
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent advances in promoting dry reforming of methane using nickel-based catalysts
Haibin
Zhu
 ,
Huichao
Chen
*,
Menghan
Zhang
,
Cai
Liang
and
Lunbo
Duan
,
Huichao
Chen
*,
Menghan
Zhang
,
Cai
Liang
and
Lunbo
Duan
School of Energy and Environment, Southeast University, Nanjing 210096, China. E-mail: hcchen@seu.edu.cn
First published on 16th February 2024
Abstract
Dry reforming of methane (DRM) is an efficient way for CO2 utilization to convert CH4 and CO2 to syngas (H2 and CO), which is an ingredient to produce hydrogen and methanol and hydrocarbons via Fischer–Tropsch process. Considerable attention is drawn to Ni-based catalyst due to its remarkable reactivity for CH4 and CO2 conversion. However, the weakness of sintering and coking restrains its industrial applications. The successful design of cost-effective, efficient and stable Ni-based catalysts would be a grand breakthrough for the technology. Herein, the thermodynamic analysis and the mechanisms of sintering and coking are presented. Then, the strategies for improving the performance of catalysts are elaborated as follows, with 1) supports improving the particle dispersion of nickels and changing the reaction process for the hindrance of coking; 2) promoters for regulating the nickel's chemical properties to enable catalytic DRM performance; 3) catalysts obtained by varied preparation methods possess different sizes and interactions. Finally, the challenges and future perspectives are highlighted.
 Haibin Zhu | Haibin Zhu is a postgraduate student in the School of Energy and Environment, Southeast University, China. His current research focuses on the development of novel catalysts for carbon capture and conversion into value-added products. |
 Huichao Chen | Huichao Chen is Associate Professor at the School of Energy and Environment at Southeast University, China. Her research interests include carbon dioxide capture and utilization and contaminated soil remediation. |
 Menghan Zhang | Menghan Zhang is a PhD student at the School of Energy and Environment, Southeast University, China. Her research interests include the development of calcium-based sorbents for CO2 capture. |
 Cai Liang | Cai Liang is Professor at the School of Energy and Environment at Southeast University, China. His main research interests focus on clean coal technology and hydrogen from waste plastics. |
 Lunbo Duan | Lunbo Duan is Professor at the School of Energy and Environment at Southeast University, China. Prof. Duan is also the vice chairman of Jiangsu Power Engineering Society. His main research interests focus on CO2 emission reduction and advanced energy materials. |
Introduction
The fast growth of human society has significantly increased fossil fuel consumption, and the excessive carbon dioxide (CO2) emission, which has passed 41 billion tons per year, has led to a variety of issues, the most prominent of which is the growing greenhouse effect.1–4 In this context, methane (CH4) is more active than CO2 in the greenhouse effect.5,6 Conceptually, inserting CO2 as a reactant into the process of fuel or chemical production could provide tangible negative emissions.7,8 Dry reforming of methane (DRM) is a promising way of CO2 utilization with the production of syngas (H2 and CO) and has been studied since 1885.5,9–11 DRM attracts interests of researchers because of the advantages of simultaneously eliminating both CO2 and CH4. The origin of the gas may be industrial emissions or biogas, predominantly consisting of methane and CO2, substances that are abundantly generated in present-day industrial process or through the anaerobic digestion or fermentation of organic matter by microbes.12,13DRM provides a feasible way for Carbon Capture, Utilization & Storage (CCUS). Taking hydrogen production as an example, currently, its global scale is more than 60 Mt per year from the process of steam reforming (CH4 + H2O → CO + 3H2, ΔHθ298K = 206 kJ mol−1).1,14 If this process is replaced by DRM (CH4 + CO2 → CO + 2H2, ΔHθ298K = 247 kJ mol−1), nearly 30 Mt per year of CO2 emissions could be removed, providing a large anthropogenic carbon sink to only 0.25 Gt per year in the current CO2 market.1,14,15 Meanwhile, DRM can also produce syngas, and these syngas components can be further processed into hydrocarbons, enabling the production of environmentally friendly fuels and valuable high-value chemicals (Fig. 1).16–18 Considering the carbon peaking and carbon neutrality proposed by the Chinese government, DRM offers a promising solution for addressing the surplus CO2 emissions while maintaining the integrity of existing infrastructure.19 Nevertheless, challenges such as the sintering of nickel metal particles and carbon deposition on the catalysts hinder its widespread commercial application.20
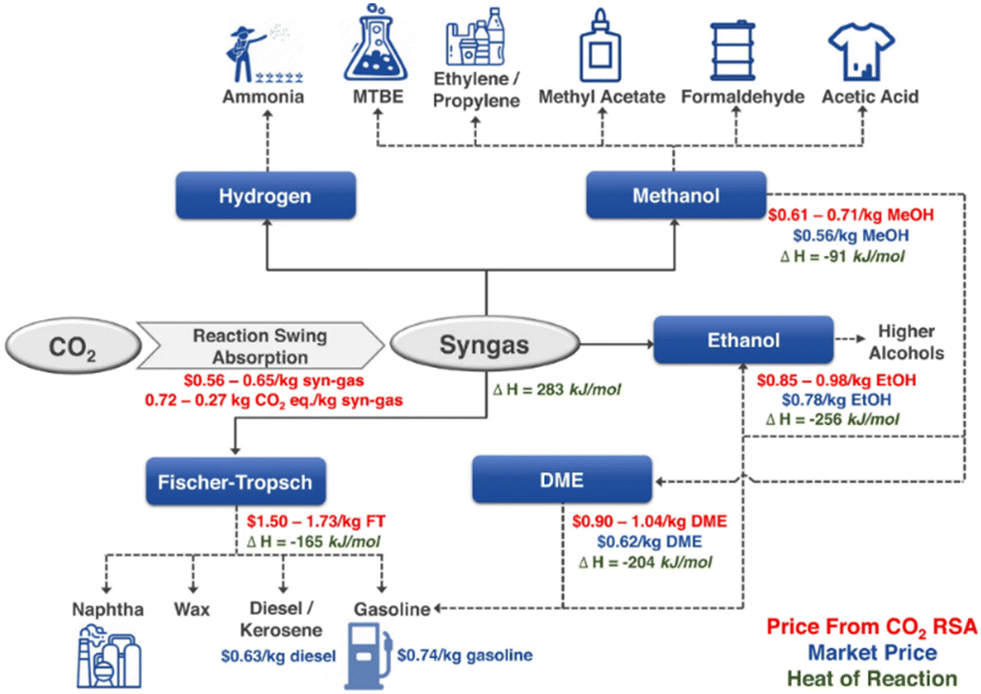 | ||
| Fig. 1 Syngas as a versatile intermediate chemical (RSA presents reaction swing absorption). Reproduced from ref. 21 with permission from Nature Communications, copyright 2022. | ||
To implement DRM in wide industrial application, it is significant to develop cost-efficient, highly active and exceptionally stable catalysts.22 Among the metal catalysts, nickel (Ni)-based catalysts are more practical because of its cost-efficiency and comparable catalytical performance over other noble metals (Ru-based and Rh-based catalysts).23–25 However, high temperature is required for the reaction as CO2 is thermodynamically stable and for the suppression of side effects as well. Thus, the catalysts suffer from sintering and rapid carbon deposition caused by high reaction temperature, which hinders its implementation at the industrial scale.
Rapid development of technology has been made and it is necessary to review the advances and challenges for further promoting the technology for wide application. Therefore, the present review focuses on the thermodynamics, reaction mechanism, and the efforts for improving the catalytic activity and stability from the aspects of supports, promoters, and synthesis methods. Furthermore, the challenges and opportunities associated with the nickel-based catalysts in DRM are proposed in the end.
Thermodynamics, reaction mechanism, sintering and carbon deposition
Thermodynamics
Fig. 2 illustrates the thermodynamic analysis of the reactions taking place under the conditions of DRM in terms of Gibbs free-energy changes (ΔGθT) obtained by the Outokumpu HSC Chemistry® program. Although the DRM reaction is mainly dominated by methane dissociation (or methane dehydrogenation, eqn (2)26 in Table 1) and reverse Boudouard reaction (or CO2 activation, eqn (3)26), some side reactions occur at the same time (eqn (5)–(9)).27–30 To obtain high conversion of CH4 and CO2 and selectivity toward syngas, the DRM temperature should be mostly above 873 K, which is not favorable for the side reactions and methane dissociation,28 reverse Boudouard reaction and reverse water-gas shift reaction (RWGS, eqn (4)28). Furthermore, the reverse Boudouard reaction and RWGS reaction are responsible for the carbon deposition because of the faster rate of methane dissociation over the reverse Boudouard reaction gap of ΔGθT.31,32 The formed carbonaceous species would remain on the active sites surface and hinder the access of CO2 and CH4 to the inner unreacted active sites.33,34 CO2 is also likely to react with H2 from the methane dissociation, according to the RWGS (eqn (4)), leading to a H2/CO ratio less than 1.0,35,36 related to the equilibrium at temperature ranges.29,35,37 To minimize the effect, high temperature for reaction and timely products removal is required.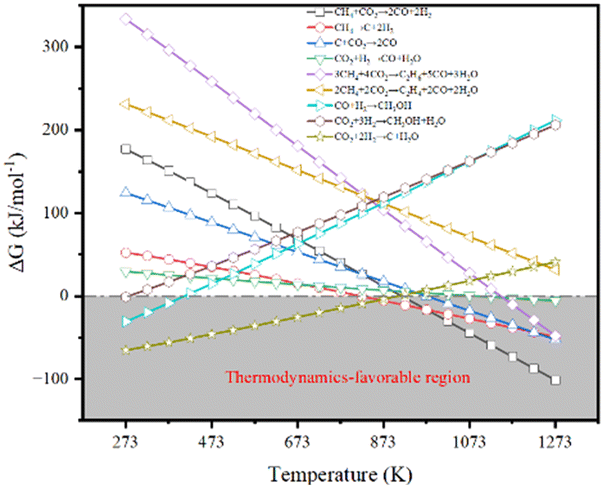 | ||
| Fig. 2 The thermodynamics of main reactions under the conditions of DRM. | ||
| Equation | Reaction | Reaction equation | ΔHθ298K (kJ mol−1) |
|---|---|---|---|
| 1 | Main reaction | CH4 + CO2 → 2CO + 2H2 | +247.016 |
| 2 | Methane dissociation | CH4 → 2H2 + C | +74.595 |
| 3 | Reverse Boudouard reaction | CO2 + C → 2CO | +174.472 |
| 4 | Reverse water-gas shift reaction | CO2 + H2 → CO + H2O | +41.138 |
| 5 | Other side reactions | 3CH4 + 4CO2 → C2H6 + 5CO + 3H2O | +434.946 |
| 6 | 2CH4 + 2CO2 → C2H4 + 2CO + 2H2O | +283.872 | |
| 7 | CO + 2H2 → CH3OH | −90.452 | |
| 8 | CO2 + 3H2 → CH3OH + H2O | −49.314 | |
| 9 | CO2 + H2 → C + H2O | −90.144 |
Reaction mechanism
There is a consensus that the methane dihydrogen on the catalysts constitutes the step that dictates the reaction rate,1 and this process follows the rules (Fig. 3): *CH4 → *CH3 + *H → *CH2 +*H → *CH + *H → *C + *H, (“*” refers Ni-based active sites) and the formed *H combines consequently to form H2.38–41 Each partially dissociated *CHx (“x” refers to 2, 3, 4) species adsorbs on Ni-based active sites, which completes its tetravalency.29 Then, the CO2 in stream reacts with *C to form CO according to the reverse Boudouard reaction, where CO2 decomposes to the surface oxygen (*O) and CO.42 During the process of reverse Boudouard reaction, *O reacts with *CHx or produces intermediates of *CHxO and *CO (*CHxO is considered as the precursors of *CO). In contrast, others hold the view that CO2 forms carbonates firstly and the carbonates are subsequently reduced by *C from methane dissociation. Thus, whether CO2 dissociates to *O and *CO or it forms carbonates first remains controversial. In general, challenges remain due to the absence of a well-established consensus regarding the intricacies of the reaction mechanism occurring on the catalytic surfaces.29,43,44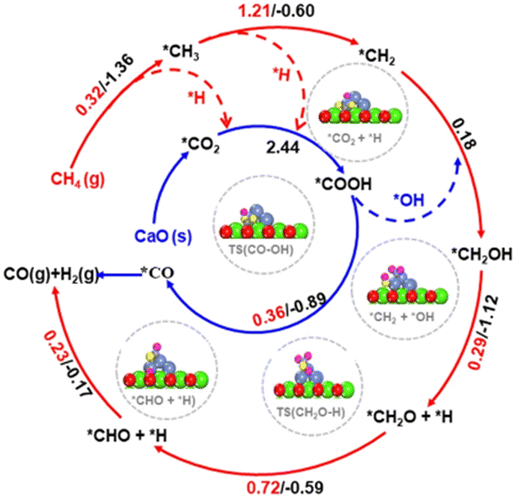 | ||
| Fig. 3 Schematic diagram of methane dry reforming reaction. The corresponding activation energy is Ea (red) and reaction energy (black) is in the unit of eV for each step. Reproduced from ref. 45 with permission from Nature Communications, copyright 2023. | ||
Sintering
When the Ni-based catalysts are employed in the DRM, especially at a high temperature, there is a strong propensity for sintering, which results in the acceleration of particles growth due to the sharply increased surface energy with decreased nickel size. Sintering inevitably leads to the loss of surface area and deactivates the catalytic activity of the catalysts.46 A fundamental understanding of Ni-based catalyst sintering is essential for the development of thermally stable catalysts.47Particles moving across the particle diffusion distance to contact with each other is the basic mechanism of sintering. Thus, increasing the particle distance on supports by using porous or mesoporous materials48,49 to achieve uniform spatial distribution50 could slow down the sintering. Different preparation methods are used for developing unique structures (i.e., core–shell structure51,52), and supports are selected for regulating the nickel metal–support interactions.53 Despite the promising approaches, diminishing the specific surface areas and increasing the resistance over mass-transfer results in decreasing conversions of CO2 and CH4. Besides, the intricate geometric modification strategies of catalysts often involve complex multi-step procedures, making them difficult to implement on the industrial scale. At the same time, there is still limited understanding regarding the quantification of the particles distance where a significant suppression of metal sintering occurs. Such fundamental knowledge is vital for developing sintering-resistant Ni-based catalysts for sustainable and economical catalytic processes.
The metal sintering mechanisms can be categorized into two types by their microscopic migration properties: (1) particle migration and coalescence (PMC), where nickel particles migrate under Brownian motion, resulting in collisions that consequently cause coalescence when two particles are close; (2) Ostwald ripening (OR), where the migration of nickel particles is propelled by a decrease in the chemical potential, with the particles released from an individual particle, dispersing across the support and adhering to adjacent ones, and culminating in the formation of larger particles (Fig. 4).46,54
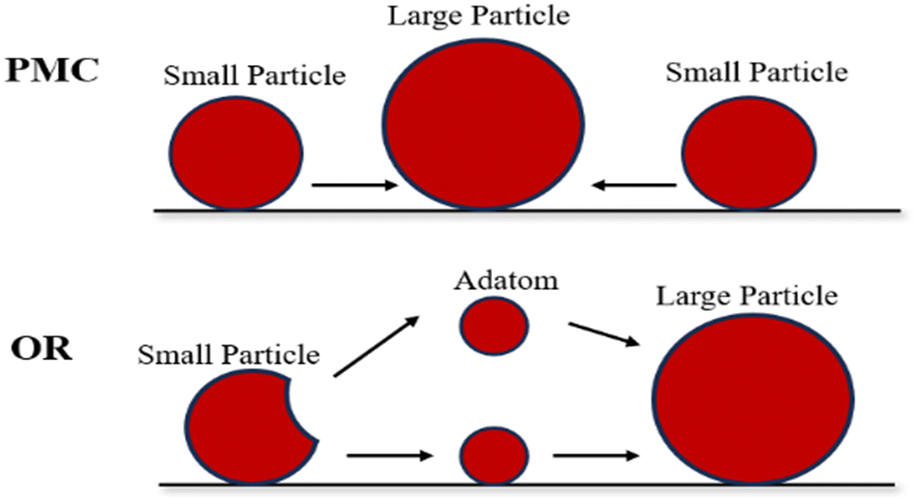 | ||
| Fig. 4 Schematic diagram of sintering process. Reproduced from ref. 55 with permission from Accounts of Chemical Research, copyright 2013. | ||
Carbon deposition
Sintering tends to expedite carbon deposition, supported by substantial evidence indicating a proclivity for coke formation on the surfaces of larger nickel particles.56–59 As coke is deposited, catalysts would become immobilized due to blockage. The carbonaceous deposits can be chemically adsorbed in the form of a strongly bonded monolayer or physically adsorbed as multiple layers, leading to the complete encasement of the metal active sites or obstructing pores. This significantly impedes the access of reactive substances to the nickel active sites.60The process of carbon deposition and transformation is shown in Fig. 5. According to the energy of carbonaceous species, three carbonaceous species can be distinguished:61,62 (I) adsorbed atomic carbon (Cα), Cα is a combination of *CHx fragments from CH4 and CO2 on the catalytic surface.63 It is the highly active atom that can mostly react with CO2 or be further gasified;22 (II) filamentous carbon (Cβ): Cβ is a type of low-activity carbon species and originates from the Cα that reacts with water, carbon dioxide and hydrogen. It would be further gasified or encapsulated into nickel microcrystals, poisoning the catalysts; (III) graphite carbon (Cγ), Cγ is transformed by Cβ at high temperature (>873 K). Among the carbonaceous species (Cα, Cβ, and Cγ), Cβ and Cγ are worthy of special attention since they are difficult to be oxidized and would destroy the catalyst structure.13,64–66
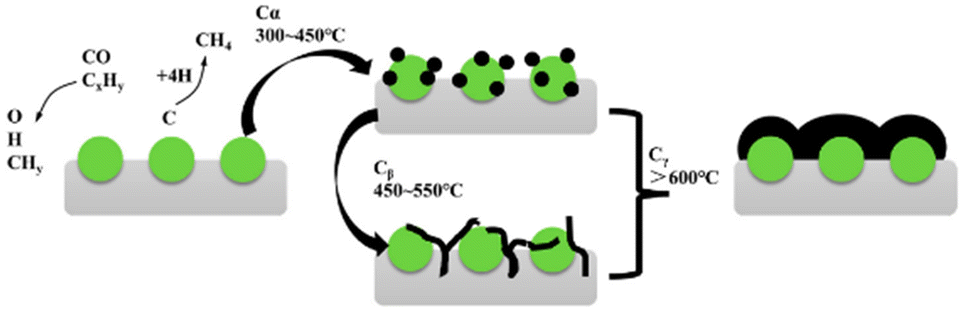 | ||
| Fig. 5 Schemic diagram of carbon formation and transformation. Reproduced from ref. 33 with permission from Atmosphere, copyright 2023. | ||
As mentioned above, catalytic sintering and inactive carbon deposition easily deactivate the catalytic performance of the catalysts. Thermodynamic analysis has shown that the DRM is occurs better at a high temperature of 873 K for reactant conversion and product selectivity, turning the ratio of CH4 and CO2 to unity.67 However, the severe temperature would lead to high consumption of energy, which is not sustainable or economic. Meanwhile, the Ni particles inevitably aggregated under severe conditions, leading to the enhancement of carbon deposition and activity reduction. Therefore, more and more researchers have researched the modification of the catalysts to maintain the DRM stability and performance at a relatively low temperature according to Le Chatelier's principle. Many strategies have been proposed, such as choosing different supports to protect the active atoms migrating by the confinement effect, regulating the features of acidity and alkalinity, adding various promoters to change the Ni valence state or switching to other reaction paths to avoid carbon formation. Novel preparation methods are implemented to obtain catalysts with different interactions between metal and supports and structures like core–shell, perovskite, and hydrotalcite. A comprehensive review of the recent advances with respect to the dry reforming of methane, especially the modification and preparation of active catalysts, is summarized.
Influencing factors on the catalytic performance of Ni-based catalysts
Support selectivity
Because of the small specific surface area and few active sites of Ni particles, normally, they are not used independently.68 The combination of Ni and supports possessing large surface area facilitates areas for nickel metal dispersion and diminishes the rate of carbon deposition.69 Commonly employed support materials consist of alumina oxide, various types of clay, zeolites and structured mesoporous silicates featuring ordered pore structures.69–71 The supports not only physically hold the Ni nanoparticles by the confinement effect caused by their uneven surface but also influence the catalytic activity and coking resistance due to their surface acid–base feature.72 Meanwhile, the strong interaction between Ni nanoparticles and supports reduces the size, enhancing the dispersion of active sites.73 Due to the high reaction temperature of DRM (usually above 700 °C), the suitable supports must have strong stability at high temperature and large specific surface area inhibiting the particles aggregation.74Table 2 summarizes the reaction conditions and CH4 and CO2 conversions by different support species.| Catalyst | Temp (°C) | Time (h) | GHSVa and feed gas radio | Conversion (%) | Carbon deposition |
|---|---|---|---|---|---|
| a GHSV refers to gas hour space velocity, which is calculated based on the Ni weight of the catalysts. | |||||
| Ni/Al2O3 | 700 | 6 | — | CH4: 87 | 0.3 gcoke−1 gcat−1 h−1 (ref. 75) |
| CH4/CO2/He = 10/10/80 | CO2: 91 | ||||
| 800 | 100 | — | CH4: 94 | 0.28 gcoke−1 gcat−1 h−1 (ref. 76) | |
| CH4/CO2/He = 1/1.05/1 | CO2: 94 | ||||
| 800 | 100 | 2500 mL g−1 h−1 | CH4: 90 | 0.015 gcoke−1 gcat−1 h−1 (ref. 77) | |
| CH4/CO2 = 1/1 | CO2: 88 | ||||
| Ni/SiO2 | 700 | 100 | 72 L g−1 h−1 | CH4: 80 | No carbon filaments by TEM78 |
| CH4/CO2/N2 = 1/1/1 | CO2: 85 | ||||
| Ni/SBA-15 | 650 | 12 | 3824 L g−1 h−1 | CH4: 77 | No structured carbon species by TEM48 |
| CH4/CO2/Ar = 0.5/0.5/9 | CO2: 77 | ||||
| 800 | 50 | 22.5 L g−1 h−1 | CH4: 85 | No small amount of carbon deposition by TEM49 | |
| CH4/CO2 = 1/1 | CO2: 91 | ||||
| Ni/MCF | 650 | 4 | 36 L g−1 h−1 | CH4: 56 | A small amount of carbon nanotubes by TEM26 |
| CH4/CO2 = 1/1 (diluted at 10% each in Ar) | CO2: 70 | ||||
| Ni/HTNT | 700 | 5 | 12 L g−1 h−1 | CH4: 50 | Carbon filaments around the Ni nanoparticles by TEM79 |
| CH4/CO2/N2 = 1/1/8 | CO2: 48 | ||||
| Ni/NaTNT | 700 | 5 | 12 L g−1 h−1 | CH4: 79 | A low level of amorphous carbon by TRO79 |
| CH4/CO2/N2 = 1/1/8 | CO2: 78 | ||||
| Ni/La2O3 | 700 | 50 | 60 L g−1 h−1 | CH4: 70 | 5.47 × 10−5 mmol gcat−1 s−1 (ref. 65) |
| CH4/CO2/N2 = 15/15/70 | CO2: 75 | ||||
| Ni–Mo/MgO | 800 | 500 | 300 L g−1 h−1 | CH4: 80 | No carbon deposited by Raman spectra1 |
| CH4/CO2/N2 = 1/1/8 | CO2: 80 | ||||
| Ni/CaO | Carbonation: 600 | 10 | The CO2 flow of 2 mL min−1 for 6 min | CH4: 88 | Filamentous carbon by TGA80 |
| DRM: 800 | The CH4 flow of 2 mL min−1 for 6 min | CO2: 59 | |||
| 650 | 1000 min | Carbonation: CO2/N2 = 10% | CH4: 96 | No carbon deposition by Raman spectra81 | |
| DRM: CH4/N2 = 5% | CO2: 96.5 | ||||
| Ni/HAP | 800 | 200 | 40 L g−1 h−1 | CH4: 93.27 | No carbon deposition by Raman spectra82 |
| CH4/CO2/N2 = 1/1/8 | CO2: 78.38 | ||||
| 800 | 200 | 30 L g−1 h−1 | CH4: 95.28 | No deposited carbon observed by TGA12 | |
| CH4/CO2/N2 = 1/1/8 | CO2: 94.94 | ||||
| Ni–Co/HAP | 750 | 60 | CH4 (20 vol%) and CO2 (20 vol%) diluted in N2 (total gas flow rate of 90 mL min−1) | CH4: 73 | Only few carbonaceous species by TEM83 |
| CO2: 79 | |||||
Among the various supports, Al2O3 and SiO2 are the most widely investigated. Researchers have synthesized numerous well-dispersed Ni/Al2O3 and Ni/SiO2via various preparation precursors and synthesis methods to increase the surface area, such as direct hydrogen calcination in the calcination process,75 solvothermal method,76 and sequential treatment method.78,84 The coke deposition level of these prepared catalysts is reduced relative to the Ni/Al2O3 synthesized by the typical co-precipitation method of a random morphology, and the coke tolerance is greatly improved relative to the precipitation catalyst, which has small and randomLy distributed pores. The different crystalline phases suggest that α-Al2O3 is a hexagonal close-packed crystal and γ-Al2O3 is a cubic compact crystal, making Al2O3 present various characteristics.85 The Ni/γ-Al2O3 not only had a large BET surface area and pore volume, facilitating the Ni dispersion on the catalysts and enhancing its anti-coking ability, but maintained high CH4 and CO2 conversions, indicating no decrease in the DRM activity even with the availability of the deposited carbon on the catalysts.77,86
However, there remain two contemporary challenges facing Ni-based catalysts with mesoporous Al2O3 and SiO2 as supports.87 Firstly, 1-dimensional nanotubes SiO2 possess small pores with low specific surface and provide an easy interface to Ni aggregation. Secondly, it is difficult to form Ni–O–Si bonds in acidic conditions required for the materials synthesis where the Ni particles have higher propensity for aggregation.88,89 To solve these issues, researchers have turned to 2-dimensional and 3-dimensional silica. SBA-15 is a type of 2-dimensional silica distinguished by high specific surface areas, large pore sizes and space to support metal catalysts. The existence of silanol groups make them excellent candidates.22,24,48 Mesocellular silica foam (MCF) is another material developed from SBA-15 to address the challenges of forming Ni–O–Si bonds.26,90 It has a three-dimensional structure with a large uniform spherical cell and an open structure that favors mass transfer.91 The Ni/SBA-15 (ref. 49) and Ni/MCF26 catalysts presented excellent resistance to sintering and carbon deposition. The nickel particles are reconstituted to form small Ni particles that are evenly dispersed. It not only allows high dispersion and stabilization of nickel metal but also enhances the homogeneity and increases the accessibility to the active sites that is contributed by their specific textural feature.26
However, these supports do not take part in the DRM reaction; both CH4 and CO2 activation sites are on the nickel atoms,92 which is called monofunctional mechanism,24 resulting in the competed reactions, and thus would finally decrease the DRM reaction rate. To avoid the inactivation by this way, researchers have turned attention to materials that make CH4 dissociation and CO2 activation occur at different sites, which is called bifunctional mechanism, where CH4 dissociation occurs on Ni active sites and CO2 activation occurs on the supports.24 Titanate nanotubes (TNTs) are famous for their high specific surface area with high aspect ratio titanate nanostructures.93 By the acid wash process, the protonated form (HTNT) can be replaced by Na atoms (NaTNT). Methane dissociation occurs on nickel sites while CO2 activation occurs on the titanate nanotube supports over Ni/HTNT and Ni/NaTNT (Fig. 6). Ni–NaTNT displayed great coking resistance. It can be explained that the Na+ could migrate from the supports to nickel particles and inhibit carbon deposition.79 Despite the replacement of Na+ improving the number of actives sites for DRM in the titanate nanotubes, it has proven that the interaction of Na+ and Ni particles may lower the conversion.79
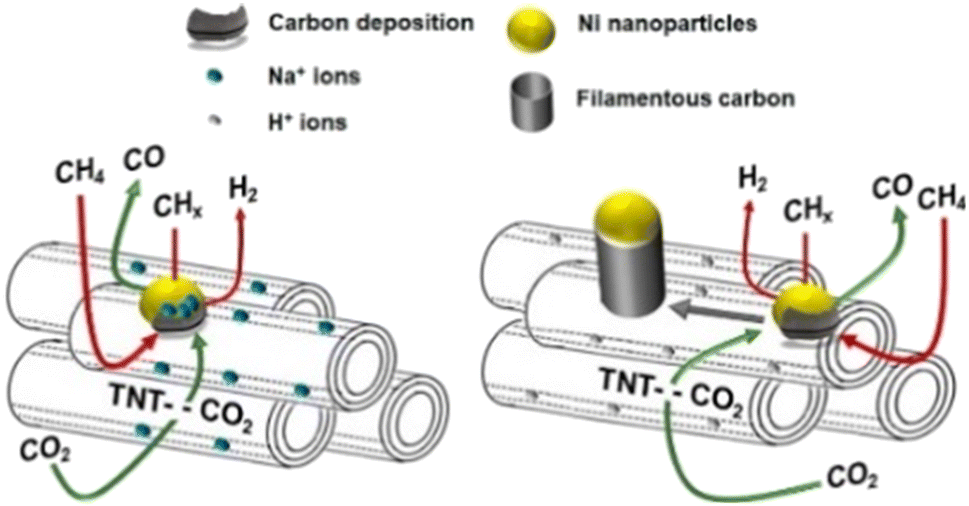 | ||
| Fig. 6 Schematic diagram of the DRM catalysed by Ni–NaTNT (left) and Ni–HTNT (right). Reproduced from ref. 79 with permission from Fuel, copyright 2019. | ||
Generally, the surface acidity and basicity also play a significant role in the performance of the catalyst for DRM.94,95 Coking is more prone to be induced by the supports with acidity, and CO2 activation is more prone to be introduced by basic supports because of the mild acidity of CO2.96 Ni/La2O3 follows the bifunctional mechanism and La2O3 reacts with CO2, accompanied by the formation of La2O2CO3, thus suppressing carbon deposition. La2O2CO3 can react with the deposited carbon to form CO and regenerate La2O3.97 To overcome the poor dispersion of active metal on the La2O3 surface, La2O2CO3 is used as the support precursor because this material would transform to La2O3 and La(OH)3 with the nanorod shape maintained in the synthesis process, which enhanced the anti-coking ability of the catalysts.58,65 La2O3 adsorbed CO2, forming La2O2CO3 (CO2 + La2O3 → La2O2CO3), and then La2O2CO3 reacted with C* from the CH4 dissociation to regenerate La2O3 with CO (La2O2CO3 + C* → CO + La2O3). By this mechanism, Ni/La2O3 would participate in the reaction with filamentous coke, inhibiting the transformation from filamentous coke to non-filamentous coke, which would cover the nickel active sites and cause a decrease in the activity and stability of DRM.98
Besides La2O3, Ni/MgO catalysts have also been widely recognized as efficient catalysts for DRM.99–102 The “Nanocatalysts On Single Crystal Edges” (NOSCE) technique was proposed according to the phenomenon that the Ni particle size was smaller in the initial reaction step and grew larger in the DRM reaction within a few moments (1 hour) but was locked on the same size after prolonged activity (500 hours).1 The modification is that the particulates move onto the high-energy step edges of the crystalline MgO (111) to form stable particles. This also prevents further sintering while eliminating the risk of MgO participation in the catalytic reaction by the coverage of high-energy step edges.103 Similarly, calcium oxide (CaO) is an excellent support because it is low-cost104,105 and has a high adsorption property toward CO2 (∼786 g of CO2 per kg of CaO (ref. 106 and 107)). The CaO support would make the catalyst size larger, but the conversion remains stable because CaO could eliminate the deposited carbon.80 Firstly, the CO2 was captured by CaO and formed CaCO3, illustrated as *CO2. Then the captured *CO2 reacts with deposited carbon on the CaO–Ni interface (*CO2 + C → 2CO), eliminating the carbon deposition. The temperature at which H2 generation from CH4 dissociation was appreciably lower than that of CO formation indicates that the interface is more inclined to activate CH4 than CO2.27 As previously mentioned, CH4 dissociation on Ni sites follows this rule: *CH4 → *CH3 + *H → *CH2 + *H → *CH + *H, then the produced *H consequently combine and form H2. But the presence of CO2 captured by CaO could make *H participate in the CO2 conversion by producing *COOH, which would dissolve into *CO and *OH. It is called *H-assisted *CO2 conversion. Subsequently, the *OH could participate in CH4 dehydrogenation through this way: *CH2 + *OH → *CH2OH → *CH2O + *H → *CHO + *H → *CO + *H. Therefore, the overflow of *OH produced in the *H-assisted *CO2 conversion can accelerate CH4 dehydrogenation, and this is named *OH-assisted CH4 dissociation. By this synergistic promotion, the coking resistance was improved significantly.34,81
Temperature-programmed reactions were employed to identify CH4 dehydrogenation and CO2 activation involved in the DRM process. Although the addition of CaO could enhance the DRM anti-coking ability over the Ni-based catalyst, the reaction ability decreases with the cycle number due to the sintering of materials. To get catalysts with anti-sintering and great performance for DRM over Ni/CaO, not only are the reduction of carbonates and CH4 dissociation but also the trade-off of the density of Ni nanoparticles and its size are required to be taken into consideration to achieve the expected adsorptive/catalytic interface as the increase in Ni nanoparticles would cause increased average sizes of Ni nanoparticles and decreased specific surface area.81,108
In addition to the supports mentioned above that enhance the performance of DRM over Ni-based catalysts by metal–support interaction, Ni particles can form a structure of solid solution by replacing the metal atoms on the supports, which contributes to homogenously dispersed Ni nanoparticles. Hydroxyapatite (HAP, Ca10(PO4)(OH)2) is a weak alkaline calcium phosphate.82,83,109 It has strong modifiable properties, where the Ni nanoparticles replace the external Ca atoms in HAP owing to its capability of forming hydroxyapatite structures over a wide Ca/P range, and the hydroxyl groups in HAP can oxidize the carbon formed by CH4 dissociation.12,110 HAP-D (HAP-D refers to the HAP inherently without Ca) facilitates a robust anchor positioning for the Ni particles and enhances the oxygen migration of hydroxyl groups in HAP. A novel scheme (Fig. 7) was proposed where CH4 dissociation is more inclined to CH3O following dehydrating to CO without coking rather than cleaving the bonds of C–H to carbon deposition. Subsequently, the oxygen vacancies generated are replenished through the dissociation of adsorbed CO2 as they are refilled with oxygen atoms. Undoubtedly, the matching between the rate of progressive cleavage of CH4 on the metal and the rate of the intermediate cleavage products oxidized by the oxygen atoms in the support plays a critical role in determining the performance. Also, the matching between the two aspects is much better for Ni/HAP without the problem of coking.111
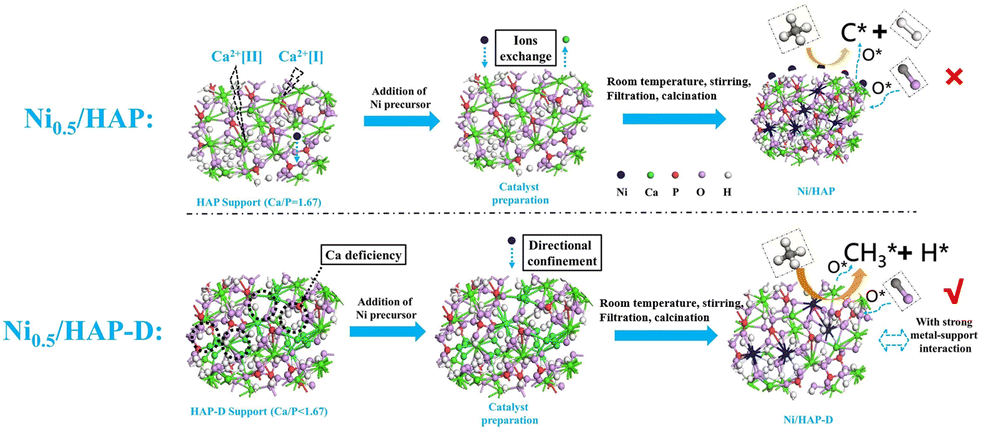 | ||
| Fig. 7 Schematic diagram of catalyst synthesis path and DRM reaction route. Reproduced from ref. 12 with permission from Fuel, copyright 2022. | ||
Promoter selectivity
The performance of Ni-based catalyst in the DRM reaction is not decided only by the Ni particles dispersed on the support but also the promoter.30,112 Depending on the species, promoters applied in DRM over Ni-based catalysts could be normally divided into metals and rare earth metallic oxides. These promoters can not only regulate the physical (changing the electronic density, maintaining small particle size in reaction, and affecting the catalytic surface acidity and basicity) and chemical (providing oxygen vacancies and enhancing the MSI) properties of the nickel particles but also participate in the process of CH4 dissociation or CO2 adsorption, thereby improving the performance or enhancing the resistance to carbon deposition.113Metals
Diverse metals are employed to enhance the catalytic activity and selectivity as well as to prevent the sintering of nickel particles.13Table 4 summarizes the reaction conditions and CH4 and CO2 conversions by different metal promoters.Metals such as Co and La have a beneficial effect on nickel-based catalysts by alloying with nickel particles, which is typically attributed to the similar surface electronical and geometric chemistry with nickel particles.13,114 Based on in situ XPS analysis, the doped metals (Co, La) donate electrons to the Ni atoms, changing the chemical properties by lowering the Ni average valence state and enhancing the CO2 dissociation process.38,115 The formed Ni–Co solid solution alloy can not only favor the reduction of the nickel species to Ni0 by increasing the active sites in the fresh Ni–Co/Al2O3–S catalyst38 but also improve the CO2 dissociation ability with a high Ni average valence state in carbonation (Fig. 8B), which would subsequently react with C* and promote H2 desorption at lower temperature (Fig. 8D). It inhibited the side RWGS reaction to give a high H2/CO ratio.38 However, in the last reaction cycles, the Co doped in the catalyst is more prone to oxidize the Ni particles to NiO, the inactive sites which decrease the catalytic activity.40 Thus, the self-oxidation process is a main challenge for the method of Co modification. Both the nickel valence state and the formation of solid solution alloy may strengthen the MSI.116 Ni–La bimetallic catalysts have been extensively researched for enhancing the carbon resistance and activity of DRM. The presence of La promotes the formation of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and Tran-2:1 nickel phyllosilicate, revealing a robust interaction with the SiO2 support. This strong interaction ensured a high reduction degree (Ni0) of Ni particles. Thus, sufficient nickel particles are exposed with basic sites in Ni–La/SiO2 interface, which favors the removal of coking, and this is the important cause for the attenuation of catalyst deactivation.117,118
:1 and Tran-2:1 nickel phyllosilicate, revealing a robust interaction with the SiO2 support. This strong interaction ensured a high reduction degree (Ni0) of Ni particles. Thus, sufficient nickel particles are exposed with basic sites in Ni–La/SiO2 interface, which favors the removal of coking, and this is the important cause for the attenuation of catalyst deactivation.117,118
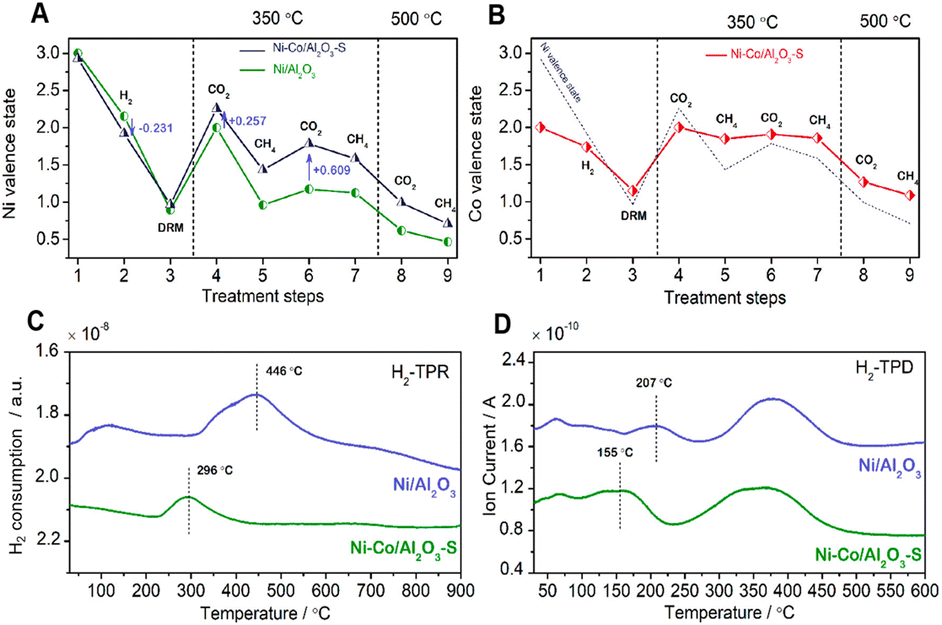 | ||
| Fig. 8 Ni average state change of (A) Ni/Al2O3 and Ni–Co/Al2O3–S (prepared by stepwise impregnation) and Co average state change of (B) Ni–Co/Al2O3–S catalyst (with Ni average state change as a reference (dashed line)) with different treatment steps: (1) the as-prepared catalysts; (2) after reduction in hydrogen at 300 °C; (3) after DRM reaction at 800 °C; (4–7) sequential treatment with CO2 (4 and 6) or CH4 (5 and 7) at 350 °C; (8 and 9) sequential treatment with CO2 or CH4 at 500 °C. H2-TPR (C) and H2-TPD (D) results of Ni/Al2O3 and Ni–Co/Al2O3–S catalysts. Reproduced from ref. 38 with permission from American Chemical Society Catalysis, copyright 2019. | ||
It is worth noting that the metals are also favorable for increasing the activity and resistance to carbon deposition by effecting the process of CO2 activation.119,120 Gadolinium (Gd) has the capacity to generate oxygen vacancies, serving as active sites to adsorb CO2. This facilitates the extraction of oxygen from CO2, thereby promoting the dynamics of the DRM reaction.121,122 The promotional effect of Gd could be contributed to the formation of Gd2O2CO3, facilitating the CO2 adsorption and activation. Along with this, Gd also maintains the size of Ni particles by further promoting the stability.123 Meanwhile, Sc in the optimum range strengthened the interaction of Ni with the support. This enhancement of basicity subsequently influences the quantity of CO2 adsorbed on the catalytic surface, thus improving the DRM performance.124
On the other hand, Ni particles are easily oxidized to NiO during the process of CO2 reduction. To slow down the deactivation, some transition metal elements are doped into the catalysts, inhibiting the oxidation of Ni sites. Taking molybdenum (Mo) for example, both CH4 and CO2 conversion of catalysts with Mo was much higher than the ones without Mo.1 According to the shifts of Ni peaks (Fig. 9), it is reasonable that there is a process of electron transfer from nickel atoms to Mo atoms within the alloy structure, revealing the strong electronic interaction between Mo and Ni.125 It indicates that the Mo shows more affinity to *O than Ni, which is consistent with the DFT results (Fig. 10). At the same time, Mo also provides unique dynamic variation of MoOx → MoCxOy, enabling efficient carbon elimination during the DRM reaction.
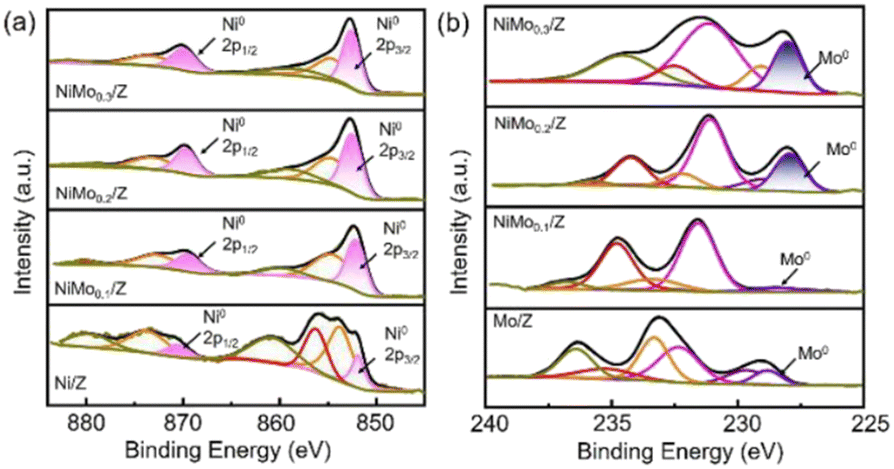 | ||
| Fig. 9 The ex situ spectra (a) Ni0 2p and (b) Mo0 3d XPS spectra of the reduced catalysts. Reproduced from ref. 125 with permission from American Chemical Society Catalysis, copyright 2021. | ||
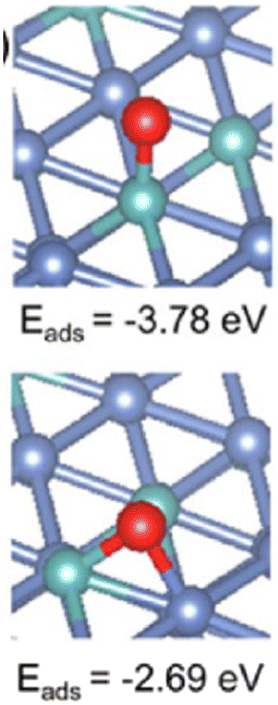 | ||
| Fig. 10 Oxygen adsorption energy on NiMo(111) (Ni: blue, Mo: green, and O: red). Reproduced from ref. 125 with permission from American Chemical Society Catalysis, copyright 2021. | ||
Besides the process of CO2 activation, the associative “formate” mechanism is also displayed for some other metals. To eliminate the carbon deposition, it is attractive to turn the CH4 dissociation intermediates of carbonate species (*CHx) to formate species (HCOO*), which can be achieved by metals like Mo (ref. 125) and Cs.126 According to the DFT calculation, the high carbon deposition resistance is attributed to the well-dispersed Cs dopants, which suppress *CH dehydrogenation (*CH → C + *H) where the *C is commonly considered as the precursor to carbon deposition, and enhance the *CH oxidation (*CH + *O → *CHO). The Cs dopants increased the energy difference to 0.1 eV between *CH dehydrogenation and*CH oxidation. This leads to more favorable *CH oxidation, whereas these two processes (*CH dehydrogenation and *CH oxidation) simultaneously occurred in the catalysts without Cs doping.
Another significant aspect of promotion is to decelerate the rate of CH4 dehydrogenation and concomitantly accelerate carbon gasification. According to DFT calculation, the promotion of Fe (ref. 127) and Pt (ref. 128) in Ni-based catalysts increases the kinetic barrier for CH4 activation, simultaneously decelerating the rate of carbon deposition. Moreover, Fe enables the nickel size into small ensembles, while the coking resistance is notably better with small particles due to the hindrance of surface carbon deposition.127,129 By the way, Fe is oxyphilic and could be oxidized by CO2 to FeO (CO2 + Fe → CO + FeO), then the carbon formed by CH4 dissociation is oxidized to CO by the lattice oxygen from FeO (C + FeO → CO + Fe) (Fig. 11).61,71,130 In a conclusion, the effect of Fe can be attributed to the following: (a) slow down the coking rate and concomitantly accelerate the carbon gasification rate; (b) enable small nickel size to change the carbon species to a type easily oxidized; (c) increase the oxyphilic property of the material.
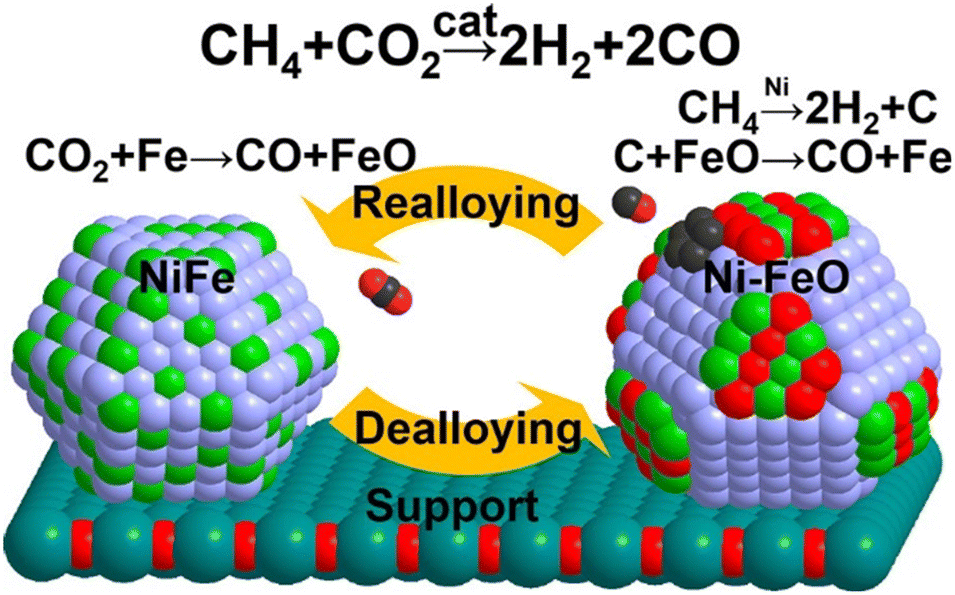 | ||
| Fig. 11 De-alloying and re-alloying behaviour of a Fe–Ni-alloyed particle during the dry reforming of methane. Reproduced from ref. 130 with permission from Journal of American Chemical Society, copyright 2017. | ||
To achieve excellent promotion, metal loadings is an important factor that must be considered. A low content would minimize the promotion, whereas a high content would cause a decrease in the conversion resulting from the coverage of active sites by the metals. Taking Fe as an example, the Fe/Ni ratio significantly determines the promotion or suppression of the catalytic activity. When the molar ratio is above 0.9, the mesoporous structure would be destroyed, thus decreasing the catalytic activity, whereas the activity of catalysts is the highest and the deactivation is the lowest when the molar ratio is 0.7.131 Besides Fe, scandium (Sc) also faces the same problem. The optimum range of Sc loading was 0.1–0.5%. An Sc content above 0.5% would cover the Ni active sites and result in a decrease in CH4 and CO2 conversion.69
In general, the scarce information concerning the role of transition metals in DRM is largely due to the absence of techniques capable of concurrently examining the alteration in electronic structure and visualizing the morphology of the active sites under the operational conditions.115 This challenge remains and inhibits the further understanding and study of synthesizing catalysts with superior properties.
Rare earth metallic oxide
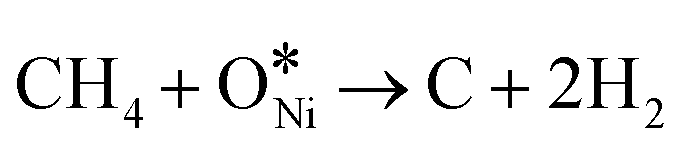
In recent years, rare earth metallic oxides as promoters have attracted attention for the enhancement in CO2 adsorption and activation to produce CO by maximizing the catalytic oxygen vacancies.129 The vacancies are normally raised from the inherent point defects on metallic oxide catalysts exposed to elevated temperatures and a reducing atmosphere.132 The oxygen vacancies are the potential sites for the adsorption and activation of CO2.133,134 The mobile oxygen on the catalyst surface can react with *C and form CO and is in turn filled by the oxygen from the adsorbed CO2 dissociation.23,39,40 The process of DRM on the Ni-based catalysts with oxygen vacancies includes the following steps (Table 3): a) oxygen migration from the rare earth metallic surface (OsurfaceCGO) to the nickel surface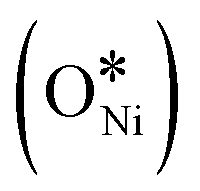 (eqn (10)); b) methane decomposition into carbon and hydrogen (H2) at the Ni sites (eqn (11)); c) methane oxidation into CO2 with the production of H2O (this process only occurs when the oxidation state of the ceria is high) (eqn (12)); d) decomposed carbon oxidation partially into CO (eqn (13)); e) oxygen migration from bulk (ObulkCGO) to surface (OsurfaceCGO) (eqn (14)); f) amorphous carbon (Camorphous) formation (eqn (15)).135–137Table 5 summarizes the reaction conditions and CH4 and CO2 conversions by different rare earth metallic oxides.
(eqn (10)); b) methane decomposition into carbon and hydrogen (H2) at the Ni sites (eqn (11)); c) methane oxidation into CO2 with the production of H2O (this process only occurs when the oxidation state of the ceria is high) (eqn (12)); d) decomposed carbon oxidation partially into CO (eqn (13)); e) oxygen migration from bulk (ObulkCGO) to surface (OsurfaceCGO) (eqn (14)); f) amorphous carbon (Camorphous) formation (eqn (15)).135–137Table 5 summarizes the reaction conditions and CH4 and CO2 conversions by different rare earth metallic oxides.
| Equation | Reaction equation |
|---|---|
| 10 |
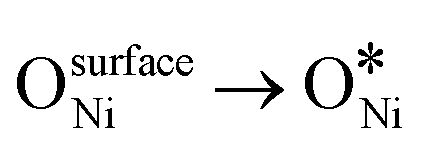
|
| 11 |
|
| 12 |
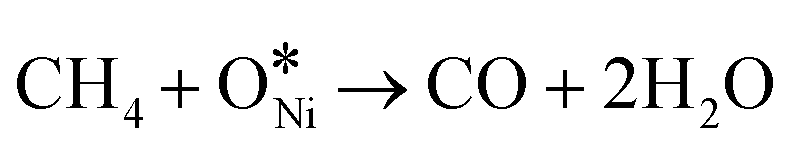
|
| 13 |
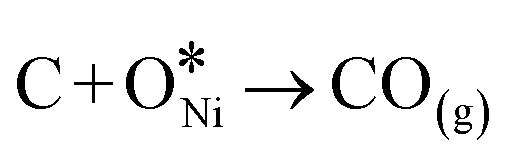
|
| 14 | ObulkCGO → OsurfaceCGO |
| 15 |
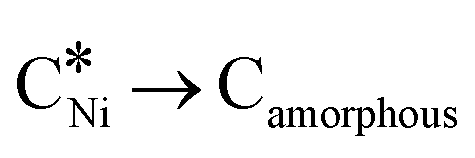
|
| 16 |

|
| Catalyst | Temp (°C) | Time (h) | GHSV and feed gas radio | Conversion (%) | Carbon deposition |
|---|---|---|---|---|---|
a GDC refers Ce0.8Gd0.2O2−δ.
b It represents catalyst surface carbon coverage radio.
c It represents La0.6Sr0.2Ti0.85Ni0.15O2.95.
d
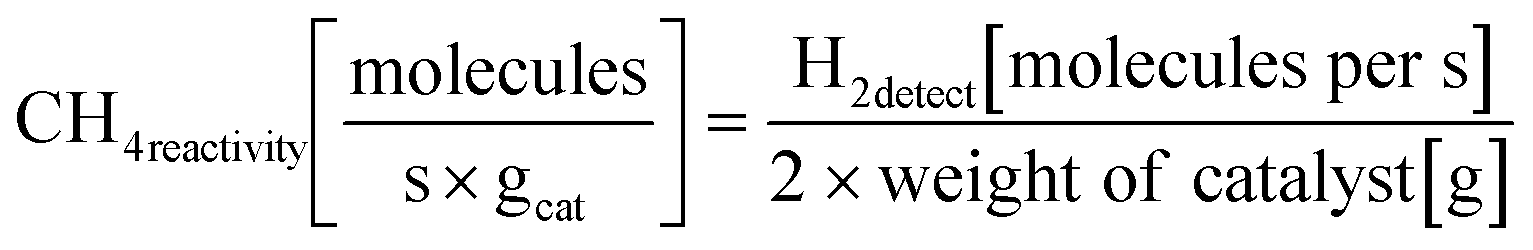 . .
|
|||||
| Ni–Co/γ-Al2O3 | 800 | 15 | Flow rate: 490 mL min−1 | CH4: 79 | Filamentous carbon formed by STEM115 |
| A gas mixture of 7% CH4 and 9.5% CO2 in N2 | CO2: 71 | ||||
| Ni–Co/Ce0.75Zr0.25O2−ð | 750 | 20 | 25 L g−1 h−1 | CH4: 60 | Low carbon deposition40 |
| CH4/CO2/N2 = 1/1 = 3 | CO2: – | ||||
| Ni–Co/Al2O3–S | 800 | 60 | 998.4 L g−1 h−1 | CH4: 92 | No surface carbonate by XPS38 |
| CH4/CO2/N2 = 1/1/8 | CO2: – | ||||
| Ni–Co–Al–Mg–O | 750 | 250 | 180 L g−1 h−1 | CH4: 0/64 mmol gcat−1 | No carbon formation by TG114 |
| CH4/CO2/N2 = 1/1/1 | |||||
| Ni–Gd/MCM | 800 | 400 min | 39 L g−1 h−1 | CH4: 87.6 | No carbon formation by TG123 |
| CH4/CO2 = 1/1 | CO2: 91.7 | ||||
| Ni–GDCa | 800 | 100 | 28800 h−1 |
CH4: 95 | Graphitic carbon by TGA121 |
| CH4/CO2/N2 = 1/1/1 | CO2: 95 | ||||
| Ni–Sc/MCM-41 | 800 | 400 min | 39 L g−1 h−1 | CH4: 85 | Carbon deposition decreased by TGA69 |
| CH4/CO2 = 1/1 | CO2: 90 | ||||
| Ni–Mo/CaO | 750 | 500 | 300 L g−1 h−1 | CH4: 81 | No filaments carbon by TEM1 |
| CH4/CO2/N2 = 50/50/20 mL min−1 | CO2: 75 | ||||
| Ni–Mo/ZSM-5 | 750 | 200 | 50 L g−1 h−1 | CH4: 90 | 16.8 mmolC molsurfaceNi−1 h−1 (ref. 125) |
| CH4/CO2 = 50/50 mL min−1 | CO2: 96 | ||||
| Ni–Cs/Al2O3 | 800 | 250 min | 72 N L g−1 h−1 | CH4: 89.8 | No carbon deposition by TEM126 |
| CH4/CO2/N2 = 1:1:2 |
CO2: 91.8 | ||||
| Ni–Fe/MgO | 800 | 100 | 86 L g−1 h−1 | CH4: 89 | 18.84 mol%b127 |
| CH4/CO2 = 1/1 | CO2: 93 | ||||
| Ni–Fe/LSTINc | 900 | 400 | 30 L g−1 h−1 | CH4 reactivity = 1.65 × 1019d | Not mentioned138 |
| CH4/CO2/He = 10:10:80 mL min−1 |
|||||
| Ni–Fe/MA | 650 | 5 | 270 L g−1 h−1 | CH4 consumption = 20.4 mmol gcat−1 min−1 | 18 wt%130 |
| CH4/CO2/N2 = 45/45/10 | |||||
| Ni–Fe/MgAl2O4 | 800 | 250 min | 1 mL s−1 | CH4: 51 | Filamentous carbon deposition by TSG71 |
| CH4/CO2 = 1/1 | — | ||||
| Ni–Pt/Ce0.8Pr0.2O2−δ | 750 | 50 | 30 L g−1 h−1 | CH4: 95 | 0.13 mg gcat−1 (ref. 128) |
| CH4/CO2/He = 20/20/60 | — | ||||
| Ni–Pt/CeZrO2 | 800 | 15 | 120 L g−1 h−1 | CH4: 93 | Carbon deposition by TGA139 |
| CH4/CO2/He = 10/10/20 mL min−1 | CO2: 96 | ||||
| Catalyst | Temp (°C) | Time (h) | GHSV and feed gas radio | Conversion (%) | Carbon deposition |
|---|---|---|---|---|---|
| a SCCM refers to the standard cubic centimeter per minute. | |||||
| Ni–CeO2/Al2O3 | 800 | 72 | CH4/CO2 = 45/45 mL min−1 | CH4: 78 | 60 mgc gcat−1 (ref. 39) |
| CO2: 81 | |||||
| Ni/CeO2 | 700 | 30 | CH4/CO2/Ar = 20/20/60 mL min−1 | CH4: 44 | Negligible amount of carbon with graphitic form by XRD149 |
| CO2: 57 | |||||
| Ni–Co/CeO2 | 700 | 5 | 1 atm, 50 sccm CH4/CO2/Ar = 1/1/3a | CH4: 70 | Carbon filaments by HCTEM140 |
| CO2: 70 | |||||
| Ni/CeO2 | 750 | 10 | 2700 L g−1 h−1 | CH4: 94 | Carbon nanofiber by SEM136 |
| A gas mixture of 50%Ar:25%CH4:25%CO2 (volume) |
CO2: – | ||||
| Ni–CeO2/MgO | 800 | 12 | 720 L g−1 h−1 | CH4: 78 | Carbon deposition suppressed by XRD150 |
| CH4/CO2/N2 = 1/1/1 | CO2: – | ||||
| Ni–CeO2/HAP | 750 | 7 | 60 L g−1 h−1 | CH4: 82 | Negligible carbon deposition by TGA151 |
| CH4/CO2/He = 10/10/30, total flow = 50 mL h−1 | CO2: 78 | ||||
| Ni/ZrO2 | 700 | 5 | 24 L g−1 h−1 | CH4: 51 | Carbon formation by TG146 |
| CH4/CO2 = 1/1 | CO2: 62 | ||||
Ceria (Ce) and ceria-containing materials have been extensively applied for DRM over Ni-based catalysts. This is attributed to their feasible reducibility and high oxygen storage capacity (OSC). They can switch between Ce3+ and Ce4+ with the variation of oxygen vacancies. They favour forming Ce3+ with the formation of oxygen vacancies at a high temperature.140 Ceria doped with lower valence state has the advantage of exhibiting high oxygen mobility and the capacity to retain its crystal structure with a significant degree of ceria reduction, which contributes to improved carbon deposition resistance.136 To analyse the activity–structure relationship in detail, a facile methane transient pulse experiment was conducted under the ambient pressure to test the impregnated Ni–Gd/CeO2 (Ni/CGO) (Fig. 12).135,141 It has founded that there are two competing oxidation processes for CO production.34 The first one is methane dehydrogenation and carbon species (C/CHx) being oxidized by the oxygen vacancies from CGO. The second one is where C/CHx is oxidized by the oxygen ions migrating from the bulk of CGO after the surface oxygen is exhausted. Thus, there is no carbon deposition if the time is long enough to allow oxygen migration. Meanwhile, a low concentration of steam can replenish the surface oxidation states, which controlled the oxygen migration and CO selectivity without influencing the ability and stability of DRM.135,136
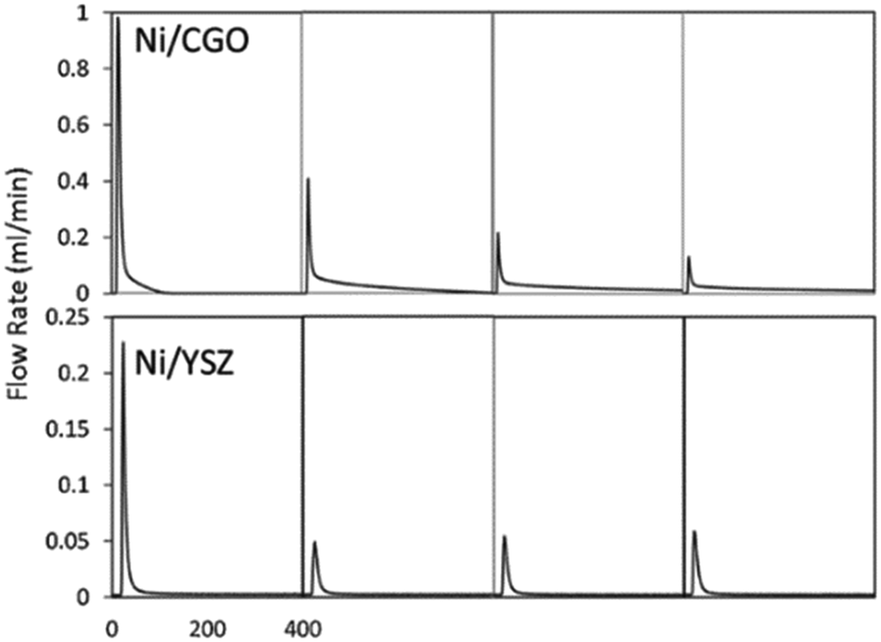 | ||
| Fig. 12 The first to fourth CO production peak of a methane pulse on Ni/CGO and on Ni/YSZ. Reproduced from ref. 135 with permission from Applied Catalysis B: Environmental, copyright 2019. | ||
ZrO2 has high stability and surface oxygen mobility.142 It has been applied in the DRM as promoter143 or support,144,145 contributing to its chemical properties of basicity, which is beneficial for the elimination of deposited carbon by the mobile oxygen from the surface oxygen vacancies.146,147 The oxygen vacancies of ZrO2 include the oxygen species (Table 6) such as the lattice oxygen (OI) and the adsorbed oxygen (OII).92,148 OII is favorable for promoting both CO2 adsorption activation and CH4 dissociation by changing the surface acidity and basicity. To increase the radio of OII/OI, the Ni/ZrO2 materials can be treated with H2 atmosphere.
| Treatment atmosphere | The lattice oxygen (OI) area (%) | The adsorbed oxygen (OII) area (%) | OII/OI |
|---|---|---|---|
| H2 | 58.57 | 41.43 | 0.71 |
| N2 | 60.25 | 39.75 | 0.66 |
| O2 | 63.61 | 36.39 | 0.57 |
Preparation
Using unconventional preparation methods or changing the reaction process to modify the interaction between metals and supports has always been a challenging issue.Impregnation
Impregnation (IM) method is an effective synthesis method. Table 7 summarizes the properties and performance of the catalysts prepared by IM. In the impregnation method, the active solid precursor is immersed in a solution of certain concentration of the active phase precursor for different periods, and then the solution is dried subsequently.152 According to the amounts of solution, the IM can be categorized into wet impregnation (WI) and incipient wetness impregnation (IWI). WI uses an excess solution while IWI uses a limited amount of solution where the volume of the solution is approximately equal or slightly lower than that of the solid.153 Due to the restricted solution diffusion in the catalyst pores, IWI produces the least dispersed metal particles, whereas WI produces more dispersed particles due to the excess solvent.12,154| Catalyst | Method | Nickel loading (wt%) | Particle size (nm) | Time (h) | CH4 conversion (%) | CO2 conversion (%) | Ref. |
|---|---|---|---|---|---|---|---|
| a HMS refers to hexagonal mesoporous silica. | |||||||
| Ni/Al2O3 | IM | 6 | 12.5 | 100 | 80 | 78 | 148 |
| Ni/Al2O3 | IM | 10 | 12.6 | 30 | 68 | 79 | 155 |
| Ni/Al2O3 | IM | 12 | 24.5 | 80 | 55 | 70 | 156 |
| Ni/Al2O3 | IM | 5 | — | 20 | 58 | 70 | 52 |
| Ni/SiO2 | IM | 5 | 6 | 20 | 82 | 89 | 52 |
| Ni-CeO2/Al2O3 | IM | 7 | 6.9 | 80 | 47 | 72 | 157 |
| Ni/SiO2 | IM | 5 | 18 | 200 | 30 | 45 | 158 |
| Ni/HMSa | IM | 5 | 16.5 | 100 | 64 | 41 | 159 |
Generally, in the catalysts prepared by IM, the nickel particles has weak MSI and may be located in the exterior pores.160 The nickel active sites are easily carried away with steam. In most instances, the catalysts are tested as control groups with other catalysts from evaporation-induced self-assembly and sol–gel methods. Some researchers also perform pretreatment before the catalysts are experimented in the DRM process. With hydrogen treatment before the calcination, the synthesized Ni/SiO2 were highly dispersed and uniformly distributed with the availability of small Ni particles with a strong MSI. This was attributed to the elimination of oxygen in the hydrogen treatment, and (phyllo)silicate structure was formed in the following calcination process.78
Evaporation-induced self-assembly
Table 8 summarizes the properties and performance of the catalysts prepared by the evaporation-induced self-assembly (EISA) method. This method involves the continuous mixing of the solution with the structure-directing agents (SDA), followed by the evaporation and the initiation of co-assembly with ordered mesopores.154Fig. 13 depicts the steps involved in the preparation of the materials.161| Catalyst | Method | Nickel loading (wt%) | Particle size (nm) | Time (h) | CH4 conversion (%) | CO2 conversion (%) | Ref. |
|---|---|---|---|---|---|---|---|
| a The materials have a core-shell structure. | |||||||
| Ni/Al2O3 | EISA | 6 | 11.2 | 100 | 94 | 90 | 162 |
| EISA | 10 | 3 | 30 | 93 | 98 | 155 | |
| EISA | 12 | 15.6 | 80 | 92 | 91 | 156 | |
| Ni@SiO2a | EISA | 5 | 4.3 | 20 | 90 | 90 | 52 |
| Ni–CeO2/Al2O3 | EISA | 7 | 5.2 | 80 | 83 | 85 | 157 |
| Ni/SiO2 | EISA | 5 | 5 | 200 | 79 | 70 | 158 |
| Ni-HMSb | EISA | 5 | 3.90 | 100 | 85 | 77 | 159 |
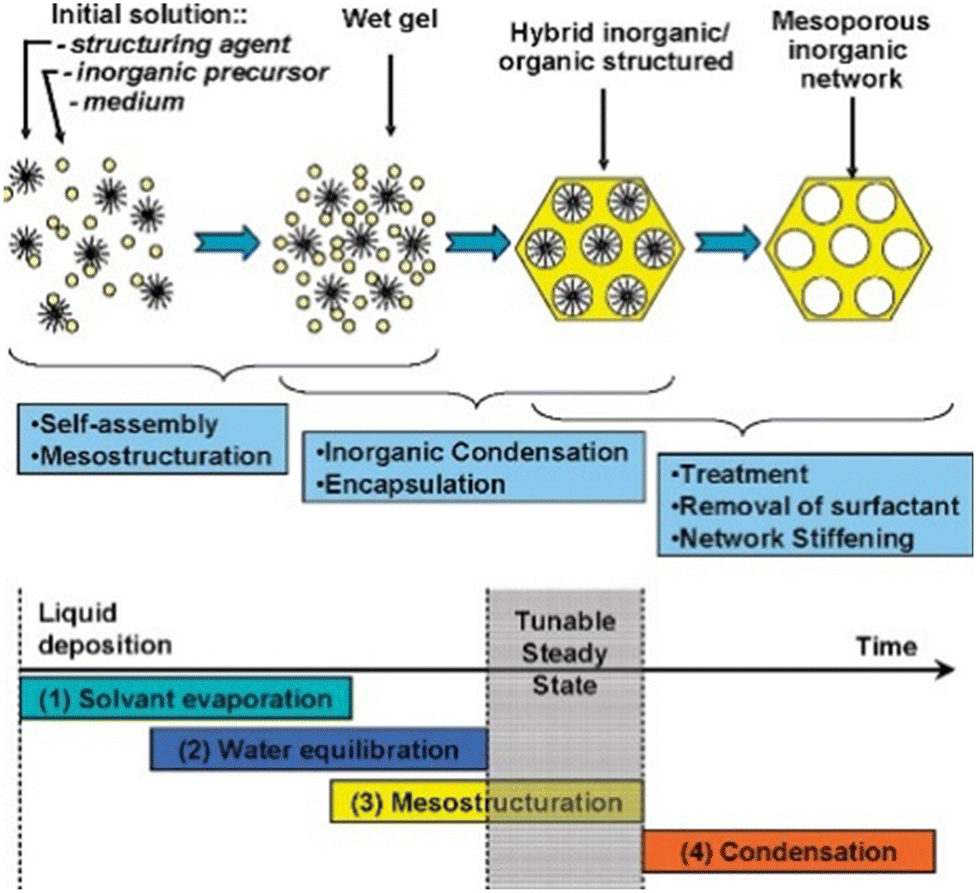 | ||
| Fig. 13 The scheme process of the EISA method. Reproduced from ref. 161 with permission from Chemical of materials, copyright 2008. | ||
The formed mesoporous catalysts are highly active and stable, with the support pore size of 3–5 nm after reaction for 30 h, whereas the pore size increased to 12 nm for the catalysts without the assistance of spray pyrolysis. The difference in size would further influence the carbon resistance, where the filamentous carbon was more difficult to form on the surface of catalysts with spray pyrolysis.155
Sol–gel method
Table 9 summarizes the properties and performance of the catalysts prepared by the sol–gel method. The sol–gel method (SG) scheme process starts by preparing a gel using precursors and complexing agents. The gel is then dried and pre-treated to form the final compounds as the catalysts (Fig. 14). In general, the particle size is large than those obtained from EISA.154 Thus, the produced catalysts are mostly applied in the situation where promoters are added. Generally speaking, the metal (CoNiAl2O4,163 Ni–Co/Al2O3–MgO–ZrO2,164 CoWNi/Al2O411), alkaline earth metal oxide (Ni–MgO/Al2O3165) and rare earth metal oxide (Ni–CeO2/Al2O3,165,166 Ni/CeO2167) could be synthesized via the sol–gel method. The detailed mechanism of the promoters has been illustrated in section 3.2.| Catalyst | Method | Nickel loading (wt%) | Particle size (nm) | Time (min) | CH4 conversion (%) | CO2 conversion (%) | Ref. |
|---|---|---|---|---|---|---|---|
| a The materials are structure of core-shell. | |||||||
| Ni/Al2O3 | SG | 10 | 35.3 | 600 | 70 | — | 166 |
| CoNiAl2O4 | SG | 10 | 14.7 | 2880 | 96.5 | 94.5 | 163 |
| Ni–Co/Al2O3–MgO–ZrO2 | 10 | 17.1 | 1440 | 58 | 97 | 164 | |
| CoWNiAl2O4 | SG | 10 | 27.6 | 2880 | 78.5 | 75 | 11 |
| Ni–MgO/Al2O3 | SG | 10 | 24.7 | 1440 | 95.0 | 63.0 | 165 |
| Ni–CeO2/Al2O3 | SG | 10 | 23.3 | 1440 | 98 | 99 | 165 |
| Ni–CeO2/Al2O3 | SG | 10 | 35.9 | 600 | 90 | 95 | 166 |
| Ni/CeO2 | SG | 15 | 11 | 700 | 22 | 28 | 167 |
| Ni@CeZrO2a | SG | 10 | 8 | 1440 | 42 | 54 | 168 |
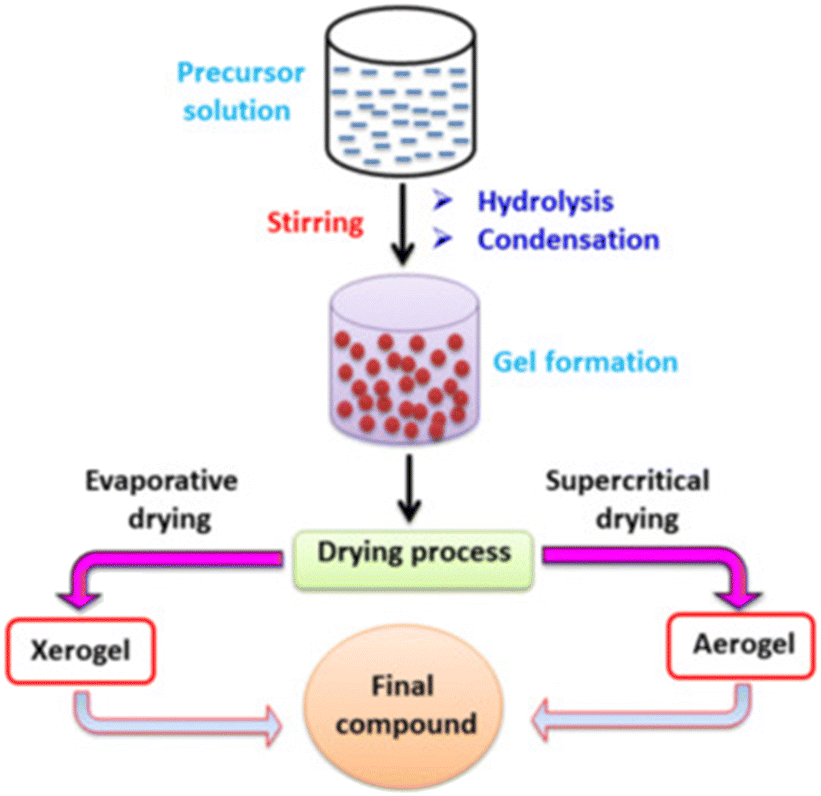 | ||
| Fig. 14 The scheme process of the sol–gel method. Reproduced from ref. 153 with permission from Journal of Environmental Chemical Engineering, copyright 2021. | ||
The plasma treatment is also used in the sol–gel method by producing catalysts of high specific areas, strong MSI, but small particle size (Fig. 15). NCA (SGP) (refers to Ni–CeO2/Al2O3 from sol–gel-plasma methods) showed great stability with the conversion dropping to 97% after 2880 min. The plasma treatment influenced the crystalline structure that lowered the growth rate of coke aggregation. Meanwhile, high energy species produced during plasma treatment acted as potent reducing agents, participating in both coking gasification and the reduction of the active phases in their oxidized state.163
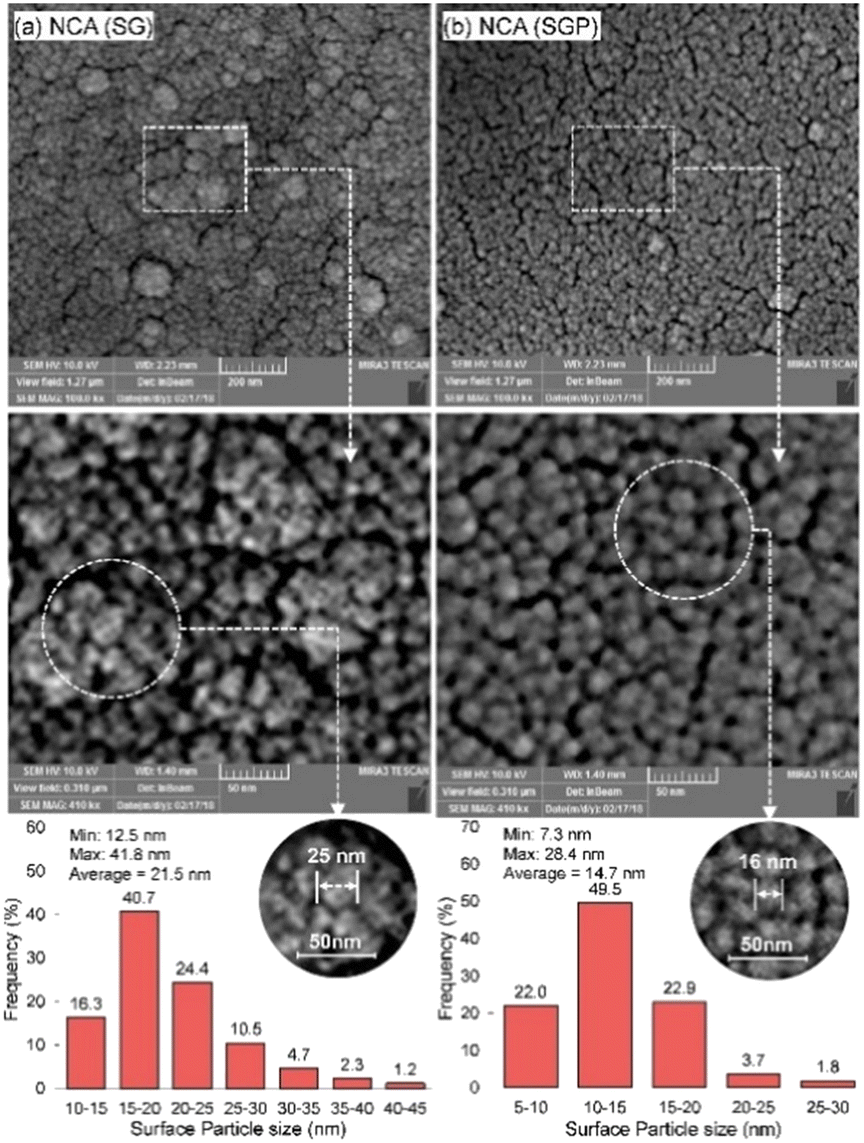 | ||
| Fig. 15 The surface particle size histogram of Co-promoted Ni-spinel nano-catalysts fabricated via sol–gel and hybrid sol–gel-plasma methods: (a) NCA (SG refers to catalysts from the sol–gel method) and (b) NCA (SGP refers to catalysts from sol–gel-plasma methods). Reproduced from ref. 163 with permission from Chemical Engineering journal, copyright 2019. | ||
Prospects
Dry reforming of methane (DRM) reaction is currently receiving renewed attentions for carbon capture, utilization & storage (CCUS) owing to its advantages of simultaneous reaction with CH4 and CO2 by forming H2 and CO, which are the ideal products for industries. This review discussed the mechanism of DRM over Ni-based catalysts, the issues and the advances of performance enhancement of the catalysts for the promotion of the technology. Owing to the fact that Ni is an active cost-effective metal for DRM, however, with propensity for sintering and carbon deposition that inhibits its performance, significant strategies have been promoted for strengthening the performance of Ni-based catalysts such as incorporating supports and doping promoters by various synthesis methods.The effects of supports on catalysts include the confinement and change of active sites. The supports with mesopores structure would improve the dispersion of active sites. It confines the Ni particles by its uneven surface and pore structure, preventing particle sintering in the DRM process. Moreover, some support species tend to promote CO2 adsorption and activation by offering more CO2 adsorption sites. These supports are mostly alkaline and widely used to suppress the CO2 adsorbed on the nickel sites but adsorb on the support sites. It avoids the competing reactions between CH4 dissociation and CO2 reduction. Also, it favors CH4 dehydrogenation to form *COOH and avoids the formation of *C (which is the precursor of deposited carbon). However, it is also worth noting that the ideal supports cannot react with the active sites, otherwise the activation would decrease.
According to the species, the promoters can be divided to two categories, namely, metals and rare earth metallic oxides, based on different functions. The influences of metallic promoters could be attributed to these aspects: (a) increasing the surface area; (b) facilitating CO2 adsorption and dissociation; (c) inhibiting the side reactions and suppressing CH* dissociation (*CH → *C + *H); (d) maintaining the Ni particle size; (e) modifying the nickel electronic properties and crystal phase. A single metal promoter incorporated into the catalysts usually comprises several of the above aspects. Rare earth metallic oxides have the distinctive characteristic of oxygen vacancies. They favour CO2 dissociation by offering more adsorbed sites, and the mobile oxygen could react with the carbonates, thus prolonging catalytic lifetime by avoiding coke deposition.
Catalysts prepared by different synthesis methods may present huge differences in the physical and chemical properties. In general, catalysts prepared by EISA and sol–gel methods are normally stable with highly dispersed and small Ni particles strongly interacting with the supports. The catalytic performance of the catalysts prepared from the EISA method largely depends on the type of structure-directing agents (SDA). The vaporization of SDA influences the MSI and consequently affects the activation and stability. On the other hand, the catalysts fabricated by the sol–gel method are featured with larger size (mostly above 10 nm) than those by the EISA and impregnation methods due to the weaker MSI. But the interaction of nickel particles and promoters would be strengthened, and active sites are more dispersed. The impregnation method is a convenient and promising one for catalyst preparation; however, the Ni particles are normally located at the exterior pores of the supports due to the main mechanism of diffusion. To enhance its performance, pre-treatments such as plasma treatment and spray pyrolysis could be used, which have been proven useful in another field.
The morphology, structure and composition are important factors that should be considered to design remarkable Ni-based catalysts. The synergistic effect of support, promoters and various synthesis methods is also an important point required for consideration.
The different performance of catalysts prepared with the same method is attributed to the different reaction conditions (synthesis method, temperature, GHSV, CH4/CO2 ratio). This confirms the important role of the reaction conditions in the overall catalytic performance. So, it's necessary to comprehensively design or prepare catalysts by considering all the factors mentioned above and not just the support and promoter separately.
To promote the technology, further research is required in the following areas.
1. Explore more materials with high specific areas or pre-treatment methods for protecting the Ni particles from sintering and carbon deposition. Supports with high specific areas and structures of core–shell or dendrimer morphology can be developed for offering strength and stability of catalysts. Modifying the commercial materials as supports or using pre-treatment such as plasma-treatment and spray pyrolysis also are significant strategies for protecting the Ni particles from sintering and carbon deposition. However, the issue of catalysts deformation or embrittlement at high temperatures still requires special attention.
2. Manipulate the interactions of metal–support and metal–metal by choosing various supports or promoters. Nickel dispersion and size, basicity, oxygen lattice lability and mobility can be influenced by these interactions and are highly beneficial for promoting the catalytic anti-sintering and anti-coking abilities. At the same time, the interaction between promoters and supports in catalysts may influence the nickel electron density and the valence state, and thus the activity and stability.
3. Quantify the critical particle distance for anti-sintering. The significance of particle spacing in the sintering process is noteworthy. However, no existing literature provides an exact quantitation and measurement of the particle spacing at which sintering would be significantly inhibited. This sort of information is vital for the development of catalysts that can effectively resist sintering.
4. Employ more advanced in situ techniques for a detailed understanding of the reaction mechanisms. The detection of reaction intermediates other than CHx species and surface carbon (Cα, Cβ, Cγ) remains elusive, and a comprehensive understanding of these unidentified intermediates would greatly contribute to elucidating the identity of crucial reaction intermediates and the detailed reaction pathway. This is essential for the further improvement of the activity and stability for Ni-based catalysts for DRM reactions.
5. Achieve DRM at medium temperatures (<800 °C). DRM in medium temperature is a challenging goal and is expected to receive grand attention. The medium temperature would suppress carbon deposition, thus improving the stability of catalysts. However, the reaction rate is also slowed down, with the side reactions occurring as well. Thus, further study is required to understand the mechanism of DRM reactions at low temperatures.
Conflicts of interest
There is no competing financial interest.Acknowledgements
This work was funded by the National Science Foundation of China – Shanxi Coal – based Low – carbon Joint Fund (U1710110).References
- Y. Song, E. Ozdemir, S. Ramesh, A. Adishev, S. Subramanian, A. Harale, M. Albuali, B. A. Fadhel, A. Jamal, D. Moon, S. H. Choi and C. T. Yavuz, Science, 2020, 367, 777–781 CrossRef CAS.
- J. Tollefson, Nature, 2017, 551, 283–283 CrossRef CAS PubMed.
- X. Jiang, Y. Jiao, C. Moran, X. W. Nie, Y. T. Gong, X. W. Guo, K. S. Walton and C. S. Song, Catal. Commun., 2019, 118, 10–14 CrossRef CAS.
- C. Figueres, C. Le Quéré, A. Mahindra, O. Bäte, G. Whiteman, G. Peters and D. Guan, Nature, 2018, 564, 27–30 CrossRef CAS PubMed.
- Q. Li, Y. Ouyang, H. Li, L. Wang and J. Zeng, Angew. Chem., Int. Ed., 2022, 61, e202108069 CrossRef CAS.
- D. T. Shindell, G. Faluvegi, D. M. Koch, G. A. Schmidt, N. Unger and S. E. Bauer, Science, 2009, 326, 716–718 CrossRef CAS PubMed.
- C. B. Field and K. J. Mach, Science, 2017, 356, 706–707 CrossRef CAS.
- C. D. Elvidge, M. D. Bazilian, M. Zhizhin, T. Ghosh, K. Baugh and F. C. Hsu, Energy Strat. Rev., 2018, 20, 156–162 CrossRef.
- L. He, Y. L. Fan, J. Bellettre, J. Yue and L. G. Luo, Renewable Sustainable Energy Rev., 2020, 119, 109589 CrossRef CAS.
- K. H. Lim, Y. F. Yue, Bella, X. Y. Gao, T. X. Zhang, F. Y. Hu, S. Das and S. Kawi, ACS Sustainable Chem. Eng., 2023, 11, 4903–4933 CrossRef CAS.
- S. M. Sajjadi, M. Haghighi, F. Rahmani and J. Eshghi, J. CO2 Util., 2022, 61, 102037 CrossRef CAS.
- J. Meng, T. Gu, W. Pan, C. Bu, J. Zhang, X. Wang, C. Liu, H. Xie and G. Piao, Fuel, 2022, 310, 122363 CrossRef CAS.
- I. V. Yentekakis, P. Panagiotopoulou and G. Artemakis, Appl. Catal., B, 2021, 296, 120210 CrossRef CAS.
- J. Rockström, O. Gaffney, J. Rogelj, M. Meinshausen, N. Nakicenovic and H. J. Schellnhuber, Science, 2017, 355, 1269–1271 CrossRef PubMed.
- S. Kim, PhD thesis, University of California, Irvine, 2021 Search PubMed.
- L. N. Chen, Z. G. Song, S. C. Zhang, C. K. Chang, Y. C. Chuang, X. X. Peng, C. Dun, J. J. Urban, J. H. Guo, J. L. Chen, D. Prendergast, M. Salmeron, G. A. Somorjai and J. Su, Science, 2023, 381, 857–861 CrossRef CAS PubMed.
- S. Bin, S. Zheyi, Z. Yun, P. Fenghongkang, Z. Kaiqing, H. Jun and L. Honglai, Huagong Jinzhan, 2022, 41, 1136–1151 Search PubMed.
- Y. B. Li, L. Zeng, G. Pang, X. E. Wei, M. H. Wang, K. Cheng, J. C. Kang, J. M. Serra, Q. H. Zhang and Y. Wang, Appl. Catal., B, 2023, 324, 122299 CrossRef CAS.
- R. Stevenson, Integr. Environ. Assess. Manage., 2021, 17, 488–489 Search PubMed.
- Z. L. Ou, Z. H. Zhang, C. L. Qin, H. Q. Xia, T. Deng, J. T. Niu, J. Y. Ran and C. F. Wu, Sustainable Energy Fuels, 2021, 5, 1845–1856 RSC.
- K. M. G. Langie, K. Tak, C. Kim, H. W. Lee, K. Park, D. Kim, W. Jung, C. W. Lee, H. S. Oh, D. K. Lee, J. H. Koh, B. K. Min, D. H. Won and U. Lee, Nat. Commun., 2022, 13, 7482 CrossRef CAS PubMed.
- Z. Qin, J. Chen, X. Xie, X. Luo, T. Su and H. Ji, Environ. Chem. Lett., 2020, 18, 997–1017 CrossRef CAS.
- D. Pakhare and J. Spivey, Chem. Soc. Rev., 2014, 43, 7813–7837 RSC.
- S. Kawi, Y. Kathiraser, J. Ni, U. Oemar, Z. Li and E. T. Saw, ChemSusChem, 2015, 8, 3556–3575 CrossRef CAS PubMed.
- L. Baharudin, N. Rahmat, N. H. Othman, N. L. Shah and S. S. A. Syed-Hassan, J. CO2 Util., 2022, 61, 102050 CrossRef CAS.
- O. Daoura, M.-N. Kaydouh, N. El-Hassan, P. Massiani, F. Launay and M. Boutros, J. CO2 Util., 2018, 24, 112–119 CrossRef CAS.
- Z. Lv, S. Chen, X. Huang and C. Qin, Curr. Opin. Green Sustainable Chem., 2023, 40, 100771 CrossRef CAS.
- Y. Ren, Y.-Y. Ma, W.-L. Mo, J. Guo, Q. Liu, X. Fan and S.-P. Zhang, Catalysts, 2023, 13, 647 CrossRef CAS.
- N. A. K. Aramouni, J. G. Touma, B. A. Tarboush, J. Zeaiter and M. N. Ahmad, Renewable Sustainable Energy Rev., 2018, 82, 2570–2585 CrossRef CAS.
- A. M. Alhassan, I. Hussain, O. A. Taialla, M. M. Awad, A. Tanimu, K. Alhooshani and S. A. Ganiyu, J. Cleaner Prod., 2023, 423, 138638 CrossRef CAS.
- K. Tomishige, D. L. Li, M. Tamura and Y. Nakagawa, Catal. Sci. Technol., 2017, 7, 3952–3979 RSC.
- W. J. Jang, D. W. Jeong, J. O. Shim, H. M. Kim, H. S. Roh, I. H. Son and S. J. Lee, Appl. Energy, 2016, 173, 80–91 CrossRef CAS.
- B. Yuan, T. Zhu, Y. W. Han, X. L. Zhang, M. D. Wang and C. Li, Atmosphere, 2023, 14, 770 CrossRef CAS.
- M. Z. Ouyang, P. Boldrin, R. C. Maher, X. L. Chen, X. H. Liu, L. F. Cohen and N. P. Brandon, Appl. Catal., B, 2019, 248, 332–340 CrossRef CAS.
- M. K. Nikoo and N. A. S. Amin, Fuel Process. Technol., 2011, 92, 678–691 CrossRef CAS.
- A. M. Gadalla and B. Bower, Chem. Eng. Sci., 1988, 43, 3049–3062 CrossRef CAS.
- R. Mansoor and M. Tahir, Energy Fuels, 2021, 35, 3675–3714 CrossRef CAS.
- Z. Wu, B. Yang, S. Miao, W. Liu, J. Xie, S. Lee, M. J. Pellin, D. Xiao, D. Su and D. Ma, ACS Catal., 2019, 9, 2693–2700 CrossRef CAS.
- A. L. A. Marinho, F. S. Toniolo, F. B. Noronha, F. Epron, D. Duprez and N. Bion, Appl. Catal., B, 2021, 281, 119459 CrossRef CAS.
- M. A. Vasiliades, C. M. Damaskinos, P. Djinović, A. Pintar and A. M. Efstathiou, Catal. Commun., 2021, 149, 106237 CrossRef CAS.
- S. Kim, J. Lauterbach and E. Sasmaz, ACS Catal., 2021, 11, 8247–8260 CrossRef CAS.
- C. C. Chong, Y. W. Cheng, M. B. Bahari, L. P. Teh, S. Z. Abidin and H. D. Setiabudi, Int. J. Hydrogen Energy, 2021, 46, 24687–24708 CrossRef CAS.
- A. S. Al-Fatesh, N. Patel, A. H. Fakeeha, M. F. Alotibi, S. B. Alreshaidan and R. Kumar, Catal. Rev.: Sci. Eng., 2023, 1–99, DOI:10.1080/01614940.2023.2211447.
- N. A. K. Aramouni, J. G. Touma, B. Abu Tarboush, J. Zeaiter and M. N. Ahmad, Renewable Sustainable Energy Rev., 2018, 82, 2570–2585 CrossRef CAS.
- B. Shao, Z. Q. Wang, X. Q. Gong, H. L. Liu, F. Qian, P. Hu and J. Hu, Nat. Commun., 2023, 14, 996 CrossRef CAS.
- P. Yin, S. L. Hu, K. Qian, Z. Y. Wei, L. L. Zhang, Y. Lin, W. X. Huang, H. F. Xiong, W. X. Li and H. W. Liang, Nat. Commun., 2021, 12, 4865 CrossRef CAS PubMed.
- F. Behafarid and B. Roldan Cuenya, Top. Catal., 2013, 56, 1542–1559 CrossRef CAS.
- O. Daoura, G. Fornasieri, M. Boutros, N. El Hassan, P. Beaunier, C. Thomas, M. Selmane, A. Miche, C. Sassoye, O. Ersen, W. Baaziz, P. Massiani, A. Bleuzen and F. Launay, Appl. Catal., B, 2021, 280, 119417 CrossRef CAS.
- X. Chen, L. Yin, K. Long, H. Sun, M. Sun, H. Wang, Q. Zhang and P. Ning, J. Energy Inst., 2020, 93, 2255–2263 CrossRef CAS.
- R. H. Ouyang, J. X. Liu and W. X. Li, J. Am. Chem. Soc., 2013, 135, 1760–1771 CrossRef CAS PubMed.
- W. Liu, L. Li, X. Zhang, Z. Wang, X. Wang and H. Peng, J. CO2 Util., 2018, 27, 297–307 CrossRef CAS.
- H. Peng, X. Zhang, L. Zhang, C. Rao, J. Lian, W. Liu, J. Ying, G. Zhang, Z. Wang, N. Zhang and X. Wang, ChemCatChem, 2017, 9, 127–136 CrossRef CAS.
- N. Gao, M. Cheng, C. Quan and Y. Zheng, Fuel, 2020, 273, 117702 CrossRef CAS.
- Q. Liu, P. Rzepka, H. Frey, J. Tripp, A. Beck, L. Artiglia, M. Ranocchiari and J. A. van Bokhoven, Mater. Today Nano, 2022, 20, 100273 CrossRef CAS.
- T. W. Hansen, A. T. Delariva, S. R. Challa and A. K. Datye, Acc. Chem. Res., 2013, 46, 1720–1730 CrossRef CAS.
- J. W. Han, J. S. Park, M. S. Choi and H. Lee, Appl. Catal., B, 2017, 203, 625–632 CrossRef CAS.
- S. B. Kim, A. A. S. Eissa, M. J. Kim, E. S. Goda, J. R. Youn and K. Lee, Catalysts, 2022, 12, 423 CrossRef CAS.
- Z. Z. Qin, J. Chen, X. L. Xie, X. Luo, T. M. Su and H. B. Ji, Environ. Chem. Lett., 2020, 18, 997–1017 CrossRef CAS.
- Z. R. Xiao, F. Hou, J. J. Zhang, Q. C. Zheng, J. S. Xu, L. Pan, L. Wang, J. J. Zou, X. W. Zhang and G. Z. Li, ACS Appl. Mater. Interfaces, 2021, 13, 48838–48854 CrossRef CAS.
- J. Á. E. L. Bomfim, J. F. S. C. Filho, T. D. Bezerra, F. C. Rangel, T. A. Simões, P. N. Romano and R. S. da Cruz, in Heterogeneous Catalysis, 2022, pp. 175–206, DOI:10.1016/b978-0-323-85612-6.00007-3.
- B. Yuan, T. Zhu, Y. Han, X. Zhang, M. Wang and C. Li, Atmosphere, 2023, 14, 770 CrossRef CAS.
- J. M. Ginsburg, J. Pina, T. El Solh and H. I. de Lasa, Ind. Eng. Chem. Res., 2005, 44, 4846–4854 CrossRef CAS.
- L. Huang, D. Li, D. Tian, L. Jiang, Z. Li, H. Wang and K. Li, Energy Fuels, 2022, 36, 5102–5151 CrossRef CAS.
- S. Helveg, J. Sehested and J. R. Rostrup-Nielsen, Catal. Today, 2011, 178, 42–46 CrossRef CAS.
- X. Li, D. Li, H. Tian, L. Zeng, Z.-J. Zhao and J. Gong, Appl. Catal., B, 2017, 202, 683–694 CrossRef CAS.
- D. D. Gong, S. S. Li, S. X. Guo, H. G. Tang, H. Wang and Y. Liu, Appl. Surf. Sci., 2018, 434, 351–364 CrossRef CAS.
- X. H. Zhao, B. Joseph, J. Kuhn and S. Ozcan, iScience, 2020, 23, 101082 CrossRef CAS PubMed.
- Q. L. Yang, G. L. Liu and Y. Liu, Ind. Eng. Chem. Res., 2018, 57, 1–17 CrossRef CAS.
- A. S. Al-Fatesh, H. Atia, J. K. Abu-Dahrieh, A. A. Ibrahim, R. Eckelt, U. Armbruster, A. E. Abasaeed and A. H. Fakeeha, Int. J. Hydrogen Energy, 2019, 44, 20770–20781 CrossRef CAS.
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710–712 CrossRef CAS.
- S. A. Theofanidis, V. V. Galvita, H. Poelman and G. B. Marin, ACS Catal., 2015, 5, 3028–3039 CrossRef CAS.
- X. Y. Gao, J. Y. Li, M. D. Zheng, S. Y. Cai, J. Y. Zhang, S. Askari, N. Dewangan, J. Ashok and S. Kawi, Process Saf. Environ. Prot., 2021, 156, 598–616 CrossRef CAS.
- Q. L. M. Ha, U. Armbruster, H. Atia, M. Schneider, H. Lund, G. Agostini, J. Radnik, H. T. Vuong and A. Martin, Catalysts, 2017, 7, 157 CrossRef.
- J. Artz, T. E. Müller, K. Thenert, J. Kleinekorte, R. Meys, A. Sternberg, A. Bardow and W. Leitner, Chem. Rev., 2018, 118, 434–504 CrossRef CAS PubMed.
- J. Juan-Juan, M. C. Román-Martínez and M. J. Illán-Gómez, Appl. Catal., A, 2009, 355, 27–32 CrossRef CAS.
- W. Y. Kim, Y. H. Lee, H. Park, Y. H. Choi, M. H. Lee and J. S. Lee, Catal. Sci. Technol., 2016, 6, 2060–2064 RSC.
- J.-K. Xu, Z.-J. Li, J.-H. Wang, W. Zhou and J.-X. Ma, Acta Phys.-Chim. Sin., 2009, 25, 253–260 CAS.
- X. Y. Gao, K. Hidajat and S. Kawi, J. CO2 Util., 2016, 15, 146–153 CrossRef CAS.
- W. F. Monteiro, M. O. Vieira, C. O. Calgaro, O. W. Perez-Lopez and R. A. Ligabue, Fuel, 2019, 253, 713–721 CrossRef CAS.
- S. C. Tian, F. Yan, Z. T. Zhang and J. G. Jiang, Sci. Adv., 2019, 5, eaav5077 CrossRef CAS PubMed.
- B. Shao, Z. Q. Wang, X. Q. Gong, H. Liu, F. Qian, P. Hu and J. Hu, Nat. Commun., 2023, 14, 996 CrossRef CAS PubMed.
- J. Meng, W. Pan, T. Gu, C. Bu, J. Zhang, X. Wang, C. Liu, H. Xie and G. Piao, Energy Fuels, 2021, 35, 19568–19580 CrossRef CAS.
- T. S. Phan, A. R. Sane, B. R. de Vasconcelos, A. Nzihou, P. Sharrock, D. Grouset and D. P. Minh, Appl. Catal., B, 2018, 224, 310–321 CrossRef CAS.
- J. Wu, L.-Y. Qiao, Z.-F. Zhou, G.-J. Cui, S.-S. Zong, D.-J. Xu, R.-P. Ye, R.-P. Chen, R. Si and Y.-G. Yao, ACS Catal., 2018, 9, 932–945 CrossRef.
- H. B. Kim and E. D. Park, Catal. Today, 2023, 411–412, 113817 CrossRef CAS.
- P. Lu, Q. X. Huang, Y. Chi and J. H. Yan, Energy Fuels, 2017, 31, 8283–8290 CrossRef CAS.
- C. F. Lv, L. L. Xu, M. D. Chen, Y. Cui, X. Y. Wen, Y. P. Li, C. E. Wu, B. Yang, Z. C. Miao, X. Hu and Q. H. Shou, Front. Chem., 2020, 8, 269 CrossRef CAS.
- S. Boujday, J. F. Lambert and M. Che, ChemPhysChem, 2004, 5, 1003–1013 CrossRef CAS.
- O. Daoura, G. Fornasieri, M. Boutros, N. El Hassan, P. Beaunier, C. Thomas, M. Selmane, A. Miche, C. Sassoye, O. Ersen, W. Baaziz, P. Massiani, A. Bleuzen and F. Launay, Appl. Catal., B, 2021, 280, 119417 CrossRef CAS.
- P. Schmidt-Winkel, W. W. Lukens, D. Y. Zhao, P. D. Yang, B. F. Chmelka and G. D. Stucky, J. Am. Chem. Soc., 1999, 121, 254–255 CrossRef CAS.
- V. Meynen, P. Cool and E. F. Vansant, Microporous Mesoporous Mater., 2009, 125, 170–223 CrossRef CAS.
- A. M. Ranjekar and G. D. Yadav, J. Indian Chem. Soc., 2021, 98, 100002 CrossRef CAS.
- B. Erjavec, R. Kaplan and A. Pintar, Catal. Today, 2015, 241, 15–24 CrossRef CAS.
- J. Ni, L. W. Chen, J. Y. Lin and S. Kawi, Nano Energy, 2012, 1, 674–686 CrossRef CAS.
- A. Abdulrasheed, A. A. Jalil, Y. Gambo, M. Ibrahim, H. U. Hambali and M. Y. S. Hamill, Renewable Sustainable Energy Rev., 2019, 108, 175–193 CrossRef CAS.
- Y. J. Chai, Y. Fu, H. Feng, W. B. Kong, C. K. Yuan, B. R. Pan, J. Zhang and Y. H. Sun, ChemCatChem, 2018, 10, 2078–2086 CrossRef CAS.
- O. Alioui, M. Badawi, A. Erto, M. A. Amin, V. Tirth, B.-H. Jeon, S. Islam, M. Balsamo, M. Virginie, B. Ernst and Y. Benguerba, Catal. Rev.: Sci. Eng., 2022, 65, 1468–1520 CrossRef.
- X. Y. Li, D. Li, H. Tian, L. Zeng, Z. J. Zhao and J. L. Gong, Appl. Catal., B, 2017, 202, 683–694 CrossRef CAS.
- X. D. Li, Y. L. Huang, Q. Zhang, C. H. Luan, V. A. Vinokurov and W. Huang, Energy Convers. Manage., 2019, 179, 166–177 CrossRef CAS.
- Y. Y. Zhan, K. Song, Z. M. Shi, C. S. Wan, J. H. Pan, D. L. Li, C. Au and L. L. Jiang, Int. J. Hydrogen Energy, 2020, 45, 2794–2807 CrossRef CAS.
- X. D. Li, Y. L. Huang, Q. Zhang, Z. J. Zuo, X. D. Wang, V. A. Vinokurov, Z. Wang and W. Huang, Fuel, 2019, 254, 115562 CrossRef CAS.
- Y. H. Wang, H. M. Liu and B. Q. Xu, J. Mol. Catal. A: Chem., 2009, 299, 44–52 CrossRef CAS.
- J. Yu, X. Wu, X. Liu, Y. Guo, J. Chu, P. Huang, J. Wu, J. Fu, Z. Wang, C. Zhao and J. Liu, Energy Fuels, 2023, 37, 16672–16687 CrossRef CAS.
- T. Setia Febriatna, P. Setyo Darmanto and F. Bagja Juangsa, Clean Energy, 2023, 7, 313–327 CrossRef.
- I. S. Omodolor, H. O. Otor, J. A. Andonegui, B. J. Allen and A. C. Alba-Rubio, Ind. Eng. Chem. Res., 2020, 59, 17612–17631 CrossRef CAS.
- J. I. Huertas, M. D. Gomez, N. Giraldo and J. Garzón, J. Chem., 2015, 2015, 1–7 CrossRef.
- J. Chen, L. Duan, Y. Ma, Y. Jiang, A. Huang, H. Zhu, H. Jiao, M. Li, Y. Hu, H. Zhou, Y. Xu, F. Donat, M. Awais Naeem and O. Kröcher, Fuel, 2023, 334, 126630 CrossRef CAS.
- Q. Chen, J. Zhang, B. Pan, W. Kong, Y. Chen, W. Zhang and Y. Sun, Chem. Eng. J., 2017, 320, 63–73 CrossRef CAS.
- J. G. Meng, T. T. Gu, W. Pan, C. S. Bu, J. B. Zhang, X. Y. Wang, C. Q. Liu, H. Xie and G. L. Piao, Fuel, 2022, 310, 122363 CrossRef CAS.
- J. M. Tang, J. G. Meng, W. Pan, T. T. Gu, Q. Zhang, J. B. Zhang, X. Y. Wang, C. S. Bu and G. L. Piao, Int. J. Hydrogen Energy, 2023, 48, 19033–19045 CrossRef CAS.
- L. Wang and F. Wang, Energy Fuels, 2022, 36, 5594–5621 CrossRef CAS.
- A. A. Ibrahim, A. S. Al-Fatesh, N. S. Kumar, A. E. Abasaeed, S. O. Kasim and A. H. Fakeeha, Catalysts, 2020, 10, 242 CrossRef CAS.
- G. J. Zhang, J. W. Liu, Y. Xu and Y. H. Sun, Int. J. Hydrogen Energy, 2018, 43, 15030–15054 CrossRef CAS.
- J. Zhang, H. Wang and A. K. Dalai, Appl. Catal., A, 2008, 339, 121–129 CrossRef CAS.
- A. Beheshti Askari, M. Al Samarai, B. Morana, L. Tillmann, N. Pfander, A. Wandzilak, B. Watts, R. Belkhou, M. Muhler and S. DeBeer, ACS Catal., 2020, 10, 6223–6230 CrossRef CAS.
- M. N. Kaydouh, N. El Hassan, A. Davidson, S. Casale, H. El Zakhem and P. Massiani, Microporous Mesoporous Mater., 2016, 220, 99–109 CrossRef CAS.
- B. Li, L. Li, K. Tomishige and X. Wang, Int. J. Hydrogen Energy, 2022, 47, 37792–37810 CrossRef CAS.
- N. D. Charisiou, L. Tzounis, V. Sebastian, S. J. Hinder, M. A. Baker, K. Polychronopoulou and M. A. Goula, Appl. Surf. Sci., 2019, 474, 42–56 CrossRef CAS.
- Z. W. Li, Y. Kathiraser, J. Ashok, U. Oemar and S. Kawi, Langmuir, 2014, 30, 14694–14705 CrossRef CAS.
- L. V. Mattos, G. Jacobs, B. H. Davis and F. B. Noronha, Chem. Rev., 2012, 112, 4094–4123 CrossRef CAS.
- H. R. Gurav, S. Dama, V. Samuel and S. Chilukuri, J. CO2 Util., 2017, 20, 357–367 CrossRef CAS.
- B. Li, X. Q. Yuan, L. Y. Li, B. T. Li, X. J. Wang and K. Tomishige, Int. J. Hydrogen Energy, 2021, 46, 31608–31622 CrossRef CAS.
- A. S. Al-Fatesh, A. Hanan, A. A. Ibrahim, A. H. Fakeeha, S. K. Singh, N. K. Labhsetwar, H. Shaikh and S. O. Qasim, Renewable Energy, 2019, 140, 658–667 CrossRef CAS.
- J. T. Niu, F. Guo, J. Y. Ran, W. J. Qi and Z. Q. Yang, Int. J. Hydrogen Energy, 2020, 45, 30267–30287 CrossRef CAS.
- X. Zhang, J. Deng, M. Pupucevski, S. Impeng, B. Yang, G. Chen, S. Kuboon, Q. Zhong, K. Faungnawakij, L. Zheng, G. Wu and D. Zhang, ACS Catal., 2021, 11, 12087–12095 CrossRef CAS.
- K. H. Oh, J. H. Lee, K. Kim, H.-K. Lee, S. W. Kang, J.-I. Yang, J.-H. Park, C. S. Hong, B.-H. Kim and J. C. Park, J. Mater. Chem. A, 2023, 11, 1666–1675 RSC.
- T. T. Zhang, Z. X. Liu, Y. A. Zhu, Z. C. Liu, Z. J. Sui, K. K. Zhu and X. G. Zhou, Appl. Catal., B, 2020, 264, 118497 CrossRef CAS.
- M. A. Vasiliades, C. M. Damaskinos, K. K. Kyprianou, M. Kollia and A. M. Efstathiou, Catal. Today, 2020, 355, 788–803 CrossRef CAS.
- B. Li, T. Li, Y. S. Xiao and Z. W. Liu, J. Rare Earths, 2023, 41, 830–838 CrossRef CAS.
- S. M. Kim, P. M. Abdala, T. Margossian, D. Hosseini, L. Foppa, A. Armutlulu, W. van Beek, A. Comas-Vives, C. Coperet and C. Muller, J. Am. Chem. Soc., 2017, 139, 1937–1949 CrossRef CAS.
- R. G. Zhang, X. Q. Guo, B. J. Wang and L. M. Ling, J. Phys. Chem. C, 2015, 119, 14135–14144 CrossRef CAS.
- M. Gerosa, C. E. Bottani, L. Caramella, G. Onida, C. Di Valentin and G. Pacchioni, J. Chem. Phys., 2015, 143, 134702 CrossRef.
- Y. X. Dai, R. Zou, T. E. Ba, J. Zhang and C. J. Liu, J. CO2 Util., 2021, 51, 101647 CrossRef CAS.
- F. Menegazzo, C. Pizzolitto, E. Ghedini, A. Di Michele, G. Cruciani and M. Signoretto, C, 2018, 4, 60 CAS.
- M. Ouyang, P. Boldrin, R. C. Maher, X. Chen, X. Liu, L. F. Cohen and N. P. Brandon, Appl. Catal., B, 2019, 248, 332–340 CrossRef CAS.
- Z. Wang, X. Shao, A. Larcher, K. Xie, D. Dong and C.-Z. Li, Catal. Today, 2013, 216, 44–49 CrossRef CAS.
- Z. Sun, C. K. Russell, K. J. Whitty, E. G. Eddings, J. Z. Dai, Y. L. Zhang, M. H. Fan and Z. Q. Sun, Prog. Energy Combust. Sci., 2023, 96, 101045 CrossRef.
- S. Joo, A. Seong, O. Kwon, K. Kim, J. H. Lee, R. J. Gorte, J. M. Vohs, J. W. Han and G. Kim, Sci. Adv., 2020, 6, eabb1573 CrossRef CAS PubMed.
- E. G. Mahoney, J. M. Pusel, S. M. Stagg-Williams and S. Faraji, J. CO2 Util., 2014, 6, 40–44 CrossRef CAS.
- H. Ay and D. Üner, Appl. Catal., B, 2015, 179, 128–138 CrossRef CAS.
- S. Q. Gao, Y. A. Li, W. Z. Guo, X. Ding, L. Zheng, L. Wu, H. L. Yan and Y. Q. Wang, Mol. Catal., 2022, 533, 112766 CrossRef CAS.
- M. Zhang, J. F. Zhang, Y. Q. Wu, J. X. Pan, Q. D. Zhang, Y. S. Tan and Y. Z. Han, Appl. Catal., B, 2019, 244, 427–437 CrossRef CAS.
- D. P. Liu, X. Y. Quek, W. N. E. Cheo, R. Lau, A. Borgna and Y. H. Yang, J. Catal., 2009, 266, 380–390 CrossRef CAS.
- Y. Lou, M. Steib, Q. Zhang, K. Tiefenbacher, A. Horváth, A. Jentys, Y. Liu and J. A. Lercher, J. Catal., 2017, 356, 147–156 CrossRef CAS.
- J. W. Hu, V. V. Galvita, H. Poelman, C. Detavernier and G. B. Marin, Appl. Catal., B, 2018, 231, 123–136 CrossRef CAS.
- M. Zhang, J. Zhang, Y. Wu, J. Pan, Q. Zhang, Y. Tan and Y. Han, Appl. Catal., B, 2019, 244, 427–437 CrossRef CAS.
- Y. Z. Ge, Y. J. Ma, R. X. Xue, F. W. Wang, P. Su, Z. J. Wang and Y. S. Li, ACS Omega, 2021, 6, 17019–17026 CrossRef CAS.
- N. Wang, K. Shen, L. H. Huang, X. P. Yu, W. Z. Qian and W. Chu, ACS Catal., 2013, 3, 1638–1651 CrossRef CAS.
- T. Herminio, M. R. Cesário, V. D. Silva, T. A. Simões, E. S. Medeiros, D. A. Macedo, H. L. Tidahy, C. Gennequin and E. Abi-Aad, Environ. Chem. Lett., 2020, 18, 895–903 CrossRef CAS.
- K.-W. Jeon, H.-M. Kim, B.-J. Kim, Y.-L. Lee, H.-S. Na, J.-O. Shim, W.-J. Jang and H.-S. Roh, Fuel Process. Technol., 2021, 219, 106877 CrossRef CAS.
- M. Akri, S. Zhao, X. Li, K. Zang, A. F. Lee, M. A. Isaacs, W. Xi, Y. Gangarajula, J. Luo, Y. Ren, Y. T. Cui, L. Li, Y. Su, X. Pan, W. Wen, Y. Pan, K. Wilson, L. Li, B. Qiao, H. Ishii, Y. F. Liao, A. Wang, X. Wang and T. Zhang, Nat. Commun., 2019, 10, 5181 CrossRef.
- W. Mui, PhD, California Institute of Technology, 2017 Search PubMed.
- O. U. Osazuwa, S. Z. Abidin, X. Fan, A. N. Amenaghawon and M. T. Azizan, J. Environ. Chem. Eng., 2021, 9, 105052 CrossRef CAS.
- S. S. Mabaleha, F. Gholizadeh and P. Kalita, Mol. Catal., 2023, 547, 113398 CrossRef CAS.
- J.-C. Seo, H. Kim, Y.-L. Lee, S. Nam, H.-S. Roh, K. Lee and S. B. Park, ACS Sustainable Chem. Eng., 2021, 9, 894–904 CrossRef CAS.
- X. Fang, C. Peng, H. Peng, W. Liu, X. Xu, X. Wang, C. Li and W. Zhou, ChemCatChem, 2015, 7, 3753–3762 CrossRef CAS.
- N. Wang, Z. X. Xu, J. Deng, K. Shen, X. P. Yu, W. Z. Qian, W. Chu and F. Wei, ChemCatChem, 2014, 6, 1470–1480 CrossRef CAS.
- J. Tian, B. Ma, S. Bu, Q. Yuan and C. Zhao, Chem. Commun., 2018, 54, 13993–13996 RSC.
- M. Wang, Q. Zhang, T. Zhang, Y. Wang, J. Wang, K. Long, Z. Song, X. Liu and P. Ning, Chem. Eng. J., 2017, 313, 1370–1381 CrossRef CAS.
- A. S. Al-Fatesh, J. Khatri, R. Kumar, V. K. Srivastava, A. I. Osman, T. S. AlGarni, A. A. Ibrahim, A. E. Abasaeed, A. H. Fakeeha and D. W. Rooney, Energy Sci. Eng., 2022, 10, 866–880 CrossRef CAS.
- C. Sanchez, C. Boissiere, D. Grosso, C. Laberty and L. Nicole, Chem. Mater., 2008, 20, 682–737 CrossRef CAS.
- Q. Ma, L. Guo, Y. Fang, H. Li, J. Zhang, T.-S. Zhao, G. Yang, Y. Yoneyama and N. Tsubaki, Fuel Process. Technol., 2019, 188, 98–104 CrossRef CAS.
- S. M. Sajjadi and M. Haghighi, Chem. Eng. J., 2019, 362, 767–782 CrossRef CAS.
- S. M. Sajjadi, M. Haghighi and F. Rahmani, J. Nat. Gas Sci. Eng., 2015, 22, 9–21 CrossRef CAS.
- S. J. Hassani Rad, M. Haghighi, A. Alizadeh Eslami, F. Rahmani and N. Rahemi, Int. J. Hydrogen Energy, 2016, 41, 5335–5350 CrossRef CAS.
- S. Aghamohammadi, M. Haghighi, M. Maleki and N. Rahemi, Mol. Catal., 2017, 431, 39–48 CrossRef CAS.
- N. Yahi, S. Menad and I. Rodríguez-Ramos, Green Processes Synth., 2015, 4, 479–486 CAS.
- A. L. A. Marinho, R. C. Rabelo-Neto, F. Epron, N. Bion, F. S. Toniolo and F. B. Noronha, Appl. Catal., B, 2020, 268, 118387 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |