ and ΔG‡ BDE values of these MR emitters are negligibly small, suggesting a substantial contribution of the intramolecular (ring-fusion) effect to the suppression of bond dissociation. However, this effect alone is insufficient, as exemplified by the much smaller BDE (ΔG‡) of 72.5 kcal mol−1 observed for 5,9-dihydro-13b-boranaphtho[3,2,1-de]anthracene (BNA).
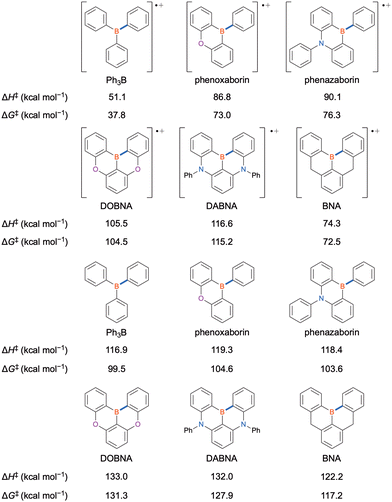 |
| | Fig. 5 Bond dissociation energies of Bsp2–C bonds (highlighted in blue) in representative compounds calculated at the B3LYP/6-31G(d) level of theory. | |
These intrinsic features of MR emitters make them ideal for practical applications, as evidenced by the fact that patents have been filed for more than 3000 related molecules in the last few years. As a result, DABNA derivatives are now widely commercialised as blue emitters in triplet–triplet annihilation (TTA)-driven fluorescent devices for smartphones and televisions.87–91 However, MR emitters have not been implemented as TADF emitters because of their rapid degradation through the long-lived T1 state in TADF-OLEDs.87 One of the most effective ways of realising reliable TADF devices is boosting the RISC of MR emitters. However, promising approaches such as the introduction of heavy atoms92–95 lead to structural weakness (e.g., small BDE of heavy atoms and strain energy), which adversely affects the device lifetime despite the increased kRISC. Another promising approach is the extension of the MR skeleton to enhance charge delocalisation and decrease ΔEST.60,74 However, most large molecules suffer from poor synthetic efficiency or require solution-phase processing and are therefore difficult to commercialise. The use of MR emitters as terminal emitters (TEs) in TADF-assisted fluorescence (TAF, i.e., hyperfluorescence)96–100 or phosphor-sensitised fluorescence (PSF) OLEDs101,102 is a more promising approach, as in this case, all the intrinsic features of MR emitters (e.g., narrowband emission, high ΦPL, small Stokes shift, high molar extinction coefficient, and horizontal dipole orientation) positively affect device properties. The precise adjustment of TE's ionisation potential and electron affinity of a TE is necessary to prevent carrier trapping in the emissive layer (comprising a sensitiser and host material), and thus maximise the characteristics of TAF- and PSF-based OLEDs, especially the device lifetime. However, adjustment based on the introduction of donor or acceptor groups leads to structural weakness. Establishing a molecular design that fulfils all of the requirements for practical applications requires an understanding of the available chemical space of MR emitters. Therefore, we herein comprehensively review organoboron-based MR emitters, highlight their synthetic strategies classified as one-pot borylation, one-shot borylation, and late-stage functionalisation, and establish material structure–photophysical property correlations to accelerate the practical application of these compounds in OLED displays and open up new applications that remain largely unexplored.103,104
Given that some MR emitters have been named differently in different papers, we list all compound names in chronological order in the tables and schemes, and the earliest reported compound name is adopted as a representative one afterwards. If the earliest paper uses compound numbers and does not provide specific compound names, we use the earliest reported compound name instead of the number. MR emitters with no names established to date are newly named, and the name is enclosed in parentheses. In addition, we list the abbreviations for general terms (Table 1) and reagents and OLED materials (Table 2).
Table 1 List of abbreviations (general terms) used in the text
| Abbreviations |
Full meaning |
Abbreviations |
Full meaning |
| acac |
Acetylacetonate |
LE |
Locally excited |
| ACQ |
Aggregation caused quenching |
3LLCT |
Ligand-to-ligand CT |
| AIE |
Aggregation-induced emission |
LUMO |
Lowest unoccupied molecular orbital |
| ArLi |
Aryl lithium species |
MR |
Multiple resonance |
| BDE |
Bond dissociation energy |
3MLCT |
Metal-to-ligand CT |
| Bpin |
Boronic acid pinacol ester |
NMR |
Nuclear magnetic resonance |
| CT |
Charge transfer |
OLED |
Organic light-emitting diode |
| CIE |
Commission internationale de l'éclairage |
PAH |
Polycyclic aromatic hydrocarbons |
| CPL |
Circularly polarised luminescence |
Φ
PL
|
Photoluminescence quantum yield, PLQY |
| D–A |
Donor–acceptor |
PL |
Photoluminescence |
| ΔEST |
S1–T1 energy gap |
PSF |
Phosphor-sensitised fluorescence |
| DFT |
Density functional theory |
RISC |
Reverse intersystem crossing |
| EPR |
Electron paramagnetic resonance |
rt |
Room temperature |
| EQE |
External quantum efficiency |
SNAr |
Nucleophilic aromatic substitution reaction |
| equiv |
Equivalent |
SMe |
Methylthio (group) |
|
f
|
Oscillator strength |
SOC |
Spin–orbit coupling |
| FMOs |
Frontier molecular orbitals |
S0 |
Singlet ground (state) |
| FWHM |
Full width at half maximum |
S1 |
The first singlet excited (state) |
| |gPL| |
Dissymmetry values |
TADF |
Thermally activated delayed fluorescence |
| HOMO |
Highest occupied molecular orbital |
τ
d
|
Delayed fluorescence lifetime |
| Hyperfluorescence |
TADF-assisted fluorescence (TAF) |
TE |
Terminal emitter |
| ICT |
Intramolecular charge-transfer |
TTA |
Triplet–triplet annihilation |
|
3ILCT |
Intraligand CT |
T1 |
The first triplet excited (state) |
| IQE |
Internal quantum efficiency |
UV-Vis |
Ultraviolet-visible |
|
k
RISC
|
RISC rate constant |
|
|
Table 2 List of abbreviations (reagents and OLED materials) used in the text
| Abbreviations |
Full meaning |
Abbreviations |
Full meaning |
| AIBN |
2-Azoisobutyronitrile |
MeI |
Iodomethane |
| AlCl3 |
Aluminium chloride |
MeOH |
Methyl alcohol |
| BBr3 |
Boron tribromide |
2-MeTHF |
2-Methyltetrahydrofuran |
| BCl3 |
Boron trichloride |
MesB(OMe)2 |
Dimethyl mesitylboronate |
| BI3 |
Boron triiodide |
MesMgBr |
Mesityl magnesium bromide |
| BCPO |
Bis-4(N-carbazolyl)phenylphosphine oxide |
mCPBA |
3-Chloroperbenzoic acid |
| Bepp2 |
Bis[2-(2-hydroxyphenyl)-pyridine] beryllium |
mCPBC |
9-(3-(9H-Carbazol-9-yl)phenyl)-9H-3,9′-bicarbazole |
| BF3·Et2O |
Boron trifluoride diethyl ether complex |
mCPCN |
9-(3-(9H-Carbazol-9-yl)phenyl)-9H-carbazole-3-carbonitrile |
| (Bpin)2 |
Bis(pinacolato)diboron |
mMTDATA |
4,4′,4′′-Tris(N-3-methylphenyl-N-phenylamino)triphenylamine |
| Bu2O |
Dibutyl ether |
NaH |
Sodium hydride |
| CBP |
4,4′-Di(9H-carbazol-9-yl)-1,1′-biphenyl |
NaHCO3 |
Sodium hydrogencarbonate |
| Co2(CO)8 |
Octacarbonyldicobalt |
NaOH |
Sodium hydroxide |
| Cs2CO3 |
Cesium carbonate |
NaOAc |
Sodium acetate |
| [Cu(CH3CN)4]PF6 |
Tetrakis(acetonitrile)copper(I) hexafluorophosphate |
NaOtBu |
Sodium tert-butoxide |
| CuCl |
Copper(I) chloride |
Na2CO3 |
Sodium carbonate |
| Cu(ClO4)2·6H2O |
Copper(II) perchlorate hexahydrate |
NECO-296 |
Bis(di-tert-butyl(3-methyl-2-butenyl)phosphine)dichloro palladium |
| CuCN |
Copper(I) cyanide |
NEtiPr2 |
N,N-Diisopropylethylamine |
| CuI |
Copper(I) iodide |
n
BuLi |
n-Butyllithium |
| 3CTF |
2,4,6-Tris(2-(3,6-di-tert-butyl-9H-carbazol-9-yl)-5-(trifluoromethyl)phenyl)-1,3,5-triazine |
NBS |
N-Bromosuccinimide |
| DBFDPO |
4,6-Bis(diphenylphosphoryl)dibenzofuran |
NMP |
N-Methyl-2-pyrrolidone |
| DBFPO (PPF) |
Dibenzo[b,d]furan-2,8-diylbis(diphenylphosphine oxide) |
NPB |
N
4,N4′-Di(naphthalen-1-yl)-N4,N4′-diphenyl-[1,1′-biphenyl]-4,4′-diamine |
| DBU |
1,8-Diazabicyclo[5.4.0]undec-7-ene |
PdCl2(amphos)2 |
Bis[di-tert-butyl(4-dimethylaminophenyl)phosphine]dichloropalladium(II) |
| 26DCzPPy |
2,6-Bis(3-(9H-carbazol-9-yl)phenyl)pyridine |
PdCl2(dppf) |
[1,1′-Bis(diphenylphosphino)ferrocene]palladium(II) dichloride |
| DDQ |
2,3-Dichloro-5,6-dicyano-1,4-benzoquinone |
PdCl2(PPh3)2 |
Bis(triphenylphosphine)palladium(II) dichloride |
| DIC-TRz |
11-(4,6-diphenyl-1,3,5-triazin-2-yl)-12-phenyl-11,12-dihydroindolo[2,3-a]carbazole |
Pd(OAc)2 |
palladium(II) acetate |
| DMAc |
N,N-Dimethylacetamide |
Pd[P(o-tol)3]2Cl2 |
Bis(tri-o-tolylphosphine)palladium(II) dichloride |
| DMAP |
4-Dimethylaminopyridine |
Pd(PPh3)4 |
Tetrakis(triphenylphosphine)palladium(0) |
| DMF |
N,N-Dimethylformamide |
Pd2(dba)3 |
Tris(dibenzylideneacetone)dipalladium(0) |
| DMFBD-TRZ |
2-(3′-(9,9-Dimethyl-9H-fluoren-2-yl)-[1,1′-biphenyl]-3-yl)-4,6-diphenyl-1,3,5-triazine |
PhBCl2 |
Dichlorophenylborane |
| DMMN |
Dimethylmalononitrile |
PhB(OH)2 |
Phenylboronic acid |
| DMIC-TRZ |
1,3-Dihydro-1,1-dimethyl-3-(3-(4,6-diphenyl-1,3,5-triazin-2-yl)phenyl)indeno-[2,1-b]-carbazole |
PhCzBCz |
9-(2-(9-Phenyl-9H-carbazol-3-yl)phenyl)-9H-3,9′-bicarbazole |
| DPEPO |
Bis[2-(diphenylphosphino)phenyl]ether oxide |
Ph2PH |
Diphenylphosphane |
| dtbpy |
4,4′-Di-tert-butyl-2,2′-bipyridyl |
Ph3B |
Triphenylborane |
| DTBPY |
2,6-Di-tert-butylpyridine |
PMMA |
Polymethylmethacrylate |
| EtOH |
Ethyl alcohol |
PMP |
1,2,2,6,6-Pentamethylpiperidine |
| NEt3 |
Triethylamine |
P(OEt)3 |
Triethyl phosphite |
| FeCl3 |
Iron(III) chloride |
PO-T2T |
2,4,6-Tris[3-(diphenylphosphinyl)phenyl]-1,3,5-triazine |
| FF |
Fluorenofluorene |
PPF (DBFPO) |
Dibenzo[b,d]furan-2,8-diylbis(diphenylphosphine oxide) |
| HBC |
Hexabenzocoronene |
PPh3 |
Triphenylphosphine |
| HBr |
Hydrogen bromide/hydrobromic acid |
PS |
Polystyrene |
| HCl |
Hydrogen chloride/hydrochloric acid |
s
BuLi |
sec-Butyllithium |
| HI |
Hydrogen iodide/hydriodic acid |
SF3TRZ |
2-(9,9′-Spirobi[fluoren]-3-yl)-4,6-diphenyl-1,3,5-triazine |
| HOAc |
Acetic acid |
SiCzCz |
9-(3-(Triphenylsilyl)phenyl)-9H-3,9′-bicarbazole |
| IF |
Indenofluorene |
SiTrzCz2 |
9,9′-(6-(3-(Triphenylsilyl)phenyl)-1,3,5-triazine-2,4-diyl)bis(9H-carbazole) |
|
i
PrOBpin |
2-Isopropoxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolane |
SnCl2 |
Tin(II) chloride |
| IrCl3 |
Iridium trichloride |
SOCl2 |
Thionyl chloride |
| [Ir(cod)(OCH3)]2 |
(1,5-Cyclooctadiene)(methoxy)iridium(I) dimer |
SPhos |
2-Dicyclohexylphosphino-2′,6′-dimethoxybiphenyl |
| KOAc |
Potassium acetate |
TBAF |
Tetrabutylammonium fluoride |
| KOtBu |
Potassium tert-butoxide |
t
Bu-Benzene |
tert-Butylbenzene |
| KPF6 |
Potassium hyxafluorophosphate |
t
BuLi |
tert-Butyllithium |
| K2CO3 |
Potassium carbonate |
[tBu3PH]BF4 |
Tri-tert-butylphosphonium tetrafluoroborate |
| K2PtCl4 |
Potassium tetrachloroplatinate(II) |
TBTA |
Tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine |
| K3PO4 |
Tripotassium phosphate |
TCTA |
Tris(4-(9H-carbazol-9-yl)phenyl)amine |
| K4[Fe(CN)6] |
Potassium ferrocyanide |
TfOH |
Trifluoromethanesulfonic acid |
| MADN |
2-Methyl-9,10-bis(naphthalen-2-yl)anthracene |
THF |
Tetrahydrofuran |
| mCBP |
3,3′-Di(9H-carbazol-9-yl)-1,1′-biphenyl |
TMS |
Trimethylsilyl |
| mCBP-CN |
3′,5-Di(9H-carbazol-9-yl)-[1,1′-biphenyl]-3-carbonitrile |
TPSS |
2,2′,7,7′-Tetraphenyl-9,9′-spirobi[thioxanthene] |
| mCP |
1,3-Di(9H-carbazol-9-yl)benzene |
XPhos |
2-Dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl |
| MeCN |
Acetonitrile |
|
|
2. One-pot borylation
As the versatile methodology for the construction of cyclic π-conjugated compounds with Bsp2 atoms connected to aromatic residues, a “one-pot borylation”105 protocol has been developed in the past decade. This protocol consists of three tandem steps, namely the lithiation of carbon–hydrogen or carbon–halogen bonds by an alkyllithium reagent, transmetalation by boron tribromide (BBr3), and tandem intramolecular bora-Friedel–Crafts reaction106 (i.e., electrophilic C–H borylation) (Scheme 1). The final cyclisation step generally proceeds in the absence of Lewis acid additives, which are generally used as efficient promoters of electrophilic C–H borylation,25,107–110 as the nucleophilicity of arene rings is increased by two electron-rich atoms (X1 and X2 in Scheme 1). Moreover, these electron-donating atoms induce the MR effect and impart photophysical properties desirable for OLED fabrication, such as narrowband emission, high ΦPL, TADF, and sufficient stability in the device. Given that the above development has inspired the design and synthesis of numerous MR-TADF optoelectronic materials, the present section overviews those prepared by one-pot borylation and classifies them according to their synthesis method. In particular, the synthesis route of one-pot borylation precursors has to be thoughtfully designed because an appropriate directing group for lithiation should be included.
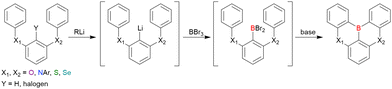 |
| | Scheme 1 General synthesis procedure of one-pot borylation. | |
2.1 DOBNA and its derivatives
2.1.1 Directed ortho-lithiation.
The one-pot borylation method was first proposed by Hatakeyama et al. (2015) and relied on directed ortho-lithiation, i.e., the ability of aromatic C–H bonds adjacent to coordinative functional groups to be lithiated in a regioselective manner.37Table 3 lists the conditions used to synthesise O,B,O-embedded DOBNA based on the selective lithiation of 1,3-diphenoxybenzene at the position between the two directing aryloxy groups. After the successive addition of n-butyllithium (nBuLi) and BBr3, the reaction mixture was supplemented with an amine and heated at 120 °C for 5 h. Screening of the reaction conditions identified the optimal amine loading as 2.0 equivalent (equiv.) (entries 1–4) and the optimal amines as N,N-diisopropylethylamine (NEtiPr2), 1,2,2,6,6-pentamethylpiperidine (PMP), and N,N-dimethyl-p-toluidine (entries 5–9). Under optimal conditions (entry 3), which are still regarded as the standard for one-pot borylation, DOBNA was obtained in an isolated yield of 62%.
Table 3 Synthesis of DOBNAvia directed ortho-lithiation in ref. 37
|
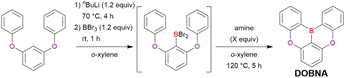
|
| Entrya |
Amine |
X
|
Yieldb [%] |
|
Reactions were carried out on a 1.0 mmol scale.
Yield determined by 1H nuclear magnetic resonance (NMR) measurements using dibromomethane as an internal standard.
Average of two experiments.
|
| 1 |
None |
0 |
16 |
| 2 |
NEtiPr2 |
1.0 |
62 |
| 3 |
NEtiPr2 |
2.0 |
71c |
| 4 |
NEtiPr2 |
3.0 |
44 |
| 5 |
NEt3 |
2.0 |
13 |
| 6 |
2,2,6,6-Tetramethylpiperidine |
2.0 |
2 |
| 7 |
PMP |
2.0 |
68 |
| 8 |
2,6-Lutidine |
2.0 |
41 |
| 9 |
N,N-Dimethyl-p-toluidine |
2.0 |
71c |
A similar procedure was used to prepare DOBNA derivatives via directed ortho-lithiation (Fig. 6 and Table 4).37,111–113 In the first step, lithiation by alkyllithiums (nBuLi, sec-butyllithium (sBuLi), and tert-butyllithium (tBuLi)) proceeded at a relatively high temperature (room temperature (rt) – 90 °C) for several hours. Subsequent borylation with BBr3 (1.2–2.0 equiv.) proceeded at rt – 50 °C, and the final intramolecular cyclisation was conducted in the presence of NEtiPr2 upon heating at 100–120 °C to afford DOBNA derivatives, including those with phenoxazine (DOBNA-Phenox) and carbazole (TMCz-BO, TMCz-3P, and TDBA-Cz) functional groups, in moderate yields. DOBNA, DOBNA-Ph-2, DOBNA-Ph-3, and DOBNA-Phenox were later named in ref. 105.
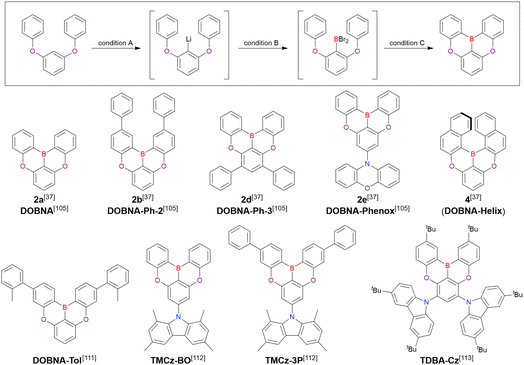 |
| | Fig. 6 DOBNA derivatives synthesised by directed ortho-lithiation. | |
Table 4 Conditions used to synthesise the compounds shown in Fig. 6
| Compound |
Condition Aa |
Condition Ba |
Condition Ca |
Solvent |
Yield [%] |
Ref. |
|
Numbers in parentheses refer to reagent loadings (equivalents).
|
|
DOBNA
|
n
BuLi (1.1)/70 °C, 4 h |
BBr3 (1.2)/40 °C, 1 h |
NEtiPr2 (2.0)/120 °C, 5 h |
t
Bu-Benzene |
62 |
37 and 105
|
|
DOBNA-Ph-2
|
n
BuLi (1.1)/70 °C, 1 h |
BBr3 (1.2)/rt, 1 h |
NEtiPr2 (2.0)/120 °C, 4 h |
o-Xylene |
54 |
37 and 105
|
|
DOBNA-Ph-3
|
t
BuLi (1.2)/rt, 12 h |
BBr3 (1.5)/50 °C, 4 h |
NEtiPr2 (2.0)/120 °C, 24 h |
t
Bu-Benzene |
42 |
37 and 105
|
|
DOBNA-Phenox
|
n
BuLi (1.2)/50 °C, 12 h |
BBr3 (1.5)/rt, 12 h |
NEtiPr2 (2.0)/120 °C, 24 h |
t
Bu-Benzene |
56 |
37 and 105
|
|
DOBNA-Helix
|
n
BuLi (1.5)/90 °C, 4 h |
BBr3 (2.0)/rt, 13 h |
NEtiPr2 (2.0)/100 °C, 24 h |
t
Bu-Benzene |
33 |
37
|
|
DOBNA-Tol
|
s
BuLi (1.2)/70 °C, 3 h |
BBr3 (1.2)/rt, 1 h |
NEtiPr2 (2.0)/120 °C, 3 h |
Xylene |
16 |
111
|
|
TMCz-BO
|
n
BuLi (1.2)/50 °C, 4 h |
BBr3 (1.5)/40 °C, 1 h |
NEtiPr2 (2.0)/120 °C, 5 h |
t
Bu-Benzene |
15 |
112
|
|
TMCz-3P
|
n
BuLi (1.2)/50 °C, 4 h |
BBr3 (1.5)/40 °C, 1 h |
NEtiPr2 (2.0)/120 °C, 5 h |
t
Bu-Benzene |
15 |
112
|
|
TDBA-Cz
|
t
BuLi (2.0)/60 °C, 2 h |
BBr3 (2.0)/rt, 1 h |
NEtiPr2 (2.0)/140 °C, 20 h |
t
Bu-Benzene |
35 |
113
|
In the case of DOBNA-Helix (Scheme 2, denoted as 4 in the original paper), which was the major product (33%) despite its sterically hindered [6]helicene substructure, borylation–cyclisation proceeded in a regioselective manner, with the less distorted isomer (“byproduct” in Scheme 2) formed in only 2% yield. This result suggests that electronic effects (i.e. HOMO distribution) are more important than steric ones under the optimised conditions of one-pot borylation.
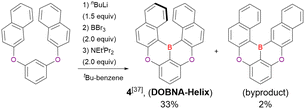 |
| | Scheme 2 Regioselectivity of borylation–cyclisation reaction. | |
2.1.2 Lithium–halogen exchange.
Although directed ortho-lithiation provides access to MR-TADF frameworks, its low reactivity and regioselectivity sometimes prevent effective borylation–cyclisation. Therefore, the development of an alternative methodology was required and borylation via lithium–halogen exchange is now widely adopted as a more practical route. This route requires the regioselective modification of 1,2,3-trisubstituted benzenes to maintain a halogen atom between the two aryloxy groups. To achieve this selectivity, nucleophilic aromatic substitution (SNAr), which proceeds preferentially on the fluorine substituent, plays an important role.
Commercially available 2-bromo-1,3-difluorobenzene is one of the most promising starting materials for one-pot borylation via lithium–halogen exchange. The first example of such synthesis was reported at the same time as the first directed ortho-lithiation method (Scheme 3).37 The double SNAr reaction of 2-bromo-1,3-difluorobenzene with [1,1-biphenyl]-3-ol furnished the bromine-containing precursor in 87% yield, with subsequent one-pot borylation under standard conditions affording 2c (later named DOBNA-Ph-1,105 or simply DOBNA-Ph114) in 74% yield.
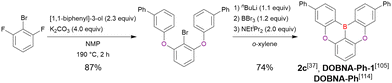 |
| | Scheme 3 Synthesis of DOBNA-Phvia lithium–halogen exchange. | |
Fig. 7 and Table 5 summarise DOBNA derivatives synthesised using lithium–halogen exchange.115–121DOBNA-Ph, B1, B2-OH, BOS, BSS, and tBuAcBO were synthesised from 1-bromo-2,6-difluorobenzene, while the other compounds in Fig. 7 were derived from 1,3-difluoro-2-iodobenzene.
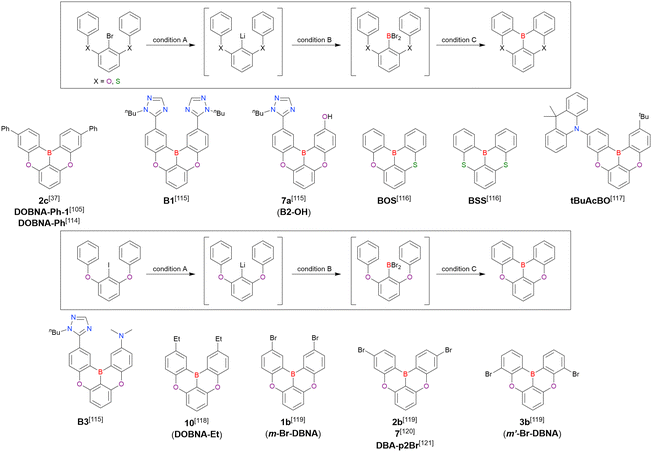 |
| | Fig. 7 DOBNA derivatives synthesised via lithium–halogen exchange. | |
Table 5 Conditions used to synthesise the compounds shown in Fig. 7
| Compound |
Condition Aa |
Condition Ba |
Condition Ca |
Solvent |
Yield [%] |
Ref. |
|
Numbers in parentheses refer to reagent loadings (equivalents).
|
|
DOBNA-Ph
|
n
BuLi (1.1)/−40 °C to rt |
BBr3 (1.2)/rt, 1 h |
NEtiPr2 (2.0)/130 °C, 3 h |
o-Xylene |
74 |
37 and 105
|
|
B1
|
t
BuLi (2.0)/−30 °C, 2 h |
BBr3 (1.2)/rt, 1 h |
NEtiPr2 (2.0)/150 °C, 24 h |
t
Bu-Benzene |
5 |
115
|
|
B2-OH
|
n
BuLi (1.2)/−30 °C, 2 h |
BBr3 (2.0)/rt, 1 h |
NEtiPr2 (2.0)/140 °C, 24 h |
t
Bu-Benzene |
22 |
115
|
|
BOS
|
n
BuLi (1.1)/50 °C, 1 h |
BBr3 (1.2)/rt, 1 h |
NEtiPr2 (2.0)/125 °C, 12 h |
m-Xylene |
25 |
116
|
|
BSS
|
n
BuLi (1.1)/50 °C, 1 h |
BBr3 (1.2)/rt, 1 h |
NEtiPr2 (2.0)/125 °C, 12 h |
m-Xylene |
40 |
116
|
|
tBuAcBO
|
n
BuLi (2.0)/rt, 2 h |
BBr3 (—)/rt, 2 h |
NEtiPr2 (—)/180 °C, 24 h |
t
Bu-Benzene |
16 |
117
|
|
B3
|
t
BuLi (2.0)/−30 °C, 2 h |
BBr3 (1.2)/rt, 1 h |
NEtiPr2 (2.0)/150 °C, 24 h |
t
Bu-Benzene |
6 |
115
|
|
DOBNA-Et
|
n
BuLi (1.1)/−78 °C, 10 min |
BBr3 (1.2)/0 °C, 1 h |
NEtiPr2 (2.0)/120 °C, 21 h |
Toluene |
30 |
118
|
|
m-Br-DBNA
|
n
BuLi (1.0)/−30 °C, 2 h |
BBr3 (1.2)/rt, 1 h |
NEtiPr2 (2.0)/140 °C, 24 h |
m-Xylene |
52 |
119
|
|
DBA-p2Br
|
n
BuLi (1.0)/−30 °C, 2 h |
BBr3 (1.2)/rt, 1 h |
NEtiPr2 (2.0)/140 °C, 24 h |
m-Xylene |
25 |
119
|
|
n
BuLi (1.1)/−30 °C, 1 h |
BBr3 (1.2)/rt, 1 h |
NEtiPr2 (2.0)/140 °C, 3 h |
t
Bu-Benzene |
37 |
120
|
|
n
BuLi (1.0)/−30 °C, 1 h |
BBr3 (1.1)/−30 °C to rt |
NEtiPr2 (5.0)/120 °C, 14 h |
t
Bu-Benzene |
72 |
121
|
|
m′-Br-DBNA
|
n
BuLi (1.0)/−30 °C, 2 h |
BBr3 (1.2)/rt, 1 h |
NEtiPr2 (2.0)/140 °C, 24 h |
m-Xylene |
40 |
119
|
Unlike directed ortho-lithiation, lithium–halogen exchange proceeded under mild conditions (−78 °C to rt). In addition to the oxygen-bridged precursor characteristic of DOBNA derivatives, sulphur-bridged ones could also be used (BOS and BSS). Although triazole functional groups were tolerated under standard conditions, the related yields were rather low (5% for B1, 22% for B2-OH, and 6% for B3). This result suggests that the coordination of Nsp2 to boron atoms can inhibit intramolecular electrophilic C–H borylation. As discussed in the next subsection, bromine-containing products can be synthesised using the same methodology.
One indicative result is the synthesis of B2-OH (Scheme 4), in which the introduction of boron was accompanied by the removal of a methoxy group under the action of BBr3, and the resulting phenoxy group was used for subsequent chemical modification (Scheme 55 in Section 4). Given that the reagents used in one-pot borylation (aryllithium species (ArLi) and BBr3) are highly reactive, one should consider functional group tolerance when designing the synthesis route.
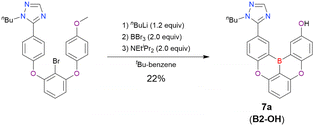 |
| | Scheme 4 Accompanying deprotection reaction (methoxy group removal) induced by BBr3. | |
2.1.3 Borylation of tetrasubstituted precursors.
As shown in the previous subsection, trisubstituted benzenes are the key starting materials for constructing DOBNA-based scaffolds. Hence, tetrasubstituted benzenes were expected to be promising starting materials for accessing more diverse MR-TADF frameworks. In an early example reported by Kwon et al. (Scheme 5),122Br-TDBA was synthesised from 1,4-dibromo-2,6-difluorobenzene in two steps. The first step, a double SNAr reaction, proceeded in high yield (80%) to afford the borylation precursor, and the second step involved one-pot borylation under standard conditions. Although the precursor contained two bromine atoms capable of reacting with nBuLi, lithium–bromine exchange mainly proceeded at the ortho-position relative to the aryloxy groups because of their directing effect. As a result, Br-TDBA was obtained in 15% yield.
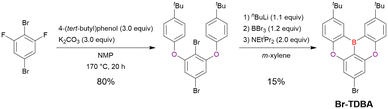 |
| | Scheme 5 Synthesis of Br-TDBA from a tetrasubstituted benzene. | |
From the viewpoint of material chemistry, bromine groups are highly important because they allow for further functionalisation, as exemplified by the tuning of photophysical properties and the addition of various functionalities through the chemical modification of bromine-containing MR-TADF frameworks (see Section 4). Therefore, various bromine-containing DOBNA derivatives have been synthesised using selective lithium–halogen exchange (Fig. 8 and Table 6).116,120,122–138 The reaction temperature of the first lithium–halogen exchange was kept relatively low (−78 °C to rt), probably to suppress the undesired exchange at the remaining bromine atom. Although the yield of Br-TDBA was relatively low (15%) in Kwon's work,122 the same product was synthesised later with up to 49% yield,129 which suggests that lithium–halogen exchange proceeds with high selectivity. The variations in product yield probably originated from the actual concentration of alkyllithium reagents or the purity of BBr3. The effect of the halogen type (Br versus I) on the reaction yield was small, as the yields of Br-TDBA prepared from Br (15–49%)122,123,125–129 and I (41%)124 derivatives were similar. Interestingly, a pyridine-containing derivative, DOBNA-Pyr, could be synthesised in much higher yield (92%), although the detailed purification and characterisation data were not reported.139
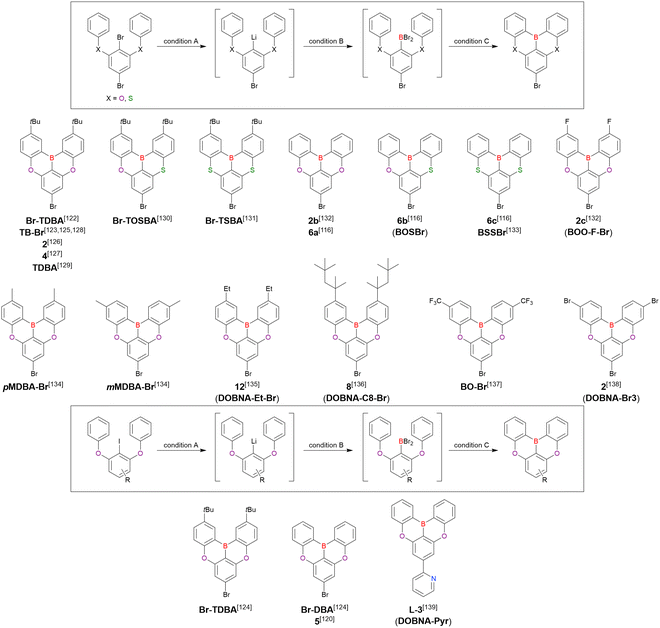 |
| | Fig. 8 DOBNA derivatives synthesised from tetrasubstituted benzenes. | |
Table 6 Conditions used to synthesise the compounds shown in Fig. 8
| Compound |
Condition Aa |
Condition Ba |
Condition Ca |
Solvent |
Yield [%] |
Ref. |
|
Numbers in parentheses refer to reagent loadings (equivalents).
Not described.
|
|
Br-TDBA
|
n
BuLi (1.1)/rt, 1 h |
BBr3 (1.2)/45 °C, 50 min |
NEtiPr2 (2.0)/120 °C, 20 h |
m-Xylene |
15 |
122
|
|
n
BuLi (1.1)/rt, 1 h |
BBr3 (1.2)/50 °C, 1 h |
NEtiPr2 (2.0)/180 °C, 10 h |
m-Xylene |
41 |
123
|
|
n
BuLi (1.1)/−20 °C, 2 h |
BBr3 (1.2)/rt, 1 h |
NEtiPr2 (2.4)/120 °C, 12 h |
m-Xylene |
41 |
124
|
|
n
BuLi (1.1)/rt, 1 h |
BBr3 (1.2)/50 °C, 1 h |
NEtiPr2 (2.0)/180 °C, 10 h |
m-Xylene |
38 |
125
|
|
n
BuLi (1.1)/0 °C, 2 h |
BBr3 (1.2)/rt, 20 min |
NEtiPr2 (0.9)/180 °C, 12 h |
o-Dichlorobenzene |
27 |
126
|
|
n
BuLi (1.0)/−30 °C, 2 h |
BBr3 (1.2)/rt, 1 h |
NEtiPr2 (2.4)/120 °C, 12 h |
m-Xylene |
43 |
127
|
|
n
BuLi (1.1)/rt, 2 h |
BBr3 (1.2)/50 °C, 50 min |
NEtiPr2 (6.0)/120 °C, 20 h |
Xylene |
19 |
128
|
|
n
BuLi (1.1)/rt, 2 h |
BBr3 (1.2)/45 °C, 1 h |
NEtiPr2 (2.0)/135 °C, 20 h |
Xylene |
49 |
129
|
|
Br-TOSBA
|
n
BuLi (0.7)/rt, 1 h |
BBr3 (0.8)/45 °C, 50 min |
NEtiPr2 (1.3)/120 °C, 20 h |
m-Xylene |
19 |
130
|
|
Br-TSBA
|
n
BuLi (0.7)/rt, 1 h |
BBr3 (0.8)/45 °C, 50 min |
NEtiPr2 (1.3)/120 °C, 20 h |
m-Xylene |
19 |
131
|
|
Br-DBA
|
n
BuLi (1.1)/−30 °C, 2 h |
BBr3 (1.2)/rt, 1 h |
NEtiPr2 (3.8)/130 °C, 20 h |
m-Xylene |
20 |
132
|
|
n
BuLi (1.1)/−30 °C, 2 h |
BBr3 (1.2)/rt, 1 h |
NEtiPr2 (3.8)/130 °C, 20 h |
m-Xylene |
49 |
116
|
|
n
BuLi (1.1)/−20 °C, 2 h |
BBr3 (1.2)/rt, 1 h |
NEtiPr2 (2.4)/120 °C, 12 h |
t
Bu-Benzene |
45 |
124
|
|
n
BuLi (1.1)/−30 °C, 1 h |
BBr3 (1.2)/rt, 1 h |
NEtiPr2 (2.0)/140 °C, 3 h |
m-Xylene |
42 |
120
|
|
BOSBr
|
n
BuLi (1.0)/−30 °C, 2 h |
BBr3 (1.2)/rt, 1 h |
NEtiPr2 (2.0)/130 °C, 20 h |
m-Xylene |
44 |
116
|
|
BSSBr
|
n
BuLi (1.0)/−30 °C, 2 h |
BBr3 (1.2)/rt, 1 h |
NEtiPr2 (2.0)/125 °C, 20 h |
m-Xylene |
40 |
116
|
|
n
BuLi (1.0)/−30 °C, 2 h |
BBr3 (1.3)/rt, 1 h |
NEtiPr2 (2.0)/125 °C, 20 h |
m-Xylene |
68 |
133
|
|
BOO-F-Br
|
n
BuLi (1.1)/−30 °C, 2 h |
BBr3 (1.2)/rt, 1 h |
NEtiPr2 (2.0)/130 °C, 20 h |
m-Xylene |
30 |
132
|
|
pMDBA-Br
|
n
BuLi (1.1)/rt, 2 h |
BBr3 (1.2)/50 °C, 50 min |
NEtiPr2 (2.0)/120 °C, 20 h |
m-Xylene |
64 |
134
|
|
mMDBA-Br
|
n
BuLi (1.1)/rt, 2 h |
BBr3 (1.2)/50 °C, 50 min |
NEtiPr2 (2.0)/120 °C, 20 h |
m-Xylene |
46 |
134
|
|
DOBNA-Et-Br
|
n
BuLi (1.0)/−78 °C, 10 min |
BBr3 (1.2)/0 °C, 0.5 h |
NEtiPr2 (2.0)/110 °C, 14 h |
Toluene |
43 |
135
|
|
DOBNA-C8-Br
|
n
BuLi (1.1)/−78 °C, 10 min |
BBr3 (1.2)/0 °C, 10 min |
NEtiPr2 (2.0)/110 °C, 14 h |
Toluene |
34 |
136
|
|
BO-Br
|
—b |
—b |
—b |
—b |
30 |
137
|
|
DOBNA-Br3
|
n
BuLi (1.1)/25 °C, 1 h |
BBr3 (1.2)/0 °C, 1 h |
NEtiPr2 (1.9)/130 °C, 12 h |
Xylene |
61 |
138
|
|
DOBNA-Pyr
|
n
BuLi (1.3)/rt, 1 h |
BBr3 (1.3)/rt, 1 h |
NEtiPr2 (2.1)/120 °C, 12 h |
m-Xylene |
92 |
139
|
2.1.4 Photophysical properties.
Unlike the MR effect of the recently reported MR-TADF frameworks, the MR effect of DOBNA derivatives has not been a research focus because of the low electron-donating ability of oxygen atoms. However, some reports investigated the photophysical properties of these derivatives in detail. Table 7 summarises λmaxPL, FWHM, ΦPL, ΔEST, and kRISC values as representative photophysical parameters of the MR-TADF emitters presented in Fig. 6–8. Compounds whose photophysical data have not been reported are excluded from Table 7. Given that ΔEST and kRISC can be estimated in several ways, the values presented herein need to be compared with caution. When grafted with diarylamine and carbazolyl groups, the DOBNA scaffold acts as an electron acceptor and induces long-range D–A-type CT emission (DOBNA-Phenox,37TMCz-BO,112TMCz-3P,112TDBA-Cz,113B3,115 and tBuAcBO117). As such emission does not fully reflect the photophysical properties derived from the MR effect, they are not discussed in this section (long-range D–A-type CT emission is reviewed in Section 4). In general, non-D–A-type DOBNA derivatives (DOBNA,37,116DOBNA-Ph,37DOBNA-Ph-2,37DOBNA-Ph-3,37B1,115BOS,116 and BSS116) feature a high transition energy (λmaxPL ≈ 400 nm, T1 = 2.9–3.0 eV), moderate ΦPL (0.60–0.80), and moderate kRISC (103–104 s−1), and are therefore widely used as host materials for the emitting layers of OLEDs, benefiting from the high T1 energy.
Table 7 Photophysical properties of the compounds shown in Fig. 6–8
| Compound |
Condition |
λ
maxPL [nm] |
FWHM [nm] |
Φ
PL [—] |
ΔEST [eV] |
k
RISC [s−1] |
Ref. |
|
Compounds showing long-range D–A-type CT emission.
See Table 2 for the full compound name.
|
|
DOBNA
|
CH2Cl2/1 wt%-PMMAb |
398/398 |
—/33 |
0.72/0.80 |
0.15/0.18 |
—/1.7× 103 |
37
|
| Toluene/1 wt%-PSb |
396/— |
30/— |
—/0.70 |
0.18/— |
—/1.1 × 104 |
116
|
|
DOBNA-Ph
|
CH2Cl2 |
410 |
— |
0.60 |
0.31 |
— |
37
|
|
DOBNA-Ph-2
|
CH2Cl2 |
410 |
— |
0.65 |
0.21 |
— |
37
|
|
DOBNA-Ph-3
|
CH2Cl2 |
436 |
— |
0.57 |
0.14 |
— |
37
|
|
B1
|
CH2Cl2/5 wt%-PMMAb |
409/407 |
—/— |
—/0.75 |
0.21/— |
—/— |
115
|
|
BOS
|
Toluene/1 wt%-PSb |
434/— |
29/— |
—/0.63 |
0.17/— |
—/6.1 × 104 |
116
|
|
BSS
|
Toluene/1 wt%-PSb |
457/— |
27/— |
—/0.58 |
0.15/— |
—/1.18 × 105 |
116
|
|
|
|
DOBNA-Phenox
|
1 wt%-PMMAb |
492 |
— |
0.92 |
0.06 |
— |
37
|
|
TMCz-BO
|
Toluene/30 wt%-PPFb |
446/467 |
—/— |
0.81/0.98 |
—/0.020 |
—/1.9 × 106 |
112
|
|
TMCz-3P
|
Toluene/30 wt%-PPFb |
455/477 |
—/— |
0.56/0.76 |
—/0.134 |
—/3.3 × 104 |
112
|
|
TDBA-Cz
|
Toluene/10 wt%-DPEPOb/neat-film |
461/456/462 |
43/46/41 |
—/1.00/0.88 |
0.14/—/— |
—/—/1.40 × 106 |
113
|
|
B3
|
CH2Cl2/5 wt%-PMMAb |
535/497 |
—/— |
—/0.51 |
0.11/— |
—/— |
115
|
|
tBuAcBO
|
Toluene/5 wt%-mCPb |
464/468 |
—/61 |
—/0.66 |
—/0.10 |
—/1.24 × 106 |
117
|
For example, OLEDs containing DOBNA-Ph and DOBNA-Ph-3 as host materials exhibited performances superior to that of 4,4′-di(9H-carbazol-9-yl)-1,1′-biphenyl (CBP) in terms of driving voltage, current efficiency, power efficiency, and external quantum efficiency at 1000 cd m−2.37 Currently, DOBNA-Ph and DOBNA-Tol are widely used as host materials for OLEDs. Importantly, the replacement of oxygen by sulphur (BOS and BSS) causes red-shifted emission (λmaxPL = 434 nm for BOO and 457 nm for BOS) and accelerated RISC (kRISC = 6.1 × 104 s−1 for BOO and 1.18 × 105 s−1 for BOS) because of the heavy-atom effect.116
2.2 DABNA and its derivatives
2.2.1 One-pot borylation of triarylamine precursors.
The one-pot borylation-based synthesis of DOBNA proposed in 2015 highlights the potential of fully cyclised π-conjugated materials containing boron and electron-rich atoms. In addition to O,B,O-embedded DOBNA-type scaffolds, O,B,S- and S,B,S-embedded frameworks can be constructed using similar procedures, as shown above. The powerful route involving an SNAr reaction is, however, not highly effective for the synthesis of triarylamine-based N,B,N-embedded scaffolds. In 2016, Hatakeyama et al. established an alternative strategy for accessing DABNA and its derivatives (Scheme 6).38
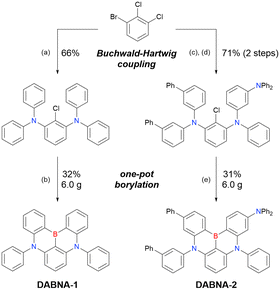 |
| | Scheme 6 Synthesis of DABNA-1 and DABNA-2. (a) HNPh2 (2.2 equiv.), tBuOK (2.5 equiv.), (AmPhos)2PdCl2 (1.0 mol%), o-xylene, 80 °C, 2 h then 120 °C, 3 h. (b) tBuLi (1.2 equiv.), tBu-benzene, 60 °C, 2 h; BBr3 (1.2 equiv.), rt, 0.5 h; NEtiPr2 (2.0 equiv.), 120 °C, 3 h. (c) HNPh(m-Ph2N-C6H4) (1.0 equiv.), tBuOK (1.5 equiv.), (AmPhos)2PdCl2 (0.5 mol%), o-xylene, 90 °C, 2.5 h. (d) HN(m-Ph-C6H4)2 (1.0 equiv.), tBuOK (1.5 equiv.), (AmPhos)2PdCl2 (1.0 mol%), o-xylene, 120 °C, 1 h. (e) tBuLi (2.0 equiv.), tBu-benzene, 60 °C, 3 h; BBr3 (2.0 equiv.), rt, 0.5 h; NEtiPr2 (2.0 equiv.), 120 °C, 1.5 h. | |
The corresponding one-pot borylation precursors were synthesised from the commercially available 1-bromo-2,3-dichlorobenzene by Buchwald–Hartwig coupling. The reaction of this starting material with diphenylamine (2.2 equiv.) afforded the key borylation precursor in 66% yield (Scheme 6, left). The chlorine atom for the subsequent lithium–halogen exchange was retained because of its sterically hindered environment. A non-symmetric intermediate could be synthesised by the sequential coupling of N1,N1,N3-triphenylbenzene-1,3-diamine and di([1,1′-biphenyl]-3-yl)amine based on the difference in reactivity between bromine and chlorine atoms (Scheme 6, right). The final one-pot borylation afforded N,B,N-embedded π-conjugated compounds, DABNA-1 and DABNA-2, in yields of 32% and 31%, respectively. In the case of DABNA-2, tandem electrophilic C–H borylation occurred at the para-position relative to the diphenylamine group, probably because of its electron-donating nature and bulkiness. These processes were robust and scalable, as confirmed by the synthesis of the target compounds in amounts of 6.0 g.
The chemical structures of derivatives possessing the DABNA skeleton and the related synthesis results are summarised in Fig. 9 and Table 8.38,88,102,104,140–149 In addition to 1-bromo-2,3-dichlorobenzene, some studies used 1,3-dibromo-2-chlorobenzene as a starting material. t-DABNA, TBE01, TBE02, and Tp-DABNA can be viewed as a straightforward expansion of DABNA-1via functional group addition. The diarylamine group of the precursor was changed to the 9,10-dihydroacridine group to obtain carbon-bridged MR-TADF frameworks (BN-DMAC, BN-DPAC, BN2). Similarly, structures bridged by oxygen (2PXZBN), nitrogen (TCZ-F-DABNA and BNIP-tBuDPAC), sulphur (2PTZBN, BN4, BN5, and helicene-BN), and selenium atoms (BNSSe and BNSeSe) were obtained. Moreover, a triangulene analogue, BN3, was synthesised using the basic one-pot borylation protocol. Tetrasubstituted starting materials (M-tDABNA, TPXZBN, and DPXZCZBN) were also used, as shown for DOBNA-type compounds. The first step (lithium–chlorine exchange) proceeded in the presence of tBuLi at high temperatures (50–80 °C), probably because of the low reactivity of chlorine groups. In most cases, the reaction yield was moderate (20–40%).
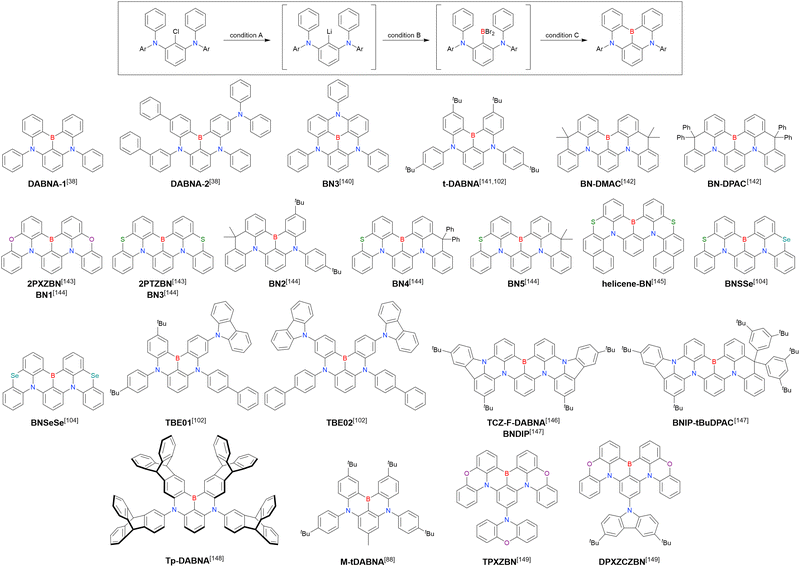 |
| | Fig. 9 DABNA-based MR-TADF emitters. | |
Table 8 Conditions used to synthesise the compounds shown in Fig. 9
| Compound |
Condition Aa |
Condition Ba |
Condition Ca |
Solvent |
Yield [%] |
Ref. |
|
Numbers in parentheses refer to reagent loadings (equivalents).
|
|
DABNA-1
|
t
BuLi (1.2)/60 °C, 2 h |
BBr3 (1.2)/rt, 0.5 h |
NEtiPr2 (2.0)/120 °C, 3 h |
t
Bu-Benzene |
32 |
38
|
|
DABNA-2
|
t
BuLi (2.0)/60 °C, 3 h |
BBr3 (2.0)/rt, 0.5 h |
NEtiPr2 (2.0)/120 °C, 1.5 h |
t
Bu-Benzene |
31 |
38
|
|
BN3
|
t
BuLi (1.0)/50 °C, 0.5 h |
BBr3 (2.1)/−45 °C |
NEtiPr2 (2.2)/reflux, 14 h |
t
Bu-Benzene |
45 |
140
|
|
t-DABNA
|
t
BuLi (2.0)/60 °C, 2 h |
BBr3 (2.0)/rt, 0.5 h |
NEtiPr2 (2.0)/100 °C, 2 h |
t
Bu-Benzene |
31 |
141
|
|
t
BuLi (1.2)/70 °C, 3 h |
BBr3 (1.5)/rt, 2 h |
NEtiPr2 (2.0)/70 °C, 12 h |
t
Bu-Benzene |
21 |
102
|
|
BN-DMAC
|
t
BuLi (2.0)/60 °C, 2 h |
BBr3 (2.0)/rt, 1 h |
NEtiPr2 (2.0)/reflux, overnight |
Mesitylene |
25 |
142
|
|
BN-DPAC
|
t
BuLi (2.0)/60 °C, 2 h |
BBr3 (2.0)/rt, 1 h |
NEtiPr2 (2.0)/reflux, overnight |
Mesitylene |
13 |
142
|
|
2PXZBN
|
t
BuLi (2.2)/60 °C, 2 h |
BBr3 (1.5)/rt, 1 h |
NEtiPr2 (2.0)/160 °C, 24 h |
Mesitylene |
23 |
143
|
|
t
BuLi (2.7)/60 °C, 2 h |
BBr3 (2.8)/rt, 0.5 h |
NEtiPr2 (2.8)/120 °C, 3 h |
t
Bu-Benzene |
32 |
144
|
|
2PTZBN
|
t
BuLi (2.2)/60 °C, 2 h |
BBr3 (1.5)/rt, 1 h |
NEtiPr2 (2.0)/160 °C, 24 h |
Mesitylene |
25 |
143
|
|
t
BuLi (2.5)/60 °C, 2 h |
BBr3 (2.5)/rt, 0.5 h |
NEtiPr2 (2.5)/120 °C, 3 h |
t
Bu-Benzene |
39 |
144
|
|
BN2
|
t
BuLi (2.5)/60 °C, 2 h |
BBr3 (2.5)/rt, 0.5 h |
NEtiPr2 (2.5)/120 °C, 3 h |
t
Bu-Benzene |
39 |
144
|
|
BN4
|
t
BuLi (2.0)/60 °C, 2 h |
BBr3 (2.0)/rt, 0.5 h |
NEtiPr2 (2.0)/120 °C, 3 h |
t
Bu-Benzene |
41 |
144
|
|
BN5
|
t
BuLi (3.0)/60 °C, 2 h |
BBr3 (3.0)/rt, 0.5 h |
NEtiPr2 (3.0)/120 °C, 3 h |
t
Bu-Benzene |
39 |
144
|
|
helicene-BN
|
t
BuLi (2.2)/60 °C, 1 h |
BBr3 (2.5)/rt, 1 h |
NEtiPr2 (1.2)/130 °C, 24 h |
t
Bu-Benzene |
15 |
145
|
|
BNSSe
|
t
BuLi (2.5)/60 °C, 5 h |
BBr3 (2.5)/rt, 0.5 h |
NEtiPr2 (2.5)/160 °C, 14 h |
Mesitylene |
21 |
104
|
|
BNSeSe
|
t
BuLi (2.5)/60 °C, 5 h |
BBr3 (2.5)/rt, 0.5 h |
NEtiPr2 (5.0)/160 °C, 14 h |
Mesitylene |
30 |
104
|
|
TBE01
|
t
BuLi (2.0)/70 °C, 3 h |
BBr3 (2.0)/rt, 2 h |
NEtiPr2 (2.4)/70 °C, 12 h |
t
Bu-Benzene |
24 |
102
|
|
TBE02
|
t
BuLi (2.0)/70 °C, 3 h |
BBr3 (2.0)/rt, 2 h |
NEtiPr2 (2.7)/70 °C, 12 h |
t
Bu-Benzene |
21 |
102
|
|
TCZ-F-DABNA
|
t
BuLi (2.5)/60 °C, 3 h |
BBr3 (1.5)/rt, 6 h |
NEtiPr2 (2.5)/130 °C, 12 h |
t
Bu-Benzene |
82 |
146
|
|
t
BuLi (2.2)/60 °C, 1 h |
BBr3 (1.5)/rt, 1 h |
NEtiPr2 (1.2)/130 °C, 24 h |
t
Bu-Benzene |
48 |
147
|
|
BNIP-tBuDPAC
|
t
BuLi (2.2)/60 °C, 1 h |
BBr3 (1.5)/rt, 1 h |
NEtiPr2 (1.2)/130 °C, 24 h |
t
Bu-Benzene |
38 |
147
|
|
Tp-DABNA
|
t
BuLi (3.0)/80 °C, 4 h |
BBr3 (3.0)/60 °C, 1 h |
NEtiPr2 (3.0)/120 °C, 12 h |
t
Bu-Benzene |
18 |
148
|
|
M-tDABNA
|
t
BuLi (1.2)/60 °C, 3 h |
BBr3 (1.2)/rt, 1 h |
NEtiPr2 (2.0)/120 °C, 12 h |
t
Bu-Benzene |
27 |
88
|
|
TPXZBN
|
t
BuLi (4.5)/60 °C, 2 h |
BBr3 (2.4)/rt, 0.5 h |
NEtiPr2 (3.8)/120 °C, 12 h |
t
Bu-Benzene |
19 |
149
|
|
DPXCZBN
|
t
BuLi (4.5)/60 °C, 2 h |
BBr3 (2.4)/rt, 0.5 h |
NEtiPr2 (3.8)/120 °C, 12 h |
t
Bu-Benzene |
15 |
149
|
Although the replacement of chlorine with bromine is the most straightforward solution to the problem of sluggish lithium–chlorine exchange, this alternative route has been underexplored because of the commercial rarity of the corresponding starting materials. The few compounds successfully prepared from tribromobenzene are listed in Fig. 10 and Table 9.150–152 The synthesis of these compounds, especially that of DTBA-BN2, is characterised by lithium–bromine exchange conditions (nBuLi, −60 °C, 1 h) that are milder than those of lithium–chlorine exchange. Thus, the choice of the halogen atom in one-pot borylation precursors should be carefully considered on the balance of reactivity and accessibility.
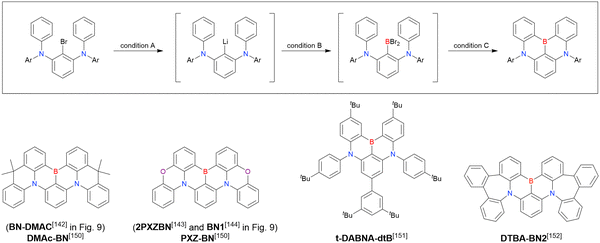 |
| | Fig. 10 DABNA derivatives synthesised by lithium–bromine exchange. | |
Table 9 Conditions used to synthesise the compounds shown in Fig. 10
| Compound |
Condition Aa |
Condition Ba |
Condition Ca |
Solvent |
Yield [%] |
Ref. |
|
Numbers in parentheses refer to reagent loadings (equivalents).
Same compound appeared in Fig. 9 and Table 8.
|
|
DMAc-BN
|
n
BuLi (1.1)/60 °C, 0.5 h |
BBr3 (1.2)/rt, 0.5 h |
NEtiPr2 (2.0)/110 °C, overnight |
Toluene |
38 |
150
|
|
PXZ-BN
|
n
BuLi (1.1)/60 °C, 0.5 h |
BBr3 (1.2)/rt, 0.5 h |
NEtiPr2 (2.0)/110 °C, overnight |
Toluene |
41 |
150
|
|
t-DABNA-dtB
|
n
BuLi (2.0)/rt, 0.5 h |
BBr3 (2.0)/rt, 6 h |
NEtiPr2 (2.0)/120 °C, overnight |
o-Xylene |
38 |
151
|
|
DTBA-BN2
|
n
BuLi (2.2)/−60 °C, 1 h |
BBr3 (1.5)/rt, 1 h |
NEtiPr2 (4.0)/120 °C, 12 h |
Toluene |
40 |
152
|
The one-pot borylation of a 1,3-diamino-5-carbazolyl-substituted precursor via directed ortho-lithiation was attempted as another route to synthesising DABNA derivatives (Scheme 7).153 The resulting compound (TBN-TPA) was identified by Huang et al. as being borylated between the two diarylamine groups. This structure was later corrected (CzDABNA-NP-TB) by Hatakeyama et al., by showing that the boron atom is inserted at the carbon between the diarylamino and carbazolyl groups.111 This behaviour can be ascribed to the less pronounced electron-donating character of carbazole groups than diarylamino groups, which can facilitate the nucleophilic reaction with nBuLi at the adjacent position. Therefore, directed ortho-lithiation can suffer from unpredictable or poor regioselectivity. In particular, the directing effects of diarylamine and carbazole groups are inferior to that of aryloxy groups because of the delocalisation of the nitrogen lone pair.
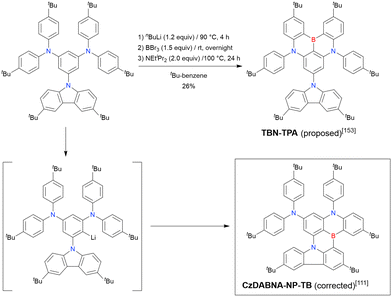 |
| | Scheme 7 Two possible chemical structures derived from the directed ortho-lithiation of a 1,3-diamino-5-carbazozyl substituted precursor. | |
2.2.2 Photophysical properties.
Although DOBNA and its derivatives were the initial examples of MR-TADF emitters, the development of the one-pot borylation protocol for DABNA derivatives in 2016 enabled the development of numerous MR-TADF emitters suitable for use in the emitting layers of OLEDs. In the original paper,38DABNA-1 and DABNA-2 were used as MR-TADF emitters in OLEDs for the first time and achieved pure blue emission at 459–467 nm with a narrow FWHM of 28 nm and an IQE of ∼100%, thus achieving a record-setting performance for blue OLEDs.
Table 10 lists the photophysical properties of DABNA-based MR-TADF emitters. The most notable feature is the sharp PL band (FWHM = 20–40 nm) and small ΔEST (0.1–0.2 eV) due to the MR effect of boron and nitrogen atoms. Moreover, the corresponding ΦPL is above 0.90 in most cases, and the average λmaxPL values (460–510 nm) are ∼50 nm higher than those of DOBNA-based emitters. This spectral shift is attributed to the higher atomic orbital energies of nitrogen compared to those of oxygen, which enhances the conjugation between the lone pair of nitrogen and the π-orbitals of the benzene ring. Note that BN3, a triangulene analogue, showed relatively low ΦPL, probably due to the small oscillator strength (0.1138).140
Table 10 Photophysical properties of the compounds shown in Fig. 9 and 10
| Compound |
Condition |
λ
maxPL [nm] |
FWHM [nm] |
Φ
PL [—] |
ΔEST [eV] |
k
RISC [s−1] |
Ref. |
|
See Table 2 for the full compound name.
Solvent was not identified.
Calculated from the reported S1 energy in eV.
Same compound named differently.
Same compound named differently.
Same compound named differently.
Same compound named differently.
|
|
DABNA-1
|
CH2Cl2/EtOH/1 wt%-mCBPa |
462/458/460 |
33/36/30 |
0.89/0.84/0.88 |
—/0.15/0.18 |
—/—/9.9 × 103 |
38
|
|
DABNA-2
|
CH2Cl2/EtOH/1 wt%-mCBPa |
470/463/469 |
34/34/28 |
0.85/0.90/0.90 |
—/0.15/0.14 |
—/—/1.48 × 104 |
38
|
|
BN3
|
CH2Cl2/1 wt%-PMMAa |
403/399 |
—/26 |
0.362/0.54 |
—/0.21 |
—/— |
140
|
|
t-DABNA
|
Solutionb/DPEPOa |
443c/— |
—/— |
—/0.85 |
0.17/— |
—/2.44 × 103 |
141
|
|
t-DABNA
|
Toluene/SiCzCz:SiTrzCz2a |
456/— |
21/— |
0.993/— |
0.21/— |
—/2.7 × 103 |
102
|
|
BN-DMAC
|
Toluene/1 wt%-mCBPa |
485/489 |
29/39 |
—/0.63 |
0.14/— |
—/9.6 × 104 |
142
|
|
DMAc-BN
|
2-MeTHF/3 wt%-mCBPa |
484/— |
33/— |
—/0.88 |
0.16/— |
—/2.4 × 104 |
150
|
|
BN-DPAC
|
Toluene/1 wt%-mCBPa |
490/493 |
30/38 |
—/0.86 |
0.11/— |
—/1.26 × 105 |
142
|
|
2PXZBN
|
Toluene/1 wt%-mCBP/PO-T2Ta |
504/515 |
34/40 |
0.78/0.84 |
0.19/0.18 |
1.03 × 105/0.56 × 105 |
143
|
|
BN1
|
Toluene |
502 |
41 |
0.61 |
0.20 |
— |
144
|
|
PXZ-BN
|
2-MeTHF/3 wt%-mCBPa |
502/— |
38/— |
—/0.90 |
0.17/— |
—/0.9 × 104 |
150
|
|
2PTZBN
|
Toluene/1 wt%-mCBP/PO-T2Ta |
510/519 |
39/44 |
0.58/0.80 |
0.15/0.14 |
2.76 × 105/1.17 × 105 |
143
|
|
BN3
|
Toluene |
511 |
46 |
0.72 |
0.16 |
1.0 × 105 |
144
|
|
BN2
|
Toluene |
475 |
34 |
0.88 |
0.20 |
— |
144
|
|
BN4
|
Toluene/3 wt%-mCPCNa |
500/522 |
43/50 |
0.88/0.96 |
0.14/— |
1.6 × 105/3.7 × 104 |
144
|
|
BN5
|
Toluene/3 wt%-mCPCNa |
497/512 |
44/49 |
0.87/0.92 |
0.14/— |
7.4 × 104/3.3 × 104 |
144
|
|
helicene-BN
|
Toluene/1 wt%-DMIC-TRZa |
520/525 |
46/48 |
—/0.98 |
0.18/0.15 |
—/4.6 × 104 |
145
|
|
BNSSe
|
Toluene/1 wt%-DMIC-TRZa |
505/520 |
39/— |
—/0.99 |
0.17/0.12 |
—/6.0 × 105 |
104
|
|
BNSeSe
|
Toluene/1 wt%-DMIC-TRZa |
502/514 |
38/— |
—/1.00 |
0.17/0.14 |
—/2.0 × 106 |
104
|
|
TBE01
|
Toluene/0.4 wt%-SiCzCz:SiTrzCz2a |
459/— |
21/— |
0.911/— |
0.16/— |
—/5.1 × 103 |
102
|
|
TBE02
|
Toluene/0.4 wt%-SiCzCz:SiTrzCz2a |
459/— |
21/— |
0.891/— |
0.14/— |
—/1.03 × 104 |
102
|
|
TCZ-F-DABNA
|
Toluene/8 wt%-PhCzBCza |
558/— |
38/— |
—/0.99 |
0.12/— |
—/7.80 × 104 |
146
|
|
BNDIP
|
Toluene/1 wt%-DMIC-TRZa |
558/584 |
38/61 |
—/0.96 |
0.13/0.09 |
—/2.3 × 104 |
147
|
|
BNIP-tBuDPAC
|
Toluene/1 wt%-DMIC-TRZa |
544/557 |
43/53 |
—/0.98 |
0.14/0.11 |
—/1.2 × 104 |
147
|
|
Tp-DABNA
|
Toluene/2 wt%-mCBP:DPEPOa |
458/463 |
19/24 |
0.96/0.99 |
0.17/0.15 |
—/5.7 × 103 |
148
|
|
M-tDABNA
|
Toluene/3 wt%-MADNa |
—/461 |
—/27 |
—/0.77 |
0.11/— |
—/— |
88
|
|
TPXZBN
|
Toluene/5 wt%-mCBPa |
502/— |
33/— |
0.91/0.99 |
0.16/— |
—/4.8 × 104 |
149
|
|
DPXCZBN
|
Toluene/5 wt%-mCBPa |
500/— |
32/— |
0.90/0.94 |
0.13/— |
—/1.11 × 105 |
149
|
|
t-DABNA-dtB
|
Toluene/3 wt%-mCBPa |
465/473 |
22/30 |
—/0.97 |
0.19/— |
—/2.08 × 104 |
151
|
|
DTBA-BN2
|
Toluene/3 wt%-26DCzPPya |
490/496 |
41/— |
0.96/0.97 |
0.13/— |
2.3 × 105/4.8 × 105 |
152
|
Table 10 reveals some trends useful for designing DABNA-based emitters. BN-DMAC, DMAc-BN, BN-DPAC, and BN2 were constructed by inserting sp3-carbon into the DABNA skeleton.142,144,150 Compared to DABNA-1, the λmaxPL bands of these materials were red-shifted (462 nm to 475–490 nm) because of the extended π-conjugation. The helical structures induced by intramolecular locking were beneficial for increasing kRISC (9.9 × 103 s−1 to 2.4 × 104–1.26 × 105 s−1).
The analysis of TCZ-F-DABNA, BNDIP, and BNIP-tBuDPAC shows that additional nitrogen atoms at ortho-positions relative to the original nitrogen atoms (i.e., at meta-positions relative to boron atoms) can extend π-conjugation and increase the bathochromic shift in PL spectra to 550 nm.146,147 Although the kRISC values are generally the same as those of DOBNA derivatives (103–104 s−1), the heavy-atom effect can increase them to >1.0 × 105 s−1 when sulphur and/or selenium atoms are introduced, as exemplified by 2PTZBN, BN4, BNSSe, and BNSeSe.104,143,144 The more distorted structures due to the long C–S or C–Se bonds can also increase the SOC to accelerate RISC.
2.3 Carbazole-based MR-TADF emitters
2.3.1 One-pot borylation of carbazole-based precursors.
As summarised in this section, the synthesis of DOBNA and DABNA derivatives have inspired the preparation of numerous MR-TADF emitters. From a synthesis viewpoint, these two frameworks are complementary, with the oxygen atoms in DOBNA scaffolds inserted via an SNAr reaction and the nitrogen atoms in DABNA scaffolds inserted via Buchwald–Hartwig coupling. Consequently, different methods of introducing nitrogen atoms are worth investigating to expand the chemical space of MR-TADF emitters. More importantly, almost all DABNA derivatives were obtained via lithium–chlorine exchange under harsh conditions because of the limited availability of starting materials. Thus, a general procedure providing access to N,B,N-embedded frameworks via lithium–bromine exchange is desired to improve functional group tolerance.
In 2020, Wang et al. reported the first MR-TADF emitter synthesised via an SNAr reaction involving carbazolyl groups (Scheme 8).154 The bis(di(tert-butyl)carbazolyl)phenylene parent skeleton was synthesised in 80% yield starting from 2-bromo-1,3-difluorobenzene, with the subsequent basic one-pot borylation affording DtBuCzB (also known as BCzBN) in 26% yield. Similar to the case of the SNAr substitution-based DOBNA skeleton construction, the carbazolyl group successfully and selectively substituted a fluorine atom, while the bromine atom was preserved for the subsequent one-pot borylation. Therefore, a bromine group could be easily introduced into the one-pot borylation precursor because of the commercial availability of 1-bromo-2,6-difluorobenzene.
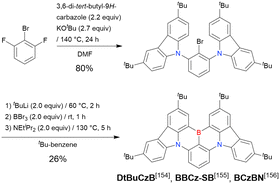 |
| | Scheme 8 Synthesis of a carbazole-based MR-TADF emitter. | |
Currently, the carbazole-based N,B,N-embedded framework (called CzBN in this review) is the one most widely used for the construction of MR-TADF emitters. The abundant methods for carbazolyl group pre-functionalisation allow numerous substructures to be attached to B,N,B-embedded frameworks (Fig. 11 and Table 11).155–165 In most cases, the para-positions of the carbazole nitrogen atoms were pre-functionalised and used in the SNAr-based one-pot borylation protocol. The highly functionalised carbazolyl subunits (e.g., those in BN-ICz-1, BN-ICz-2, and BN-DICz) were also completely constructed before being merged into the central phenyl ring. Even an aza[7]helicene subunit could be introduced into the MR-TADF framework (3-C2, 3-Cs, 4-C1).
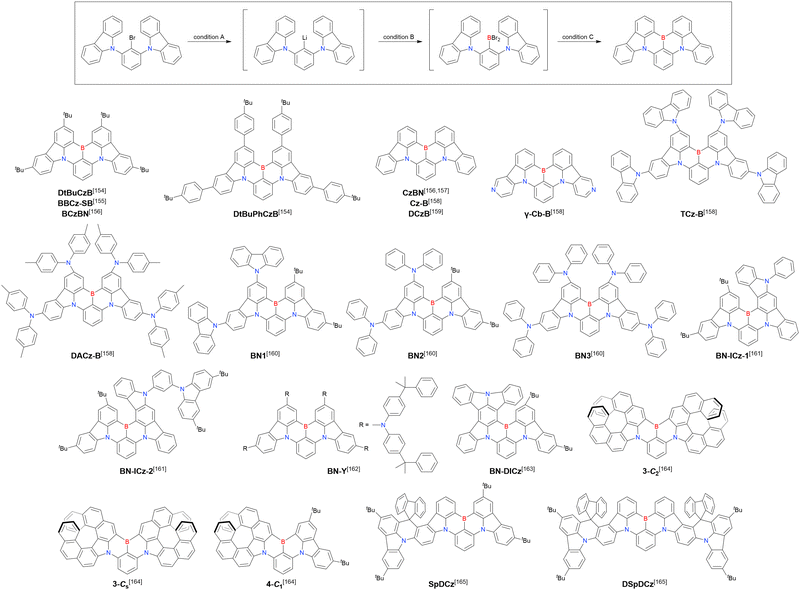 |
| | Fig. 11 Carbazole moiety-containing MR-TADF emitters. | |
Table 11 Conditions used to synthesise the compounds shown in Fig. 11
| Compound |
Condition Aa |
Condition Ba |
Condition Ca |
Solvent |
Yield [%] |
Ref. |
|
Numbers in parentheses refer to reagent loadings (equivalents).
|
|
DtBuCzB
|
t
BuLi (2.0)/60 °C, 2 h |
BBr3 (2.0)/rt, 1 h |
NEtiPr2 (2.0)/130 °C, 5 h |
t
Bu-Benzene |
26 |
154
|
|
n
BuLi (1.7)/rt, 4 h |
BBr3 (1.7)/rt, 12 h |
NEtiPr2 (2.7)/170 °C, 24 h |
t
Bu-Benzene |
24 |
155
|
|
n
BuLi (1.2)/rt, 2 h |
BBr3 (1.2)/rt, 0.5 h |
NEtiPr2 (2.0)/120 °C, 3 h |
t
Bu-Benzene |
47 |
156
|
|
DtBuPhCzB
|
t
BuLi (2.0)/60 °C, 2 h |
BBr3 (2.0)/rt, 1 h |
NEtiPr2 (2.0)/130 °C, 5 h |
t
Bu-Benzene |
17 |
154
|
|
CzBN
|
n
BuLi (1.2)/rt, 2 h |
BBr3 (1.2)/rt, 0.5 h |
NEtiPr2 (2.0)/120 °C, 3 h |
t
Bu-Benzene |
41 |
156
|
|
n
BuLi (1.2)/60 °C, 2 h |
BBr3 (1.2)/rt, 1 h |
NEtiPr2 (2.0)/110 °C, 8 h |
Toluene |
29 |
157
|
|
n
BuLi (1.5)/rt, 2 h |
BBr3 (1.5)/rt, 2 h |
NEtiPr2 (2.0)/110 °C, 24 h |
Toluene |
37 |
158
|
|
t
BuLi (2.0)/60 °C, 2 h |
BBr3 (2.0)/rt, 1 h |
NEtiPr2 (2.0)/130 °C, 6 h |
t
Bu-Benzene |
20 |
159
|
|
γ-Cb-B
|
n
BuLi (2.0)/rt, 2 h |
BBr3 (2.0)/rt, 2 h |
PMP (3.0)/170 °C, 24 h |
t
Bu-Benzene |
12 |
158
|
|
TCz-B
|
n
BuLi (1.1)/0 °C, 1 h |
BBr3 (1.2)/rt, 2 h |
NEtiPr2 (2.0)/140 °C, 18 h |
o-Xylene |
56 |
158
|
|
DACz-B
|
n
BuLi (1.1)/0 °C, 1 h |
BBr3 (1.2)/rt, 2 h |
NEtiPr2 (2.0)/170 °C, 32 h |
t
Bu-Benzene |
12 |
158
|
|
BN1
|
t
BuLi (2.0)/60 °C, 2 h |
BBr3 (2.0)/rt, 1 h |
NEtiPr2 (2.0)/130 °C, 5 h |
t
Bu-Benzene |
21 |
160
|
|
BN2
|
t
BuLi (2.0)/60 °C, 2 h |
BBr3 (2.0)/rt, 1 h |
NEtiPr2 (2.0)/130 °C, 5 h |
t
Bu-Benzene |
41 |
160
|
|
BN3
|
t
BuLi (2.0)/60 °C, 2 h |
BBr3 (2.0)/rt, 1 h |
NEtiPr2 (2.0)/130 °C, 5 h |
t
Bu-Benzene |
35 |
160
|
|
BN-ICz-1
|
n
BuLi (3.0)/60 °C, 24 h |
BBr3 (3.0)/rt, 0.5 h |
NEtiPr2 (4.5)/120 °C, 5 h |
t
Bu-Benzene |
23 |
161
|
|
BN-ICz-2
|
n
BuLi (3.0)/60 °C, 24 h |
BBr3 (3.0)/rt, 0.5 h |
NEtiPr2 (4.5)/120 °C, 5 h |
t
Bu-Benzene |
22 |
161
|
|
BN-Y
|
t
BuLi (2.0)/40 °C, 2 h |
BBr3 (1.0)/rt, 1 h |
NEtiPr2 (2.0)/120 °C, 8 h |
t
Bu-Benzene |
82 |
162
|
|
BN-DICz
|
n
BuLi (3.0)/60 °C, 24 h |
BBr3 (3.0)/rt, 0.5 h |
NEtiPr2 (4.5)/120 °C, 5 h |
t
Bu-Benzene |
22 |
163
|
|
3-C2/3-Cs
|
n
BuLi (2.5)/rt, 4 h |
BBr3 (3.0)/rt, 15 h |
NEtiPr2 (3.5)/175 °C, 24 h |
o-Dichlorobenzene |
18 |
164
|
|
4-C1
|
n
BuLi (2.3)/rt, 4 h |
BBr3 (3.0)/rt, 15 h |
NEtiPr2 (3.5)/175 °C, 24 h |
o-Dichlorobenzene |
19 |
164
|
|
SpDCz
|
n
BuLi (1.1)/40 °C, 1 h |
BBr3 (3.1)/50 °C, 3 h |
NEtiPr2 (3.1)/125 °C, 20 h |
m-Xylene |
65 |
165
|
|
DSpDCz
|
n
BuLi (1.1)/40 °C, 1 h |
BBr3 (3.1)/50 °C, 3 h |
NEtiPr2 (3.1)/125 °C, 20 h |
m-Xylene |
58 |
165
|
In addition to di-carbazolyl frameworks, a combination of carbazolyl and chalcogen groups was easily achieved using sequential nucleophilic substitutions followed by basic one-pot borylation (Fig. 12, Table 12).94,166–171 Oxygen-bridged (CzBO, BON-D0, BON-D1, BON-D2, CzBNO, and SOBN) and sulphur-bridged (CzBS and Cz-BSN) compounds were successfully prepared from 2-bromo-1,3-difluorobenzene. Additionally, selenium-bridged compounds were demonstrated, which was not addressed in previous subsections. The corresponding precursors were synthesised from 2-bromo-3-fluoroaniline using a Sandmeyer-type reaction (CzBSe)94 or from 2-bromo-1,3-difluorobenzene using an SNAr reaction (Cz-BSeN).169
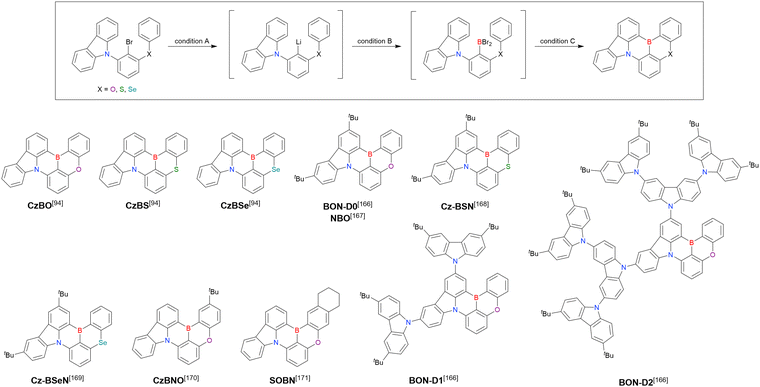 |
| | Fig. 12 MR-TADF emitters synthesised by sequential SNAr reactions. | |
Table 12 Conditions used to synthesise the compounds shown in Fig. 12
| Compound |
Condition Aa |
Condition Ba |
Condition Ca |
Solvent |
Yield [%] |
Ref. |
|
Numbers in parentheses refer to reagent loadings (equivalents).
|
|
CzBO
|
n
BuLi (1.1)/−30 °C, 1 h |
BBr3 (1.2)/rt, 2 h |
NEtiPr2 (2.0)/140 °C, 12 h |
t
Bu-Benzene |
28 |
94
|
|
CzBS
|
n
BuLi (1.1)/−30 °C, 1 h |
BBr3 (1.2)/rt, 2 h |
NEtiPr2 (2.0)/140 °C, 12 h |
t
Bu-Benzene |
36 |
94
|
|
CzBSe
|
n
BuLi (1.1)/−30 °C, 1 h |
BBr3 (1.2)/rt, 2 h |
NEtiPr2 (2.0)/140 °C, 12 h |
t
Bu-Benzene |
32 |
94
|
|
BON-D0
|
n
BuLi (1.1)/60 °C, 2 h |
BBr3 (1.2)/rt, 0.5 h |
NEtiPr2 (2.0)/120 °C, 5 h |
m-Xylene |
31 |
166
|
|
n
BuLi (1.1)/60 °C, 2 h |
BBr3 (1.2)/rt, 0.5 h |
NEtiPr2 (2.0)/110 °C, overnight |
Toluene |
17 |
167
|
|
Cz-BSN
|
n
BuLi (1.1)/60 °C, 2 h |
BBr3 (1.2)/rt, 1 h |
NEtiPr2 (2.0)/130 °C, 24 h |
o-Xylene |
20 |
168
|
|
Cz-BSeN
|
n
BuLi (1.1)/50 °C, 2 h |
BBr3 (1.2)/−30 °C, 0.5 h |
NEtiPr2 (2.0)/130 °C, 48 h |
o-Xylene |
15 |
169
|
|
CzBNO
|
t
BuLi (2.0)/60 °C, 2 h |
BBr3 (2.0)/rt, 1 h |
NEtiPr2 (2.0)/140 °C, 24 h |
t
Bu-Benzene |
32 |
170
|
|
SOBN
|
n
BuLi (4.0)/90 °C, 2 h |
BBr3 (4.0)/rt, 1 h |
NEtiPr2 (11)/120 °C, 12 h |
t
Bu-Benzene |
20 |
171
|
|
BON-D1
|
n
BuLi (1.0)/60 °C, 2 h |
BBr3 (1.2)/rt, 0.5 h |
NEtiPr2 (2.0)/120 °C, 5 h |
m-Xylene |
23 |
166
|
|
BON-D2
|
n
BuLi (1.0)/60 °C, 2 h |
BBr3 (1.2)/rt, 0.5 h |
NEtiPr2 (2.0)/120 °C, 5 h |
m-Xylene |
22 |
166
|
Carbazole-based MR-TADF emitters derived from tetrasubstituted benzene can be accessed in a similar manner as DOBNA- and DABNA-based ones (Fig. 13, 14 and Tables 13, 14).172–187 Although a methoxy group was deprotected by BBr3 in the one-pot borylation procedure (Scheme 4), the methylthio (SMe) group was preserved under the same conditions (BN(p)SCH3, Fig. 13). Bromine-containing compounds suitable for subsequent chemical modification were also obtained (BNCz-Br and BNCz-Br-Cz2, Fig. 14).
At this point, one should mention some indicative results. In 2023, the synthesis of indole-fused MR-TADF frameworks was reported (Scheme 9).188 The first attempt to obtain the non-substituted indole derivative InBN failed in the borylation part, probably because the reactive β-position of the indole unit inhibited the desired reaction during the electrophilic borylation step. Hence, methyl-protected precursors were prepared and smoothly converted into indole-based MR-TADF emitters (TMInBN, Mes-InBN, Cz-InBN, and TCz-InBN) with yields of 8–12%.
 |
| | Fig. 13 Carbazole-based MR-TADF emitters synthesised from tetrasubstituted benzene (1). | |
 |
| | Fig. 14 Carbazole-based MR-TADF emitters synthesised from tetrasubstituted benzene (2). | |
Table 13 Conditions used to synthesise the compounds shown in Fig. 13
| Compound |
Condition Aa |
Condition Ba |
Condition Ca |
Solvent |
Yield [%] |
Ref. |
|
Numbers in parentheses refer to reagent loadings (equivalents).
|
|
2F-BN
|
t
BuLi (3.0)/60 °C, 2 h |
BBr3 (3.0)/rt, 0.5 h |
NEtiPr2 (4.8)/120 °C, 5 h |
t
Bu-Benzene |
25 |
172
|
|
3F-BN
|
t
BuLi (3.0)/60 °C, 2 h |
BBr3 (3.0)/rt, 0.5 h |
NEtiPr2 (4.8)/120 °C, 5 h |
t
Bu-Benzene |
31 |
172
|
|
4F-BN
|
t
BuLi (3.0)/60 °C, 2 h |
BBr3 (3.0)/rt, 0.5 h |
NEtiPr2 (4.8)/120 °C, 5 h |
t
Bu-Benzene |
26 |
172
|
|
TW-BN
|
t
BuLi (2.0)/60 °C, 2 h |
BBr3 (2.0)/rt, 1 h |
NEtiPr2 (2.0)/130 °C, 6 h |
t
Bu-Benzene |
32 |
173
|
|
t
BuLi (2.0)/60 °C, 2 h |
BBr3 (2.0)/rt, 1 h |
NEtiPr2 (2.0)/130 °C, 5 h |
t
Bu-Benzene |
35 |
174
|
|
TPh-BN
|
t
BuLi (2.0)/60 °C, 2 h |
BBr3 (2.0)/rt, 1 h |
NEtiPr2 (2.0)/130 °C, 6 h |
t
Bu-Benzene |
25 |
173
|
|
pCz-BN
|
t
BuLi (2.0)/60 °C, 2 h |
BBr3 (2.0)/rt, 1 h |
NEtiPr2 (2.0)/130 °C, 6 h |
t
Bu-Benzene |
22 |
173
|
|
mCz-BN
|
t
BuLi (2.0)/60 °C, 2 h |
BBr3 (2.0)/rt, 1 h |
NEtiPr2 (2.0)/130 °C, 6 h |
t
Bu-Benzene |
20 |
173
|
|
TCzBN-DPF
|
t
BuLi (2.0)/60 °C, 2 h |
BBr3 (2.0)/rt, 1 h |
NEtiPr2 (2.0)/130 °C, 5 h |
t
Bu-Benzene |
34 |
174
|
|
TCzBN-oPh
|
t
BuLi (2.0)/60 °C, 2 h |
BBr3 (2.0)/rt, 1 h |
NEtiPr2 (2.0)/130 °C, 5 h |
t
Bu-Benzene |
38 |
174
|
|
BN-Ph-OH
|
n
BuLi (1.5)/70 °C, 2 h |
BBr3 (1.8)/rt, 0.5 h |
NEtiPr2 (2.5)/120 °C, 10 h |
t
Bu-Benzene |
20 |
175
|
|
BN-PhN(CH3)2
|
n
BuLi (1.5)/70 °C, 2 h |
BBr3 (1.8)/rt, 0.5 h |
NEtiPr2 (2.5)/120 °C, 10 h |
t
Bu-Benzene |
18 |
175
|
|
Tip-DtCzB
|
t
BuLi (2.0)/60 °C, 2 h |
BBr3 (2.0)/rt, 1 h |
NEtiPr2 (2.0)/130 °C, 6 h |
t
Bu-Benzene |
38 |
159
|
|
tDPA-DtCzB
|
t
BuLi (2.0)/60 °C, 2 h |
BBr3 (2.0)/rt, 1 h |
NEtiPr2 (2.0)/130 °C, 6 h |
t
Bu-Benzene |
25 |
159
|
|
BN-AC
|
t
BuLi (2.9)/90 °C, 2 h |
BBr3 (2.9)/rt, 2 h |
NEtiPr2 (8.0)/130 °C, 24 h |
t
Bu-Benzene |
44 |
176
|
|
BN-PXZ
|
t
BuLi (2.9)/90 °C, 2 h |
BBr3 (2.9)/rt, 2 h |
NEtiPr2 (8.0)/130 °C, 24 h |
t
Bu-Benzene |
59 |
176
|
|
BN-PZ
|
t
BuLi (2.9)/90 °C, 2 h |
BBr3 (2.9)/rt, 2 h |
NEtiPr2 (8.0)/130 °C, 24 h |
t
Bu-Benzene |
34 |
176
|
|
p-1-PCzBN
|
t
BuLi (4.0)/80 °C, 2 h |
BBr3 (4.5)/rt, 40 min |
NEtiPr2 (4.0)/125 °C, 17 h |
t
Bu-Benzene |
92 |
177
|
|
m-1-PCzBN
|
t
BuLi (4.0)/80 °C, 2 h |
BBr3 (4.5)/rt, 40 min |
NEtiPr2 (4.0)/125 °C, 17 h |
t
Bu-Benzene |
45 |
177
|
|
BN(p)SCH3
|
t
BuLi (3.0)/60 °C, 2 h |
BBr3 (3.0)/rt, 0.5 h |
NEtiPr2 (5.0)/120 °C, 5 h |
Mesitylene |
21 |
178
|
Table 14 Conditions used to synthesise the compounds shown in Fig. 14
| Compound |
Condition Aa |
Condition Ba |
Condition Ca |
Solvent |
Yield [%] |
Ref. |
|
Numbers in parentheses refer to reagent loadings (equivalents).
|
|
CzBNCz
|
n
BuLi (1.2)/60 °C, 2 h |
BBr3 (1.2)/rt, 2 h |
NEtiPr2 (2.4)/110 °C, 10 h |
Toluene |
34 |
157
|
|
TCz-BN
|
t
BuLi (3.0)/60 °C, 2 h |
BBr3 (3.0)/rt, 0.5 h |
NEtiPr2 (4.8)/120 °C, 5 h |
t
Bu-Benzene |
22 |
172
|
|
m-Cz-BNCz
|
t
BuLi (2.0)/60 °C, 2 h |
BBr3 (2.0)/rt, 1 h |
NEtiPr2 (2.0)/130 °C, 6 h |
t
Bu-Benzene |
15 |
179
|
|
CzBNNa
|
n
BuLi (1.2)/60 °C, 2 h |
BBr3 (1.5)/rt, 2 h |
NEtiPr2 (2.2)/110 °C, 10 h |
Toluene |
28 |
180
|
|
CzBNpyr
|
n
BuLi (1.2)/60 °C, 2 h |
BBr3 (1.5)/rt, 2 h |
NEtiPr2 (2.3)/110 °C, 10 h |
Xylene |
35 |
180
|
|
p-TBNCz
|
t
BuLi (2.0)/60 °C, 4 h |
BBr3 (2.0)/rt, 6 h |
NEtiPr2 (4.0)/120 °C, 24 h |
t
Bu-Benzene |
52 |
181
|
|
BNCz-Br
|
n
BuLi (1.1)/−0 °C to rt |
BBr3 (2.0)/rt, 0.5 h |
NEtiPr2 (2.0)/180 °C, 12 h |
Mesitylene |
74 |
182
|
|
n
BuLi (1.1)/60 °C, 2 h |
BBr3 (1.2)/rt, 0.5 h |
NEtiPr2 (2.0)/120 °C, 5 h |
m-Xylene |
55 |
183
|
|
n
BuLi (1.1)/−60 °C, 1 h |
BBr3 (1.5)/rt, 1 h |
NEtiPr2 (4.0)/120 °C, 8 h |
Toluene |
42 |
184
|
|
n
BuLi (1.1)/rt, 3 h |
BBr3 (1.5)/rt, 3 h |
NEtiPr2 (2.3)/160 °C, 24 h |
t
Bu-Benzene |
31 |
185
|
|
n
BuLi (1.1)/−78 °C, 3 h |
BBr3 (1.5)/rt, 1 h |
NEtiPr2 (4.0)/130 °C, 8 h |
Toluene |
20 |
186
|
|
n
BuLi (1.2)/rt, 1 h |
BBr3 (1.5)/rt, 1 h |
NEtiPr2 (4.0)/130 °C, 10 h |
Xylene |
16 |
187
|
|
BNCz-Br-Cz2
|
n
BuLi (1.1)/−60 °C, 1 h |
BBr3 (1.5)/rt, 1 h |
NEtiPr2 (4.0)/120 °C, 8 h |
Toluene |
45 |
184
|
 |
| | Scheme 9 Synthesis of indole-based MR-TADF emitters reported in ref. 188. | |
As described in Section 2.1.3, the synthesis of bromine-substituted MR-TADF skeletons is an intriguing topic. In particular, sulphur- and selenium-containing compounds are favourable for improving the TADF character. Br-Cz-BSN and Cz-BSeN-Br were successfully synthesised as heavy-atom-containing MR-type building blocks (Scheme 10).168,169 Similar to the synthesis of bromo-containing DOBNA derivatives (Section 2.1.3), the commercial availability of the starting material, 1,4-dibromo-2,6-difluorobenzene, and the regioselectivity of aryloxy group-directed lithium–halogen exchange make these compounds easily accessible. Given that the remaining bromo group facilitates subsequent chemical modification, these compounds can be versatile platforms for the synthesis of various types of functional emitters.
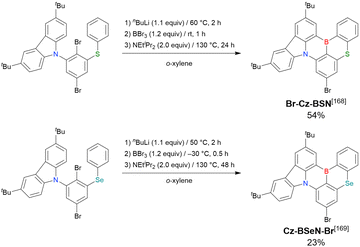 |
| | Scheme 10 Synthesis of sulphur- and selenium-containing MR-TADF emitters. | |
A one-pot multiple cyclisation method for synthesising highly substituted MR-TADF molecules via a combination of imino nitrogen-centred radical-based cyclisation and borylation–cyclisation was proposed in 2020 (Scheme 11).189 The cyano-substituted starting material was prepared by Suzuki–Miyaura coupling, and AZA-BN was obtained in 20% yield under optimal conditions (3.0 equiv. of nBuLi and 4.8 equiv. of BBr3). Lower reagent ratios resulted in failed boron introduction, which indicated that the addition of nBuLi to the CN group occurred prior to the substitution of the chlorine atom. In addition, other lithium reagents such as phenyllithium and tBuLi did not give the desired product.
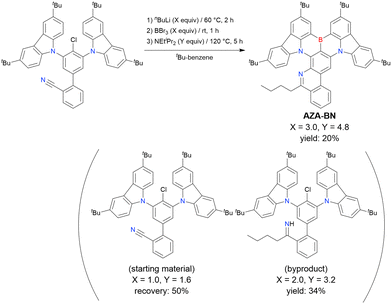 |
| | Scheme 11 Synthesis of aza-fused MR-TADF emitters reported in ref. 189. | |
Recently, a novel boron-bridged compound, DBCz-Mes, was synthesised from a dibrominated precursor (Scheme 12).91 Two boron atoms were simultaneously introduced using 2.4 equiv. of nBuLi and BBr3, and the reactive B–Br site remaining after intramolecular cyclisation was trapped by a mesityl Grignard reagent (MesMgBr) to afford DBCz-Mes in a total yield of 23%, which is as high as those of other general one-pot borylation protocols.
 |
| | Scheme 12 Synthesis of the boron-bridged compound reported in ref. 91. | |
Although tri- and tetrasubstituted MR-TADF emitters can be easily accessed from commercially available starting materials, the synthesis of penta- and hexa-substituted N,B,N-embedded frameworks require multi-step chemical modification towards borylation precursors (Scheme 13 and Table 15).155,190–194BBCz-Y, BBCz-G, and BN-2Cz were conveniently obtained through the simultaneous introduction of multiple carbazolyl groups. Highly cyclised compounds, NBNP, VTCzBN, TCz-VTCzBN, and PPZ-BN, were synthesised in a stepwise manner using intramolecular cyclisation. Finally, the synthesis of TRZCzPh-BNCz, TRZTPh-BNCz, and TPh-BNCz involved iodination after the introduction of carbazolyl groups via a substitution reaction. The final one-pot borylation step was achieved via directed ortho-lithiation or lithium–halogen exchange under standard conditions.
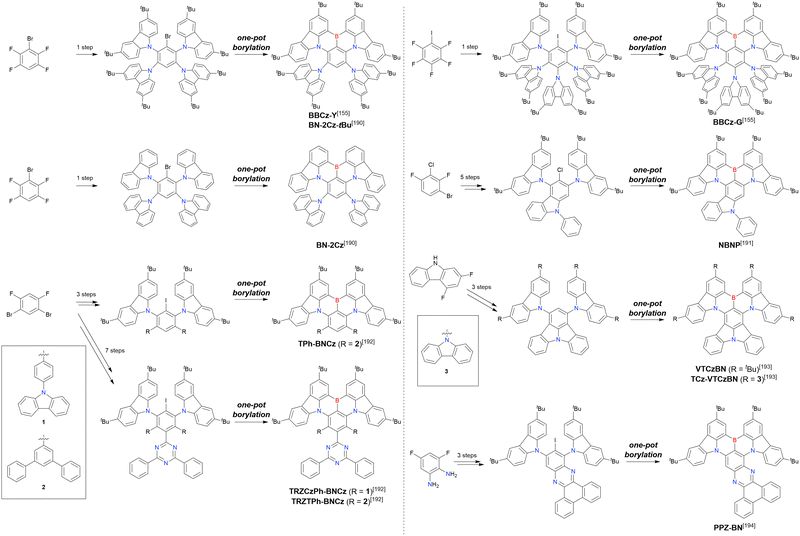 |
| | Scheme 13 Penta- and hexa-substituted MR-TADF emitters. | |
Table 15 Conditions used to synthesise the compounds shown in Scheme 13
| Compound |
Condition Aa |
Condition Ba |
Condition Ca |
Solvent |
Yield [%] |
Ref. |
|
Numbers in parentheses refer to reagent loadings (equivalents).
|
|
BBCz-Y
|
n
BuLi (1.5)/rt, 4 h |
BBr3 (1.5)/rt, 12 h |
NEtiPr2 (2.4)/170 °C, 24 h |
t
Bu-Benzene |
28 |
155
|
|
n
BuLi (2.1)/rt, 4 h |
BBr3 (2.1)/rt, 2 h |
NEtiPr2 (2.1)/160 °C, 6 h |
t
Bu-Benzene |
15 |
190
|
|
BBCz-G
|
n
BuLi (1.5)/rt, 4 h |
BBr3 (1.5)/rt, 12 h |
NEtiPr2 (2.4)/170 °C, 24 h |
t
Bu-Benzene |
23 |
155
|
|
BN-2Cz-tBu
|
n
BuLi (2.1)/rt, 4 h |
BBr3 (2.1)/rt, 2 h |
NEtiPr2 (2.1)/160 °C, 6 h |
t
Bu-Benzene |
17 |
190
|
|
NBNP
|
n
BuLi (3.0)/70 °C, 1 h |
BBr3 (4.0)/rt, 0.5 h |
NEtiPr2 (2.9)/130 °C, 20 h |
t
Bu-Benzene |
14 |
191
|
|
TPh-BNCz
|
n
BuLi (1.5)/rt, 1 h |
BBr3 (1.5)/rt, 1 h |
NEtiPr2 (4.0)/180 °C, 12 h |
o-Dichlorobenzene |
37 |
192
|
|
TRZCzPh-BNCz
|
n
BuLi (1.5)/rt, 1 h |
BBr3 (1.5)/rt, 1 h |
NEtiPr2 (4.0)/180 °C, 12 h |
o-Dichlorobenzene |
35 |
192
|
|
TRZTPh-BNCz
|
n
BuLi (1.5)/rt, 1 h |
BBr3 (1.5)/rt, 1 h |
NEtiPr2 (4.0)/180 °C, 12 h |
o-Dichlorobenzene |
43 |
192
|
|
VTCzBN
|
t
BuLi (4.0)/70 °C, 1 h |
BBr3 (4.4)/rt, 0.5 h |
NEtiPr2 (3.7)/130 °C, 24 h |
t
Bu-Benzene |
15 |
193
|
|
TCz-VTCzBN
|
t
BuLi (4.0)/70 °C, 1 h |
BBr3 (4.4)/rt, 0.5 h |
NEtiPr2 (3.7)/130 °C, 24 h |
t
Bu-Benzene |
10 |
193
|
|
PPZ-BN
|
n
BuLi (3.0)/rt, 1 h |
BBr3 (3.0)/rt, 5 h |
NEtiPr2 (4.5)/120 °C, 12 h |
Xylene |
21 |
194
|
2.3.2 Photophysical properties.
As summarised above, the construction of MR-TADF frameworks via SNAr reactions is currently the most common procedure because of their uncomplicated nature and the ready availability of the starting materials (1-bromo-2,6-difluorobenzene and its family). Additionally, coupling partners with carbazole- and chalcogen-containing substituents can also be endowed with diverse structures through pre-functionalisation. Therefore, the resulting MR-TADF emitters show diverse photophysical properties (Tables 16 and 17). As discussions of specific examples are outside the scope of this review, we briefly summarise the overall trends.
Table 16 Photophysical properties of the compounds shown in Fig. 11
| Compound |
Condition |
λ
maxPL [nm] |
FWHM [nm] |
Φ
PL [—] |
ΔEST [eV] |
k
RISC [s−1] |
Ref. |
|
Same compound named differently.
See Table 2 for the full compound name.
Same compound named differently.
|
|
DtBuCzB
|
Toluene/1 wt%-mCBPb |
481/493 |
22/35 |
0.91/0.88 |
0.13/— |
—/— |
154
|
|
BBCz-SB
|
Toluene/2 wt%-mCBPb |
489/490 |
23/25 |
0.98/0.98 |
0.15/— |
1.4 × 104/2.0 × 104 |
155
|
|
BCzBN
|
CH2Cl2/EtOH/toluene/neat film |
497/—/—/517·571 |
—/—/—/— |
—/—/0.863/— |
—/0.11/—/— |
—/—/1.48 × 104/— |
156
|
|
DtBuPhCzB
|
Toluene/1 wt%-mCBPb |
496/508 |
21/43 |
0.97/0.90 |
0.09/— |
—/— |
154
|
|
CzBN
|
CH2Cl2/EtOH/toluene/neat film |
479/—/—/502·570 |
—/—/—/— |
—/—/0.871/— |
—/0.12/—/— |
—/—/1.69 × 104/— |
156
|
|
CzBN
|
Toluene/1 wt%-mCBPb |
473/485 |
25/32 |
0.91/0.99 |
0.15/— |
—/1.24 × 104 |
157
|
|
Cz-B
|
Toluene/1 wt%-mCBPb |
477/484 |
25/30 |
0.98/0.97 |
0.14/— |
1.2 × 104/2.7 × 104 |
158
|
|
DCzB
|
Toluene/1 wt%-PhCzBCzb |
474/484 |
24/36 |
0.81/0.78 |
0.13/— |
—/5.0 × 103 |
159
|
|
γ-Cb-B
|
Toluene/1 wt%-oCBPb |
460/461 |
23/30 |
0.83/0.89 |
0.12/— |
3.1 × 104/5.8 × 104 |
158
|
|
TCz-B
|
Toluene/1 wt%-mCBPb |
512/517 |
27/31 |
1.00/0.89 |
0.09/— |
2.0 × 104/1.3 × 104 |
158
|
|
DACz-B
|
Toluene/1 wt%-mCBPb |
572/576 |
34/44 |
0.87/0.87 |
0.14/— |
1.8 × 104/1.0 × 104 |
158
|
|
BN1
|
Toluene/1 wt%-mCBPb |
496/499 |
23/38 |
0.99/0.93 |
0.11/— |
—/1.9 × 105 |
160
|
|
BN2
|
Toluene/1 wt%-mCBPb |
534/538 |
30/41 |
0.98/0.89 |
0.13/— |
—/1.5 × 105 |
160
|
|
BN3
|
Toluene/1 wt%-mCBPb |
562/563 |
30/44 |
0.98/0.86 |
0.09/— |
—/1.4 × 105 |
160
|
|
BN-ICz-1
|
Toluene/3 wt%-3CTF:mCBPb |
521/522 |
21/24 |
0.992/0.98 |
0.22/— |
2.9 × 104/— |
161
|
|
BN-ICz-2
|
Toluene/3 wt%-3CTF:mCBPb |
521/522 |
22/24 |
0.983/0.97 |
0.18/— |
6.4 × 104/— |
161
|
|
BN-Y
|
Toluene |
567 |
34 |
0.95 |
0.12 |
— |
162
|
|
BN-DICz
|
Toluene/3 wt%-mCBPb |
533/534 |
20/32 |
0.994/— |
0.26/— |
7.8 × 104/— |
163
|
|
3-C2
|
Toluene/2-MeTHF |
543/544 |
23/— |
0.72/— |
—/0.35 |
—/— |
164
|
|
3-Cs
|
Toluene/2-MeTHF |
546/546 |
23/— |
0.78/— |
—/0.35 |
—/— |
164
|
|
4-C1
|
Toluene/2-MeTHF |
520/523 |
28/— |
0.85/— |
—/0.32 |
—/— |
164
|
|
SpDCz
|
Toluene/1 wt%-mCPb |
498/498 |
26/29 |
—/0.97 |
0.14/— |
—/3.1 × 105 |
165
|
|
DSpDCz
|
Toluene/1 wt%-mCPb |
505/505 |
25/31 |
—/0.99 |
0.12/— |
—/3.0 × 105 |
165
|
| Compound |
Condition |
λ
maxPL [nm] |
FWHM [nm] |
Φ
PL [—] |
ΔEST [eV] |
k
RISC [s−1] |
Ref. |
|
See Table 2 for the full compound name.
Same compound named differently.
Same compound named differently.
Taken from the EL spectrum.
Same compound named differently.
|
|
CzBO
|
Toluene/1 wt%-mCBPa |
445/448 |
26/29 |
0.98/0.99 |
0.15/0.16 |
—/9.0 × 103 |
94
|
|
CzBS
|
Toluene/1 wt%-mCBPa |
471/472 |
28/30 |
0.99/0.98 |
0.11/0.14 |
4.0 × 105/2.2 × 105 |
94
|
|
CzBSe
|
Toluene/1 wt%-mCBPa |
477/479 |
33/34 |
0.98/0.98 |
0.12/0.15 |
1.5 × 108/1.8 × 108 |
94
|
|
BON-D0
|
Toluene/5 wt%-mCPa |
450/— |
23/— |
—/0.85 |
0.18/— |
—/6.73 × 104 |
166
|
|
NBO
|
Toluene/3 wt%-mCBPa |
448/458 |
25/45 |
—/0.99 |
—/0.103 |
—/1.2 × 104 |
167
|
|
BON-D1
|
Toluene/5 wt%-mCPa |
476/— |
28/— |
—/0.94 |
0.14/— |
—/7.72 × 104 |
166
|
|
BON-D2
|
Toluene/5 wt%-mCPa |
472/— |
31/— |
—/0.98 |
0.10/— |
—/9.81 × 104 |
166
|
|
Cz-BSN
|
Toluene/1 wt%-PSa |
476/— |
24/— |
—/0.82 |
0.13/— |
—/9.6 × 104 |
168
|
|
Cz-BSeN
|
Toluene/1 wt%-PSa |
479/— |
30/— |
—/0.87 |
0.15/— |
—/7.5 × 106 |
169
|
|
CzBNO
|
Toluene/3 wt%-26DCzPPya |
443/450 |
23/30 |
—/0.96 |
0.21/— |
—/3.47 × 104 |
170
|
|
SOBN
|
Toluene/5 wt%-26DCzPPya |
449/— |
22/— |
0.78/0.82 |
0.19/— |
—/1.4 × 104 |
171
|
|
2F-BN
|
Toluene/6 wt%-mCPBCa |
494/— |
24/— |
—/0.887 |
0.16/— |
—/2.2 × 104 |
172
|
|
3F-BN
|
Toluene/6 wt%-mCPBCa |
499/— |
24/— |
—/0.834 |
0.08/— |
—/3.9 × 104 |
172
|
|
4F-BN
|
Toluene/6 wt%-mCPBCa |
496/— |
25/— |
—/0.914 |
0.11/— |
—/4.4 × 104 |
172
|
|
TW-BN
|
Toluene/3 wt%-mCBPa |
486/485 |
20/25 |
0.86/0.92 |
0.12/— |
—/7.56 × 103 |
173
|
|
TCzBN-TMPh
|
Toluene/1 wt%-SF3TRZa |
486/487 |
23/25 |
—/0.94 |
0.12/— |
—/1.83 × 104 |
174
|
|
TPh-BN
|
Toluene/3 wt%-mCBPa |
492/495 |
21/28 |
0.91/0.94 |
0.09/— |
—/1.43 × 104 |
173
|
|
pCz-BN
|
Toluene/3 wt%-mCBPa |
491/496 |
21/30 |
0.90/0.95 |
0.15/— |
—/6.42 × 103 |
173
|
|
mCz-BN
|
Toluene/3 wt%-mCBPa |
495/494 |
21/29 |
0.85/0.88 |
0.14/— |
—/1.64 × 104 |
173
|
|
TCzBN-DPF
|
Toluene/1 wt%-SF3TRZa |
491/495 |
24/26 |
—/0.969 |
0.13/— |
—/3.92 × 104 |
174
|
|
TCzBN-oPh
|
Toluene/1 wt%-SF3TRZa |
489/491 |
23/24 |
—/0.961 |
0.14/— |
—/1.62 × 104 |
174
|
|
BN-Ph-OH
|
Toluene/3 wt%-mCBPa |
485/494 |
26/— |
—/0.80 |
0.14/— |
—/2.85 × 104 |
175
|
|
BN-PhN(CH3)2
|
Toluene/3 wt%-mCBPa |
486/500 |
24/— |
—/0.71 |
0.14/— |
—/8.10 × 104 |
175
|
|
Tip-DtCzB
|
Toluene/1 wt%-PhCzBCza |
477/486 |
19/29 |
0.96/0.95 |
0.13/— |
—/9.2 × 103 |
159
|
|
tDPA-DtCzB
|
Toluene/1 wt%-PhCzBCza |
470/484 |
21/30 |
0.86/0.85 |
0.11/— |
—/2.45 × 104 |
159
|
|
BN-AC
|
Toluene/15 wt%-mCBPa |
479/484 |
26/— |
—/0.94 |
0.14/— |
—/1.1 × 105 |
176
|
|
BN-PXZ
|
Toluene/15 wt%-mCBPa |
480/486 |
28/— |
—/0.97 |
0.12/— |
—/7.5 × 105 |
176
|
|
BN-PZ
|
Toluene/15 wt%-mCBPa |
634/610 |
82/— |
—/0.98 |
0.03/— |
—/1.85 × 106 |
176
|
|
p-1-PCzBN
|
Toluene/4 wt%-26DCzPPya |
489/495d |
25/26d |
—/0.92 |
0.14/— |
—/7.0 × 104 |
177
|
|
m-1-PCzBN
|
Toluene/4 wt%-26DCzPPya |
502/500d |
27/30d |
—/0.95 |
0.09/— |
—/6.3 × 104 |
177
|
|
CzBNCz
|
Toluene/1 wt%-mCBPa |
465/470 |
22/26 |
0.92/0.95 |
0.18/— |
—/7.6 × 103 |
157
|
|
TCz-BN
|
Toluene |
477 |
22 |
— |
— |
— |
172
|
|
m-Cz-BNCz
|
Toluene/solid/1 wt%-PhCzBCza |
519/521/520 |
38/39/47 |
0.97/—/0.92 |
0.08/—/— |
—/—/8.6 × 105 |
179
|
|
CzBNNa
|
Toluene/1 wt%-mCBPa |
483/487 |
25/29 |
0.78/0.98 |
0.15/— |
—/3.07 × 104 |
180
|
|
CzBNpyr
|
Toluene/1 wt%-mCBPa |
480/485 |
25/29 |
0.85/0.90 |
0.15/— |
—/— |
180
|
|
p-TBNCz
|
Toluene/1 wt%-PhCzBCza |
484/490 |
23/29 |
0.92/0.92 |
0.12/— |
—/1.4 × 104 |
181
|
|
TMInBN
|
Toluene/1 wt%-mCPBCa |
475/480d |
23/31d |
0.66/— |
0.39/— |
4.6 × 104/— |
188
|
|
Mes-InBN
|
Toluene/1 wt%-mCPBCa |
475/479d |
23/40d |
0.70/— |
0.40/— |
4.6 × 104/— |
188
|
|
Cz-InBN
|
Toluene/1 wt%-mCPBCa |
470/470d |
22/30d |
0.66/— |
0.41/— |
5.9 × 104/— |
188
|
|
TCz-InBN
|
Toluene/1 wt%-mCPBCa |
470/473d |
22/45d |
0.69/— |
0.42/— |
5.4 × 104/— |
188
|
|
AZA-BN
|
Toluene/4 wt%-mCBPa |
522/526 |
28/36 |
0.997/0.94 |
0.18/— |
—/7.53 × 103 |
189
|
|
DBCz-Mes
|
Toluene/1 wt%-mCBPa |
452/452 |
14/16 |
0.99/0.98 |
0.14/— |
2.9 × 104/— |
91
|
|
BBCz-Y
|
Toluene/2 wt%-mCBPa |
549/549 |
42/48 |
0.85/0.90 |
0.14/— |
1.0 × 105/1.1 × 105 |
155
|
|
BN-2Cz-tBu
|
Toluene |
543 |
— |
0.84 |
— |
1.39 × 105 |
190
|
|
BBCz-G
|
Toluene/2 wt%-mCBPa |
517/519 |
34/50 |
0.90/0.99 |
0.14/— |
1.8 × 105/1.9 × 105 |
155
|
|
BN-2Cz-tBu
|
Toluene |
522 |
— |
0.70 |
— |
1.83 × 105 |
190
|
|
NBNP
|
Toluene/4 wt%-mCBPa |
500/502d |
29/33d |
—/0.93 |
0.09/— |
—/3.0 × 105 |
191
|
|
TRZCzPh-BNCz
|
Toluene/3 wt%-CBPa |
514/516 |
34/37d |
0.930/0.976 |
0.13/— |
2.13 × 106/8.8 × 105 |
192
|
|
TRZTPh-BNCz
|
Toluene/3 wt%-CBPa |
513/516 |
29/33d |
0.947/0.990 |
0.11/— |
1.55 × 106/7.5 × 105 |
192
|
|
TPh-BNCz
|
Toluene |
509 |
33 |
— |
— |
— |
192
|
|
VTCzBN
|
Toluene/4 wt%-26DCzPPya |
496/— |
34/— |
—/0.98 |
0.06/— |
—/1.0 × 106 |
193
|
|
TCz-VTCzBN
|
Toluene/4 wt%-26DCzPPya |
521/— |
29/— |
—/0.98 |
<0.01/— |
—/0.9 × 106 |
193
|
|
PPZ-BN
|
Toluene/0.5 wt%-CBPa |
613/619 |
48/59 |
0.88/0.82 |
0.25/— |
2.2 × 104/— |
194
|
First, compared to that of DABNA derivatives, λmaxPL shifts to longer wavelengths. For example, the simplest carbazole-based MR-TADF compound, CzBN, exhibits a PL maximum at 479 nm in CH2Cl2 solution, which is red-shifted by 17 nm relative to that of DABNA-1.156 This change is attributed to the extension of π-conjugation and the increase in the ICT localisation area for a more efficient MR effect. Overall, simple carbazole-based MR-TADF emitters cover the PL colour range of sky-blue, green, and yellow, with blue emission remaining difficult to achieve, except for examples utilising pyridine units (γ-CB-B158) or oxygen bridges (CzBO,94BON-D0,166 and CzBNO170). The carbazole unit is more rigid and planar than carbon-, oxygen-, sulphur-, and selenium-bridged diarylamino units, which are included in some compounds listed in Fig. 9 and 10. This is beneficial for obtaining small FWHMs (generally 20–30 nm) in the solution state. On the other hand, planarity often induces intermolecular interactions with the host material and causes PL spectrum broadening (10–20 nm). To overcome this drawback, peripheral bulky substituents have been introduced. For example, dicarbazole-based symmetric compounds, TCzBN-DPF,174TCzBN-oPh,174p-1-PCzBN,177m-1-PCzBN,177DBCz-Mes,91 and TRZCzPh-BNCz,192 successfully suppressed spectral broadening within 3 nm, even in host materials. In addition, the highly planar carbazole-based MR skeleton is disadvantageous for accelerating RISC (kRISC typically below 1.0 × 105 s−1). Thus, TAF and PSF OLEDs are often fabricated to achieve high EL performance.97–100,102
Second, electron-donating groups at the para-position of nitrogen (also the meta-position of boron) cause a drastic bathochromic shift in the PL spectra. Tables 16 and 17 list numerous examples of PL wavelength tuning; generally, λmaxPL can be increased to >500 nm by introducing one or more electron-donating groups. In some cases, PL wavelengths exceeding 550 nm (572 nm for DACz-B,158 562 nm for BN3,160 567 nm for BN-Y,162 and 613 nm for PPZ-BN194) were realised through peripheral functionalisation. The other photophysical parameters, namely FWHM, ΦPL, ΔEST, and kRISC, are comparable to those of DABNA-type MR-TADF emitters.
2.4 Other routes to MR-TADF emitters based on one-pot borylation
2.4.1 Combination of two complementary reactions.
Previous subsections overviewed the synthetic routes to DOBNA, DABNA, and CzBN frameworks. The merging of SNAr and Buchwald–Hartwig coupling reactions is another powerful route used to access other MR-TADF skeletons. In 2021, Yang et al. reported the sequential construction of a carbazole-triarylamine-fused compound, DPACzBN1, starting from 1-bromo-2-chloro-3-fluorobenzene (Scheme 14).195 The successive introduction of 9H-carbazole and diphenylamine moieties afforded the key intermediate in 49% yield (two steps). Although the following one-pot borylation required harsh conditions because of the sluggishness of lithium–chlorine exchange, the N,B,N-embedded skeleton, DPACzBN1, was formed in 13% yield.
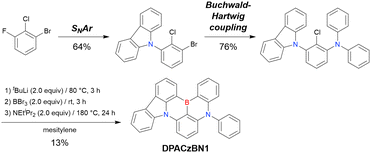 |
| | Scheme 14 Example of sequential MR-TADF precursor construction reported in ref. 195. | |
MR-TADF emitters synthesised by similar protocols are summarised in Fig. 15 and Table 18.103,147,170,195–199 As all compounds were derived from 1-bromo-2-chloro-3-fluorobenzene, the final borylation step was conducted under harsh conditions. The phenothiazine group could be introduced using both SNAr (PTZBN1)196 and Buchwald–Hartwig coupling (Cz-PTZ-BN)197,198 reactions.
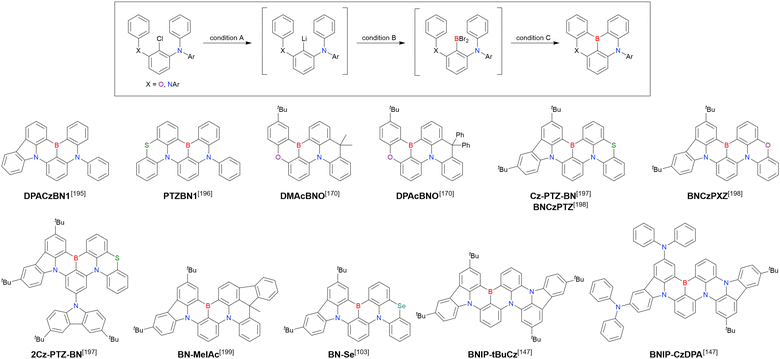 |
| | Fig. 15 MR-TADF emitters synthesised by sequential electrophilic aromatic substitution and Buchwald–Hartwig coupling reactions. | |
Table 18 Conditions used to synthesise the compounds shown in Fig. 15
| Compound |
Condition Aa |
Condition Ba |
Condition Ca |
Solvent |
Yield [%] |
Ref. |
|
Numbers in parentheses refer to reagent loadings (equivalents).
|
|
DPACzBN1
|
t
BuLi (2.0)/80 °C, 3 h |
BBr3 (2.0)/rt, 3 h |
NEtiPr2 (2.0)/180 °C, 24 h |
Mesitylene |
13 |
195
|
|
PTZBN1
|
t
BuLi (2.4)/60 °C, 2 h |
BBr3 (1.5)/rt, 1 h |
NEtiPr2 (2.0)/160 °C, 24 h |
Mesitylene |
25 |
196
|
|
DMAcBNO
|
t
BuLi (2.0)/60 °C, 2 h |
BBr3 (2.0)/rt, 1 h |
NEtiPr2 (2.0)/140 °C, 24 h |
t
Bu-Benzene |
25 |
170
|
|
DPAcBNO
|
t
BuLi (2.0)/60 °C, 2 h |
BBr3 (2.0)/rt, 1 h |
NEtiPr2 (2.0)/140 °C, 24 h |
t
Bu-Benzene |
32 |
170
|
|
Cz-PTZ-BN
|
t
BuLi (2.0)/60 °C, 2 h |
BBr3 (2.0)/rt, 1 h |
NEtiPr2 (2.0)/150 °C, 6 h |
t
Bu-Benzene |
22 |
197
|
|
t
BuLi (2.0)/60 °C, 2 h |
BBr3 (2.0)/rt, 1 h |
NEtiPr2 (2.0)/150 °C, 8 h |
t
Bu-Benzene |
20 |
198
|
|
BNCzPXZ
|
t
BuLi (2.0)/60 °C, 2 h |
BBr3 (2.0)/rt, 1 h |
NEtiPr2 (2.0)/150 °C, 8 h |
t
Bu-Benzene |
18 |
198
|
|
2Cz-PTZ-BN
|
t
BuLi (2.0)/60 °C, 2 h |
BBr3 (2.0)/rt, 1 h |
NEtiPr2 (2.0)/150 °C, 6 h |
t
Bu-Benzene |
16 |
197
|
|
BN-MelAc
|
t
BuLi (2.0)/60 °C, 2 h |
BBr3 (2.0)/rt, 1 h |
NEtiPr2 (2.0)/150 °C, 24 h |
Mesitylene |
35 |
199
|
|
BN-Se
|
t
BuLi (2.0)/60 °C, 3 h |
BBr3 (2.0)/rt, 1 h |
NEtiPr2 (2.0)/150 °C, 12 h |
t
Bu-Benzene |
34 |
103
|
|
BNIP-tBuCz
|
t
BuLi (2.2)/60 °C, 1 h |
BBr3 (1.5)/rt, 1 h |
NEtiPr2 (1.2)/130 °C, 24 h |
t
Bu-Benzene |
54 |
147
|
|
BNIP-CzDPA
|
t
BuLi (2.2)/60 °C, 1 h |
BBr3 (1.5)/rt, 1 h |
NEtiPr2 (1.2)/130 °C, 24 h |
t
Bu-Benzene |
40 |
147
|
2.4.2 Late-stage bromination.
The first DOBNA framework was synthesised via directed ortho-lithiation (see Section 2.1.1). Subsequently, borylation via lithium–halogen exchange became the most common one-pot borylation procedure. In this case, the halogen atom required for borylation is already contained in the starting material, and electron-rich substituents are sequentially introduced using the remaining key halogen atoms. As an alternative route, bromination just before borylation was attempted in some reports (Scheme 15 and Table 19).191,200,201 The ortho-position of the two carbazolyl groups was easily brominated by Br2 or N-bromosuccinimide (NBS), probably because of the directing effect of these electron-donating groups. In the synthesis of NBO and LTCz-BN, bromination and subsequent one-pot borylation were conducted without purification. Although the synthesis of tCzphB-ph and tCzphB-Fl consisted of two independent steps, the first bromination by NBS proceeded in an almost quantitative yield. This late-stage bromination procedure can facilitate the synthesis of complex structures, as it helps to avoid the use of difficult-to-obtain starting materials such as highly substituted benzenes.
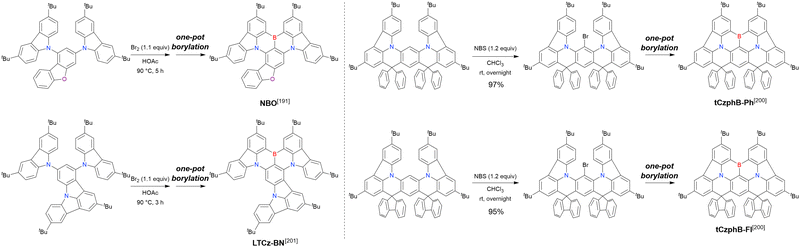 |
| | Scheme 15 MR-TADF emitters synthesised via late-stage bromination. | |
Table 19 Conditions used to synthesise the compounds shown in Scheme 15
| Compound |
Condition Aa |
Condition Ba |
Condition Ca |
Solvent |
Yield [%] |
Ref. |
|
Numbers in parentheses refer to reagent loadings (equivalents).
|
|
NBO
|
n
BuLi (3.0)/70 °C, 1 h |
BBr3 (4.0)/rt, 0.5 h |
NEtiPr2 (2.9)/130 °C, 18 h |
t
Bu-Benzene |
12 (2 steps) |
191
|
|
tCzphB-Ph
|
n
BuLi (1.5)/−40 °C, 2 h |
BBr3 (2.9)/rt, overnight |
NEtiPr2 (2.9)/130 °C, 4 h |
t
Bu-Benzene |
61 |
200
|
|
tCzphB-Fl
|
n
BuLi (1.5)/−40 °C, 2 h |
BBr3 (2.9)/rt, overnight |
NEtiPr2 (2.9)/130 °C, 4 h |
t
Bu-Benzene |
55 |
200
|
|
LTCz-BN
|
t
BuLi (4.0)/70 °C, 1 h |
BBr3 (4.2)/rt, 0.5 h |
NEtiPr2 (3.2)/130 °C, 24 h |
t
Bu-Benzene |
10 (2 steps) |
201
|
2.4.3 Stepwise borylation and cyclisation.
MR-TADF frameworks are generally constructed by one-pot methods with BBr3 as a boron source. In some reports, an alternative stepwise procedure was applied (Fig. 16 and Table 20).202–204 In this procedure, the key intermediate possessing a boronic acid pinacol ester (Bpin) group is synthesised in high or moderate yield by the sequential addition of nBuLi and 2-isopropoxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (iPrOBpin) to a bromine-containing precursor, with the subsequent intramolecular bora-Friedel–Crafts reaction affording fully cyclised target compounds.
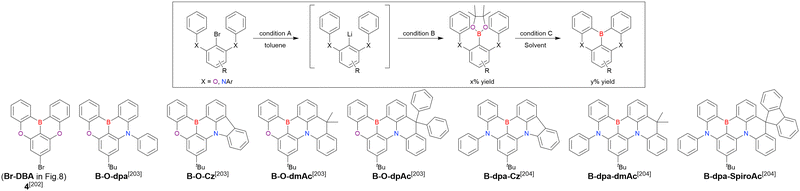 |
| | Fig. 16 MR-TADF emitters synthesised by stepwise borylation–cyclisation. | |
Table 20 Conditions used to synthesise the compounds shown in Fig. 16
| Compound |
Condition Aa |
Condition Ba |
x [%] |
Condition Ca |
Solvent |
y [%] |
Ref. |
|
Numbers in parentheses refer to reagent loadings (equivalents).
|
|
Br-DBA
|
n
BuLi (—)/rt, 2 h |
i
PrOBpin (1.2)/rt, 2 h |
— |
AlCl3 (4.8)/100 °C, 3 h |
Toluene |
37 (2 steps) |
202
|
|
B-O-dpa
|
n
BuLi (1.2)/−20 °C, 1 h |
i
PrOBpin (3.0)/rt, 1 h |
65 |
AlCl3 (4.0), NEtiPr2 (5.0)/110 °C, 2 h |
Chlorobenzene |
43 |
203
|
|
B-O-Cz
|
n
BuLi (1.5)/−20 °C, 1 h |
i
PrOBpin (3.9)/rt, 1 h |
76 |
AlCl3 (4.0), NEtiPr2 (5.0)/110 °C, 2 h |
Chlorobenzene |
47 |
203
|
|
B-O-dmAc
|
n
BuLi (2.5)/−20 °C, 1 h |
i
PrOBpin (4.5)/rt, 1 h |
49 |
AlCl3 (4.0), NEtiPr2 (5.0)/110 °C, 2 h |
Chlorobenzene |
36 |
203
|
|
B-O-dpAc
|
n
BuLi (2.5)/−20 °C, 1 h |
i
PrOBpin (5.0)/rt, 1 h |
— |
AlCl3 (6.0), NEtiPr2 (2.4)/110 °C, 2 h |
Chlorobenzene |
33 (2 steps) |
203
|
|
B-dpa-Cz
|
n
BuLi (2.0)/−20 °C, 1 h |
i
PrOBpin (4.0)/rt, 1 h |
65 |
AlCl3 (5.0), NEtiPr2 (2.0)/110 °C, 2 h |
Chlorobenzene |
57 |
204
|
|
B-dpa-dmAc
|
n
BuLi (2.5)/−20 °C, 1 h |
i
PrOBpin (4.5)/rt, 1 h |
84 |
AlCl3 (3.0), NEtiPr2 (1.0)/110 °C, 2 h |
Toluene |
25 |
204
|
|
B-dpa-SpiroAc
|
n
BuLi (2.8)/−20 °C, 1 h |
i
PrOBpin (5.0)/rt, 1 h |
44 |
AlCl3 (5.0), NEtiPr2 (2.0)/110 °C, 2 h |
Toluene |
50 |
204
|
The intermediate can be either isolated (B-O-dpa, B-O-Cz, B-O-dmAc, B-dpa-Cz, B-dpa-dmAc, and B-dpa-SpiroAc) or used without purification (Br-DBA and B-O-dpAc). Notably, Br-DBA was also synthesised by the basic one-pot borylation method (Fig. 8 and Table 6) with a comparable total yield (∼40%).120,124
2.4.4 Photophysical properties.
The photophysical properties of the compounds discussed in Section 2.4 are summarised in Table 21. Although a wide variety of structures are included, their properties are in line with previous discussions in Sections 2.1.4, 2.2.2, and 2.3.2.
| Compound |
Condition |
λ
maxPL [nm] |
FWHM [nm] |
Φ
PL [—] |
ΔEST [eV] |
k
RISC [s−1] |
Ref. |
|
See Table 2 for the full compound name.
Same compound named differently.
|
|
DPACzBN1
|
Toluene/3 wt%-26DCzPPya |
469/479 |
23/31 |
—/0.98 |
—/0.11 |
—/1.2 × 104 |
195
|
|
PTZBN1
|
Toluene/2 wt%-26DCzPPya |
490/497 |
41/44 |
—/0.98 |
—/0.16 |
—/1.11 × 105 |
196
|
|
DMAcBNO
|
Toluene/3 wt%-26DCzPPya |
462/470 |
34/41 |
—/0.99 |
0.23/— |
—/1.40 × 104 |
170
|
|
DPAcBNO
|
Toluene/3 wt%-26DCzPPya |
462/468 |
33/39 |
—/0.98 |
0.19/— |
—/1.75 × 104 |
170
|
|
Cz-PTZ-BN
|
Toluene/3 wt%-PhCzBCza |
510/524 |
37/50 |
0.84/0.91 |
0.11/— |
—/8.1 × 104 |
197
|
|
BNCzPTZ
|
Toluene/15 wt%-PhCzBCza |
509/— |
42/— |
0.84/0.91 |
0.09/— |
—/1.54 × 105 |
198
|
|
BNCzPXZ
|
Toluene/7 wt%-PhCzBCza |
508/— |
36/— |
0.88/0.94 |
0.13/— |
—/2.31 × 104 |
198
|
|
2Cz-PTZ-BN
|
Toluene/3 wt%-PhCzBCza |
505/512 |
38/48 |
0.87/0.96 |
0.09/— |
—/1.05 × 105 |
197
|
|
BN-MelAc
|
Toluene/2-MeTHF/1 wt%-DMIC-TRZa |
497/—/503 |
30/30/33 |
—/—/0.96 |
—/0.11/— |
—/—/6.3 × 104 |
199
|
|
BN-Se
|
Toluene/1 wt%-DMIC-TRZa |
502/505 |
42/44 |
0.99/0.98 |
0.08/— |
1.6 × 106/1.2 × 106 |
103
|
|
BNIP-tBuCz
|
Toluene/1 wt%-DMIC-TRZa |
564/570 |
60/61 |
—/0.96 |
0.08/0.13 |
—/0.9 × 104 |
147
|
|
BNIP-CzDPA
|
Toluene/1 wt%-DMIC-TRZa |
583/585 |
49/57 |
—/0.95 |
0.06/0.12 |
—/0.9 × 104 |
147
|
|
NBO
|
Toluene/4 wt%-mCBPa |
487/— |
27/— |
—/0.92 |
0.12/— |
—/9.3 × 105 |
191
|
|
tCzphB-Ph
|
Toluene/2 wt%-TPSSa |
523/527 |
21/23 |
—/0.98 |
—/0.04 |
—/— |
200
|
|
tCzphB-Fl
|
Toluene/2 wt%-TPSSa |
531/535 |
21/25 |
—/0.93 |
—/0.04 |
—/— |
200
|
|
LTCz-BN
|
Toluene/5 wt%-mCBPa |
497/— |
27/— |
—/0.93 |
0.10/— |
—/8.3 × 105 |
201
|
|
B-O-dpa
|
Toluene/10 wt%-DPEPOa |
433/— |
28/— |
—/0.86 |
0.18/— |
—/0.83 × 104 |
203
|
|
B-O-Cz
|
Toluene/10 wt%-DPEPOa |
441/— |
27/— |
—/0.94 |
0.15/— |
—/4.02 × 104 |
203
|
|
B-O-dmAc
|
Toluene/10 wt%-DPEPOa |
461/— |
38/— |
—/0.91 |
0.11/— |
—/1.82 × 104 |
203
|
|
B-O-dpAc
|
Toluene/10 wt%-DPEPOa |
463/— |
38/— |
—/0.94 |
0.06/— |
—/3.12 × 104 |
203
|
|
B-dpa-Cz
|
Toluene/3 wt%-mCPBa |
466/469 |
27/33 |
—/0.94 |
0.15/— |
—/2.74 × 104 |
204
|
|
B-dpa-dmAc
|
Toluene/3 wt%-mCPBa |
472/476 |
31/38 |
—/0.98 |
0.16/— |
—/4.48 × 104 |
204
|
|
B-dpa-SpiroAc
|
Toluene/3 wt%-mCPBa |
472/476 |
31/38 |
—/0.92 |
0.14/— |
—/4.97 × 104 |
204
|
2.5 Simultaneous introduction of two boron atoms
2.5.1 One nitrogen and two boron atoms.
In 2019, Hatakeyama et al. developed a boron-enriched MR-TADF material, azadiboranaphthoanthracene (ADBNA), which contains two boron atoms and one nitrogen atom and corresponds to DABNA-1 inverted through the interchange of boron and nitrogen positions (Fig. 17a).205 The one-pot borylation of the tribrominated starting material afforded an intermediate possessing two B–Br bonds, which was then reacted with the desired Grignard reagents to afford ADBNA derivatives. One year later, Wang et al. reported a different route to synthesise ADBNA-based frameworks (Fig. 17b).206 The key intermediate, Pre-ADBNA, was prepared in 65% yield by treating the lithiated starting material with dimethyl mesitylboronate (MesB(OMe)2) (2.0 equiv.) and subsequently reacted with BBr3 and ArLi to afford cyclised B,N,B-embedded compounds. Notably, both symmetric and non-symmetric products were obtained by controlling the reagent amounts. When 1.0 equiv. of BBr3 and 1.2 equiv. of ArLi were used, the B–Ar group was introduced only at the acyclic site of Pre-ADBNA. However, treatment with 5.0 equiv. of BBr3 for a longer time and the use of 2.4 equiv. of ArLi resulted in the replacement of the remaining B-Mes site of Pre-ADBNA with an additional B–Ar group.
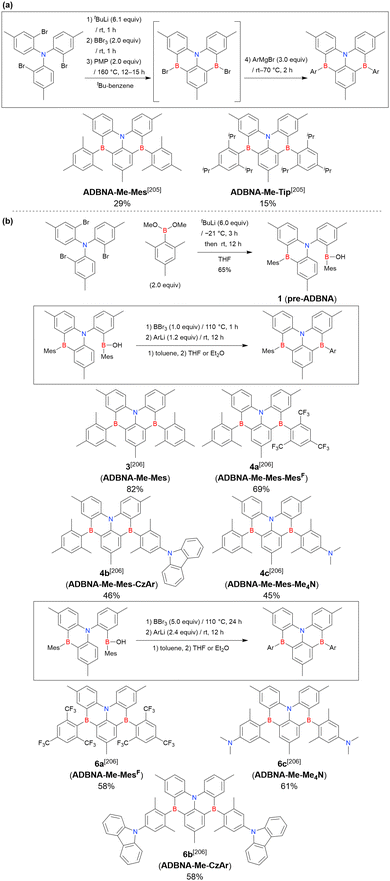 |
| | Fig. 17 Synthesis of boron-bridged compounds reported in ref. 205 and 206. | |
2.5.2 One-pot double borylation.
Finally, the simultaneous introduction of two fully cyclised boron atoms using the basic one-pot borylation procedure are discussed. In 2020, Yasuda et al. reported the synthesis of a doubly borylated compound, BBCz-R (Scheme 16), starting from 1-bromo-2,6-difluorobenzene.155 One equivalent of 3,6-di-tert-butylcarbazole was introduced by the standard SNAr reaction. Subsequently, a similar type of reaction with 2,8-di-tert-butyl-5,11-dihydroindolo[3,2-b]carbazole was conducted at the remaining fluorine group to form the dibrominated precursor. The final one-pot borylation step did not proceed regioselectively, i.e., a regioisomer (BBCz-Y-II, 7% yield) was obtained in addition to the desired BBCz-R (5% yield).
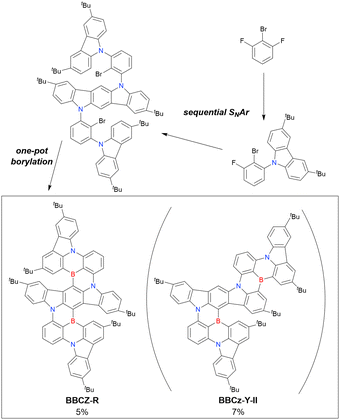 |
| | Scheme 16 Synthesis of doubly borylated compound from 1-bromo-2,6-difluorobenzene reported in ref. 155. | |
The procedure proposed by Yasuda et al. is now one of the most common strategies for one-pot double borylation (Fig. 18 and Table 22).117,155,163,167,207,208 The sequential SNAr reactions of 2-bromo-1,6-difluorobenzene (first mono-substitution, then dimerisation) afforded diverse doubly borylated compounds. The data presented in ref. 207 (SBON and SBSN) suggest that protonation after lithium–bromine exchange is a common side reaction and can affect the yield of the final product. The possible proton sources are residual water in the solvents, HBr contained in BBr3, protons generated during C–H borylation, and so on.
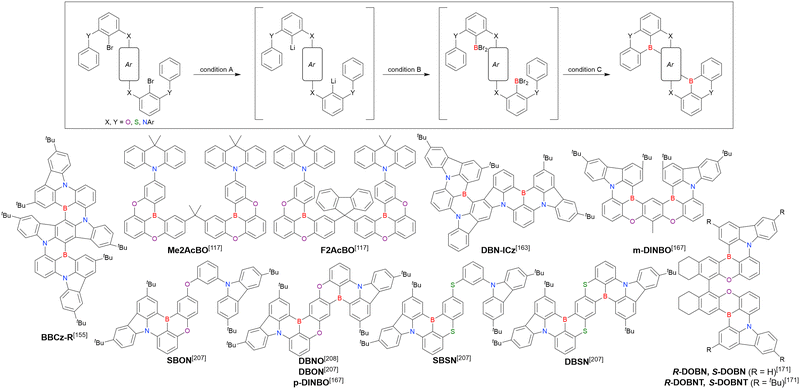 |
| | Fig. 18 MR-TADF emitters synthesised by one-pot double borylation. | |
Table 22 Conditions used to synthesise the compounds shown in Fig. 18
| Compound |
Condition Aa |
Condition Ba |
Condition Ca |
Solvent |
Yield [%] |
Ref. |
|
Numbers in parentheses refer to reagent loadings (equivalents).
|
|
BBCz-R
|
n
BuLi (3.0)/rt, 4 h |
BBr3 (3.0)/rt, 12 h |
NEtiPr2 (4.6)/170 °C, 24 h |
t
Bu-Benzene |
5 |
155
|
|
Me2AcBO
|
t
BuLi (4.0)/rt, 2 h |
BBr3 (—)/rt, 2 h |
NEtiPr2 (—)/180 °C, 24 h |
t
Bu-Benzene |
8 |
117
|
|
F2AcBO
|
t
BuLi (4.0)/rt, 2 h |
BBr3 (—)/rt, 2 h |
NEtiPr2 (—)/180 °C, 24 h |
t
Bu-Benzene |
16 |
117
|
|
DBN-ICz
|
n
BuLi (6.0)/60 °C, 24 h |
BBr3 (6.0)/rt, 0.5 h |
NEtiPr2 (9.0)/120 °C, 5 h |
t
Bu-Benzene |
36 |
163
|
|
m-DINBO
|
n
BuLi (2.3)/60 °C, 2 h |
BBr3 (3.1)/rt, 2 h |
NEtiPr2 (4.1)/170 °C, 24 h |
t
Bu-Benzene |
22 |
167
|
|
SBON
|
n
BuLi (3.9)/80 °C, 5 h |
BBr3 (7.8)/rt, 3 h |
NEtiPr2 (6.1)/190 °C, 16 h |
t
Bu-Benzene |
16 |
207
|
|
DBNO
|
t
BuLi (4.0)/0 °C, 6 h |
BBr3 (2.1)/rt, 2 h |
NEtiPr2 (5.0)/120 °C, 24 h |
t
Bu-Benzene |
16 |
208
|
|
n
BuLi (3.9)/80 °C, 5 h |
BBr3 (7.8)/rt, 3 h |
NEtiPr2 (6.1)/190 °C, 16 h |
t
Bu-Benzene |
10 |
207
|
|
n
BuLi (2.3)/60 °C, 2 h |
BBr3 (3.1)/rt, 2 h |
NEtiPr2 (4.1)/170 °C, 48 h |
t
Bu-Benzene |
2 |
167
|
|
SBSN
|
n
BuLi (4.0)/80 °C, 5 h |
BBr3 (8.1)/rt, 3 h |
NEtiPr2 (6.4)/190 °C, 16 h |
t
Bu-Benzene |
24 |
207
|
|
DBSN
|
n
BuLi (4.0)/80 °C, 5 h |
BBr3 (8.1)/rt, 3 h |
NEtiPr2 (6.4)/190 °C, 16 h |
t
Bu-Benzene |
12 |
207
|
|
S-DOBN
|
n
BuLi (4.0)/90 °C, 2 h |
BBr3 (4.0)/rt, 1 h |
NEtiPr2 (11)/120 °C, 12 h |
t
Bu-Benzene |
10 |
171
|
|
S-DOBNT
|
n
BuLi (4.0)/90 °C, 2 h |
BBr3 (4.0)/rt, 1 h |
NEtiPr2 (11)/120 °C, 12 h |
t
Bu-Benzene |
12 |
171
|
Scheme 17 presents another versatile route to synthesise doubly borylated MR-TADF emitters.209 This procedure starts from 1,4-dibromo-2,3,5,6-tetrafluorobenzene and affords the di-brominated precursor through multiple SNAr reactions. A hexa-substituted doubly N,B,N-embedded framework is then constructed by subsequent one-pot borylation. This protocol was adopted in some studies to prepare the desired MR-TADF frameworks (Fig. 19 and Table 23).210–214 In addition to tetra-carbazolyl substituted compounds (R-BN, R-TBN, R-TPhBN, and BNS), the combination of a carbazolyl group and chalcogen bridge (BNO1, BNO2, BNO3, DBNS, DBNS-tBu, and BNNO) was also achieved. As mentioned in the following subsection, these species possess para-positioned boron and electron-donating atoms, which is beneficial for obtaining long-wavelength fluorescence.
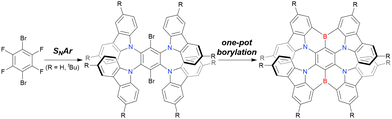 |
| | Scheme 17 Synthesis of a doubly borylated compound from 1,4-dibromo-2,3,5,6-tetrafluorobenzene. | |
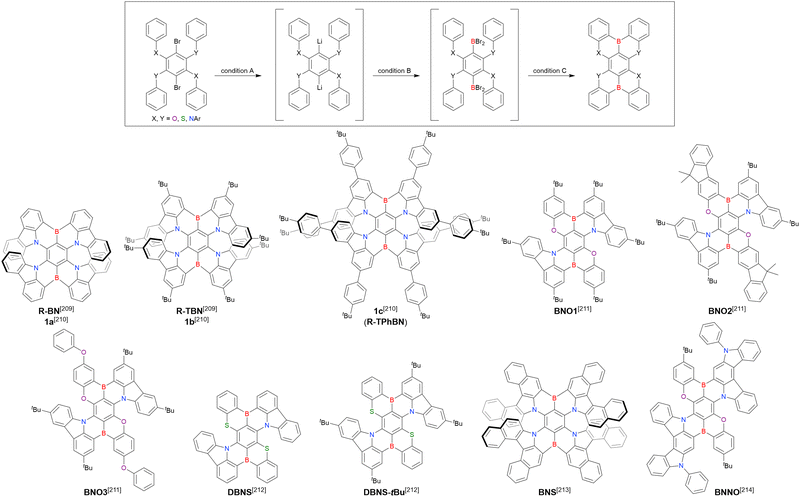 |
| | Fig. 19 Double-borylated MR-TADF emitters synthesised from hexa-substituted benzene. | |
Table 23 Conditions used to synthesise the compounds shown in Fig. 19
| Compound |
Condition Aa |
Condition Ba |
Condition Ca |
Solvent |
Yield [%] |
Ref. |
|
Numbers in parentheses refer to reagent loadings (equivalents).
|
|
R-BN
|
n
BuLi (6.1)/60 °C, 24 h |
BBr3 (12)/rt, 0.5 h |
NEtiPr2 (18)/120 °C, 5 h |
t
Bu-Benzene |
38 |
209
|
|
n
BuLi (4.0)/rt, 12 h |
BBr3 (4.0)/rt, 24 h |
NEtiPr2 (4.0)/180 °C, 72 h |
o-Dichlorobenzene |
53 |
210
|
|
R-TBN
|
n
BuLi (6.1)/60 °C, 24 h |
BBr3 (12)/rt, 0.5 h |
NEtiPr2 (18)/120 °C, 5 h |
t
Bu-Benzene |
40 |
209
|
|
n
BuLi (4.0)/rt, 12 h |
BBr3 (4.0)/rt, 24 h |
NEtiPr2 (4.0)/180 °C, 72 h |
o-Dichlorobenzene |
25 |
210
|
|
R-TPhBN
|
n
BuLi (4.0)/rt, 12 h |
BBr3 (4.0)/rt, 24 h |
NEtiPr2 (4.0)/180 °C, 72 h |
o-Dichlorobenzene |
23 |
210
|
|
BNO1
|
n
BuLi (—)/60 °C, 8 h |
BBr3 (—)/rt, 1 h |
NEtiPr2 (—)/170 °C, 24 h |
Mesitylene |
34 |
211
|
|
BNO2
|
n
BuLi (—)/60 °C, 8 h |
BBr3 (—)/rt, 1 h |
NEtiPr2 (—)/170 °C, 24 h |
Mesitylene |
38 |
211
|
|
BNO3
|
n
BuLi (—)/60 °C, 8 h |
BBr3 (—)/rt, 1 h |
NEtiPr2 (—)/170 °C, 24 h |
Mesitylene |
31 |
211
|
|
DBNS
|
n
BuLi (2.5)/rt, 4 h |
BBr3 (2.5)/rt, 2 h |
NEtiPr2 (3.9)/170 °C, 24 h |
t
Bu-Benzene |
30 |
212
|
|
DBNS-tBu
|
n
BuLi (2.5)/rt, 4 h |
BBr3 (2.5)/rt, 2 h |
NEtiPr2 (3.5)/170 °C, 24 h |
t
Bu-Benzene |
16 |
212
|
|
BNS
|
n
BuLi (4.0)/60 °C, 2 h |
BBr3 (4.0)/rt, 6 h |
NEtiPr2 (4.0)/180 °C, 48 h |
o-Dichlorobenzene |
22 |
213
|
|
BNNO
|
n
BuLi (6.0)/60 °C, 2 h |
BBr3 (6.0)/rt, 0.5 h |
NEtiPr2 (10)/135 °C, 8 h |
Xylene |
30 |
214
|
Other synthesis routes for doubly borylated compounds are summarised in Fig. 20 and Table 24.92,155,181,215,216 Overall, the reaction yields are lower than those observed for single borylation. Regioselectivity in electrophilic C–H borylation should be considered because poor selectivity causes the formation of undesired isomers after one-pot borylation. For example, the precursors of m-DBCz and DBTN-2 have similar chemical structures except for tBu groups on the central two benzene rings, which results in different regioselectivity in one-pot borylation. Although double lithiation at the same benzene ring is a good way to avoid the regioselectivity problem, the reaction yields remain low (BBCz-DB,155BSBS-N1,92BOBO-Z,215BOBS-Z,215 and BSBS-Z215). Moreover, the simultaneous introduction of three or more boron atoms has not been achieved to date.
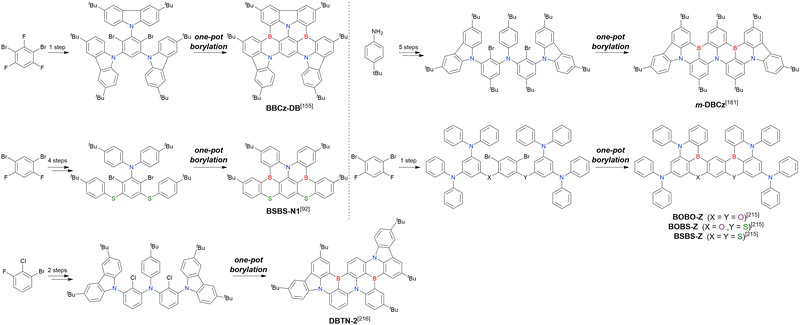 |
| | Fig. 20 Double-borylated MR-TADF emitters not included in Fig. 18 and 19. | |
Table 24 Conditions used to synthesise the compounds shown in Fig. 20
| Compound |
Condition Aa |
Condition Ba |
Condition Ca |
Solvent |
Yield [%] |
Ref. |
|
Numbers in parentheses refer to reagent loadings (equivalents).
|
|
BBCz-DB
|
n
BuLi (2.5)/rt, 4 h |
BBr3 (2.5)/rt, 12 h |
NEtiPr2 (3.8)/170 °C, 24 h |
t
Bu-Benzene |
36 |
155
|
|
m-DBCz
|
t
BuLi (4.0)/0 °C, 12 h |
BBr3 (3.0)/rt, 6 h |
PMP (6.0)/160 °C, 24 h |
t
Bu-Benzene |
13 |
181
|
|
BSBS-N1
|
t
BuLi (4.8)/rt, 4 h |
BBr3 (3.0)/rt, 12 h |
PMP (4.0)/170 °C, 24 h |
t
Bu-Benzene |
18 |
92
|
|
BOBO-Z
|
n
BuLi (3.0)/60 °C, 2 h |
BBr3 (3.0)/rt, 2 h |
PMP (4.5)/160 °C, 24 h |
t
Bu-Benzene |
20 |
215
|
|
BOBS-Z
|
n
BuLi (4.4)/60 °C, 2 h |
BBr3 (2.4)/rt, 2 h |
PMP (4.0)/160 °C, 24 h |
t
Bu-Benzene |
20 |
215
|
|
BSBS-Z
|
n
BuLi (4.4)/60 °C, 2 h |
BBr3 (2.4)/rt, 2 h |
PMP (4.0)/160 °C, 24 h |
t
Bu-Benzene |
14 |
215
|
|
DBTN-2
|
t
BuLi (2.5)/60 °C, 6 h |
BBr3 (2.5)/rt, 6 h |
NEtiPr2 (2.5)/120 °C, 12 h |
t
Bu-Benzene |
5 |
216
|
2.5.3 Photophysical properties.
Table 25 lists the photophysical properties of doubly borylated MR-TADF emitters, showing that compounds possessing one nitrogen and two boron atoms (ADBNA derivatives) exhibit photophysical parameters comparable to those of DABNA derivatives, except for the red-shifted emission wavelength. This difference in λmaxPL was attributed to the FMO energy levels. Although both HOMO and LUMO energy levels of ADBNA are lower than those of DABNA-1, the larger decrease in the LUMO energy level of ADBNA results in a smaller HOMO–LUMO energy gap.205 In the case of compounds obtained by one-pot double borylation, λmaxPL largely depends on the relative position of boron and electron-donating atoms (or groups). If two electron-donating atoms are placed in the para-position, λmaxPL substantially increases to >600 nm (e.g., 615 nm for BBCz-R).155 In particular, given that the compounds in Fig. 19 were obtained from 1,4-dibromo-2,3,5,6-tetrafluorobenzene, para-type substitution was inevitable and led to orange or red emission with λmaxPL > 600 nm (660–662 nm for R-BN,209,210 684–692 nm for R-TBN,209,210 696 nm for R-TPhBN,210 605 nm for BNO1,211 609 nm for BNO2,211 616 nm for BNO3,211 631 nm for DBNS,212 641 nm for DBNS-tBu,212 737 nm for BNS,213 and 637 nm for BNNO214). On the other hand, two electron-donating atoms in a meta-relationship had a smaller effect on the PL wavelength, resulting in λmaxPL = 440–520 nm.
Table 25 Photophysical properties of the compounds shown in Fig. 17–20
| Compound |
Condition |
λ
maxPL [nm] |
FWHM [nm] |
Φ
PL [—] |
ΔEST [eV] |
k
RISC [s−1] |
Ref. |
|
See Table 2 for the full compound name.
See Scheme 27.
Same compound named differently.
|
|
ADBNA-Me-Mes
|
CH2Cl2/1 wt%-DOBNA-OArb |
484/482 |
38/33 |
0.79/0.89 |
—/0.19 |
—/7.58 × 103 |
205
|
| CH2Cl2/1 wt%-PMMAa |
482/485 |
—/— |
0.71/0.84 |
—/0.18 |
—/— |
206
|
|
ADBNA-Me-Tip
|
CH2Cl2/1 wt%-DOBNA-OArb |
482/479 |
36/34 |
0.76/0.88 |
—/0.20 |
—/8.98 × 103 |
205
|
|
ADBNA-Me-Mes-MesF
|
CH2Cl2/1 wt%-PMMAa |
485/488 |
—/— |
0.88/0.91 |
—/0.19 |
—/— |
206
|
|
ADBNA-Me-Mes-CzAr
|
CH2Cl2/1 wt%-PMMAa |
483/485 |
—/— |
0.82/0.91 |
—/0.18 |
—/— |
206
|
|
ADBNA-Me-Mes-Me4N
|
CH2Cl2/1 wt%-PMMAa |
477·609/487 |
—/— |
0.19/0.26 |
—/0.17 |
—/— |
206
|
|
ADBNA-Me-MesF
|
CH2Cl2/1 wt%-PMMAa |
487/491 |
—/— |
0.85/0.93 |
—/0.19 |
—/— |
206
|
|
ADBNA-Me-CzAr
|
CH2Cl2/1 wt%-PMMAa |
486/487 |
—/— |
0.81/0.86 |
—/0.17 |
—/— |
206
|
|
ADBNA-Me-Me4N
|
CH2Cl2/1 wt%-PMMAa |
481·601/495 |
—/— |
0.18/0.24 |
—/0.13 |
—/— |
206
|
|
BBCz-R
|
Toluene/2 wt%-mCBPa |
615/619 |
21/27 |
0.89/0.79 |
0.19/— |
1.2 × 104/1.2 × 104 |
155
|
|
Me2AcBO
|
Toluene/5 wt%-mCPa |
474/470 |
—/57 |
—/0.99 |
—/0.05 |
—/1.13 × 106 |
117
|
|
F2AcBO
|
Toluene/5 wt%-mCPa |
478/467 |
—/59 |
—/0.97 |
—/0.05 |
—/1.38 × 106 |
117
|
|
DBN-ICz
|
Toluene/3 wt%-mCBPa |
542/545 |
18/23 |
0.986/— |
0.20/— |
0.7 × 104/— |
163
|
|
m-DINBO
|
Toluene/3 wt%-mCBPa |
456/466 |
17/22 |
—/0.94 |
—/0.060 |
—/3.1 × 104 |
167
|
|
p-DINBO
|
Toluene/3 wt%-mCBPa |
500/512 |
19/45 |
—/0.96 |
—/0.061 |
—/1.4 × 104 |
167
|
|
DBON
|
Toluene/4 wt%-mCBPa |
505/— |
20/— |
—/0.98 |
0.13/— |
—/0.8 × 105 |
207
|
|
DBNO
|
Toluene/1 wt%-PhCzBCza |
500/508 |
19/28 |
0.96/0.84 |
0.17/— |
—/3.0 × 104 |
208
|
|
SBON
|
Toluene/4 wt%-mCBPa |
463/— |
24/— |
—/0.74 |
0.16/— |
—/0.5 × 105 |
207
|
|
SBSN
|
Toluene/4 wt%-mCBPa |
489/— |
27/— |
—/0.76 |
0.10/— |
—/1.5 × 105 |
207
|
|
DBSN
|
Toluene/4 wt%-mCBPa |
553/— |
28/— |
—/0.98 |
0.13/— |
—/1.9 × 105 |
207
|
|
R-DOBN
|
Toluene/5 wt%-26DCzPPya |
453/— |
21/— |
0.83/0.91 |
0.14/— |
—/1.6 × 104 |
171
|
|
R-DOBNT
|
Toluene/5 wt%-26DCzPPya |
459/— |
21/— |
0.89/0.96 |
0.12/— |
—/0.8 × 104 |
171
|
|
R-BN
|
Toluene |
662 |
38 |
1.00 |
0.18 |
6.7 × 104 |
209
|
| Toluene |
660 |
— |
1.00 |
0.22 |
7.3 × 104 |
210
|
|
R-TBN
|
Toluene |
692 |
38 |
1.00 |
0.16 |
2.5 × 104 |
209
|
| Toluene |
684 |
— |
0.99 |
0.21 |
2.3 × 104 |
210
|
|
R-TPhBN
|
Toluene |
696 |
— |
0.90 |
0.18 |
3.0 × 104 |
210
|
|
BNO1
|
Toluene/1 wt%-DMIC-TRZa |
605/610 |
32/35 |
0.96/— |
—/0.25 |
—/— |
211
|
|
BNO2
|
Toluene/1 wt%-DMIC-TRZa |
609/618 |
32/37 |
0.95/— |
—/0.27 |
—/— |
211
|
|
BNO3
|
Toluene/1 wt%-DMIC-TRZa |
616/624 |
33/38 |
0.96/— |
—/0.26 |
—/— |
211
|
|
DBNS
|
CH2Cl2 |
631 |
40 |
0.80 |
0.20 |
2.1 × 105 |
212
|
|
DBNS-tBu
|
CH2Cl2 |
641 |
39 |
0.85 |
0.19 |
2.2 × 105 |
212
|
|
BNS
|
Toluene |
737 |
— |
0.82 |
0.14 |
8.1 × 103 |
213
|
|
BNNO
|
Toluene |
637 |
32 |
0.95 |
0.09 |
1.4 × 104 |
214
|
|
BBCz-DB
|
Toluene/2 wt%-mCBPa |
466/471 |
16/26 |
0.93/0.91 |
0.15/— |
1.9 × 104/2.2 × 104 |
155
|
|
m-DBCz
|
Toluene/1 wt%- PhCzBCza |
541/547 |
32/35 |
0.95/0.97 |
0.04/— |
—/1.65 × 105 |
181
|
|
BSBS-N1
|
Toluene/2 wt%-mCBPa |
473/478 |
21/24 |
0.59/0.89 |
0.13/0.14 |
1.5 × 106/1.9 × 106 |
92
|
|
BOBO-Z
|
Toluene/3 wt%-mCBPa |
441/445 |
15/18 |
0.76/0.64 |
0.15/0.10 |
0.7 × 105/0.7 × 105 |
215
|
|
BOBS-Z
|
Toluene/3 wt%-mCBPa |
453/457 |
21/24 |
0.94/0.93 |
0.16/0.12 |
4.3 × 105/8.6 × 105 |
215
|
|
BSBS-Z
|
Toluene/3 wt%-mCBPa |
460/464 |
20/22 |
0.93/0.88 |
0.14/0.12 |
8.8 × 105/1.6 × 106 |
215
|
|
DBTN-2
|
Toluene/2.5 wt%-SF3TRZa |
512/— |
20/— |
—/1.00 |
—/0.06 |
—/1.7 × 105 |
216
|
The relative position of the two boron atoms strongly influences the TADF properties. When two boron atoms are placed in a meta-position, a decrease in ΔEST and an increase in kRISC can be expected because the enlarged π-plane enhances long-range electronic delocalisation.74 Wang et al. evaluated the effect of double borylation by comparing p-TBNCz and m-DBCz (Fig. 21)181 and showed that with an increase in the number of boron atoms, ΔEST decreased, whereas kRISC increased. On the other hand, two para-positioned boron atoms do not improve the TADF properties because the MR effect is partially broken by the undesired π-extension. In this case, the positive effect of enhanced long-range electronic delocalisation is negated, and ΔEST (0.1–0.2 eV) and kRISC (<1.0 × 105 s−1) remain moderate. In particular, BNO1, BNO2, and BNO3 showed a very weak TADF character owing to the para-positioned boron atoms and highly planar structures.211
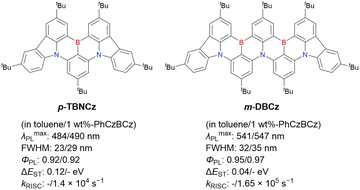 |
| | Fig. 21 Photophysical parameters of p-TBNCz and m-DBCz reported in ref. 181. | |
2.6 Summary
Numerous MR-TADF compounds have been synthesised since the first report of one-pot borylation in 2015. Although the basic protocol for one-pot borylation has been established, the synthesis route for borylation precursors needs to be tailored according to their structures. The obtained compounds usually show TADF properties and small FWHMs, benefiting from the MR effect. In addition, the results shown in Fig. 21 induce strong interest in introducing two or more boron atoms into MR-TADF frameworks. However, as discussed above, the simultaneous introduction of multiple boron atoms using one-pot protocols remains challenging. The further development of multiple borylation methodologies should enable the development of superior materials with MR-TADF characteristics.
3. One-shot borylation
Since Hatakeyama et al. proposed the molecular design of boron-embedded MR compounds with DOBNA and DABNA frameworks and their synthesis protocol (one-pot borylation), numerous corresponding derivatives have been prepared using the same methodology, as described in Section 2. One-pot borylation generally relies on metal–boron exchange followed by intramolecular tandem bora-Friedel–Crafts reactions. Therefore, in many cases, the borylation precursor should possess a halogen (Cl, Br, or I) atom at the desired borylation position to provide a scaffold for metal–halogen exchange. However, the ability of halogen groups to engage in cross-couplings often interferes with the desired reactions or increases the number of steps in precursor synthesis. In addition, molecules with multiple boron atoms are difficult to synthesise because one-pot borylation proceeds via organometallic species. Indeed, MR compounds containing more than three boron atoms have not been synthesised by one-pot borylation. Thus, despite its power for the synthesis of boron-embedded MR compounds, one-pot borylation is by no means a panacea.
This section focuses on one-shot borylation, which has been established in the last five years as a complementary method to one-pot borylation and involves successive intra- and intermolecular bora-Friedel–Crafts reactions converting multiple C–H bonds into C–B bonds in one shot. Unlike the one-pot borylation protocol, the one-shot borylation does not require functional groups such as halogens and features easier precursor synthesis, enabling the synthesis of MR compounds containing multiple (up to eight) boron atoms. This section overviews the one-shot borylation protocol and discusses its selectivity and the optical properties of the resulting compounds. In addition, synthesis protocols involving not only C–H but also N–H borylation and a recently reported sequential one-pot/one-shot borylation protocol are presented.
3.1 One-shot (C–H) borylation
In 2018, Hatakeyama et al. demonstrated the potential utility of one-shot borylation protocols.217 In the two-step synthesis of B4N3-doped nanographene B4, the crucial step is the quadruple borylation of triarylamines that converts 11 C–H bonds into C–B bonds in one shot. In addition, the judicious choice of the Brønsted base enables the selective double and triple borylation of triarylamines. The borylation precursor, N1,N1,N3,N3,N5,N5-hexakis(4-methylphenyl)-1,3,5-benzenetriamine, was obtained from 1,3,5-tribromobenzene by Buchwald–Hartwig coupling in 98% yield, and the subsequent one-shot quadruple borylation proceeded in the presence of 12 equiv. of boron triiodide (BI3) under reflux conditions (bath temperature = 200 °C) to afford the target compound B4 in 35% isolated yield and a small amount of B3 as a byproduct (Scheme 18). Notably, B4 was synthesised using only the highly reactive BI3 and not other boron sources such as boron trichloride (BCl3) and BBr3. Selective double and triple borylations were also achieved under optimal conditions. In the presence of 5.0 equiv. of BI3 and 2.0 equiv. of Ph3B, selective double borylation occurred under reflux conditions (bath temperature = 190 °C) to afford B2 in 76% yield. In contrast, triple borylation occurred at elevated temperatures (bath temperature = 200 °C) in 1,2,4-trichlorobenzene as a high-boiling-point solvent to afford B3 in 45% yield. In addition, the double one-shot borylation of a fluorine-bearing triarylamine proceeded under similar conditions to afford B2-F in 58% yield (Scheme 18). Notably, the use of Ph3B as a Brønsted base was crucial for B3 formation. The addition of Ph3B reduced the concentration of HI via its retro-Friedel–Crafts reaction. As a result, adequate HI concentrations were maintained in situ, and the Friedel–Crafts/retro-Friedel–Crafts reaction of the products was not inhibited to selectively give the thermodynamic product, i.e., B3. The reduction in HI concentration simultaneously increased the yield by suppressing competitive C–N bond cleavage. Indeed, the reaction in the absence of Ph3B selectively afforded B3 in significantly reduced yield (13%). In contrast, other Brønsted bases such as NEtiPr2 and N,N-dimethyltoluidine did not induce the formation of B3. Amine-type Brønsted bases effectively captured HI and therefore hindered the Friedel–Crafts/retro-Friedel–Crafts reaction of the products to afford only B2 as the kinetic product (44–54%). Hence, the trapping of Brønsted acids (e.g., HBr and HI) generated during the reaction is one of the general strategies for suppressing side reactions and controlling the selectivity of one-shot borylation. The resulting B,N-doped nanographenes (B2–B4 and B2-F) showed pure deep blue fluorescence and small ΔEST values (0.14–0.18 eV) (Table 26).
 |
| | Scheme 18 Synthesis of B2, B3, B4, and B2-F. Conditions and reagents: (i) BI3 (12 equiv.), o-dichlorobenzene, reflux (bath temperature: 200 °C), 12 h, (ii) BI3 (5.0 equiv.), Ph3B (2.0 equiv.), o-dichlorobenzene, reflux (bath temperature: 190 °C), 20 h, (iii) BI3 (5.0 equiv.), Ph3B (2.0 equiv.), 1,2,4-trichlorobenzene, 200 °C, 20 h. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Not detected in ref. 217. Not detected in ref. 217. | |
Table 26 Photophysical properties of the compounds shown in Scheme 18
| Compound |
Condition |
λ
maxPL [nm] |
FWHM [nm] |
Φ
PL
|
ΔEST [eV] |
k
RISC [s−1] |
|
See Table 2 for the full compound name.
|
|
B2
|
CH2Cl2/1 wt%-PMMAa |
461/455 |
39/32 |
0.19/0.68 |
0.12/0.18 |
—/4.1 × 104 |
|
B3
|
CH2Cl2/1 wt%-PMMAa |
442/441 |
50/34 |
0.13/0.47 |
0.31/0.15 |
—/— |
|
B4
|
CH2Cl2/1 wt%-PMMAa |
449/450 |
22/38 |
0.21/0.61 |
0.19/0.15 |
—/— |
|
B2-F
|
1 wt%-PMMAa |
467 |
44 |
0.57 |
0.15 |
—/— |
The selectivity of one-shot borylation can be attributed to several factors, as discussed below. Hatakeyama et al. reported the synthesis of carbazole-based DABNA analogues using one-shot borylation (Scheme 19).111 The one-shot borylation of the precursor bearing one carbazolyl and two di-p-tolylamino groups proceeded at 120 °C in the presence of 2.0 equiv. of BBr3 to afford CzDABNA-NP-M/TB in 63% yield. Notably, no regioisomers were observed. Moreover, the double borylation of the same substrate was achieved at 140 °C using 4.0 equiv. of BBr3. Two boron atoms were selectively introduced at ortho-positions with respect to the carbazolyl group to afford CzB2-M/TB in 72% yield. According to density functional theory (DFT) calculations, the HOMO energy levels of the borylation precursor, CzDABNA-NP-M/TB, and CzB2-M/TB (−4.84, −4.77, and −4.73 eV, respectively) are sufficiently high for electrophilic C–H borylation. The HOMO of the borylation precursor is mostly localised at para-positions relative to the di-p-tolylamino groups (red circles, Fig. 22), which is attributed to their stronger electron-donating nature compared to that of the carbazolyl group. This indicates that the initial borylation predominantly occurs at the carbon atom between the 9-carbazolyl and di-p-tolylamino groups to afford CzDABNA-NP-M/TB. In other words, the initial borylation of the one-shot borylation protocol occurs at the para-position with respect to the most electron-donating substituent and is followed by tandem bora-Friedel–Crafts reactions affording the kinetic product. The second borylation of CzDABNA-NP-M/TB can proceed at two sites, namely the ortho-positions relative to the di-p-tolylamino group. However, the p-tolyl groups (blue circles, Fig. 22) perpendicular to the central benzene ring prevent BBr3 from approaching the reactive carbon owing to steric hindrance.
 |
| | Scheme 19 Synthesis of CzDABNA-NP-M/TB and CzB2-M/TB reported in ref. 111. | |
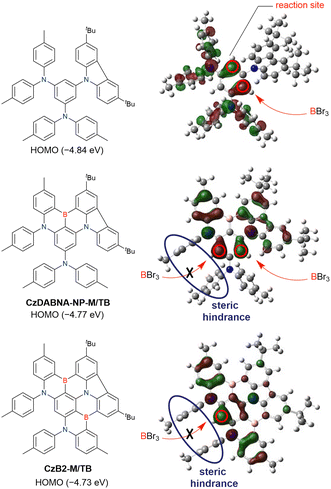 |
| | Fig. 22 Highest occupied Kohn–Sham orbitals (HOMOs) of the compounds shown in Scheme 19 calculated at the B3LYP/6-31G(d) level of theory. | |
In contrast, the carbazolyl group is positioned horizontally relative to the central benzene ring, allowing for easy access by BBr3. Consequently, the second borylation predominantly occurs at the carbon atom located between the 9-carbazolyl and di-p-tolylamino groups to afford CzB2-M/TB. In brief, the selectivity of one-shot borylation is determined by both electronic and steric factors.
The synthesis of narrowband deep-blue emissive compounds BN1–BN3 by Yang et al. are a good example illustrating the kinetic and thermodynamic selectivity of one-shot borylation (Scheme 20 and Table 27).218 The one-shot borylation was performed with 12 equiv. of BBr3 at 180 °C and mainly occurred at the carbon atom in the central benzene ring at the para-position with respect to the 2-carbazolylmesitylamino group (where the HOMO was primarily localised) to afford mono-borylated BN1 in 68% yield. In contrast, the mono-borylated regioisomer BN2 was obtained in 39% yield as the major product at a higher reaction temperature (210 °C).
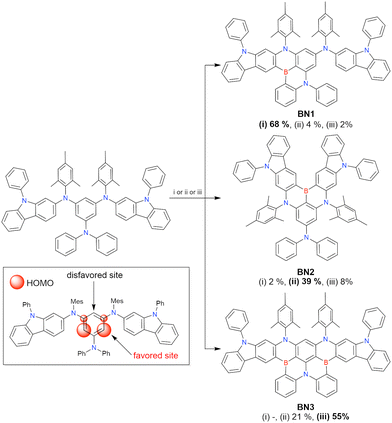 |
| | Scheme 20 Synthesis of BN1–BN3. Conditions and reagents: (i) BBr3 (12 equiv.), o-dichlorobenzene, 180 °C, 20 h; then NEtiPr2, (ii) BBr3 (12 equiv.), o-dichlorobenzene, 210 °C, 20 h; then NEtiPr2, (iii) BBr3 (24 equiv.), o-dichlorobenzene, 210 °C, 20 h; then NEtiPr2 (ref. 218). | |
Table 27 Photophysical properties of the compounds shown in Scheme 20
| Compound |
Condition |
λ
maxPL [nm] |
FWHM [nm] |
Φ
PL
|
ΔEST [eV] |
k
RISC [s−1] |
|
See Table 2 for the full compound name.
|
|
BN1
|
Toluene/1 wt%-DBFPOa |
454/458 |
18/22 |
—/0.91 |
0.20/0.15 |
—/1.3 × 104 |
|
BN2
|
Toluene/1 wt%-DBFPOa |
464/467 |
15/22 |
—/0.93 |
0.16/0.13 |
—/2.6 × 104 |
|
BN3
|
Toluene/1 wt%-DBFPOa |
456/458 |
17/21 |
—/0.98 |
0.15/0.12 |
—/2.55 × 105 |
Although the borylated position of BN2 does not contribute to the precursor HOMO and is therefore a disfavoured site, DFT calculations suggest that BN2 is 2.2 kcal mol−1 more stable than BN1. Hence, initial borylation is assumed to occur at the same position as in the case of BN1, with a subsequent retro-Friedel–Crafts reaction involving in situ generated HBr affording BN2 as the thermodynamic product. In addition, double one-shot borylation proceeded in the presence of an elevated amount of BBr3 (24 equiv.) at 210 °C to give BN3 in 55% yield as the kinetic product.
The synthesis of O,B,N-embedded MR compounds by Wong et al. also describe the selectivity of one-shot borylation (Scheme 21).219 Based on the above description, initial borylation should occur at the para-position relative to the diphenylamino group. Indeed, the one-shot borylation performed using 10 equiv. of BBr3 at room temperature occurred at this position, and the subsequent intramolecular bora-Friedel–Crafts reaction in the presence of NEtiPr2 as a Brønsted base afforded OBN in 42% yield. The base reduced the concentration of in situ generated HBr and prevented the retro-Friedel–Crafts reaction to promote the formation of the kinetic product OBN. In contrast, one-shot borylation performed at higher temperature (160 °C) using 10 equiv. of BBr3 selectively furnished NBN in 35% yield, i.e., boron was introduced at the para-position with respect to the phenoxy group. Given that this group is a weaker electron donor than the diphenylamine group and the para-position relative to this group is therefore unfavourable for borylation, NBN was concluded to be the thermodynamic product. In addition, double one-shot borylation affording ODBN in 17% yield was achieved by increasing the amount of BBr3 (to 20 equiv.) and the reaction temperature (to 210 °C). Importantly, controlling the borylation temperature, number of boron equivalents, and/or Brønsted base addition enables the highly selective synthesis of kinetic/thermodynamic products.
 |
| | Scheme 21 Synthesis of OBN, NBN, and ODBN in ref. 219. | |
Various MR emitters have been synthesised based on the abovementioned selectivity. One-shot borylation has also been applied to the synthesis of DABNA-type compounds (Scheme 22).111,220–223 An overview of these compounds and their borylation precursors shows that 1,3,5-trisubstituted benzenes are the preferred substrates, as they help to prevent undesired reactions. As mentioned above, one-shot borylation prefers to proceed at carbons where the HOMO is localised (ortho- and para-positions relative to the electron-donating group). However, in the case of 1,3-disubstituted benzene-type substrates, the borylation proceeds at the 4- or 6-positions rather than the sterically hindered 2-position. BBr3 is the preferred boron source for their one-shot borylation, with no uses of BCl3 and BI3 reported to date. The synthesis of hexa-tBu-substituted DABNA (DABNA-NP-TB, t-DAB-DPA, or 3tPAB) has been independently reported by Hatakeyama et al.,111 Lee et al.,220 and Wang et al.221 However, the corresponding yields varied widely. In the last two reports, the high reaction temperature was believed to facilitate the retro-Friedel–Crafts reaction involving the in situ generated HBr and induce deborylation and tBu group cleavage. In addition, all three substituents on the central benzene ring need not be electron-donating (e.g., amino and phenoxy) groups; for example, one of them can be a PAH.
 |
| | Scheme 22 Synthesis of DABNA analogues by one-shot borylation. | |
The MR compounds BP-2DPA and DBP-4DPA, which have a perylene moiety instead of one diphenylamino group in the DABNA-NP core, were synthesised by Kwon et al. in yields of 39% and 37%, respectively, using one-shot borylation (Scheme 23).224 Although many DABNA derivatives exhibit blue MR-TADF (e.g., PAB: λmaxPL = 449 nm in toluene),221BP-2DPA and DBP-4DPA feature significantly red-shifted emission (λmaxPL = 599 nm and 605 nm in toluene, respectively), as perylene is a highly conjugated planar polycyclic aromatic system. On the other hand, BP-2DPA and DBP-4DPA do not exhibit TADF character due to the weak MR effect attributed to the non-MR perylene skeleton. Hence, BP-2DPA and DBP-4DPA were utilised as TEs for hyperfluorescence devices. The bonding/anti-bonding character of the FMOs of perylene remained, although weakened due to the fusion with the DABNA skeleton, and the shoulder peaks originating from the vibronic coupling between the S1–S0 electronic transition and stretching vibrations were observed in the PL and EL spectra.
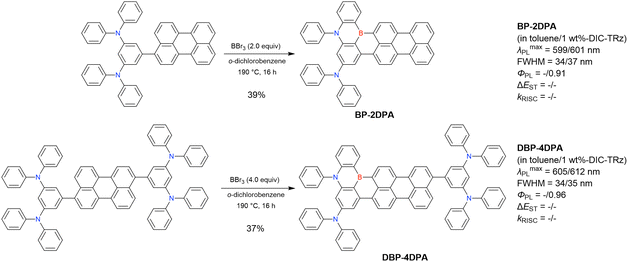 |
| | Scheme 23 Synthesis of the perylene-fused BN compounds by one-shot borylation reported in ref. 224. | |
Electron-donating groups other than diarylamino moieties are also available, including carbazolyl, acridyl, azetidyl, and ether groups. In particular, numerous carbazole-fused CzDABNA analogues were synthesised by one-shot borylation (Scheme 24).111,195,225–229 When the 9-carbazolyl group is used as an electron-donating group, one-shot borylation proceeds based on the selectivity described above, as this group is a weaker electron donor than the diarylamino group. BBr3 is used as the boron source in most cases, but BI3 is also available. Carbazole ring incorporation is often used for emission wavelength tuning, as it results in the extension of the π-conjugated system and, hence, in red-shifted emission.
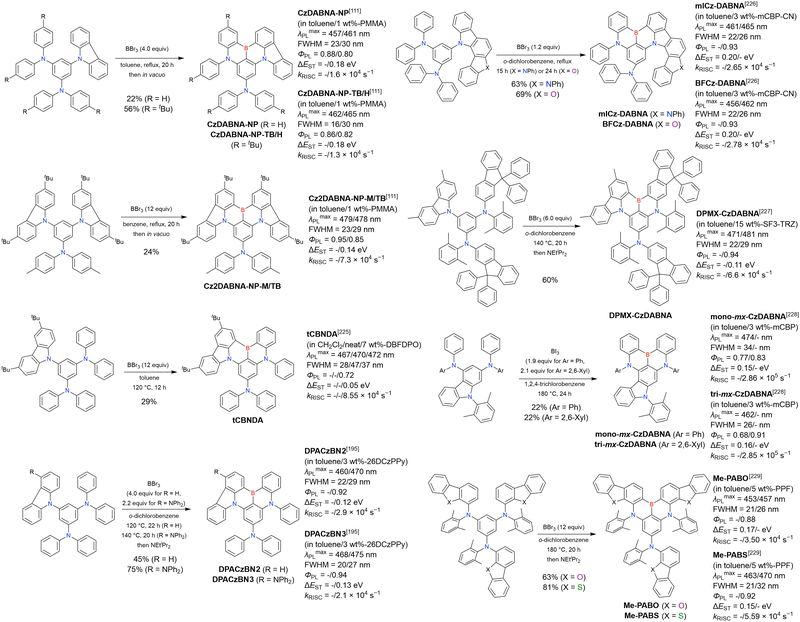 |
| | Scheme 24 Synthesis of carbazole-based DABNA analogues by one-shot borylation. | |
According to Wang et al., the one-shot borylation of a 9,10-dihydro-9,9-dimethylacridine-substituted substrate with 4.0 equiv. of BBr3 proceeded at the para-position relative to the diarylamino group to give tDMAC-BN in 32% yield, whereas that of a 9,10-dihydro-9,9-diphenylacridine-substituted substrate proceeded at the para-position relative to the acridine moiety to afford tDPAC-BN in 41% yield (Scheme 25).230 Similarly, Wang et al. reported that the 9,10-dihydro-9,9-dimethylacridine moiety-containing borylated compound Ph-DMAC-BN was obtained in 44% yield and with similar selectivity under almost identical conditions.231 When the 9,10-dihydroacridine moiety is used as an electron-donating group, the substituent at its 9-position seems to determine the selectivity of one-shot borylation.
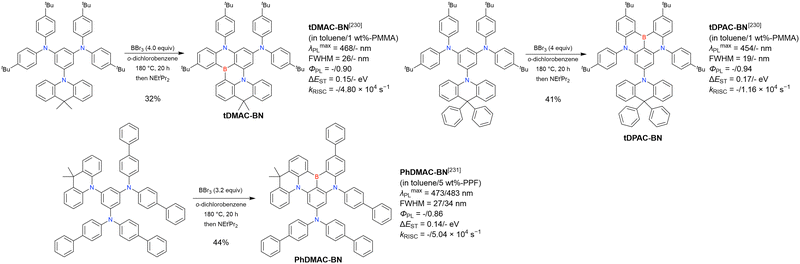 |
| | Scheme 25 Synthesis of acridine-based DABNA analogues by one-shot borylation. | |
Yang et al. attempted the one-shot borylation of the phenothiazine 5,5-dioxide-substituted substrate.196 The one-shot borylation proceeded in the presence of 5.0 equiv. of BBr3 to afford the borylation product PTZBN3 in 11% yield and the deoxygenated product PTZBN2 was obtained in 15% yield (Scheme 26). Although some S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond cleavage was observed, the sulfone moiety was found to be tolerant to one-shot borylation conditions to some extent.
O bond cleavage was observed, the sulfone moiety was found to be tolerant to one-shot borylation conditions to some extent.
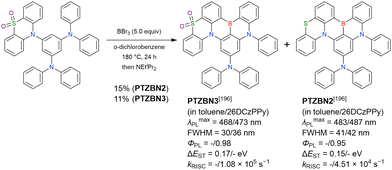 |
| | Scheme 26 Synthesis of the phenothiazine-based DABNA analogues by one-shot borylation. | |
One-shot borylation is also feasible for precursors bearing oxygenated electron-donating groups (e.g., aryloxy). In most cases, DOBNA derivatives are synthesised by one-pot borylation, but can also be prepared using the one-shot borylation of 1,3,5-trisubstituted benzene-type substrates. Hatakeyama et al. synthesised DOBNA-OAr in 91% yield as a host material for blue OLEDs by the one-shot borylation of three-fold symmetric 1,3,5-triaryloxybenzene in the presence of 4.0 equiv. of BI3 followed by heat treatment in acetic acid (HOAc) (Scheme 27).60
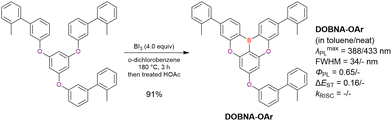 |
| | Scheme 27 Synthesis of DOBNA-OAr by one-shot borylation in ref. 60. | |
Heat treatment in acetic acid is an important procedure for increasing the yield of borylation reactions, especially when using a large excess of the boron source. As one-shot borylation proceeds at carbons contributing to the HOMO (often at the para-position relative to the electron-donating group), borylation at undesired peripheral positions may occur as a side reaction. Heat treatment in acetic acid removes the non-fused boron introduced at the periphery by excess BI3. This technique was used to synthesise 2B-DTACrs by Zysman-Colman et al.232 In contrast to the mono-borylated 1B-DTACrs, which was synthesised using 4.1 equiv. of BBr3, the di-borylated 2B-DTACrs was synthesised using 10 equiv. of BBr3 followed by acetic acid treatment (Scheme 28).
 |
| | Scheme 28 Synthesis of 1B-DTACrs and 2B-DTACrs reported in ref. 232. | |
Furthermore, the conditions of one-shot borylation allow for the retention of convertible halogens such as chlorine and bromine as well as for late modification (Scheme 29). Lee and Hong reported that six chlorine or bromine atoms were maintained even upon treatment with 36 equiv. of BBr3 in o-dichlorobenzene at 200 °C, which afforded mono-borylated compounds Cl-MR and Br-MR in yields of 56% and 63%, respectively.233 In the synthesis of DBT-BN, DBF-DBN, and DBT-DBN by Wu and Lan et al., the chlorine atoms in the borylation precursor were partially exchanged for bromine during one-shot borylation with BBr3. However, this did not present a problem, as these halogen atoms were consumed in the subsequent Suzuki–Miyaura coupling.234
 |
| | Scheme 29 Synthesis of BN compounds bearing halogen atoms by one-shot borylation. | |
One-shot borylation is also a powerful method for achieving double borylation (Scheme 30). For example, the one-shot double borylation of a precursor synthesised from a commercially available compound in three steps was carried out in the presence of 6.0 equiv. of BI3 at 180 °C to afford the sterically protected OAB-ABP-1 in 18% yield.235 Note that the mesityl and methyl groups suppressed the undesirable borylation at the carbon atom contributing to the HOMO. Duan et al. used one-shot double borylation to prepare DABNA-incorporated calix[4]arene C-BN in 30% yield.236 Furthermore, You et al. used one-shot double borylation at 190 °C in the presence of 26 equiv. of BBr3 to synthesise DTBA-B2N3 in 26% yield as an MR emitter by introducing a medium (azepine) ring to suppress aggregation-caused quenching (ACQ).152 Kwon et al. amalgamated the conventional DOBNA and DABNA structures, synthesising TPD4PA and tBu-TPD4PA in yields of 23% and 26%, respectively, via one-shot double borylation in the presence of 4.0 equiv. of BBr3.237 One-shot double borylation is often performed using a large excess of BBr3 or BI3. The thus obtained B2 compounds are expected to exhibit enhanced MR effects and smaller ΔEST than B1 MR compounds because of the highly extended π-skeleton of the former.
 |
| | Scheme 30 Double one-shot borylation reactions. | |
In 2019, Hatakeyama et al. reported ν-DABNA as a new scaffold for narrowband MR-TADF emitters.60ν-DABNA was synthesised from commercially available compounds in three steps including palladium-catalysed C–N coupling and one-shot borylation. The one-shot borylation of the precursor with 4.0 equiv. of BBr3 at 180 °C afforded ν-DABNA in 36% yield (Scheme 31). According to DFT calculations, the boron atoms were selectively introduced at the carbon atoms primarily contributing to the HOMO. Additionally, the small number of substituents at the central benzene ring and its steric accessibility are important for achieving highly selective borylation. Compared to DABNA-1,38ν-DABNA exhibited a substantially higher kRISC (2.0 × 105 s−1) and was used to realise an OLED with an excellent EQE, minimum efficiency roll-off (34.4/32.8/26.0% at 15/100/1000 cd m−2, respectively), and ultrapure blue emission (FWHM = 18 nm). Therefore, ν-DABNA would replace DABNA as the benchmark for MR compounds. Following the success of ν-DABNA, various related derivatives have been synthesised. Hatakeyama et al. reported ν-DABNA-O-Me, which can be viewed as ν-DABNA with an oxygen atom instead of the nitrogen atom.238 The one-shot double borylation of a chlorine-bearing precursor was carried out with a stoichiometric amount of BI3 at 90 °C to obtain the desired intermediate in 51% yield, and the subsequent cross-coupling with bis(3,5-dimethylphenyl)amine at 130 °C afforded ν-DABNA-O-Me in 59% yield. In view of its lower atomic orbital energy, the oxygen atom restricts the π-conjugation of the HOMO rather than that of the LUMO. The replacement of nitrogen with oxygen affects the HOMO energy level more than it does the LUMO energy level. As a result, ν-DABNA-O-Me exhibits a larger HOMO–LUMO gap and displays a deeper blue emission (465 nm) than ν-DABNA. The emission maximum of ν-DABNA (469 nm) is slightly red-shifted relative to that of DABNA-1 (459 nm) because of π-extension. As a result, the Commission Internationale de l’Eclairage (CIE) coordinates (0.12, 0.11) deviated from the requirements defined by the National Television System Committee (0.14, 0.08). Therefore, the replacement of nitrogen by oxygen is a powerful strategy for achieving optimal emission wavelengths. Kwon et al. used a similar concept to synthesise NO-DBMR, where the different nitrogen position in ν-DABNA was replaced by oxygen.239 The final double one-shot borylation was carried out using 4.2 equiv. of BBr3 to afford NO-DBMR in 39% yield. Fluorination and methylation are also used to realise blue-shifted emission. Kwon et al. also obtained m-ν-DABNA, 4F-ν-DABNA, and 4F-m-ν-DABNA in yields of 32%, 45%, and 51%, respectively, by one-shot borylation with 4.0 equiv. of BBr3 from a substrate with a fluorinated or methylated benzene ring-substituted nitrogen.240 Other approaches to derivative design involve the introduction of bulky substituents to prevent luminescence peak broadening due to excimer formation in the aggregated state and enhance the horizontal orientation. Based on this strategy, Kwon et al. synthesised tBu-ν-DABNA in 31% yield by introducing tBu groups into ν-DABNA,134 while Yoo et al. synthesised o-Tol-ν-DABNA-Me in 42% yield via one-shot borylation with BBr3 by changing the substituent on the nitrogen to o-tolyl or xylyl.241 As mentioned above, the introduction of the carbazole scaffold extends the π-conjugated system and leads to red-shifted emission. The same is true for ν-DABNA derivatives, as exemplified by carbazole-fused Cz-DBMR and Π-CzBN synthesised by Kwon et al. and Zhang et al., respectively.239,242
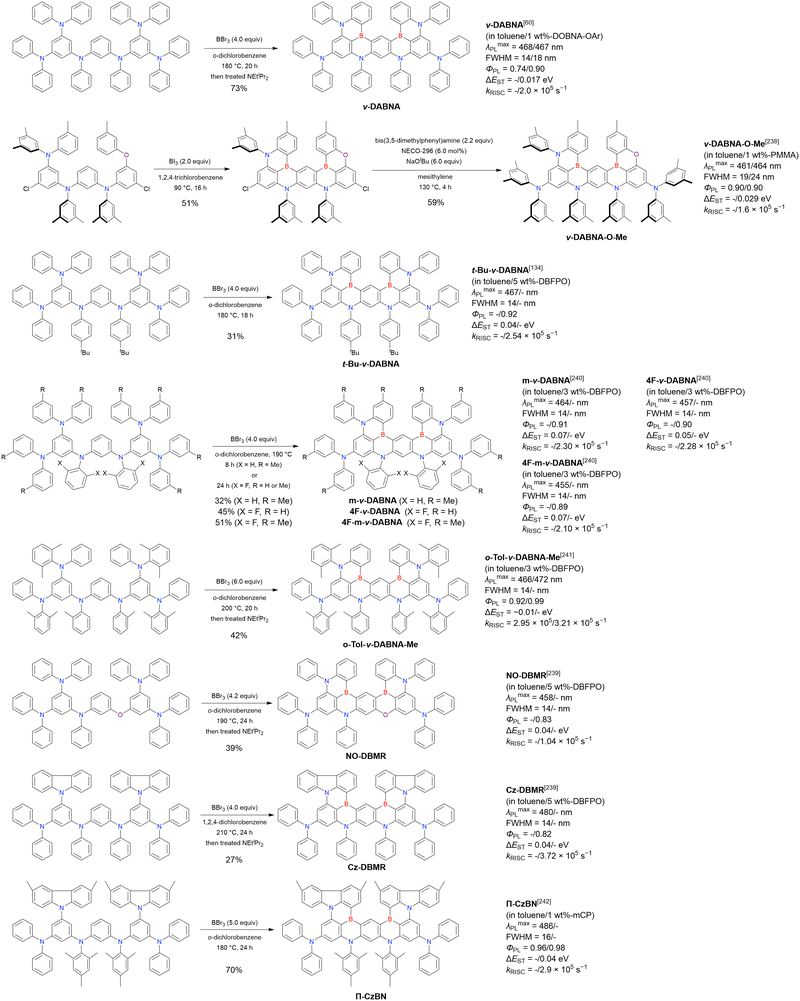 |
| | Scheme 31 Synthesis of ν-DABNA and its analogues by one-shot borylation. | |
In 2022, Hatakeyama et al. synthesised an expanded B,N-embedded heterohelicene, V-DABNA-Mes, as a representative new member of the DABNA series by triple one-shot borylation.243 The one-shot triple borylation was achieved using 32 equiv. of BBr3 in an autoclave at 180 °C to afford V-DABNA-Mes in 44% yield (Scheme 32). The steric protection provided by the mesityl groups suppresses both overborylation and borylation at undesired reactive sites. Notably, a significant drop in the yield of V-DABNA-Mes (3%) was observed under standard reflux conditions in a flask because of the decreased concentration of BBr3 in the reaction mixture. The one-step construction of such giant helix structures is not only synthetically useful but also an important as a design strategy for the emitters of high-performance OLEDs, as the enhanced MR effect and extended π-helix structure led to small FWHM (13 nm) and large kRISC (4.22 × 105 s−1) in toluene. This facile and scalable method was applied to unsubstituted and fluorine-substituted compounds, and triple one-shot borylation with 16 equiv. of BBr3 gave V-DABNA and V-DABNA-F in 11% and 35% yields, respectively.244
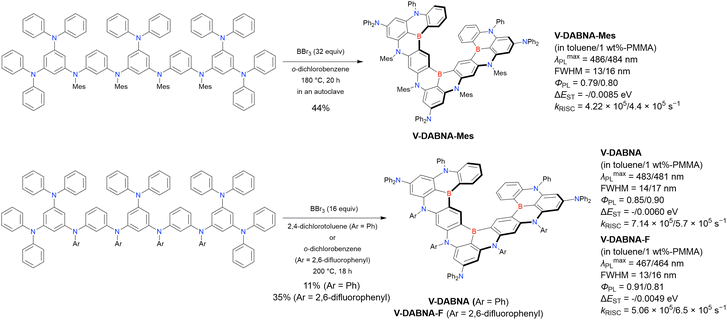 |
| | Scheme 32 Synthesis of the V-DABNA series reported in ref. 243 and 244. | |
Hatakeyama et al. achieved quadruple, sextuple, and octuple borylation to construct the largest MR-core structures, which comprised B,N-embedded nonacene (CzB4), tridecacene (CzB6), and heptadecacene (CzB8) frameworks, respectively.245 The key to success was the judicious choice of the borylating reagent, namely BI3 combined with 2,6-di-tert-butylpyridine (DTBPY), and long alkyl-substituted carbazolyl groups as boron-trapping groups, which suppressed the decrease in HOMO energy and insolubilisation associated with borylation. The one-shot multiple borylation with 16–32 equiv. of BI3 and 12–24 equiv. of DTBPY (as a bulky Brønsted base) proceeded at 150 °C to afford CzB4, CzB6, and CzB8 in yields of 90%, 86%, and 83%, respectively. Thus, each electrophilic C–H borylation proceeded in >99% yield, which is the highest efficiency reported to date for C–H borylation reactions (Scheme 33). The thus obtained large MR compounds exhibited ultra-narrowband green TADF with FWHM = 12–16 nm. This achievement suggests that carbazolyl groups may enable the construction of larger acene frameworks by multiple borylation.
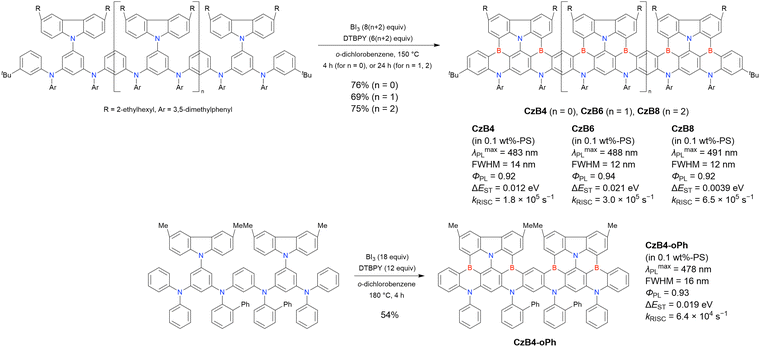 |
| | Scheme 33 Synthesis of CzB4–CzB8 and CzB4-oPh by one-shot quadruple, sextuple, and octuple borylation reported in ref. 245. | |
Thus, one-shot borylation is a powerful synthesis method for developing boron-embedded MR compounds, especially for the introduction of multiple boron atoms, and is complementary to one-pot borylation. The selectivity of this reaction is mainly determined by electronic and steric requirements; therefore, a deep understanding of these requirements is useful for MR emitter design. It should be noted that one-shot borylation is not a panacea, allowing the synthesis of only a small fraction of the vast chemical space of MR compounds.
3.2 One-shot (N–H) borylation
This subsection focuses on MR compounds with B–N bonds synthesised by amine-directed one-shot borylation (N–H borylation). B–N covalent bonds were first used to formally replace the isoelectronic CC bonds by Dewar in 1958.25 Although the corresponding B,N-doped PAHs are being intensively researched because of the unique optical and electronic properties imparted by dipole B–N bonds,26–33 highly B–N-doped PAHs do not exhibit MR-TADF and exhibit relatively large FWHMs.246,247 In 2022, Duan et al. used the amine-directed formation of B–N bonds to demonstrate the preparation of MR compounds m[B-N]N1 and m[B-N]N2.248 Amine-directed double borylation106 occurred in chlorobenzene at 150 °C in the presence of 4.0 equiv. of BBr3 and 4.0 equiv. of base to produce m[B-N]N1 and m[B-N]N2 in yields of 90% and 92%, respectively (Scheme 34). Notably, the two boron atoms were introduced at ortho-positions relative to the nitrogen atoms, and other byproducts including regioisomers and mono-borylated compounds were not detected. While m[B-N]N1 and m[B-N]N2 displayed narrowband pure emission with FWHM = 26–32 nm and ΦPL >90% but exhibited slightly larger ΔEST (0.13–0.15 eV) and smaller kRISC compared with other B2 compounds. This indicated that the MR effect was weakened because of carbazole moieties connecting the central benzene ring via C–C bonds.
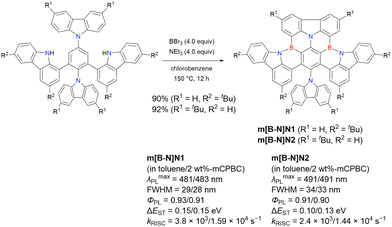 |
| | Scheme 34 Synthesis of m[B-N]N1 and m[B-N]N2 by the amine-directed one-shot borylation reported in ref. 248. | |
The same group used this facile and scalable amine-directed double borylation method to prepare a standard para-B–π–B skeleton modified by the stepwise replacement of nitrogen atoms with oxygen atoms (Scheme 35).249 The resulting compounds featured a wide λmaxPL range of 442–545 nm while maintaining narrow FWHM (19–26 nm) and high luminescence efficiency (Φ > 0.84). On the other hand, these para-B–π–B compounds exhibited large ΔEST (0.26–0.44 eV) among the B2 compounds and did not show TADF behaviour because the MR effect is weakened when the two boron atoms are in the para-position with respect to each other, and also because the carbazole moiety, which forms a C–C bond with the central benzene ring, has a weak MR effect.
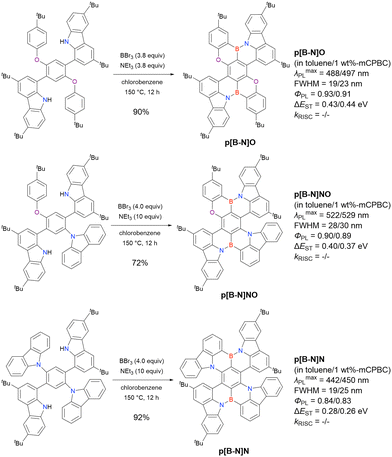 |
| | Scheme 35 Synthesis of B–N bond-embedded MR compounds reported in ref. 249. | |
Not only the synthesis of tricoordinate boron-based MR-TADF compounds, but also those of tetracoordinate boron-based MR-TADF compounds containing B–N bonds have been achieved using amine-directed one-shot borylation. Wang et al. reported tetracoordinate boron-containing MR-TADF emitters based on a C,N,C- or N,N,N-chelating pincer ligand.250 Among them, N,N,N-chelating compounds BN2, Tcz-BN2, and BN3 were synthesised using amine-directed one-shot borylation (Scheme 36). The amine-directed one-shot borylation of the N,N,N-ligand bearing one pyridine moiety and two N–H bonds occurred in the presence of dichlorophenylborane (PhBCl2) as a boron source to afford BN2 and TCz-BN2, while the subsequent Scholl reaction of BN2 gave BN3. Unlike the emission spectra of conventional MR-compounds, the emission spectra of BN2 and TCz-BN2 were broad (FWHM = 97 and 108 nm, respectively) because of the large structural differences between the ground and excited states derived from tetracoordinate boron. In addition, BN2 and TCz-BN2 exhibited TADF character, although the boron orbitals are not involved in the MR structure and the MR effect is derived from the pyridine-containing skeleton.
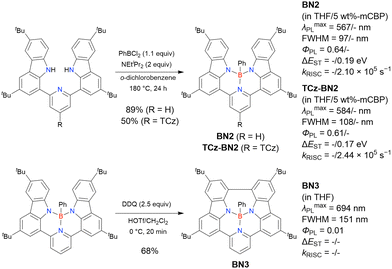 |
| | Scheme 36 Synthesis of tetracoordinate boron-containing MR compounds by one-shot borylation reported in ref. 250. | |
In brief, amine-directed borylation is a facile and scalable method for synthesising B–N bond-bearing PAHs, although they often display no MR effects. The study of MR compounds bearing B–N bonds is currently in its early stage.
3.3 Sequential multiple borylation
One-shot borylation is a facile method for developing π-extended MR compounds containing multiple boron atoms. However, because this reaction proceeds at the most sterically accessible carbon atom contributing to the HOMO,251 the chemical space of boron-based MR compounds remains largely unexplored. To address this issue, Hatakeyama et al. proposed a new synthesis method for boron-based MR compounds, namely sequential multiple borylation, which involves one-pot borylation, Buchwald–Hartwig coupling, and one-shot borylation.252 This method was used to synthesise an ultrapure green MR emitter (ω-DABNA) as a proof-of-concept (Scheme 37). The omega-shaped MR framework containing three boron atoms and four nitrogen atoms could not be accessed using conventional borylation protocols such as one-pot or one-shot borylation. This structure, featuring three boron atoms introduced into the same benzene ring, could not be synthesised by one-pot borylation because electron repulsion destabilises the trilithiated intermediate formed in the course of this reaction. In the case of one-shot borylation, triple borylation could not proceed because the HOMO is not localised at the third borylation position after the introduction of two boron atoms. The initial precursor, which has four chlorine atoms and one bromine atom, was prepared in two steps from a commercially available compound through iterative Buchwald–Hartwig coupling. The first key step in sequential multiple borylation, i.e., the one-pot borylation of the initial precursor, was the construction of a DABNA skeleton retaining four chlorine atoms. Lithium–bromine exchange was followed by a bora-Friedel–Crafts reaction in the presence of 2.0 equiv. of BBr3, which afforded a DABNA derivative bearing four chlorine atoms in 42% yield. The second key step, i.e., the introduction of four diphenylamino groups into the DABNA derivative, was achieved by Buchwald–Hartwig coupling with diphenylamine, which afforded the final borylation precursor in 88% yield. The third key step was the one-shot borylation. Importantly, the HOMO of the final borylation precursor was mainly localised on the DABNA skeleton, and regioselective borylation reactions were therefore expected. Indeed, in the presence of DTBPY, the one-shot double borylation with 8.0 equiv. of BI3 afforded ω-DABNA in 50% yield. In the absence of DTBPY, a significant drop in yield (4%) was observed, and a considerable amount of the precursor (80%) was recovered. This indicated that the sterically hindered DTBPY selectively captured the in situ generated HI to promote borylation and suppress deborylation via the retro-bora-Friedel–Crafts reaction. In addition, the final one-shot borylation with BBr3 was carried out at 180 °C to afford ω-DABNA in a yield of 23%, which is lower than that obtained with BI3 as the boron source (50%) because of the lower reactivity of BBr3.
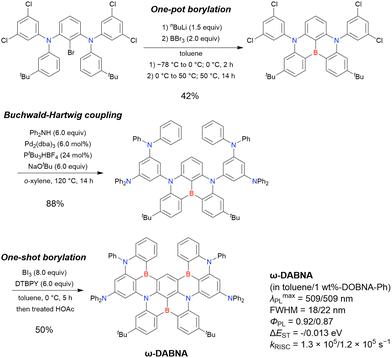 |
| | Scheme 37 Synthesis of ω-DABNA by sequential multiple borylation reported in ref. 252. | |
At about the same time as Hatakeyama et al., Zysman-Calman et al. independently reported the synthesis of ladder-type deep-blue MR compounds NBONacene and MesBDIDOBNA-N using a protocol which can be categorized as sequential multiple borylations (Scheme 38).253,254 One-pot borylation afforded pMDBA-Br in 73% yield, with subsequent coupling giving the final borylation precursors in 47% and 57% yields, respectively. One-shot borylation was performed with excess BBr3, and the R2BBr species generated in situ were trapped with a Grignard reagent to afford the target compounds, NBONacene and MesB-DIDOBNA-N, in 45% and 74% yields, respectively. Although the final borylation step does not construct a fully fused structure around the introduced boron atom, these synthesis methods also seem to be sequential multiple borylation protocols.
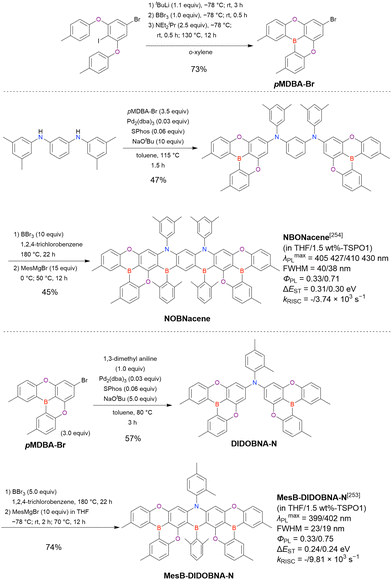 |
| | Scheme 38 Synthesis of NOBNacene and MesB-DIDOBNA-N by sequential multiple borylation. | |
Sequential multiple borylation can be useful for the synthesising MR-TADF materials, especially those bearing multiple boron atoms at the same aromatic ring, and thus has the potential to become a new standard protocol for MR emitter synthesis.
3.4 Summary
One-shot borylation converts multiple C–H/N–H bonds into C–B/N–B bonds to construct the boron-embedded π-conjugated skeletons in one shot. This is a powerful synthesis method for producing π-extended MR compounds. Furthermore, a sequential multiple borylation protocol that elaborately combines one-shot borylation and one-pot borylation was recently developed to expand the chemical space of MR compounds. Since detailed discussion of the photophysical properties of each obtained compound is beyond the scope of this review, we summarise the photophysical data for the related compounds in Table 28, and provide brief structure–property correlations. Basic relationships, such as the dependence of the maximum emission wavelength on the electron-donating abilities of the substituents, can be explained in a similar way to that of compounds obtained by one-pot borylation (see Sections 2.2.2 and 2.3.2). Most π-extended MR compounds with multiple boron atoms obtained by one-shot borylation show a small ΔEST and large kRISC because of enhanced short-range CT and long-range electronic delocalisation in the excited states.74 Although π-extensions with 1,2-azaborine, heterole, and perylene result in materials with poor TADF properties because of the partial cancelation of the MR effect, the rigid and π-extended framework provides a narrow FWHM, which is suitable for use as a TE for hyperfluorescence or PSF OLEDs. Thus, one-shot borylation has contributed significantly to the development of excellent emitters for OLEDs. However, the accessible chemical space is still limited because this reaction can only proceed at sterically unhindered carbons with a certain HOMO distributions. Therefore, it is desirable to develop new methodologies to further expand the chemical space.
Table 28 Photophysical properties of the compounds shown in Section 3
| Compound |
Condition |
λ
maxPL [nm] |
FWHM [nm] |
Φ
PL [—] |
ΔEST [eV] |
k
RISC [s−1] |
Ref. |
|
See Table 2 for the full compound name.
Same compound named differently.
See Scheme 3.
|
|
B2
|
CH2Cl2/1 wt%-PMMAa |
461/455 |
39/32 |
0.19/0.68 |
0.12/0.18 |
—/4.1 × 104 |
217
|
|
B3
|
CH2Cl2/1 wt%-PMMAa |
442/441 |
50/34 |
0.13/0.47 |
0.31/0.15 |
—/— |
217
|
|
B4
|
CH2Cl2/1 wt%-PMMAa |
449/450 |
22/38 |
0.21/0.61 |
0.19/0.15 |
—/— |
217
|
|
B2-F
|
1 wt%-PMMAa |
467 |
44 |
0.57 |
0.15 |
— |
217
|
|
CzDABNA-NP-M/TB
|
Toluene/1 wt%-PMMAa |
465/468 |
23/28 |
0.82/0.86 |
—/0.18 |
—/1.1 × 104 |
111
|
|
CzB2-M/TB
|
Toluene/1 wt%-PMMAa |
473/491 |
23/34 |
0.72/0.88 |
—/0.11 |
—/2.4 × 104 |
111
|
|
BN1
|
Toluene/1 wt%-DBFPOa |
454/458 |
18/22 |
—/0.91 |
0.20/0.15 |
—/1.3 × 104 |
218
|
|
BN2
|
Toluene/1 wt%-DBFPOa |
464/467 |
15/22 |
—/0.93 |
0.16/0.13 |
—/2.6 × 104 |
218
|
|
BN3
|
Toluene/1 wt%-DBFPOa |
456/458 |
17/21 |
—/0.98 |
0.15/0.12 |
—/2.55 × 105 |
218
|
|
OBN
|
Toluene/10 wt%-DBFPOa |
425/— |
30/46 |
0.85/0.50 |
0.20/— |
—/1.3 × 104 |
219
|
|
NBN
|
Toluene/10 wt%-DBFPOa |
440/— |
29/43 |
0.90/0.71 |
0.12/— |
—/2.9 × 104 |
219
|
|
ODBN
|
Toluene/10 wt%-DBFPOa |
429/— |
26/55 |
0.93/0.86 |
0.19/— |
—/2.1 × 104 |
219
|
|
DABNA-NP-M
|
1 wt%-PMMAa |
460 |
29 |
0.88 |
0.17 |
1.4 × 104 |
111
|
|
DABNA-NP-TB
|
1 wt%-PMMAa |
453 |
26 |
0.83 |
0.17 |
1.4 × 104 |
111
|
|
t-DAB-DPA
|
3 wt%-mCBP:mCBP-CNa |
— |
— |
0.94 |
— |
3.97 × 104 |
220
|
|
PAB
|
Toluene/3 wt%-mCPa |
449/453 |
23/— |
—/0.61 |
—/0.06 |
—/6.95 × 104 |
221
|
|
2tPAB
|
Toluene/3 wt%-mCPa |
456/457 |
26/— |
—/0.67 |
—/0.08 |
—/6.34 × 104 |
221
|
|
3tPAB
|
Toluene/3 wt%-mCPa |
456/458 |
23/— |
—/0.75 |
—/0.10 |
—/5.74 × 104 |
221
|
|
mBP-DABNA-Me
|
Toluene/5 wt%-mCP:DPEPOa |
464/467 |
—/— |
—/0.97 |
0.12/0.13 |
—/1.95 × 104 |
222
|
|
pBP-DABNA-Me
|
Toluene/5 wt%-mCP:DPEPOa |
458/462 |
15/22 |
—/0.98 |
0.11/0.18 |
—/6.85 × 104 |
223
|
|
BP-2DPA
|
Toluene/1 wt%-DIC-TRza |
599/601 |
34/37 |
—/0.91 |
—/— |
—/— |
224
|
|
DBP-4DPA
|
Toluene/1 wt%-DIC-TRza |
605/612 |
34/35 |
—/0.96 |
—/— |
—/— |
224
|
|
CzDABNA-NP
|
Toluene/1 wt%-PMMAa |
457/461 |
23/30 |
0.88/0.80 |
—/0.18 |
—/1.6 × 104 |
111
|
|
CzDABNA-NP-TB/H
|
Toluene/1 wt%-PMMAa |
462/465 |
16/30 |
0.86/0.82 |
—/0.18 |
—/1.3 × 104 |
111
|
|
Cz2DABNA-NP-M/TB
|
Toluene/1 wt%-PMMAa |
479/478 |
23/29 |
0.95/0.85 |
—/0.14 |
—/7.3 × 104 |
111
|
|
tCBNDA
|
CH2Cl2/neat/7 wt%-DBFDPOa |
467/470/472 |
28/47/37 |
—/—/0.72 |
—/—/0.05 |
—/—/8.55 × 104 |
225
|
|
DPACzBN2
|
Toluene/3 wt%-26DCzPPya |
460/470 |
22/29 |
—/0.92 |
—/0.12 |
—/2.9 × 104 |
195
|
|
DPACzBN3
|
Toluene/3 wt%-26DCzPPya |
468/475 |
20/27 |
—/0.94 |
—/0.13 |
—/2.1 × 104 |
195
|
|
mICz-DABNA
|
Toluene/3 wt%-mCBP-CNa |
461/465 |
22/26 |
—/0.93 |
0.20/— |
—/2.65 × 104 |
226
|
|
BFCz-DABNA
|
Toluene/3 wt%-mCBP-CNa |
456/462 |
22/26 |
—/0.93 |
0.20/— |
—/2.78 × 104 |
226
|
|
DPMX-CzDABNA
|
Toluene/15 wt%-SF3-TRZa |
471/481 |
22/29 |
—/0.94 |
—/0.11 |
—/6.6 × 104 |
227
|
|
mono-mx-CzDABNA
|
Toluene/3 wt%-mCBPa |
474/— |
34/— |
0.77/0.83 |
0.15/— |
—/2.86 × 105 |
228
|
|
tri-mx-CzDABNA
|
Toluene/3 wt%-mCBPa |
462/— |
26/— |
0.68/0.91 |
0.16/— |
—/2.85 × 105 |
228
|
|
Me-PABO
|
Toluene/5 wt%-PPFa |
453/457 |
21/26 |
—/0.88 |
0.17/— |
—/3.50 × 104 |
229
|
|
Me-PABS
|
Toluene/5 wt%-PPFa |
463/470 |
21/32 |
—/0.92 |
0.15/— |
—/5.59 × 104 |
229
|
|
tDMAC-BN
|
Toluene/1 wt%-PMMAa |
468/— |
26/— |
—/0.90 |
0.15/— |
—/4.80 × 104 |
230
|
|
tDPAC-BN
|
Toluene/1 wt%-PMMAa |
454/— |
19/— |
—/0.94 |
0.17/— |
—/1.16 × 104 |
230
|
|
PhDMAC-BN
|
Toluene/5 wt%-PPFa |
473/483 |
27/34 |
—/0.86 |
0.14/— |
—/5.04 × 104 |
231
|
|
PTZBN3
|
Toluene/26DCzPPya |
468/473 |
30/36 |
—/0.98 |
0.17/— |
—/1.08 × 105 |
196
|
|
PTZBN2
|
Toluene/26DCzPPya |
483/487 |
41/42 |
—/0.95 |
0.15/— |
—/4.51 × 104 |
196
|
|
DOBNA-Oar
|
Toluene/neat |
388/433 |
34/— |
0.65/— |
0.16/— |
—/— |
60
|
|
1B-DTACrs
|
Toluene/5 wt%-mCBPa |
438/443 |
27/28 |
0.99/0.83 |
0.22/0.21 |
—/— |
232
|
|
2B-DTACrs
|
Toluene/5 wt%-mCBPa |
443/448 |
21/24 |
0.64/0.74 |
0.16/0.16 |
—/1.3 × 105 |
232
|
|
Cl-MR
|
Toluene/10 wt%-DPEPOa |
460/474 |
27/36 |
—/0.85 |
0.14/0.13 |
—/9.8 × 104 |
233
|
|
Br-MR
|
Toluene/10 wt%-DPEPOa |
460/474 |
27/36 |
—/0.76 |
0.14/0.13 |
—/5.9 × 105 |
233
|
|
DBT-BN
|
Toluene |
494 |
22 |
— |
— |
— |
234
|
|
DBF-DBN
|
Toluene/2 wt%-CBPa |
514/529 |
22/— |
—/0.90 |
0.24/— |
—/1.1 × 105 |
234
|
|
DBT-DBN
|
Toluene/2 wt%-CBPa |
516/525 |
19/— |
—/0.90 |
0.25/— |
—/7.4 × 105 |
234
|
|
OAB-ABP-1
|
Toluene/1 wt%-PMMAa |
503/506 |
31/34 |
>0.99/0.90 |
—/0.12 |
—/4.0 × 104 |
235
|
|
CzB2-M/P
|
1 wt%-PMMAa |
504 |
39 |
0.87 |
0.06 |
5.1 × 104 |
111
|
|
Cz2B2-M/TB
|
Toluene/1 wt%-PMMAa |
469/483 |
24/38 |
0.70/0.88 |
—/0.11 |
—/3.1 × 104 |
111
|
|
C-BN
|
Toluene |
450 |
19 |
1.00 |
0.21 |
1.8 × 104 |
236
|
|
TPD4PA
|
Toluene/3 wt%-mCBP-CNa |
445/— |
19/— |
—/0.88 |
0.05/— |
—/2.51 × 105 |
237
|
|
tBu-TPD4PA
|
Toluene/3 wt%-mCBP-CNa |
451/— |
19/— |
—/0.90 |
0.06/— |
—/2.44 × 105 |
237
|
|
DTBN-B2N3
|
Toluene/3 wt%-26DCzPPya |
471/475 |
23/— |
0.97/0.96 |
0.10/— |
1.6 × 106/1.2 × 106 |
152
|
|
ν-DABNA
|
Toluene/1 wt%-DOBNA-Oar |
468/467 |
14/18 |
0.74/0.90 |
—/0.017 |
—/2.0 × 105 |
60
|
|
ν-DABNA-O-Me
|
Toluene/1 wt%-PMMAa |
461/464 |
19/24 |
0.90/0.90 |
—/0.029 |
—/1.6 × 105 |
238
|
|
t-Bu-ν-DABNA
|
Toluene/5 wt%-DBFPOa |
467/— |
14/— |
—/0.92 |
0.04/— |
—/2.54 × 105 |
134
|
|
m-ν-DABNA
|
Toluene/3 wt%-DBFPOa |
464/— |
14/— |
—/0.91 |
0.07/— |
—/2.30 × 105 |
240
|
|
4F-ν-DABNA
|
Toluene/3 wt%-DBFPOa |
457/— |
14/— |
—/0.90 |
0.05/— |
—/2.28 × 105 |
240
|
|
4F-m-ν-DABNA
|
Toluene/3 wt%-DBFPOa |
455/— |
14/— |
—/0.89 |
0.07/— |
—/2.10 × 105 |
240
|
|
o-Tol-ν-DABNA-Me
|
Toluene/3 wt%-DBFPOa |
466/472 |
14/— |
0.92/0.99 |
∼0.01/— |
2.95 × 105/3.21 × 105 |
241
|
|
NO-DBMR
|
Toluene/5 wt%-DBFPOa |
458/— |
14/— |
—/0.83 |
0.04/— |
—/1.04 × 105 |
239
|
|
Cz-DBMR
|
Toluene/5 wt%-DBFPOa |
480/— |
14/— |
—/0.82 |
0.04/— |
—/3.72 × 105 |
239
|
|
Π-CzBN
|
Toluene/1 wt%-mCPa |
486/— |
16/— |
0.96/0.98 |
—/0.04 |
—/2.9 × 105 |
242
|
|
V-DABNA-Mes
|
Toluene/1 wt%-PMMAa |
486/484 |
13/16 |
0.79/0.80 |
—/0.0085 |
4.22 × 105/4.4 × 105 |
243
|
|
V-DABNA
|
Toluene/1 wt%-PMMAa |
483/481 |
14/17 |
0.85/0.90 |
—/0.0060 |
7.14 × 105/5.7 × 105 |
244
|
|
V-DABNA-F
|
Toluene/1 wt%-PMMAa |
467/464 |
13/16 |
0.91/0.81 |
—/0.0049 |
5.06 × 105/6.5 × 105 |
244
|
|
CzB4
|
0.1 wt%-PSa |
483 |
14 |
0.92 |
0.012 |
1.8 × 105 |
245
|
|
CzB6
|
0.1 wt%-PSa |
488 |
12 |
0.94 |
0.021 |
3.0 × 105 |
245
|
|
CzB8
|
0.1 wt%-PSa |
491 |
12 |
0.92 |
0.0039 |
6.5 × 105 |
245
|
|
CzB4-oPh
|
0.1 wt%-PSa |
478 |
16 |
0.93 |
0.019 |
6.4 × 104 |
245
|
|
m[B-N]N1
|
Toluene/2 wt%-mCPBCa |
481/483 |
29/28 |
0.93/0.91 |
0.15/0.15 |
3.8 × 103/1.59 × 104 |
248
|
|
m[B-N]N2
|
Toluene/2 wt%-mCPBCa |
491/491 |
34/33 |
0.91/0.90 |
0.10/0.13 |
2.4 × 103/1.44 × 104 |
248
|
|
p[B-N]O
|
Toluene/1 wt%-mCPBCa |
488/497 |
19/23 |
0.93/0.91 |
0.43/0.44 |
—/— |
249
|
|
p[B-N]NO
|
Toluene/1 wt%-mCPBCa |
522/529 |
28/30 |
0.90/0.89 |
0.40/0.37 |
—/— |
249
|
|
p[B-N]N
|
Toluene/1 wt%-mCPBCa |
442/450 |
19/25 |
0.84/0.83 |
0.28/0.26 |
—/— |
249
|
|
BN2
|
THF/5 wt%-mCBPa |
567/— |
97/— |
0.64/— |
—/0.19 |
—/2.10 × 105 |
250
|
|
TCz-BN2
|
THF/5 wt%-mCBPa |
584/— |
108/— |
0.61/— |
—/0.17 |
—/2.44 × 105 |
250
|
|
BN3
|
THF |
694 |
151 |
0.01 |
—/— |
—/— |
250
|
|
ω-DABNA
|
Toluene/1 wt%-DOBNA-Phc |
509/509 |
18/22 |
0.92/0.87 |
—/0.013 |
1.3 × 105/1.2 × 105 |
252
|
|
NBONacene
|
THF/1.5 wt%-TSPO1a |
305 427/410 430 |
40/38 |
0.33/0.71 |
0.31/0.30 |
—/3.74 × 103 |
254
|
|
MesB-DIDOBNA-N
|
THF/1.5 wt%-TSPO1a |
399/402 |
23/19 |
0.33/0.75 |
0.24/0.24 |
—/9.81 × 103 |
253
|
4. Late-stage functionalisation
As shown in previous sections, the photophysical properties of MR compounds, including their TADF characteristics, can be tuned over a wide range through the introduction of appropriate substituents. The routes to compounds with planar tricoordinate boron can be classified into those with early- and late-stage functionalisation relative to borylation–cyclisation. The embedded boron centre sometimes limits certain types of post-reactions, e.g., some nucleophiles form tetra-coordinated species while strongly acidic conditions promote retro-Friedel–Crafts reactions. Moreover, certain functional groups inhibit borylation, e.g., Lewis bases such as pyridyl groups can coordinate borylation reagents such as BBr3, especially in the case of one-shot borylation. Thus, compounds with certain functional groups require appropriate synthesis approaches. In addition, late-stage functionalisation is advantageous for developing a series of derivatives and comparing their properties, even if these compounds can be synthesised using both schemes. Indeed, the first example of donor-functionalised DOBNA (DOBNA-Phenox) was synthesised by one-pot borylation (Fig. 6),37,105 while a wide variety of donor units have recently been developed using late-stage functionalisation. This section discusses compounds prepared using post-borylation functionalisation to understand the factors determining the success of a given approach and therefore covers a wide variety of properties and structures. In particular, the DOBNA scaffold, which holds some distinct merits besides the MR effect with short-range CT, is frequently incorporated as an acceptor unit into D–A-type TADF molecules showing long-range CT. Furthermore, this relatively stable scaffold can be used to synthesise B-doped PAHs with electronic structures largely modulated by heterocycle fusion. In contrast, the functionalisation of the N,B,N-embedded CzBN structure mainly aims to improve the MR effect and OLED performances. The general synthesis procedures are summarised in Schemes 39–41 and 45, which cover most of the compounds appearing in this section. Thus, the structures and properties of these compounds are separately summarised in the following figures and tables. Meanwhile, certain reaction types such as nucleophilic addition and oxidation are discussed in additional schemes in each case.
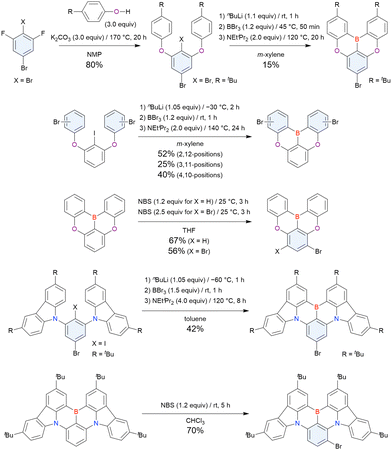 |
| | Scheme 39 Synthesis of bromo-substituted DOBNA and CzBN. The reaction conditions are reported in ref. 122, 119, 256, 184 and 258. | |
4.1 Preparation of key intermediates bearing Br and Bpin groups
In the case of DOBNA, late-stage substitution is possible at 2, 3, 4, 6, and 7-positions via bromo-functionalisation (Fig. 23). In particular, 7-bromo DOBNA derivatives, which can be prepared as shown in Scheme 39, are often subjected to Buchwald–Hartwig coupling to introduce arylamine electron donors.122 The tBu groups at the peripheral 2,12-positions of the terminal phenyl rings reduce aggregation in the solid state. In addition, alkyl groups result in acceptor strengths lower than that of the unsubstituted DOBNA core (R = H), thus inducing a blue shift of the emission spectrum while increasing ΔEST when a donor unit is connected to 7-position.202,255 Regarding borylation, lithium–halogen exchange might preferentially occur at the ortho-bromo atom with respect to the oxygen atoms despite the presence of two bromo atoms. Compounds prepared via lithium–bromine exchange were obtained in yields of up to ∼50%,127,129 and the yields were not improved with lithium–iodine exchange using iodine compounds (X = I).120,124 Dibromo derivatives substituted at the terminal phenyl rings were synthesised in a similar manner in moderate yields.119 On the other hand, 6-monobromo and 6,8-dibromo derivatives were prepared by the bromination of DOBNA with NBS because of the large HOMO coefficients at 6- and 8-positions (Fig. 23).256 The HOMO distribution also indicated the difficulty of brominating DOBNA at 7-position after borylation, which explains the different synthesis strategy depicted in Scheme 39. Most CzBN derivatives used for late-stage reactions bear four tBu groups at 2,5,15,18-positions except for a very limited number of examples because these substituents hinder side reactions and increase the solubility of products.162,257 CzBN derivatives mono-brominated at 10- and 9-positions were obtained using the approach employed to access brominated DOBNA derivatives.184,258 Similarly, intermediates with various cyclised configurations and heteroatom bridges used for late-stage reactions were obtained by essentially identical methods (Fig. 24).116,130,131,133,168,169 DOBNA and CzBN derivatives bearing boronic ester groups, which engage in Suzuki–Miyaura coupling with aryl halides, can be synthesised from brominated derivatives via Miyaura–Ishiyama borylation or iridium-catalysed direct aromatic C–H (Hartwig–Miyaura) borylation (Scheme 40).118,135,185,259–261 An asymmetric bridge with N and O atoms was also demonstrated.95
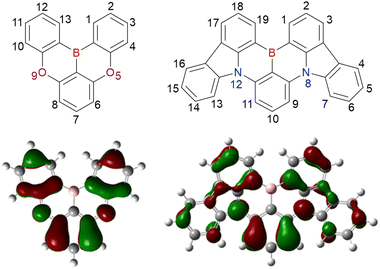 |
| | Fig. 23 Atomic position numbers and HOMO distribution for DOBNA and CzBN. | |
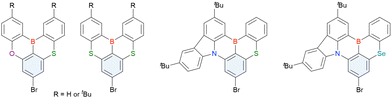 |
| | Fig. 24 Brominated derivatives with S- and Se-bridged structures. | |
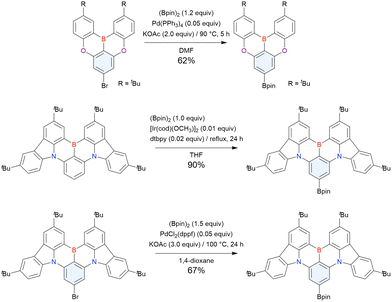 |
| | Scheme 40 Synthesis of Bpin-substituted DOBNA and CzBN. The reaction conditions are reported in ref. 259, 260, and 185. | |
4.2 Donor-functionalised DOBNA-based materials
The DOBNA structure is useful for developing OLED host materials with high triplet energies as well as D–A-type TADF molecules with efficient spin–flip processes.37,112 The introduction of arylamine (e.g., carbazole-, diphenylamine-, 9,10-dihydroacridine- and phenoxazine-based) donors by Buchwald–Hartwig coupling affords highly efficient D–A-type TADF emitters (Scheme 41). We emphasise that one-pot borylation is also useful for developing simple donor-functionalised DOBNA derivatives (Fig. 25 and 26). As mentioned above, the advantages of utilising DOBNA structures are the easy derivation of deep blue and relatively sharp emission owing to the high excited-state energies and rigid structures compared to those of acceptors such as sulfones used in other deep blue emitters, although the emission colour can be tuned to orange-red by changing donor strength. The combination of the DOBNA structure and donors featuring spacers, such as the phenyl group, synthesised by Suzuki–Miyaura coupling leads to weak D–A interactions including through-space CT interactions and results in ultradeep blue (<450 nm) emitter and host materials.259,262,263 The Buchwald–Hartwig amination of DOBNA derivatives typically proceeds in the presence of Pd2(dba)3 (0.01–0.2 equiv.), [tBu3PH]BF4 (0.015–0.3 equiv.), and NaOtBu (1–4 equiv.) in aromatic solvents at >100 °C, although SPhos and XPhos are sometimes used as ligands. Suzuki–Miyaura coupling typically proceeds under the standard conditions of Pd(PPh3)4 (0.02–0.1 equiv.) and aqueous K2CO3 (2–3 equiv.). The yields of these reactions are generally sufficiently high (40–80%).
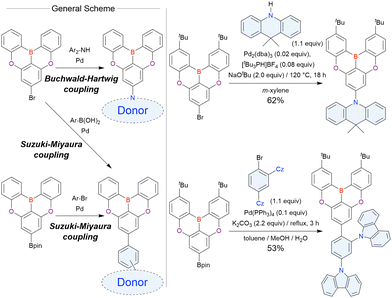 |
| | Scheme 41 Late-stage introduction of donor groups into DOBNA. The right panel shows the specific reaction conditions appearing in ref. 122 and 259. | |
 |
| | Fig. 25 DOBNA-based D–A-type TADF materials with carbazole-based donors at 7-position synthesised by late-stage functionalisation except TMCz-BO. | |
 |
| | Fig. 26 DOBNA-based D–A-type TADF materials with 9,10-dihydroacridine-, phenoxazine- and 5,10-dihydrophenazine-based donors at 7-position synthesised by late-stage functionalisation except DOBNA-Phenox. | |
The HOMO and LUMO energies of DOBNA (about −6.0 and −3.0 eV, respectively) are deeper than those of standard donor units such as di-tBu-carbazole and 9,10-dihydro-9,9-dimethylacridine. Hence, the functionalised DOBNA derivatives in Fig. 25 and 26 show CT excited states where the HOMO and LUMO are well separated in the substituted donor and DOBNA moieties, respectively, as is commonly supported by DFT calculations. However, according to Tables 29 and 30, the FWHMs of these compounds are smaller than those typically observed for D–A-type TADF materials, probably because of a certain contribution of the MR effect. In addition, a markedly high ΦPL, small ΔEST, and high kRISC were simultaneously achieved.
Table 29 Photophysical properties of the materials shown in Fig. 25
| Compound |
Condition |
λ
maxPL [nm] |
FWHM [nm] |
Φ
PL [—] |
ΔEST [eV] |
k
RISC [s−1] |
Ref. |
|
See Table 2 for the full compound name.
Taken from the EL spectrum.
|
|
TMCz-BO
|
Toluene/30 wt%-PPFb |
446/467 |
—/— |
0.81/0.98 |
—/0.020 |
—/1.9 × 106 |
112
|
|
TB-tCz
|
Toluene/neat-film |
415/433 |
45/44b |
0.53/0.41 |
0.23/0.20 |
—/0.85 × 106 |
125
|
|
TB-tPCz
|
Toluene/neat-film |
422/445 |
46/42b |
0.56/0.52 |
0.17/0.15 |
—/1.05 × 106 |
125
|
|
BFCz-TDBA
|
Toluene/20 wt%-mCPa |
393/424b |
36/41b |
0.84/0.58 |
0.24/— |
—/0.91 × 106 |
129
|
|
ICz-TDBA
|
Toluene/20 wt%-mCPa |
416/431b |
49/45b |
0.84/0.63 |
0.19/— |
—/1.27 × 106 |
129
|
|
PIdCz-TDBA
|
Toluene/20 wt%-mCPa |
424/433b |
43/43b |
0.88/0.55 |
0.20/— |
—/1.14 × 106 |
129
|
|
DBA-BFICz
|
Toluene/25 wt%-DBFPOa |
446/476b |
—/64b |
—/0.97 |
0.17/— |
—/3.50 × 105 |
264
|
|
DBA-BTICz
|
Toluene/25 wt%-DBFPOa |
445/476b |
—/64b |
—/0.95 |
0.29/— |
—/6.30 × 105 |
264
|
|
DBA-DI
|
Toluene/30 wt%-mCBP-can |
467/— |
—/— |
—/0.95 |
0.03/— |
—/6.21 × 106 |
202
|
|
TDBA-DI
|
Toluene/20 wt%-DBFPOa |
456/— |
55/65b |
—/0.99 |
0.11/0.05 |
—/1.08 × 106 |
122
|
|
TB-3Cz
|
Toluene/neat-film |
413/433 |
—/— |
0.52/0.97 |
—/— |
—/2.54 × 105 |
123
|
|
TB-3tBuCz
|
Toluene/10 wt%-26DCzPPya |
425/425b |
46/46b |
—/1.00 |
0.05/— |
—/2.70 × 105 |
128
|
|
M3CzB
|
Toluene/20 wt%-DBFPOa |
445/478b |
—/— |
—/0.93 |
0.14/— |
—/1.40 × 105 |
255
|
|
3CzTB
|
Toluene/20 wt%-DBFPOa |
433/470b |
—/— |
—/0.88 |
0.23/— |
—/1.02 × 105 |
255
|
|
TB-P3Cz
|
Toluene/neat-film |
433/460 |
—/— |
0.51/0.92 |
—/— |
—/2.46 × 105 |
123
|
|
TB-DACz
|
Toluene/neat-film |
493/494 |
—/— |
0.52/0.89 |
—/— |
—/1.35 × 105 |
123
|
|
BO-tCzPhICz
|
Toluene/30 wt%-mCPa |
440/444 |
52/54 |
—/0.93 |
—/0.08 |
—/3.44 × 106 |
265
|
|
BO-tCzDICz
|
Toluene/30 wt%-mCPa |
438/451 |
50/50 |
—/0.77 |
—/0.15 |
—/2.09 × 106 |
265
|
|
TDBA-TPDICz
|
Toluene/20 wt%-DBFPOa |
447/462b |
—/58b |
—/0.86 |
0.41/— |
—/1.15 × 105 |
266
|
|
BNB-m
|
Toluene/10 wt%-mCPa |
504/502 |
60/72b |
—/1.00 |
—/0.03 |
—/1.60 × 105 |
267
|
|
BNB-p
|
Toluene/10 wt%-mCPa |
529/518 |
64/74b |
—/0.86 |
—/0.11 |
—/9.88 × 104 |
267
|
|
1BOICz
|
Toluene/20 wt%-mCPBCa |
527/534 |
85/101b |
0.90/1.00 |
—/0.05 |
—/1.12 × 106 |
137
|
|
2BOICz
|
Toluene/20 wt%-mCPBCa |
518/528 |
83/91b |
0.89/1.00 |
—/0.06 |
—/1.18 × 106 |
137
|
|
2BOICz-tBu
|
Toluene/20 wt%-mCPBCa |
—/467b |
—/— |
—/— |
—/0.25 |
—/— |
137
|
Table 30 Photophysical properties of the materials shown in Fig. 26
| Compound |
Condition |
λ
maxPL [nm] |
FWHM [nm] |
Φ
PL [—] |
ΔEST [eV] |
k
RISC [s−1] |
Ref. |
|
See Table 2 for the full compound name.
Taken from the EL spectrum.
|
|
DOBNA-Phenox
|
1 wt%-PMMAb/20 wt%-CBPa |
492/504b |
/— |
0.92/— |
0.06/— |
—/— |
37
|
|
TDBA-Ac
|
Toluene/20 wt%-DBFPOa |
458/— |
50/55b |
—/0.93 |
0.06/— |
—/0.99 × 106 |
122
|
|
TDBA-DPAC
|
Toluene/20 wt%-DPEPOa |
444/450b |
52/60b |
0.88/0.88 |
0.04/0.02 |
—/1.23 × 106 |
127
|
|
OBO-I
|
Toluene/20 wt%-PPFa |
439/451 |
51/60b |
0.47/0.81 |
—/— |
—/1.1 × 106 |
120
|
|
DTBA-PAS
|
Toluene/20 wt%-DPEPOa |
427/435b |
50/54 |
0.35/0.92 |
—/— |
—/1.70 × 106 |
127
|
|
TDBA-SAF
|
Toluene/20 wt%-DPEPOa |
450/456b |
47/55b |
—/0.90 |
—/0.11 |
—/2.1 × 106 |
124
|
|
OBOtSAc
|
Toluene/10 wt%-DPEPOa |
452/454 |
50/50b |
—/0.97 |
—/0.06 |
—/5.59 × 105 |
268
|
|
tBuOBOtSAc
|
Toluene/10 wt%-DPEPOa |
446/450 |
48/48b |
—/0.90 |
—/0.08 |
—/4.74 × 105 |
268
|
|
MH
|
Toluene/10 wt%-PPFa |
477/473b |
—/58b |
—/0.99 |
−0.01/— |
—/1.52 × 106 |
126
|
|
MP
|
Toluene/10 wt%-PPFa |
472/470b |
—/57b |
—/0.98 |
0.02/— |
—/0.58 × 106 |
126
|
|
PP
|
Toluene/10 wt%-PPFa |
475/471b |
—/57b |
—/0.80 |
0.20/— |
—/— |
126
|
|
TDBA-SBA
|
Toluene/20 wt%-DBFPOa |
450/467b |
—/55b |
—/0.89 |
0.01/— |
—/1.37 × 106 |
269
|
|
DBA-SAB
|
Toluene/20 wt%-DPEPOa |
460/472b |
52/62b |
—/0.87 |
—/0.07 |
—/1.9 × 106 |
124
|
|
2TDBA-SBA
|
Toluene/20 wt%-DBFPOa |
448/475b |
—/56b |
—/0.87 |
0.01/— |
—/1.35 × 106 |
269
|
|
PXZBO1
|
Toluene/20 wt%-DBFPOa |
506/507 |
—/— |
—/0.94 |
0.01/— |
—/2.42 × 106 |
270
|
|
PXZBO2
|
Toluene/20 wt%-DBFPOa |
572/552 |
—/— |
—/0.97 |
0.01/— |
—/1.49 × 106 |
270
|
|
PzDBA
|
Toluene/5 wt%-TCTAa:Bepp2a |
610/595b |
—/— |
—/0.85 |
—/0.05 |
—/0.84 × 106 |
271
|
|
PzTDBA
|
Toluene/5 wt%-TCTAa:Bepp2a |
599/576b |
—/— |
—/1.00 |
—/0.06 |
—/1.19 × 106 |
271
|
The seminal work on this series of compounds was reported by Kwon et al. in 2019,122 and numerous following papers described a wide variety of donor units. The emission wavelengths seemed to be linearly correlated with the strength of the D–A interactions. For example, the emission of DBA-BFICz was blue-shifted relative to that of DBA-DI with a stronger donor,202,264 which enabled the fabrication of hyperfluorescence devices using ν-DABNA.60 In addition, the emission of di-tBu-substituted DOBNA derivatives was blue-shifted by ∼10 nm relative to that of unsubstituted DOBNA derivatives, as represented by the TDBA-DIversusDBA-DI case in Table 29 and the OBO-IversusDTBA-PAS case in Table 30 despite the reports from different research groups.120,122,127,202 Stronger D–A interactions also seem to reduce ΔEST, as shown by the comparison between M3CzB and 3CzTB,255 although comparing materials reported by different research groups may be more difficult because of the different criteria used to extract S1 and T1 energies from the spectra. Compounds having relatively strong donors with phenoxazine subunits (DOBNA-Phenox, BNB-m, BNB-p, PXZBO1, and PXZBO2) or strong electron-withdrawing CF3 groups attached to the DOBNA structure (1BOICz and 2BOICz) showed emission peaks at >500 nm,137,267,270i.e., in the region where the hyperfluorescence system using green MR emitters is also operational.163 In addition, A–D–A-type TADF compounds, PzDBA and PzTDBA, which represent a very rare configuration in the TADF family of materials, were developed using a 5,10-dihydrophenazine unit and showed orange to red emission.271 However, these green to red emitters have relatively large FWHM values.
The distinct photophysical features of these materials are their quite short delayed-fluorescence lifetime (τd), weak concentration quenching, good solubility, and high molecular orientational factor. For D–A-type TADF materials, τd typically ranges from several to a few hundred microseconds and is generally shorter than that of MR-type TADF materials. In contrast, some of the materials in Fig. 25 and 26, e.g., TMCz-BO, ICz-TDBA, DBA-BFICz, TDBA-DI, DBA-DI, BO-tCzPhICz, BO-tCzDICz, 1BOICz, 2BOICz, TDBA-Ac, TDBA-SBA, DBA-SAB, and 2TDBA-SBA, showed nanosecond-scale τd values.112,122,124,129,137,202,264,265,269 As shown in Tables 29 and 30, relatively high doping concentrations are generally selected for these compounds. In particular, TB-3Cz, TB-P3Cz, and TB-DACz showed very high ΦPL values of >80% in the neat-film state.123 As a result, aggregation-induced emission (AIE) properties were also investigated for these compounds as well as for TB-tCz and TB-tPCz.123,125 Meanwhile, the introduction of silicon suppressed the broadening of the emission spectra at high doping concentrations.127 The solubility was also good despite the relatively planar nature of the DOBNA structure. For some compounds, namely TB-tCz, TB-tPCz, BFCz-TDBA, ICz-TDBA, PIdCz-TDBA, TB-3Cz, TB-3tBuCz, TB-P3Cz, TB-DACz, BO-tCzPhICz, and BO-tCzDICz, solution-processed OLEDs were fabricated, and high EQEs (>10%) and excellent CIEy values (<0.1) were achieved.123,125,128,129,265 The high EQEs of these compounds were ascribed to not only the high ΦPL but also high degree of horizontal dipole orientation. In general, rigid and linear molecular shapes are favourable for increasing horizontal molecular orientation during vacuum deposition.272 Therefore, linear molecules such as TDBA-DI, 3CzTB, BNB-m, 1BOICz, 2BOICz, TDBA-SAF, DBA-SAB, OBOtSAc, tBuOBOtSAc, TDBA-SBA, 2TDBA-SBA, PzDBA, and PzTDBA are oriented horizontally, as the high aspect ratios of the molecular-shape result in high orientation factors, which follow the order of TDBA-SAF < DBA-SAB, 1BOICz < 2BOICz, OBOtSAc < tBuOBOtSAc, and TDBA-SBA < 2TDBA-SBA.122,124,137,255,267–269,271
The compounds shown in Fig. 27 are slightly different from those in Fig. 25 and 26. Although BCz, BBFCz, and BICz have CT excited states between donor and acceptor units, BCz also has an LE feature within the MR core according to the calculated S1, while the emission spectrum of BICz has a vibrational shoulder.259 Although the donor strength is higher for BICz than for BBFCz, the related emission maxima are similar (Table 31). This behaviour is different from that in the BFCz-TDBAversusICz-TDBA case (Fig. 25), indicating a different nature of D–A interactions. However, these compounds also have the advantages of short τd (∼1 μs), weak concentration quenching with high ΦPL in the neat film, AIE, and solution processability. BCzTC features a D–A interaction through a σ-linker, which results in a very weak CT interaction. Therefore, this compound was used as a bipolar host for a basic TADF material (t4CzIPN), with BDTC adopted as a reference unipolar host.262AC-BO, QAC-BO, and Cz-BO were synthesised as shown in Scheme 42.263 Although the organoboron atom generally acts as a Lewis acid, the Lewis acidity of the boron centre in the DOBNA scaffold might be decreased by the π-electrons of co-planar oxygen and become weaker than that of the carbonyl group. These compounds have through-space CT and thus exhibit deep blue emission. The emission properties of tBOSi and tBOSiCz should be similar to that of 7-phenyl-substituted DOBNA; moreover, the triphenylsilane moiety inhibits intermolecular interactions and thus helps to preserve the deep blue emission in the solid state.273 The combination of a carbonyl-based MR structure (QAO) and DOBNA, named BOQAO and synthesised by Suzuki–Miyaura coupling (Scheme 41), should be critically different from the other abovementioned compounds because the DOBNA structure does not function as an acceptor owing to its LUMO level being shallower than that of QAO.274 Thus, the photophysical properties of BOQAO are similar to those of QAO, although a weak long-range intramolecular CT is involved.
 |
| | Fig. 27 DOBNA derivatives with substituents at 7-position synthesised by late-stage functionalisation. | |
Table 31 Photophysical properties of the materials shown in Fig. 27
| Compound |
Condition |
λ
maxPL [nm] |
FWHM [nm] |
Φ
PL [—] |
ΔEST [eV] |
k
RISC [s−1] |
Ref. |
|
See Table 2 for the full compound name.
|
|
BCz
|
Toluene/neat-film |
405/414 |
25/37 |
0.26/0.61 |
—/0.40 |
—/— |
259
|
|
BBFCz
|
Toluene/neat-film |
405/423 |
25/51 |
0.44/0.86 |
0.26/0.32 |
—/0.90 × 106 |
259
|
|
BICz
|
Toluene/neat-film |
407/431 |
49/53 |
0.21/0.76 |
0.15/0.18 |
—/1.10 × 106 |
259
|
|
BDTC
|
Toluene/neat-film |
403/416, 439 |
—/— |
—/— |
0.28/— |
—/— |
262
|
|
BCzTC
|
Toluene/neat-film |
368, 404/414 |
—/— |
—/— |
0.24/— |
—/— |
262
|
|
AC-BO
|
Toluene/10 wt%-PMMAa |
—/446 |
—/57 |
—/0.77 |
0.13/— |
—/0.23 × 106 |
263
|
|
QAC-BO
|
Toluene/10 wt%-PMMAa |
—/428 |
—/49 |
—/0.83 |
0.20/— |
—/1.61 × 107 |
263
|
|
Cz-BO
|
Toluene/10 wt%-PMMAa |
—/411 |
—/35 |
—/0.89 |
0.29/— |
—/— |
263
|
|
tBOSi
|
Toluene/5 wt%-mCPCNa |
408/414 |
28/28 |
0.51/0.81 |
0.26/— |
—/— |
273
|
|
tBOSiCz
|
Toluene/5 wt%-mCPCNa |
408/414 |
28/28 |
0.52/0.85 |
0.21/— |
—/— |
273
|
|
BOQAO
|
Toluene/5 wt%-CBPa |
474/487 |
28/33 |
—/0.99 |
0.22/— |
—/2.4 × 104 |
274
|
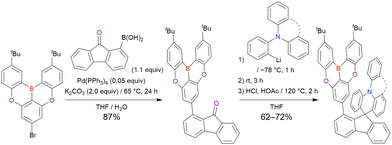 |
| | Scheme 42 Late-stage introduction of donor groups for through-space CT. | |
DOBNA derivatives substituted at positions other than 7-position are summarised in Fig. 28 and Table 32. The molecules described by Wang et al. and Wu et al. were developed in 2019.119,256 The emission of compounds with two donors at the terminal phenyl rings was red-shifted relative to that of 7-substituted compounds (m-Ac-DBNAversusTDBA-Ac; DBA-CzversusTB-tCz).113,119,122,125 This class of compounds also showed high ΦPL in the aggregated states including neat films (>90% for p-AC-DBNA), short τd (920 ns for DBA-DTMCz), and highly oriented alignment (>0.8 for DBA-DTMCz and OBO-II).119–121 Notably, the EQE of the device with a neat OBO-II-based emissive layer reached 23.1%. In addition, the hyperfluorescence device with ν-DABNA doped into a DBA-DTMCz-based emissive layer showed an EQE of 43.9%, which is the highest value reported to date for such devices. Meanwhile, the three isomers of AC-DBNA display temperature-dependent emission and mechanochromism. In a series of OBA derivatives, phenoxazine derivatives (OBA-O and OBA-BrO) showed better ΦPL and EQE values as well as emission maxima at shorter wavelengths than phenothiazine derivatives (OBA-S and OBA-BrS).256 However, OBA-BrS showed the shortest τd of 810 ns. Bromo-substituted derivatives showed elevated kRISC values owing to the heavy-atom effect.
 |
| | Fig. 28 DOBNA-based D–A-type TADF materials with substituents at 2, 3, 4 and 6-positions synthesised by late-stage functionalisation. | |
Table 32 Photophysical properties of the materials shown in Fig. 28
| Compound |
Condition |
λ
maxPL [nm] |
FWHM [nm] |
Φ
PL [—] |
ΔEST [eV] |
k
RISC [s−1] |
Ref. |
|
See Table 2 for the full compound name.
Taken from the EL spectrum.
|
|
m-AC-DBNA
|
CH2Cl2/5 wt%-BCPOa/neat-film |
569/492/514 |
—/—/— |
0.58/0.89/0.73 |
—/0.01/— |
—/2.61 × 105/3.03 × 105 |
119
|
|
p-AC-DBNA
|
CH2Cl2/5 wt%-BCPOa/neat-film |
557/496/516 |
—/—/— |
0.96/0.96/0.94 |
—/0.01/— |
—/11.7 × 105/8.33 × 105 |
119
|
|
m′-AC-DBNA
|
CH2Cl2/5 wt%-BCPOa/neat-film |
568/498/518 |
—/—/— |
0.51/0.87/0.70 |
—/0.03/— |
—/8.44 × 105/3.98 × 105 |
119
|
|
DBA-DmICz
|
Toluene/30 wt%-DBFPOa/neat-film |
448/485b/— |
58/68b/— |
—/0.95/0.83 |
0.12/—/— |
—/0.62 × 106/— |
121
|
|
DBA-DTMCz
|
Toluene/30 wt%-DBFPOa/neat-film |
455/479b/— |
53/58b/— |
—/0.99/0.86 |
0.02/—/— |
—/2.10 × 106/— |
121
|
|
DBA-Cz
|
Toluene/10 wt%-DPEPOa/neat-film |
447/450/494 |
38/47/75 |
—/0.90/0.52 |
0.03/—/— |
—/—/4.6 × 105 |
113
|
|
OBO-II
|
Toluene/20 wt%-PPFa/neat-film |
450/465/476 |
53/58b/— |
0.48/0.98/0.73 |
—/—/— |
—/9.2 × 105/5.0 × 105 |
120
|
|
OBA-O
|
Toluene/10 wt%-mCPa |
444/450 |
—/— |
0.76/0.84 |
—/0.09 |
—/4.28 × 105 |
256
|
|
OBA-S
|
Toluene/10 wt%-mCPa |
456/470 |
—/— |
0.63/0.75 |
—/0.09 |
—/2.76 × 105 |
256
|
|
OBA-BrO
|
Toluene/10 wt%-mCPa |
470/476 |
—/— |
0.84/0.92 |
—/0.04 |
—/8.97 × 105 |
256
|
|
OBA-BrS
|
Toluene/10 wt%-mCPa |
478/470 |
—/— |
0.53/0.55 |
—/0.07 |
—/8.41 × 105 |
256
|
The synthesis and properties of DOBNA-based polymers are summarised in Scheme 43 and Table 33. After the introduction of the vinyl group by the Stille coupling of 7-brominated DOBNA derivatives with tributylvinylstannane or by their Suzuki–Miyaura coupling with (para-vinylphenyl)boronic acid under palladium catalysis, co-polymers with donor units were synthesised by free radical polymerisation using 2,2′-azobis(isobutyronitrile) (AIBN) as an initiator.116,132,275 Compounds of the PBO series showed through-space D–A interactions, which resulted in PBO-TB-5 and AC-BO showing similar emission maxima (Fig. 27).132,263 Although the ΦPL values of the polymers were lower than those of small molecules, sufficiently small ΔEST values were achieved. The electron-donating tBu and electron-withdrawing F groups attached to the DOBNA scaffold modulated D–A interactions in a similar manner to that observed for small molecules in Fig. 25 and 26. The emission maxima of PBO derivatives shifted to longer wavelengths (by ∼10 nm) with increasing DOBNA molar fraction. On the other hand, the effect of this parameter on the emission spectra is more obvious for the ABP series.275 In view of the absence of intramolecular through-space CT, which was confirmed in solution-phase emission spectra, the intermolecular D–A interactions in the film state caused the appearance of a new CT emission peak, resulting in a large variation of the CT/LE ratio upon going from ABP91 to ABP55. Solution-processed OLEDs were fabricated using PBO derivatives as the emitter or ABP derivatives as the host for t4CzIPN, and EQEs of up to 15.0 and 22.2%, respectively, were achieved.
 |
| | Scheme 43 Synthesis of DOBNA-based polymers reported in ref. 132, 275, and 116. | |
Table 33 Photophysical properties of the materials shown in Scheme 43a
| Compound |
Condition |
λ
maxPL [nm] |
FWHM [nm] |
Φ
PL [—] |
ΔEST [eV] |
Ref. |
|
k
RISC values were not reported.
|
|
PBO-TB-5
|
Neat-film |
444 |
— |
0.32 |
0.09 |
132
|
|
PBO-TB-10
|
Neat-film |
453 |
— |
0.37 |
0.09 |
132
|
|
PBO-H-5
|
Neat-film |
448 |
— |
0.44 |
0.06 |
132
|
|
PBO-H-10
|
Neat-film |
455 |
— |
0.48 |
0.07 |
132
|
|
PBO-F-5
|
Neat-film |
471 |
— |
0.65 |
0.01 |
132
|
|
PBO-F-10
|
Neat-film |
480 |
— |
0.70 |
0.02 |
132
|
|
ABP91
|
Neat-film |
407 |
— |
— |
0.30 |
275
|
|
ABP73
|
Neat-film |
412, 436 |
— |
— |
0.29 |
275
|
|
ABP55
|
Neat-film |
414, 442 |
— |
— |
0.29 |
275
|
|
PS-BOO
|
Toluene |
398 (LE) |
33 |
0.73 |
— |
116
|
|
PS-BOS
|
Toluene |
435 (LE) |
31 |
0.65 |
— |
116
|
|
PS-BSS
|
Toluene |
456 (LE) |
30 |
0.59 |
— |
116
|
|
PAc-BOO
|
Toluene |
457 (CT) |
63 |
0.44 |
— |
116
|
|
PAc-BOS
|
Toluene |
434 (LE), 475 (CT) |
62 |
0.56 |
— |
116
|
|
PAc-BSS
|
Toluene |
455 (LE) |
30 |
0.60 |
— |
116
|
Wang et al. developed S-bridged DOBNA analogues (BOS and BSS) and investigated the photophysical properties of the parent structures and their polymers.116 The introduction of sulphur reduced the energy gap by increasing HOMO and decreasing LUMO energies, thus resulting in deep blue emission with emission maxima of 434 nm (BOS) and 457 nm (BSS) without any substitutions. These maxima were red-shifted by 38–61 nm relative to that of unsubstituted DOBNA (BOO) at 396 nm. Importantly, the S-bridged structure preserved the narrow FWHMs (30, 29, and 27 nm for BOO, BOS and BSS, respectively). Moreover, kRISC increased by one order of magnitude because of the enhancement of SOC via the heavy-atom effect of sulphur.55 As a result, the delayed fluorescence ratio in total emission increased from 4% for BOO to 81% and 90% for BOS and BSS, respectively. Polystyrene (PS)-based polymers (PS-BOO, PS-BOS, and PS-BSS) showed emission similar to that of their parent DOBNA analogues. Interestingly, donor-based polymers showed different emission profiles because of the formation of CT excited states. The emission of PAc-BOO was broad, dependent on solvent polarity, and red-shifted by 59 nm compared to that of PS-BOO, which was assigned to long-range CT. In contrast, PAc-BSS showed only one narrow emission peak corresponding to the LE emission of MR BSS with short-range CT. PAc-BOS showed two (LE and CT) emission bands. This feature was clearly correlated with the increase in the HOMO energy of BOO, BOS, and BSS, and the small offset of HOMO levels between donor and acceptor units hindered CT formation. The OLED with PAc-BSS showed the highest EQE of 13.1% for blue TADF polymers with a CIEy of <0.15.
Small molecules with S-bridged DOBNA analogues and donor units connected at 7-position were subsequently developed (Fig. 29 and Table 34). Similar to the polymer case mentioned above, the higher HOMO levels of S-bridged structures resulted in red-shifted and LE-dominated emission. PhCz-TOSBA and TPA-TOSBA showed similar emission spectra in toluene, whereas the emission of TPA-TOSBA with a stronger donor exhibited partial long-range CT character.130 For TSBA-Cz and TSBA-PhCz, both S1 and T1 states were LE-dominant, resulting in substantially similar photophysical properties based on the MR effect.131 In BSS-Cz, BSS-PA, BSS-SpAc, and BSS-Ac, the HOMO levels of donor units become shallower in the order of Cz < PA < SpAc < Ac.133 Similar to TSBA-Cz, BSS-Cz showed LE-dominated emission with a narrow FWHM of 28 nm because of the small HOMO offset. Notably, the emission wavelength was red-shifted by the incorporation of tBu groups, which differed from the CT emission in D–A-type molecules in Fig. 25 and 26, suggesting pure MR emission. Although no CT between BSS and 9,10-dihydro-9,9-dimethylacridine was observed in the PAc-BSS polymer, BSS-SpAc and BSS-Ac showed long-range CT emission in addition to LE emission.
 |
| | Fig. 29 S-Bridged DOBNA analogues synthesised by late-stage functionalisation. | |
Table 34 Photophysical properties of the materials shown in Fig. 29
| Compound |
Condition |
λ
maxPL [nm] |
FWHM [nm] |
Φ
PL [—] |
ΔEST [eV] |
k
RISC [s−1] |
Ref. |
|
See Table 2 for the full compound name.
|
|
PhCz-TOSBA
|
Toluene/10 wt%-26DCzPPya |
444/454 |
32/41 |
—/0.61 |
0.23/— |
—/4.08 × 104 |
130
|
|
TPA-TOSBA
|
Toluene/10 wt%-26DCzPPya |
447/467 |
34/67 |
—/0.62 |
0.36/— |
—/1.91 × 104 |
130
|
|
TSBA-Cz
|
Toluene/10 wt%-26DCzPPya |
463/469 |
30/40 |
—/0.80 |
0.21/— |
—/5.29 × 104 |
131
|
|
TSBA-PhCz
|
Toluene/10 wt%-26DCzPPya |
470/478 |
36/40 |
—/0.80 |
0.18/— |
—/7.62 × 104 |
131
|
|
BSS-Cz
|
Toluene/1 wt%-PSa |
455/457 |
28/29 |
0.86/— |
0.14/— |
—/1.5 × 105 |
133
|
|
BSS-PA
|
Toluene/1 wt%-PSa |
454/— |
35/— |
0.66/— |
0.13/— |
—/1.6 × 105 |
133
|
|
BSS-SpAc
|
Toluene/1 wt%-PSa |
456, 505/— |
101/— |
0.89/— |
0.07/— |
—/1.5 × 105, 3.0 × 105 |
133
|
|
BSS-Ac
|
Toluene/1 wt%-PSa |
456, 513/— |
105/— |
0.90/— |
0.04/— |
—/1.5 × 105, 5.2 × 105 |
133
|
Scheme 44 shows some examples of the fused π-extension of DOBNA derivatives. The synthesis of Ph-OBNA involved Suzuki–Miyaura coupling followed by cyclisation and N-aryl coupling reactions.276 Although Ph-OBNA showed TADF properties, its planar structure led to excimer formation at high concentrations and ACQ. Another fused DOBNA derivative, DOBDiKTa, has a carbonyl-based MR structure (QAO, here called DiKTa) composed of two merged MR fragments.277 Although DOBDiKTa is composed of the same units as BOQAO in Fig. 27, the fused benzene is substituted by the electron-withdrawing boron atom and carbonyl group in a meta relationship as well as the electron-donating nitrogen and oxygen atoms in the same relationship, effectively merging the MR effect. The synthesis of DOBDiKTa involved two coupling reactions followed by saponification and two-fold intramolecular Friedel–Crafts acrylation promoted by SnCl2 as a Lewis acid. The emission maximum of DOBDiKTa was slightly blue-shifted relative to that of QAO. In addition, the TADF properties including ΦPL, ΔEST, and kRISC were superior to those of each individual MR unit.
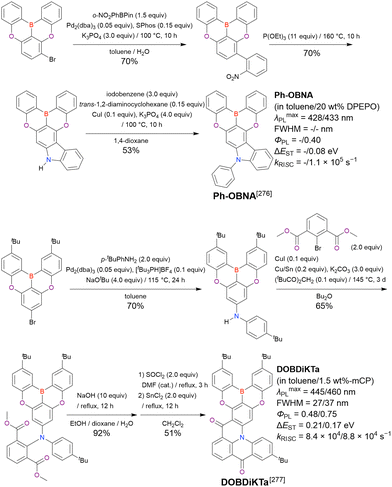 |
| | Scheme 44 Late-stage ring-fusion of DOBNA derivatives. | |
4.3 Functionalised CzBN
The late-stage synthesis strategies for CzBN derivatives are fundamentally identical to those for DOBNA derivatives, as shown in Scheme 45 corresponding to Scheme 41. Similar to the DOBNA case, one-pot borylation is seldom selected for synthesising some donor-substituted CzBN derivatives but may still be useful given the reports of exactly the same compounds prepared by both methods (vide infra). The standard reaction conditions for the Buchwald–Hartwig amination of CzBN derivatives are Pd2(dba)3 (0.02–0.08 equiv.), [tBu3PH]BF4 (0.08–0.32 equiv.), and NaOtBu (2–4 equiv.) in aromatic solvents at >80 °C. The Suzuki–Miyaura coupling conditions are broadly variable, with a representative example being Pd(PPh3)4 (∼0.05 equiv.) and K2CO3 (∼2 equiv.) in THF/H2O at reflux. As most materials discussed here are based on 3,6-di-tert-butyl-9H-carbazole, the photophysical properties of the tetra-tBu CzBN skeleton (originally named DtBuCzB, but referred to here as BCzBN to correspond to the substituted CzBN) are the basis for evaluating substitution effects. Typical emission maximum, FWHM, ΦPL, ΔEST, and kRISC values for BCzBN in toluene are 481–484 nm, 22–23 nm, 91–98%, 0.13–0.15 eV, and 1.4–3.0 × 104 s−1, respectively.154,257,278 In general, the MR effect with narrow emission is maintained even for relatively strong donor substituents because the HOMO of CzBN is shallower than that of DOBNA. Changes in the excited-state nature upon going from MR with short-range CT to long-range D–A-type CT could be observed by comparing phenoxazine-substituted derivatives, with the HOMO levels of the CzBN moiety slightly decreasing upon going from CzBN3 to CzBN1 (Fig. 30 and Table 35).257 Thus, CzBN1 showed two emission bands, namely a sharp band at 471 nm and a broad band at 533 nm. Although CzBN3 (aka BN-PXZ176 in Section 2 and BNCz-PXZ) exhibited a small FWHM comparable to that of BCzBN in toluene, a new broad band at ∼600 nm emerged in polar solvents.187 In contrast, BNCz-DMAC (aka BN-AC in Section 2)176 and BNCz-SAF with weaker donors showed a single narrow emission band even in polar solvents with a small positive solvatochromic effect, which is a common feature of MR materials. These results indicate that the HOMO of BCzBN was very slightly shallower than that of the phenoxazine moiety. Despite the negligible change in the shape of the emission spectrum upon the introduction of diarylamine donors, these substituents affected the HOMO–LUMO gap of the CzBN core and the electronic properties of the related derivatives. The HOMO of CzBN has no coefficient on the atom in 10-position (Fig. 23), unlike the corresponding LUMO. The opposite HOMO–LUMO behaviour is observed at 9- and 11-positions. Thus, the introduction of electron-donating substituents such as arylamines directly at 10-position, which is para-position with respect to the B atom, increases the energy gap. In fact, the emission spectra of CzBN3, BNCz-DMAC, BNCz-SAF, and SPBAC-tCzBN were slightly blue-shifted relative to that of BCzBN.187,257,279 In contrast, the emission spectra were slightly red-shifted upon the introduction of electron-withdrawing substituents (Fig. 31 and Table 36). Note that the introduction of electron-donating arylamines with a phenyl spacer caused a red shift because of the contribution of long-range CT characteristics (vide infra, e.g., o-SPAC-tCzBN).279 The important advantage of introducing substituents is the incorporation of intersegmental CT triplet states such as the T2 level, which accelerates RISC. This level is silently induced without perturbing the lowest excited singlet states. Thus, the emission properties of, e.g., CzBN3 and SPAC-tCzBN are substantially similar to those of BCzBN, while the kRISC values of these two compounds exceed that of BCzBN by about one order of magnitude. The emission of a heptagonal tribenzo[b,d,f]azepine derivative, TBA-BCz-BN, is further blue-shifted relative to that of carbazole and diphenylamine derivatives.111,172,186DCz-BSN and DCz-BSeN synthesised based on this strategy and featuring asymmetric S- and Se-bridged CzBN structures, respectively, exhibit high-colour-purity emission in the deep blue region and large krisc.168,169 Compounds bearing electron-donating groups with a phenyl spacer, namely BNCz-pTPA, BNCz-mTPA, o-SPAC-tCzBN, S-Cz-BN, D-Cz-BN, BN-CP1, BN-CP2, BN-PhOH, and BN-PhOCH3, showed slightly red-shifted emission, while some compounds with relatively strong donors, namely BNCz-pTPA, BNCz-mTPA, and o-SPAC-tCzBN, exhibited higher kRISC because of the distinct contribution of the long-range CT triplet state.175,278–281 Although the basic photophysical properties of BN-PhOH and BN-PhOCH3 are not significantly different, the synthesis of methoxy-substituted compounds might not be straightforward because of demethylation by BBr3; however, the SMe group can be preserved.175,178 Thus, the methoxy derivative was synthesised using the Williamson ether synthesis in the presence of sodium hydride (Scheme 46).175
 |
| | Scheme 45 General scheme for the late-stage functionalisation of CzBN by Buchwald–Hartwig and Suzuki–Miyaura couplings. | |
 |
| | Fig. 30 CzBN derivatives with donor substituents at the 10-position and related chalcogen-bridged compounds synthesised by late-stage functionalisation. | |
Table 35 Photophysical properties of the compounds shown in Fig. 30
| Compound |
Condition |
λ
maxPL [nm] |
FWHM [nm] |
Φ
PL [—] |
ΔEST [eV] |
k
RISC [s−1] |
Ref. |
|
See Table 2 for the full compound name.
Taken from the EL spectrum.
|
|
CzBN1
|
Toluene/5 wt%-DMIC-TRZa |
471, 533/523b |
78/75b |
0.95/0.95 |
0.13/— |
2.06 × 106/1.3 × 106 |
257
|
|
CzBN2
|
Toluene/5 wt%-DMIC-TRZa |
477, 522/481b |
75/75b |
0.97/0.97 |
0.14/— |
7.82 × 105/4.4 × 105 |
257
|
|
CzBN3
|
Toluene/5 wt%-DMIC-TRZa |
478/487b |
21/27b |
0.99/0.98 |
0.15/— |
3.27 × 105/2.8 × 105 |
257
|
|
BNCz-PXZ
|
Toluene/3 wt%-mCBPa |
477/482 |
23/34 |
—/0.84 |
0.12/— |
—/3.11 × 105 |
187
|
|
BNCz-DMAC
|
Toluene/3 wt%-mCBPa |
478/484 |
22/26 |
—/0.83 |
0.13/— |
—/1.20 × 106 |
187
|
|
BNCz-SAF
|
Toluene/3 wt%-mCBPa |
478/484 |
23/25 |
—/0.86 |
0.15/— |
—/7.4 × 105 |
187
|
|
SPAC-tCzBN
|
Toluene/10 wt%-PhCzBCza |
481/480b |
20/24b |
—/0.83 |
0.13/— |
—/0.9 × 105 |
279
|
|
SPBAC-tCzBN
|
Toluene/10 wt%-PhCzBCza |
480/480b |
20/25b |
—/0.89 |
0.13/— |
—/1.2 × 105 |
279
|
|
TBA-BCz-BN
|
Toluene/3 wt%-mCBPa |
468/472 |
22/28 |
—/0.86 |
0.14/— |
—/— |
186
|
|
DCz-BSN
|
Toluene/1 wt%-PSa |
463/462 |
26/— |
—/0.88 |
0.12/— |
—/1.04 × 105 |
168
|
|
DCz-BSeN
|
Toluene/1 wt%-PSa |
472/467 |
28/— |
—/0.93 |
0.14/— |
—/8.8 × 106 |
169
|
|
BNCz-pTPA
|
Toluene/1 wt%-mCBPa |
487/490b |
22/29b |
0.95/— |
0.11/— |
—/4.45 × 105 |
280
|
|
BNCz-mTPA
|
Toluene/1 wt%-mCBPa |
489/490b |
22/30b |
0.92/— |
0.12/— |
—/2.29 × 105 |
280
|
|
o-SPAC-tCzBN
|
Toluene/10 wt%-PhCzBCza |
490/492b |
21/26b |
—/0.86 |
0.11/— |
—/1.1 × 105 |
279
|
|
S-Cz-BN
|
Toluene/1 wt%-mCBPa/neat-film |
490/488b/— |
23/26b/40 |
0.94/0.95/0.47 |
0.16/—/— |
1.8 × 104/2.9 × 104/— |
278
|
|
D-Cz-BN
|
Toluene/1 wt%-mCBPa/neat-film |
490/488b/— |
22/24b/26 |
0.98/0.98/0.54 |
0.14/—/— |
1.8 × 104/3.0 × 104/— |
278
|
|
BN-CP1
|
Toluene/1 wt%-mCBPa/neat-film |
490/496/502 |
23/25/28 |
—/0.93/0.40 |
0.12/—/— |
—/1.56 × 104/— |
281
|
|
BN-CP2
|
Toluene/1 wt%-mCBPa/neat-film |
490/496/521 |
23/26/48 |
—/0.91/0.25 |
0.13/—/— |
—/1.43 × 104/— |
281
|
|
BN-PhOH
|
Toluene/3 wt%-mCBPa |
485/494 |
26/32b |
0.80/— |
0.14/— |
—/2.85 × 104 |
175
|
|
BN-PhOCH3
|
Toluene/3 wt%-mCBPa |
485/494 |
26/29b |
0.78/— |
0.15/— |
—/2.98 × 104 |
175
|
|
SF1BN
|
Toluene/2 wt%-mCBPa |
493/492b |
23/28b |
0.90/0.93 |
0.13/— |
—/9.12 × 104 |
282
|
|
SF3BN
|
Toluene/2 wt%-mCBPa |
493/492b |
25/28b |
0.86/0.90 |
0.15/— |
—/3.31 × 104 |
282
|
|
BN-36Cz-BN
|
Toluene/5 wt%-DMIC-TRZa |
488/497 |
22/33 |
—/0.65 |
0.125/— |
—/1.6 × 105 |
283
|
|
BN-27Cz-BN
|
Toluene/5 wt%-DMIC-TRZa |
489/499 |
22/32 |
—/0.66 |
0.124/— |
—/0.80 × 105 |
283
|
|
PCzBN1
|
Toluene/neat-film |
421, 487/430, 452, 491 |
25/33 |
—/0.58 |
—/0.14 |
—/8.2 × 104 |
284
|
|
PCzBN3
|
Toluene/neat-film |
421, 487/423, 451, 496 |
25/42 |
—/0.51 |
—/0.13 |
—/8.6 × 104 |
284
|
|
PCzBN5
|
Toluene/neat-film |
421, 488/501 |
25/43 |
—/0.43 |
—/0.13 |
—/2.18 × 105 |
284
|
|
D1-BNN
|
Toluene/10 wt%-mCPa |
474/483b |
21/26 |
—/0.68 |
0.15/— |
—/1.55 × 103 |
183
|
|
D2-BNN
|
Toluene/10 wt%-mCPa |
473/478b |
21/24 |
—/0.80 |
0.16/— |
—/2.35 × 103 |
183
|
|
D3-BNN
|
Toluene/10 wt%-mCPa |
473/477b |
21/24 |
—/0.92 |
0.17/— |
—/3.94 × 103 |
183
|
|
BSeN-CZ
|
Toluene/1 wt%-PSa |
476/— |
26/30 |
—/0.76 |
0.12/— |
—/8.5 × 106 |
285
|
|
BSeN-DCZ
|
Toluene/1 wt%-PSa |
475/— |
26/29 |
—/0.79 |
0.12/— |
—/9.0 × 106 |
285
|
|
BSeN-TCZ
|
Toluene/1 wt%-PSa |
474/— |
26/30 |
—/0.85 |
0.12/— |
—/9.1 × 106 |
285
|
 |
| | Fig. 31 CzBN derivatives with acceptor substituents at 10-position synthesised by late-stage functionalisation. | |
Table 36 Photophysical properties of the materials shown in Fig. 31
| Compound |
Condition |
λ
maxPL [nm] |
FWHM [nm] |
Φ
PL [—] |
ΔEST [eV] |
k
RISC [s−1] |
Ref. |
|
See Table 2 for the full compound name.
Taken from the EL spectrum.
|
|
2PyBN
|
Toluene/1 wt%-DMIC-TRZa |
499/507 |
21/26b |
—/0.94 |
—/0.02 |
—/7.7 × 104 |
182
|
|
3PyBN
|
Toluene/1 wt%-DMIC-TRZa |
490/497 |
23/27b |
—/0.90 |
—/0.03 |
—/8.7 × 104 |
182
|
|
4PyBN
|
Toluene/1 wt%-DMIC-TRZa |
495/506 |
24/27b |
—/0.86 |
—/0.02 |
—/9.5 × 104 |
182
|
|
DtCzB-DPTRZ
|
Toluene/3 wt%-PhCzBCza |
521/536 |
24/43 |
0.94/0.87 |
0.18/— |
—/0.10 × 104 |
261
|
|
DtCzB-TPTRZ
|
Toluene/3 wt%-PhCzBCza |
501/520 |
27/41 |
0.97/0.95 |
0.11/— |
—/1.08 × 104 |
261
|
|
DtCzB-PPm
|
Toluene/3 wt%-PhCzBCza |
499/510 |
25/36 |
0.96/0.94 |
0.11/— |
—/1.02 × 104 |
261
|
|
DtCzB-CNPm
|
Toluene/3 wt%-PhCzBCza |
515/540 |
36/45 |
0.93/0.87 |
0.15/— |
—/0.14 × 104 |
261
|
|
BN-R
|
Toluene/1 wt%-NPBa:DMFBD-TRZa |
624/620 |
46/49 |
0.94/0.90 |
0.11/— |
—/1.1 × 104 |
162
|
|
TCzBN-BP
|
Toluene/3 wt%-SF3TRZa |
497/508 |
29/33 |
—/0.98 |
0.14/0.09 |
—/7.7 × 104 |
287
|
|
TCzBN-FP
|
Toluene/5 wt%-SF3TRZa |
516/542 |
37/56 |
—/0.75 |
0.17/0.14 |
—/5.1 × 104 |
287
|
|
BN-XTO
|
Toluene/5 wt%-DMIC-TRZa |
—/515 |
26/33 |
—/0.93 |
—/0.08 |
—/3.0 × 104 |
288
|
|
BN-STO
|
Toluene/5 wt%-DMIC-TRZa |
—/517 |
29/34 |
—/0.96 |
—/0.13 |
—/1.2 × 105 |
288
|
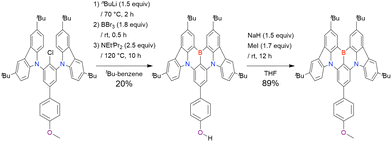 |
| | Scheme 46 Synthesis of a methoxy-substituted compound. | |
MR emitters generally suffer from strong molecular aggregation owing to their planar structures and thus experience substantial deterioration of emission properties at high doping concentrations. In particular, concentration quenching-induced decreases in ΦPL and Dexter energy transfer-induced triplet loss are serious issues in the corresponding OLEDs. Thus, MR emitter-based OLEDs are usually fabricated using very low doping concentrations such as 1 wt%. The structural modifications of MR emitters also aim to suppress aggregation. Among the available methods, the steric wrapping strategy is particularly efficient. For example, the ΦPL of a 30 wt% BCzBN-doped film was less than 50%, while doping with 30 wt% S-Cz-BN and D-Cz-BN maintained high ΦPL values of >80–90%.278 In addition, spectral broadening was found to be negligible for D-Cz-BN even in the neat-film. This trend was also confirmed by the comparison between BN-CP1 and BN-CP2 simultaneously reported by different groups.281 Two derivatives with the more electrically neutral spiro-9,9′-bifluorene moiety, namely SF1BN and SF3BN, were examined to prove the influence of steric hindrance on photophysical properties.282 Although SF3BN, D-Cz-BN, and BN-CP1 were synthesised by routine Suzuki–Miyaura coupling (Scheme 45), SF1BN had to be synthesised by a different method (Scheme 47) because the bulky fluorene moiety prevented the catalytic cycle of this coupling.
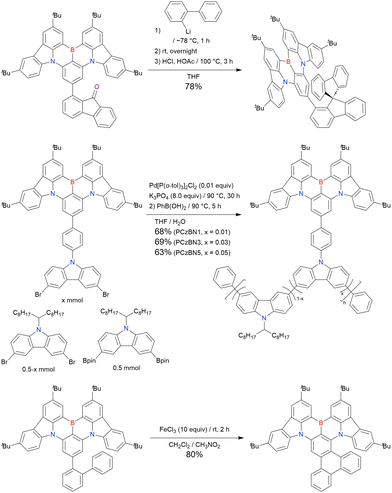 |
| | Scheme 47 Sequential synthesis of CzBN derivatives involving Suzuki–Miyaura coupling in Scheme 45. Top: Nucleophilic addition of 2-lithio-1,1′-biphenyl to a carbonyl group followed by intramolecular condensation-cyclisation. Middle: Polymerisation reaction. The dibromo precursor was prepared from CzBN-Bpin and 3,6-dibromo-9-(4-iodophenyl)-9H-carbazole. Bottom: Oxidative Scholl coupling. | |
Solution-processable BCzBN derivatives were developed using N-branched-alkyl carbazoles. Small molecules with two BCzBN moieties, BN-36Cz-BN and BN-27Cz-BN, showed relatively weak ACQ.283 A polycarbazole-based MR polymer (PCzBN) was prepared by grafting the BCzBN moiety as a pendant (Scheme 47).284 MR dendrimers were also developed by introducing up to third-generation carbazole dendrons at the periphery of the BCzBN skeleton.183,285 Compounds based on a third-generation carbazole dendron (D3-BNN and BSeN-TCZ) showed higher ΦPL than those based on a first-generation dendron (D1-BNN and BSeN-Cz). Concentration quenching and emission spectrum broadening were also suppressed when the third-generation dendron was used at high doping concentrations.
Compounds with acceptor moieties at 10-position are summarised in Fig. 31 and Table 36. The presence of pyridine-type substructures may disorder the tandem lithiation-borylation-annulation and one-shot electrophilic C–H borylation. Some works also reported these attempts and the failure of the corresponding reactions.182,261 Thus, compounds 2PyBN, 3PyBN, 4PyBN, DtCzB-DPTRZ, DtCzB-TPTRZ, DtCzB-PPm, and DtCzB-CNPm were prepared by late-stage reactions, with the synthesis of the PyBN series involving CuCl-assisted Suzuki–Miyaura coupling.286 Note that in pyridyl DOBNA synthesis, the pyridine-functionalised precursor underwent lithiation-borylation to afford the desired product in good yield.139 Although all of these pyridine-based BCzBN derivatives exhibited red-shifted emission compared to that of the parent BCzBN, the intramolecular hydrogen-bonding in 2PyBN and DtCzB-DPTRZ caused molecular rigidity, enhanced conjugation, and more strongly red-shifted emission. The replacement of the tBu group by a bis[4-(2-phenyl-2-propyl)phenyl]amine unit in BN-R substantially increased the HOMO energy, affording a red emitter with a narrow spectrum.162 The bulky donor moiety also helps to suppress aggregation and enhance solubility. The carbonyl moiety may also cause unpredictable side reactions during borylation. Thus, some compounds have been developed using late-stage reactions. Similar to the case of relatively strong donors as substituents,187 long-range CT became dominant with relatively strong acceptors (TCzBN-BPversusTCzBN-FP).287 In addition to the management of intersegmental CT triplet states, the heavy-atom effect further increased kRISC in BN-STO.288
BCzBN derivatives with donors and acceptors at 9-position (Fig. 32 and Table 37) were synthesised through bromination using NBS (Scheme 39) followed by Suzuki–Miyaura coupling.258 Polycyclisation extended at 9- and 10-positions formed a triphenylene core in BN-TP (Fig. 33 and Table 38), which was difficult to obtain from triphenylene as the starting material. Alternatively, this species could be synthesised from BCzBNvia a three-step process including the Hartwig-Miyaura C–H borylation, Suzuki–Miyaura coupling, and the Scholl reaction in the presence of FeCl3 (Scheme 47).289 The large HOMO coefficient at 9-position was conducive to intramolecular cyclisation. Nitrogen-containing compounds BN-TP-N1–4 were synthesised using the same approach, with the construction of phenyl-pyridine moieties in BT-TP-N2 and BT-TP-N3 performed using a different sequence.290
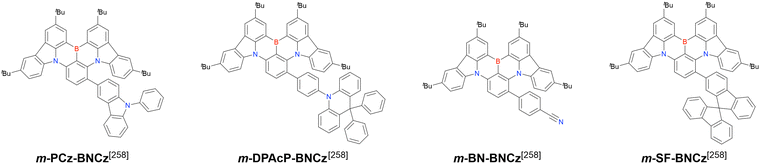 |
| | Fig. 32 CzBN derivatives with substituents at 9-position synthesised by late-stage functionalisation. | |
Table 37 Photophysical properties of the materials shown in Fig. 32
| Compound |
Condition |
λ
maxPL [nm] |
FWHM [nm] |
Φ
PL [—] |
ΔEST [eV] |
k
RISC [s−1] |
Ref. |
|
See Table 2 for the full compound name.
|
|
m-PCz-BNCz
|
Toluene/3 wt%-PhCzBCza |
494/500 |
25/33 |
0.97/0.98 |
0.15/— |
—/1.29 × 104 |
258
|
|
m-DPAcP-BNCz
|
Toluene/3 wt%-PhCzBCza |
491/498 |
26/34 |
0.97/0.97 |
0.14/— |
—/1.16 × 104 |
258
|
|
m-BN-BNCz
|
Toluene/3 wt%-PhCzBCza |
488/496 |
25/32 |
0.93/0.95 |
0.16/— |
—/0.93 × 104 |
258
|
|
m-SF-BNCz
|
Toluene/3 wt%-PhCzBCza |
491/498 |
25/32 |
0.96/0.98 |
0.16/— |
—/1.16 × 104 |
258
|
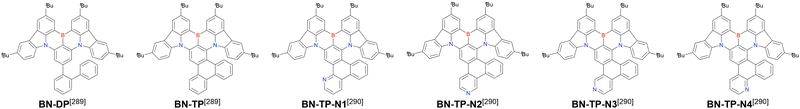 |
| | Fig. 33 CzBN derivatives with fused π-extension synthesised by late-stage functionalisation. | |
Table 38 Photophysical properties of the compounds shown in Fig. 33
| Compound |
Condition |
λ
maxPL [nm] |
FWHM [nm] |
Φ
PL [—] |
ΔEST [eV] |
k
RISC [s−1] |
Ref. |
|
See Table 2 for the full compound name.
|
|
BN-DP
|
Toluene |
490/— |
23/— |
0.94/— |
0.16/— |
—/— |
289
|
|
BN-TP
|
Toluene/3 wt%-/10 wt%-PhCzBCza |
523/529/537 |
34/36/39 |
0.96/0.96/0.92 |
0.14/—/— |
—/2.09 × 104/2.26 × 104 |
289
|
|
BN-TP-N1
|
Toluene/3 wt%-PhCzBCza |
526/534 |
33/43 |
0.94/0.95 |
0.17/— |
—/1.01 × 104 |
290
|
|
BN-TP-N2
|
Toluene/3 wt%-PhCzBCza |
528/535 |
33/43 |
0.92/0.91 |
0.19/— |
—/0.83 × 104 |
290
|
|
BN-TP-N3
|
Toluene/3 wt%-PhCzBCza |
519/526 |
32/39 |
0.99/0.97 |
0.15/— |
—/1.70 × 104 |
290
|
|
BN-TP-N4
|
Toluene/3 wt%-PhCzBCza |
520/530 |
32/42 |
0.98/0.97 |
0.14/— |
—/1.90 × 104 |
290
|
The HOMO and LUMO of BN-TP extend to each benzene ring connected to 9- and 10-positions, respectively, and the whole molecular orbital preserves the alternating distribution characteristic of MR materials. Thus, the conventional HOMO and LUMO distributions of PAHs including triphenylene showing uniform bonding and anti-bonding between carbon atoms were effectively modulated with the help of the MR fragment. The extension of π-conjugation onto the triphenylene part in BN-TP resulted in an emission peak that was red-shifted relative to that of BN-DP. In addition, nitrogen atoms anchored at positions contributing to the LUMO afforded further red-shifted emission in BN-TP-N1 and BN-TP-N2. In contrast, blue-shifted (ultrapure green) emission was observed for BN-TP-N3 and BN-TP-N4. The dimerisation of BCzBN also extended the conjugation of MR cores, resulting in green emitters.185 As shown in Scheme 48, the synthesis of p-CzB involved the frequently used standard Suzuki–Miyaura coupling, while m-CzB was synthesised in moderate yield by oxidative coupling using Cu(ClO4)2·6H2O as the oxidant, as this type of reaction was favoured by the large HOMO distribution at 9- and 11-positions.
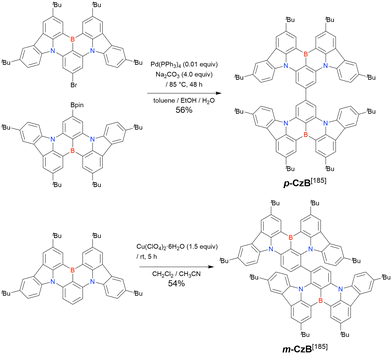 |
| | Scheme 48 Synthesis of a CzBN dimer by oxidative coupling. | |
The introduction of cyano groups at the para-position of boron resulted in a bathochromic shift to give MR materials with green to red emission (Table 39). Interestingly, several methods have been used to introduce cyano groups by substituting halogen atoms in late-stage functionalisation (Scheme 49). Treatment with CuCN in refluxing DMF (Rosenmund–von Braun reaction) is the most simple and widely used method, and has been applied for the synthesis of CN-BCz-BN.184 Meanwhile, CNCz-BNCz, which has two additional carbazole rings, was synthesised through lithiation with nBuLi followed by transnitrilation with dimethylmalononitrile (DMMN).291 Lithiation after the introduction of boron might cause relatively low yields. Palladium-catalysed cyanation with K4[Fe(CN)6] as an environmentally benign reagent was used to synthesise a DABNA derivative, where the MR core was prepared by one-shot borylation.114 The combination of one-shot borylation and late-stage cyanation through the substitution of halogen atoms enables the further development of MR emitters over the full colour range.
Table 39 Photophysical properties of the materials shown in Schemes 48–50
| Compound |
Condition |
λ
maxPL [nm] |
FWHM [nm] |
Φ
PL [—] |
ΔEST [eV] |
k
RISC [s−1] |
Ref. |
|
See Table 2 for the full compound name.
|
|
p-CzB
|
Toluene/1 wt%-mCBP-can |
505/513 |
34/41 |
0.85/0.80 |
0.17/0.14 |
4.2 × 104/4.6 × 104 |
185
|
|
m-CzB
|
Toluene/1 wt%-mCBP-can |
515/517 |
35/40 |
0.90/0.85 |
0.13/0.09 |
1.2 × 105/2.3 × 105 |
185
|
|
CN-BCz-BN
|
Toluene |
496 |
21 |
— |
— |
— |
184
|
|
CNCz-BNCz
|
Toluene/3 wt%-CBPa |
581/582 |
42/45 |
0.90/0.96 |
0.18/— |
4.2 × 105/— |
184
|
|
ν-DABNA-CN-Me
|
Toluene/1 wt%-PMMAa |
496/503 |
17/27 |
0.86/0.74 |
—/0.1 |
1.0 × 105/— |
114
|
|
BN(p)SCH3
|
Toluene/3 wt%-26DCzppya |
479/488 |
24/38 |
—/0.92 |
0.12/— |
—/6.4 × 104 |
178
|
|
BN(p)SOCH3
|
Toluene/3 wt%-26DCzppya |
482/490 |
22/33 |
—/0.71 |
0.16/— |
—/3.0 × 104 |
178
|
|
BN(p)SO2CH3
|
Toluene/3 wt%-26DCzppya |
490/496 |
21/29 |
—/0.99 |
0.20/— |
—/3.5 × 104 |
178
|
|
TCzBN-S
|
Toluene/8 wt%-SF3TRZa |
488/498 |
25/32 |
—/0.98 |
0.14/0.13 |
—/1.4 × 105 |
292
|
|
TCzBN-SO
|
Toluene/8 wt%-SF3TRZa |
494/508 |
27/31 |
—/0.98 |
0.14/0.14 |
—/5.6 × 104 |
292
|
|
tCBNDADPO
|
CH2Cl2/10 wt%-/30 wt%-DBFDPOa |
466/472/472 |
26/38/44 |
—/0.79/0.99 |
—/——/0.04 |
—/1.75 × 104/2.43 × 104 |
293
|
|
tCBNDASPO
|
CH2Cl2/20 wt%-/30 wt%-DBFDPOa |
467/472/472 |
28/32/32 |
—/0.92/0.79 |
—/0.04/— |
—/4.55 × 104/3.82 × 104 |
225
|
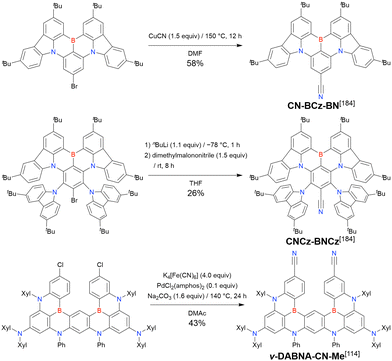 |
| | Scheme 49 Late-stage introduction of cyano groups. | |
The late-stage synthesis of oxidised sulphur and phosphorus species are summarised in Scheme 50. As mentioned above, BN(p)SCH3 was synthesised via tandem lithiation-borylation-annulation from a BCzBN precursor with a pre-installed SMe group178 and then treated with 3-chloroperbenzoic acid (mCPBA) to afford the corresponding sulfoxide (BN(p)SOCH3) and sulfone (BN(p)SO2CH3). Diphenyl sulphide and diphenyl sulfone derivatives (TCzBN-S and TCzBN-SO, respectively) were prepared by Suzuki–Miyaura coupling with the corresponding bromide.292 The electron-donating SMe group caused blue-shifted emission, while electron-withdrawing sulfoxide and sulfone moieties yielded red-shifted emission. The kRISC values of BN(p)SCH3 and TCzBN-S exceeded those of the oxidised compounds because of the differences in ΔEST. A phosphine oxide derivative, tCBNDADPO, was designed as an ambipolar self-host.293 The host segments were integrated into the MR core without involving a CT excited state. The synthesis of this compound included bromination by NBS on the diphenylamine unit, palladium-catalysed P–C coupling, and oxidation with H2O2. The ΦPL of tCBNDADPO with a high doping concentration of 30 wt% was 99% in the doped film. The comparison of compounds with/without phosphorylation was reported in a different paper.225
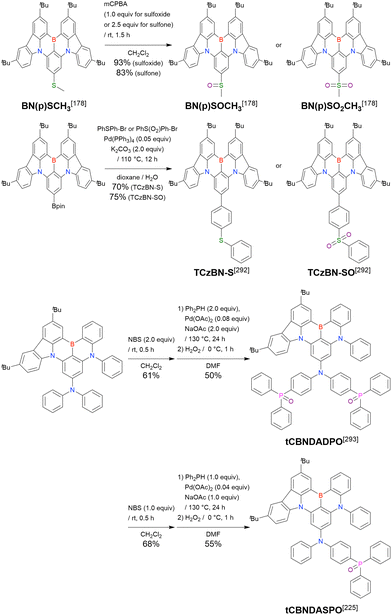 |
| | Scheme 50 Late-stage introduction of sulfoxide, sulfone, and phosphine oxide groups. | |
Late-stage reactions allow for the introduction of various functionalities. The chiral MR compounds shown in Fig. 34 were synthesised using the coupling reactions presented in Scheme 45.260,294 The effects of π-framework extension on the MR properties were basically same as those for the materials mentioned above. Additional chiral units of [2.2]paracyclo(1,4)carbazolophane (Czp) and octahydro-binaphthol (OBN) participated in the circularly polarised luminescence (CPL) process. Thus, the |gPL| factors of Czp-tBuCzB (1.6 × 10−3) and Czp-POAB (1.4 × 10−3) were similar to those of other CP-TADF molecules with the Czp moiety.295 Similarly, the CPL properties of OBN-2CN-BN and OBN-4CN-BN were similar to those reported in the literature.296
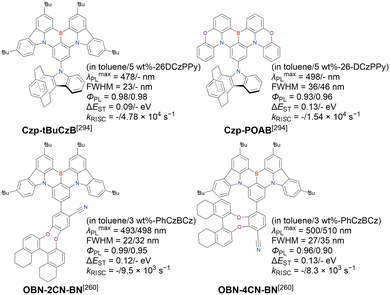 |
| | Fig. 34 CzBN derivatives showing CPL. | |
4.4 Metal complexes
Phosphorescent metal complexes based on the DOBNA scaffold have been reported; e.g., a ligand based on DOBNA-pyridine was synthesised by the borylation of 2-(4-bromo-3,5-diphenoxyphenyl)pyridine (Scheme 51 and Table 40). Phosphorescent iridium(III) complexes with the acetylacetonate (acac) ligand, [Ir(C^N)2(acac)], were prepared by well-established methods.139 The emission maximum of IrBBacac having two DOBNA-based ligands (638 nm) was largely red-shifted relative to that of the corresponding complex without boron atoms (544 nm). The good rigidity and planarity of the DOBNA-based ligand contributed to the small FWHM. The combination with a N-embedded ligand (IrBNacac) showed a slightly blue-shifted emission maximum at 523 nm and a narrower FWHM.297 The different types of ligands led to the T1 level with several characters including a metal-to-ligand CT transition (3MLCT), CT transition from the N-embedded ligand to the B-embedded ligand (3LLCT), and intraligand CT (3ILCT). Platinum(II) complexes, PtBacac and PtBbpy, were also synthesised by standard methods using the same ligand.298 The gold(I) coordination strategy aimed to enhance the RISC efficiency for MR emission because of the large spin–orbit coupling constant. The MR structure of BCzBN for green emission and that of the newly developed BNO for blue emission were covalently linked to Au-containing N-heterocyclic carbene (Au-NHC) via a Au–Caryl bond. Five bulky NHC ancillary ligands strongly bonded to gold(I). The complexes were synthesised by a coupling reaction between Bpin-functionalised MR cores and (NHC)AuCl (Scheme 51).95,299 The emission wavelengths of gold complexes were slightly red-shifted compared to those without the Au-NHC functionality. The high ΦPL and narrow FWHM were maintained despite the large kRISC.
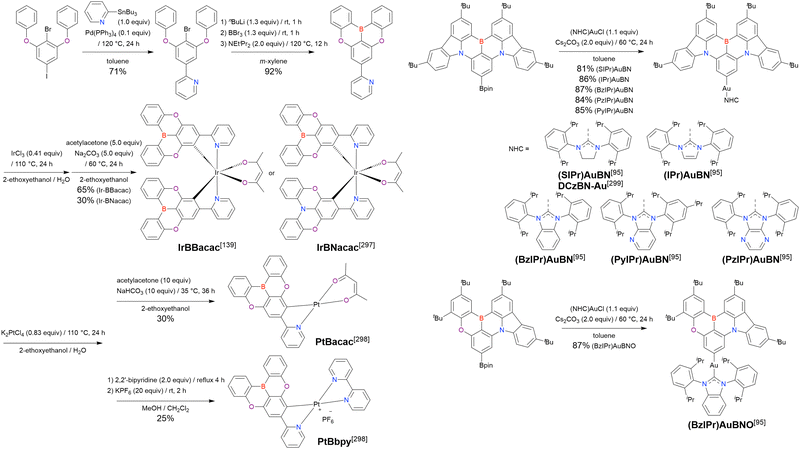 |
| | Scheme 51 Synthesis of DOBNA- and CzBN-based metal complexes. | |
Table 40 Photophysical properties of the compounds shown in Scheme 51
| Compound |
Condition |
λ
maxPL [nm] |
FWHM [nm] |
Φ
PL [—] |
ΔEST [eV] |
k
RISC [s−1] |
Ref. |
|
See Table 2 for the full compound name.
Not applicable because of phosphorescent materials.
|
|
IrBBacac
|
CH2Cl2/10 wt%-CBPa:mMTDATAa |
638/639 |
—/53 |
0.21/0.34 |
NAb/NAb |
NAb/NAb |
139
|
|
IrBNacac
|
CH2Cl2/1 wt%-PSa |
625/623 |
—/50 |
—/0.35 |
NAb/NAb |
NAb/NAb |
297
|
|
PtBacac
|
CH2Cl2/1 wt%-PSa |
578/578 |
—/— |
0.30/0.45 |
NAb/NAb |
NAb/NAb |
298
|
|
PtBbpy
|
CH2Cl2/1 wt%-PSa |
—/588 |
—/— |
—/0.12 |
NAb/NAb |
NAb/NAb |
298
|
|
(SIPr)AuBN
|
THF/2 wt%-PMMAa |
511/515 |
30/37 |
0.88/0.92 |
—/— |
—/— |
95
|
|
DCzBN-Au
|
Toluene/1 wt%-mCPa |
514/508 |
28/39 |
—/0.95 |
0.09/0.13 |
—/2.3 × 107 |
299
|
|
(IPr)AuBN
|
THF/MeCN/2 wt%-PMMAa |
511/—/514 |
31/—/37 |
0.83/—/0.90 |
—/—/— |
—/3.3 × 104/— |
95
|
|
(BzIPr)AuBN
|
THF/MeCN/2 wt%-PMMAa |
511/—/513 |
30/—/37 |
0.86/—/0.91 |
—/—/— |
—/5.0 × 104/— |
95
|
|
(PyIPr)AuBN
|
THF/MeCN/2 wt%-PMMAa |
511/—/514 |
30/—/37 |
0.93/—/0.88 |
—/—/— |
—/3.2 × 104/— |
95
|
|
(PzIPr)AuBN
|
THF/2 wt%-PMMAa |
510/514 |
30/37 |
0.78/0.87 |
—/— |
—/— |
95
|
|
(BzIPr)AuBNO
|
THF/MeCN/2 wt%-PMMAa |
471/—/473 |
30/—/37 |
0.89/—/0.84 |
—/—/— |
—/1.1 × 104/— |
95
|
4.5 B-doped fused PAHs
The introduction of heteroatoms, especially boron, oxygen, and nitrogen, into fused PAHs affords novel π-conjugated systems showing dramatically different optoelectronic properties despite their structural similarities. Recent progress in the development of the bottom-up synthesis of nanographenes using solution-based techniques holds great promise for accelerating the evolution of heteroatom incorporation. Late-stage synthesis using pre-prepared MR structures such as DOBNA and BCzBN is advantageous for constructing extended π-conjugation in a short number of steps. We have not summarised photophysical properties for the following compounds described in Schemes 52–56 since they do not show any TADF characteristics.
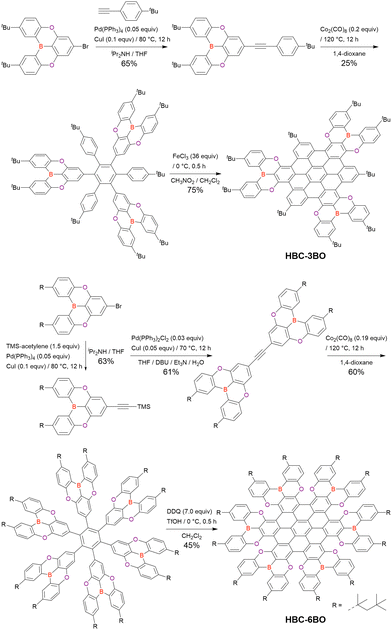 |
| | Scheme 52 Synthesis of DOBNA-based nanographene reported in ref. 136. | |
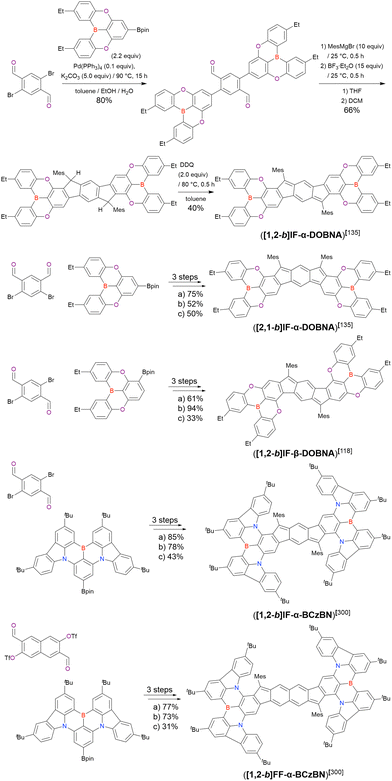 |
| | Scheme 53 Synthesis of DOBNA-based biradicaloids. | |
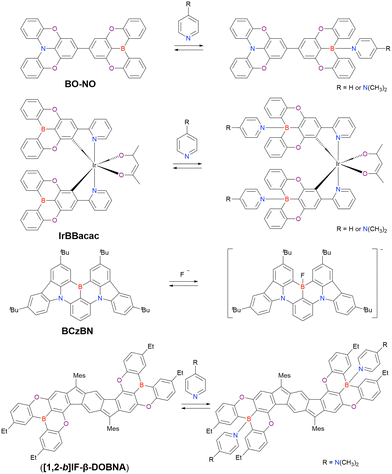 |
| | Scheme 54 Lewis acid–base adducts of DOBNA and CzBN structures. | |
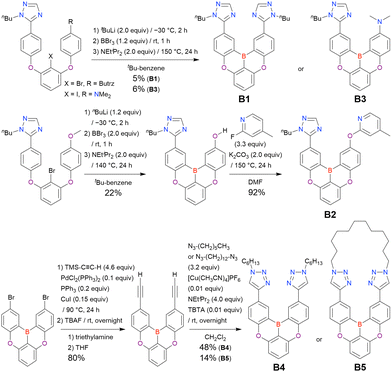 |
| | Scheme 55 Synthesis of multi-stimuli responsive materials. | |
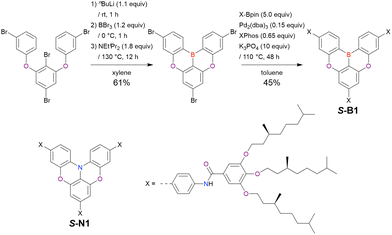 |
| | Scheme 56 Late-stage introduction of peripheral gallic amides with (S)-3,7-dimethyloctyl chains for supramolecular copolymers. | |
Hexabenzocoronene-based molecules are examples of DOBNA-fused systems (Scheme 52).136 Chemical structures having multiple helical regions and distorted configurations affect the electronic properties. Intermediates with alkyne moieties were synthesised by Sonogashira–Hagihara coupling using DOBNA derivatives. The subsequent cyclotrimerisation of these alkyne derivatives catalysed by Co2(CO)8 yielded hexa-aryl precursors.
Intramolecular cyclodehydrogenation was performed using FeCl3 for HBC-3BO and 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) and trifluoromethanesulfonic acid (TfOH) for HBC-6BO to complete the reaction. For HBC-6BO, the branched alkyl chains were used to enhance solubility. The absorption/emission maxima of tBu-HBC, HBC-3BO, and HBC-6BO were located at 360/493, 438/529 and 514/602 nm, respectively, showing that the introduction of the DOBNA scaffold resulted in a substantial red-shift. ΦPL also increased after the incorporation of the DOBNA moiety (from 5.8% to 18–20%).
Certain PAHs have a unique open-shell singlet biradical character. Indenofluorenes (IFs) and their π-extended derivatives show moderate-to-high biradical characters, while B-containing biradicaloids might be more challenging to synthesise because of their high reactivities. Therefore, rigid and stable DOBNA and CzBN structures were merged with IFs to afford, e.g., DOBNA and CzBN-fused s- and as-indacenes. The central core structures are based on indeno[1,2-b]fluorene, indeno[2,1-b]fluorene, and fluoreno[1,2-b]fluorene, which are herein referred to as [1,2-b]IF, [2,1-b]IF, and [1,2-b]FF, respectively (Scheme 53). The corresponding synthesis process involved three steps including Suzuki–Miyaura coupling, nucleophilic addition of mesityl magnesium bromide to yield a diol, intramolecular Friedel–Crafts alkylation induced by treatment with BF3·Et2O, and oxidative dehydrogenation with DDQ.118,135,300 The characterisation of [1,2-b]IF-α-DOBNA and [2,1-b]IF-α-DOBNA using NMR, X-ray single-crystal analysis, electron paramagnetic resonance (EPR), ultraviolet-visible (UV-vis) absorption, and cyclic voltammetry measurements confirmed the significant contributions of the open-shell biradical form, which exceeded those for the corresponding indenofluorenes. The isomeric compound [1,2-b]IF-β-DOBNA showed clearly resolved NMR signals, a very weak EPR signal, and a wider energy gap, which indicated a much smaller biradical character. N,B,N-substituted species showed a higher biradical character than O,B,O-substituted ones according to theoretical calculations. Thus, [1,2-b]IF-α-BCzBN and [1,2-b]FF-α-BCzBN showed significantly red-shifted absorption maxima at 803 and 876 nm, respectively, as well as small energy gaps. These results demonstrate that MR structures greatly influence the open-shell biradical nature of PAHs.
4.6 Lewis acid–base adducts
The vacant p-orbital of boron is responsible for Lewis acidity, although the structural constraint of boron atoms in the planar π-conjugated systems with N,O heteroatom bridges in MR structures lowers this acidity. Indeed, DOBNA and CzBN derivatives can interact with Lewis bases such as pyridine, 4-dimethylaminopyridine (DMAP), and fluoride ions (Scheme 54). A compound with a DOBNA and N-embedded dioxygen-bridged structure, BO-NO, showed yellow emission in the polar CH2Cl2.301 The emission intensity decreased upon the addition of pyridine, while the addition of DMAP induced the emergence of a new emission band in the blue region and decreased the intensity of the emission band in the yellow region. The binding constants of BO-NO towards pyridine and DMAP were estimated as 27 and 4.2 × 104 M−1, respectively, which are much lower than those of planar fused trinaphthylboranes.302 The phosphorescence of IrBBacac showed a similar behaviour.139 The emission peak lost intensity and shifted to longer wavelengths (from 647 to 667 nm) upon the addition of pyridine. In contrast, DMAP addition caused an emission blue shift and intensity increase.
The two Lewis bases, pyridine and DMAP, coordinated at different positions, and π–π interactions between two DMAP molecules were observed in the crystal structure. As BCzBN shows intense emission with a narrow FWHM and high ΦPL, as well as good chemical and thermal stability, it is well suited for the construction of turn-off-type fluorescent probes. The interactions between the boron centre and the fluoride ion upon the addition of tetrabutylammonium fluoride (TBAF) resulted in the formation of a non-fluorescent complex and, hence, substantial emission quenching.303 Consequently, boron-based MR materials hold great promise for the highly sensitive, selective, and reversible sensing of fluoride ions. Lewis acid–base coordination not only changed optical properties, but also enhanced the open-shell biradical character118,135
N-Heterocycle-tethered DOBNA structures were investigated as stimuli-responsive materials.115 Although compounds B1 and B3 were synthesised by one-pot borylation, the synthesis of B2 involved an additional SNAr reaction. The syntheses of B4 and B5 involved Sonogashira–Hagihara cross-coupling followed by a click reaction (Scheme 55). Compounds B1–B3 having an n-butyl-1H-1,2,4-triazole scaffold exhibited emission intensity enhancement at elevated temperatures in solution, while compounds B4 and B5 containing an n-hexyl-1H-1,2,3-triazole scaffold showed fluorescence quenching under the same conditions. The thermally responsive emission of B1–B3 was caused by the dynamic switching of intermolecular B–N interactions. In addition, these compounds exhibited remarkable mechanochromism in the solid state. The formation of B–N pairs was also used to develop supramolecular copolymers.138 The peripheries of the DOBNA core accommodated three amide units as H-bonding supramolecular motifs, which were introduced by late-stage coupling to give S-B1 (Scheme 56). Co-facial B–N contacts in the copolymer with S-N1 facilitated the formation of an exciplex with TADF properties, followed by circularly polarised emission. Overall, the high stability of bridged triarylboranes is of key importance, enabling a wide variety of applications.
4.7 Summary
Late-stage functionalisation is a useful method for developing a wide variety of structures, as photophysical properties can be finely tuned by the attachment of appropriate donor and acceptor moieties. Moreover, this method enables the utilisation of MR fragments as building blocks and thus allows one to expand the application scope of MR materials by gaining additional functionality and incorporation into systems with extended π-conjugation.
5. Ladder-type boron-based MR compounds
Most boron-based MR-TADF compounds introduced in the previous sections possess boron atoms at the ring junction, featuring planarity constraints, i.e., planarised triaryl boron-based compounds.37 On the other hand, some B,N-based PAHs, in which boron atoms are not fully constrained, also exhibit MR effects. These compounds are synthesised using methods closely resembling one-pot or one-shot borylation, as discussed in the present section. The incorporation of tricoordinate boron atoms into the framework of linear acenes is an effective strategy for achieving the properties of the parent π-conjugated systems, which are inaccessible in carbon-only acenes. In 2006, Kawashima et al. reported a series of ladder-type fused azaborines in which the boron and nitrogen atoms are located in para-positions with respect to each other.304 Zysman-Colman et al. reported B,N-doped heptacene α-3BNOH, where the boron and nitrogen atoms are located in para-positions with respect to each other.305 The triple electrophilic C–H borylation of N1-(3-(di-p-tolylamino)phenyl)-N1,N3,N3-tri-p-tolylbenzene-1,3-diamine occurred in the presence of 18 equiv. of BBr3 and afforded a tri-B–Br intermediate that was subsequently hydrolysed to give α-3BNOH in 68% yield (Scheme 57, top). Note that the process up to the point of hydrolysis is the same as that of one-shot borylation. The thus obtained α-3BNOH exhibited narrow deep blue emission with CIE coordinates of (0.17, 0.01) and presented an attractive combination of TADF and TTA in a single material. α-3BNMes was also synthesized by trapping the tri-B–Br intermediate with a Grignard reagent instead of hydrolysing the former (Scheme 57, bottom).306α-3BNMes showed a narrow blue emission at 442 nm (FWHM = 30 nm) that was red-shifted compared with that of α-3BNOH because of the replacement of the strongly mesomerically electron-donating hydroxyl groups with inductively electron-withdrawing mesityl substituents. It is noteworthy that BN-doped heptacene α-3BNMes exhibited TADF behaviour in contrast to BN-doped pentacene reported by Kawashima et al.304 due to the small ΔEST (0.28 eV) attributed to the highly π-extended structure.
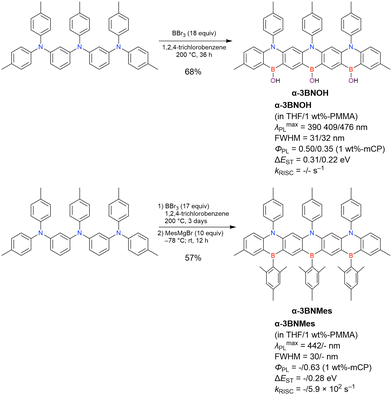 |
| | Scheme 57 Synthesis of α-3BNOH and α-3BNMes by triple electrophilic C–H borylation. | |
Duan et al. prepared carbazole-fused 1,4-diazaborine type MR-TADF compounds with nonplanar boron atoms on the periphery.307 Starting with mono-brominated bis(N-carbazolyl)benzene, the one-pot sequence of lithium–bromine exchange, borylation, intramolecular bora-Friedel–Crafts reaction, and trapping with a Grignard reagent afforded mono-borylated BIC-mCz and BIC-pCz in yields of 35% and 33%, respectively (Scheme 58). In the same manner, double-borylated mDBIC and pDBIC were obtained from a dibrominated precursor in 29% and 49% yields, respectively. Notably, the process up to trapping with the Grignard reagent is identical to the one-pot borylation protocol, the only difference being the presence of a non-condensed substituent on the boron centre. The above borylated compounds displayed deep blue to pure green MR emission at 426–532 nm with a FWHM of 27–38 nm. These ladder-type boron-based MR compounds exhibit larger ΔEST and smaller kRISC due to their relatively small MR effects.
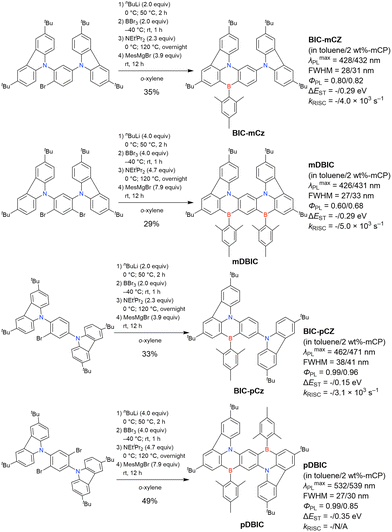 |
| | Scheme 58 Synthesis of BIC-mCz, BIC-pCz, mDBIC, and pDBIC. | |
6. Conclusions
This review summarises the synthetic routes for boron-based MR emitters. For single or double borylation, one-pot borylation is the first choice because of its high regioselectivity. Commercially available polyhalobenzenes containing fluorine, chlorine, bromine, and iodine atoms are efficiently converted to one-pot borylation precursors via SNAr reactions and Buchwald–Hartwig couplings with complementary site (halogen) selectivity. However, diarylamines cannot be introduced via SNAr reactions, which has severely limited the chemical space of MR emitters. Moreover, double borylation is generally low-yielding and therefore impractical. For multiple borylation, one-shot borylation is an advantageous method but requires a judicious choice of precursors and reaction conditions to achieve kinetic or thermodynamic regioselectivity control. The combination of one-pot and one-shot borylations, i.e., sequential multiple borylation, is a promising method for constructing novel MR skeletons with multiple boron atoms. The late-stage functionalisation of MR emitters is useful for tuning their emission wavelengths, increasing their kRISC values, or imparting new functions (e.g., chiroptical, sensing, and stimuli-responsive properties). Although these protocols have achieved impressive results for the last several years, the practical application of MR emitters is still limited to their use as blue-fluorescent emitters in OLED displays. Therefore, to design MR emitters not only exhibiting specific properties but also complying with all practical requirements, especially high stability during device operation, the current chemical space needs to be well understood and further expanded by developing new synthetic protocols. Furthermore, the expansion of the chemical space will break through unconscious limits of one's imagination and pave the way for practical applications beyond OLEDs.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by JST CREST (Grant Number JPMJCR22B3), JSPS Grants-in-Aid for Transformative Research Areas (Condensed Conjugation, 20H05863), Grants-in-Aid for Scientific Research(B) (21H02019), Grants-in-Aid for Scientific Research(C) (23K04879), and Grant-in-Aid for Research Activity Start-up (23K19243 and 23K19245).
Notes and references
- F. Jäkle, Coord. Chem. Rev., 2006, 250, 1107–1121 Search PubMed.
- A. Wakamiya and S. Yamaguchi, Pure Appl. Chem., 2006, 78, 1413–1424 Search PubMed.
- Z. M. Hudson and S. Wang, Acc. Chem. Res., 2009, 42, 1584–1596 Search PubMed.
- C. R. Wade, A. E. J. Broomsgrove, S. Aldridge and F. P. Gabbai, Chem. Rev., 2010, 110, 3958–3984 Search PubMed.
- F. Jäkle, Chem. Rev., 2010, 110, 3985–4022 Search PubMed.
- A. Escande and M. J. Ingleson, Chem. Commun., 2015, 51, 6257–6274 Search PubMed.
- A. Wakamiya and S. Yamaguchi, Bull. Chem. Soc. Jpn., 2015, 88, 1357–1377 Search PubMed.
- Y. Ren and F. Jäkle, Dalton Trans., 2016, 45, 13996–14007 Search PubMed.
- L. Ji, S. Griesbeck and T. B. Marder, Chem. Sci., 2017, 8, 846–863 Search PubMed.
- G. Turkoglu, M. E. Cinar and T. Ozturk, Molecules, 2017, 22, 1522 Search PubMed.
- E. von Grotthuss, A. John, T. Kaese and M. Wagner, Asian J. Org. Chem., 2018, 7, 37–53 Search PubMed.
- S. K. Mellerup and S. Wang, Chem. Soc. Rev., 2019, 48, 3537–3549 Search PubMed.
- H. Helten, Chem. – Asian J., 2019, 14, 919–935 Search PubMed.
- J. Huo, H. Wang, S. Li, H. Shi, Y. Tang and B. Z. Tang, Chem. Rec., 2020, 20, 556–569 Search PubMed.
- X. Yin, J. Liu and F. Jäkle, Chem. – Eur. J., 2021, 27, 2973–2986 Search PubMed.
- S. M. Berger, M. Ferger and T. B. Marder, Chem. – Eur. J., 2021, 27, 7043–7058 Search PubMed.
- J. Shi, Z. Ran, F. Peng, M. Chen, L. Li, L. Ji and W. Huang, J. Mater. Chem. C, 2022, 10, 9165–9191 Search PubMed.
- S. M. Berger and T. B. Marder, Mater. Horiz., 2022, 9, 112–120 Search PubMed.
- M. Wang and C.-H. Zhao, Chem. Rec., 2022, 22, e202100199 Search PubMed.
- J. Han, Y. Chen, N. Li, Z. Huang and C. Yang, Aggregate, 2022, 3, e182 Search PubMed.
- R. Wang, C.-S. Lee and Z. Lu, J. Organomet. Chem., 2023, 984, 122564 Search PubMed.
- J. Doty, B. Babb, P. Grisdale, M. Glogowski and J. Williams, J. Organomet. Chem., 1972, 38, 229–236 Search PubMed.
- C. D. Entwistle and T. B. Marder, Angew. Chem., Int. Ed., 2002, 41, 2927–2931 Search PubMed.
- C. D. Entwistle and T. B. Marder, Chem. Mater., 2004, 16, 4574–4585 Search PubMed.
- M. J. S. Dewar, V. P. Kubba and R. Pettit, J. Chem. Soc., 1958, 3073–3076 Search PubMed.
- Z. Liu and T. B. Marder, Angew. Chem., Int. Ed., 2008, 47, 242–244 Search PubMed.
- M. J. D. Bosdet and W. E. Piers, Can. J. Chem., 2009, 87, 8–29 Search PubMed.
- P. G. Campbell, A. J. V. Marwitz and S.-Y. Liu, Angew. Chem., Int. Ed., 2012, 51, 6074–6092 Search PubMed.
- M. M. Morgan and W. E. Piers, Dalton Trans., 2016, 45, 5920–5924 Search PubMed.
- J.-Y. Wang and J. Pei, Chin. Chem. Lett., 2016, 27, 1139–1146 Search PubMed.
- G. Belanger-Chabot, H. Braunschweig and D. K. Roy, Eur. J. Inorg. Chem., 2017, 4353–4368 Search PubMed.
- Z. X. Giustra and S.-Y. Liu, J. Am. Chem. Soc., 2018, 140, 1184–1194 Search PubMed.
- X. Chen, D. Tan and D.-T. Yang, J. Mater. Chem. C, 2022, 10, 13499–13532 Search PubMed.
- I. Shin, H. N. Lim and W. P. Hong, Synthesis, 2022, 570–588 Search PubMed.
- Z. Zhou, A. Wakamiya, T. Kushida and S. Yamaguchi, J. Am. Chem. Soc., 2012, 134, 4529–4532 Search PubMed.
- M. Hirai, N. Tanaka, M. Sakai and S. Yamaguchi, Chem. Rev., 2019, 119, 8291–8331 Search PubMed.
- H. Hirai, K. Nakajima, S. Nakatsuka, K. Siren, J. Ni, S. Nomura, T. Ikuta and T. Hatakeyama, Angew. Chem., Int. Ed., 2015, 54, 13581–13585 Search PubMed.
- T. Hatakeyama, K. Shiren, K. Nakajima, S. Nomura, S. Nakatsuka, K. Kinoshita, J. Ni, Y. Ono and T. Ikuta, Adv. Mater., 2016, 28, 2777–2781 Search PubMed.
- C. A. Parker and C. G. Hatchard, Trans. Faraday Soc., 1961, 57, 1894–1904 Search PubMed.
- A. Endo, M. Ogasawara, A. Takahashi, D. Yokoyama, Y. Kato and C. Adachi, Adv. Mater., 2009, 21, 4802–4806 Search PubMed.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 Search PubMed.
- C. W. Tang and S. A. Vanslyke, Appl. Phys. Lett., 1987, 51, 913–915 Search PubMed.
- M. A. Baldo, D. F. O’Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151–154 Search PubMed.
- S. M. Suresh, D. Hall, D. Beljonne, Y. Olivier and E. Zysman-Colman, Adv. Funct. Mater., 2020, 30, 1908677 Search PubMed.
- H. J. Kim and T. Yasuda, Adv. Opt. Mater., 2022, 10, 2201714 Search PubMed.
- R. K. Konidena and K. R. Naveen, Adv. Photonics Res., 2022, 3, 2200201 Search PubMed.
- T. Fan, Y. Zhang, D. Zhang and L. Duan, Chem. – Eur. J., 2022, 28, e202104624 Search PubMed.
- K. R. Naveen, H. I. Yang and J. H. Kwon, Commun. Chem., 2022, 5, 149 Search PubMed.
- K. R. Naveen, P. Palanisamy, M. Y. Chae and J. H. Kwon, Chem. Commun., 2023, 59, 3685–3702 Search PubMed.
- Y. Xu, Q. Wang, X. Song, Y. Wang and C. Li, Chem. – Eur. J., 2023, 29, e202203414 Search PubMed.
- L. Xiaofeng, Z. Dongdong, D. Lian and Z. Yuewei, Front. Chem., 2023, 11, 1198404 Search PubMed.
- H.-Z. Li, F.-M. Xie, Y.-Q. Li and J.-X. Tang, J. Mater. Chem. C, 2023, 11, 6471–6511 Search PubMed.
- C. Lv, X. Wang, Q. Zhang and Y. Zhang, Mater. Chem. Front., 2023, 7, 2809–2827 Search PubMed.
- F. Santoro, A. Lami, R. Improta, J. Bloino and V. Barone, J. Chem. Phys., 2008, 128, 224311 Search PubMed.
- I. S. Park, K. Matsuo, N. Aizawa and T. Yasuda, Adv. Funct. Mater., 2018, 28, 1802031 Search PubMed.
- S. Park, Y. Xiong, R.-H. Kim, P. Elvikis, M. Meitl, D.-H. Kim, J. Wu, J. Yoon, C.-J. Yu, Z. Liu, Y. Huang, K.-C. Hwang, P. Ferreira, X. Li, K. Choquette and J. A. Rogers, Science, 2009, 325, 977–981 Search PubMed.
- V. L. Colvin, M. C. Schlamp and A. P. Alivisatos, Nature, 1994, 370, 354–357 Search PubMed.
- S. Coe, W. K. Woo, M. Bawendi and V. Bulovic, Nature, 2002, 420, 800–803 Search PubMed.
-
T. Sato, K. Tokunaga, N. Iwahara, K. Shizu and K. Tanaka, in The Jahn–Teller Effect, ed. H. Koppel, D. Yarkony and H. Barentzen, Springer, Springer Series in Chemical Physics, 2009, vol. 97, pp. 99–129 Search PubMed.
- Y. Kondo, K. Yoshiura, S. Kitera, H. Nishi, S. Oda, H. Gotoh, Y. Sasada, M. Yanai and T. Hatakeyama, Nat. Photonics, 2019, 13, 678–682 Search PubMed.
- M. G. Prais, D. F. Heller and K. F. Freed, Chem. Phys., 1974, 6, 331–352 Search PubMed.
- Y. Lei, H. Guo, J. Wang and R. Jia, Dalton Trans., 2019, 48, 5064–5071 Search PubMed.
- L. Liu, Q. Wei, Y. Cheng, H. Ma, S. Xiong and X. Zhang, J. Mater. Chem. C, 2020, 8, 5839–5846 Search PubMed.
- K. Goushi, K. Yoshida, K. Sato and C. Adachi, Nat. Photonics, 2012, 6, 253–258 Search PubMed.
- Q. Zhang, J. Li, J. Shizu, S. Huang, S. Hirata, H. Miyazaki and C. Adachi, J. Am. Chem. Soc., 2012, 134, 14706–14709 Search PubMed.
- J.-L. Brédas, D. Beljonne, V. Coropceanu and J. Cornil, Chem. Rev., 2004, 104, 4971–5004 Search PubMed.
- M. K. Etherington, J. Gibson, H. F. Higginbotham, T. J. Penfold and A. P. Monkman, Nat. Commun., 2016, 7, 13680 Search PubMed.
- P. K. Samanta, D. Kim, V. Coropceanu and J.-L. Brédas, J. Am. Chem. Soc., 2017, 139, 4042–4051 Search PubMed.
- H. Noda, X.-K. Chen, H. Nakanotani, T. Hosokai, M. Miyajima, N. Notsuka, Y. Kashima, J.-L. Brédas and C. Adachi, Nat. Mater., 2019, 18, 1084–1090 Search PubMed.
- B. Wex and B. R. Kaafarani, J. Mater. Chem. C, 2017, 5, 8622–8653 Search PubMed.
- X.-K. Chen, D. Kim and J.-L. Brédas, Acc. Chem. Res., 2018, 51, 2215–2224 Search PubMed.
- J. Eng and T. J. Penfold, Chem. Rec., 2020, 20, 831–856 Search PubMed.
- H. Nakanotani, Y. Tsuchiya and C. Adachi, Chem. Lett., 2021, 50, 938–948 Search PubMed.
- A. Pershin, D. Hall, V. Lemaur, J.-C. Sancho-Garcia, L. Muccio-li, E. Zysman-Colman, D. Beljonne and Y. Olivier, Nat. Commun., 2019, 10, 597 Search PubMed.
- H. Nakanotani, T. Furukawa, T. Hosogai, T. Hatakeyama and C. Adachi, Adv. Opt. Mater., 2017, 5, 1700051 Search PubMed.
- C.-C. Yan, X.-D. Wang and L.-S. Liao, Adv. Sci., 2022, 9, 2200525 Search PubMed.
- M. Mamada, S. Maedera, S. Oda, T. B. Nguyen, H. Nakanotani, T. Hatakeyama and C. Adachi, Mater. Chem. Front., 2023, 7, 259–266 Search PubMed.
- D. Yokoyama, J. Mater. Chem., 2011, 21, 19187–19202 Search PubMed.
- J. Frischeisen, D. Yokoyama, A. Endo, C. Adachi and W. Brütting, Org. Electron., 2011, 12, 809–817 Search PubMed.
- W. Brütting, J. Frischeisen, T. D. Schmidt, B. J. Scholz and C. Mayr, Phys. Status Solidi A, 2013, 210, 44–65 Search PubMed.
- T. D. Schmidt, T. Lampe, M. R. D. Sylvinson, P. I. Djurovich, M. E. Thompson and W. Brütting, Phys. Rev. Applied., 2017, 8, 037001 Search PubMed.
- T. Northey and T. J. Penfold, Org. Electron., 2018, 59, 45–48 Search PubMed.
- I. Kim, K. H. Cho, S. O. Jeon, W.-J. Son, D. Kim, Y. M. Rhee, I. Jang, H. Choi and D. S. Kim, JACS Au, 2021, 1, 987–997 Search PubMed.
- X. Wu, B.-K. Su, D.-G. Chen, D. Liu, C.-C. Wu, Z.-X. Huang, T.-C. Lin, C.-H. Wu, M. Zhu, E. Y. Li, W.-Y. Hung, W. Zhu and P.-T. Chou, Nat. Photonics, 2021, 15, 780–786 Search PubMed.
- K. Shizu and H. Kaji, Commun. Chem., 2022, 5, 53 Search PubMed.
- S. Schmidbauer, A. Hohenleutner and B. König, Adv. Mater., 2013, 25, 2114–2129 Search PubMed.
- J. Lee, C. Jeong, T. Batagoda, C. Coburn, M. E. Thompson and S. R. Forrest, Nat. Commun., 2017, 8, 15566 Search PubMed.
- H. Lim, S.-J. Woo, Y. H. Ha, Y.-H. Kim and J.-J. Kim, Adv. Mater., 2022, 34, 2100161 Search PubMed.
- T. B. Nguyen, H. Nakanotani and C. Adachi, Adv. Opt. Mater., 2022, 10, 2200704 Search PubMed.
- H. Jiang, P. Tao and W.-Y. Wong, ACS Mater. Lett., 2023, 5, 822–845 Search PubMed.
- X. Wang, L. Wang, G. Meng, X. Zeng, D. Zhang and L. Duan, Sci. Adv., 2023, 9, eadh1434 Search PubMed.
- M. Nagata, H. Min, E. Watanabe, H. Fukumoto, Y. Mizuhata, N. Tokitoh, T. Agou and T. Yasuda, Angew. Chem., Int. Ed., 2021, 60, 20280–20285 Search PubMed.
- S. M. Pratik, V. Coropceanu and J.-L. Brédas, ACS Mater. Lett., 2022, 4, 440–447 Search PubMed.
- I. S. Park, H. Min and T. Yasuda, Angew. Chem., Int. Ed., 2022, 61, e202205684 Search PubMed.
- S. Cai, G. S. M. Tong, L. Du, G. K.-M. So, F.-F. Hung, T.-L. Lam, G. Cheng, H. Xiao, X. Chang, Z.-X. Xu and C.-M. Che, Angew. Chem., Int. Ed., 2022, 61, e202213392 Search PubMed.
- H. Nakanotani, T. Higuchi, T. Furukawa, K. Masui, K. Morimoto, M. Numata, H. Tanaka, Y. Sagara, T. Yasuda and C. Adachi, Nat. Commun., 2014, 5, 4016 Search PubMed.
- D. Zhang, X. Song, A. J. Gillett, B. H. Drummond, S. T. E. Jones, G. Li, H. He, M. Cai, D. Credgington and L. Duan, Adv. Mater., 2020, 32, 1908355 Search PubMed.
- C.-Y. Chan, M. Tanaka, Y.-T. Lee, Y.-W. Wong, H. Nakanotani, T. Hatakeyama and C. Adachi, Nat. Photonics, 2021, 15, 203–207 Search PubMed.
- S. O. Jeon, K. H. Lee, J. S. Kim, S.-G. Ihn, Y. S. Chung, J. W. Kim, H. Lee, S. Kim, H. Choi and J. Y. Lee, Nat. Photonics, 2021, 15, 208–215 Search PubMed.
- M. Mamada, H. Katagiri, C.-Y. Chan, Y.-T. Lee, K. Goushi, H. Nakanotani, T. Hatakeyama and C. Adachi, Adv. Funct. Mater., 2022, 32, 2204352 Search PubMed.
- P. Heimel, A. Mondal, F. May, W. Kowalsky, C. Lennartz, D. Andrienko and R. Lovrincic, Nat. Commun., 2018, 9, 4990 Search PubMed.
- E. Kim, J. Park, M. Jun, H. Shin, J. Baek, T. Kim, S. Kim, J. Lee, H. Ahn and S. Kim, Sci. Adv., 2022, 8, eabq1641 Search PubMed.
- X. Cao, K. Pan, J. Miao, X. Lv, Z. Huang, F. Ni, X. Yin, Y. Wei and C. Yang, J. Am. Chem. Soc., 2022, 144, 22976–22984 Search PubMed.
- Y. X. Hu, J. Miao, T. Hua, Z. Huang, Y. Qi, Y. Zou, Y. Qiu, H. Xia, H. Liu, X. Cao and C. Yang, Nat. Photonics, 2022, 16, 803–810 Search PubMed.
- S. Oda and T. Hatakeyama, Bull. Chem. Soc. Jpn., 2021, 94, 950–960 Search PubMed.
- T. Hatakeyama, S. Hashimoto, S. Seki and M. Nakamura, J. Am. Chem. Soc., 2011, 133, 18614–18617 Search PubMed.
- A. D. Grosso, R. G. Pritchard, C. A. Muryn and M. J. Ingleson, Organometallics, 2010, 29, 241–249 Search PubMed.
- N. Ishida, T. Moriya, T. Goya and M. Murakami, J. Org. Chem., 2010, 75, 8709–8712 Search PubMed.
- S. A. Iqbal, J. Pahl, K. Yuan and M. J. Ingleson, Chem. Soc. Rev., 2020, 49, 4564–4591 Search PubMed.
- S. Rej and N. Chatani, Angew. Chem., Int. Ed., 2022, 61, e202209539 Search PubMed.
- S. Oda, W. Kumano, T. Hama, R. Kawasumi, K. Yoshiura and T. Hatakeyama, Angew. Chem., Int. Ed., 2021, 60, 2882–2886 Search PubMed.
- J. U. Kim, I. S. Park, C.-Y. Chan, M. Tanaka, Y. Tsuchiya, H. Nakanotani and C. Adachi, Nat. Commun., 2020, 11, 1765 Search PubMed.
- J. Han, Z. Huang, J. Miao, Y. Qiu, Z. Xie and C. Yang, Chem. Sci., 2022, 13, 3402–3408 Search PubMed.
- S. Oda, T. Sugitani, H. Tanaka, K. Tabata, R. Kawasumi and T. Hatakeyama, Adv. Mater., 2022, 34, 2201778 Search PubMed.
- G. Meng, T. Peng, Y. Shi, H. Li, X. Wang, X. Yin, D.-T. Yang, S. Wang and N. Wang, J. Mater. Chem. C, 2020, 8, 7749–7754 Search PubMed.
- F. Chen, L. Zhao, X. Wang, Q. Yang, W. Li, H. Tian, S. Shao, L. Wang, X. Jing and F. Wang, Sci. China: Chem., 2021, 64, 547–551 Search PubMed.
- Y. Chen, N. Li, Z. Huang, G. Xie and C. Yang, Chem. Eng. J., 2022, 430, 133078 Search PubMed.
- X. Tian, J. Guo, W. Sun, L. Yuan, C. Dou and Y. Wang, Chem. – Eur. J., 2022, 28, e202200045 Search PubMed.
- G. Meng, X. Chen, X. Wang, N. Wang, T. Peng and S. Wang, Adv. Opt. Mater., 2019, 7, 1900130 Search PubMed.
- I. S. Park, H. Min, J. U. Kim and T. Yasuda, Adv. Opt. Mater., 2021, 9, 2101282 Search PubMed.
- H. Lee, R. Braveenth, S. Muruganantham, C. Y. Jeon, H. S. Lee and J. H. Kwon, Nat. Commun., 2023, 14, 419 Search PubMed.
- D. H. Ahn, S. W. Kim, H. Lee, I. J. Ko, D. Karthik, J. Y. Lee and J. H. Kwon, Nat. Photonics, 2019, 13, 540–546 Search PubMed.
- H. J. Kim, M. Godumala, S. K. Kim, J. Yoon, C. Y. Kim, H. Park, J. H. Kwon, M. J. Cho and D. H. Choi, Adv. Opt. Mater., 2020, 8, 1902175 Search PubMed.
- H. Lim, H. J. Cheon, S.-J. Woo, S.-K. Kwon, Y.-H. Kim and J.-J. Kim, Adv. Mater., 2020, 32, 2004083 Search PubMed.
- H. J. Kim, H. Kang, J.-E. Jeong, S. H. Park, C. W. Koh, C. W. Kim, H. Y. Woo, M. J. Cho, S. Park and D. H. Choi, Adv. Funct. Mater., 2021, 31, 2102588 Search PubMed.
- R. Pei, H. Liu, C. Zhou, J. Miao and C. Yang, J. Mater. Chem. C, 2021, 9, 17136–17142 Search PubMed.
- H.-J. Tan, G.-X. Yang, Y.-L. Deng, C. Cao, J.-H. Tan, Z.-L. Zhu, W.-C. Chen, Y. Xiong, J.-X. Jian, C.-S. Lee and Q.-X. Tong, Adv. Mater., 2022, 34, 2200537 Search PubMed.
- Y. Xie, L. Hua, Z. Wang, Y. Liu, S. Ying, Y. Liu, Z. Ren and S. Yan, Sci. China: Chem., 2023, 66, 826–836 Search PubMed.
- B. Chen, C. Liao, D. Li, H. Liu and S. Wang, J. Mater. Chem. C, 2023, 11, 8767–8775 Search PubMed.
- H. Gao, S. Shen, Y. Qin, G. Liu, T. Gao, X. Dong, Z. Pang, X. Xie, P. Wang and Y. Wang, J. Phys. Chem. Lett., 2022, 13, 7561–7567 Search PubMed.
- H. Gao, Z. Li, Z. Pang, Y. Qin, G. Liu, T. Gao, X. Dong, S. Shen, X. Xie, P. Wang, C.-S. Lee and Y. Wang, ACS Appl. Mater. Interfaces, 2023, 15, 5529–5537 Search PubMed.
- F. Chen, J. Hu, X. Wang, S. Shao, L. Wang, X. Jing and F. Wang, Sci. China: Chem., 2020, 63, 1112–1120 Search PubMed.
- Y. Chang, Y. Wu, X. Wang, W. Li, Q. Yang, S. Wang, S. Shao and L. Wang, Chem. Eng. J., 2023, 451, 138545 Search PubMed.
- K. R. Naveen, H. Lee, R. Braveenth, D. Karthik, K. J. Yang, S. J. Hwang and J. H. Kwon, Adv. Funct. Mater., 2021, 32, 2110356 Search PubMed.
- J. Guo, Y. Yang, C. Dou and Y. Wang, J. Am. Chem. Soc., 2021, 143, 18272–18279 Search PubMed.
- J. Guo, T. Zhang, Z. Li, K. Ye, Y. Wang and C. Dou, Chem. Commun., 2023, 59, 2644–2647 Search PubMed.
- G. Meng, H. Dai, Q. Wang, J. Zhou, T. Fan, X. Zeng, X. Wang, Y. Zhang, D. Yang, D. Ma, D. Zhang and L. Duan, Nat. Commun., 2023, 14, 2394 Search PubMed.
- B. Adelizzi, P. Chidchob, N. Tanaka, B. A. G. Lamers, S. C. J. Meskers, S. Ogi, A. R. A. Palmans, S. Yamaguchi and E. W. Meijer, J. Am. Chem. Soc., 2020, 142, 16681–16689 Search PubMed.
- Q. Li, C. Shi, M. Huang, X. Wei, H. Yan, C. Yang and A. Yuan, Chem. Sci., 2019, 10, 3257–3263 Search PubMed.
- S. Nakatsuka, H. Gotoh, K. Kinoshita, N. Yasuda and T. Hatakeyama, Angew. Chem., Int. Ed., 2017, 56, 5087–5090 Search PubMed.
- S. H. Han, J. H. Jeong, J. W. Yoo and J. Y. Lee, J. Mater. Chem. C, 2019, 7, 3082–3089 Search PubMed.
- P. Jiang, L. Zhan, X. Cao, X. Lv, S. Gong, Z. Chen, C. Zhou, Z. Huang, F. Ni, Y. Zou and C. Yang, Adv. Opt. Mater., 2021, 9, 2100825 Search PubMed.
- T. Hua, L. Zhan, N. Li, Z. Huang, X. Cao, Z. Xiao, S. Gong, C. Zhou, C. Zhong and C. Yang, Chem. Eng. J., 2021, 426, 131169 Search PubMed.
- X. Wu, J.-W. Huang, B.-K. Su, S. Wang, L. Yuan, W.-Q. Zheng, H. Zhang, Y.-X. Zheng, W. Zhu and P.-T. Chou, Adv. Mater., 2022, 34, 2105080 Search PubMed.
- W. Yang, N. Li, J. Miao, L. Zhan, S. Gong, Z. Huang and C. Yang, CCS Chem., 2022, 4, 3463–3471 Search PubMed.
- Y.-C. Cheng, X.-C. Fan, F. Huang, X. Xiong, J. Yu, K. Wang, C.-S. Lee and X.-H. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202212575 Search PubMed.
- W. Yang, J. Miao, F. Hu, Y. Zou, C. Zhong, S. Gong and C. Yang, Adv. Funct. Mater., 2023, 33, 2213056 Search PubMed.
- H. Mubarok, A. Amin, T. Lee, J. Jung, J.-H. Lee and M. H. Lee, Angew. Chem., Int. Ed., 2023, 62, e202306879 Search PubMed.
- J.-J. Hu, X.-F. Luo, Y.-P. Zhang, M.-X. Mao, H.-X. Ni, X. Liang and Y.-X. Zheng, J. Mater. Chem. C, 2022, 10, 768–773 Search PubMed.
- G. Liu, H. Sasabe, K. Kumada, A. Matsunaga, H. Katagiri and J. Kido, J. Mater. Chem. C, 2021, 9, 8308–8313 Search PubMed.
- J. Park, K. J. Kim, J. Lim, T. Kim and J. Y. Lee, Adv. Mater., 2022, 34, 2108581 Search PubMed.
- B. Lei, Z. Huang, S. Li, J. Liu, Z. Bin and J. You, Angew. Chem., Int. Ed., 2023, 62, e202218405 Search PubMed.
- X. Liang, Z.-P. Yan, H.-B. Han, Z.-G. Wu, Y.-X. Zheng, H. Meng, J.-L. Zuo and W. Huang, Angew. Chem., Int. Ed., 2018, 57, 11316–11320 Search PubMed.
- Y. Xu, Z. Cheng, Z. Li, B. Liang, J. Wang, J. Wei, Z. Zhang and Y. Wang, Adv. Opt. Mater., 2020, 8, 1902142 Search PubMed.
- M. Yang, I. S. Park and T. Yasuda, J. Am. Chem. Soc., 2020, 142, 19468–19472 Search PubMed.
- S. Xu, Q. Yang, Y. Zhang, H. Li, Q. Xue, G. Xie, M. Gu, J. Jin, L. Huang and R. Chen, Chin. Chem. Lett., 2021, 32, 1372–1376 Search PubMed.
- Y.-T. Lee, C.-Y. Chan, M. Tanaka, M. Mamada, U. Balijapalli, Y. Tsuchiya, H. Nakanotani, T. Hatakeyama and C. Adachi, Adv. Electron. Mater., 2021, 7, 2001090 Search PubMed.
- M. Yang, S. Shikita, H. Min, I. S. Park, H. Shibata, N. Amanokura and T. Yasuda, Angew. Chem., Int. Ed., 2021, 60, 23142–23147 Search PubMed.
- X. Yan, Z. Li, Q. Wang, Y. Qu, Y. Xu and Y. Wang, J. Mater. Chem. C, 2022, 10, 15408–15415 Search PubMed.
- Y. Qi, W. Ning, Y. Zou, X. Cao, S. Gong and C. Yang, Adv. Funct. Mater., 2021, 31, 2102017 Search PubMed.
- Y. Zhang, G. Li, L. Wang, T. Huang, J. Wei, G. Meng, X. Wang, X. Zeng, D. Zhang and L. Duan, Angew. Chem., Int. Ed., 2022, 61, e202202380 Search PubMed.
- X. Cai, Y. Xu, Y. Pan, L. Li, Y. Pu, X. Zhuang, C. Li and Y. Wang, Angew. Chem., Int. Ed., 2023, 62, e202216473 Search PubMed.
- Y. Zhang, J. Wei, L. Wang, T. Huang, G. Meng, X. Wang, X. Zeng, M. Du, T. Fan, C. Yin, D. Zhang and L. Duan, Adv. Mater., 2023, 35, 2209396 Search PubMed.
- F. Zhang, F. Rauch, A. Swain, T. B. Marder and P. Ravat, Angew. Chem., Int. Ed., 2023, 62, e202218965 Search PubMed.
- B. Du, K. Zhang, P. Wang, X. Wang, S. Wang, S. Shao and L. Wang, J. Mater. Chem. C, 2023, 11, 9578–9585 Search PubMed.
- J. Liu, L. Chen, X. Wang, Q. Yang, L. Zhao, C. Tong, S. Wang, S. Shao and L. Wang, Macromol. Rapid Commun., 2022, 43, e2200079 Search PubMed.
- G. Liu, H. Sasabe, K. Kumada, H. Arai and J. Kido, Chem. – Eur. J., 2022, 28, e202201605 Search PubMed.
- Q. Li, Y. Wu, X. Wang, Q. Yang, J. Hu, R. Zhong, S. Shao and L. Wang, Chem. – Eur. J., 2022, 28, e202104214 Search PubMed.
- Q. Li, Y. Wu, Q. Yang, S. Wang, S. Shao and L. Wang, ACS Appl. Mater. Interfaces, 2022, 14, 49995–50003 Search PubMed.
- J. Han, Z. Huang, X. Lv, J. Miao, Y. Qiu, X. Cao and C. Yang, Adv. Opt. Mater., 2022, 10, 2102092 Search PubMed.
- Z.-P. Yan, L. Yuan, Y. Zhang, M.-X. Mao, X.-J. Liao, H.-X. Ni, Z.-H. Wang, Z. An, Y.-X. Zheng and J.-L. Zuo, Adv. Mater., 2022, 34, 2204253 Search PubMed.
- Y. Zhang, D. Zhang, J. Wei, Z. Liu, Y. Lu and L. Duan, Angew. Chem., Int. Ed., 2019, 58, 16912–16917 Search PubMed.
- F. Liu, Z. Cheng, L. Wan, Z. Feng, H. Liu, H. Jin, L. Gao, P. Lu and W. Yang, Small, 2022, 18, e2106462 Search PubMed.
- F. Huang, X.-C. Fan, Y.-C. Cheng, H. Wu, Y.-Z. Shi, J. Yu, K. Wang, C.-S. Lee and X.-H. Zhang, Mater. Horiz., 2022, 9, 2226–2232 Search PubMed.
- W. Xue, H. Yan, Y. He, L. Wu, X. Zhang, Y. Wu, J. Xu, J. He, C. Yan and H. Meng, Chem. – Eur. J., 2022, 28, e202201006 Search PubMed.
- Y.-H. He, F.-M. Xie, H.-Z. Li, K. Zhang, Y. Shen, F. Ding, C.-Y. Wang, Y.-Q. Li and J.-X. Tang, Mater. Chem. Front., 2023, 7, 2454–2463 Search PubMed.
- X.-F. Luo, H.-X. Ni, X. Liang, D. Yang, D. Ma, Y.-X. Zheng and J.-L. Zuo, Adv. Opt. Mater., 2023, 11, 2203002 Search PubMed.
- Y. Li, W. Li, J. Hu, X. Yao, L. Hua, W. Cai, S. Shi, C. Zhang, Z. Liu, S. Li, X. Chen, Z. Sun, Z. Ren, M.-C. Tang, G. Wei and Z. Fei, Adv. Opt. Mater., 2023, 11, 2300298 Search PubMed.
- Y. Xu, C. Li, Z. Li, Q. Wang, X. Cai, J. Wei and Y. Wang, Angew. Chem., Int. Ed., 2020, 59, 17442–17446 Search PubMed.
- Y.-T. Lee, C.-Y. Chan, M. Tanaka, M. Mamada, K. Goushi, X. Tang, Y. Tsuchiya, H. Nakanotani and C. Adachi, Adv. Opt. Mater., 2022, 10, 2200682 Search PubMed.
- X. Cai, Y. Pu, C. Li, Z. Wang and Y. Wang, Angew. Chem., Int. Ed., 2023, 62, e202304104 Search PubMed.
- Y. Zou, M. Yu, J. Miao, T. Huang, S. Liao, X. Cao and C. Yang, Chem. Sci., 2023, 14, 3326–3331 Search PubMed.
- J.-P. Liu, L. Chen, L. Zhao, C.-Y. Tong, S.-M. Wang, S.-Y. Shao and L.-X. Wang, Chin. J. Polym. Sci., 2023, 41, 802–810 Search PubMed.
- Y. Liu, X. Xiao, Y. Ran, Z. Bin and J. You, Chem. Sci., 2021, 12, 9408–9412 Search PubMed.
- M. Yang, R. K. Konidena, S. Shikita and T. Yasuda, J. Mater. Chem. C, 2023, 11, 917–922 Search PubMed.
- X. Xiao, B. Lei, D. Wu and Z. Bin, Chem. Commun., 2023, 59, 6556–6559 Search PubMed.
- Q. Wu, J. Li, D. Liu, Y. Mei, B. Liu, J. Wang, M. Xu and Y. Li, Dyes Pigm., 2023, 217, 111421 Search PubMed.
- C.-Z. Du, Y. Lv, H. Dai, X. Hong, J. Zhou, J.-K. Li, R.-R. Gao, D. Zhang, L. Duan and X.-Y. Wang, J. Mater. Chem. C, 2023, 11, 2469–2474 Search PubMed.
- Y. Zhang, D. Zhang, J. Wei, X. Hong, Y. Lu, D. Hu, G. Li, Z. Liu, Y. Chen and L. Duan, Angew. Chem., Int. Ed., 2020, 59, 17499–17503 Search PubMed.
- Y. Wei, K. Pan, X. Cao, Y. Li, X. Zhou and C. Yang, CCS Chem., 2022, 4, 3852–3863 Search PubMed.
- X.-F. Luo, H.-X. Ni, H.-L. Ma, Z.-Z. Qu, J. Wang, Y.-X. Zheng and J.-L. Zuo, Adv. Opt. Mater., 2022, 10, 2102513 Search PubMed.
- Y. Liu, X. Xiao, Z. Huang, D. Yang, D. Ma, J. Liu, B. Lei, Z. Bin and J. You, Angew. Chem., Int. Ed., 2022, 61, e202210210 Search PubMed.
- X.-F. Luo, S.-Q. Song, H.-X. Ni, H. Ma, D. Yang, D. Ma, Y.-X. Zheng and J.-L. Zuo, Angew. Chem., Int. Ed., 2022, 61, e202209984 Search PubMed.
- H. Chen, T. Fan, G. Zhao, D. Zhang, G. Li, W. Jiang, L. Duan and Y. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202300934 Search PubMed.
- Y. Qiu, H. Xia, J. Miao, Z. Huang, N. Li, X. Cao, J. Han, C. Zhou, C. Zhong and C. Yang, ACS Appl. Mater. Interfaces, 2021, 13, 59035–59042 Search PubMed.
- T. Hua, J. Miao, H. Xia, Z. Huang, X. Cao, N. Li and C. Yang, Adv. Funct. Mater., 2022, 32, 2201032 Search PubMed.
- F. Liu, Z. Cheng, Y. Jiang, L. Gao, H. Liu, H. Liu, Z. Feng, P. Lu and W. Yang, Angew. Chem., Int. Ed., 2022, 61, e202116927 Search PubMed.
- X. Xiong, Y.-C. Cheng, K. Wang, J. Yu and X.-H. Zhang, Mater. Chem. Front., 2023, 7, 929–936 Search PubMed.
- Y. Yang, N. Li, J. Miao, X. Cao, A. Ying, K. Pan, X. Lv, F. Ni, Z. Huang, S. Gong and C. Yang, Angew. Chem., Int. Ed., 2022, 61, e202202227 Search PubMed.
- J. Liu, Y. Zhu, T. Tsuboi, C. Deng, W. Lou, D. Wang, T. Liu and Q. Zhang, Nat. Commun., 2022, 13, 4876 Search PubMed.
- X.-F. Luo, H.-X. Ni, L. Shen, L. Wang, X. Xiao and Y.-X. Zheng, Chem. Commun., 2023, 59, 2489–2492 Search PubMed.
- D. H. Ahn, J. H. Maeng, H. Lee, H. Yoo, R. Lampande, J. Y. Lee and J. H. Kwon, Adv. Opt. Mater., 2020, 8, 2000102 Search PubMed.
- J. Park, J. Lim, J. H. Lee, B. Jang, J. H. Han, S. S. Yoon and J. Y. Lee, ACS Appl. Mater. Interfaces, 2021, 13, 45798–45805 Search PubMed.
- J. Park, J. Moon, J. Lim, J. Woo, S. S. Yoon and J. Y. Lee, J. Mater. Chem. C, 2022, 10, 12300–12306 Search PubMed.
- S. Oda, B. Kawakami, R. Kawasumi, R. Okita and T. Hatakeyama, Org. Lett., 2019, 21, 9311–9314 Search PubMed.
- J. A. Knöller, G. Meng, X. Wang, D. Hall, A. Pershin, D. Beljonne, Y. Olivier, S. Laschat, E. Zysman-Colman and S. Wang, Angew. Chem., Int. Ed., 2020, 59, 3156–3160 Search PubMed.
- X.-F. Luo, H.-X. Ni, A.-Q. Lv, X.-K. Yao, H.-L. Ma and Y.-X. Zheng, Adv. Opt. Mater., 2022, 10, 2200504 Search PubMed.
- X. Cai, J. Xue, C. Li, B. Liang, A. Ying, Y. Tan, S. Gong and Y. Wang, Angew. Chem., Int. Ed., 2022, 61, e202200337 Search PubMed.
- Y. Zhang, D. Zhang, T. Huang, A. J. Gillett, Y. Liu, D. Hu, L. Cui, Z. Bin, G. Li, J. Wei and L. Duan, Angew. Chem., Int. Ed., 2021, 60, 20498–20503 Search PubMed.
- J.-K. Li, X.-Y. Chen, Y.-L. Guo, X.-C. Wang, A. C.-H. Sue, X.-Y. Cao and X.-Y. Wang, J. Am. Chem. Soc., 2021, 143, 17958–17963 Search PubMed.
- Y. Zou, J. Hu, M. Yu, J. Miao, Z. Xie, Y. Qiu, X. Cao and C. Yang, Adv. Mater., 2022, 34, 2201442 Search PubMed.
- Y. Wang, K. Zhang, F. Chen, X. Wang, Q. Yang, S. Wang, S. Shao and L. Wang, Chin. J. Chem., 2022, 40, 2671–2677 Search PubMed.
- J.-K. Li, M.-Y. Zhang, L. Zeng, L. Huang and X.-Y. Wang, Angew. Chem., Int. Ed., 2023, 62, e202303093 Search PubMed.
- T. Fan, M. Du, X. Jia, L. Wang, Z. Yin, Y. Shu, Y. Zhang, J. Wei, D. Zhang and L. Duan, Adv. Mater., 2023, 35, 2301018 Search PubMed.
- I. S. Park, M. Yang, H. Shibata, N. Amanokura and T. Yasuda, Adv. Mater., 2022, 34, 2107951 Search PubMed.
- X.-C. Fan, K. Wang, Y.-Z. Shi, Y.-C. Cheng, Y.-T. Lee, J. Yu, X.-K. Chen, C. Adachi and X.-H. Zhang, Nat. Photonics, 2023, 17, 280–285 Search PubMed.
- K. Matsui, S. Oda, K. Yoshiura, K. Nakajima, N. Yasuda and T. Hatakeyama, J. Am. Chem. Soc., 2018, 140, 1195–1198 Search PubMed.
- X. Lv, J. Miao, M. Liu, Q. Peng, C. Zhong, Y. Hu, X. Cao, H. Wu, Y. Yang, C. Zhou, J. Ma, Y. Zou and C. Yang, Angew. Chem., Int. Ed., 2022, 61, e202201588 Search PubMed.
- J. Jin, C. Duan, H. Jiang, P. Tao, H. Xu and W.-Y. Wong, Angew. Chem., Int. Ed., 2023, 62, e202218947 Search PubMed.
- J. H. Kim, W. J. Chung, J. Kim and J. Y. Lee, Mater. Today Energy, 2021, 21, 100792 Search PubMed.
- Y. Wang, Y. Duan, R. Guo, S. Ye, K. Di, W. Zhang, S. Zhuang and L. Wang, Org. Electron., 2021, 97, 106275 Search PubMed.
- H. J. Cheon, Y.-S. Shin, N.-H. Park, J.-H. Lee and Y.-H. Kim, Small, 2022, 18, e2107574 Search PubMed.
- H.-J. Cheon, S.-J. Woo, S.-H. Baek, J.-H. Lee and Y.-H. Kim, Adv. Mater., 2022, 34, 2207416 Search PubMed.
- K. R. Naveen, S. J. Hwang, H. Lee and J. H. Kwon, Adv. Electron. Mater., 2022, 8, 2101114 Search PubMed.
- J. Bian, S. Chen, L. Qiu, N. Zhang, J. Zhang, C. Duan, C. Han and H. Xu, Research, 2022, 2022, 9838120 Search PubMed.
- H. Lee, R. Braveenth, J. D. Park, C. Y. Jeon, H. S. Lee and J. H. Kwon, ACS Appl. Mater. Interfaces, 2022, 14, 36927–36935 Search PubMed.
- Y.-N. Hu, X.-C. Fan, F. Huang, Y.-Z. Shi, H. Wang, Y.-C. Cheng, M.-Y. Chen, K. Wang, J. Yu and X.-H. Zhang, Adv. Opt. Mater., 2023, 11, 2202267 Search PubMed.
- E. Ravindran, H. E. Baek, H. W. Son, J. H. Park, Y.-H. Kim and M. C. Suh, Adv. Funct. Mater., 2023, 33, 2213461 Search PubMed.
- K. Di, R. Guo, Y. Wang, Y. Lv, H. Su, Q. Zhang, B. Yang and L. Wang, J. Mater. Chem. C, 2023, 11, 6429–6437 Search PubMed.
- Y. Wang, K. Di, Y. Duan, R. Guo, L. Lian, W. Zhang and W. L. Wang, Chem. Eng. J., 2022, 431, 133221 Search PubMed.
- Y. Wang, R. Guo, A. Ying, K. Di, L. Chen, H. Gu, S. Liu, Y. Duan, H. Su, S. Gong and L. Wang, Adv. Opt. Mater., 2023, 11, 2202034 Search PubMed.
- C.-Y. Chan, S. Madayanad Suresh, Y.-T. Lee, Y. Tsuchiya, T. Matulaitis, D. Hall, A. M. Z. Slawin, S. Warriner, D. Beljonne, Y. Olivier, C. Adachi and E. Zysman-Colman, Chem. Commun., 2022, 58, 9377–9380 Search PubMed.
- Y. Lee and J.-I. Hong, J. Mater. Chem. C, 2022, 10, 11855–11861 Search PubMed.
- M. Wang, Z. Fu, R. Cheng, J. Du, T. Wu, Z. Bin, D. Wu, Y. Yang and J. Lan, Chem. Commun., 2023, 59, 5126–5129 Search PubMed.
- N. Ikeda, S. Oda, R. Matsumoto, M. Yoshioka, D. Fukushima, K. Yoshiura, N. Yasuda and T. Hatakeyama, Adv. Mater., 2020, 32, 2004072 Search PubMed.
- T. Fan, Y. Zhang, L. Wang, Q. Wang, C. Yin, M. Du, X. Jia, G. Li and L. Duan, Angew. Chem., Int. Ed., 2022, 61, e202213585 Search PubMed.
- K. R. Naveen, H. Lee, L. H. Seung, Y. H. Jung, C. P. Keshavananda Prabhu, S. Muruganantham and J. H. Kwon, Chem. Eng. J., 2023, 451, 138498 Search PubMed.
- H. Tanaka, S. Oda, G. Ricci, H. Gotoh, K. Tabata, R. Kawasumi, D. Beljonne, Y. Olivier and T. Hatakeyama, Angew. Chem., Int. Ed., 2021, 60, 17910–17914 Search PubMed.
- K. R. Naveen, J. H. Oh, H. Lee and J. H. Kwon, Angew. Chem., Int. Ed., 2023, 62, e202306768 Search PubMed.
- K. Rayappa Naveen, H. Lee, R. Braveenth, K. Joon Yang, S. Jae Hwang and J. Hyuk Kwon, Chem. Eng. J., 2022, 432, 134381 Search PubMed.
- H. S. Kim, H. J. Cheon, D. Lee, W. Lee, J. Kim, Y.-H. Kim and S. Yoo, Sci. Adv., 2023, 9, eadf1388 Search PubMed.
- F. Huang, X.-C. Fan, Y.-C. Cheng, H. Wu, X. Xiong, J. Yu, K. Wang and X.-H. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202306413 Search PubMed.
- S. Oda, B. Kawakami, Y. Yamasaki, R. Matsumoto, M. Yoshioka, D. Fukushima, S. Nakatsuka and T. Hatakeyama, J. Am. Chem. Soc., 2022, 144, 106–112 Search PubMed.
- S. Oda, B. Kawakami, M. Horiuchi, Y. Yamasaki, R. Kawasumi and T. Hatakeyama, Adv. Sci., 2023, 10, 2205070 Search PubMed.
- Y. Sano, T. Shintani, M. Hayakawa, S. Oda, M. Kondo, T. Matsushita and T. Hatakeyama, J. Am. Chem. Soc., 2023, 145, 11504–11511 Search PubMed.
- S. Nakatsuka, N. Yasuda and T. Hatakeyama, J. Am. Chem. Soc., 2018, 140, 13562–13565 Search PubMed.
- D. T. Yang, T. Nakamura, Z. He, X. Wang, A. Wakamiya, T. Peng and S. Wang, Org. Lett., 2018, 20, 6741–6745 Search PubMed.
- G. Meng, H. Dai, T. Huang, J. Wei, J. Zhou, X. Li, X. Wang, X. Hong, C. Yin, X. Zeng, Y. Zhang, D. Yang, D. Ma, G. Li, D. Zhang and L. Duan, Angew. Chem., Int. Ed., 2022, 61, e202207293 Search PubMed.
- G. Meng, H. Dai, J. Zhou, T. Huang, X. Zeng, Q. Wang, X. Wang, Y. Zhang, T. Fan, D. Yang, D. Ma, D. Zhang and L. Duan, Chem. Sci., 2023, 14, 979–986 Search PubMed.
- G. Meng, L. Liu, Z. He, D. Hall, X. Wang, T. Peng, X. Yin, P. Chen, D. Beljonne, Y. Olivier, E. Zysman-Colman, N. Wang and S. Wang, Chem. Sci., 2022, 13, 1665–1674 Search PubMed.
- S. Oda, K. Ueura, B. Kawakami and T. Hatakeyama, Org. Lett., 2020, 22, 700–704 Search PubMed.
- S. Uemura, S. Oda, M. Hayakawa, R. Kawasumi, N. Ikeda, Y.-T. Lee, C.-Y. Chan, Y. Tsuchiya, C. Adachi and T. Hatakeyama, J. Am. Chem. Soc., 2023, 145, 1505–1511 Search PubMed.
- S. Madayanad Suresh, L. Zhang, T. Matulaitis, D. Hall, C. Si, G. Ricci, A. M. Z. Slawin, S. Warriner, D. Beljonne, Y. Olivier, I. D. W. Samuel and E. Zysman-Colman, Adv. Mater., 2023, 35, 2300997 Search PubMed.
- S. Madayanad Suresh, L. Zhang, D. Hall, C. Si, G. Ricci, T. Matulaitis, A. M. Z. Slawin, S. Warriner, Y. Olivier, I. D. W. Samuel and E. Zysman-Colman, Angew. Chem., Int. Ed., 2023, 62, e202215522 Search PubMed.
- D. Karthik, D. H. Ahn, J. H. Ryu, H. Lee, J. H. Maeng, J. Y. Lee and J. H. Kwon, J. Mater. Chem. C, 2020, 8, 2272–2279 Search PubMed.
- D. Song, Y. Yu, L. Yue, D. Zhong, Y. Zhang, X. Yang, Y. Sun, G. Zhou and Z. Wu, J. Mater. Chem. C, 2019, 7, 11953–11963 Search PubMed.
- Z. Huang, H. Xie, J. Miao, Y. Wei, Y. Zou, T. Hua, X. Cao and C. Yang, J. Am. Chem. Soc., 2023, 145, 12550–12560 Search PubMed.
- Q. Wang, Y. Xu, T. Yang, J. Xue and Y. Wang, Adv. Mater., 2023, 35, 2205166 Search PubMed.
- J. Hwang, H. Kang, J.-E. Jeong, H. Y. Woo, M. J. Cho, S. Park and D. H. Choi, Chem. Eng. J., 2021, 416, 129185 Search PubMed.
- Y. Xu, Q. Wang, X. Cai, C. Li and Y. Wang, Adv. Mater., 2021, 33, 2100652 Search PubMed.
- Y. Xu, C. Li, Z. Li, J. Wang, J. Xue, Q. Wang, X. Cai and Y. Wang, CCS Chem., 2022, 4, 2065–2079 Search PubMed . (Previous citation CCS Chem., 2021, 3, 2077).
- D. W. Lee, J. Hwang, H. J. Kim, H. Lee, J. M. Ha, H. Y. Woo, S. Park, M. J. Cho and D. H. Choi, ACS Appl. Mater. Interfaces, 2021, 13, 49076–49084 Search PubMed.
- Z. Zhao, C. Zeng, X. Peng, Y. Liu, H. Zhao, L. Hua, S.-J. Su, S. Yan and Z. Ren, Angew. Chem., Int. Ed., 2022, 61, e202210864 Search PubMed.
- R. Braveenth, H. Lee, J. D. Park, K. J. Yang, S. J. Hwang, K. R. Naveen, R. Lampande and J. H. Kwon, Adv. Funct. Mater., 2021, 31, 2105805 Search PubMed.
- J. Hwang, C. W. Koh, J. M. Ha, H. Y. Woo, S. Park, M. J. Cho and D. H. Choi, ACS Appl. Mater. Interfaces, 2021, 13, 61454–61462 Search PubMed.
- J. H. Maeng, D. H. Ahn, H. Lee, Y. H. Jung, D. Karthik, J. Y. Lee and J. H. Kwon, Dyes Pigm., 2020, 180, 108485 Search PubMed.
- J. Wang, J. Miao, C. Jiang, S. Luo, C. Yang and K. Li, Adv. Opt. Mater., 2022, 10, 2201071 Search PubMed.
- Y. Lee and J.-I. Hong, Adv. Opt. Mater., 2021, 9, 2100406 Search PubMed.
- T. Hua, Y.-C. Liu, C.-W. Huang, N. Li, C. Zhou, Z. Huang, X. Cao, C.-C. Wu and C. Yang, Chem. Eng. J., 2022, 433, 133598 Search PubMed.
- H. Xie, Z. Huang, N. Li, T. Hua, J. Miao and C. Yang, J. Mater. Chem. C, 2022, 10, 11239–11245 Search PubMed.
- D. Karthik, Y. H. Jung, H. Lee, S. Hwang, B.-M. Seo, J.-Y. Kim, C. W. Han and J. H. Kwon, Adv. Mater., 2021, 33, 2007724 Search PubMed.
- Y. Watanabe, H. Sasabe and J. Kido, Bull. Chem. Soc. Jpn., 2019, 92, 716–728 Search PubMed.
- X.-Q. Gan, Z.-M. Ding, D.-H. Liu, W.-Q. Zheng, B. Ma, H. Zhang, X. Chang, L. Wang, Y. Liu, X. Wu, S.-J. Su and W. Zhu, Adv. Opt. Mater., 2023, 11, 2300195 Search PubMed.
- Y.-J. Yu, S.-N. Zou, C.-C. Peng, Z.-Q. Feng, Y.-K. Qu, S.-Y. Yang, Z.-Q. Jiang and L.-S. Liao, J. Mater. Chem. C, 2022, 10, 4941–4946 Search PubMed.
- M. Godumala, J. Hwang, H. Kang, J.-E. Jeong, A. K. Harit, M. J. Cho, H. Y. Woo, S. Park and D. H. Choi, ACS Appl. Mater. Interfaces, 2020, 12, 35300–35310 Search PubMed.
- E. Y. Park, J. H. Park, Y.-H. Kim and M. C. Suh, J. Mater. Chem. C, 2022, 10, 4705–4716 Search PubMed.
- S. Wu, L. Zhang, J. Wang, A. Kumar Gupta, I. D. W. Samuel and E. Zysman-Colman, Angew. Chem., Int. Ed., 2023, 62, e202305182 Search PubMed.
- Y. Zhang, J. Wei, D. Zhang, C. Yin, G. Li, Z. Liu, X. Jia, J. Qiao and L. Duan, Angew. Chem., Int. Ed., 2022, 61, e202113206 Search PubMed.
- X. Song, S. Shen, S. Zou, F. Guo, Y. Wang, S. Gao and Y. Zhang, Chem. Eng. J., 2023, 467, 143557 Search PubMed.
- G. Chen, J. Wang, W.-C. Chen, Y. Gong, N. Zhuang, H. Liang, L. Xing, Y. Liu, S. Ji, H.-L. Zhang, Z. Zhao, Y. Huo and B. Z. Tang, Adv. Funct. Mater., 2023, 2211893 Search PubMed.
- P. Jiang, J. Miao, X. Cao, H. Xia, K. Pan, T. Hua, X. Lv, Z. Huang, Y. Zou and C. Yang, Adv. Mater., 2022, 34, 2106954 Search PubMed.
- Y.-K. Qu, D.-Y. Zhou, F.-C. Kong, Q. Zheng, X. Tang, Y.-H. Zhu, C.-C. Huang, Z.-Q. Feng, J. Fan, C. Adachi, L.-S. Liao and Z.-Q. Jiang, Angew. Chem., Int. Ed., 2022, 61, e202201886 Search PubMed.
- T. Wang, X. Yin, X. Cao and C. Yang, Angew. Chem., Int. Ed., 2023, 62, e202301988 Search PubMed.
- T. Wang, Y. Zou, Z. Huang, N. Li, J. Miao and C. Yang, Angew. Chem., Int. Ed., 2022, 61, e202211172 Search PubMed.
- L. Yang, P. Wang, K. Zhang, S. Wang, S. Shao and L. Wang, Dyes Pigm., 2023, 216, 111371 Search PubMed.
- J. Z. Deng, D. V. Paone, A. T. Ginnetti, H. Kurihara, S. D. Dreher, S. A. Weissman, S. R. Stauffer and C. S. Burgey, Org. Lett., 2009, 11, 345–347 Search PubMed.
- F. Huang, X.-C. Fan, Y.-C. Cheng, Y. Xie, S. Luo, T. Zhang, H. Wu, X. Xiong, J. Yu, D.-D. Zhang, X.-K. Chen, K. Wang and X.-H. Zhang, Adv. Opt. Mater., 2023, 11, 2202950 Search PubMed.
- Y. Hu, J. Miao, C. Zhong, Y. Zeng, S. Gong, X. Cao, X. Zhou, Y. Gu and C. Yang, Angew. Chem., Int. Ed., 2023, 62, e202302478 Search PubMed.
- Y. Xu, Q. Wang, J. Wei, X. Peng, J. Xue, Z. Wang, S.-J. Su and Y. Wang, Angew. Chem., Int. Ed., 2022, 61, e202204652 Search PubMed.
- Q. Wang, Y. Xu, T. Huang, Y. Qu, J. Xue, B. Liang and Y. Wang, Angew. Chem., Int. Ed., 2023, 62, e202301930 Search PubMed.
- Y. Xu, C. Li, Z. Li, J. Wang, J. Xue, Q. Wang, X. Cai and Y. Wang, J. Am. Chem. Soc., 2015, 137, 9481–9488 Search PubMed.
- F. Huang, Y.-C. Cheng, H. Wu, X. Xiong, J. Yu, X.-C. Fan, K. Wang and X.-H. Zhang, Chem. Eng. J., 2023, 465, 142900 Search PubMed.
- J. Bian, S. Chen, L. Qiu, R. Tian, Y. Man, Y. Wang, S. Chen, J. Zhang, C. Duan, C. Han and H. Xu, Adv. Mater., 2022, 34, 2110547 Search PubMed.
- X.-J. Liao, D. Pu, L. Yuan, J. Tong, S. Xing, Z.-L. Tu, J.-L. Zuo, W.-H. Zheng and Y.-X. Zheng, Angew. Chem., Int. Ed., 2023, 62, e202217045 Search PubMed.
- N. Sharma, E. Spuling, C. Mattern, W. Li, O. Fuhr, Y. Tsuchiya, C. Adachi, S. Brase, I. Samuel and E. Zysman-Colman, Chem. Sci., 2019, 10, 6689–6696 Search PubMed.
- Z.-G. Wu, H.-B. Han, Z.-P. Yan, X.-F. Luo, Y. Wang, Y.-X. Zheng, J.-L. Zuo and Y. Pan, Adv. Mater., 2019, 31, 1900524 Search PubMed.
- M. Wu, N. Li, C. Shi, J. Song, R. Zeng, F. Li, Q. Li, A. Yuan and C. Yang, Inorg. Chem. Front., 2023, 10, 1262–1269 Search PubMed.
- C. Shi, F. Li, Q. Li, W. Zhao, Y. Cao, Q. Zhao and A. Yuan, Inorg. Chem., 2021, 60, 525–534 Search PubMed.
- J. Wang, N. Li, C. Zhong, J. Miao, Z. Huang, M. Yu, Y. X. Hu, S. Luo, Y. Zou, K. Li and C. Yang, Adv. Mater., 2023, 35, 2208378 Search PubMed.
- J. Guo, Z. Li, X. Tian, T. Zhang, Y. Wang and C. Dou, Angew. Chem., Int. Ed., 2023, 62, e202217470 Search PubMed.
- G. Li, X. Liu, M. Wu, R. Zeng, Q. Li, A. Yuan and C. Shi, Dyes Pigm., 2022, 208, 110805 Search PubMed.
- K. Matsuo, S. Saito and S. Yamaguchi, J. Am. Chem. Soc., 2014, 136, 12580–12583 Search PubMed.
- Y. Qi, X. Cao, Y. Zou and C. Yang, J. Mater. Chem. C, 2021, 9, 1567–1571 Search PubMed.
- T. Agou, J. Kobayashi and T. Kawashima, Org. Lett., 2006, 8, 2241–2244 Search PubMed.
- S. M. Suresh, E. Duda, D. Hall, Z. Yao, S. Bagnich, A. M. Z. Slawin, H. Bässler, D. Beljonne, M. Buck, Y. Olivier, A. Köhler and E. Zysman-Colman, J. Am. Chem. Soc., 2020, 142, 6588–6599 Search PubMed.
- K. Stavrou, S. Madayanad Suresh, D. Hall, A. Danos, N. A. Kukhta, A. M. Z. Slawin, S. Warriner, D. Beljonne, Y. Olivier, A. Monkman and E. Zysman-Colman, Adv. Opt. Mater., 2022, 10, 2200688 Search PubMed.
- X. Wang, Y. Zhang, H. Dai, G. Li, M. Liu, G. Meng, X. Zeng, T. Huang, L. Wang, Q. Peng, D. Yang, D. Ma, D. Zhang and L. Duan, Angew. Chem., Int. Ed., 2022, 61, e202206916 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: List of compounds. See DOI: https://doi.org/10.1039/d3cs00837a |
| ‡ These authors equally contributed to this work. |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Masahiro
Hayakawa‡
,
Masahiro
Hayakawa‡
































































































