
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
2-Pyridonates: a versatile ligand platform in 3d transition metal coordination chemistry and catalysis
Andrey
Fedulin†
and
Axel
Jacobi von Wangelin
 *
*
Dept. of Chemistry, University of Hamburg, Martin Luther King Pl. 6, 20146 Hamburg, Germany. E-mail: axel.jacobi@uni-hamburg.de
First published on 15th November 2023
Abstract
Aminoalcohols are an easily available, highly diverse, and inexpensive class of ligands with numerous applications in biological and technological contexts of metal–ligand mediated processes. Among them, 2-pyridonates exhibit especially intriguing stereoelectronic features that have enabled a versatile coordination chemistry. Learning from the natural role model of [Fe]-hydrogenase, 3d-transition metal complexes with pyridonate ligands have recently been developed for powerful catalytic transformations. This review illustrates the general properties of pyridonate ligands and their key roles in 3d transition metal catalysts.
1 Introduction
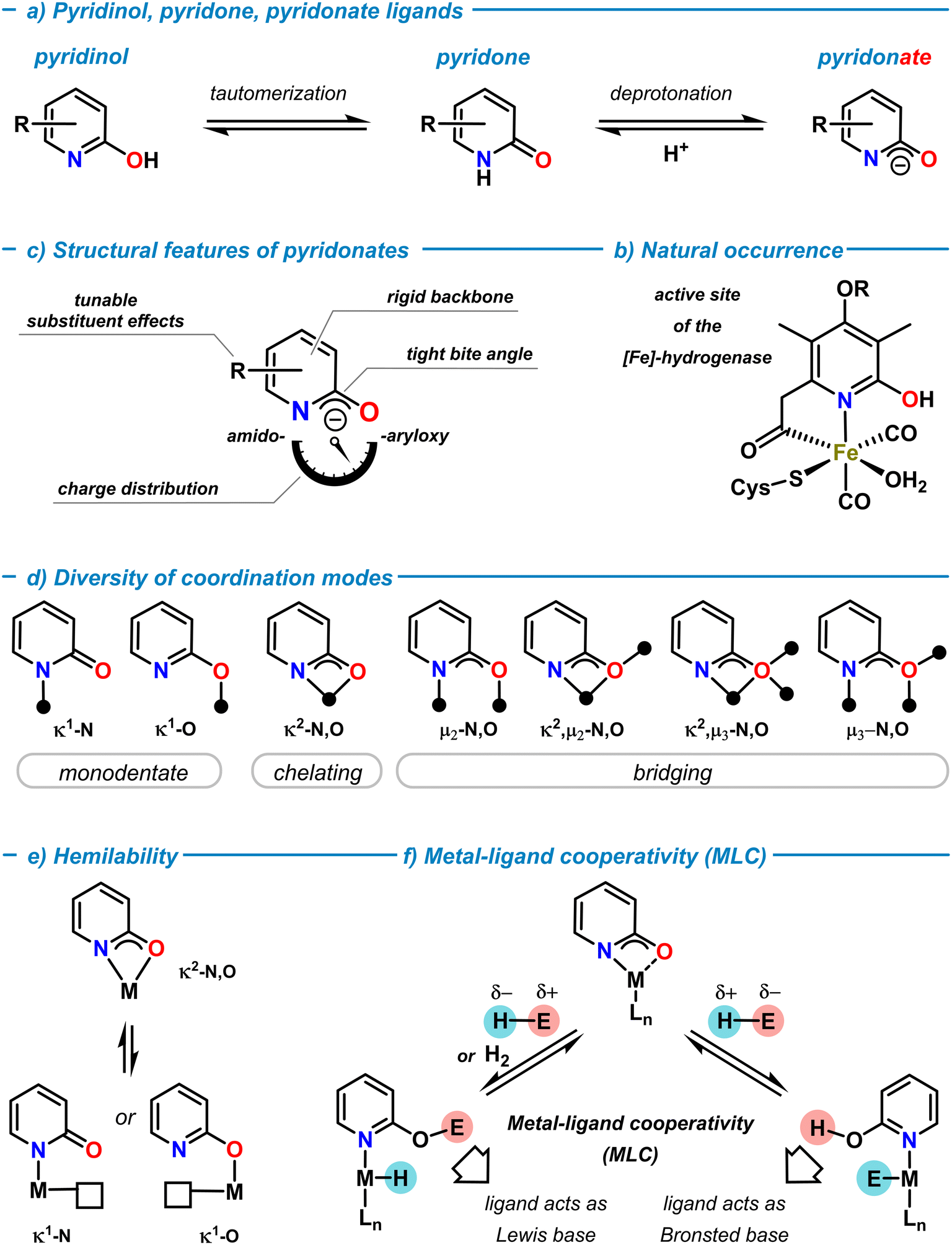
Rational ligand design that taps uncharted territories of the structures, properties, and reactivities of metal complexes is a key challenge in modern coordination chemistry and catalysis.1 Since Alfred Werner's conceptual work on coordination chemistry (Nobel Prize 1913), the ability of ligands to modulate coordination compounds and access new chemical spaces and properties has been developed to great maturity over the past decades.2 Systematic variations of the nature of ligand backbones, donor functions, core and peripheral substituents, chelation and bridging modes, conformational and configurational properties, and various other stereo-electronic effects can enable precise control over local and global structures, physicochemical properties, and distinct reactivity profiles for both the ligands and the resultant metal complexes. The scientific prospects of such seemingly unlimited degrees of freedom has led to an ever-growing number of ligand frameworks that were designed to support molecular metal complexes for very diverse applications. Critical evaluations of the resultant efficacies for the targeted use and considerations of availability, access, robustness, toxicity, sustainability, convenience, etc. have enabled the identification of “privileged” ligand families.In the arsenal of “successful” ligands in coordination chemistry, N,O-ligands based on the 2-pyridonates exhibit an especially versatile set of properties that can be finely tuned due to their modular building principle. As a neutral molecule, 2-pyridone is an N-heterocycle that is in tautomeric equilibrium with 2-pyridinol (or 2-hydroxypyridine) both in solution and solid state (Scheme 1a).3–5 The solution structure highly depends on solvent polarity and ring substituents, with polar solvents and electron-withdrawing substituents in the 6-position favoring the pyridone form. The 2-pyridone tautomer is a cyclic amide (lactam) with high C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O double bond character. In contrast, the 2-pyridinol tautomer constitutes an aromatic pyridine ring with a 2-hydroxy substituent. Deprotonation of 2-pyridone (pKa ∼ 17, in DMSO)6 provides the 2-pyridonate anion with delocalized negative charge between the N- and O-atoms (Scheme 1a). Both, the neutral tautomers and their corresponding anionic bases can act as ligands in main group element and transition metal complexes. The combination of the Brønsted and Lewis acid/base properties of the ligand and the properties of the metal ion may enable cooperative behaviour of metal pyridon(at)es (vide infra).7 The metal–pyridinol motif has a natural role in the active site of the [Fe]-hydrogenase metalloenzyme (Scheme 1b).8 The natural occurrence of such, and related, transition metal oxypyridine complexes and their mechanistic role in biocatalytic pathways has been serving as a great inspiration for the development of biomimetic models and related metal complexes.9,10 2-Pyridonates are LX-type donors and important members of the 1,3-N,O-hetero-bidentate ligand family (Scheme 1c).11 The inherent presence of hard oxygen and nitrogen donor atoms with multiple electron lone pairs in close proximity, the ambiphilicity of the pyridine function (as an N-donor and electron-deficient π-system), and the softer properties of the delocalized negative charge between N- and O-atoms make for a rich diversity of σ- and π-bonding motifs in coordination compounds. The adoption of structures bearing more N-centered amido or O-centered aryloxy resonance character is highly depending on the coordinated metal ion, substituent effects and the choice of solvents (Scheme 1b). The fixation of the nitrogen donor within the six-membered ring confers high conformational rigidity to the conjugated NCO motif and a tight bite angle in chelation or μ-bridging structures. The facile adoption and dynamic interconversion of diverse coordination motifs in metal complexes is an especially remarkable feature of 2-pyridonate complexes (Scheme 1d).12,13 Monometallic κ1-N and κ1-O modes, chelation (κ2-N,O), and several bridging modes (μ2,μ3-N,O) are known, as well as many combinations of these coordination patterns with up to three coordinated metal ions per ligand. There are no stringent correlations between the nature of metal ions, the bond lengths, and the resultant bonding modes. This is partially due to a lack of comparable series of metal pyridonate complexes without further binding motifs that potentially could override the preferences of the pyridonate ligand. It appears that the coordination motifs of pyridonate ligands are very susceptible to the local environments including the metal ions, coordination numbers, the nature of neighboring ligand motifs, ring substitutions, and possibly even crystal packing interactions. As an example, the analysis of X-ray diffraction data of closely related ruthenium pyridonate complexes (without further donor functions in the ligands) provided the following picture: a C–O bond distance of 1.27 Å was indicative of the pyridone form in a monodentate κ1-N coordination14 (vs. 1.25 Å in free 2-pyridone).15 The pyridonate in chelating κ2-N,O coordination gave an elongated C–O bond distance of 1.30–1.35 Å due to the increased single bond character of the 2-oxypyridine motif.16,17 The bridging μ2-N,O coordination was characterized by an intermediate C–O bond length of 1.30 Å and could be interpreted as a hybrid between the two tautomeric forms.18 The C–C bonds within the pyridonate rings showed alternating bond length patterns (1.36–1.39–1.35–1.43 Å) in the κ1-N binding mode to Ru,14 which approached an average value in the chelation mode (1.39–1.39–1.42–1.41 Å).17 In a series of copper pyridonate complexes, IR spectroscopy exhibited distinct absorptions of the terminal and bridging κ1-O bound pyridonates (1648 cm−1) and the μ2-O bound pyridonate (1620 cm−1).19
O double bond character. In contrast, the 2-pyridinol tautomer constitutes an aromatic pyridine ring with a 2-hydroxy substituent. Deprotonation of 2-pyridone (pKa ∼ 17, in DMSO)6 provides the 2-pyridonate anion with delocalized negative charge between the N- and O-atoms (Scheme 1a). Both, the neutral tautomers and their corresponding anionic bases can act as ligands in main group element and transition metal complexes. The combination of the Brønsted and Lewis acid/base properties of the ligand and the properties of the metal ion may enable cooperative behaviour of metal pyridon(at)es (vide infra).7 The metal–pyridinol motif has a natural role in the active site of the [Fe]-hydrogenase metalloenzyme (Scheme 1b).8 The natural occurrence of such, and related, transition metal oxypyridine complexes and their mechanistic role in biocatalytic pathways has been serving as a great inspiration for the development of biomimetic models and related metal complexes.9,10 2-Pyridonates are LX-type donors and important members of the 1,3-N,O-hetero-bidentate ligand family (Scheme 1c).11 The inherent presence of hard oxygen and nitrogen donor atoms with multiple electron lone pairs in close proximity, the ambiphilicity of the pyridine function (as an N-donor and electron-deficient π-system), and the softer properties of the delocalized negative charge between N- and O-atoms make for a rich diversity of σ- and π-bonding motifs in coordination compounds. The adoption of structures bearing more N-centered amido or O-centered aryloxy resonance character is highly depending on the coordinated metal ion, substituent effects and the choice of solvents (Scheme 1b). The fixation of the nitrogen donor within the six-membered ring confers high conformational rigidity to the conjugated NCO motif and a tight bite angle in chelation or μ-bridging structures. The facile adoption and dynamic interconversion of diverse coordination motifs in metal complexes is an especially remarkable feature of 2-pyridonate complexes (Scheme 1d).12,13 Monometallic κ1-N and κ1-O modes, chelation (κ2-N,O), and several bridging modes (μ2,μ3-N,O) are known, as well as many combinations of these coordination patterns with up to three coordinated metal ions per ligand. There are no stringent correlations between the nature of metal ions, the bond lengths, and the resultant bonding modes. This is partially due to a lack of comparable series of metal pyridonate complexes without further binding motifs that potentially could override the preferences of the pyridonate ligand. It appears that the coordination motifs of pyridonate ligands are very susceptible to the local environments including the metal ions, coordination numbers, the nature of neighboring ligand motifs, ring substitutions, and possibly even crystal packing interactions. As an example, the analysis of X-ray diffraction data of closely related ruthenium pyridonate complexes (without further donor functions in the ligands) provided the following picture: a C–O bond distance of 1.27 Å was indicative of the pyridone form in a monodentate κ1-N coordination14 (vs. 1.25 Å in free 2-pyridone).15 The pyridonate in chelating κ2-N,O coordination gave an elongated C–O bond distance of 1.30–1.35 Å due to the increased single bond character of the 2-oxypyridine motif.16,17 The bridging μ2-N,O coordination was characterized by an intermediate C–O bond length of 1.30 Å and could be interpreted as a hybrid between the two tautomeric forms.18 The C–C bonds within the pyridonate rings showed alternating bond length patterns (1.36–1.39–1.35–1.43 Å) in the κ1-N binding mode to Ru,14 which approached an average value in the chelation mode (1.39–1.39–1.42–1.41 Å).17 In a series of copper pyridonate complexes, IR spectroscopy exhibited distinct absorptions of the terminal and bridging κ1-O bound pyridonates (1648 cm−1) and the μ2-O bound pyridonate (1620 cm−1).19
 | ||
| Scheme 1 Coordination bonding motifs of the 2-pyridone/2-pyridonate ligand platform. | ||
Rapid interconversion between different coordination modes in solution can often be observed. This dynamic behaviour via intermediates with lower coordination numbers delineates another important property of pyridonate ligands: hemilability.20 Chelating pyridonate ligands can readily change their coordination modes by dissociation of one of the donor arms. The resultant vacant coordination site may induce distinct reactivities in the vicinity of the pyridonate or with active participation of the pyridonate. For pyridonates, hemilability has been mostly observed as changes between κ2-N,O and κ1-N or κ1-O coordinations (Scheme 1e).21 The pyridonate backbone contains four positions for covalent functionalization that can be individually addressed by selective synthesis methods. 2-Pyridone, 6-methyl-2-pyridone, and 6-choro-2-pyridone are commercially available. Numerous protocols of cross-coupling reactions with halopyridine derivatives are available in the literature.22 The installation of a 2-oxy function is easily achieved by nucleophilic aromatic substitution. The incorporation of additional ligand moieties via backbone functionalization further increases the versatility of the pyridonate ligand platform. For example, metalation and subsequent substitution at the 6-methyl substituent has proven a versatile strategy of introduction of further donor functions.23 By careful choice of the substituents, a huge chemical space of metal complexes with finely tuned bonding motifs, various stereoelectronic properties, structures, stabilities, and distinct reactivities can be accessed. This versatility and diversity renders pyridonates to entertain an extremely rich coordination chemistry ranging from mononuclear complexes to high-nuclearity clusters and coordination polymers.
Apart from the use as a spectator ligand platform supporting metal-centered reactivity, pyridonates may be actively involved in the bond breaking and bond forming events of reaction mechanisms. Such synergetic bond activation of substrates is described in modern literature as metal–ligand cooperation (MLC) and constitutes an important feature of many catalytic transformations.24,25 For example, the metal-bound oxygen (κ2-N,O) or the peripheral oxygen donor (κ1-N) of a 2-pyridonato metal complex can act as a Lewis base and mediate bond cleavage of an E–H substrate molecule in cooperation with the neighboring metal center (Scheme 1f). Depending on the polarity of an E–H bond, three distinct pathways could operate: i) Brønsted acid–base reaction between a sufficiently acidic E–H and the pyridonate ligand to give a pyridinol ligand (pyO-H) and metal-E bonding. Subsequent transfer of the E moiety to electrophilic substrates may be accompanied by a proton transfer from pyO-H. ii) An unpolar E–H molecule such as H2 may undergo heterolysis by metal–ligand cooperativity to result in a metal hydride species M–H coordinated by a pyridinol ligand pyO-H. Such scenario is often observed in transfer hydrogenation, dehydrogenation, or hydrogen borrowing mechanisms. iii) If E–H is bears high hydridicity (e.g. a borane or silane), cooperative metal–ligand bond cleavage results in a metal hydride and pyO-E ligand. Such intermediates can be found as active catalyst species in hydroelementation reactions. It is important to note that the thermodynamic driving forces of either mechanism include not only the individual bond strengths between metal, N, O, E, and H atoms but also the degree of conjugation and aromaticity of the pyridine ligand backbone.26,27
In conclusion, the wide variety of κ- and μ-binding modes make pyridonate ligands highly attractive building blocks for the synthesis of diverse coordination compounds, polynuclear clusters and materials. The facile and modular incorporation of various structural and stereoelectronic properties such as basicity, hemilability, metal–ligand cooperativity, second coordination sphere effects, and further donor functions have prompted several applications to sophisticated metal complex design and homogeneous catalysis. While pyridonate complexes of heavier transition metals have been studied for many years, combinations of 3d transition metals and pyridonate ligand derivatives have very recently enabled new insights intro structure–activity relationships. From biomimetic models of the [Fe]-hydrogenase to active hydro-elementation catalysts and electro-catalytic applications to water splitting processes, this review is intended to shine light onto the most important developments in the area of 3d metal pyridonate catalysis.
2 Noble metal complexes
Historically, molecular pyridonate complexes were first investigated with noble metals, i.e. Ir, Rh, Ru, Pd. The recent advances in the design and applications of novel 3d transition metal pyridonates were greatly inspired by the pioneering work on their heavier homologues that resulted in novel classes of coordination compounds, clusters, and polymers. For example, diruthenium pyridonates constitute important members of the family of paddle–wheel complexes with metal–metal bonds.28,29 Selected noble metal complexes with catalytic applications are highlighted in Scheme 2. One of the most prominent examples of a molecular noble metal pyridone catalyst species is the half-sandwich Ir(III) complex 1 that was reported by Yamaguchi in 2007.30 This κ1-N-2-pyridinol complex exhibited good catalytic activity in the dehydrogenation of various secondary alcohols with TONs up to 700,30,31 and in the reversible (de)hydrogenation of 1,2,3,4-tetrahydroquinolines.32 The related bi(κ-N-2-pyridonate) iridium(III) complex 2 by Fujita and Yamaguchi showed good conversion in the hydrogenation of CO2 to formate33 and methanol34 as well as dehydrogenation of formic acid.35 The planar N,N,N-pincer type bis(2-hydroxy-6-iminopyridyl)-isoindoline ruthenium complex 3 containing two hydroxypyridine motifs was prepared by Szymczak and employed in the hydroboration of organic nitriles.36 Recently, the Rh-N-heterocyclic carbene complex 4 bearing an anionic 2-pyridonate was shown to effect facile linear alkyne dimerizations.37 Addition of bis(trifluoromethyl)pyridonates) to Pd(OAc)2 leads to high activity of undirected C–H olefination and carboxylation with various arene, heteroarene and alkene substrates (κ1-N ligated complex 5 was structurally characterized).38 Pyridonate metal complexes have been successfully applied to various hydrofunctionalization reactions. The intermediate metal hydride complexes formed by H–E bond cleavage reactions of various hydrogen-bearing substrates such as dihydrogen, main group element hydrides (e.g. silanes, boranes), or hydrocarbons (e.g. alcohols, alkynes, arenes). Cooperativity of the metal center and the pyridonate ligand was found to be critical for high catalytic performance.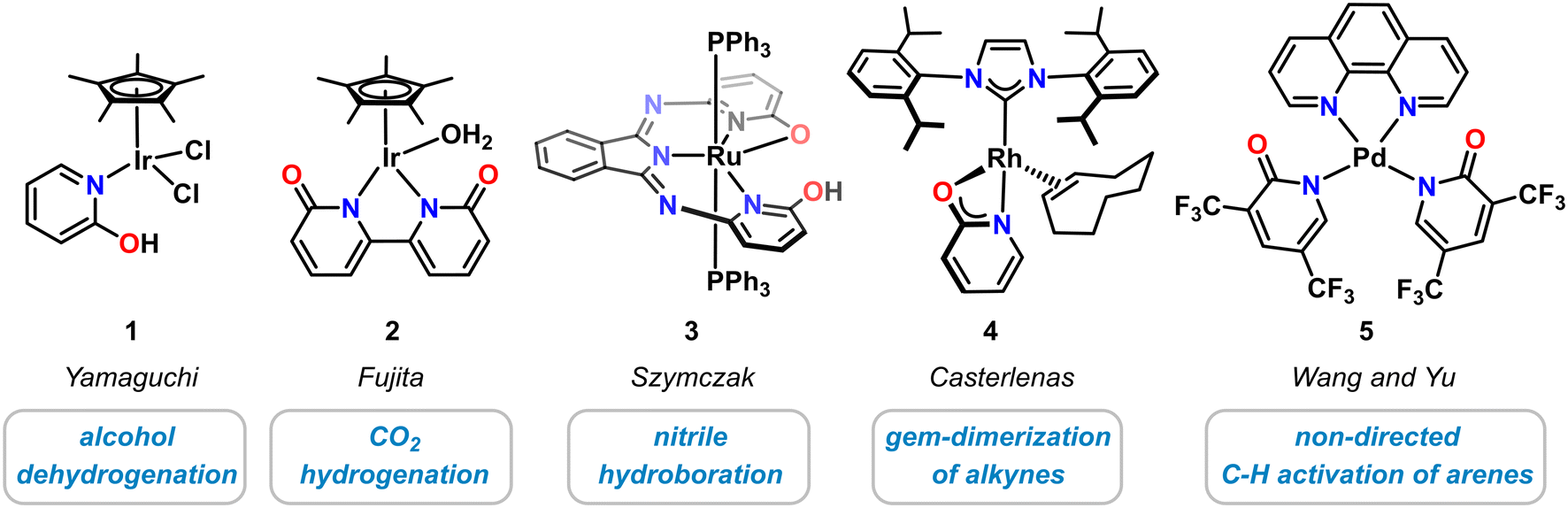 | ||
| Scheme 2 Selected examples of noble metal pyridonate catalysts for hydrofunctionalizations. | ||
3 3d transition metal complexes
The diverse modes of pyridonate coordination have enabled the synthesis of various dinuclear and polynuclear 3d transition metal complexes,13 such as the single molecular magnet 6 (ref. 39) or the dinuclear paddle-wheel complex 7.40 Much fewer examples of mononuclear complexes were reported, such as the cobalt(II) pyridonate catalyst 8 (ref. 41) and the biomimetic hydrogenase model complex 9 (ref. 42) (Scheme 3).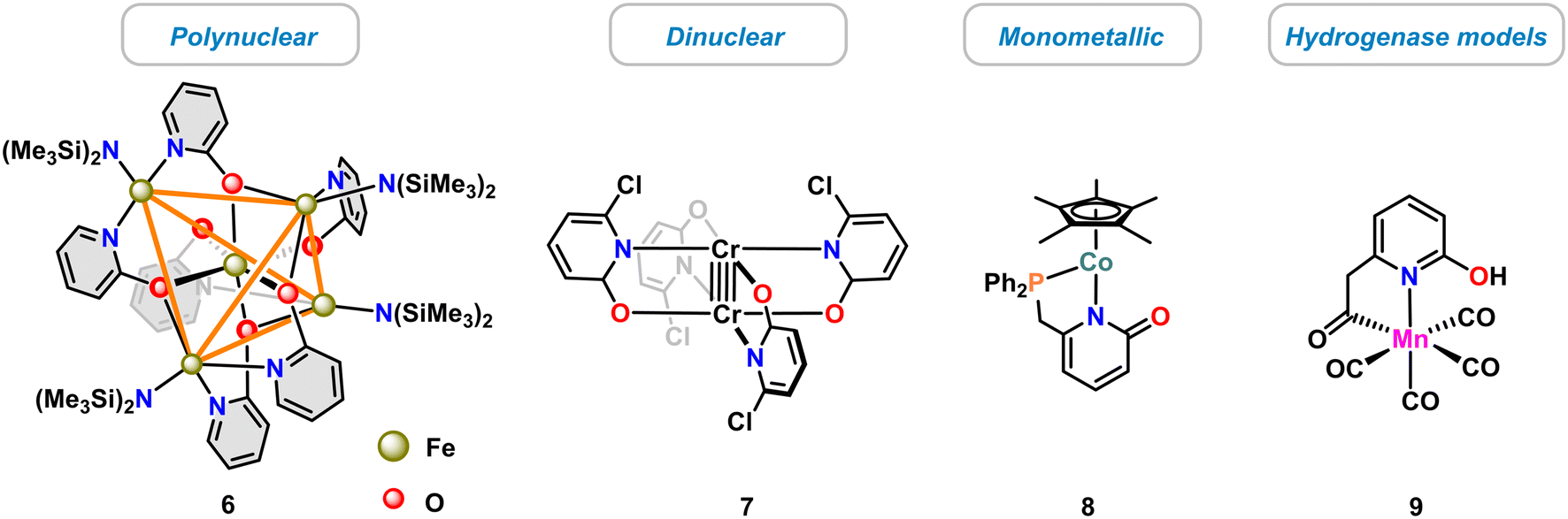 | ||
| Scheme 3 Selected structural classes of 3d transition metal pyridonate complexes. | ||
Applications as catalysts have only recently been recognized. In the following, recent advances of molecular 3d metal pyridonates for catalytic applications will be discussed. Prominent examples are given for metals of the 3d row from Ti to Cu. Prior to that, detailed insight is provided into the structure and activity of the biological role model of many pyridinol/pyridonate catalysts, the [Fe]-hydrogenase.
3.1 The [Fe] hydrogenase
Hydrogenases are a class of metalloenzymes that reversibly catalyze the splitting of H2 and its generation.8 Three types of hydrogenases with 3d transition metal centers are known: [NiFe]-, [FeFe]- and [Fe]-hydrogenases. Unlike the bimetallic [NiFe]- and [FeFe]-hydrogenases, the [Fe]-hydrogenase contains only one redox-innocent metal ion and lacks the prototypical iron–sulfur cluster unit.43 The [Fe]-hydrogenase was isolated from the methanogenic archaea methanobacter marburgensis when grown under nickel-limited conditions.44 The active center of the metalloenzyme was first crystallographically characterized in 2008 by Shima and coworkers,45 and the activated state recently solved at 1.08 Å atomic resolution.46 The active site of the [Fe]-hydrogenase involves an iron-guaninylpyridinol cofactor (FeGP) that is a low-spin octahedral Fe(II) ion ligated by two CO ligands, cysteine, water, and the 6-carbonylmethyl-pyridin-2-ol derivative of a 5-phosphorylated guanosine nucleotide (Scheme 4, top). The pyridinol ligand coordinates to Fe in a bidentate fashion through the terminal acyl function and the pyridine-N, with the pendent OH group being a free Brønsted acid motif that is not coordinated to the metal. The guanosine monophosphate (GMP) is attached at the para-position of the pyridine. The [Fe]-hydrogenase is involved in one step of the catalytic methanogenesis pathway that converts CO2 and H2 to methane in a multi-step process (Scheme 4, bottom). It enables the reversible heterolytic cleavage of H2 to a formal H+/H− pair where the hydride is stereoselectively transferred to the biological C1-building block methenyltetrahydro-methanopterin (CH-H4MPT+) to form the aminal-type methylene-H4MPT (CH2-H4MPT). Further reductive transformations ultimately lead to the formation of methane as the metabolic product in hypoxic conditions.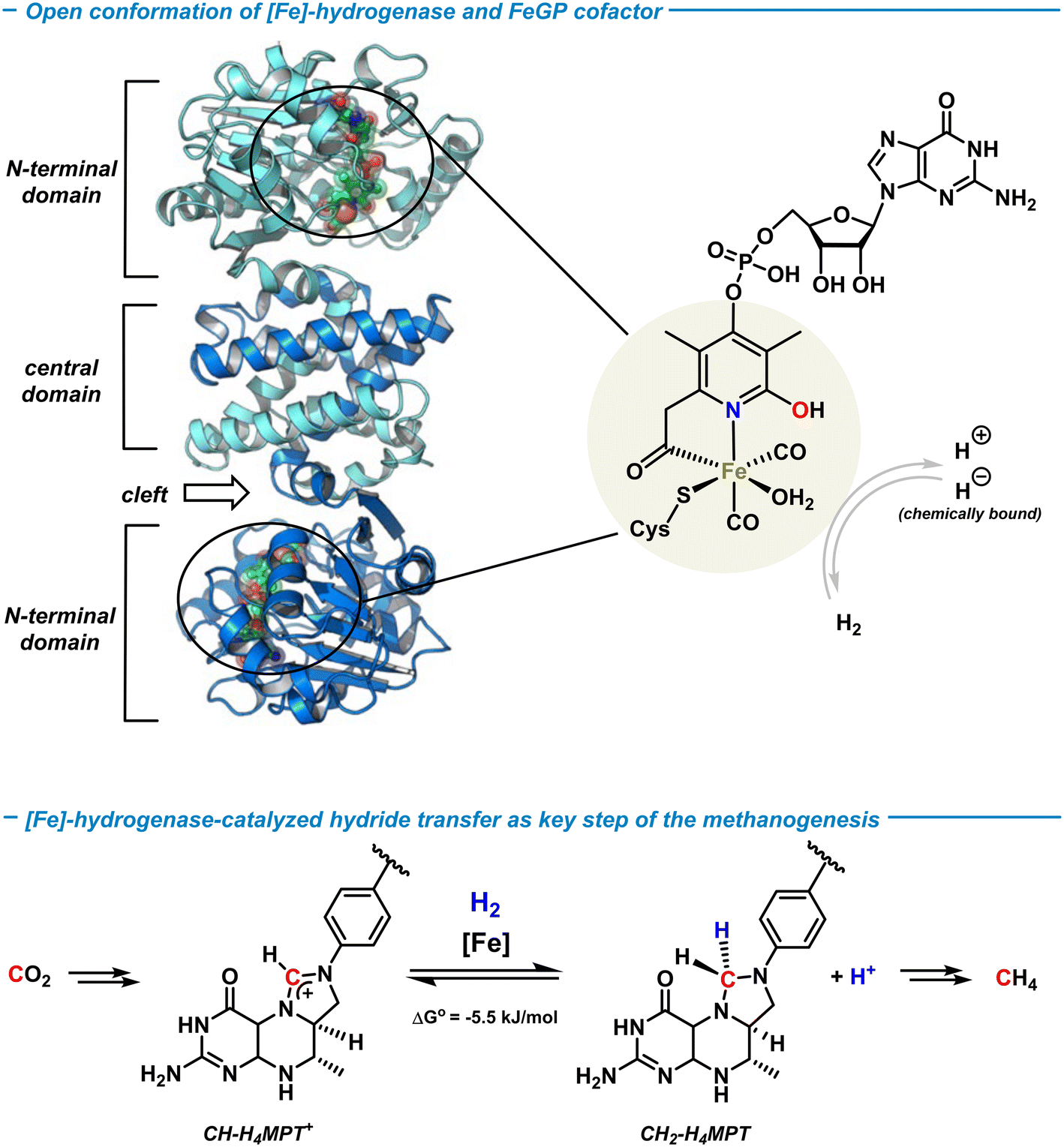 | ||
| Scheme 4 Structure of the [Fe]-hydrogenase metalloenzyme and the FeGP cofactor (top). Catalytic hydrogenation by [Fe]-hydrogenase in the biological methanogenesis (bottom). | ||
The [Fe]-hydrogenase metalloenzyme is composed of two N-terminal peptide domains and one central domain that contains the active cleft accessible for the substrate (Scheme 4, top). The catalytic cleft can adopt open and closed conformations along with structural reorganization of neighboring domains. The conformational change was proposed to be essential for the hydride transfer onto the substrate. Based on crystal structure analyses and theoretical studies, an operating mechanism involving iron-pyridonate cooperativity was suggested (Scheme 5).46 The open form of the metalloenzyme I binds the substrate CH-MPT+ which effects closure of the catalytic cleft and dissociation of a water ligand from the Fe center. Deprotonation of the pyridinol in this penta-coordinate intermediate II affords the active species III that binds H2 diffusing through short protein gas channels. The molecular H2-ligand in IV undergoes heterolytic splitting assisted by the proximal oxy-anionic base of the pyridonate ligand to provide an Fe–H complex coordinated by the pyridinol ligand (V). Hydride transfer onto the electrophilic CH-H4MPT+ substrate results in formation of the aminal derivative CH2-H4MPT, opening of the catalytic cleft, release of CH2-H4MPT, and regeneration of the coordinatively saturated complex I by association of H2O.
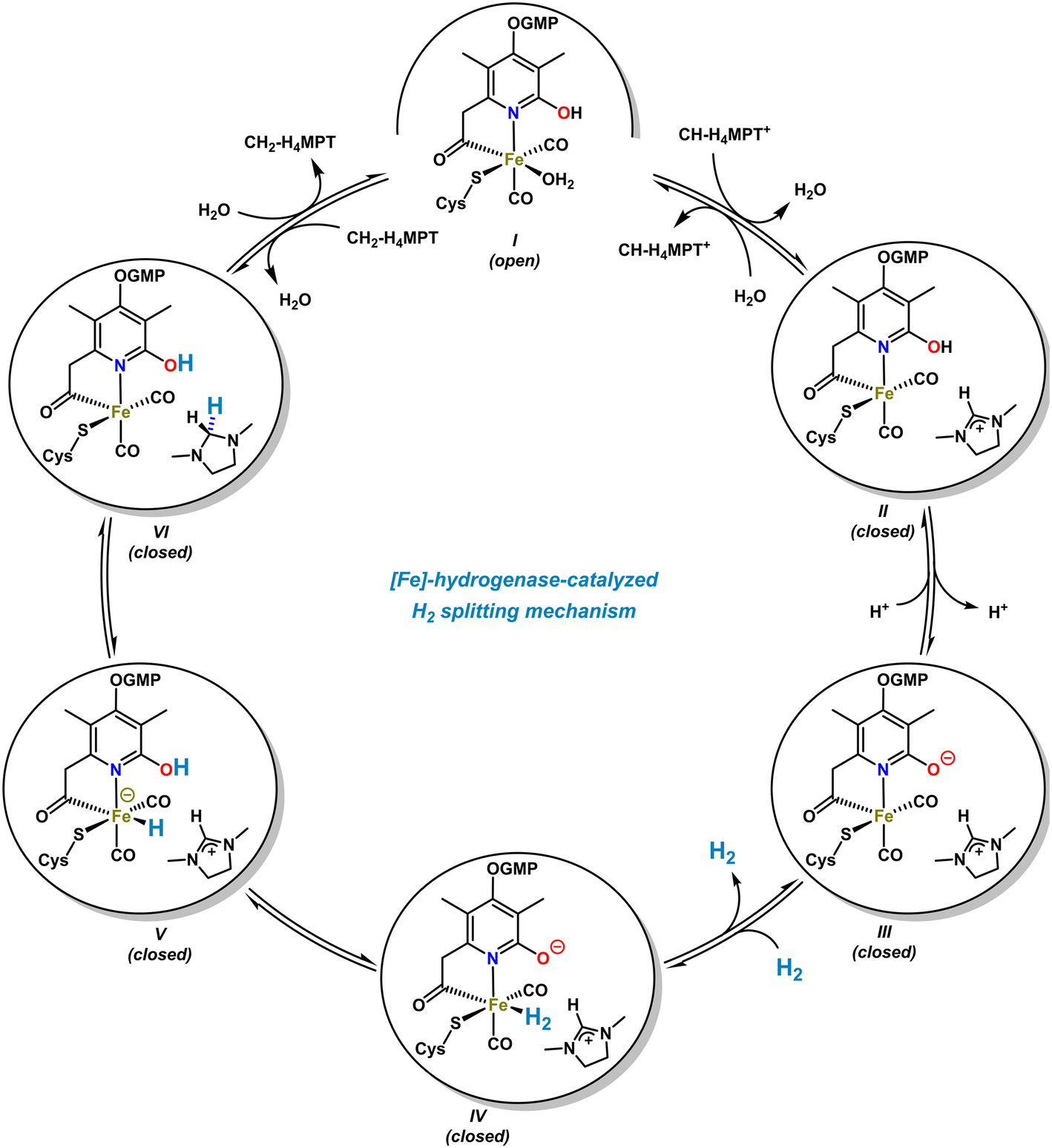 | ||
| Scheme 5 Proposed catalytic cycle of the [Fe]-hydrogenase-catalyzed H2-splitting and stereoselective hydride transfer. | ||
3.2 Synthetic models of the [Fe]-hydrogenase
The relative simplicity of the molecular structure of the [Fe]-hydrogenase active site, its capability of operating under low H2 pressure in aqueous media, and the cooperativity between metal center and ligand periphery have attracted great attention to the design of various artificial analogues. Several structural and functional biomimetic complexes were prepared over the past decades. The studies of applications to catalytic H2 activations and hydrogenation reactions were greatly inspired by the metalloenzymatic role model.9 Among the rich collection of hydrogenase models, many emulated the specific fac-C,N,S coordination environment using a wide variety of ligand architectures.47–49 Synthetic models containing pyridonate/pyridinol ligand motifs were developed by the groups of Hu and Song, respectively (Scheme 6, top). The first [Fe]-hydrogenase model complex containing a 2-pyridone motif was reported by Hu and coworkers.50 Complex 10, obtained from the reaction of sodium 6-methyl-2-pyridonate with Fe(CO)3(PPh3)I2, contains a pseudo-octahedral Fe(II) with κ2-N,O coordination of the 6-methyl-pyridonate ligand (Scheme 6, top). Substitution of the iodide with bulky thiolates resulted in rapid decomposition at room temperature in solution.51 The first synthetic model complexes bearing a truncated version of the natural 6-carbonylmethylpyridin-2-ol ligand were reported in 2014 by Hu and coworkers.52 Treatment of lithiated 2-t-butoxy-6-methylpyridine with Fe(CO)5 installed the acetyliron motif; subsequent iodination (to 11a), deprotection (to 11b), and thiolation afforded the penta-coordinate biomimetic complexes 12a–e (Scheme 6, middle). Despite being the structurally most related models of the iron-guaninyl-pyridinol cofactor (FeGP) site, extensive studies were halted by their low stability in solution. Incorporation of the 2-pyridinol-containing complex 11b into the [Fe]-hydrogenase apoenzyme (lacking the natural FeGP cofactor) imparted the reconstructed semi-synthetic hydrogenase with enzymatic activity, whereas its O-methylated analogue was inactive.53 These findings supported the notion of the essential role of the 2-hydroxy function in the second coordination sphere for enzymatic H2 heterolysis. In 2014, Song et al. prepared the related hydrogenase model complex 14 containing a biomimetic hydroxypyridyl acetyl ligand and an S,N-chelating co-ligand.54 Despite the significant structural resemblance to the native FeGP coordination sphere, none of the synthetic model complexes 10–13 were active towards H2 cleavage or catalytic hydrogenation. The lack of activity of 11b and 12 was rationalized by their rapid decomposition in the presence of base. For higher stability, the diphosphine ligand Et2P-CH2N(Me)CH2-PEt2 bearing an internal amine base was incorporated into complex 14.55 In contrast to the previous models, 14 displayed good activity in the H2 heterolysis, as documented by H/D scrambling and HD formation. However, 14 was poorly active in the hydrogenation of 4-fluorobenzaldehyde (50 bar, 4 h, r.t., TON ∼ 1) and rapidly decomposed (Scheme 6, bottom).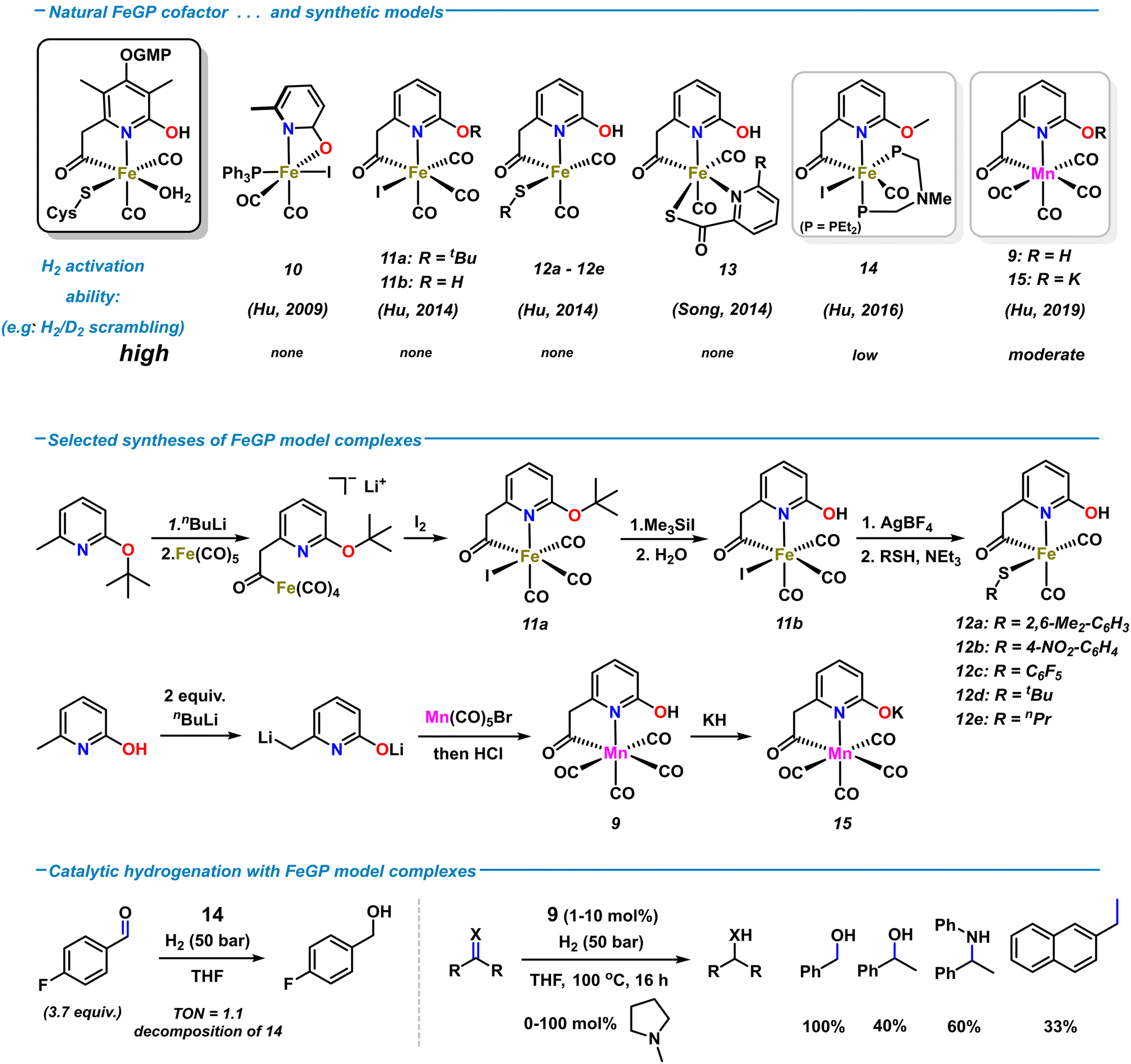 | ||
| Scheme 6 Selected examples of synthetic [Fe]-hydrogenase models reported by Hu and Song (top). Syntheses of model complexes 9, 11, 12, 15 (middle). Applications of hydrogenase model complexes to catalytic hydrogenations (bottom). | ||
Metal substitution in synthetic FeIIGP models was investigated with the isoelectronic Mn(I) ion. The first manganese complex 9 was obtained by reaction of 6-methyl-2-pyridone with 2 equiv. nBuLi and subsequent addition to Mn(CO)5Br (Scheme 6, middle).42 Complex 9 crystallized in octahedral geometry with the C,N-chelating 2-hydroxypyridyl-6-methylcarbonyl ligand and four CO ligands. Mixtures of 9 and amine bases and the isolated deprotonated metallate 15 were competent in H2 heterolysis, respectively, again documenting the critical role of the proximal oxy function of the ligand. 9 and 15 were also active as hydrogenation catalyst for some carbonyl derivatives and much less so for alkenes (1 mol% 9, 20 mol% 2-methylpyrrolidine, 50 bar H2, THF, 80 °C; see Scheme 6, bottom). Although the catalytic activity of the synthetic Mn models of FeGP were far inferior to state-of-the-art Mn(I) hydrogenation catalysts,569 (with base) displayed the highest activities so far as functional mimetics of the [Fe]-hydrogenase. Reconstruction of the [Fe]-hydrogenase apoenzyme with 9 instead of the native cofactor resulted in a semi-synthetic [Mn]-hydrogenase that achieved far higher molar activities than the best semi-synthetic Fe analogues.42
In conclusion, the recent studies of FeGP structure elucidation and the preparation of synthetic models of FeGP showed that the hydroxypyridine ligand is crucial to metal ion coordination and heterolytic H2 activation by metal–ligand cooperativity (MLC). The early examples of synthetic Fe(II) models demonstrated high levels of structural resemblance to the native cofactor of the [Fe]-hydrogenase but exhibited no catalytic hydrogenation activity.
3.3 Titanium
Among other 1,3-bifunctional ligands with N,O- and N,N-chelating behaviour, 2-pyridonates were utilized by Schafer and coworkers for the stabilization of electrophilic early-transition metal complexes. The distorted octahedral titanium κ2-pyridonate complex 16 was prepared from Ti(NMe2)4/2 equiv. 3-phenyl-2-pyridone and applied to the intramolecular hydroaminoalkylation (HAA) of primary aminoalkenes (Scheme 7).57 High chemoselectivity in favour of the hydroaminoalkylation (HAA) over the hydroamination (HA) pathway was observed. The authors proposed the formation of a dinuclear μ-imidotitanium intermediate which undergoes CH-activation in α-position to the amine and subsequent alkene insertion.57,58 On the contrary, the terminal imidotitanium complex is believed to undergo intermolecular [2 + 2] cycloaddition resulting in an overall hydroamination process. The κ2-coordination mode of the pyridonate ligand most likely favours the active μ-imido species thus determining hydroaminoalkylation selectivity. Similar titanium pyridonate complexes were successfully applied as initiators in ring-opening polymerization reactions.59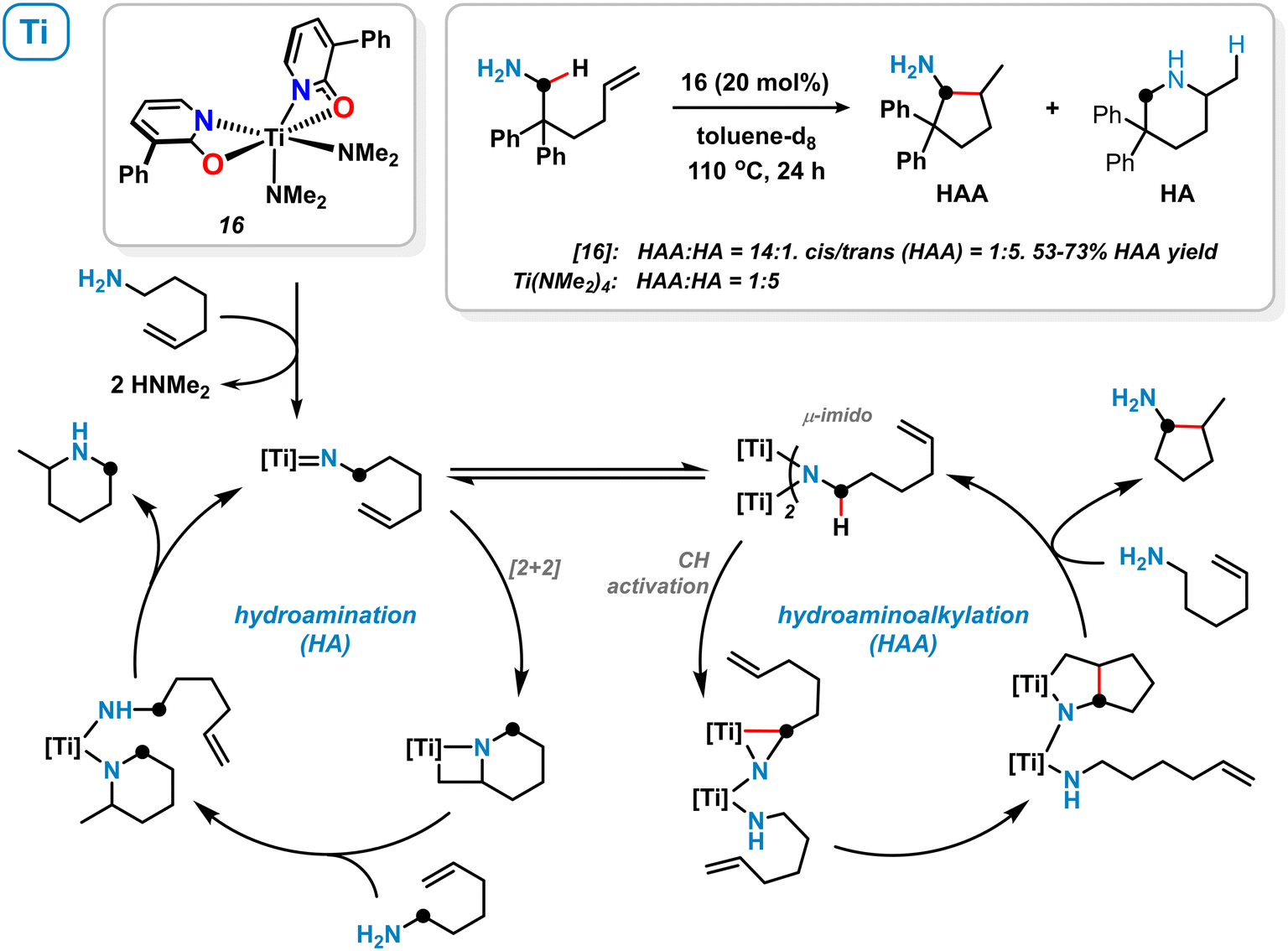 | ||
| Scheme 7 Titanium pyridonate complex 16 and its catalytic activity in the hydroaminoalkylation (HAA) and hydroamination (HA). | ||
3.4 Vanadium
Very recently, Schafer et al. also reported the synthesis and reactivity of vanadium(III) pyridonates.60,61 Complex 17 could be accessed from (THF)VIII(Mes)3 (ref. 61) and VIV(NMe2)4 (ref. 60) by reaction with 6-methylpyridone (Scheme 8, top). The synthesis from the VIV precursor involves protolytic ligand exchange and reduction with in situ generated HNMe2 acting as reductant. Complex 17 is a C2-symmetric dimer adopting κ2-N,O and μ2-O binding modes of the pyridonate ligands. Strong donors such as 4-dimethylaminopyridine (DMAP) afford monomeric complexes.60 Complex 17 was employed as catalysts in the reductive deoxygenative coupling of benzyl alcohols to hydrocarbons. Key catalyst intermediates were isolated with diphenylmethanol as model substrate and a reaction mechanism involving V(III) and V(IV) states was proposed (Scheme 8, bottom). Ligand-assisted deprotonation of the substrate and subsequent homolytic C–O cleavage affords an oxovanadium(IV) complex. The resultant benzhydryl radical undergoes homo-dimerization. Another substrate molecule acts as sacrificial reductant to give benzophenone and the catalytically active V(III) complex 17.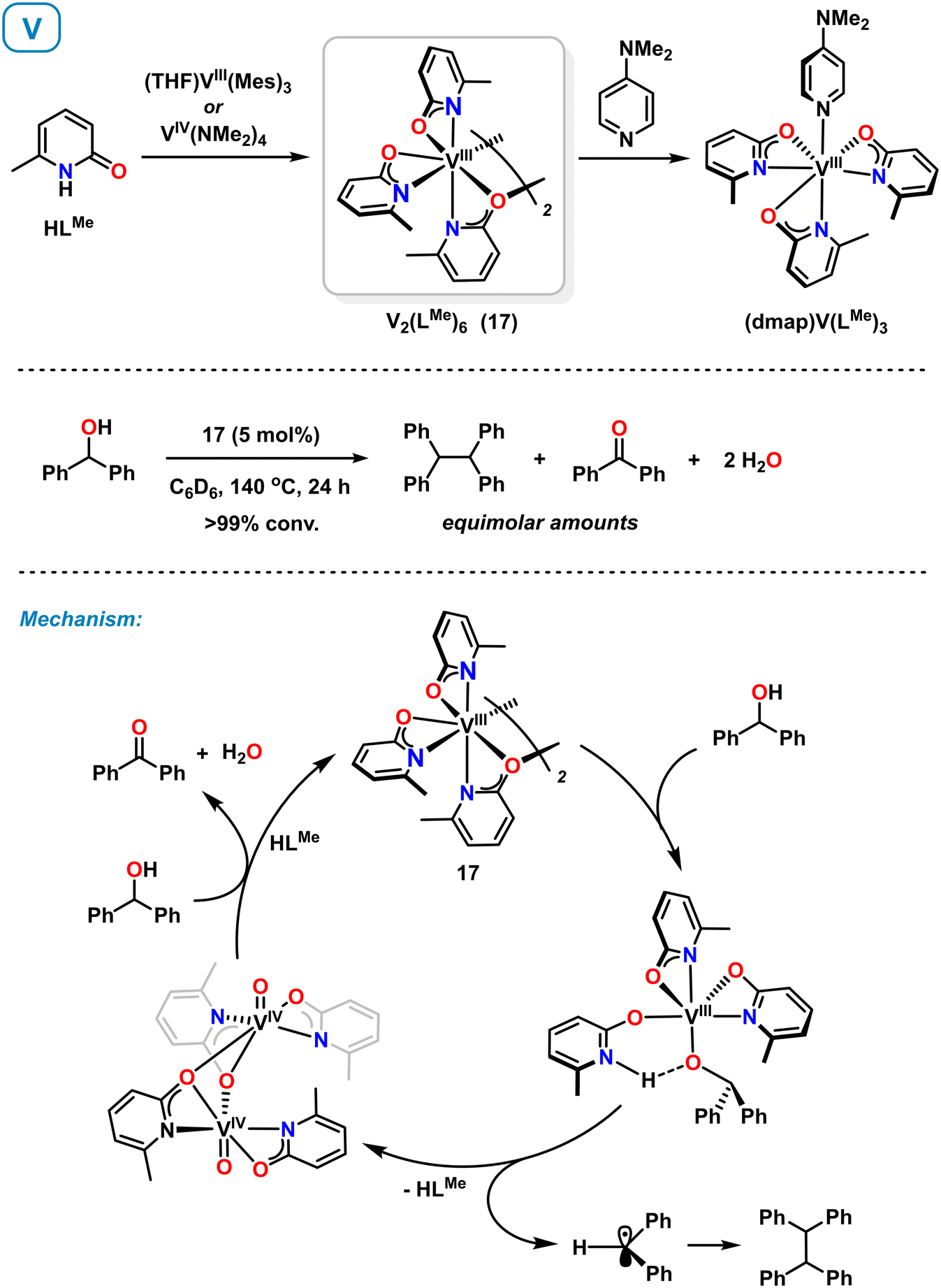 | ||
| Scheme 8 Synthesis of vanadium pyridonates (top). Mechanism of the catalytic reductive dimerization of alcohols with isolated intermediates shown (bottom). | ||
3.5 Manganese
A manganese-containing mimic of the [Fe]-hydrogenase active site was reported that exhibited moderate activity in hydrogen activation (see complex 9 in Scheme 6). A related complex bearing an extended heteroaromatic ligand backbone was prepared by Khusnutdinova et al. in 2017 (ref. 62) (Scheme 9, top). The Mn(I) tricarbonyl complex 18 with the planar tetra-donor functionalized ligand 6,6′-dihydroxy-2,2′-bipyridine showed good catalytic activity in the hydrogenation of carbon dioxide at rather mild conditions.62 At 0.15 mol% catalyst loading, 65 °C and 60 bar CO2/H2, the clean formation of formate was observed in the presence of DBU (TON > 6000). With dimethylamine, reduction to the formamide was achieved using the same catalyst. Shortly after, the same group reported the catalytic transfer hydrogenation of aldehydes, ketones, imines and heterocycles with pre-catalyst 18 and co-catalytic amounts of KOtBu.63 Both catalytic reduction methods were postulated to involve metal–ligand cooperativity of the manganese–hydroxypyridine motif within 18. Consistently, ligands bearing H or OMe in the ortho-position gave no activity in the CO2 hydrogenation and very poor performance in the carbonyl transfer hydrogenations. Further studies by DFT calculations64 and deuterium labelling experiments63 led to the postulation of a catalytic reaction mechanism that proceeds through deprotonation of the 2-OH group by an external base (DBU or KOtBu), bromide dissociation from the catalyst, followed by heterolytic H2 (or iPrOH) splitting across the 1,4-MnNCO motif. The resultant hydro-catalyst species may undergo facile reduction of CX bond substrates. A closely related Mn(I) bipyridine complex was reported by Xia and coworkers (Scheme 9, middle).65 One of the peripheral OH functions was replaced with a pyridine motif to give an unsymmetrical ligand containing one 2-pyridinol moiety (19). Complex 19 was active in the transfer hydrogenation of carbonyl compounds in the presence of base (0.5 mol% catalyst, 20 mol% KOtBu, 85 °C, 24 h, with 2-propanol) and in the acceptorless dehydrogenative condensation of alcohols in the presence of amines. A very related synthesis of pyrimidines and quinolines was realized under similar conditions with a benzimidazole-tethered pyridonate (20) complex by Kundu et al. (Scheme 9, middle).66
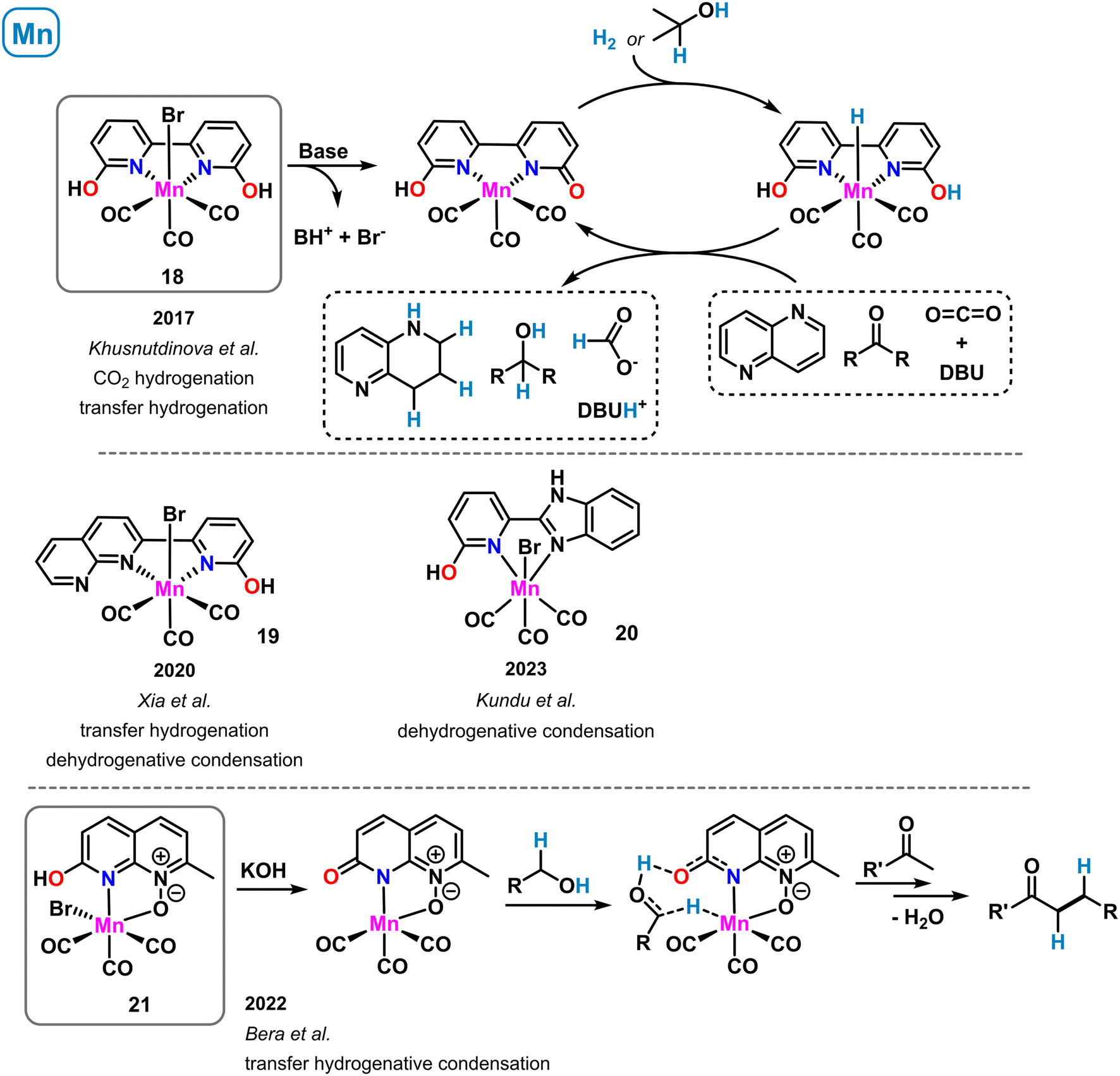 | ||
| Scheme 9 Structurally related manganese complexes in catalytic dehydrogenations and transfer hydrogenations operating via metal–ligand cooperativity. | ||
The concept of proton-responsive pyridonate manganese complexes was also applied to the alkylation of ketones via a hydrogen-borrowing mechanism by Bera et al.67 A manganese carbonyl complex 21 bearing a 1,8-naphthyridine-N-oxide ligand showed good activity in the synthesis of arylethylketones from benzyl alcohols and methylketones via a sequence of benzyl alcohol oxidation to benzaldehyde, condensation with the ketone, and formal 1,4-hydrogenation by the H2-loaded catalyst species, a hydroxypyridine manganese hydride (Scheme 9, bottom).
3.6 Cobalt
A series of monomeric cobalt pyridonate was described by Winpenny in 1996.68 For example, the cobalt(II) complex 22 bearing two κ2-N,O bound pyridonate ligands was prepared from the reaction of Co(OAc)2 with 2 equiv. sodium 6-methyl-2-pyridonate in the presence of phenanthroline (Scheme 10, top).68 The heteroleptic complexes bearing N,N-chelating ligands and pyridonates were characterized by single crystal analyses and paramagnetic NMR spectroscopy. The latter technique documented that the NMR resonances of the diimine derivatives correlated well with the basicities of the N-bound pyridonates. The purely oxygen-coordinated cobalt(II) complex 23 with planar geometry of four H-bridged 2-hydroxypyridine ligands was reported by Chen et al.69 Crystal structure analysis of this cationic complex exhibited a 3D network with channels occupied by the perchlorate anions. The cobalt-analogue (24) of the earlier reported, water-soluble pentamethylcyclopentadienyl-iridium(III) complex 2 containing the 6,6′-dihydroxy-2,2′-bipyridine ligand was reported by Fujita and coworkers.70 Complex 24 was active in the hydrogenation of CO2, although with very low activity (TON 1.3) which was attributed to its thermal instability under reaction conditions. A DFT study documented that the ligand-assisted heterolytic H2 cleavage was the rate-limiting step.71 The decrease of catalytic activity of Ir > Rh > Co was rationalized by the lower back-donation in the intermediate metal–(H2) complex. The calculations also indicated the critical role of the hydro-form of ligand in the hydride transfer to lowering the LUMO of CO2.71 The half-sandwich Cp*Co(II) complex 8 bearing the phosphinomethyl pyridonate ligand was introduced by Wang and coworkers.41 The bidentate P,N-ligand binds the metal center in a five-membered chelate while the 2-carbonyl function remains free. This structural motif was utilized in an interesting sequential catalysis that converts terminal alkynes to E,Z-dienes with highly selective syn-insertion in both steps (Scheme 10, center). With 3 mol% 8, dimerization of terminal alkynes delivers (E)-enynes as intermediate products that – upon addition of ammonia-borane – are reduced to (E,Z)-dienes. Based on DFT calculation and mechanistic experiments, two sequential catalytic cycles were proposed: the dimerization reaction is believed to proceed through pyridonate-assisted metalation deprotonation to afford an alkynylcobalt–pyridone complex, that was isolated and structurally characterized. Subsequent 1,2-cis-carbometallation of a second molecule alkyne gives an (E)-vinylcobalt intermediate and protolysis by the pendant pyridone releases the (E)-enyne. The second cycle involves BH3-coordination to the Lewis basic 2-oxy function of the pyridonate and metal–ligand cooperative B–H bond cleavage. The resultant Co(III)–H may react with the (E)-1,3-enyne via 1,2-cis-hydrometallation, and proton transfer from the coordinated ammonia provides the (E,Z)-dienes (Scheme 10, bottom). | ||
| Scheme 10 Selected examples of molecular 2-pyridonatocobalt complexes (top). Stereoselective catalytic dimerization/semi-hydrogenation with catalyst 8 (bottom). | ||
3.7 Nickel
The first example of a mononuclear nickel pyridonate complex was structurally characterized by López et al. in 1998 (Scheme 11, top).72 The distorted square-pyramidal cation 25, containing a κ-N,O-bound 2-pyridonate and the tripodal co-ligand 2,4,4,9-tetramethyl-1,5,9-triazacyclododec-1-ene, was characterized by 1H NMR spectroscopy (paramagnetic shifts, in acetone-d6), and pyridonate resonances were assigned by NOESY. Jones et al. reported the synthesis of the related quinolinate complex 26 supported by tris(3,5-dimethylpyrazolylborate) and documented its catalytic activity in the acceptorless dehydrogenation of alcohols.73 The key role of the 2-OH function for catalysis was proven and a mechanism involving metal–ligand cooperativity of the concerted alcohol dehydrogenation, and the intermediacy of a nickel hydride proposed (Scheme 11, center).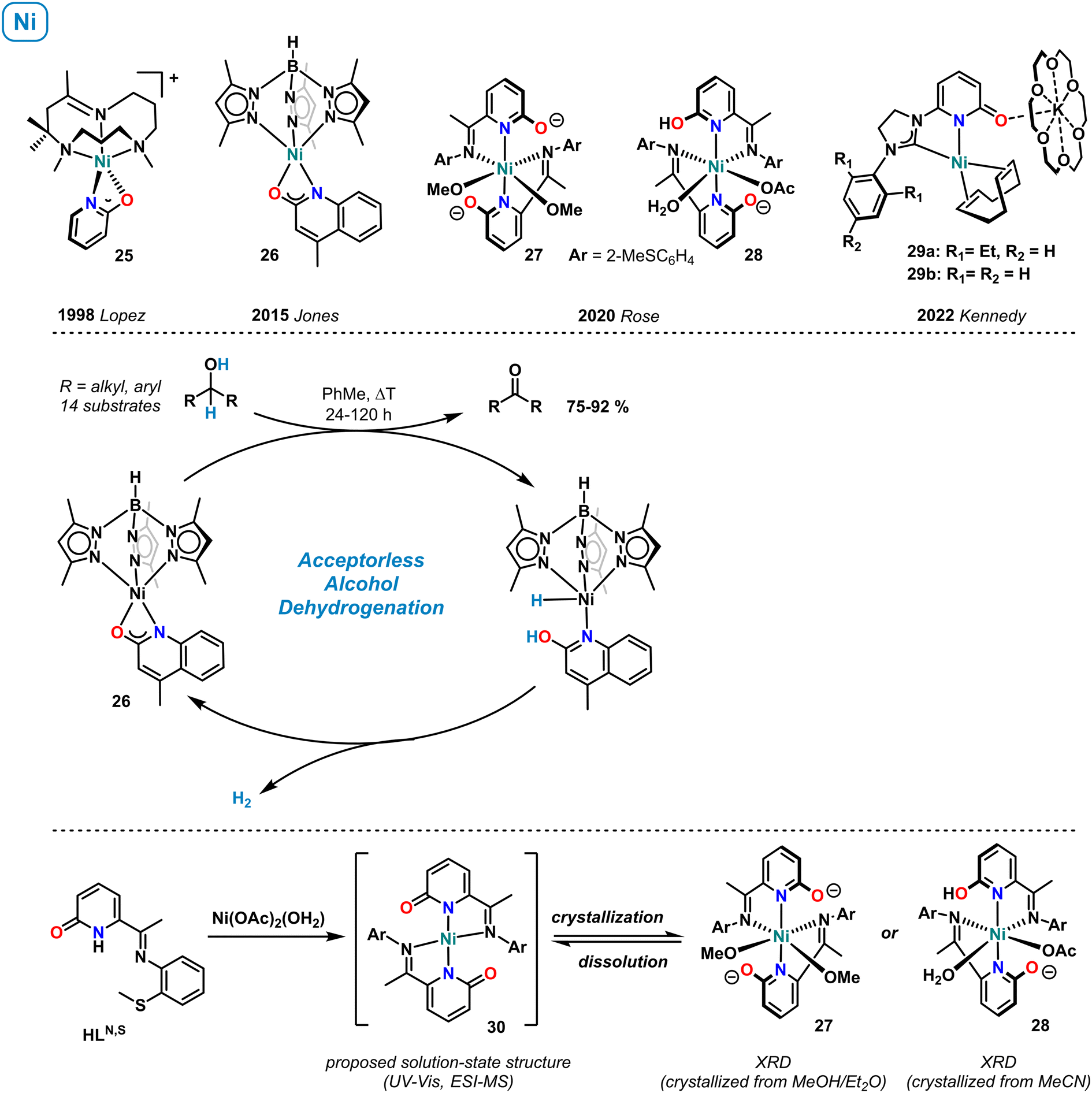 | ||
| Scheme 11 Selected examples of molecular nickel pyridonate complexes (top). Acceptorless alcohol dehydrogenation catalyzed by 26 (center). Solution and solid-state structures of nickel pyridonates 27, 28, and 30 (bottom). | ||
In 2020, Rose and coworkers prepared pyridonate complexes bearing an additional imine function in 6-position. Mononuclear and dinuclear structures were obtained.74 The octahedral nickel complexes 27 (crystallization by slow diffusion of Et2O into MeOH) and 28 (crystallization by slow evaporation of MeCN) showed chelating coordination of the N,N-motif, while the oxy function remained free. Both pyridonates in complex 27 and one pyridonate in 28 are formally anionic and exhibit relatively long C–O distances which are in full accord with the rare pyridinolate (2-O−) resonance form. Interestingly, UV-vis and high-resolution mass spectra suggested that the predominant species in solution is the square-planar complex 30. This work nicely illustrates the diverse coordination properties of pyridonate complexes, here in solution and solid-state. In 2022, the group of Kennedy reported anionic nickel(0) complexes such as 29a and 29b bearing C,N-bidentate carbene-pyridone ligands.75 These complexes were prepared from Ni(COD)2 and the NHC-pyridonate in the presence of 18-crown-6. The K(18-c-6) cation formed a contact ion pair through the 2-oxy function of the pyridonate. Complex 29a showed high catalytic activity in the Markovnikov-selective hydroboration of styrenes with pinacolborane (HBPin). Recently, complex 29b was shown to catalyzed the double hydroboration of nitriles to amines (HBpin, r.t.).76
3.8 Copper
Upon simple mixing of equimolar amounts of Cu(II) salts and the bipyridine derivative 6,6′-dihydroxy-2,2′-bipyridine (H2L) in basic water, bipyridonate copper(II) complexes can be prepared such as the monomeric complex 31 (pH > 12), coordination polymers of the formula [Cu(HL)(μ2-OH)] (pH ∼ 11), and related species. Such in situ generated copper complexes enabled effective electrocatalytic oxygen evolution under low applied overpotential of only 640 mV. The proposed catalytically active intermediate 31 involves a hydroxypyridine motif that actively facilitates the PCET steps around the Cu center, similar to the tyrosinyl radical in the photosystem II (Scheme 12, top right).77 A detailed DFT study by Li, Chen and coworkers was performed on the mechanism of this homogeneous water oxidation catalysis (WOC).78 Two equally rapid pathways were described, which both operate via metal–ligand cooperativity: the co-substrate mechanism forms an O–O bond between the Cu–O· terminal oxyl radical and the adjacent oxyanionic ligand. In the hydroxyl coupling mechanism, O–O bond formation proceeds by reaction of a coordinated Cu–OH and a second-coordination sphere OH group. Metal–ligand cooperativity of this catalytic water oxidation was proven. Both, the copper ion and the hydroxypyridine ligand engage in water coordination/H-bonding and the 1e-redox events (overall 2e-redox). A more detailed study of O–O bond formation using first-principles metadynamics simulations was published recently by Mallik and co-workers.79 An alternative mechanism via Cu-complex decomposition and formation of a Cu thin film on the electrode was also proposed.80 Papish and coworkers showed that reaction of CuSO4 with 6,6′-dihydroxy-2,2′-bipyridine in ethylene glycol led to the formation of the well-defined Cu[N]4 complex 32a that occurs in different protonation states depending on pH (Scheme 12, top).81 Complexes 32 displayed moderate activity in the electrocatalytic water oxidation at even lower overpotential (447 mV) than the related postulated 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 adduct 31. The presence of the ligand–OH groups were shown to be essential for catalytic activity. Highest activity was recorded in basic media (pH > 12), so that the fully deprotonated aqua-complex 32c was suggested to be catalytically competent, with further support derived from EPR and 2D-HYSCORE spectra. Moreover, the authors proposed that intramolecular hydrogen bonding between copper-coordinated water and the proximal pyridonate-oxo atoms in 32c (or related species) may engage in the stabilization of key intermediates of the underlying proton-transfer steps of the proton-coupled electron transfer (PCET) mechanism.
:1 adduct 31. The presence of the ligand–OH groups were shown to be essential for catalytic activity. Highest activity was recorded in basic media (pH > 12), so that the fully deprotonated aqua-complex 32c was suggested to be catalytically competent, with further support derived from EPR and 2D-HYSCORE spectra. Moreover, the authors proposed that intramolecular hydrogen bonding between copper-coordinated water and the proximal pyridonate-oxo atoms in 32c (or related species) may engage in the stabilization of key intermediates of the underlying proton-transfer steps of the proton-coupled electron transfer (PCET) mechanism.
 | ||
| Scheme 12 Copper complexes featuring the 6,6′-dihydroxy-2,2′-bipyridine ligand for electrocatalytic water oxidation (top). DFT-derived proposal of the O–O bond-forming transition state operating through metal ligand cooperativity (top). Copper(II) fluoride coordination and nitrite reduction with tri(pyridonato)copper complexes (center). Bimetallic methylpyridonate complexes with zwitterionic and σ-bond character of the Cu–Al interaction (bottom). | ||
A series of copper complexes with the tripodal ligand tris(6-hydroxypyrid-2-ylmethyl)amine (H3thpa) were reported by Szymczak and coworkers (Scheme 12, center).82–84 In the cationic copper(II) complex 33, metal-bound fluoride is stabilized by hydrogen-bonding interactions with the pendent OH group of the 2-pyridinols.82 Upon one electron reduction, the Cu–F bond is cleaved and adduct 34 formed, where the dissociated fluoride anion is exclusively coordinated in the second coordination sphere by H-bonding to the hydroxypyridine ligands.83 This complex underwent stoichiometric reduction of the nitrite Ph3Si(ONO) to NO gas and the Cu(II) species 35.84
A new type of dinuclear CuAl complexes bearing bridging pyridonate ligands were prepared from Me3Al and Cu(I) precursors. The resultant heterobimetallic μ2-κ-N-Cu,κ-O-Al pyridonates were studied for the electronic nature of the Cu–Al bond that could adopt 2c1e/2c2e σ-bond character (36) as well as a non-bonding zwitterionic state (37).85
4 Summary and conclusions
Despite its molecular simplicity, the 2-pyridon(at)e ligand platform displays a rich coordination chemistry in structurally diverse polynuclear clusters, polymers (μn coordination) or mononuclear metal complexes (κ1, κ2 coordination). These structural modes can be combined with various modes of reactivity, such as hemilability, metal–ligand cooperativity, redox activity, and proton responsivity. The metal–pyridinol motif has a natural role model in the active site of the [Fe]-hydrogenase metalloenzyme. These conditions render pyridonates a highly versatile ligand class for the development of novel metal complexes with catalytic applications in the context of small-molecule activations.To date, most examples of catalytically active pyridonates are noble metal complexes (Ir, Ru, Pd). Only recently have 3d transition metal pyridonates been studied toward their catalytic applications to molecular transformations. Early transition metal pyridonates (Ti, V) showed catalytic activity in polar carbon–nitrogen (hydroaminoalkylation) and polar carbon–carbon (deoxygenative coupling) forming reactions as well as polymerizations. Interestingly, no literature precedents of mononuclear chromium pyridonates are known to date. The later 3d transition metal pyridonates (Mn–Cu) were shown to display catalytic activities in several reductive transformations including hydrogenation, hydrofunctionalization, and isomerization. Copper complexes were mostly studied for their electrocatalytic water oxidation abilities. Several models of the [Fe]-hydrogenase were reported based on Fe and Mn complexes bearing pyridonate derivatives.
In contrast to many conventional ligands used in industry and academia, (e.g. phosphines, carbenes), pyridonates do not contain notoriously expensive, rare or redox-sensitive moieties so that broad variations and scientific explorations could entail. The general 2-hydroxypyridine platform allows for highly selective manipulations by the introduction of further substituents. Common strategies include the incorporation of additional donor groups into the 6-position of the pyridone and the fusion of two pyridonate fragments into 6,6′-bipyridone scaffolds. The availability of additional chelation motifs favors mononuclear complexes and induces κ1-N bonding motif of the pyridonate and a free oxy function that may engage in substrate activation, proton response, or redox. Pyridonates in κ1-N bonding mode resemble the active center of the [Fe]-hydrogenase.
The toolbox of 3d metal pyridonate is only half-full. The recent literature reports on 3d metal pyridonate catalysts documented the key role of metal–ligand cooperativity, hemilability, and proton-response to facilitate catalytic transformations. Further applications of these ligand properties in the exploration of new chemical space are easily foreseen and will have especially strong impacts on ionic and polar reaction mechanisms in aqueous or protic media, such as (transfer)hydrogenations, hydrofunctionalizations, redox reactions, electrocatalysis, hydrogen borrowing processes, water-splitting, and small-molecule activations (e.g. CO2, O2, H2O, ROH, NH3, RNH2, N2, R2BH, etc.). Despite the recent progress, several challenges reside in this area of research. The molecular design and effective application of 3d-transition metal catalysts will certainly require further insight into the solution structures and their dynamic interconversion under reaction conditions by rapid spectroscopic techniques, such as (paramagnetic NMR or solution-phase X-ray absorption methods). Beyond the few biomimetic complexes of the [Fe]-hydrogenase active site, their catalytic activities are mostly poor. The application of chiral ligands is still in its infancy; the facile incorporation of proximal N-, O-, P-, or C-based donor functions into the pyridonate platform offers a rapid entry into chiral ligand design for asymmetric catalytic applications. Apart from the handful electrocatalytic reactions of copper(II) complexes, the ability of 3d metal pyridonates to act as electron reservoirs has not been effectively tapped. Combinations of redox-active pyridonate ligands with redox-active 3d transition metal ions hold great potential for the utilization of metal–ligand cooperativity in challenging redox processes.
Conflicts of interest
There are no conflicts to declare.References
- R. J. Lundgren and M. Stradiotto, Ligand Design in Metal Chemistry: Reactivity and Catalysis, 2016, pp. 1–14 Search PubMed.
- E. C. Constable and C. E. Housecroft, Chem. Soc. Rev., 2013, 42, 1429–1439 RSC.
- R. H. Cox and A. A. Bothner-By, J. Phys. Chem., 1969, 73, 2465–2468 CrossRef CAS.
- J. Frank and A. R. Katritzky, J. Chem. Soc., Perkin Trans. 2, 1976, 1428–1431 RSC.
- M. J. Cook, A. R. Katritzky, L. G. Hepler and T. Matsui, Tetrahedron Lett., 1976, 17, 2685–2688 CrossRef.
- F. G. Bordwell, Acc. Chem. Res., 1988, 21, 456–463 CrossRef CAS.
- D. B. Burks, M. Vasiliu, D. A. Dixon and E. T. Papish, J. Phys. Chem. A, 2018, 122(8), 2221–2231 CrossRef CAS PubMed.
- C. Wang, Z. Lai, G. Huang and H.-J. Pan, Chem. – Eur. J., 2022, 28, e202201499 CrossRef CAS PubMed.
- C. M. Moore, E. W. Dahl and N. K. Szymczak, Curr. Opin. Chem. Biol., 2015, 25, 9–17 CrossRef CAS PubMed.
- M. J. Rose, [Fe]-Hydrogenase (Hmd): Insights From Enzyme Structure, Spectroscopy and Synthetic Models, in Comprehensive Coordination Chemistry III, Elsevier, 2021, pp. 757–771 Search PubMed.
- S. A. Ryken, P. R. Payne and L. L. Schafer, in Ligand Design in Metal Chemistry: Reactivity and Catalysis, 2016, pp. 364–405 Search PubMed.
- M. W. Drover, J. A. Love and L. L. Schafer, Chem. Soc. Rev., 2017, 46, 2913–2940 RSC.
- J. M. Rawson and R. E. P. Winpenny, Coord. Chem. Rev., 1995, 139, 313–374 CrossRef CAS.
- E. P. Kelson and P. P. Phengsy, J. Chem. Soc., Dalton Trans., 2000, 4023–4024 RSC.
- H. W. Yang and B. M. Craven, Acta Crystallogr., Sect. B: Struct. Sci., 1998, 54, 912–920 CrossRef.
- W. Clegg, M. Berry and C. D. Garner, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1980, 36, 3110–3112 CrossRef.
- P. Laruerta, J. Latorre, M. Sanau, F. A. Cotton and W. Schwotzer, Polyhedron, 1988, 7, 1311–1316 CrossRef.
- W. Clegg, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1980, 36, 3112–3114 CrossRef.
- K. C. Shortsleeves, M. M. Turnbull, C. B. Seith, E. N. Tripodakis, F. Xiao, C. P. Landee, L. N. Dawe, D. Garrett, G. D. De Delgado and B. M. Foxman, Polyhedron, 2013, 64, 110–121 CrossRef CAS.
- J. C. Jeffrey and T. B. Rauchfuss, Inorg. Chem., 1979, 18, 2658–2666 CrossRef CAS.
- J. M. Clarkson and L. L. Schafer, Inorg. Chem., 2017, 56, 5553–5566 CrossRef CAS PubMed.
- F. Jiang, M. Achard, T. Roisnel, V. Dorcet and C. Bruneau, Eur. J. Inorg. Chem., 2015, 4312–4317 CrossRef CAS.
- The literature on pyridine cross-coupling reactions is vast. See for example: T. Gallagher, C. Smith, C. Hirschhäuser, G. Malcolm and D. Nasrallah, Synlett, 2014, 25, 1904–1908 CrossRef.
- J. R. Khusnutdinova and D. Milstein, Angew. Chem., Int. Ed., 2015, 54, 12236–12273 CrossRef CAS PubMed.
- M. R. Elsby and R. T. Baker, Chem. Soc. Rev., 2020, 49, 8933–8987 RSC.
- T. P. Gonçalves, I. Dutta and K. W. Huang, Chem. Commun., 2021, 57, 3070–3082 RSC.
- T. Shimbayashi and K. I. Fujita, Catalysts, 2020, 10, 635 CrossRef CAS.
- J. F. Berry and M. D. Roy, 2-Aminopyridine and Related Ligands to Support Metal-Metal Bonded Compounds, in Comprehensive Coordination Chemistry III, ed. E. C. Constable, G. Parkin and L. Que, Jr., Elsevier, Amsterdam, 3rd edn, 2021, vol. 1, pp. 406–427 Search PubMed.
- M. J. Trenerry, C. M. Wallen, T. R. Brown, S. V. Park and J. F. Berry, Nat. Chem., 2021, 13, 1221–1227 CrossRef CAS PubMed.
- K. I. Fujita, N. Tanino and R. Yamaguchi, Org. Lett., 2007, 9, 109–111 CrossRef CAS PubMed.
- A. M. Royer, T. B. Rauchfuss and D. L. Gray, Organometallics, 2010, 29, 6763–6768 CrossRef CAS.
- R. Yamaguchi, C. Ikeda, Y. Takahashi and K. I. Fujita, J. Am. Chem. Soc., 2009, 131, 8410–8412 CrossRef CAS PubMed.
- W. H. Wang, J. F. Hull, J. T. Muckerman, E. Fujita and Y. Himeda, Energy Environ. Sci., 2012, 5, 7923–7926 RSC.
- K. I. Fujita, R. Kawahara, T. Aikawa and R. Yamaguchi, Angew. Chem., Int. Ed., 2015, 54, 9057–9060 CrossRef CAS.
- M. Iguchi, H. Zhong, Y. Himeda and H. Kawanami, Chem. – Eur. J., 2017, 23, 17788–17793 CrossRef CAS.
- J. B. Geri and N. K. Szymczak, J. Am. Chem. Soc., 2015, 137, 12808–12814 CrossRef CAS.
- M. Galiana-Cameo, A. Urriolabeitia, E. Barrenas, V. Passarelli, J. J. Pérez-Torrente, A. Di Giuseppe, V. Polo and R. Castarlenas, ACS Catal., 2021, 11, 7553–7567 CrossRef CAS.
- P. Wang, P. Verma, G. Xia, J. Shi, J. X. Qiao, S. Tao, P. T. W. Cheng, M. A. Poss, M. E. Farmer, K. S. Yeung and J. Q. Yu, Nature, 2017, 551, 489–493 CrossRef CAS PubMed.
- A. Fedulin, S. K. Gupta, I. Rüter, F. Meyer and A. Jacobi von Wangelin, Inorg. Chem., 2022, 61, 6149–6159 CrossRef CAS.
- F. A. Cotton, W. H. Ilsley and W. Kaim, Inorg. Chem., 1980, 19, 1453–1457 CrossRef CAS.
- X. Zhuang, J. Y. Chen, Z. Yang, M. Jia, C. Wu, R. Z. Liao, C. H. Tung and W. Wang, Organometallics, 2019, 38, 3752–3759 CrossRef CAS.
- H. J. Pan, G. Huang, M. D. Wodrich, F. F. Tirani, K. Ataka, S. Shima and X. Hu, Nat. Chem., 2019, 11, 669–675 CrossRef CAS.
- S. Shima and U. Ermler, Eur. J. Inorg. Chem., 2011, 963–972 CrossRef CAS.
- R. K. Thauer, Microbiology, 1998, 144, 2377–2406 CrossRef CAS PubMed.
- S. Shima, O. Pilak, S. Vogt, M. Schick, M. S. Stagni, W. Meyer-Klaucke, E. Warkentin, R. K. Thauer and U. Ermler, Science, 2008, 321, 572–575 CrossRef CAS.
- G. Huang, T. Wagner, M. D. Wodrich, K. Ataka, E. Bill, U. Ermler, X. Hu and S. Shima, Nat. Catal., 2019, 2, 537–543 CrossRef CAS.
- S. A. Kerns and M. J. Rose, Acc. Chem. Res., 2020, 53, 1637–1647 CrossRef CAS PubMed.
- P. J. Turrell, J. A. Wright, J. N. T. Peck, V. S. Oganesyan and C. J. Pickett, Angew. Chem., Int. Ed., 2010, 49, 7508–7511 CrossRef CAS.
- P. J. Turrell, A. D. Hill, S. K. Ibrahim, J. A. Wright and C. J. Pickett, Dalton Trans., 2013, 42, 8140–8146 RSC.
- B. V. Obrist, D. Chen, A. Ahrens, V. Schünemann, R. Scopelliti and X. Hu, Inorg. Chem., 2009, 48, 3514–3516 CrossRef CAS PubMed.
- D. Chen, A. Ahrens-Botzong, V. Schünemann, R. Scopelliti and X. Hu, Inorg. Chem., 2011, 50, 5249–5257 CrossRef CAS.
- B. Hu, D. Chen and X. Hu, Chem. – Eur. J., 2014, 20, 1677–1682 CrossRef CAS PubMed.
- S. Shima, D. Chen, T. Xu, M. D. Wodrich, T. Fujishiro, K. M. Schultz, J. Kahnt, K. Ataka and X. Hu, Nat. Chem., 2015, 7, 995–1002 CrossRef CAS PubMed.
- L. C. Song, F. Q. Hu, G. Y. Zhao, J. W. Zhang and W. W. Zhang, Organometallics, 2014, 33, 6614–6622 CrossRef CAS.
- T. Xu, C. J. M. Yin, M. D. Wodrich, S. Mazza, K. M. Schultz, R. Scopelliti and X. Hu, J. Am. Chem. Soc., 2016, 138, 3270–3273 CrossRef CAS PubMed.
- P. Schlichter and C. Werlé, Synthesis, 2021, 54, 517–534 Search PubMed.
- E. Chong and L. L. Schafer, Org. Lett., 2013, 15, 6002–6005 CrossRef CAS PubMed.
- J. A. Bexrud, P. Eisenberger, D. C. Leitch, P. R. Payne and L. L. Schafer, J. Am. Chem. Soc., 2009, 131, 2116–2118 CrossRef CAS PubMed.
- S. A. Ryken and L. L. Schafer, Acc. Chem. Res., 2015, 48, 2576–2586 CrossRef CAS PubMed.
- S. E. Griffin, O. V. Adamczyk and L. L. Schafer, Dalton Trans., 2022, 51, 14654–14663 RSC.
- S. E. Griffin and L. L. Schafer, Inorg. Chem., 2020, 59, 5256–5260 CrossRef CAS PubMed.
- A. Dubey, L. Nencini, R. R. Fayzullin, C. Nervi and J. R. Khusnutdinova, ACS Catal., 2017, 7, 3864–3868 CrossRef CAS.
- A. Dubey, S. M. W. Rahaman, R. R. Fayzullin and J. R. Khusnutdinova, ChemCatChem, 2019, 11, 3844–3852 CrossRef CAS.
- X. Yan, H. Ge and X. Yang, Catal. Sci. Technol., 2018, 8, 5735–5739 RSC.
- C. Zhang, B. Hu, D. Chen and H. Xia, Organometallics, 2019, 38, 3218–3226 CrossRef CAS.
- S. Nandi, I. Borthakur, K. Ganguli and S. Kundu, Organometallics, 2023, 42, 1793–1802 CrossRef CAS.
- K. Patra, R. A. Laskar, A. Nath and J. K. Bera, Organometallics, 2022, 41, 1836–1846 CrossRef CAS.
- A. J. Blake, L. M. Gilby, S. Parsons, J. M. Rawson, D. Reed, G. A. Solan and R. E. P. Winpenny, J. Chem. Soc., Dalton Trans., 1996, 3575–3581 RSC.
- M. L. Tong, Z. J. Lin, W. Li, S. L. Zheng and X. M. Chen, Cryst. Growth Des., 2002, 2, 443–448 CrossRef CAS.
- Y. M. Badiei, W. H. Wang, J. F. Hull, D. J. Szalda, J. T. Muckerman, Y. Himeda and E. Fujita, Inorg. Chem., 2013, 52, 12576–12586 CrossRef CAS PubMed.
- C. Hou, J. Jiang, S. Zhang, G. Wang, Z. Zhang and Z. Ke, ACS Catal., 2014, 4(9), 2990–2997 CrossRef CAS.
- M. D. Santana, G. García, A. Rufete, G. Sánchez, M. C. R. De Arellano and G. López, Inorg. Chem. Commun., 1998, 1, 267–269 CrossRef CAS.
- S. Chakraborty, P. E. Piszel, W. W. Brennessel and W. D. Jones, Organometallics, 2015, 34, 5203–5206 CrossRef CAS.
- S. T. Goralski, T. A. Manes, S. E. A. Lumsden, V. M. Lynch and M. J. Rose, Organometallics, 2020, 39, 1070–1079 CrossRef CAS.
- M. Afandiyeva, A. A. Kadam, X. Wu, W. W. Brennessel and C. R. Kennedy, Organometallics, 2022, 41, 3014–3023 CrossRef CAS.
- M. Afandiyeva, X. Wu, W. W. Brennessel, A. A. Kadam and C. R. Kennedy, Chem. Commun., 2023, 59, 13450–13453 RSC.
- T. Zhang, C. Wang, S. Liu, J.-L. Wang and W. Lin, J. Am. Chem. Soc., 2014, 136, 273–281 CrossRef CAS PubMed.
- Q. Y. Mao, Y. J. Pang, X. C. Li, G. J. Chen and H. W. Tan, ACS Catal., 2019, 9, 8798–8809 CrossRef CAS.
- K. R. Gorantla and B. S. Mallik, J. Phys. Chem. A, 2023, 127, 3788–3795 CrossRef CAS PubMed.
- M. S. Ali Akbari, S. Nandy, K. H. Chae, R. Bikas, A. Kozakiewicz-Piekarz and M. M. Najafpour, Langmuir, 2023, 39, 5542–5553 CrossRef CAS.
- D. L. Gerlach, S. Bhagan, A. A. Cruce, D. B. Burks, I. Nieto, H. T. Truong, S. P. Kelley, C. J. Herbst-Gervasoni, K. L. Jernigan, M. K. Bowman, S. Pan, M. Zeller and E. T. Papish, Inorg. Chem., 2014, 53, 12689–12698 CrossRef CAS.
- C. M. Moore, D. A. Quist, J. W. Kampf and N. K. Szymczak, Inorg. Chem., 2014, 53, 3278–3280 CrossRef CAS.
- C. M. Moore and N. K. Szymczak, Chem. Commun., 2015, 51, 5490–5492 RSC.
- C. M. Moore and N. K. Szymczak, Chem. Sci., 2015, 6, 3373–3377 RSC.
- O. J. García de Jesus, A. Mondragón-Díaz, B. Donnadieu and M. Á. Muñoz-Hernández, Inorg. Chem., 2023, 62, 2518–2529 CrossRef PubMed.
Footnote |
| † Current address: Dept. of Chemistry, University of Regensburg, 93040 Regensburg, Germany. |
| This journal is © The Royal Society of Chemistry 2024 |