
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBoron enabled bioconjugation chemistries
Mengmeng
Zheng
,
Lingchao
Kong
and
Jianmin
Gao
 *
*
Department of Chemistry, Merkert Chemistry Center, Boston College, 2609 Beacon Street, Chestnut Hill, MA 02467, USA. E-mail: jianmin.gao@bc.edu
First published on 30th October 2024
Abstract
Novel bioconjugation reactions have been heavily pursued for the past two decades. A myriad of conjugation reactions have been developed for labeling molecules of interest in their native context as well as for constructing multifunctional molecular entities or stimuli-responsive materials. A growing cluster of bioconjugation reactions were realized by tapping into the unique properties of boron. As a rare element in human biology, boronic acids and esters exhibit remarkable biocompatibility. A number of organoboron reagents have been evaluated for bioconjugation, targeting the reactivity of either native biomolecules or those incorporating bioorthogonal functional groups. Owing to the dynamic nature of B–O and B–N bond formation, a significant portion of the boron-enabled bioconjugations exhibit rapid reversibility and accordingly have found applications in the development of reversible covalent inhibitors. On the other hand, stable bioconjugations have been developed that display fast kinetics and significantly expand the repertoire of bioorthogonal chemistry. This contribution presents a summary and comparative analysis of the recently developed boron-mediated bioconjugations. Importantly, this article seeks to provide an in-depth discussion of the thermodynamic and kinetic profiles of these boron-enabled bioconjugations, which reveals structure–reactivity relationships and provides guidelines for bioapplications.
 Mengmeng Zheng | Mengmeng Zheng obtained her PhD from Nanjing University in 2016. She was a postdoctoral fellow at the University of South Florida from 2016 to 2019, focusing on the discovery of cyclic peptidomimetic ligands. In the fall of 2019, she joined the Gao group in the Department of Chemistry of Boston College as a post-doctoral researcher. Her research focuses on developing novel strategies for constructing phage-displayed peptide libraries that incorporate covalent warheads as binding enhancers. |
 Lingchao Kong | Lingchao Kong obtained his Master's degree from Brandeis University in 2017. After a few years of experience in industry, Lingchao returned to graduate school and joined the Gao group at Boston College. His current research focuses on the design, synthesis, and characterization of lysine-targeting covalent warheads, both reversible and irreversible, and their application in the discovery of covalent inhibitors. |
 Jianmin Gao | Jianmin Gao received his PhD from Stanford University and completed his post-doctoral training at the Scripps Research Institute. In 2007, he started his independent career at Boston College, where he remains as a Professor of Chemistry. The Gao group strives to innovate molecular design strategies to allow ligand discovery for “undruggable” biomolecules. The invention of novel biocompatible reactions and their implementation in pertinent biological systems have been a central theme of Gao's research. In particular, the Gao group has been a leader in the use of reversible covalent chemistry to target lysines and the creation of reversible covalent phage libraries. |
1. Introduction
In the past two decades, the field of bioconjugation chemistry has seen tremendous advances, yielding many elegant conjugation reactions that can be implemented in biological milieu. It culminated with the 2022 Nobel Prize awarded to Professors Carolyn Bertozzi, Barry Sharpless and Morten Meldal for their seminal work developing click chemistry1 and bioorthogonal chemistry.2,3 Bioconjugation refers to chemical reactions that covalently link two molecules, of which one or both are biomolecules such as proteins, nucleic acids, carbohydrates, and lipids. Although the field initiated with native biomolecules, bioconjugations are increasingly done on engineered biomolecules with nonnatural motifs. The biological origin of the client molecules demands biocompatibility in terms of reaction conditions as well as reagent stability in water. Specifically, bioconjugations need to be performed in complete or predominant aqueous solutions of near neutral pH – such conditions are necessary to maintain the integrity and function of pertinent biomolecules. Bioconjugation reactions have been developed to modify or track molecules of interest under various contexts, ranging from engineering recombinant proteins as therapeutics, to cell and animal studies, and even further to clinical studies on human patients. A bioconjugation reaction would ideally benefit from the following attributes: (1) it can be carried out under physiological conditions (neutral aqueous media and ambient temperature), (2) it produces high yields and minimal side products, and (3) it can be chemoselective and, even better, target and site selective to allow precision labelling of a target molecule in complex biological mixtures. The relative importance of these attributes is expected to vary according to specific end applications. For example, chemoselectivity is essential for activity-based protein profiling,4 where target specificity is not an intended goal. In contrast, to study protein homeostasis in cells and living organisms, target-specific labelling of the protein of interest is critical. We will discuss these different levels of selectivity in the context of individual reactions and applications throughout this review.A significant and growing sub-collection of these bioconjugation reactions involves the use of organoboron reagents. Although present in the human diet, boron exists in the human body primarily as boric acid and has not been extensively studied in human physiology.5 In sharp contrast, synthetic organoboron compounds have attracted remarkable attention in medicinal chemistry. Several organoboron compounds have received approval for clinical use from the U.S. Food and Drug Administration (FDA),6 including the anticancer drug bortezomib and antibacterial agent vaborbactam (Fig. 1). These molecules function by binding their protein target via reversible covalent bond formation. This covalent binding and inhibition mechanism represents a unique form of bioconjugation, which has inspired additional and creative use of organoboron compounds to modify and conjugate with biomolecules.
 | ||
| Fig. 1 Organoboron drugs that function via covalent conjugation to proteins. (A) Bortezomib in complex with human 20S proteasome as an example to illustrate the mechanism of covalent inhibition (PDB: 5LF3). (B) Chemical structure of bortezomib and several other FDA-approved drugs that contain reactive boron. | ||
Inspired by the covalent mechanism of the organoboron drugs, much effort has been paid to explore the use of boron in bioconjugation reactions.7–10 Various organoboron reagents have been synthesized and investigated for their conjugation with endogenous nucleophiles and functional groups have been designed to achieve bioorthogonal conjugations. We will discuss these reactions and their applications with special attention given to the most notable advances of the past decade or so. For earlier developments, we will direct the readers to pertinent review articles published previously. It is worth noting upfront that many boron-enabled bioconjugations exhibit reversibility, which is desirable for certain applications, yet less optimal for others where stable conjugation is required. Hence, tuning the reversibility has turned out to be a recurring theme of boron mediated conjugation reactions. Nevertheless, the dynamic nature of boron-mediated chemistries renders a unique collection of bioconjugation reactions, which will be summarized and comparatively analysed in this contribution. We also note that our discussions herein will emphasize the design principles and mechanistic insights to highlight the uniqueness of the reactions. Pertinent biological applications will be noted, but not described in detail, to highlight the types of applications uniquely enabled by these bioconjugation reactions.
2. Conjugation with hydroxyls
2.1 Boronate ester mediated binding of biomolecules
Boronic acids (BAs) and boronate esters (BEs) represent the dominant classes of organoboron reagents used in bioconjugation reactions. Boronate esters, also referred to as boronic esters depending on their ionization status, depict esters formed between a BA and a hydroxyl group or diol. A new BE can also result from the exchange of a BE with an alcohol. A distinctive feature of BE formation in water is its rapid reversibility: a BE of monomeric alcohols and even some diols can undergo hydrolysis on the time scale of seconds under physiological conditions.11 The Ka or Kd (= 1/Ka) value of such a fast thermodynamic equilibrium can be used to describe the BA's binding affinity or strength to a hydroxyl substrate. This binding strength is pH dependent,12 as both the BA reactant and the BE product can exist as a mixture of neutral and anionic forms, an equilibrium dictated by their respective pKa's (Fig. 2(A)).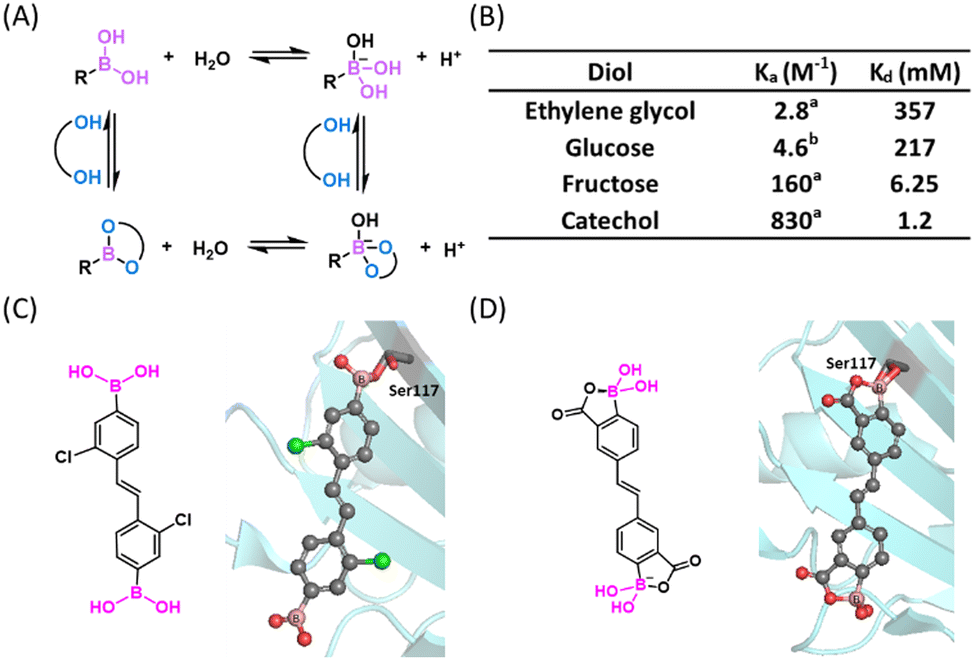 | ||
| Fig. 2 Boronate ester (BE) formation of diols. (A) Complexation mechanism of BA with diol. (B) The Ka and Kd values of diols with phenylboronic acid at pH 7.4. aKa value in water, cited from ref. 13. The Ka value in buffer is expected to be even smaller. bKa value in pH 7.4, 0.10 M phosphate buffer, cited from ref. 14. (C) A transthyretin aggregation inhibitor showing a sp2 hybridized boronic ester upon conjugation to the protein (PDB: 5U4F). (D) Structure (free and protein bound) 2-carboxy-PBA that shows improved stability against oxidation (PDB: 6U0Q). Colour scheme used for all small molecule crystal structures: C: gray; N: blue; O: red; B: pink; S: yellow; H: white; Cl: green. H atoms omitted for clarity in the crystal structures of this figure. | ||
The importance of BE formation has long been recognized in medicinal chemistry. For example, BE formation underlies the working mechanism of the boron-based anticancer drugs, bortezomib and ixazomib (Fig. 1). Bortezomib reached clinical use in 2003 and works by targeting the β5-subunit of the 20S proteasome.15,16 The X-ray analysis revealed a covalent bond between boron and a threonine side chain oxygen, which renders an anionic boron centre with a tetrahedral geometry.17 Ixazomib, approved in 2015 as the first oral proteasome inhibitor,18 also targets the β5-subunit of the 20S proteasome. Other boron based covalent drugs, such as tavaborole as an antifungal agent19 and vaborbactam as a β-lactamase inhibitor, function by covalently binding to tRNA or serine residues respecitvely.20,21 While these boron therapeutics give anionic BEs with a sp3 hybridized boron, a series of stilbene BA derivatives were found to inhibit transthyretin amyloidosis via formation of a sp2-hybridized BE (Fig. 2(C)), highlighting the geometric versatility of BEs towards covalent inhibition.13 The working mechanism of these BA-based covalent inhibitors may appear counter-intuitive as BA conjugation with an isolated alcohol in water is typically unfavourable in terms of thermodynamics due to water competition – even ethylene glycol as a diol does not conjugate with phenylboronic acid (PBA) until reaching molar concentrations (Fig. 2(B)).22 However, inside a drug binding pocket of a protein where water is excluded, BE formation can provide favourable free energy for binding. Importantly, the boronate ester formed presents a structural mimic of the tetrahedral intermediate of peptide bond hydrolysis, hence it can be stabilized by and form readily within the target protein. Furthermore, proper formation of a B–O bond confers exquisite selectivity to the organoboron drugs for binding their intended target over other structurally similar proteins.
With the aim of using organoboron compounds as drugs, their oxidative vulnerability has been somewhat of a concern. For example, unsubstituted PBA can go through oxidation by endogenous reactive oxidative species (ROS) in minutes. In fact, the vulnerability of BA to oxidation has been cleverly utilized to create ROS sensors23 as well as ROS triggered drug release systems,24 although these topics will not be the focus of this review. To mitigate oxidative vulnerability, Raines and co-workers found that a 2-carboxy substituent slowed down the oxidation of PBA by four orders of magnitude, yet the 2-carboxy PBA (2-CPBA) motif is still capable of forming boronate esters as demonstrated by a crystal structure of transthyretin with a covalent ligand (Fig. 2(D)).25
Another prominent application of BE formation concerns carbohydrate recognition and sensing. Carbohydrate molecules often display one or more 1,2 or 1,3-diol functionalities, which are known to conjugate with a BA to form a corresponding cyclic BE. To minimize ring strain, this kind of cyclic ester favours a tetrahedral boron centre, which affords a lowered pKa value of the Lewis acid. This kind of BE formation was first reported by Lorand and Edwards in 1959.22 This simple bioconjugation reaction has been developed into one of the most powerful tools for the recognition and quantification of various carbohydrates under physiological conditions, leading to the creation of “boronolectin”, a term describing a boron analogue of sugar binding proteins (lectins).14 As the BA-diol conjugation is greatly dependent on the diol substrate, the preorganization and orientation of the two –OH groups exerts a significant impact on the binding affinities of a diol to a BA. For example, fructose binds a PBA with ∼10 times higher affinity than glucose and catechol binds PBA with another order of magnitude greater affinity than fructose.26 The use of BA/diol complexation for carbohydrate sensing has been extensively reviewed,27–33 hence, will not be discussed in detail here. Note that this pH reversible BE conjugation has been adapted to design responsive materials as well.34,35
2.2 Boronate ester mediated stable conjugations
While the reversibility of BE conjugation proved useful for biomolecular binding and sensing, stable (irreversible) BE formation has been sought after in the realm of bioorthogonal conjugation. To improve the stability of BE conjugates under biological conditions, efforts were made to optimize both reactants. While installing various substituents on PBA only afforded modest changes of BE stability,12 the Schepartz group reported a bis-PBA pro-fluorescent rhodamine molecule, which recognizes a tetraserine motif (SSPGSS) with a sub-micromolar Kd (452 ± 106 nM).36 This rhodamine-derived bis-PBA exhibits cell membrane permeability and turn-on fluorescence when subjected to conjugation with tetraserine tagged proteins in live cells (Fig. 3(A)). Another major development came from Hall and co-workers, who reported that nopoldiol, a highly strained diol, can conjugate with aryl BAs to give BEs with much improved stability (Fig. 3(B)).37 Specifically, a 2-methyl substituted PBA was found to conjugate with nopoldiol with fast ligation kinetics (kon = 7.7 ± 0.5 M−1 s−1), yielding a product with high thermodynamic stability as well (Ka = 1.2 × 105 M−1). Based on these values, one can estimate a koff of 6.4 × 10−5 s−1, which indicates a half-life of several hours for the BE conjugate, in sharp contrast to the instant reversibility of typical BEs. Thermodynamically speaking, the Ka value mentioned above is 2–3 orders of magnitude greater than that of PBA–fructose (Ka: 102–103 M−1, Fig. 2). Comparative studies of PBA and nopoldiol structural variants showed that the 2-methyl substituent of PBA and the long alkyl group of nopoldiol are important for the observed high affinity of binding. Remarkably, the presence of fructose or glucose had little influence on nopoldiol-2-methyl PBA conjugation even at above serum concentrations. Hall and co-workers further demonstrated efficient nopoldiol ligation to proteins under physiologic conditions. Specifically, a 2-methyl PBA moiety was installed on model proteins, namely albumin and thioredoxin, using cysteine-maleimide chemistry. Subjecting the PBA modified proteins to a fluorophore labelled nopoldiol resulted in fluorescence labelling of the proteins. Consistent with the high BE stability, the nopoldiol labelled proteins were found to survive SDS-PAGE as well as LC–MS analysis. | ||
| Fig. 3 Stable BE conjugation. (A) Conjugation of a rhodamine-derived bis-BPA to a tetraserine tag gives a fluorescence product. (B) Highly strained nopoldiol promotes BE formation. (C) Bifunctional nopoldiol with a thiosemicarbazide elicits irreversible conjugation with 2-APBA. (D) Crystallographic analysis confirms the formation of a model tetracyclic adduct in (C); CCDC ID: VEDWUA; H atoms omitted for clarity in the crystal structure of this figure. | ||
To further improve conjugate stability for in vivo applications, the Hall group designed and optimized a bifunctional reagent, a thiosemicarbazide-functionalized nopoldiol (Fig. 3(C)). This bifunctional molecule was found to readily conjugate with 2-acetyl-PBA (2-APBA), yielding a rate constant of 9 M−1 s−1 as measured by NMR spectroscopy.38 As will be discussed in detail later, 2-APBA together with its aldehyde analogue, 2-FPBA (2-formyl-PBA), have been recently found to be highly versatile reagents for bioconjugations, owing to the cooperative action of the BA and the carbonyl moieties.7,39 Hall and co-workers postulated that a thiosemicarbazide-functionalized nopoldiol could form a hydrazone linkage with 2-APBA in addition to the nopoldiol BE. The authors showcased the use of this reaction for site selective protein labelling on HEK293T cells. Briefly, 2-APBA was installed onto the extracellular domain of the β2-adrenergic receptor using the SNAP-tag technology. Efficient fluorescence labelling of the cell surface was observed after the 2-APBA decorated cells were incubated with a bifunctional nopoldiol carrying a fluorescein label. More recently, the team performed further structural analysis of the adduct by X-ray crystallography and 11B NMR, which revealed a tetracyclic hydrazine-boronate adduct (Fig. 3(D)). This multicyclic adduct structure is believed to protect the BE from hydrolysis and explains the apparent irreversibility of this reaction.40 This stable BE conjugation was shown to be applicable in live animals: the 2-APBA warhead was implanted into tissues through intradermal injection of an NHS ester derivative of 2-APBA. The locally injected 2-APBA was found to retain a fluorescently labelled nopoldiol, allowing fluorescence imaging of the injection site in live mice.
It has been long recognized that aromatic diols like catechol bind PBA with much greater affinity than aliphatic diols (Fig. 2(B)).41–43 Modelling after the binding mode of PBA and catechol, Stolowitz and co-workers discovered that the conjugation of salicylhydroxamic acid (SHA) and PBA gave a six-membered ring complex (Fig. 4(A)).44 Jaffrey and co-workers have carefully characterized the reaction and shown that the conjugation proceeds remarkably quickly, giving complete conjugation within ∼5 min at a 6 mM reactant concentration.45 Unfortunately, no explicit measurement of the dissociation kinetics was carried out for the SHA–PBA complex. However, the protein immobilization studies by Stolowitz and co-workers show that protein immobilized on agarose through SHA–PBA conjugation could not be eluted using neutral buffers, indicating extremely slow dissociation of the SHA–PBA complex under neutral conditions. Importantly, the conjugate was found to exhibit pH dependent boron hybridization in 11B-NMR. At neutral pH, a single species was observed with sp3 hybridization, while under acidic conditions (pH < 5), multiple peaks were observed corresponding to sp2 and sp3 boron centres as well as that of PBA. This observation suggests possible hydrolysis of the conjugate under acidic conditions. Indeed, the binding constant Kd of PBA/salicyhydroxamic acid at pH 7.4 was found to be 5.6 × 10−5 M and increased to 0.25 M at pH 4.5, indicating a dramatically different thermodynamic stability of the conjugate under neutral versus acidic conditions.44 Capitalizing on the high conjugate stability at neutral pH, Jaffrey and co-workers demonstrated the use of a SHA dimer to assemble bioactive peptide dimers that incorporate a PBA moiety (Fig. 4(B)).45 On the other hand, the acid-triggered reversibility of this bioconjugation has been applied to realize protein immobilization and purification,46,47 enzyme activity control,48 and even targeted gene deliveries.49,50
 | ||
| Fig. 4 pH-controlled conjugation of PBA and SHA. (A) Illustration showing distinctive stability of the conjugate at neutral vs. acidic pH. (B) Illustration of a peptide dimerizer based on PBA–SHA bioorthogonal conjugation. | ||
3. Conjugation with amines
3.1 Iminoboronate-mediated binding of amines
Amino groups are highly abundant in biomolecules with protein lysines a prominent example. However, there are few examples of direct conjugation of BAs with amines under physiologic conditions. In neutral aqueous media, B–N bond formation is thermodynamically unfavourable for at least two reasons: (1) most amino groups exist as an ammonium ion (non-nucleophilic) under physiologic conditions; (2) the B–N bond is prone to hydrolysis due to the high concentration of water. Nevertheless, intramolecular B–N coordination has been documented in the literature. In particular, ortho-aminomethyl-phenylboronic acid (AMPBA) has been reported to adopt a closed conformation due to B–N interaction (Fig. 5(A)),51 although a detailed mechanistic study by Anslyn and co-workers showed that the AMPBA motif predominantly adopts an ion-pair structure (cationic N and anionic B) instead of direct B–N coordination.52 The dative bond character was found to increase slightly when going from a tertiary to a secondary and a primary amine. The ion pair structure, resulting from dynamic water molecule insertion, is nevertheless consistent with a strong interaction between boronic acid and a proximal nitrogen. Analogous to the closed structure of AMPBA, an ortho imine substituent of PBA can forge a strong B–N interaction as well to give iminoboronates (Fig. 5(B)). Again, the elegant mechanistic study by Anslyn and co-workers concluded that an iminoboronate can form in aprotic solvents, yet in protic solvent, it predominantly adopts a solvent-inserted ion pair structure.53 In the following discussions, we use the term iminoboronate to describe complexes with strong B–N interactions without differentiating a direct B–N interaction from an ion pair. | ||
| Fig. 5 Thermodynamic and kinetic considerations of iminoboronate formation. (A) Illustration of intramolecular B–N coordination within an AMPBA motif. (B) A bioconjugation reaction of primary amines and 2-FPBA/2-APBA yields iminoboronates. (C) Imine formation of non-borylated benzaldehyde/acetophenone is thermodynamically unfavourable. (D) A (10×) dilution of a preformed iminoboronate monitored by UV-vis absorption. The UV-vis absorption right after dilution (blue) overlaps with a preequilibrated sample at the same concentration (green), indicating an instantaneous equilibrium of iminoboronate formation. | ||
An iminoboronate complex was first reported by Dunn et al. in 1968, in which 2-FPBA dissolved in benzene was found to conjugate with an amine and further with a catechol to give an iminoboronate catechol ester.54 Thereafter, the James group55,56 and the Nitschke group57 adapted this three component assembly chemistry to achieve resolution of chiral amines and enable subcomponent self-assembly to give macrocyclic compounds. While these earlier reports describe iminoboronate formation in organic solvents, the Gois group58 and Yatsimirsky group59 in 2012 separately reported that, in aqueous media, 2-FPBA can conjugate with proteins and aminosugars at low millimolar concentrations. In particular, Gois and co-workers presented careful NMR and mass spectrometry characterization of the iminoboronate conjugates of small molecule amines (including a protected lysine), peptides as well as model proteins. Interestingly, 2-APBA was found to form iminoboronates with comparable efficiency to its aldehyde analogue. The team further noted that the resulting iminoboronates can be reversed upon the addition of fructose, dopamine, and glutathione through competitive mechanisms. Using this chemistry, Gois and co-workers reported the conjugation of poly ethylene glycol (PEG) to insulin, which can be reversed upon fructose addition. They also prepared a paclitaxel conjugate with folic acid, which showed greater anticancer potency against NCI-H460 cells than the paclitaxel precursor before conjugation.60
As a part of our search for covalent binding mechanisms similar to the reversible BE formation of bortezomib, we reported in 2015 that the iminoboronate formation of 2-APBA is rapidly reversible under physiologic conditions without the need for competitive binders, such as fructose, dopamine, and glutathione.61,62 In other words, the iminoboronate formation in aqueous media is under thermodynamic control: the forward and backward reactions both proceed with fast kinetics and the reaction rapidly reaches an equilibrium (Fig. 5(B)). Indeed, instantaneous re-equilibrium was observed after dilution of a concentrated 2-APBA-lysine solution (Fig. 5(D)), indicating the iminoboronate conjugate dissociates on the time scale of seconds or shorter.62 The rapid equilibrium of iminoboronate formation can be perhaps best rationalized by the prospect that B–N coordination would activate the imine for hydrolysis. In contrast, the dissociation of a salicylaldehyde–lysine conjugate is much slower and happens on the time scale of ∼30 min (unpublished data of the Gao lab). Treating the iminoboronate formation as an equilibrium, we experimentally determined the dissociation constant (Kd) of 2-APBA binding lysine ε-amine to be ∼10 mM,62 which compares much more favourably (by several orders of magnitude) than the imine formation of non-boron substituted benzaldehyde or acetophenone (Fig. 5(C)).63
The fast, thermodynamically controlled, conjugation of 2-FPBA/APBA with amines made them appealing as a warhead to “bind” amine-presenting molecules covalently. This train of thought coincided with the increasing interest in the development of reversible covalent inhibitors. Within the past two decades, covalent inhibition of disease-driving proteins has delivered a remarkable number of life-saving drugs.64,65 However, covalent drugs often encounter idiosyncratic toxicity, presumably due to off-target binding and/or permanent modification of proteins, which in turn can elicit undesirable immune responses. These problems may be circumventable by reversible covalent inhibitors,66,67 which can minimize off-target binding and avoid permanent modification of proteins as well. While reversible covalent binding of cysteine thiol has been accomplished using a nitrile or alpha-cyanoacrylamide warhead (Fig. 6(A)),68 reversible covalent binding of lysines has been elusive until recently.
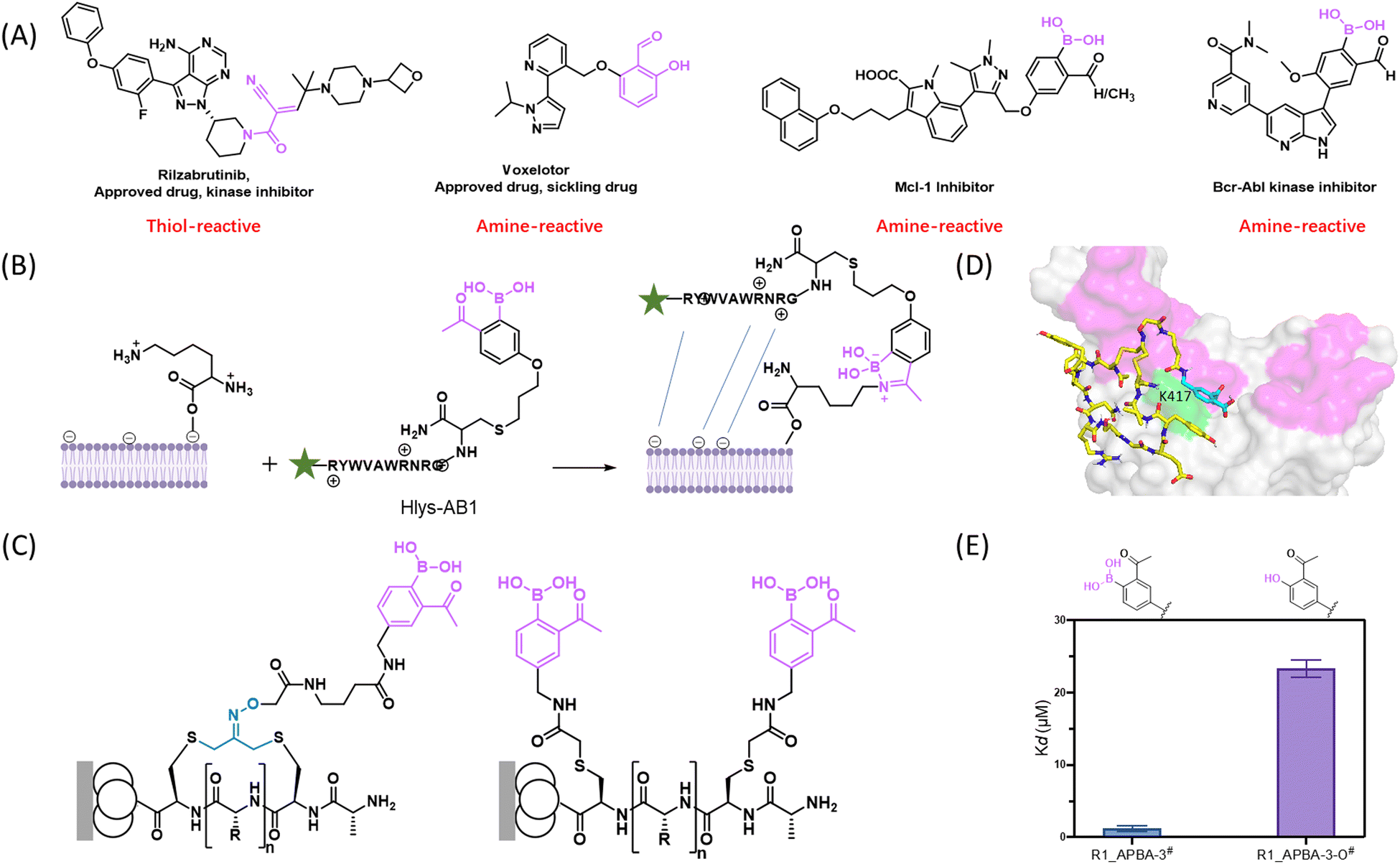 | ||
| Fig. 6 Reversible covalent inhibitors and probes enabled by iminoboronate chemistry. (A) Examples of small molecule based reversible covalent inhibitors that target a thiol or an amine. (B) Reversible covalent labelling of Gram-positive bacteria enabled by the iminoboronate formation of Lys-PG. (C) APBA-presenting phage libraries. (D) Computational modelling of the peptide ligand R1_APBA-3 showing its covalent conjugation to K417 of SARS-CoV2 spike protein. (E) Comparison of R1_APBA-3 and R1_APBA-3-O in terms of their spike protein binding affinity. | ||
Our exploration of 2-APBA as an amine-binding motif paralleled in time the development of the amine-targeted covalent drug voxelotor, formerly known as GBT440,69 which reached clinical use in late 2019 to treat sickle cell diseases. This covalent drug harbours a salicylaldehyde moiety as an amine-binding warhead (Fig. 6(B)). Our group first demonstrated the use of 2-APBA as a lysine-binding motif in 2015 towards the development of molecular probes for a bacterial lipid Lys-PG, which is a key player in the immune evasion mechanisms of bacterial pathogens including S. aureus.70 Specifically, we linked an APBA-presenting non-natural amino acid AB1 to a cationic peptide Hlys. The resulting peptide Hlys-AB1 was demonstrated to label S. aureus cells at sub-micromolar concentrations even in the presence of bovine serum.61 Furthermore, in collaboration with the Lapi group, we showed that a radiolabelled Hlys-AB1 allowed facile detection of S. aureus infection in live mice with an implanted thigh infection.71 This result highlights the feasibility and promise of using iminoboronate chemistry in live animals. Mechanistically speaking, Hlys-AB1 binds Lys-PG through a combination of electrostatic interactions and the covalent binding by iminoboronate formation (Fig. 6(C)). The peptide probe's binding to the bacteria can be readily reversed by washing, as expected for the facile reversibility of the iminoboronate linkage.
To streamline the discovery of reversible covalent probes and inhibitors for various biological targets, our group has developed phage-displayed peptide libraries that incorporate 2-APBA as a warhead (Fig. 6(D)). Phage display is a powerful technology that allows facile screening of billions of distinct peptide sequences with their identity encoded by the phage genome.72 Using chemoselective thiol-conjugation chemistries, we have devised an APBA-dimer library on a linear peptide scaffold73 as well as a cyclic peptide library displaying a single 2-APBA warhead.74 More recently, we have extended our phage library collection to include a double warhead library that displays 2-APBA together with a α-cyanoacrylamide that react with a lysine and cysteine respectively.75
We have applied these covalent binding libraries to discover molecular probes for bacterial pathogens,73,76 as well as reversible covalent ligands for challenging proteins that have frustrated small molecule inhibition.74,75 Screening of the APBA-dimer library has yielded sub-micromolar binders for a number of bacterial pathogens. Comparative studies of control peptides showed the essential role of the 2-APBA warhead in the peptides’ binding of the bacterial cells. Integrating such peptide probes into graphene based sensor platforms has allowed sensitive detection of bacterial strains that show specific antibiotic resistance phenotypes.77 On a different front, we have investigated the potential of the APBA-displaying libraries for revealing covalent ligands that inhibit challenging proteins, which have frustrated small molecule binding. 2-APBA-displaying cyclic peptide libraries were screened against a bacterial protein ligase sortase A (SrtA) as well as the spike protein of SARS-CoV-2. The screens yielded multiple peptide hits that bind the target proteins with a single digit to sub micromolar potency. The covalent binding mechanism was unambiguously demonstrated using mass spectrometry-based peptide mapping experiments. For example, the peptide ligand R1_APBA-3, which we identified for the spike protein, was found to covalently bind K417 of the protein (Fig. 7(B)). Importantly, we have assessed the energetic contribution of the iminoboronate formation by comparing R1_APBA-3 to a close structural analogue (R1_APBA-3-O) that replaces the boronic acid moiety with an -OH. In sharp contrast to APBA, the 2-acetylphenol moiety is incapable of covalently binding lysine to give imines. The peptide's binding affinity to spike was found to be at least 20 times greater for R1_APBA-3 over the deborylated analogue (Fig. 7(C)), highlighting the energetic significance of iminoboronate formation for the peptide's binding of the target protein.
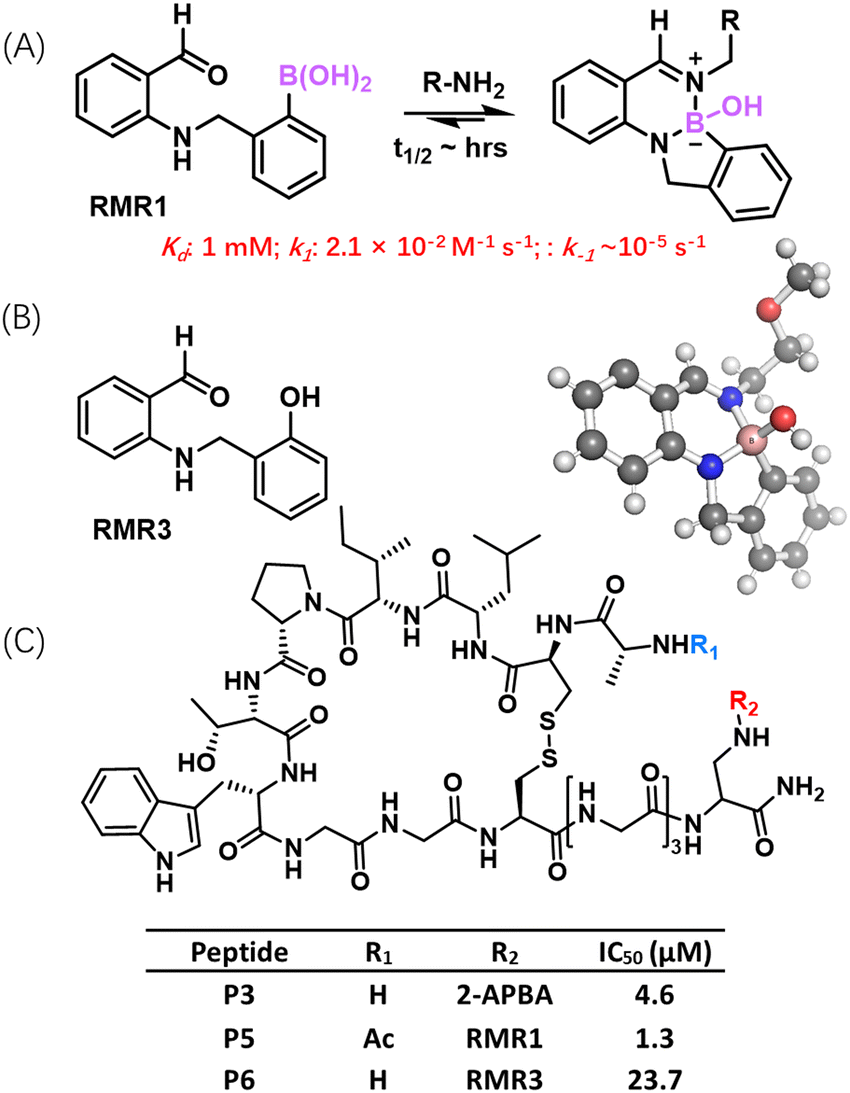 | ||
| Fig. 7 RMR1–amine conjugation. (A) RMR1 reversible conjugation with amine to give a diazaborine hydrate conjugate, the identity of which is confirmed by a crystal structure (CCDC ID: MARRAC) shown below. (B) Chemical structure of RMR3, a structural analogue that fails to bind amine covalently. (C) RMR1 modified peptide inhibitor of sortase showing greater potency than the peptide variants carrying either RMR3 or 2-APBA. | ||
In parallel to our work on peptide-based reversible covalent ligands, several groups explored the use of iminoboronate-forming warheads to develop small molecule-based reversible covalent inhibitors. In particular, Su and co-workers in 2016 functionalized known indole acid-based Mcl-1 protein inhibitors with 2-carbonyl PBA warheads (Fig. 6(A)), which enabled iminoboronate formation with a non-catalytic K234 residue and resulted in 20–50 times better binding affinity.78 More recently, Yao and co-workers reported the structure-based design of reversible covalent Bcr-Abl kinase inhibitors based on 2-FPBA chemistry (Fig. 6(A)). The designed inhibitors showed potent inhibition by reacting with the catalytic residue K271. Co-crystal structures of inhibitor-ABL complex confirmed the covalent interaction of the 2-carbonyl PBA warhead with the lysine residue.79
3.2 Diazaborine mediated binding of amines
To expand the collection of reversible covalent warheads that target amines, our group recently developed a new warhead RMR1, which elicits reversible covalent conjugation with amines to give a diazaborine hydrate conjugate (Fig. 7(A)).80 The structural design of RMR1 installs an AMPBA motif to the ortho position of benzaldehyde. The full and autonomous reversibility of this diazaborine formation reaction was confirmed via dilution experiments monitored by NMR and LC–MS. Under physiologic conditions, RMR1 covalently binds a lysine with ∼10 times greater potency than 2-APBA or 2-FPBA (1 mM vs. 10 mM Kd). Importantly, in sharp contrast to the rapid dissociation of iminoboronates, the diazaborine conjugates of RMR1 dissociate much more slowly, displaying half-life values on the multi-hour time scale. The forward reaction rate of the RMR1-lysine conjugation was found to be 2.1 × 10−2 M−1 s−1, which is significantly slower than that of iminoboronate formation. This slower forward reaction is perhaps not surprising given that the BA moiety in RMR1 is further removed from the aldehyde group in comparison to 2-FPBA. Consequently, the BA of RMR1 does not provide as much activating effect for the aldehyde to form imines. Nevertheless, RMR3 (Fig. 7(B)), a close structural analogue of RMR1, failed to give any conjugation with lysine, indicating the structural importance of the AMPBA motif. As discussed earlier, the AMPBA motif favours a closed conformation due to N–B coordination (Fig. 5(A)), which would place the boron centre close to the carbonyl in RMR1 to facilitate its conjugation to form imines. This hypothesis is supported by our DFT calculations that revealed a potential reaction trajectory towards diazaborine formation.Intrigued by the distinct properties of RMR1 covalent binding of lysine, we comparatively investigated RMR1 and 2-APBA for their use as a warhead to enable reversible covalent inhibitors. To this end, we respectively incorporated RMR1 and 2-APBA into a cyclic peptide that binds to sortase A with modest (17 μM) potency. Both 2-APBA and RMR1 significantly enhanced the peptide's potency for sortase inhibition, with the RMR1 peptide P5 yielded superior potency than the 2-APBA variant P3 (1.3 vs. 4.6 μM) (Fig. 7(C)). This is consistent with the greater potency of RMR1 for lysine binding. Importantly, peptide P5 also afforded sortase A inhibition on live S. aureus cells with a minimally compromised potency (IC50: 2.9 uM), indicating minimal off-target binding of this peptide on cell surfaces. Furthermore, the P5-induced sortase inhibition appears to be long lasting – efficient sortase inhibition was observed even 6 hours after the unbound inhibitors were washed away. This long-lasting inhibition, which presumably originates from the slow dissociation of the diazaborine conjugate, presents an appealing yet underexplored attribute of covalent drugs.
3.3 Amine-specific conjugation with β-boryl alkynone
Both the iminoboronate and diazaborine chemistries elicit lysine amine-specific conjugation due to the imine-based reaction mechanism. Lysine-specific conjugations are in fact quite rare, albeit highly desirable, due to the greater nucleophilicity of cysteine thiols. Loh and co-workers recently reported a novel amine-specific conjugation.81 Specifically, a β-boryl alkynone was found to readily react with amine to give an oxoboracycle stabilized by an exocyclic C–N double bond, yet minimal reactivity was seen towards thiols (Fig. 8(A)), in sharp contrast to the non-borylated alkynone control, which predominantly reacted with cysteine thiols (Fig. 8(B)). When subjected to N-terminal cysteine, the β-boryl alkynone afforded exclusively the amine-addition product. Furthermore, the team demonstrated peptide and protein conjugation, which was accomplished with excellent yields over 2–4 h. Note the β-boryl alkynone elicited conjugate exhibits robust stability and conjugation appears to be irreversible, differing from the iminoboronate and diazaborine hydrate chemistries described earlier in this section.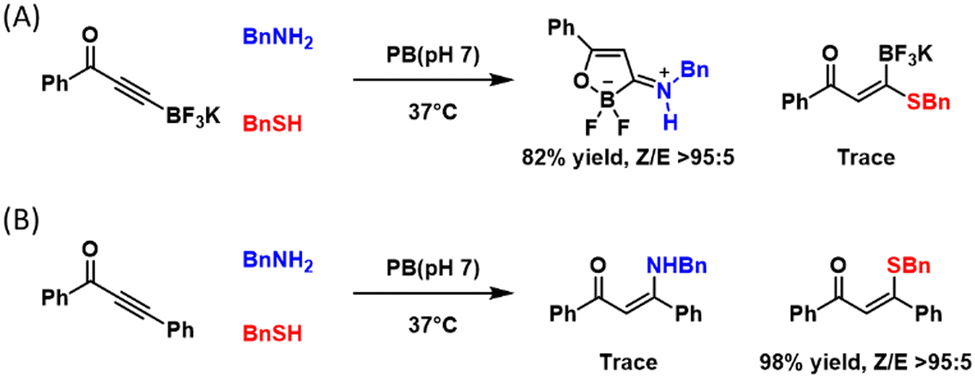 | ||
| Fig. 8 Amine-selective conjugation of β-boryl alkynone (A). In sharp contrast, the non-borylated control molecule (B) shows overwhelming preference for thiols. | ||
4. Conjugation with N-terminal cysteines
Biocompatible conjugation chemistries of N-terminal cysteines (NCys) have received significant attention as the 1,2-aminothiol functionality of an NCys presents a unique handle for site-specific modification of proteins. As NCys is rarely seen in endogenous proteins, NCys-specific conjugation can be considered a form of bioorthogonal conjugation. Previously known methods for NCys conjugation include the use of thioesters in native chemical ligation82 and the use of 2-cyanobenzothiazole (CBT) to mimic luciferin biosynthesis.83 Recently, a number of new NCys-selective reactions have been developed,84,85 a few of which involve clever use of boron and exhibit fast reaction kinetics and superb selectivity.4.1 NCys conjugation via TzB formation
In 2016, our group86 and the Gois group87 independently reported the fast conjugation of 2-FPBA to NCys, leading to a thiazolidine boronate (TzB) complex. In this reaction, 2-FPBA rapidly conjugates with NCys to give an iminoboronate intermediate, which sets up an intramolecular thiol addition reaction to form a thiazolidine ring (Fig. 9(A)). The conjugation was found to be remarkably fast and the reaction rate constants were found to be in the order of 102 and 103 M−1 s−1.86–88 The N atom of the thiazolidine ring further coordinates with the boron centre to thermodynamically stabilize the conjugate. Indeed, when a TzB conjugate was examined in aqueous solution over time, no change in its NMR signature was observed, in contrast to the slow but clear degradation of a thiazolidine control.86 Despite the thermodynamic stability, we found that a preformed TzB complex did exchange with another NCys-presenting molecule and the exchange completed over the time course of two hours.86 This finding indicates a modest kinetic stability of the TzB complex. A similar observation was noted by the Gois group, where a TzB complex was found to exchange with an oxyamine over 1 hour.87 Consistently, Spicer and co-workers reported a Kd of 4.9 μM and k−1 of 3.0 × 10−3 s−1, determined using a FRET assay.88 Comparatively, the TzB complex appears to show faster dissociation than the corresponding thiazolidine.86 This can be perhaps rationalized by the N–B coordination in the TzB structure, which has the potential to facilitate the dissociative reaction pathways, similar to the fastened hydrolysis of the iminoboronates. Paradoxically, the thermodynamic stability as evidenced by the unchanged NMR signature over time can be explained by the equilibrium (fast dissociation as well as reformation) of this conjugation reaction.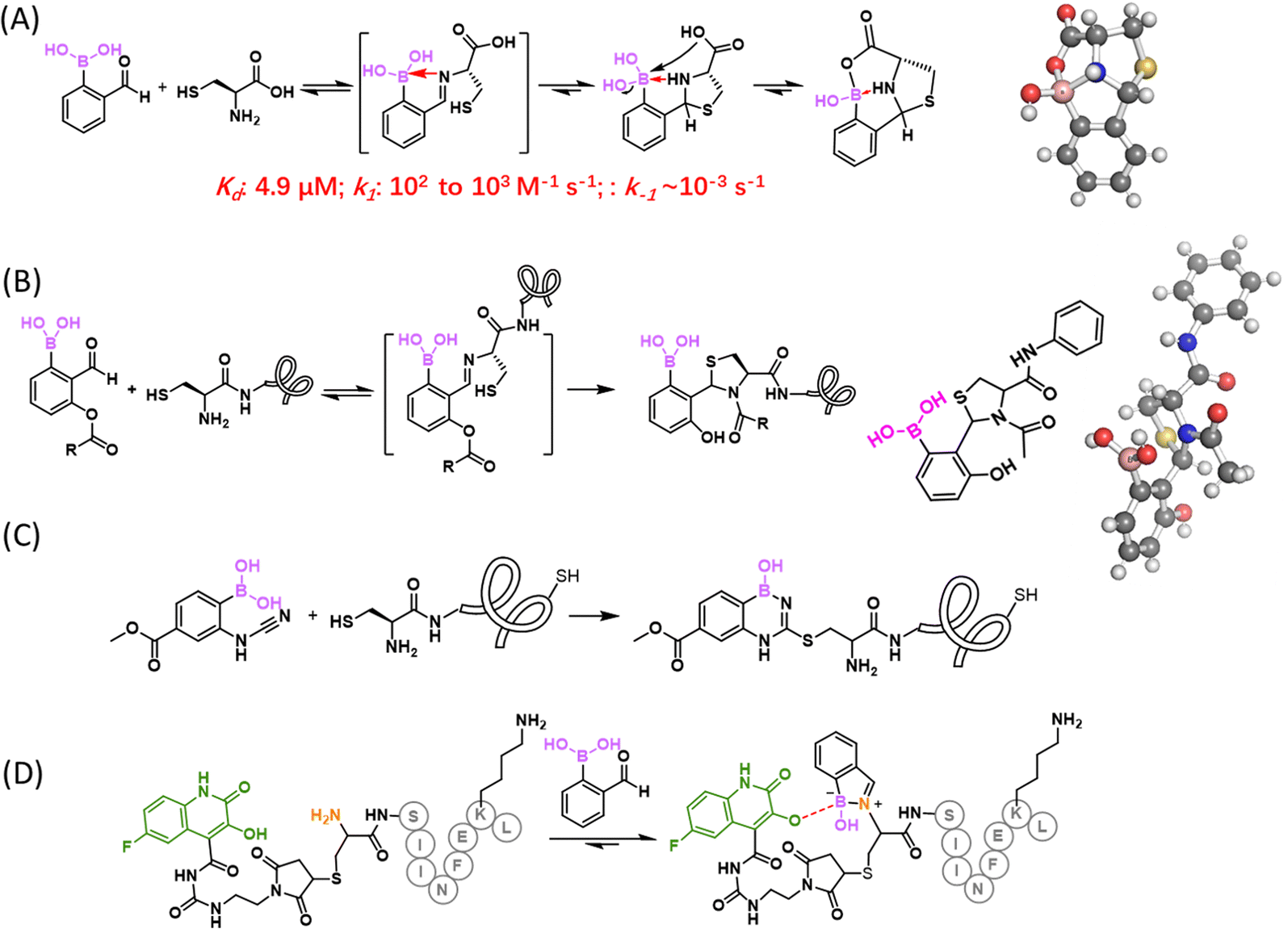 | ||
| Fig. 9 Boron enabled NCys conjugation chemistries. (A) Conjugation of 2-FPBA with NCys gives a TzB complex with modest kinetic stability. A crystal structure (CCDC ID: OKUMUE01) shown on the right revealed an additional cyclization between the –COOH of NCys and boron (colour scheme used: C: gray; N: blue; O: red; B: pink; S: yellow; H: white). The relevance of this cyclization to NCys bearing peptides/proteins is currently unclear. (B) Stable NCys conjugation through TzB-mediated acyl transfer that gives a stable N-acyl-thiazolidine. The structure (CCDC ID: MAGMEQ) of an N-acetyl-thiazolidine is shown on the right. (C) NCys bioconjugation with a 2-boronyl phenylcyanamide. (D) Reaction of 3HQ-modified peptide with 2-FPBA that gives a stabilized iminoboronate. | ||
In comparison to previously known NCys conjugation chemistries, the 2-FPBA elicited TzB formation exhibits exquisite selectivity towards NCys, due to its imine-based reaction mechanism. However, for the purpose of labelling and tracking biomolecules, the modest kinetic stability of the TzB conjugate can be problematic. With the aim of developing stable conjugation reactions, we have explored two NCys analogues, namely 2,3-diaminoproprioic acid (Dap)89 and tris base.90 Both yielded rapidly reversible conjugates except that of Tris with 2-APBA, which displayed a half-life of 115 h. A better solution to the conjugate instability issue came from a TzB-mediated acyl transfer reaction that gives an N-acyl-thiazolidine as a final product.91 In comparison to thiazolidines or TzB conjugates, N-acyl thiazolidines exhibit robust kinetic stability (Fig. 9(B)), with no dissociation observed over a range of pH values. Satisfyingly, the N-acyl-thiazolidine formation maintains fast reaction kinetics and the excellent NCys selectivity of the TzB chemistry. We demonstrated the utility of this reaction by labelling multiple model proteins that bear an engineered NCys residue. Site-specific labelling of these proteins was readily achieved by using low micromolar concentrations of a 6-acyloxy-FPBAs (e.g., KL42/72, Fig. 9(B)) that installs either an acetyl group or a biotin moiety. This powerful NCys conjugation chemistry was further applied to modify phage libraries with an engineered NCys on pIII, the carrier protein of library peptides.
A related but different idea of stabilizing a thiazoline conjugate was reported by Wang and co-workers.92 In their work, 2-FPBA was found to rapidly conjugate with 2-aminobenzothiol to give a thiazolidine conjugate, which undergoes oxidation to give a stable benzothiazole product. Although not directly applicable to NCys modification, this efficient and stable conjugation may be useful as bioorthogonal reactions.
4.2 Additional NCys conjugation reactions
Gois and co-workers recently reported a new NCys bioconjugation with the formation of benzodiazaborines (Fig. 9(C)).93 Cyanamides are known to react with a cysteine thiol to generate a thiourea-like adduct, which in diluted aqueous solutions undergoes rapid hydrolysis to form urea. To prevent this hydrolysis, a BA moiety was installed onto the ortho position of a phenyl cyanamide, which captures the thiourea intermediate and stabilizes it in the form of a benzodiazaborine (Fig. 9(C)). This BA substituted phenylcyanamide was found to react with cysteine with a pseudo first-order reaction rate of a 3.44 × 10−5 s−1, but no reaction with acetylcysteine was observed at all. These results highlight the NCys selectivity of this reaction over internal cysteines. Furthermore, the team performed DFT computation based mechanistic studies and found that the amino group of a free cysteine or NCys initiates the hydrogen transfer from the thiol group, which explains the absence of reactivity with internal cysteines. When incubated with 10 eq. of 2-boronyl phenylcyanamide, a bombesin peptide was found to give a single N-terminal modification, even though the peptide carries both an NCys and internal cysteine. The team further showed that, following the NCys conjugation, the internal cysteine could be alkylated by a maleimide to generate double modified peptides.Another nice contribution of the Gois group describes selective iminoboronate formation at a peptide's N-terminus even when the peptides do have lysine as potential competitors (Fig. 9(D)).94 Briefly, the authors installed 3-hydroxyquinolin-2(1H)-one (3HQ) onto a NCys side chain of a peptide through thiol–maleimide conjugation. Upon iminoboronate formation at the N-terminus, the OH group of 3HQ can forge a B–O bond and stabilize the charged boronate species by coordinating its carbonyl group to the boron atom, stabilizing the 2-FPBA conjugation at the N-terminus. As a demonstration of its utility, a 3HQ-modified c-ovalbumin peptide, harbouring an NCys and a lysine, was subjected to 10 eq. of 2-FPBA, which gave only iminoboronate formation at the N-terminus. When subjected to a large excess of 2-FPBA (1000 eq.), both the N-terminus and lysine residue were modified. However, as expected, the iminoboronate formation at lysine ε-amino groups dissociated completely upon dilution.
5. Iminoboronate-inspired bioorthogonal conjugations
While the discussions above largely focused on conjugation reactions of native proteins, organoboron compounds have been developed to enable bioorthogonal conjugations, in which both reactants are designed and biological entities. When incorporated into a biomolecule of interest, these designer functionalities allow modifications of the target biomolecule in a site-specific manner.5.1 Stabilized iminoboronates
The iminoboronate formation, while serving as a powerful binding mechanism, is nevertheless unsuitable for labelling and tracking biomolecules due to the rapid reversibility. Luckily, there have been some clever approaches developed to give stable iminoboronates. For example, the Gois group examined the reaction of 2-APBA with aminophenols as bidentate ligands (Fig. 10(A)).95 Specifically, the aminophenol 10-I reacted with 2-APBA to give an N,O-iminoboronate (10-II), which features an additional B–O bond that can provide extra stability to the iminoboronate conjugate. Indeed, the N,O-iminoboronate conjugate was found to give less than 10% hydrolysis even after 2 days in a physiological buffer. The slow dissociation is in sharp contrast to the instantaneous dissociation of non-stabilized iminoboronates. Consistent with the improved stability, the N,O-iminoboronates were found to go through slow exchange (5 days, Fig. 10(B)). Despite the remarkable stability at neutral pH, the N,O-iminoboronate exhibits fast hydrolysis (half-life of several hours) under acidic conditions. This low pH triggered dissociation has been applied to achieve selective delivery of fluorophores into cancer cells that overexpress folate receptors (Fig. 10(C)).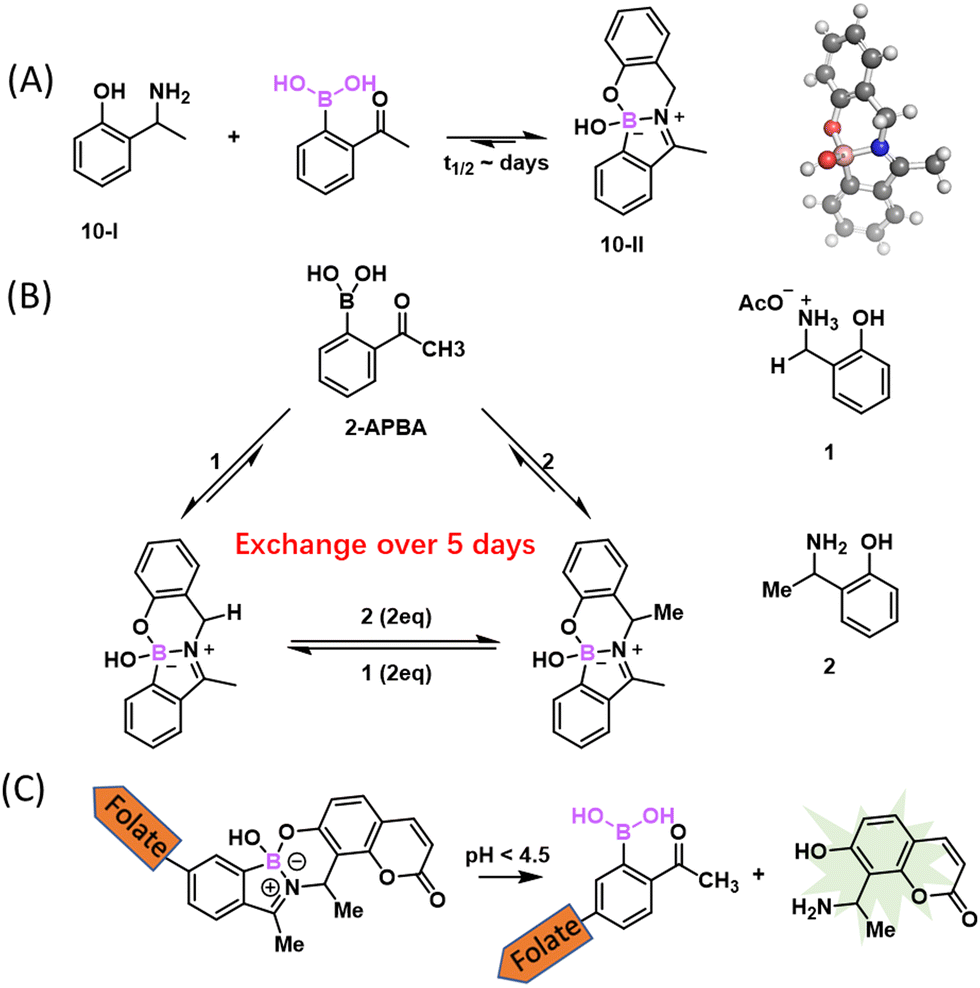 | ||
| Fig. 10 Stabilized iminoboronates. (A) Reaction of 2-APBA with aminophenol with improved stability. Conjugate structure confirmed by crystallography (CCDC ID: BIJDOQ). (B) N,O-iminoboronates undergo slow exchange over the time scale of days. (C) A folate derivative of N,O-iminoboronates selectively delivers amino coumarin to MDA-MB-231 human breast cancer cells, due the acidity-triggered dissociation. | ||
5.2 Reaction with α-nucleophiles
To achieve stable bioorthogonal conjugations, the iminoboronate chemistry has been extended to α-nucleophiles, such as oxyamines and hydrazines/hydrazides, which give oximes and hydrozones/acylhydrazones as products. It is well known that the conjugation of α-nucleophiles with a carbonyl results in biorthogonality. However, the slow reaction kinetics96 at neutral pH had greatly limited their applications until recently when boron-accelerated variants of these venerable conjugation reactions were invented.In 2015, several papers were published independently describing that an ortho-boronic acid substituent greatly accelerates the conjugation of aryl carbonyls and α-nucleophiles. Specifically, the Gillingham group reported that 2-FPBA conjugated with benzylhydroxylamine in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio under neutral conditions (Fig. 11(A)) with a rate constant over 103 M−1 s−1.97 The reaction rate is several orders of magnitude higher than that of typical oxime ligations even with aniline catalysis.96 Though an oxime–oxyamine exchange experiment, the dissociation rate constant (k−1) of the oxime product of 2-FPBA was determined to be 4.21 × 10−5 s−1, which corresponds to a half-life of 6.5 hours, comparable to typical oximes without an ortho-BA substituent.98 These kinetic parameters were later corroborated by other research groups.88,99 This fast conjugation was found to be remarkably bioorthogonal, encountering little interference by glutathione, sucrose, lysozyme or even blood serum. The team also examined 2-APBA, the methyl ketone analogue of 2-FPBA. 2-APBA was also found to rapidly conjugate with benzyloxyamine. The resulting product was unfortunately not explicitly assessed for its thermodynamic and kinetic stability. Our group in parallel examined the conjugation of 2-APBA with several α-nucleophiles.62 Interestingly, we found that 2-APBA conjugates with acetohydrazide, benzhydrazide, as well as an alkoxyamine in a rapidly reversible manner (Fig. 11(A) and (B)). Dilution experiments indicated a rapid equilibrium that could be reached over seconds and potentially shorter. The dissociation constant (Kd) was estimated to be 0.6 × 10−3 M for acetohydrazide and 1.4 × 10−5 M for an alkoxyamine. Consistently, our 11B-NMR studies revealed a broad and upfield shifted boron peak for the 2-APBA oxime, indicating the formation of iminoboronate structures, which presumably underlie the fast dissociation of the oxime product of 2-APBA.
:1 ratio under neutral conditions (Fig. 11(A)) with a rate constant over 103 M−1 s−1.97 The reaction rate is several orders of magnitude higher than that of typical oxime ligations even with aniline catalysis.96 Though an oxime–oxyamine exchange experiment, the dissociation rate constant (k−1) of the oxime product of 2-FPBA was determined to be 4.21 × 10−5 s−1, which corresponds to a half-life of 6.5 hours, comparable to typical oximes without an ortho-BA substituent.98 These kinetic parameters were later corroborated by other research groups.88,99 This fast conjugation was found to be remarkably bioorthogonal, encountering little interference by glutathione, sucrose, lysozyme or even blood serum. The team also examined 2-APBA, the methyl ketone analogue of 2-FPBA. 2-APBA was also found to rapidly conjugate with benzyloxyamine. The resulting product was unfortunately not explicitly assessed for its thermodynamic and kinetic stability. Our group in parallel examined the conjugation of 2-APBA with several α-nucleophiles.62 Interestingly, we found that 2-APBA conjugates with acetohydrazide, benzhydrazide, as well as an alkoxyamine in a rapidly reversible manner (Fig. 11(A) and (B)). Dilution experiments indicated a rapid equilibrium that could be reached over seconds and potentially shorter. The dissociation constant (Kd) was estimated to be 0.6 × 10−3 M for acetohydrazide and 1.4 × 10−5 M for an alkoxyamine. Consistently, our 11B-NMR studies revealed a broad and upfield shifted boron peak for the 2-APBA oxime, indicating the formation of iminoboronate structures, which presumably underlie the fast dissociation of the oxime product of 2-APBA.
 | ||
| Fig. 11 Fast bioconjugation of 2-FPBA/APBA with α-nucleophiles. (A) 2-FPBA and 2-APBA both elicit fast bioconjugation yet give rise to conjugates of differing stabilities. (B) Acetohydrazide conjugates with 2-FPBA and 2-APBA to give a rapid equilibrium. (C) Conjugation of phenylhydrazine with 2-FPBA and 2-APBA yielding diazaborines with varied potential for exchange. (D) and (E) Stable diazaborines were obtained from semicarbazide and sulfohydrazide respectively. (F) Crystal structures showing that acetohydrazide and semicarbazide yield conjugates of distinct geometries of the boron centre. CCDC IDs: MEHKUI (left), TAZGEJ (middle) and TAZGIN (right). Colour scheme used: C: gray; N: blue; O: red; B: pink; S: yellow; H: white. | ||
In contrast to the hydrazides and alkoxyamine, the conjugation of 2-APBA with phenylhydrazine yielded a more stable product, with near quantitative conjugation formation at sub-micromolar concentrations of reagents and dissociation only quantifiable under exchange conditions.62 At that time, we were puzzled by the chemical basis of the superior conjugate stability afforded by phenylhydrazine. An important paper100 as well as a patent application101 came from the Bane group describing that an aryl hydrazine rapidly conjugated with 2-FPBA to give a stable diazaborine heterocycle (Fig. 11(C)). Similar findings were also reported by the Gillingham group.102,103 Both groups presented evidence for a two-step mechanism for the diazaborine formation, which involves a fast bimolecular conjugation (>103 M−1 s−1) followed by an intramolecular cyclization. A rate constant of 15 × 10−3 and 2.5 × 10−3 s−1 was reported for the cyclization by the Bane and Gillingham groups respectively. These data indicate a relatively long lived hydrazone intermediate with a half-life on the scale of minutes. In light of these findings, we suspect the stable conjugation of 2-APBA and phenylhydrazine that we observed is likely due to the formation of a diazaborine product, although we failed to confirm the conjugate structure via crystallography at the time of our initial publication.62 Interestingly, no long-living intermediate was observed with our investigation of 2-APBA conjugations, which suggests a faster cyclization step to give diazaborines.
A number of experiments speak to the stability of the diazaborine conjugate. Bane and co-workers demonstrated protein labelling using the conjugation of 2-FPBA and a phenylhydrazine derivative, the product of which survived SDS-PAGE analysis.100 Gillingham and co-workers examined the potential exchange of a pre-formed diazaborine compound with 2-FPBA, in which no exchange was observed for 24 h.102 A detailed kinetics study by Spicer and co-workers showed that a pre-formed diazaborine did exchange with a hydrazine (3.7 mM), although very slowly (Fig. 11(C)). They estimated the dissociate rate of 2.6 × 10−6 s−1, which corresponds to a t1/2 of 4–5 days for the diazaborine to dissociate and exchange.88 The diazaborine conjugate of 2-APBA appeared to exchange faster,62 which reached equilibrium over 4 h (with 1 μM exchanging hydrazine) indicating a more kinetically labile diazaborine formed by 2-APBA. Nevertheless, the diazaborine conjugates exhibited great stability in the absence of α-nucleophiles for exchange.104
As phenylhydrazines are prone to oxidation and can be cytotoxic, it is desirable to find more biofriendly alternatives. To this end, semicarbazide and thiosemicarbazide were found to conjugate with 2-APBA/FPBA to give diazaborines (Fig. 11(D) and (F)).104 We note that such reactivity of (thio)semicarbazide had been predicted by Bane and co-workers in their 2015 patent application.101 Our results show that semicarbazide reacted with both 2-FPBA and 2-APBA efficiently to give a diazaborine heterocycle, whose structure is unambiguously confirmed by X-ray crystallography (Fig. 11(F)). Similar to phenylhydrazine, the conjugation of semicarbazide proceeds with a rate constant of >103 M−1 s−1. In comparison, thiosemicarbazide showed compromised yields reacting with 2-APBA, although it conjugated with 2-FPBA efficiently. As 2-FPBA is known to conjugate with free cysteines to form a quasi-stable TzB complex,86 2-APBA is arguably a better conjugation partner with semicarbazide for bioorthogonal applications. Indeed, the diazaborine formation of 2-APBA was found to proceed without interference by blood serum or cell lysates.104 More recently, Bandyopadhyay and co-workers have expanded the diazaborine-forming chemistry of 2-FPBA/APBA to sulfonyl hydrazide (Fig. 11(E)),105 which elicits quantitative conjugation with both 2-FPBA and 2-APBA to give stable diazaborine products.
Although the diazaborine-forming chemistry had been documented much earlier in pure organic solvents,54,106,107 the discovery that it can proceed under phycological conditions is quite exciting and has enabled a number of applications in chemical biology and protein engineering. For example, in addition to protein labelling, the versatile diazaborine conjugation has been used to construct DNA-encoded libraries (Fig. 12(A)),108 as well as to devise fluorogenic conjugation reactions (Fig. 12(B)).102,109 Furthermore, 2-APBA-semicarbazide conjugation has been applied to label live bacterial cells. Specifically, we synthesized D-amino acids that incorporate either 2-APBA or semicarbazide on their side chains (Fig. 12(C)).104,109,110 These D-amino acids were incorporated into the peptidoglycan of bacterial cells via a cell wall remodelling mechanism.111 Then these amino acids served as an anchor to conjugate with a suitable partner to enable fluorescence labelling of the bacterium. Again, due to the fast reaction kinetics and superb biorthogonality, such cell labelling experiments can be completed in minutes with low micromolar concentrations of reagents. Finally, the diazaborines have been found to be prone to oxidation by endogenous reactive oxidative species, such as peroxide and oxynitrite, which has been capitalized to design drug delivery and release systems.39,99,112
 | ||
| Fig. 12 Applications of the diazaborine chemistry. (A) Constructing DNA encoded libraries via diazaborine conjugation. (B) Fluorogenic conjugation reactions based on diazaborine formation. (C) Application of diazaborine formation for live bacterial cell labelling. | ||
Other than aromaticity, Bane and workers reported the use of α-aminohydrazide to achieve stable conjugation with 2-FPBA (Fig. 13(A)).113 This design is inspired by the discovery that 2-FPBA can conjugate with acetohydrazide to form an unstable B–N heterocycle that harbours a tetrahedron boron centre.113 Pending an amino group on the α-carbon of acetohydrazide elicits additional cyclization (via the formation of another N–B bond), which stabilizes the diazaborine conjugate. The formation of the tricyclic conjugate was confirmed by X-ray crystallography and shown to be stable for several weeks under both acidic and basic conditions. The Bane group demonstrated that this conjugation could be used for fluorescence protein labelling and the labelled proteins survived SDS-PAGE gel analysis. More recently, the Bane group applied this 2-FPBA-α-amino-hydrazide conjugation to create site-specifically modified antibodies.114 They also demonstrated that the reaction could be used in combination with other bioorthogonal reactions, such as the strain-promoted azide–alkyne cycloaddition and tetrazine–transcyclooctene (Tz–TCO) ligation to allow simultaneous triple labelling of antibodies. Following a similar design principle, Bane and co-workers described that installing a β-hydroxyl group could also stabilize a acetohydrazide's conjugate with 2-FPBA (Fig. 13(B)).115 The postulated tricyclic structure of the stable conjugate was confirmed by X-ray crystallography and found to show little degradation in aqueous solution over 24 hours. However, when mixed with hydrazine, the conjugate of β-hydroxyl hydrazide was converted to the conjugate of hydrazine over the time course of ∼4 hours.
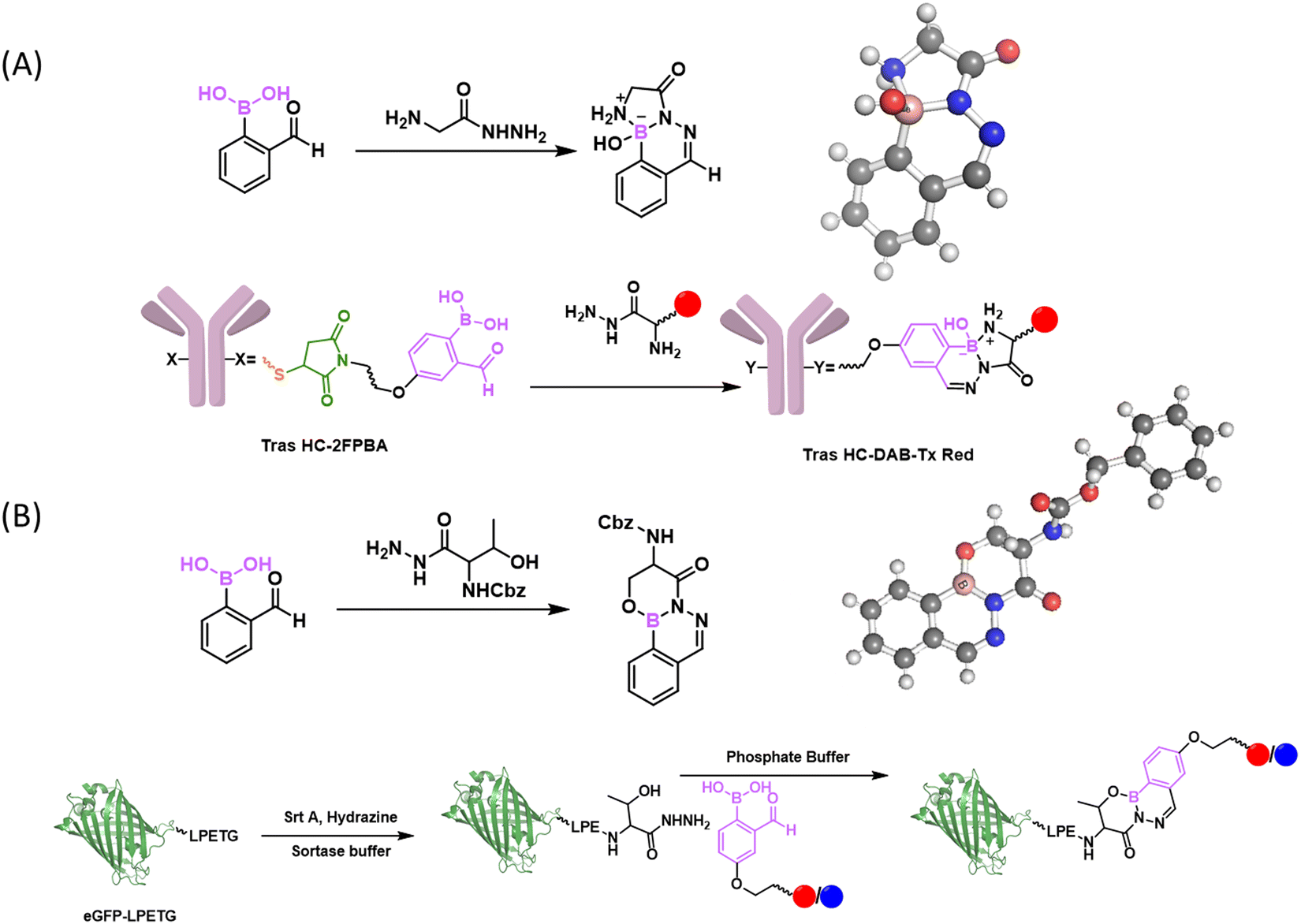 | ||
| Fig. 13 Stabilizing the conjugates of hydrazides. (A) Hydrazide conjugate stabilized by an α-amino substituent. The conjugate structure was confirmed by crystallography (CCDC ID: MEHKIW). This strategy has been demonstrated for antibody labelling. (B) Hydrazide conjugates stabilized by β-hydroxyl substituent. The conjugate structure was confirmed by crystallography (CCDC ID: EPOLUT). This strategy has been implemented for protein C-terminal conjugation, where a C-terminal Thr hydrazide can be generated via sortase mediated ligation with hydrazine. Red and blue dots represent fluorophore or drug payloads. | ||
5.3 Three component reactions
As described earlier in the section, an iminoboronate could conjugate with diols to give three-component assembled products, as initially described by Dunn et al. in 1968.54 This three-component conjugation was later applied by James and co-workers to resolve stereoisomers in organic solvents. More recently, such reactions have been adapted into an aqueous phase to construct functional micelles and hydrogels (Fig. 14(A)).116,117 These applications take advantage of the induced reversibility of the conjugate products by acidification or addition of GSH. | ||
| Fig. 14 Three component reactions. (A) Three-component coupling reaction to assemble crosslinked G-quadruplex based hydrogels. (B) Condensation reaction of 2-FPBA, a hydroxylamine and a catechol for peptide modification. (C) Conjugation reaction of PBA, 1,2-aminoalcohol and ortho-carbonyl-phenol to construct bioactive molecules of varied stability. | ||
Anslyn and co-workers have recently reported a new three component conjugation, which resembles the conjugation reaction above, but replaces the amine with an N-hydroxyamine (Fig. 14(B)).118 The assembly mechanism was examined by the order of addition of the three components. When 2-FPBA combined with hydroxylamine first, the conjugate will not react with catechol. The hydroxylamine addition is irreversible and proved stable over 24 h over a wide range of pH values (1–13) and temperatures up to 50 °C. The formation of a cyclic oxime boronate was confirmed by X-ray analysis which showed B–O bond formation between the hydroxylamine oxygen and boron. The author demonstrated that this three-component assembly is compatible with canonical oxime formation to achieve dual labelling of peptides that incorporate a catechol and an aldehyde as a reactive handle respectively.
In the same year, the Gois group developed a one-pot three-component condensation reaction with a phenylboronic acid, an aminophenol and a 2-hydoxy-acetaphenone as reactants (Fig. 14(C)).119 This simple protocol readily yielded the conjugates with up to 90% yield without the need of chromatographic isolation. While the condensation reaction was carried out in organic solvent (e.g., CH3CN, DMSO), the resulting boronate complexes exhibited high stability with a half-life of 38 h at pH 4.8 or in human plasma, and only dissociated in the presence of GSH with a half-life of 7 h. It is interesting to note that salicylaldehyde was found to elicit more efficient conjugation, which however yielded products of significantly lower stability. The team applied this condensation reaction to assemble a conjugate bearing a PEG unit, a folic acid targeting unit and the cytotoxic drug bortezomib as a bioactive payload. This drug delivery complex showed high selectivity in vitro against folate-positive MDA-MB-231 cells with nanomolar IC50 values. In addition, they generated an innovative fluorophore scaffold, dubbed boronic-acid derived salicylidenehydrazone (BASHY), by this robust multicomponent reaction. This efficient one-pot reaction enabled a straightforward construction of fluorophores with tuneable photophysical properties. They also incorporated an extended polymethine chain in the BASHY complex to yield Cy-BASHY dyes that are capable of efficient red-emitting and two-photon absorption. These fluorescent dyes have shown promise for biological imaging applications.120–124
6. Deborylative cross-coupling reactions
As exemplified by the Nobel Prize winning Suzuki coupling reaction,125 organoboron reagents occupy a prominent position in synthetic organic chemistry due to their utilization in cross coupling reactions. Not surprisingly, these metal-catalysed cross coupling reactions have been adapted for bioconjugation under physiological conditions.6.1 Suzuki–Miyaura coupling on proteins
In 2005, the Hamachi group first reported a Suzuki–Miyaura coupling reaction on proteins, which was accomplished in aqueous solution using Na2PdCl4 as a catalyst. Specifically, a synthetic WW-domain that incorporated a p-iodophenylalanine residue was found to couple with several aryl boronic acids with 40–90% yields.126 The Davis group developed a new catalyst that enabled highly efficient Suzuki coupling on peptides and proteins in pure aqueous solutions. Specifically, the group found that the sodium salt of 2-amino-4,6-dihydroxypyrimidine formed a water soluble complex with Pd(OAc)2. The resulting catalyst promoted coupling of peptides and proteins bearing an aryl iodide with various boronic acids with >95% yields (Fig. 15(A)).127 This initial report of Davis and co-workers introduced the aryl iodide group into proteins via cysteine modification. Their follow-up publications achieved the incorporation of aryl halides into recombinant proteins via genetic code expansion, which in turn allowed Suzuki coupling on recombinant proteins as well as live cells.128,129 The efficient conjugation on live cells is quite remarkable even though the boronic acids as well as catalyst were used at (sub to low) millimolar concentrations. Another clever application of this efficient Suzuki coupling reaction on recombinant proteins allowed facile generation of various glycoforms of the glucosyltransferase enzyme, glycogenin.130 The ready access to these glycoforms allowed a detailed analysis of the glycogen biosynthesis initiation with unprecedented precision. On related fronts, the enhanced Suzuki coupling has been implemented to enable peptide ligation131 as well as radioactive labelling of peptides and proteins.132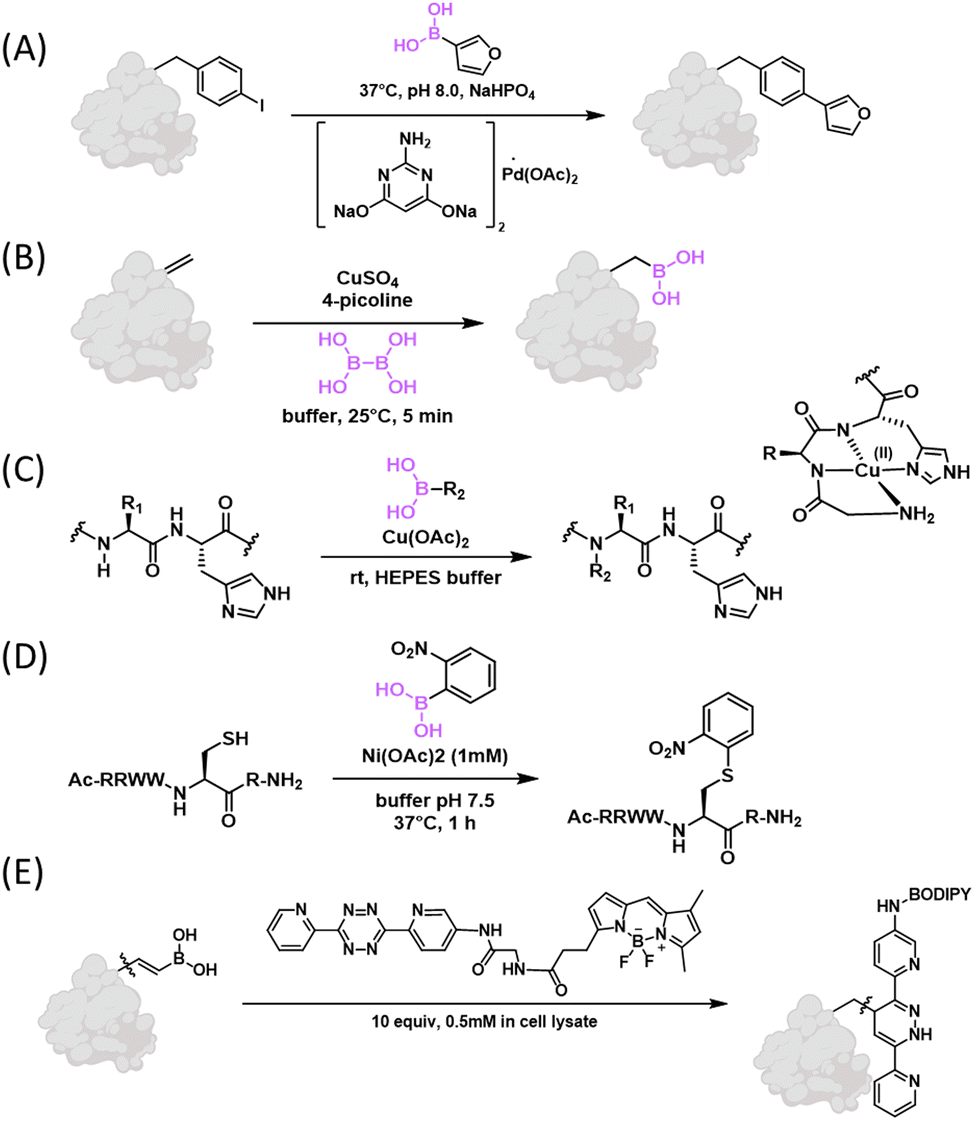 | ||
| Fig. 15 Deborylative bioconjugation reactions. (A) Suzuki–Miyaura coupling reaction on proteins. (B) Incorporation of boronoalanine (Bal) into proteins using dehydroalanine (Dha) as a precursor. (C) Histidine-directed backbone NH modification under Cu2+ catalysis. (D) Nickel catalysed cysteine thiol arylation using ortho-substituted aryl boronic acid. (E) Vinylboronic acid participated in the Carboni–Lindsey reaction. | ||
Contrary to introducing aryl iodide into proteins, the Schultz group in 2008 reported the incorporation of p-borono-phenylalanine into proteins via genetic code expansion (Fig. 15(B)).133 Schultz and co-workers further demonstrated the feasibility of Suzuki coupling of this genetically encoded phenylboronic acid. Note that this 2008 publication reported a modest yield (30%) and the need for high temperature (70 °C) for the Suzuki coupling. This less ideal result is presumably because the work was ahead of its time – the above-mentioned new catalyst for aqueous Suzuki coupling was not invented yet in 2008. In addition to genetic encoding of boronophenylalanine, Davis and co-workers recently developed an elegant approach to incorporate boronoalanine (Bal) into proteins (Fig. 15(C)).134 Specifically, a Bal residue was installed by borylation of a dehydroalanine (Dha) residue under biocompatible conditions. Bal-incorporated proteins were examined for a myriad of properties. Particularly noteworthy in the context of this review is the observation that Bal-presenting proteins bind nopoldiol and fructose with similar potencies as phenylboronic acid. Interestingly, a mCherry variant with Bal incorporated was found to bind mammalian cell surface polyols at 10 μM concentrations, although the exact nature of this binding event is not clear. We note that, in the same year, Roelfes and co-workers interpedently described this methodology for Dha-to-Bal transformation and applied it to a number of peptide natural products.135 More recently, a desulfurative borylation protocol was developed by Walczak and coworkers, which allowed direct transformation of a cysteine thiol directly into Bal in peptides and proteins.136
6.2 Metal-catalysed conjugation with native protein nucleophiles
To explore the potential of organic boronic acids for native protein modification, Ball and co-workers examined the role of various metal catalysts and this line of efforts revealed several new conjugation reactions of proteins.137 For example, histidine-directed arylation/alkenylation of backbone NH was achieved under Cu2+ catalysis (Fig. 15(B)).138 The Chan–Lam like reaction exhibits remarkable site-selectivity and modifies the backbone amide NH preceding a histidine residue. This histidine directed selectivity can be rationalized by the formation of an ATCUN-like copper complex that activates the backbone amide (Fig. 15(B)). Ball and co-workers further reported a pyroglutamate–histidine (Glp–His) dipeptide motif that is particularly amenable to the copper catalysed coupling with aryl/alkenyl boronic acids.139 This peptide motif underwent aryl/alkenyl boronic acids (1 mM) over the time course of minutes under copper (0.3 mM) catalysis. The Glp–His motif can be engineered into recombinant proteins through glutaminyl cyclase processing a N-terminal QH dipeptide, which enables facile and site-specific labelling of recombinant proteins. The copper mediated backbone modification was found to be quite sensitive to sterics – ortho-substituted aryl BA or even cis-alkenyl BA afforded no backbone NH modification. Taking advantage of this finding, Ball and co-workers developed a method for cysteine thiol arylation with arylboronic acid (Fig. 15(C)).140 Specifically, ortho-nitro substituted aryl boronic acid readily reacted with cysteine thiols under Ni2+ catalysis. The reaction completes over 10 min with 2 mM 2-nitrophenyl boronic acid and 1 mM of nickel salt. Combining these two distinctive reactivities, the Ball group devised a dual boronic acid reagent that allowed conjugation to a cysteine and then a Glp–His motif sequentially.141 Finally, tyrosine-selective conjugation chemistry was realized using an ortho-amide substituted aryl boronic acid under Rh3+ catalysis, which resulted in a tyrosine–Rh complex with remarkable stability.1426.3 Vinylboronic acid for iEDDA conjugation
The Bonger group in 2016 identified vinylboronic acid as a new non-strained alkene that reacted with 3,6-dipyridyl-s-tetrazines following the mechanism of inversed electron demand Diels–Alder (iEDDA) reaction (Fig. 15(D)).143 An optimized vinylboronic acid gave a second-order rate constant of 27 M−1 s−1 in aqueous solution at room temperature, which is one order of magnitude faster than the commonly used tetrazine reactive partner, norbornene (2.2 M−1 s−1). The proposed mechanism suggests that a boric acid is released immediately after the iEDDA cycloaddition, followed by dinitrogen release by a retro-Diels–Alder reaction. The author further showed that the vinylboronic acid moiety is compatible with cellular components, stable in cell lysate, and applicable for protein modifications. Recently, Bonger and co-workers demonstrated that this biorthogonal chemistry is suitable for protein labelling in live cells,144 as well as for prodrug design with tetrazine triggered drug release.1457. Conclusion
This contribution aims to present an up-to-date summary of the novel bioconjugation reactions enabled by the use of organoboron species. To this end, several classes of bioconjugation reactions have been discussed that tap into the biocompatibility as well as the unique reactivities of boronic acids and esters. Whenever applicable, we provide an in-depth discussion of the reaction mechanisms with the intention to inspire organic chemists and chemical biologists to further expand the repertoire of the organoboron enabled bioconjugations.In conclusion, we highlight several unique features of boron enabled bioconjugation chemistries: (1) the facile B–N and B–O bond formation has given rise to a family of fast conjugation chemistries, with many reactions displaying a second order reaction rate constant over 1000 M−1 s−1. The fast reaction kinetics ensures efficient conjugation even at low micromolar concentrations of reactants; (2) the dynamic nature of the B–N and B–O bonds renders reversibility to a number of the organoboron mediated bioconjugations. Such dynamic conjugation reactions in turn give rise to a collection of rare but powerful tools to covalently “bind” native biomolecules. This feature, discussed in detail in the context of boronate ester formation as well as the use of iminoboronate chemistry to bind amines, is rarely seen in other families of bioconjugation reactions; (3) the majority of the organoboron elicited bioconjugations exhibit remarkable tunability. The thermodynamic and kinetic stability of the conjugation products can be systematically tuned to fit the needs of specific biological applications. This manifests in the successful development of stimuli responsive molecules and materials122 for various applications including protein purification and delivery of drugs. Overall, introducing boron into biological settings has resulted in a rapidly increasing collection of bioconjugation reactions, which display unique properties absent from other family of bioconjugations. We expect that further exploration into this domain will yield more diverse and useful tools for biological research and therapeutic innovations. To this end, the focus of future research could be on the suitability for in vivo applications. While much excitement has been demonstrated with test tube studies such as protein labelling, only a few limited in vivo studies have been reported to date,40,71 which demonstrate the potential applicability of 2-APBA mediated conjugation in living animals. More and systematic studies on this front will yield valuable insight into the potential, as well as challenges that need to be overcome, for boron-enabled conjugation chemistries.
Data availability
The current submission is a review article and hence no new research data were generated. Any cited data can be found in the relevant citations. When crystal structures are presented, PDB or CCDC ID is provided.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Gao group acknowledges the financial support provided by the National Institute of General Medical Sciences (GM152005), the National Science Foundation (CHE-2204078), and the Ono Pharma Foundation.References
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 Search PubMed.
- C. R. Bertozzi, Acc. Chem. Res., 2011, 44, 651–653 Search PubMed.
- J. A. Prescher and C. R. Bertozzi, Nat. Chem. Biol., 2005, 1, 13–21 Search PubMed.
- Y. Liu, M. P. Patricelli and B. F. Cravatt, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 14694–14699 Search PubMed.
- I. Uluisik, H. C. Karakaya and A. Koc, J. Trace Elem. Med. Biol., 2018, 45, 156–162 Search PubMed.
- B. C. Das, N. K. Nandwana, S. Das, V. Nandwana, M. A. Shareef, Y. Das, M. Saito, L. M. Weiss, F. Almaguel, N. S. Hosmane and T. Evans, Molecules, 2022, 27, 2615 Search PubMed.
- S. Cambray and J. Gao, Acc. Chem. Res., 2018, 51, 2198–2206 Search PubMed.
- B. Akgun and D. G. Hall, Angew. Chem., Int. Ed., 2018, 57, 13028–13044 Search PubMed.
- J. P. M. António, R. Russo, C. P. Carvalho, P. M. S. D. Cal and P. M. P. Gois, Chem. Soc. Rev., 2019, 48, 3513–3536 Search PubMed.
- S. Chatterjee, E. V. Anslyn and A. Bandyopadhyay, Chem. Sci., 2021, 12, 1585–1599 Search PubMed.
- Y. Furikado, T. Nagahata, T. Okamoto, T. Sugaya, S. Iwatsuki, M. Inamo, H. D. Takagi, A. Odani and K. Ishihara, Chem. – Eur. J., 2014, 20, 13194–13202 Search PubMed.
- W. L. A. Brooks, C. C. Deng and B. S. Sumerlin, ACS Omega, 2018, 3, 17863–17870 Search PubMed.
- T. P. Smith, I. W. Windsor, K. T. Forest and R. T. Raines, J. Med. Chem., 2017, 60, 7820–7834 Search PubMed.
- C. F. Dai, A. Sagwal, Y. F. Cheng, H. J. Peng, W. X. Chen and B. H. Wang, Pure Appl. Chem., 2012, 84, 2479–2498 Search PubMed.
- M. P. Curran and K. McKeage, Drugs, 2009, 69, 859–888 Search PubMed.
- R. C. Kane, A. T. Farrell, R. Sridhara and R. Pazdur, Clin. Cancer Res., 2006, 12, 2955–2960 Search PubMed.
- M. Groll, C. R. Berkers, H. L. Ploegh and H. Ovaa, Structure, 2006, 14, 451–456 Search PubMed.
- M. Shirley, Drugs, 2016, 76, 405–411 Search PubMed.
- B. E. Elewski, R. Aly, S. L. Baldwin, R. F. González Soto, P. Rich, M. Weisfeld, H. Wiltz, L. T. Zane and R. Pollak, J. Am. Acad. Dermatol., 2015, 73, 62–69 Search PubMed.
- T. Akama, S. J. Baker, Y.-K. Zhang, V. Hernandez, H. Zhou, V. Sanders, Y. Freund, R. Kimura, K. R. Maples and J. J. Plattner, Bioorg. Med. Chem. Lett., 2009, 19, 2129–2132 Search PubMed.
- M. Z. H. Kazmi, O. M. Schneider and D. G. Hall, J. Med. Chem., 2023, 66, 13768–13787 Search PubMed.
- J. P. Lorand and J. O. Edwards, J. Org. Chem., 1959, 24, 769–774 Search PubMed.
- J. Chan, S. C. Dodani and C. J. Chang, Nat. Chem., 2012, 4, 973–984 Search PubMed.
- B. Gatin-Fraudet, R. Ottenwelter, T. Le Saux, S. Norsikian, M. Pucher, T. Lombes, A. Baron, P. Durand, G. Doisneau, Y. Bourdreux, B. I. Iorga, M. Erard, L. Jullien, D. Guianvarc’h, D. Urban and B. Vauzeilles, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2107503118 Search PubMed.
- B. J. Graham, I. W. Windsor, B. Gold and R. T. Raines, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2013691118 Search PubMed.
- J. Yan, G. Springsteen, S. Deeter and B. H. Wang, Tetrahedron, 2004, 60, 11205–11209 Search PubMed.
- T. D. James, K. R. A. S. Sandanayake and S. Shinkai, Angew. Chem., Int. Ed. Engl., 1996, 35, 1910–1922 Search PubMed.
- S. Jin, Y. Cheng, S. Reid, M. Li and B. Wang, Med. Res. Rev., 2010, 30, 171–257 Search PubMed.
- X. Wu, Z. Li, X.-X. Chen, J. S. Fossey, T. D. James and Y.-B. Jiang, Chem. Soc. Rev., 2013, 42, 8032–8048 Search PubMed.
- S. D. Bull, M. G. Davidson, J. M. H. van den Elsen, J. S. Fossey, A. T. A. Jenkins, Y.-B. Jiang, Y. Kubo, F. Marken, K. Sakurai, J. Zhao and T. D. James, Acc. Chem. Res., 2013, 46, 312–326 Search PubMed.
- W. Zhai, X. Sun, T. D. James and J. S. Fossey, Chem. – Asian J., 2015, 10, 1836–1848 Search PubMed.
- G. T. Williams, J. L. Kedge and J. S. Fossey, ACS Sens., 2021, 6, 1508–1528 Search PubMed.
- R. J. Grams, W. L. Santos, I. R. Scorei, A. Abad-García, C. A. Rosenblum, A. Bita, H. Cerecetto, C. Viñas and M. A. Soriano-Ursúa, Chem. Rev., 2024, 124, 2441–2511 Search PubMed.
- W. L. Brooks and B. S. Sumerlin, Chem. Rev., 2016, 116, 1375–1397 Search PubMed.
- G. S. Han and D. W. Domaille, J. Mater. Chem. B, 2022, 10, 6263–6278 Search PubMed.
- T. L. Halo, J. Appelbaum, E. M. Hobert, D. M. Balkin and A. Schepartz, J. Am. Chem. Soc., 2009, 131, 438–439 Search PubMed.
- B. Akgun and D. G. Hall, Angew. Chem., Int. Ed., 2016, 55, 3909–3913 Search PubMed.
- B. Akgun, C. Li, Y. Hao, G. Lambkin, R. Derda and D. G. Hall, J. Am. Chem. Soc., 2017, 139, 14285–14291 Search PubMed.
- J. G. Haggett and D. W. Domaille, Chem. – Eur. J., 2024, 30, e202302485 Search PubMed.
- S. Palvai, J. Bhangu, B. Akgun, C. T. Moody, D. G. Hall and Y. Brudno, Bioconjugate Chem., 2020, 31, 2288–2292 Search PubMed.
- G. Springsteen and B. Wang, Tetrahedron, 2002, 58, 5291–5300 Search PubMed.
- R. Pizer and L. Babcock, Inorg. Chem., 1977, 16, 1677–1681 Search PubMed.
- A. Gennari, C. Gujral, E. Hohn, E. Lallana, F. Cellesi and N. Tirelli, Bioconjugate Chem., 2017, 28, 1391–1402 Search PubMed.
- M. L. Stolowitz, C. Ahlem, K. A. Hughes, R. J. Kaiser, E. A. Kesicki, G. Li, K. P. Lund, S. M. Torkelson and J. P. Wiley, Bioconjugate Chem., 2001, 12, 229–239 Search PubMed.
- S. B. Shin, R. D. Almeida, G. Gerona-Navarro, C. Bracken and S. R. Jaffrey, Chem. Biol., 2010, 17, 1171–1176 Search PubMed.
- M. M. Zegota, T. Wang, C. Seidler, D. Y. Wah Ng, S. L. Kuan and T. Weil, Bioconjugate Chem., 2018, 29, 2665–2670 Search PubMed.
- J. P. Wiley, K. A. Hughes, R. J. Kaiser, E. A. Kesicki, K. P. Lund and M. L. Stolowitz, Bioconjugate Chem., 2001, 12, 240–250 Search PubMed.
- D. Y. W. Ng, M. Arzt, Y. Wu, S. L. Kuan, M. Lamla and T. Weil, Angew. Chem., Int. Ed., 2014, 53, 324–328 Search PubMed.
- S. Moffatt, S. Wiehle and R. J. Cristiano, Hum. Gene Ther., 2005, 16, 57–67 Search PubMed.
- S. Moffatt, S. Wiehle and R. J. Cristiano, Gene Ther., 2006, 13, 1512–1523 Search PubMed.
- B. E. Collins, P. Metola and E. V. Anslyn, Supramol. Chem., 2013, 25, 79–86 Search PubMed.
- B. E. Collins, S. Sorey, A. E. Hargrove, S. H. Shabbir, V. M. Lynch and E. V. Anslyn, J. Org. Chem., 2009, 74, 4055–4060 Search PubMed.
- B. M. Chapin, P. Metola, V. M. Lynch, J. F. Stanton, T. D. James and E. V. Anslyn, J. Org. Chem., 2016, 81, 8319–8330 Search PubMed.
- H. E. Dunn, J. C. Catlin and H. R. Snyder, J. Org. Chem., 1968, 33, 4483–4486 Search PubMed.
- Y. Pérez-Fuertes, A. M. Kelly, A. L. Johnson, S. Arimori, S. D. Bull and T. D. James, Org. Lett., 2006, 8, 609–612 Search PubMed.
- E. Galbraith, A. M. Kelly, J. S. Fossey, G. Kociok-Kohn, M. G. Davidson, S. D. Bull and T. D. James, New J. Chem., 2009, 33, 181–185 Search PubMed.
- M. Hutin, G. Bernardinelli and J. R. Nitschke, Chem. – Eur. J., 2008, 14, 4585–4593 Search PubMed.
- P. M. Cal, J. B. Vicente, E. Pires, A. V. Coelho, L. F. Veiros, C. Cordeiro and P. M. Gois, J. Am. Chem. Soc., 2012, 134, 10299–10305 Search PubMed.
- N. J. Gutierrez-Moreno, F. Medrano and A. K. Yatsimirsky, Org. Biomol. Chem., 2012, 10, 6960–6972 Search PubMed.
- P. M. Cal, R. F. Frade, C. Cordeiro and P. M. Gois, ACS Chem. Biol., 2016, 11(2), 319–323 Search PubMed.
- A. Bandyopadhyay, K. A. McCarthy, M. A. Kelly and J. Gao, Nat. Commun., 2015, 6, 6561 Search PubMed.
- A. Bandyopadhyay and J. Gao, Chem. – Eur. J., 2015, 21, 14748–14752 Search PubMed.
- J. Crugeiras, A. Rios, E. Riveiros, T. L. Amyes and J. P. Richard, J. Am. Chem. Soc., 2008, 130, 2041–2050 Search PubMed.
- L. Boike, N. J. Henning and D. K. Nomura, Nat. Rev. Drug Discovery, 2022, 21, 881–898 Search PubMed.
- J. Singh, R. C. Petter, T. A. Baillie and A. Whitty, Nat. Rev. Drug Discovery, 2011, 10, 307–317 Search PubMed.
- A. Bandyopadhyay and J. Gao, Curr. Opin. Chem. Biol., 2016, 34, 110–116 Search PubMed.
- J. Gao and V. Nobile, in Annual Reports in Medicinal Chemistry, ed. R. A. Ward and N. P. Grimster, Academic Press, 2021, vol. 56, pp. 75–94 Search PubMed.
- M. Gehringer and S. A. Laufer, J. Med. Chem., 2019, 62, 5673–5724 Search PubMed.
- D. Oksenberg, K. Dufu, M. P. Patel, C. Chuang, Z. Li, Q. Xu, A. Silva-Garcia, C. Zhou, A. Hutchaleelaha, L. Patskovska, Y. Patskovsky, S. C. Almo, U. Sinha, B. W. Metcalf and D. R. Archer, Br. J. Haematol., 2016, 175, 141–153 Search PubMed.
- C. Slavetinsky, S. Kuhn and A. Peschel, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2017, 1862, 1310–1318 Search PubMed.
- T. A. Aweda, Z. F. B. Muftuler, A. V. F. Massicano, D. Gadhia, K. A. McCarthy, S. Queern, A. Bandyopadhyay, J. Gao and S. E. Lapi, Contrast Media Mol. Imaging, 2019, 2019, 3149249 Search PubMed.
- G. P. Smith and V. A. Petrenko, Chem. Rev., 1997, 97, 391–410 Search PubMed.
- K. A. McCarthy, M. A. Kelly, K. Li, S. Cambray, A. S. Hosseini, T. van Opijnen and J. Gao, J. Am. Chem. Soc., 2018, 140, 6137–6145 Search PubMed.
- M. Zheng, F.-J. Chen, K. Li, R. M. Reja, F. Haeffner and J. Gao, J. Am. Chem. Soc., 2022, 144, 15885–15893 Search PubMed.
- M. Zheng and J. Gao, ACS Chem. Biol., 2023, 18, 2259–2266 Search PubMed.
- M. Kelly, S. Cambray, K. A. McCarthy, W. Wang, E. Geisinger, J. Ortiz-Marquez, T. van Opijnen and J. Gao, ACS Infect. Dis., 2020, 6, 2410–2418 Search PubMed.
- N. Kumar, W. Wang, J. C. Ortiz-Marquez, M. Catalano, M. Gray, N. Biglari, K. Hikari, X. Ling, J. Gao, T. van Opijnen and K. S. Burch, Biosens. Bioelectron., 2020, 156, 112123 Search PubMed.
- G. Akçay, M. A. Belmonte, B. Aquila, C. Chuaqui, A. W. Hird, M. L. Lamb, P. B. Rawlins, N. Su, S. Tentarelli, N. P. Grimster and Q. Su, Nat. Chem. Biol., 2016, 12, 931–936 Search PubMed.
- D. Quach, G. Tang, J. Anantharajan, N. Baburajendran, A. Poulsen, J. L. K. Wee, P. Retna, R. Li, B. Liu, D. H. Y. Tee, P. Z. Kwek, J. K. Joy, W.-Q. Yang, C.-J. Zhang, K. Foo, T. H. Keller and S. Q. Yao, Angew. Chem., Int. Ed., 2021, 60, 17131–17137 Search PubMed.
- R. M. Reja, W. Wang, Y. Lyu, F. Haeffner and J. Gao, J. Am. Chem. Soc., 2022, 144, 1152–1157 Search PubMed.
- S. Teng, E. W. H. Ng, Z. Zhang, C. N. Soon, H. Xu, R. Li, H. Hirao and T.-P. Loh, Sci. Adv., 2023, 9, eadg4924 Search PubMed.
- P. E. Dawson, T. W. Muir, I. Clark-Lewis and S. B. Kent, Science, 1994, 266, 776–779 Search PubMed.
- H. Ren, F. Xiao, K. Zhan, Y. P. Kim, H. Xie, Z. Xia and J. Rao, Angew. Chem., Int. Ed., 2009, 48, 9658–9662 Search PubMed.
- A. Istrate, M. B. Geeson, C. D. Navo, B. B. Sousa, M. C. Marques, R. J. Taylor, T. Journeaux, S. R. Oehler, M. R. Mortensen, M. J. Deery, A. D. Bond, F. Corzana, G. Jiménez-Osés and G. J. L. Bernardes, J. Am. Chem. Soc., 2022, 144, 10396–10406 Search PubMed.
- X. Zheng, Z. Li, W. Gao, X. Meng, X. Li, L. Y. P. Luk, Y. Zhao, Y.-H. Tsai and C. Wu, J. Am. Chem. Soc., 2020, 142, 5097–5103 Search PubMed.
- A. Bandyopadhyay, S. Cambray and J. Gao, Chem. Sci., 2016, 7, 4589–4593 Search PubMed.
- H. Faustino, M. J. S. A. Silva, L. F. Veiros, G. J. L. Bernardes and P. M. P. Gois, Chem. Sci., 2016, 7, 5052–5058 Search PubMed.
- N. C. Rose, A. V. Sanchez, E. F. Tipple, J. M. Lynam and C. D. Spicer, Chem. Sci., 2022, 13, 12791–12798 Search PubMed.
- K. Li, C. Weidman and J. Gao, Org. Lett., 2018, 20, 20–23 Search PubMed.
- K. Li, M. A. Kelly and J. Gao, Org. Biomol. Chem., 2019, 17, 5908–5912 Search PubMed.
- K. Li, W. Wang and J. Gao, Angew. Chem., Int. Ed., 2020, 59, 14246–14250 Search PubMed.
- A. B. Draganov, K. Wang, J. Holmes, K. Damera, D. Wang, C. Dai and B. Wang, Chem. Commun., 2015, 51, 15180–15183 Search PubMed.
- R. Padanha, R. A. N. Cavadas, P. Merino, J. P. M. António and P. M. P. Gois, Org. Lett., 2023, 25, 5476–5480 Search PubMed.
- R. Russo, R. Padanha, F. Fernandes, L. F. Veiros, F. Corzana and P. M. P. Gois, Chem. – Eur. J., 2020, 26, 15226–15231 Search PubMed.
- R. M. R. M. Lopes, A. E. Ventura, L. C. Silva, H. Faustino and P. M. P. Gois, Chem. – Eur. J., 2018, 24, 12495–12499 Search PubMed.
- A. Dirksen, T. M. Hackeng and P. E. Dawson, Angew. Chem., Int. Ed., 2006, 45, 7581–7584 Search PubMed.
- P. Schmidt, C. Stress and D. Gillingham, Chem. Sci., 2015, 6, 3329–3333 Search PubMed.
- J. Kalia and R. T. Raines, Angew. Chem., Int. Ed., 2008, 47, 7523–7526 Search PubMed.
- G. S. Han and D. W. Domaille, Org. Biomol. Chem., 2021, 19, 4986–4991 Search PubMed.
- O. Dilek, Z. Lei, K. Mukherjee and S. Bane, Chem. Commun., 2015, 51, 16992–16995 Search PubMed.
- S. B. Tuttle, O. Dilek and K. Mukherjee, Patent application, US20150329568A1, 2015 Search PubMed.
- C. J. Stress, P. J. Schmidt and D. G. Gillingham, Org. Biomol. Chem., 2016, 14, 5529–5533 Search PubMed.
- D. Gillingham, Org. Biomol. Chem., 2016, 14, 7606–7609 Search PubMed.
- A. Bandyopadhyay, S. Cambray and J. Gao, J. Am. Chem. Soc., 2017, 139, 871–878 Search PubMed.
- A. Chowdhury, S. Chatterjee, A. Kushwaha, S. Nanda, T. J. Dhilip Kumar and A. Bandyopadhyay, Chem. – Eur. J., 2023, 29, e202300393 Search PubMed.
- M. J. S. Dewar and R. C. Dougherty, J. Am. Chem. Soc., 1962, 84, 2648–2649 Search PubMed.
- P. Tschampel and H. R. Snyder, J. Org. Chem., 1964, 29, 2168–2172 Search PubMed.
- P. Cai, L. A. Schneider, C. Stress and D. Gillingham, Org. Lett., 2021, 23, 8772–8776 Search PubMed.
- S. Cambray, A. Bandyopadhyay and J. Gao, Chem. Commun., 2017, 53, 12532–12535 Search PubMed.
- S. H. Klass, L. E. Sofen, Z. F. Hallberg, T. A. Fiala, A. V. Ramsey, N. S. Dolan, M. B. Francis and A. L. Furst, Chem. Commun., 2021, 57, 2507–2510 Search PubMed.
- T. J. Lupoli, H. Tsukamoto, E. H. Doud, T. S. Wang, S. Walker and D. Kahne, J. Am. Chem. Soc., 2011, 133, 10748–10751 Search PubMed.
- J. P. M. António, J. I. Carvalho, A. S. André, J. N. R. Dias, S. I. Aguiar, H. Faustino, R. M. R. M. Lopes, L. F. Veiros, G. J. L. Bernardes, F. A. da Silva and P. M. P. Gois, Angew. Chem., Int. Ed., 2021, 60, 25914–25921 Search PubMed.
- H. Gu, T. I. Chio, Z. Lei, R. J. Staples, J. S. Hirschi and S. Bane, Org. Biomol. Chem., 2017, 15, 7543–7548 Search PubMed.
- T. I. Chio, H. Gu, K. Mukherjee, L. N. Tumey and S. L. Bane, Bioconjugate Chem., 2019, 30, 1554–1564 Search PubMed.
- H. Gu, S. Ghosh, R. J. Staples and S. L. Bane, Bioconjugate Chem., 2019, 30, 2604–2613 Search PubMed.
- Y. Li, Y. Liu, R. Ma, Y. Xu, Y. Zhang, B. Li, Y. An and L. Shi, ACS Appl. Mater. Interfaces, 2017, 9, 13056–13067 Search PubMed.
- R. Ma, C. Zhang, Y. Liu, C. Li, Y. Xu, B. Li, Y. Zhang, Y. An and L. Shi, RSC Adv., 2017, 7, 21328–21335 Search PubMed.
- M. K. Meadows, E. K. Roesner, V. M. Lynch, T. D. James and E. V. Anslyn, Org. Lett., 2017, 19, 3179–3182 Search PubMed.
- F. M. F. Santos, A. I. Matos, A. E. Ventura, J. Gonçalves, L. F. Veiros, H. F. Florindo and P. M. P. Gois, Angew. Chem., Int. Ed., 2017, 56, 9346–9350 Search PubMed.
- F. M. F. Santos, J. N. Rosa, N. R. Candeias, C. P. Carvalho, A. I. Matos, A. E. Ventura, H. F. Florindo, L. C. Silva, U. Pischel and P. M. P. Gois, Chem. – Eur. J., 2016, 22, 1631–1637 Search PubMed.
- V. G. Jiménez, F. M. F. Santos, S. Castro-Fernández, J. M. Cuerva, P. M. P. Gois, U. Pischel and A. G. Campaña, J. Org. Chem., 2018, 83, 14057–14062 Search PubMed.
- F. M. F. Santos, Z. Domínguez, M. M. Alcaide, A. I. Matos, H. F. Florindo, N. R. Candeias, P. M. P. Gois and U. Pischel, ChemPhotoChem, 2018, 2, 1038–1045 Search PubMed.
- S. Baldo, P. Antunes, J. F. Felicidade, F. M. F. Santos, J. F. Arteaga, F. Fernandes, U. Pischel, S. N. Pinto and P. M. P. Gois, ACS Med. Chem. Lett., 2022, 13, 128–133 Search PubMed.
- J. Felicidade, F. M. F. Santos, J. F. Arteaga, P. Remón, R. Campos-González, H.-C. Nguyen, F. Nájera, F. Boscá, D. Y. W. Ng, P. M. P. Gois and U. Pischel, Chem. – Eur. J., 2023, 29, e202300579 Search PubMed.
- N. Miyaura, K. Yamada and A. Suzuki, Tetrahedron Lett., 1979, 20, 3437–3440 Search PubMed.
- A. Ojida, H. Tsutsumi, N. Kasagi and I. Hamachi, Tetrahedron Lett., 2005, 46, 3301–3305 Search PubMed.
- J. M. Chalker, C. S. C. Wood and B. G. Davis, J. Am. Chem. Soc., 2009, 131, 16346–16347 Search PubMed.
- C. D. Spicer and B. G. Davis, Chem. Commun., 2011, 47, 1698–1700 Search PubMed.
- C. D. Spicer, T. Triemer and B. G. Davis, J. Am. Chem. Soc., 2012, 134, 800–803 Search PubMed.
- M. K. Bilyard, H. J. Bailey, L. Raich, M. A. Gafitescu, T. Machida, J. Iglésias-Fernández, S. S. Lee, C. D. Spicer, C. Rovira, W. W. Yue and B. G. Davis, Nature, 2018, 563, 235–240 Search PubMed.
- T.-K. Lee, B. Manandhar, K. J. Kassees and J.-M. Ahn, J. Org. Chem., 2020, 85, 1376–1384 Search PubMed.
- Z. Gao, V. Gouverneur and B. G. Davis, J. Am. Chem. Soc., 2013, 135, 13612–13615 Search PubMed.
- E. Brustad, M. L. Bushey, J. W. Lee, D. Groff, W. Liu and P. G. Schultz, Angew. Chem., Int. Ed., 2008, 47, 8220–8223 Search PubMed.
- T. A. Mollner, P. G. Isenegger, B. Josephson, C. Buchanan, L. Lercher, D. Oehlrich, D. F. Hansen, S. Mohammed, A. J. Baldwin, V. Gouverneur and B. G. Davis, Nat. Chem. Biol., 2021, 17, 1245–1261 Search PubMed.
- R. H. de Vries, J. H. Viel, O. P. Kuipers and G. Roelfes, Angew. Chem., Int. Ed., 2021, 60, 3946–3950 Search PubMed.
- R. Jing, W. C. Powell, K. J. Fisch and M. A. Walczak, J. Am. Chem. Soc., 2023, 145, 22354–22360 Search PubMed.
- Z. T. Ball, Acc. Chem. Res., 2019, 52, 566–575 Search PubMed.
- J. Ohata, M. B. Minus, M. E. Abernathy and Z. T. Ball, J. Am. Chem. Soc., 2016, 138, 7472–7475 Search PubMed.
- J. Ohata, Y. Zeng, L. Segatori and Z. T. Ball, Angew. Chem., Int. Ed., 2018, 57, 4015–4019 Search PubMed.
- K. Hanaya, J. Ohata, M. K. Miller, A. E. Mangubat-Medina, M. J. Swierczynski, D. C. Yang, R. M. Rosenthal, B. V. Popp and Z. T. Ball, Chem. Commun., 2019, 55, 2841–2844 Search PubMed.
- M. K. Miller, M. J. Swierczynski, Y. Ding and Z. T. Ball, Org. Lett., 2021, 23, 5334–5338 Search PubMed.
- J. Ohata, M. K. Miller, C. M. Mountain, F. Vohidov and Z. T. Ball, Angew. Chem., Int. Ed., 2018, 57, 2827–2830 Search PubMed.
- S. Eising, F. Lelivelt and K. M. Bonger, Angew. Chem., Int. Ed., 2016, 55, 12243–12247 Search PubMed.
- S. Eising, N. G. A. van der Linden, F. Kleinpenning and K. M. Bonger, Bioconjugate Chem., 2018, 29, 982–986 Search PubMed.
- L. P. W. M. Lelieveldt, S. Eising, A. Wijen and K. M. Bonger, Org. Biomol. Chem., 2019, 17, 8816–8821 Search PubMed.
| This journal is © The Royal Society of Chemistry 2024 |