
Modulating stereoselectivity and catalytic efficiency of carbenoid reactions catalysed by self-sufficient P450s†
Binhao
Wang
,
Cuiping
You
,
Guochao
Xu
 * and
Ye
Ni
*
* and
Ye
Ni
*
Key Laboratory of Industrial Biotechnology, Ministry of Education, School of Biotechnology, Jiangnan University, Wuxi 214122, Jiangsu, China. E-mail: guochaoxu@jiangnan.edu.cn; yni@jiangnan.edu.cn
First published on 8th December 2023
Abstract
Self-sufficient P450s are versatile biocatalysts. Among six self-sufficient P450 backbones, two variants from Tepidiphilus thermophilus exhibited a high TOF value of 1.5 min−1 (P450TT-C385S) and excellent diastereoselectivity of deE = 95.4% (P450TT-C385H). Molecular simulation was performed to reveal the different catalytic mechanisms among the self-sufficient P450 backbones. This work could provide useful information for exploring challenging unnatural reactions.
Carbenoid reactions, as powerful reactions, can effectively construct fundamental building blocks of organic molecules, including C–C, C–Si, C–N bonds, etc.1 They could be used to construct complex organic molecules, introduce functional groups, achieve reaction selectivity, and exhibit reaction diversity. Thereby they provide possibilities to expand the catalytic capabilities of nature for a sustainable chemical industry.2 Traditional carbene reactions are derived from transition metal-catalyzed reactions (such as Rh, Ru, Pd, Co, Fe). In 2012, Arnold et al. discovered that heme containing proteins could catalyse styrene cyclopropanation when a diazo carbene precursor is provided (Fig. 1a). This enzymatic catalysis of carbene reactions was previously unknown.3 The biocatalytic carbene transfer reactions demonstrated by these heme containing proteins are often complementary to the state-of-the-art processes based on small-molecule transition-metal catalysts.3 This highlights that engineered biocatalysts are valuable addition to the toolbox of synthetic chemists.3 Moreover, with asymmetric three-dimensional structures, heme containing proteins can provide precise chiral synthesis while creating unnatural enzymatic reactions. The biocatalytic platform offers an exciting opportunity to tackle challenging problems in modern synthetic chemistry and selective catalysis.4
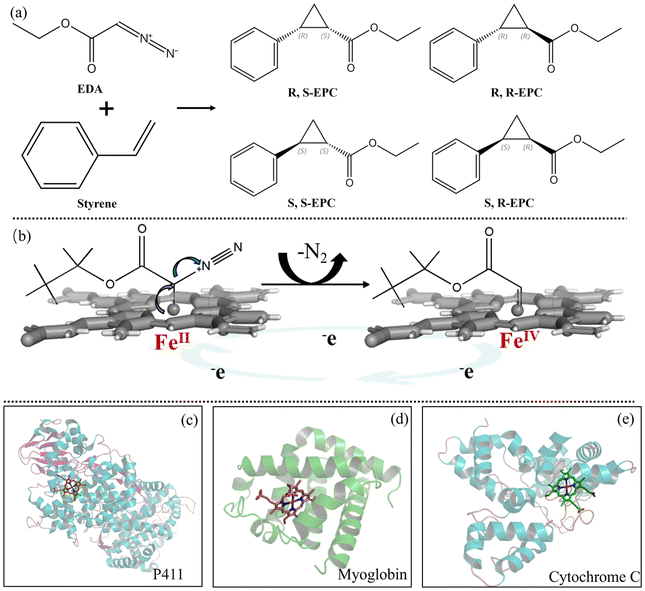 | ||
| Fig. 1 Carbene reaction and the protein structures of three reported enzyme backbones with excellent performance in carbene reactions. (a) Styrene cyclopropanation as the carbenoid model reaction. (b) Formation of active intermediates in the carbene reaction. (c) P411 (PDB: 4H23). (d) Myoglobin (PDB: 1A6K). (e) Cytochrome C (PDB: 1YNR). | ||
P450 monooxygenases are important biocatalysts possessing vital functions,5 most typically represented by their ability to catalyse a series of challenging reactions. Active iron–oxo intermediate compound I is required for the oxidative activity of P450s, strikingly similar to the carbenoid active intermediate.6 P450s gain electrons from NAD(P)H and delocalize N2 as a leaving group to generate carbene-reactive intermediates and thus catalyse the generation of carbenes (Fig. 1b).7 In addition, a series of heme proteins such as myoglobin, lactococcal multidrug resistance regulator and cytochrome C have also been shown to catalyse carbenoid reactions (Fig. 1c–e).8 Compared with other heme proteins, self-sufficient P450s allow efficient electron transfer through a polypeptide chain linking the heme domain and redox partner (e.g., flavoenzymes and iron–sulfur). There is no need to search for suitable redox partners to establish a functional system. Moreover, the reductase domain can furnish electrons to other enzymes in chimeric fusion proteins, while the P450 domain can be engineered for high activity and selectivity.9 Some of them, derived from extreme environments, also exhibit stability even under harsh conditions.10 Introducing artificial enzymes of transition metals is expensive and of low economic value compared with heme containing enzymes, which depend on cheap metallic iron. Therefore, exploring efficient self-sufficient P450 backbones with high enantioselectivity for synthesizing carbene stereoisomers is of great importance for discovering challenging unnatural reactions, and also could provide guidance to engineering existing P450s to extend their boundaries for natural reactions. Herein, six self-sufficient P450s were evaluated using the carbenoid model reaction of cyclopropanation of ethyl diazoacetate (EDA) (Fig. 1a). The molecular basis of carbenoid reactions catalysed by different self-sufficient P450 backbones was investigated.
Here, using three well studied enzymes with excellent catalytic performance in carbenoid reactions as probes (P411-CIS derived from Priestia megaterium,11 myoglobin derived from Physeter catodon12 and cytochrome C derived from Rhodothermus marinus13), genome mining was performed. Six P450s derived from Tepidiphilus thermophilus (P450TT also known as CYP116B46 (ref. 14)), Jhaorihella thermophiles (P450JT also known as CYP116B63 (ref. 14)), Aspergillus fumigatus (CYP505X identified by R. Weis et al.15), Myceliophthora thermophila (CYP505A30 identified by G. J. Baker et al.16), Bacillus methanolicus (CYP152K6 identified by Hazel M. Girvan et al.17), and Collariella virescens (CviUPO also known as rCviUPO18) were chosen as candidate enzymes. The phylogenetic tree was employed to analyse the relationship between P450BM3, myoglobin (also with activity in the carbene reaction) and six self-sufficient P450s. Among them, CviUPO is the most closely related to myoglobin. CYP505X originates from a common ancestral branch with P450BM3. Similarly, P450TT and P450JT are derived from an ancestral branch with myoglobin, specifically, they belong to two different subfamilies. P450JT was observed to exhibit better catalytic activity than P450TT (Fig. 2a), reflecting the functional differences that arose from the specific differentiation of CYP116 during evolution. Among them, CYP152K6 has the furthest relationship with others. P450s have several highly conserved regions which could be associated with their secondary structures.19 As expected, despite low degrees of sequence identity, the different P450s share a few conserved key catalytic residues and a common fold (Fig. 2b). The conservative domains (90–290 amino acids) of the six P450s are highly similar. Composed of a hydrophobic amino acid, they can enhance the stability of the inner core (Fig. 2b). The conserved catalytic domain sequences of the six P450s provide possibilities to catalyse carbenoid reactions. The performance of the six P450s was evaluated in carbenoid model reactions.
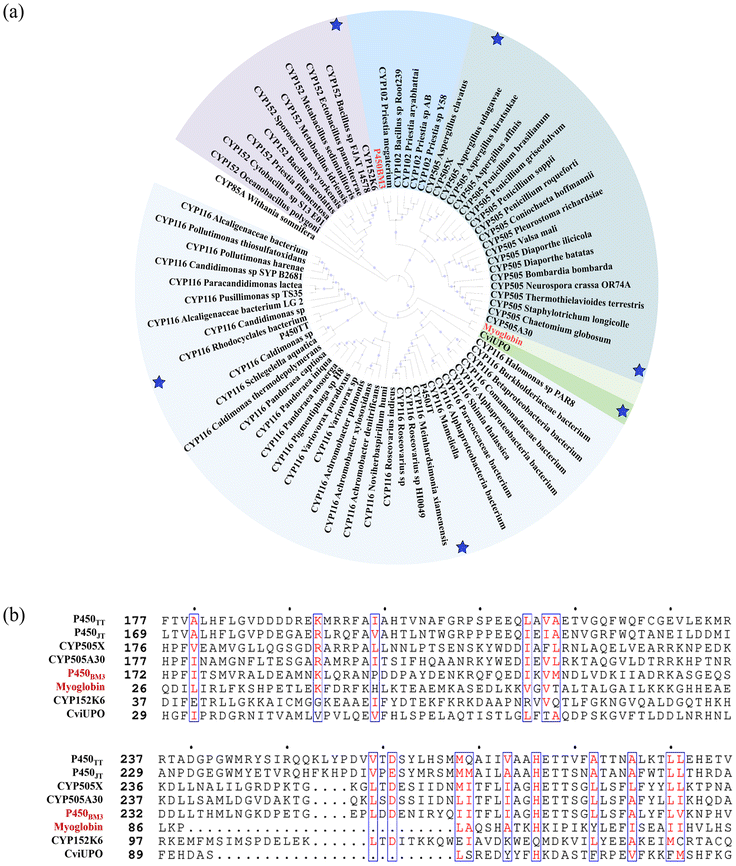 | ||
| Fig. 2 Phylogenetic tree and sequence alignment for P450BM3, myoglobin and six self-sufficient P450s. (a) Phylogenetic tree. Blue stars: six self-sufficient P450s. (b) Sequence alignment. The alignment shows the implicitly conserved catalytic domain (blue box). | ||
The cyclopropanation activity of P450s is highly dependent on the electronic properties of the proximal ligand.8,20 To increase the redox potential for reducing the resting FeIII-heme to FeII-heme, the vigorous electron donor cystine at the axial ligand of the heme cofactor was substituted with a weaker electron donor serine or histidine. Herein, the six self-sufficient P450s were evaluated with the carbenoid mode reaction of styrene and EDA as substrates. Under assay conditions, crude enzymes of the six P450 backbones could catalyse the carbenoid model reaction (Fig. S7–S13†). Based on the carbenoid reaction performances of the different self-sufficient P450 crude enzymes (Fig. S7–S13†), enzymes with high TOF potential (according to detection responses) and excellent stereoselectivity were purified for further research. Initially, the turnover frequencies of the model carbenoid reaction among seven candidates were compared using purified P450s. Furthermore, P411/P450-CIS11 and two other backbones (myoglobin12 and cytochrome C13) were chosen as positive controls (Fig. 3), which have been reported with high turnover frequencies and stereoselectivity in carbenoid reactions. Under assay conditions, the TOF ratio of P411-CIS and P450-CIS was approximately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.3, consistent with their reported conversion rate ratio,11 and was higher than that of the cytochrome C backbone. However, the turnover frequencies of the two enzymes were lower than that of the myoglobin backbone. Notably, under the same conditions, the seven tested self-sufficient P450s had higher conversion rates than all four positive controls. Among all the tested purified enzymes, P450TT-C385S showed the highest TOF (1.5 min−1), which is 4.3 times higher than that of P411-CIS (0.35 min−1), and 3 times higher than that of P450JT-WT with the 2nd highest TOF value (1.2 min−1). It is worth noting that both P450TT-C385S and P450JT-C377S displayed higher TOF than that substituted with His. CYP505X-WT displayed a moderate TOF value (0.95 min−1) compared with P450JT-WT and P450TT-C385S. All the TOF values of purified P450s were consistent with their UV response in crude enzyme reactions (Fig. S7†).
:1.3, consistent with their reported conversion rate ratio,11 and was higher than that of the cytochrome C backbone. However, the turnover frequencies of the two enzymes were lower than that of the myoglobin backbone. Notably, under the same conditions, the seven tested self-sufficient P450s had higher conversion rates than all four positive controls. Among all the tested purified enzymes, P450TT-C385S showed the highest TOF (1.5 min−1), which is 4.3 times higher than that of P411-CIS (0.35 min−1), and 3 times higher than that of P450JT-WT with the 2nd highest TOF value (1.2 min−1). It is worth noting that both P450TT-C385S and P450JT-C377S displayed higher TOF than that substituted with His. CYP505X-WT displayed a moderate TOF value (0.95 min−1) compared with P450JT-WT and P450TT-C385S. All the TOF values of purified P450s were consistent with their UV response in crude enzyme reactions (Fig. S7†).
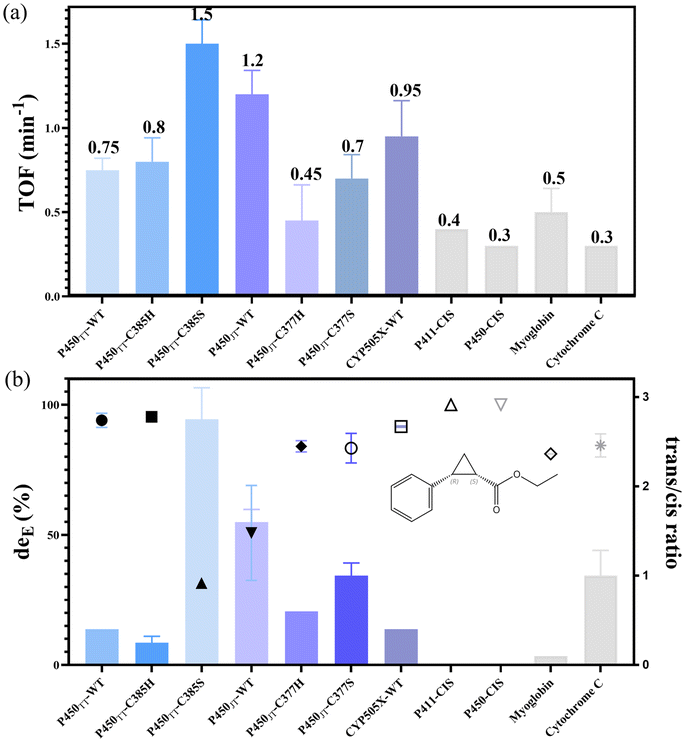 | ||
Fig. 3 Comparison of turnover frequencies (TOFs) and stereoselectivity of the carbenoid model reaction catalysed by different purified P450s. (a) Turnover frequencies. (b) Diastereoselectivity and trans/cis ratio. Dots: diastereoselectivity. Columns: trans/cis ratio. Reactions were performed with 10 μM enzyme, 10 mM styrene, 20 mM EDA, and 10 mM Na2S2O4 in 100 mM Tris·HCl (pH = 8.0). Stereoselectivity was determined by ultra-performance convergence chromatography 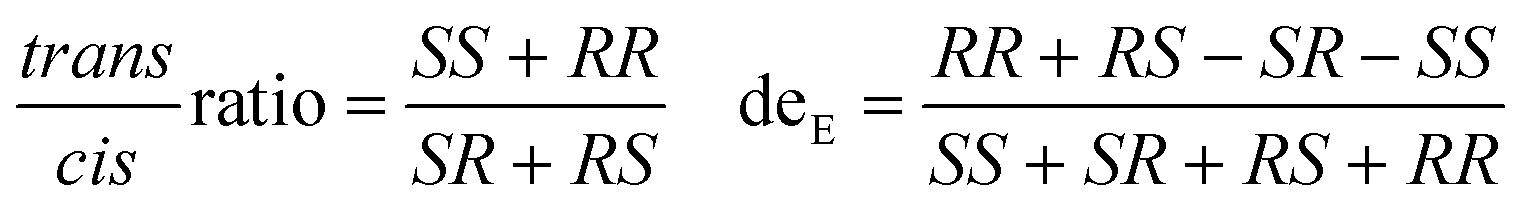 . . | ||
Some proteins in E. coli exhibit significant background in crude enzyme reactions, particularly for poorly expressed or unstable P450s.21 As carbene transfer biocatalysts, the stereoselectivity is regarded as a critical character. Under assay conditions, purified P450-CIS displayed excellent diastereoselectivity as P411-CIS (99.9%), dominated by R,S-ethyl 2-phenylcyclopropane carboxylate (R,S-EPC) (Table S4†). Among all the tested purified enzymes, almost perfect diastereoselectivity was observed with P450TT-C385H (95.4%), P450TT-WT (94.0%), and CYP505X-WT (91.6%). Slightly inferior to the above three enzymes, P450JT-C377H (84.0%) and P450JT-C377S (83.3%) showed impressive diastereoselectivity performance (Table S4†). P450JT-WT (50.7%) and P450TT-C385S (31.5%) displayed relatively poor diastereoselectivity. An almost excellent trans/cis ratio was also observed in P450TT-C385H (0.2), which was slightly inferior than that of P411-CIS (0) and P450-CIS (0) (Fig. 3b). Notably, all the enzymes produced a cis-stereoisomer relatively rich in (R,S)-EPC, while the trans-stereoisomer was rich in (R,R)-EPC. Among them, P450TT-WT, P450TT-C385H, P450JT-C377H, and CYP505X-WT all displayed excellent cis-selectivity (eecis = 99.9%) while maintaining high deE values. Differently, P450JT-C377S had a moderate enantioselectivity (deE = 83.3%, eecis = 93.5%, eetrans = 51.9%) (Fig. 3b, Table S4†). Although the myoglobin backbone had a higher turnover frequency than P411-CIS, the diastereoselectivity of myoglobin (81.1%) was inferior to that of P411-CIS (99.9%) and the other self-sufficient P450s (P450TT-WT, P450TT-C285H, CYP505X-WT) tested (Fig. 3b). Compared with the tested P450s, cytochrome C displayed only moderate turnover frequency (0.30 min−1) and diastereoselectivity (deE = 84.4%). It is speculated that the complete electron transport chain of self-sufficient P450s could provide more vigorous catalytic power to allow higher turnover frequencies.
With the determined and refined crystal structure, P450BM3 and P450TT could be used as excellent model proteins to investigate the influence of intramolecular electron transport chains in the carbenoid model reaction. The edge-to-edge straight-line distances of FMN-heme (P450BM3) and [2Fe–2S]-heme (P450TT) are 18.6 Å and 25.3 Å, respectively.22 Several residues between [2Fe–2S] and heme of P450TT may contribute to the electron delivery path (Fig. 4a).22 These residues predominantly consist of acidic residues (E723, E729) and basic residues (R388, R718). Notably, a pattern of alternating positively charged (arginine) and negatively charged (glutamine) amino acids was observed in adjacent positions. These residues play a crucial role in facilitating smooth electron transfer by providing redox potentials along their main chains. Furthermore, it was observed that certain polar residues (C385, Q725, S726), which lack aromatic groups, exhibited unexpectedly low dependence on distance.21 This phenomenon can be attributed to the specific configurations of their side chains.21 In P450TT, the efficient peptide electron transfer chains might play a more important role than the “domain close” interaction observed in P411-CIS, which relies on FMN–heme domain interaction. Consequently, a higher catalytic activity (TOF) was observed with P450TT (Fig. 4a).
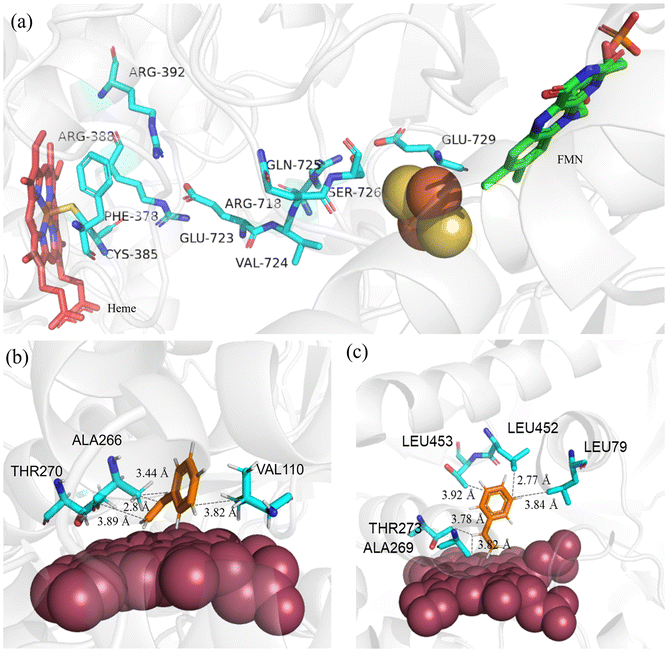 | ||
| Fig. 4 Molecular mechanism analysis. (a) Residues proposed by Guo et al. that may have contributed to long-range electron transfer between [2Fe–2S] and heme of P450TT (PDB ID: 2HPD). Molecular docking of substrate styrene with (b) P450JT and (c) CYP505X. Red sphere: heme cofactor. Blue sticks: residues interacting with substrates. Black dashed lines: interaction distances. | ||
The interactions of heme-iron and nearby residues were analysed (Fig. S14a–f†). It was found that a cluster of hydrophobic residues is located near the heme-iron, facilitating strong hydrophobic interactions at the core. These residues collectively create a compact hydrophobic pocket that allows the insertion of the heme cofactor in the interior of the pocket.23 Moreover, multiple nonpolar and aromatic residues were also observed in the active site and substrate channel. These residues have been proved to dominate the cone-shaped active site and help P450s accept hydrophobic substrates (Fig. S14a–f†).24,25 For CviUPO, the substrate binds heme with high affinity in a hydrophobic pocket, which is composed of eighteen nonpolar residues, including P21 (3.98 Å), A22 (3.17 Å), L48 (3.26 Å) and F52 (2.98 Å). In addition to the salt bridge between R97 (5.08 Å) and the carbonyl group that stabilizes the porphyrin conformation (also observed in most members of the CYP116 subfamily26), a number of residues were observed to engage in hydrogen bonds with heme, including E90 (2.56 Å), H91 (1.85 Å), L95 (1.63 Å), S96 (2.30 Å) and R97 (3.34 Å). Additionally, H91 may also coordinate with heme through an imidazole moiety.26 All these interactions provide CviUPO with excellent catalytic performance in the carbenoid model reaction, despite the low soluble protein expression observed (Fig. S2†).
P450JT exhibited relatively poor diastereoselectivity compared with CYP505X. To understand the mechanisms of stereoselectivity in the carbenoid model reaction, molecular simulation of CYP505X (deE = 91.6%) and P450JT (deE = 50.7%) complexes was conducted (Fig. 4b and c). For P450JT, hydrophobic interactions with the substrate were generated by three nonpolar residues (V110, A266, T270). All the interactions between the substrate and surrounding residues were observed to occur within a distance range of 2.8–3.9 Å. In addition, all of them were observed in an unbalanced manner around styrene (only one force acting on the benzene ring portion, while the rest were distributed on the same side of the substrate) (Fig. 4b). Unbalanced forces with the hydrophobic substrate resulted in a “tilted” conformation in P450JT (Fig. 4b). By contrast, a more “vertical” conformation was observed in CYP505X, benefiting from more uniform forces with the substrate (Fig. 4c). All the hydrophobic interactions with the substrate are formed with five residues (L79, A269, T273, L452 and T453). Five forces were evenly distributed in the benzene ring and olefin, resulting in a relatively “vertical” form where the substrate could “stand” on the porphyrin. The morphology of the substrate binding to the active site of enzymes leads to the difference in stereoselectivity of the final product. Enhanced cis-selectivity could be obtained when the terminal olefin of the substrate approaches the porphyrin at a perpendicular angle in the carbenoid model reaction, consistent with those reported by others.27
Conclusions
In conclusion, excellent potential carbenoid reaction backbones (CYP505X) and mutants with high TOF (P450TT-C385S) and excellent stereoselectivity (P450TT-C385H) were obtained based on their catalytic performances. The study of electron transfer pathways between P450TT and P450BM3 revealed that more efficient electron transfer can lead to increased catalytic activity in the carbenoid model reaction. In addition, the influence of the heme axial key residue and hydrophobic environment of the heme pocket on the carbenoid model reaction was also investigated. Substituting the crucial axial residue with a weaker electron donor enhanced the catalytic activities in carbenoid model reactions for most P450s. However, in the case of P450JT, P450JT-WT exhibited higher catalytic activity (TOF) than variants P450JT-C377H and P450JT-C377S. For CviUPO, although poor protein expression was observed, the strong hydrophobic interaction in the core region contributes to its surprising catalytic performance. Based on molecular simulation analysis of P450JT and CYP505X complexes with the substrate, a perpendicular conformation of the terminal olefin of the substrate in the porphyrin is conducive to better cis-selectivity in carbenoid model reactions. This work provides promising self-sufficient P450s for carbenoid reactions and instructive information for exploring the application of enzymatic carbenoid reactions.Author contributions
B. W., G. X. and Y. N. conceived and designed the project. B. W. conducted enzyme screening, characterization and mutation. C. Y. performed stereoselectivity analysis. B. W. carried out the computation analysis. B. W., G. X. and Y. N. wrote the manuscript. G. X. and Y. N. obtained financial support for the project leading to this publication.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the National Key R&D Program (2021YFC2102700) and the National Natural Science Foundation of China (22078127, 22077054) for the financial support.Notes and references
- F. H. Arnold, Q. Rev. Biophys., 2015, 48, 404–410 CrossRef CAS PubMed.
- F. H. Arnold, Angew. Chem., Int. Ed., 2018, 57, 4143–4148 CrossRef CAS PubMed.
- Z. Liu and F. H. Arnold, Curr. Opin. Biotechnol., 2021, 69, 43–51 CrossRef CAS PubMed.
- Y. Yang and F. H. Arnold, Acc. Chem. Res., 2021, 54, 1209–1225 CrossRef CAS PubMed.
- V. B. Urlacher and M. Girhard, Trends Biotechnol., 2019, 37, 882–897 CrossRef CAS PubMed.
- T. K. Hyster and F. H. Arnold, Isr. J. Chem., 2015, 55, 14–20 CrossRef CAS.
- P. C. E. Moody and E. L. Raven, Acc. Chem. Res., 2018, 51, 427–435 CrossRef CAS PubMed.
- P. Srivastava, H. Yang, K. Ellis-Guardiola and J. C. Lewis, Nat. Commun., 2015, 6, 7789 CrossRef CAS PubMed.
- L. L. Zhang, Z. Z. Xie, Z. W. Liu, S. Y. Zhou, L. X. Ma, W. D. Liu, J. W. Huang, T. P. Ko, X. Q. Li, Y. C. Hu, J. Min, X. J. Yu, R. T. Guo and C. C. Chen, Nat. Commun., 2020, 11, 2676 CrossRef CAS PubMed.
- D. Correddu, G. Di Nardo and G. Gilardi, Trends Biotechnol., 2021, 39, 1184–1207 CrossRef CAS PubMed.
- Z. J. Wang, H. Renata, N. E. Peck, C. C. Farwell, P. S. Coelho and F. H. Arnold, Angew. Chem., Int. Ed., 2014, 53, 6810–6813 CrossRef CAS PubMed.
- M. Bordeaux, V. Tyagi and R. Fasan, Angew. Chem., Int. Ed., 2015, 54, 1744–1748 CrossRef CAS PubMed.
- S. B. J. Kan, X. Huang, Y. Gumulya, K. Chen and F. H. Arnold, Nature, 2017, 552, 132–136 CrossRef CAS PubMed.
- M. Tavanti, J. L. Porter, S. Sabatini, N. J. Turner and S. L. Flitsch, ChemCatChem, 2018, 10, 1042–1051 CrossRef CAS.
- R. Weis, M. Winkler, M. Schittmayer, S. Kambourakis, M. Vink, J. D. Rozzell and A. Glieder, Adv. Synth. Catal., 2009, 351, 2140–2146 CrossRef CAS.
- G. J. Baker, H. M. Girvan, S. Matthews, K. J. McLean, M. Golovanova, T. N. Waltham, S. E. J. Rigby, D. R. Nelson, R. T. Blankley and A. W. Munro, ACS Omega, 2017, 2, 4705–4724 CrossRef CAS PubMed.
- H. M. Girvan, H. Poddar, K. J. McLean, D. R. Nelson, K. A. Hollywood, C. W. Levy, D. Leys and A. W. Munro, J. Inorg. Biochem., 2018, 188, 18–28 CrossRef CAS PubMed.
- D. Linde, E. Santillana, E. Fernández-Fueyo, A. González-Benjumea, J. Carro, A. Gutiérrez, A. T. Martínez and A. Romero, Antioxidants, 2022, 11, 891 CrossRef CAS PubMed.
- I. Matsunaga, A. Ueda, N. Fujiwara, T. Sumimoto and K. Ichihara, Lipids, 1999, 34, 841–846 CrossRef CAS PubMed.
- J. A. Mcintosh, T. Heel, A. R. Buller, L. Chio and F. H. Arnold, J. Am. Chem. Soc., 2015, 137, 13861–13865 CrossRef CAS PubMed.
- F. Polo, S. Antonello, F. Formaggio, C. Toniolo and F. Maran, J. Am. Chem. Soc., 2005, 127, 492–493 CrossRef CAS PubMed.
- L. Zhang, Z. Xie, Z. Liu, S. Zhou, L. Ma, W. Liu, J. W. Huang, T. P. Ko, X. Li, Y. Hu, J. Min, X. Yu, R. T. Guo and C. C. Chen, Nat. Commun., 2020, 11, 2676 CrossRef CAS PubMed.
- C. Y. Li, G. Anuraga, C. P. Chang, T. Y. Weng, H. P. Hsu, H. D. K. Ta, P. F. Su, P. H. Chiu, S. J. Yang, F. W. Chen, P. H. Ye, C. Y. Wang and M. D. Lai, J. Exp. Clin. Cancer Res., 2023, 42, 22 CrossRef CAS PubMed.
- M. Tavanti, J. L. Porter, C. W. Levy, J. R. G. Castellanos, S. L. Flitsch and N. J. Turner, Biochem. Biophys. Res. Commun., 2018, 501, 846–850 CrossRef CAS PubMed.
- J. C. Aschenbrenner, A. C. Ebrecht, C. Tolmie, M. S. Smit and D. J. Opperman, Catal. Sci. Technol., 2021, 11, 7359–7367 RSC.
- A. Ciaramella, G. Catucci, G. Gilardi and G. Di Nardo, Int. J. Biol. Macromol., 2019, 140, 577–587 CrossRef CAS PubMed.
- S. J. Thompson, M. R. Brennan, S. Y. Lee and G. Dong, Chem. Soc. Rev., 2018, 47, 929–981 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cy01258a |
| This journal is © The Royal Society of Chemistry 2024 |