
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolten multi-phase catalytic system comprising Li–Zn alloy and LiCl–KCl salt for nitrogen fixation and ammonia synthesis at ambient pressure†
Xian
Meng‡
,
Jian
Liu‡
 ,
Zujian
Tang
,
Bingxu
Xi
,
Pu
Yan
,
Xingran
Wang
,
Kecheng
Cao
,
Bo
Yang
* and
Xiaofei
Guan
*
,
Zujian
Tang
,
Bingxu
Xi
,
Pu
Yan
,
Xingran
Wang
,
Kecheng
Cao
,
Bo
Yang
* and
Xiaofei
Guan
*
School of Physical Science and Technology, ShanghaiTech University, 393 Huaxia Middle Road, Shanghai, 201210, China. E-mail: yangbo1@shanghaitech.edu.cn; guanxf@shanghaitech.edu.cn
First published on 26th March 2024
Abstract
Ammonia (NH3) is one of the most important synthetic inorganic commodities. The current industrial NH3 production is dominated by the Haber–Bosch process with high energy cost and CO2 emission as well as the need for large-scale centralized operation. Liquid metals and molten salts have recently emerged as promising catalytic materials for NH3 synthesis. Herein, we present a molten system comprising Li–Zn alloy and eutectic LiCl–KCl salt for effective NH3 synthesis at 400 °C and 1 bar. The 70 mol% Li–Zn liquid alloy activates N2 dissociation more easily than the pure liquid Zn and the 60 mol% Li–Sn liquid alloy. Effective N2 fixation by the liquid Li–Zn alloy is followed by the hydrogenation of Li3N dissolved in the molten salt above. For the first time, this work reports a volcano-type relationship between the Li3N concentration in the molten salt and the NH3 synthesis rate when feeding H2 to the molten salt. Ab initio molecular dynamics simulations suggest that, within this system, both N2 cleavage and Li3N hydrogenation are quite reactive. Through combined experiments and simulations, this work unravels the molecular mechanisms of nitrogen fixation and ammonia synthesis in the liquid alloy–salt catalytic system, and also demonstrates effective strategies for improving the ammonia synthesis rate. Such a hybrid molten catalytic system offers a promising solution for distributed NH3 production with low energy cost and CO2 emission.
1. Introduction
The ammonia (NH3) industry plays a significant role in the world economy.1 More than 80% of synthetic NH3 is used as a raw material for the production of agricultural fertilizer,2 which is a key to food security. Small fractions of NH3 are used as a refrigerant, cleaning agent, and pharmaceutical feedstock.3 In addition, NH3 has recently re-emerged as an important and carbon-free energy carrier due to its advantages including high energy density (3 kW h kg−1), high hydrogen content (∼18 wt%), facile storage and transportation, and the infrastructure maturity.4,5Currently, the industrial production of NH3 still relies on the Haber–Bosch process, a technology invented over a century ago.6 Hydrocarbon steam reforming of high emissions is often utilized to provide H2, although renewable H2 from water electrolysis is under heavy investigation as a new supply.1,7,8 The Haber–Bosch process combines H2 with N2 separated from air in a high-temperature (400–600 °C) and high-pressure (100–300 bar) reactor containing iron-based catalysts to produce NH3. The reaction is expressed as
| N2 + 3H2 → 2NH3 | (1) |
Extensive studies have been conducted over the past decades with the goal of producing NH3 under milder conditions. Compared with first-generation iron-based catalysts, ruthenium-based catalysts synthesize NH3 at lower temperatures (300–450 °C) and pressures (40–150 bar), but their applications are constrained by the high cost and severe H2 poisoning.5,12–14 To improve the catalytic performance of transition metals, it is important to modify the nitrogen adsorption energy or even circumvent the scaling relation exerted on them.15–24 For example, Wang et al. employed LiH as the second active center for hydrogenation to intervene in TM-mediated catalysis and achieved high catalytic activities at only 150–350 °C and 1 bar.16 Peng et al. have reported a RuCo dual single-atom active catalyst to decouple the sites for N2 activation from those for the desorption of N-containing intermediate species, and realized a high synthesis rate of NH3 at merely 200 °C and 10 bar.19 In addition, the ternary intermetallic electride LaCoSi and the related dual-site mechanism studies have been reported for effective ammonia synthesis with N2 dissociation on the Co site and N hydrogenation on the La site, circumventing the scaling relation.25–28
The search for non-transition metal catalysts with high activity, stability, and cost effectiveness represents another important research direction. Kawamura and Taniguchi have reported a route using sodium (Na) melt as an effective catalyst for NH3 synthesis from N2 and H2 at 500–590 °C under atmospheric pressure.29 The function of the liquid Na was to ionize N2 and dissolve nitrogen.30 In addition, Li has been reported to spontaneously split the triple bond in N2 to form Li3N under ambient conditions.31 Li3N can be protonated by H2O to form NH3 and LiOH; the LiOH product can be electrolyzed to regenerate Li, closing the loop.32 However, the step of LiOH electrolysis remains a challenge mainly due to the consumption of Li by LiOH.33,34 Solid Li-based alloys have also gained significant research attention for thermocatalytic NH3 synthesis.35,36 For example, Yamaguchi et al. reported NH3 synthesis from N2 and H2 using a solid Li–Sn alloy below 400 °C under ambient pressure.37 However, the solid Li–Sn pseudo-catalyst suffered from performance degradation after the first cycle. Meanwhile, the hydrogenation of the intermediates (e.g., LiNH2, Li2NH) was limited by both thermodynamics and kinetics.38 The reaction kinetics between Li2NH and H2 for NH3 synthesis might be improved by the addition of Li2O as a scaffold material.39 Recently, Tang et al. reported an innovative Li-based loop for NH3 synthesis in a liquid catalytic system comprising a Li–Sn alloy and LiCl–KCl salt.40 The materials used were earth abundant and did not need complicated preparation. The liquid alloy and molten salt self-segregated by density into two distinct layers due to their immiscibility.33,41 The Li-based loop consisted of three simultaneous steps: (i) N2 fixation by Li in the liquid alloy to produce Li3N, (ii) the hydrogenation of Li3N dissolved in the molten salt to form NH3 and LiH, and (iii) LiH decomposition to regenerate Li. The liquid catalytic system took advantage of the floating and dissolution of Li3N into the molten salt and circumvented the scaling relation exerted on Li, leading to effective NH3 synthesis. In place of alkali metals, alkaline earth metals might also be candidate materials for fixing N2 due to their high reactivity and low cost.32,40,42 However, the mechanisms for nitrogen fixation and ammonia synthesis remained elusive, and the operation temperature of the liquid alloy–salt catalytic systems reported was relatively high (e.g., 510 °C for the system with a Li–Sn liquid alloy and LiCl–KCl molten salt). For a fixed pressure, lowering the operating temperature is critically important for increasing the equilibrium molar fraction of the NH3 product based on the thermodynamic analysis.40,43 The goal of unlocking the full potential of the liquid alloy–salt catalytic systems for NH3 synthesis motivates the investigations, particularly unveiling the mechanisms of NH3 formation and release.
It is necessary to identify the ideal materials for the liquid alloy–salt catalytic systems to achieve high performance. Based on the previous work,40 several criteria are proposed here, including: (i) spontaneous reaction of N2 with the reactive metal to form a metal nitride when the alloy (or metal) is exposed to N2, (ii) facile transport (e.g., flotation, diffusion) of the metal nitride from the alloy (or metal) to the salt, (iii) easy dissolution and ionization of the metal nitride in the salt, (iv) spontaneous reaction between the nitride ion and the proton source to produce NH3; a metal hydride or salt is also formed depending on the type of proton source, (v) the easy release or collection of NH3 from the catalytic system, and (vi) effective conversion of the metal hydride or salt formed in (iv) back to the reactive metal. According to the phase diagram,44 the Li–Zn alloy of appropriate composition remains liquid at relatively low temperatures (Fig. S1a†). In addition, Li–Zn has a greater Li activity than Li–Sn for a fixed Li content,45 indicating a greater capacity for N2 fixation. Based on these properties, the liquid Li–Zn is investigated as a promising alloy candidate in the liquid catalytic system for operating at lower temperatures.
Fig. 1a shows a schematic of the liquid alloy–salt catalytic system. N2 and H2 are fed through the bubbling tubes to the bottoms of the Li–Zn liquid alloy and LiCl–KCl molten salt, respectively. N2 is fixed by Li in the Li–Zn liquid alloy to form Li3N (reaction (2) and Fig. 1b). Driven by the buoyancy and also the rising bubbles, Li3N floats up to the molten salt and dissolves into Li+ and N3− because the highly ionic molten salt has enough attraction to break down the Li3N compound (reaction (3)).46,47 The N3− ions react with H2 to form NH3 and H− (reaction (4) and Fig. 1c). Reaction (4) occurs stepwise via the formation of the imide ion (NH2−) and the amide ion (NH2−), as expressed by reactions (4a)–(4c).48 In the presence of Zn, a small portion of the LiH product (a combination of Li+ and H−) might decompose to regenerate Li that alloys with Zn at the alloy/salt interface (reaction (5)).40,49,50 In addition, NH3 can also be produced by bimolecular decomposition of NH2− (reaction (6) and Fig. 1c).48 The standard molar Gibbs free energy change of the relevant reactions in the Li–N–H system are shown in Fig. S1b.† We systematically study the roles of multiple variables in nitrogen fixation and ammonia synthesis, and gain a deeper understanding of the process through combined experiments and modeling. The catalytic system integrates the functions of the Li–Zn liquid alloy and the LiCl–KCl molten salt, and is demonstrated for effective NH3 synthesis at relatively low temperatures and ambient pressure.
| N2 + 6Li(Zn) → 2Li3N | (2) |
| Li3N → 3Li+ + N3− | (3) |
| N3− + 3H2 → NH3 + 3H− | (4) |
| N3− + H2 → NH2− + H− | (4a) |
| NH2− + H2 → NH2− + H− | (4b) |
| NH2− + H2 → NH3 + H− | (4c) |
| 2LiH → 2Li(Zn) + H2 | (5) |
| 2NH2− → NH2− + NH3 | (6) |
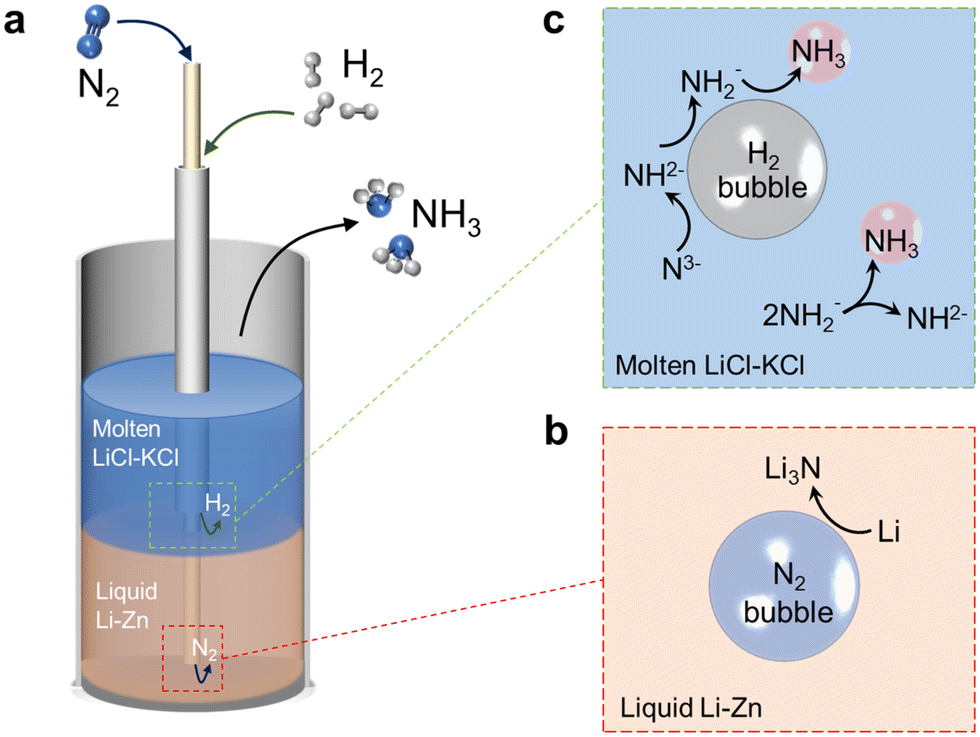 | ||
| Fig. 1 (a) Schematic illustration of the liquid catalytic system comprising the Li–Zn alloy and LiCl–KCl eutectic salt for NH3 synthesis from N2 and H2 inside an alumina bubble-column reactor. (b) N2 fixation by Li in the liquid Li–Zn alloy to form Li3N at the N2 bubble/alloy interface. (c) The N3− ions in the molten salt react with H2 to produce NH2−, NH2− and NH3, successively, at the H2 bubble/salt interface. Additionally, the bimolecular decomposition of NH2− for producing NH3 and NH2− is also shown. | ||
2. Experimental section
2.1. Pretreatment of the Li–Zn mixture and the LiCl–KCl eutectic salt
The volume expansion caused by the Li–Zn alloying is relatively small.51 However, considering the large difference in the melting point and the density between Li and Zn, solid Li and Zn were pretreated to prepare the Li–Zn mixture, which would make the complete alloying easier in the subsequent high-temperature experiments. The desired amounts of Li strips and Zn granules were mixed in a small alumina crucible (OD: 14 mm, ID: 12 mm, height: 44 mm) that was placed in a bigger alumina container (OD: 80 mm, ID: 76 mm, height: 100 mm) with an alumina cover. The system was heated at 260 °C for 2 hours on a hot plate (IKA, RCT Basic) inside an Ar-filled glovebox. The phases of the pre-treated Li–Zn mixture were determined to be LiZn and Li by X-ray diffraction (Fig. S3†).To remove the moisture, the LiCl or KCl powders were transferred to an alumina crucible (OD: 80 mm, ID: 76 mm, height: 100 mm) in an electric furnace, and heated at 300 °C for 12 hours under vacuum, during which Ar was flowed into the system at 20 cm3 min−1. The pre-treated salt was collected and stored in the Ar-filled glovebox. LiCl and KCl were weighted with a molar ratio of 0.592![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.408 and fully mixed using a vortex mixer (Vortex-Genie 2, Scientific Industries, Inc.) for 10 minutes to obtain the LiCl–KCl eutectic mixture. The actual amounts of metals and salts used for different experimental conditions are shown in Table S2.†
:0.408 and fully mixed using a vortex mixer (Vortex-Genie 2, Scientific Industries, Inc.) for 10 minutes to obtain the LiCl–KCl eutectic mixture. The actual amounts of metals and salts used for different experimental conditions are shown in Table S2.†
2.2. Bubble column reactors for NH3 synthesis
Bubble column reactors were assembled using one-end-closed alumina tubes (OD: 18 mm; ID: 13 mm; height: 400 mm) and Swagelok Ultra-Torr™ vacuum fittings, as shown in Fig. S2.† The pretreated Li–Zn mixture and LiCl–KCl eutectic mixture were transferred sequentially to the alumina tube and sealed by vacuum fittings. The assembly was done in an Ar-filled glovebox with O2 and H2O levels less than 10 ppm.Inside a vertical electric furnace, the reactor fed with Ar (20 cm3 min−1) was first maintained at 300 °C for 12 hours under vacuum to remove the moisture. Afterwards, the temperature was ramped to 500 °C, maintained for 30 min, and then cooled to 400 °C under Ar atmosphere to obtain the Li–Zn liquid alloy (height: ∼5 cm) and LiCl–KCl eutectic molten salt (height: ∼5 cm). The density of the 70 mol% Li–Zn alloy (2.33 g cm−3) is greater than that of LiCl–KCl eutectic salt (1.69 g cm−3) at 400 °C,52–54 so the alloy was below the salt. Then, pure N2 (5 cm3 min−1) and H2 (15 cm3 min−1) were introduced into the reactor in either a separate or mixed way. The flow rates of N2 and H2 were regulated using mass flow controllers (MFC; Omega Engineering Inc., Part No.: FMA5508A).
The NH3 product in the effluent gas was absorbed by 0.05 mol L−1 H2SO4 aqueous solution (10 mL) which was replaced every two hours to determine the NH3 synthesis rate. A colorimetric indicator method was coupled with ultraviolet visible spectroscopy (UV-Vis) for quantification of NH3 trapped in the H2SO4 aqueous solution based on the NH3 standard calibration curve.40 An Agilent Cary 5000 UV-Vis–NIR spectrophotometer was used to obtain the relationship between the absorbance and the wavelength.
2.3. Material characterization
X-ray diffraction (XRD) with Cu Kα radiation and a θ–2θ diffraction geometry was used to determine the crystalline phases of the solidified alloy and salt (Bruker, D8 Advance, USA). The crystalline phases were identified with the International Centre for Diffraction Data (ICDD) powder diffraction file (PDF)-4+ database. The sample preparation was carried out in an Ar-filled glovebox, and polyimide tape was used to seal the sample to prevent oxidation during the sample transfer and XRD measurement. Chemical elements on the alloy or salt surface were analyzed using X-ray photoelectron spectroscopy (XPS) with an ESCALAB™ 250Xi XPS microprobe (Thermo Fisher Scientific, USA). The NIST XPS database was used for chemical state identification.55 All binding energies were calibrated by referencing to the C 1s peak of adventitious carbon at 284.8 eV, similar to previous reports.56,57 The sample was prepared in an Ar-filled glovebox and then transferred to the XPS sample chamber using a portable sample transfer-bin.Raman spectroscopy was used to characterize the samples with an Andor Shamrock SR-500i Raman system (×50 objective lens, ×600 grating, and 532 nm laser excitation). The surface morphologies and elemental distribution were studied using scanning electron microscopy (SEM) and energy dispersive X-ray spectroscopy (EDS) on a JEOL SEM IT-500HR/LA. The transmission electron microscopy (TEM) images, scanning transmission electron microscopy (STEM) images and electron energy-loss spectroscopy (EELS) results were collected using a 300 kV aberration-corrected transmission electron microscope (JEOL GrandARM-300F) equipped with a double spherical corrector with a Gatan Oneview IS camera. The sample was first ground with a pestle and mortar into powders, some of which were transferred to a copper grid and then fixed in a holder for characterization. The sample preparation was all done inside an Ar-filled glovebox.
3. Results and discussion
3.1. N2 fixation by the Li–Zn liquid alloy
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N triple bond.40 We first investigated the reaction of N2 with the Li–Zn alloy inside a bubble column reactor as illustrated in Fig. 2a. The pre-treated 70 mol% Li–Zn mixture inside the reactor was heated to 500 °C and then cooled to operation temperature (400 °C) under Ar atmosphere to obtain the liquid alloy with a height of approximately 5 cm. Then, N2 was introduced through an alumina bubbling tube to the liquid alloy bottom at 5 cm3 min−1 for 1 hour. The reactor was subsequently cooled to room temperature and dismantled for material characterization. Fig. 2b shows the XRD patterns of the samples collected from the solidified alloy column. Sample 1 was collected from the wall close to the top of the alloy column, sample 2 was collected right from the top, whereas sample 3 was collected from the lower portion. Based on the XRD pattern for sample 1, the dominant phase was identified to be high-crystallinity α-Li3N with preferred orientations and a small amount of the LiZn intermetallic phase.58 No characteristic peaks for LiZnN or Zn3N2 were observed.47,59 The presence of α-Li3N was consistent with the XPS characterization results (Fig. 2c),60,61 validating reaction (2). The XRD pattern for sample 2 shows the diffraction peaks for both α-Li3N and LiZn;58 yet, it's noted that the crystallinity seemed not as high as that in sample 1. The XRD results for sample 2 were consistent with the SEM and EDS results (Fig. S4†). The XRD pattern for sample 3 didn't show the diffraction peaks for Li3N; the N 1s XPS result in Fig. 2c didn't exhibit the characteristic peaks either, indicating the absence of Li3N. Li3N (density: 1.296 g cm−3 at room temperature)62 floated to the top of the liquid alloy due to the density difference and phase immiscibility.40 It's noted that, based on the Li–Zn phase diagram44 (Fig. S1a†), the cooling of the 70 mol% Li–Zn alloy would transform to a mixture of LiZn and Li. However, the amount of Li was below the XRD detection limit because N2 fixation consumed some Li in the Li–Zn alloy.
N triple bond.40 We first investigated the reaction of N2 with the Li–Zn alloy inside a bubble column reactor as illustrated in Fig. 2a. The pre-treated 70 mol% Li–Zn mixture inside the reactor was heated to 500 °C and then cooled to operation temperature (400 °C) under Ar atmosphere to obtain the liquid alloy with a height of approximately 5 cm. Then, N2 was introduced through an alumina bubbling tube to the liquid alloy bottom at 5 cm3 min−1 for 1 hour. The reactor was subsequently cooled to room temperature and dismantled for material characterization. Fig. 2b shows the XRD patterns of the samples collected from the solidified alloy column. Sample 1 was collected from the wall close to the top of the alloy column, sample 2 was collected right from the top, whereas sample 3 was collected from the lower portion. Based on the XRD pattern for sample 1, the dominant phase was identified to be high-crystallinity α-Li3N with preferred orientations and a small amount of the LiZn intermetallic phase.58 No characteristic peaks for LiZnN or Zn3N2 were observed.47,59 The presence of α-Li3N was consistent with the XPS characterization results (Fig. 2c),60,61 validating reaction (2). The XRD pattern for sample 2 shows the diffraction peaks for both α-Li3N and LiZn;58 yet, it's noted that the crystallinity seemed not as high as that in sample 1. The XRD results for sample 2 were consistent with the SEM and EDS results (Fig. S4†). The XRD pattern for sample 3 didn't show the diffraction peaks for Li3N; the N 1s XPS result in Fig. 2c didn't exhibit the characteristic peaks either, indicating the absence of Li3N. Li3N (density: 1.296 g cm−3 at room temperature)62 floated to the top of the liquid alloy due to the density difference and phase immiscibility.40 It's noted that, based on the Li–Zn phase diagram44 (Fig. S1a†), the cooling of the 70 mol% Li–Zn alloy would transform to a mixture of LiZn and Li. However, the amount of Li was below the XRD detection limit because N2 fixation consumed some Li in the Li–Zn alloy.
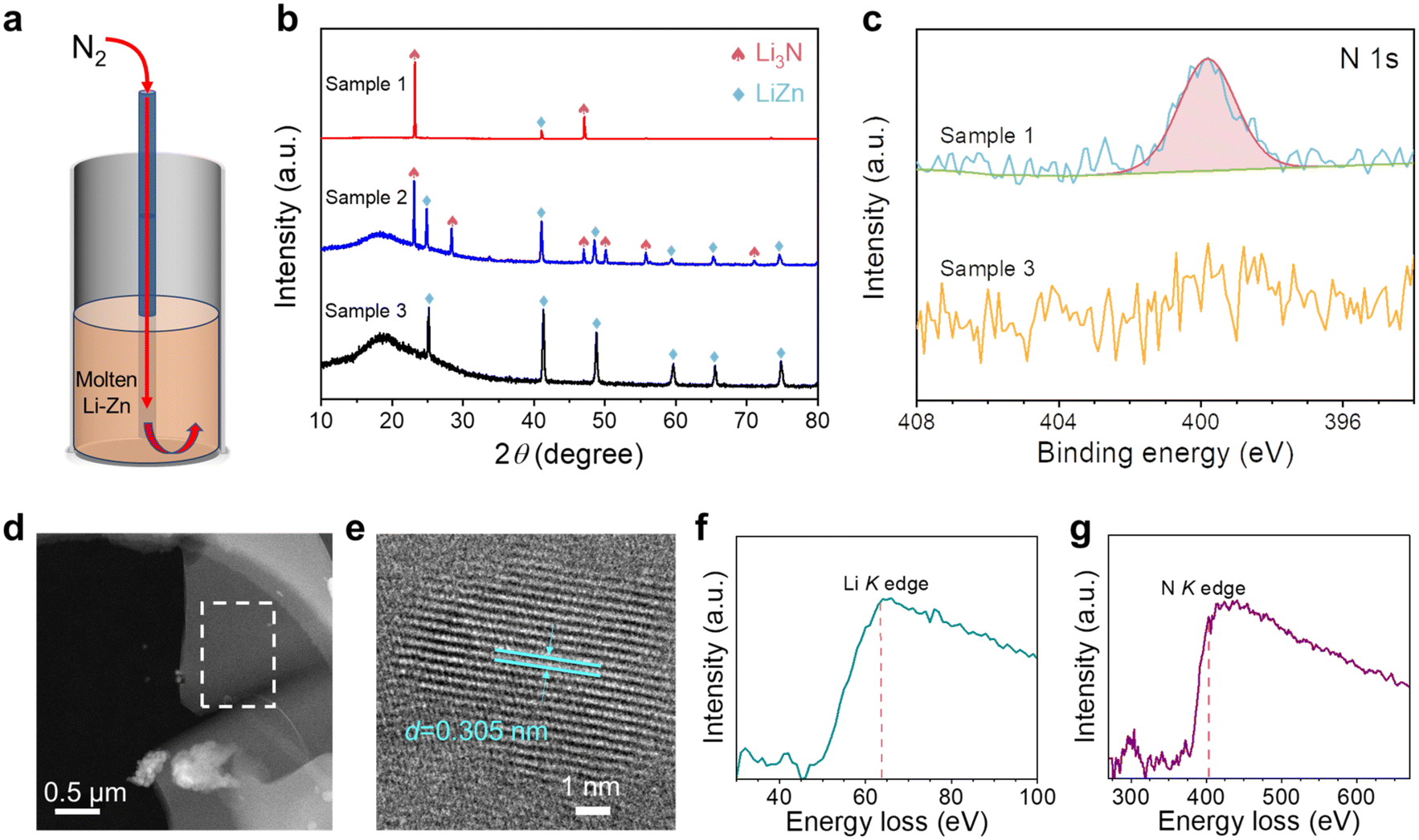 | ||
| Fig. 2 (a) Schematic of the reactor containing the liquid Li–Zn alloy (height: ∼5 cm) bubbled with N2 (5 cm3 min−1). (b) XRD patterns of the three samples collected from different places of the solidified alloy column. The characteristic peaks for α-Li3N (PDF No. 76-0820) and LiZn intermetallic (PDF No. 03-0954) were indexed. The wide signal at ∼18° observed for samples 2 and 3 was attributed to the polyimide film cover. (c) High-resolution N 1s XPS results of sample 1 and sample 3. (d) STEM image for sample 1 on a copper grid. (e) High-resolution TEM image of sample 1 with the (100) interplanar spacing identified to be 0.305 nm. (f) The Li K-edge and (g) the N K-edge EELS spectra for the area enclosed by the white dashed rectangle in (d). | ||
To analyze the local structure and elemental composition, scanning transmission electron microscopy (STEM) and electron energy-loss spectroscopy (EELS) were used to characterize sample 1. The STEM images show the particles of sample 1 after grinding (Fig. 2d and S5†). Fig. 2e shows the lattice-resolved TEM image of sample 1, and the interplanar spacing (d) of 0.305 nm was indexed to the (100) planes of α-Li3N. Fig. 2f and g confirmed the presence of Li and N based on their K-shell ionization edges at 63 eV and 403 eV, respectively.63,64
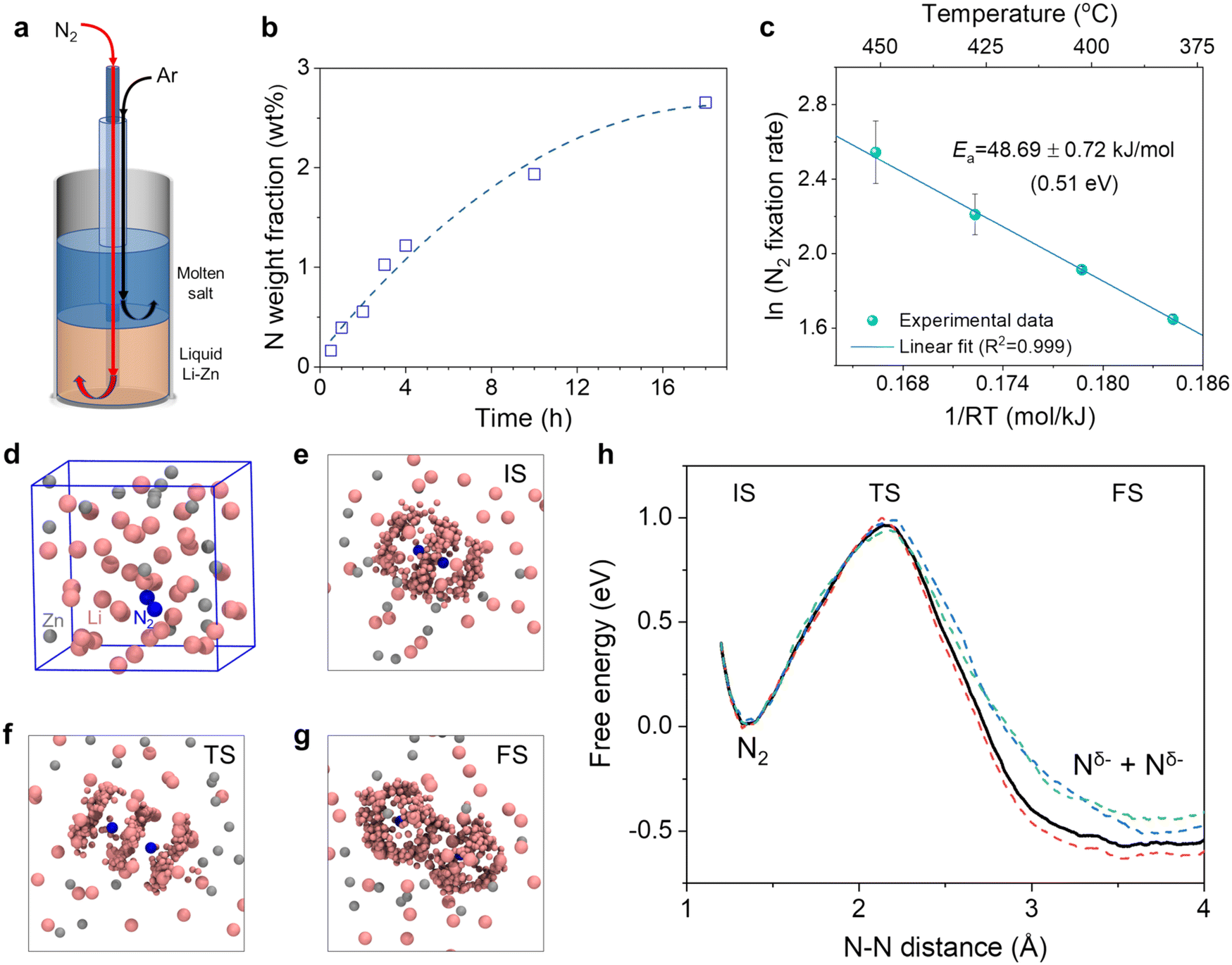 | ||
| Fig. 3 Kinetics and AIMD simulation for N2 fixation by the Li–Zn liquid alloy. (a) Schematic diagram of the reactor containing 70 mol% Li–Zn alloy (height: 5 cm) in the bottom bubbled with N2 (5 cm3 min−1) and the eutectic LiCl–KCl molten salt (height: 5 cm) on the top bubbled with Ar (15 cm3 min−1). (b) Dependence of the nitrogen weight fraction in the salt on the reaction time. The dashed line served as a guide for the eyes. (c) Arrhenius plot for the N2 fixation rate in the temperature range of 380–450 °C. (d) Snapshot of the model system after a pre-relax simulation for 5 ps. The cubic box represents the periodic unit cell of the infinite bulk system. Color code: pink, Li; gray, Zn; blue, N. (e–g) The local environments of the initial, transition, and final states (IS, TS, and FS) during N2 cleavage, respectively. The statistical distribution of Li and Zn near the two nitrogen atoms was illustrated by superimposed snapshots with smaller spheres. (h) The free energy profile as a function of the N–N distance during the N2 cleavage process, as denoted by the black bold line. The work profiles of 3 slow-growth trajectories are also shown, as denoted by the dashed lines in color. | ||
Fig. 3b shows the nitrogen weight fraction in the salt by analyzing five solidified samples from each experiment and then taking the mass-weighted average (see detailed data in Table S3†). In each experiment, the relatively homogeneous distribution of nitrogen in the solidified salt was consistent with the dissolution and ionization of Li3N in the molten salt (reaction (3)). The tiny amount of nitrogen in the impurities of the commercial LiCl and KCl was subtracted (Fig. S7†).65 The N weight fraction in the salt increased with the reaction time (Fig. 3b). Yet, the increase gradually slowed down, likely because the reaction of Li with N2 reduced the Li molar content in the alloy (e.g., from 70% to 66.81% for the 18-hour experiment). The Li activity declined exponentially with the decreasing content,45 and accordingly the ability of the liquid alloy for N2 fixation gradually became weaker. In addition, the Li content decrease shifts the alloy composition towards the liquidus line based on the Li–Zn phase diagram (Fig. S1a†) and might change the physicochemical properties (e.g., viscosity and surface tension) of the liquid alloy. Besides, foaming and channel formation in the liquid alloy could also be possible. Their effects on N2 fixation will be investigated in future studies.
To evaluate the temperature dependence of the N2 fixation rate, we performed a series of experiments at different temperatures from 380 to 450 °C using the setup shown in Fig. 3a. Each experiment last for 2 hours. The nitrogen fixation rate by the 70 mol% Li–Zn liquid alloy was calculated by dividing the total amount of Li3N in the salt above by the reaction time (Note S1 and Fig. S8†). Fig. 3c shows the Arrhenius plot of the natural logarithm of the N2 fixation rate vs. 1/RT, where R is the gas constant (8.31 J K−1 mol−1). The value of the apparent activation energy (Ea) for N2 fixation by the liquid Li–Zn was determined to be approximately 48.69 kJ mol−1, equivalent to 0.51 eV. Such a low energy barrier was mainly attributed to the high activity of Li for N2 activation and fixation,35 as the overlap between the Zn d orbital and the Li s orbital was relatively small,66 as will be discussed in section 3.1.3. To investigate the contribution of Zn to N2 fixation and the subsequent NH3 synthesis, we performed another experiment with a liquid catalytic system comprising pure Zn (height: 5 cm) and eutectic molten LiCl–KCl on the top (height: 5 cm) at 500 °C (Fig. S9†). The results suggest that the liquid Zn itself has negligible activity for N2 fixation.
To examine the reactivity of N2 cleavage in liquid 70 mol% Li–Zn alloy, a randomly mixed Li42Zn18 model was built in a periodic cubic cell with the edge length = 11.2 Å. Then, a N2 molecule was placed in the cell, and a pre-relax simulation was performed for 5 ps, allowing the liquid catalyst around the N2 molecule to find the favorable arrangements. Fig. 3d shows the structure obtained, suggesting that the distributions of the Li and Zn atoms were not entirely random after the pre-relax simulation (Fig. S10a and b†). Further AIMD simulations were performed under the experimental condition of 400 °C. Fig. 3h shows the free energy change for N2 cleavage to two Nδ− as a function of the N–N distance. The value of δ was calculated to be 3.3 ± 0.2 based on the Bader charge analysis.68 The activation free energy (Ga) is defined as the energy difference between the transition state (TS) and the initial state (IS), and the reaction free energy (ΔG) is defined as the energy difference between the final state (FS) and the IS. The N2 cleavage in the liquid Li–Zn alloy showed a Ga of 1.0 eV and ΔG of −0.5 eV, which are much lower than the cleavage in the gas phase.69 The results suggest that the N2 cleavage would be favorable in the liquid Li–Zn than in the gas. There is a discrepancy between the theoretical Ga and experimental values from the Arrhenius plot. The discrepancy may have two main origins: firstly, the slope of the Arrhenius plot is the activation enthalpy actually, rather than the activation free energy; and secondly, the dissolution of N2 in the liquid alloy was not taken into account in the theoretical model. In spite of this, the main catalytic mechanisms can still be captured through theoretical simulations. The local environment near N2 in the IS, TS and FS is illustrated in Fig. 3e–g, respectively. From these figures we found that, in each stage of N2 cleavage, only Li atoms in the Li–Zn alloy interact with the N atoms, while Zn atoms rarely appear in the vicinity of the N atom. The results indicate that the N2 cleavage in the Li–Zn liquid alloy is assisted by the Li atoms, and the Zn atoms mainly act as a solvent for Li. Notably, Zn was added to the experimental system mainly based on the following considerations. First, the addition of Zn increases the density of the liquid alloy. Thus, the liquid alloy could stay beneath the molten salt (Fig. 1a and 3a), and the Li3N formed by the reaction between N2 and Li in the alloy could float to the molten salt. Second, adding Zn can reduce the activity of Li, decreasing the corrosiveness to the alumina reactor.
To investigate the impact of Zn addition on the Li activity in the 70 mol% Li–Zn liquid alloy, we conducted an analysis of the projected density of states (PDOS) in the Li42Zn18 model. The PDOS analysis (Fig. S10c and d†) indicates a reduced overlap of the d orbital of Zn and the s orbital of Li, consistent with previous findings,66 despite the differences in their modeling system where the Li atoms were aggregated. These computational results demonstrate that in the Li–Zn system, the charge transfer from Zn to Li atoms is minimal, and as a result, the activity of Li is not significantly hindered for N2 fixation.
We also examined the reactivity of N2 cleavage in liquid pure Zn (Note S2, Fig. S10e and f†), liquid pure Li (Note S2, Fig. S10g and h†), and liquid 60 mol% Li–Sn alloy (Note S3 and Fig. S11†) with AIMD simulations. A comparison of the results suggests that the N2 cleavage in the pure liquid Zn is difficult due to the high activation energy of almost 4 eV. The liquid 70 mol% Li–Zn with a Ga of 1 eV is more reactive than the liquid 60 mol% Li–Sn with a Ga of 1.7 eV for N2 activation. The N2 cleavage in pure liquid Li shows a Ga of 0.8 eV, which is only 0.2 eV smaller than that in the 70 mol% Li–Zn. It can be inferred that the activity mainly originates from the high electron-donating ability of Li, and the 70 mol% Li–Zn liquid alloy still maintains the high activity of Li for N2 fixation.
3.2. The reaction of Li3N with H2 for NH3 synthesis in the molten salt
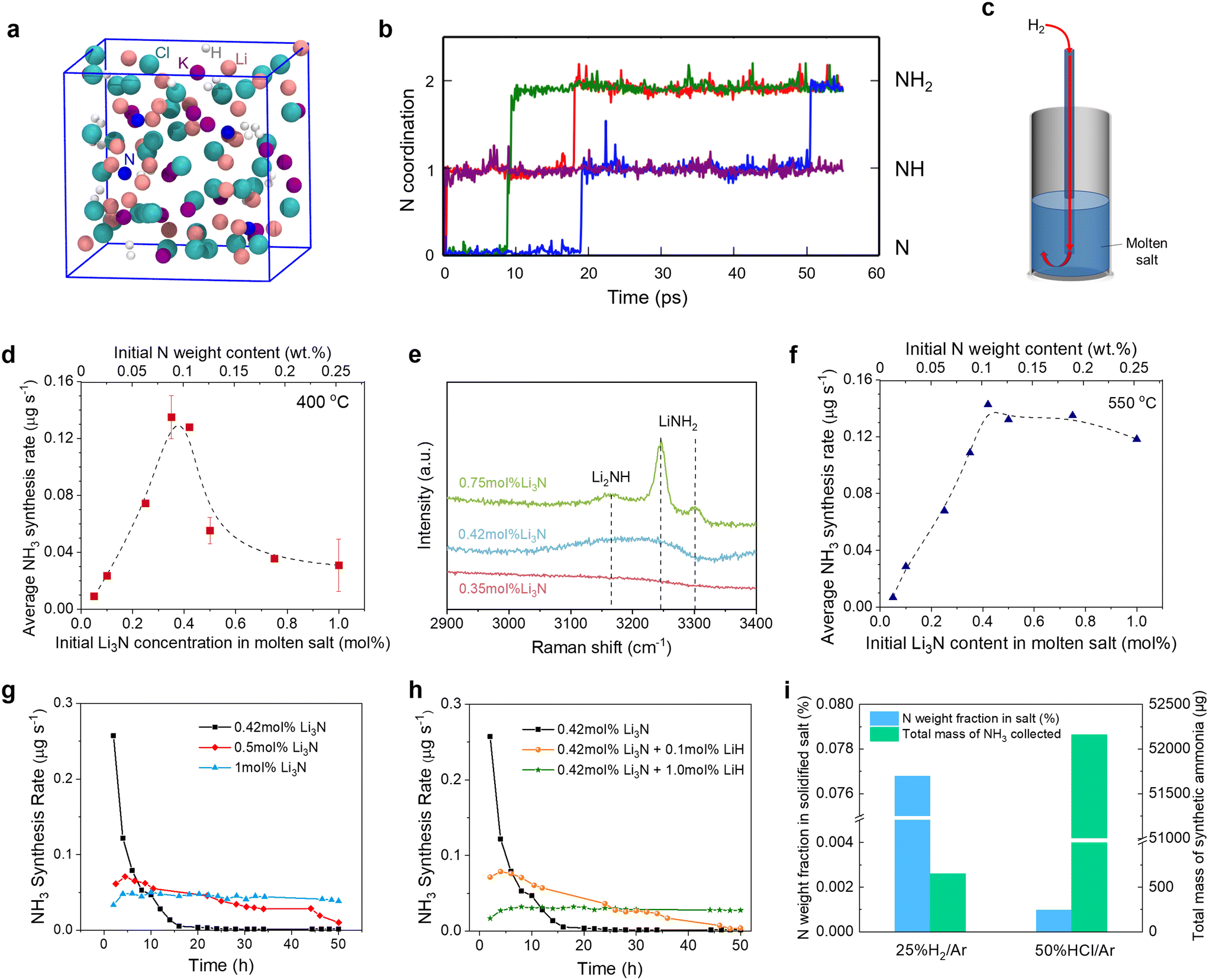 | ||
| Fig. 4 The reaction of Li3N with H2 for NH3 synthesis in the molten salt. (a) A snapshot of the model system of Li3N hydrogenation reaction in the molten LiCl–KCl at the end of the AIMD simulation. Color code: pink, Li; purple, K; cyan, Cl; blue, N; white, H. (b) The trajectory of the coordination number of each of the four N atoms in the AIMD simulation. Different colors were used for these trajectories. (c) Schematic diagram of the experimental setup for bubbling H2 (15 cm3 min−1) to the eutectic LiCl–KCl molten salt (height: 5 cm) containing Li3N. The dependence of the average NH3 synthesis rate on the Li3N content in the molten salt in the experiments carried out for 8 hours at (d) 400 °C and (f) 550 °C. The dashed lines in (d) and (f) serve as guides for the eyes. (e) Raman spectra in the N–H stretching region of the solidified salt samples collected from the experiments that originally contained 0.35, 0.42, and 0.75 mol% Li3N at 400 °C. The time dependence of the NH3 synthesis rate measured for the 50-hour experiments of bubbling H2 into (g) the eutectic LiCl–KCl molten salts containing varying amounts of Li3N (0.42 mol%, 0.5 mol%, and 1.0 mol%) and (h) the eutectic LiCl–KCl molten salts containing 0.42 mol% Li3N and varying amounts of LiH (0%, 0.1 mol%, and 1.0 mol%). (i) The N weight fractions in the solidified salts and the total amounts of NH3 collected in the experiments of feeding 25% H2/Ar or 50% HCl/Ar to the melts containing 0.5 mol% Li3N at 500 °C. | ||
From Fig. 4b, it is obvious that the two N3− ions were readily hydrogenated to NH2− within only hundreds of femtoseconds (fs), suggesting that the models composed of N3− and H2 were quite reactive. After 55 picoseconds (ps) of the AIMD simulation, the formation of three NH2− and one NH2− was observed, as shown in Fig. 4a and b. No NH3 was observed in the simulation, probably because the simulation time was still shorter than the experimental condition. The hydrogenation to NH3 may need a much longer time which is beyond the computational capacities. Moreover, no dehydrogenation processes occurred in the simulations. Therefore, we predict that the hydrogenation of Li3N in the molten salt is reactive under the experimental conditions, and occurs stepwise as described by reactions (4a)–(4c). It's also noted that, through simulation, we found that some nitrogen ions are bound to the surrounding Li ions, making it difficult for these nitrogen ions to come into contact and react with H2. The results suggest that it's vital to separate the Li+ and the nitrogen ions dissolved in the molten salt to accelerate NH3 synthesis.
| N3− + NH3 → NH2− + NH2− | (7) |
Using the setup shown in Fig. 4c, we also investigated the relation between the average NH3 synthesis rate and the Li3N concentration at 550 °C, a much higher temperature than the target operation temperature in this work (400 °C). The average NH3 synthesis rate vs. the Li3N concentration curve obtained at 550 °C also exhibited a volcano-like plot (Fig. 4f); yet, the changing trend was not exactly the same as that at 400 °C. In the low Li3N concentration regime (0–0.4 mol%), the NH3 synthesis rate for both temperatures was close. However, in the high Li3N concentration regime (0.4–1 mol%), the average NH3 synthesis rate at 550 °C decreased only slightly due to the following reasons. Reaction (7) became less thermodynamically favorable as the temperature increased from 400 °C to 550 °C (Fig. S1b†). In addition, the increased temperature would increase the kinetics of reaction (4), although reaction (4) became slightly unfavorable. Overall, the temperature increase exhibited a promoting effect for NH3 synthesis in the high Li3N concentration regime.
The time dependence of the NH3 synthesis rate was also investigated in 50-hour experiments with H2 (15 cm3 min−1) bubbled into the eutectic LiCl–KCl of different Li3N concentrations (0.42, 0.5, and 1.0 mol%) at 400 °C and ambient pressure (Fig. 4g). In the experiment with 0.42 mol% Li3N, the NH3 synthesis rate decreased rapidly over the first 10 hours but the decreasing trend gradually slowed down, because the consumption of Li3N and the accumulation of LiH made reaction (4) more difficult to proceed.40 In the experiment with the Li3N concentration raised to 0.5 mol%, the NH3 synthesis rate increased in the beginning, reached a peak value (0.071 μg s−1) at approximately 4.5 hours, and then gradually decreased. A delayed emergence of the peak for the NH3 synthesis rate was due to the absorption of NH3 (reaction (7)), consistent with previous studies.48,79
When the Li3N concentration in the molten salt was further increased to 1.0 mol%, the NH3 synthesis rate initially increased during the first 4 hours and then remained at a relatively stable value (Fig. 4g), indicating a subtle balance between various reactions. First, the high Li3N concentration not only favored NH3 production (reaction (4)) but also led to a strong absorption of NH3 (reaction (7)). In addition, based on reactions (4c) and (6), the accumulation of LiNH2 would increase the thermodynamic driving force to promote NH3 production (reaction (6) was validated as shown in Fig. S13†). Eventually, the reaction products in the molten salt might be transformed into Li2NH, LiNH2, and LiH. Similar to Li3N, LiH can absorb NH3 as expressed by reaction (8), which is invertible to reaction (4c),80 although the thermodynamic driving force for NH3 absorption by LiH was smaller than that by Li3N (Fig. S1b†).
| LiH + NH3 → LiNH2 + H2 | (8) |
| 3HCl + Li3N → NH3 + 3LiCl | (9) |
3.3. The reaction of LiH with N2 for NH3 synthesis in the molten salt
LiH is a byproduct of the reaction between H2 and Li3N in the molten salt based on reaction (4). The accumulation of LiH in the molten salt has been identified to reduce the thermodynamic driving force for NH3 synthesis.40 The solid LiH is known to have a high thermodynamic stability due to the presence of a strong ionic bond, and its decomposition to Li and H2 typically requires high temperatures above 700 °C.81 To destabilize the LiH dissolved in the molten salt, previous work has proposed using a third element (e.g., Sn) to alloy with Li upon LiH decomposition. This method of alloying lowered the energy barrier for LiH decomposition; however, LiH decomposition was still a thermodynamically unspontaneous reaction at the operation temperatures. The effectiveness of Li recovery from LiH decomposition with this method remains to be critically determined.To evaluate the difficulty of LiH decomposition in the presence of the Li–Zn alloy (reaction (5)), we used AIMD to simulate the reverse reaction and investigate the reactivity of H2 with the 70 mol% Li–Zn alloy. A randomly mixed Li42Zn18 system was built in a periodic cubic cell with the edge length = 11.2 Å, and a H2 molecule was placed inside, as shown in Fig. 5a. The AIMD simulation was performed at 400 °C. Fig. 5b shows the free energy change for H2 cleavage to two H− as a function of the H–H distance. The Ga for H2 cleavage in the liquid Li–Zn alloy was merely 0.1 eV, much smaller than that in the gas phase. On the other hand, the Ga for LiH decomposition to form H2 and Li–Zn alloy was identified to be merely 1 eV, validating reaction (5). Nevertheless, the results verified that LiH decomposition was unspontaneous; even with the presence of the Li–Zn alloy, the energy barrier of its decomposition was still much greater than that of its reverse reaction.
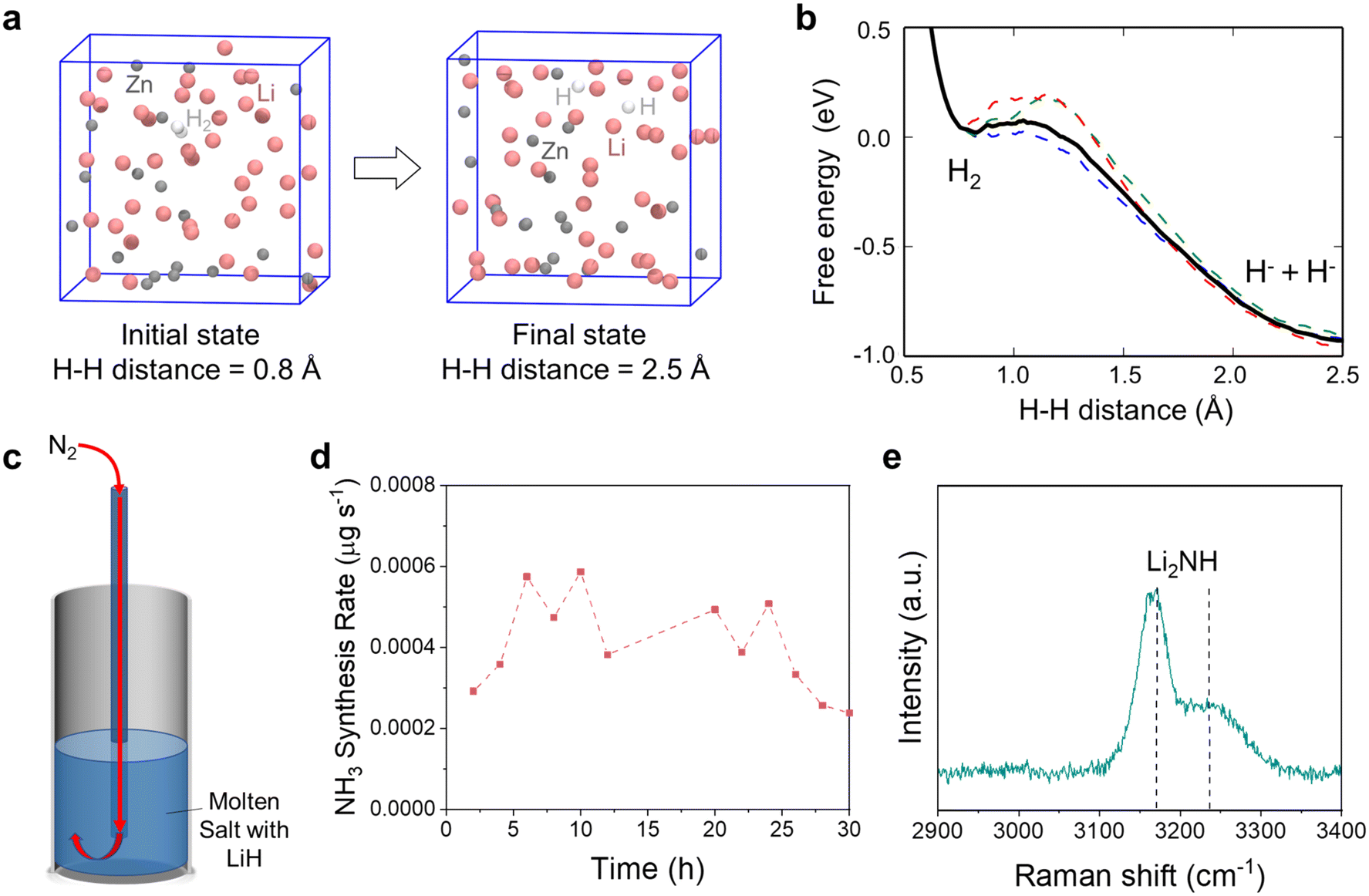 | ||
| Fig. 5 (a) Snapshots of the initial state and the final state of the model system where the cubic box represents the periodic unit cell of the infinite bulk system. Color code: pink, Li; gray, Zn; white, H. (b) The free energy profile as a function of the H–H distance during the H2 cleavage process, as denoted by the black bold line. The work profiles of 3 slow-growth trajectories are also shown, as denoted by the dashed lines in color. (c) Schematic diagram of the experimental setup for bubbling N2 (5 cm3 min−1) to the eutectic LiCl–KCl molten salt (height: 5 cm) containing 3 mol% LiH. (d) The NH3 synthesis rate measured for the experiment illustrated in (c) over 30 hours at 400 °C. (e) Raman spectrum of the solidified salt sample from the experiment showing characteristic peaks for Li2NH. | ||
Given the difficulty of LiH thermal decomposition, here we investigated the reaction of LiH in the molten salt with N2. The eutectic LiCl–KCl mixture containing 3 mol% LiH was first heated to 400 °C in Ar, and then fed with N2 (15 cm3 min−1) to the bottom of the molten salt (Fig. 5c). Fig. 5d shows that the NH3 synthesis rate was relatively small over 30 hours at 400 °C, indicating that the reaction of LiH with N2 contributed little to NH3 synthesis. Nevertheless, the solidified salt contained approximately 0.05 wt% of nitrogen based on the UV-Vis characterization (Fig. S15†), almost three times greater than that in the commercial LiCl–KCl salt (Fig. S7†), indicating nitrogen fixation by LiH. Raman spectroscopy of the solidified salt shows characteristic peaks at 3170 cm−1 and 3230 cm−1 for Li2NH (Fig. 5e),77 indicating that the major product of the reaction between LiH and N2 was Li2NH (reaction (10)). The degree of nitridation of LiH dissolved in the molten LiCl–KCl salt was calculated to be 13.18% at 400 °C (Note S5†). The dissolution of LiH in the molten salt enabled a gas–liquid reaction between LiH and N2 in the bubbles. Unlike the gas–solid reaction between N2 and solid LiH, the Li2NH product dissolved into the molten salt without forming a blocking layer to hinder further reaction. In addition, Li2NH may be further utilized by reacting with H2 to form NH3 (reactions (4b) and (4c)), but the resulting NH3 synthesis rate was supposed to be limited as these reactions were thermodynamically unfavorable (Fig. S1b†). In principle, Li2NH in the molten salt may also be converted to NH3 by using HCl gas as an alternative and more effective proton source (reaction (11)), as discussed in section 3.2.3.
| 4LiH + N2 → 2Li2NH + H2 | (10) |
| Li2NH + 2HCl → 2LiCl + NH3 | (11) |
3.4. NH3 synthesis in the liquid alloy–salt catalytic system
The combined experiments and simulations above have shown that the Li–Zn alloy is effective for fixing N2, and the hydrogenation of Li3N dissolved in the molten salt is reactive. We then investigated the effects of multiple variables on NH3 synthesis from N2 and H2 in the molten catalytic system comprising the Li–Zn alloy and the LiCl–KCl salt.N bonds and the short residence time of the bubbles in the alloy column (5 cm).5 Increasing the temperature generally promoted the kinetics of nitrogen fixation. Second, the ΔG° of the reaction between Li3N and NH3 (reaction (7)) became less negative with the increase of temperature (Fig. S1b†), weakening the NH3 absorption and increasing the NH3 release. As shown in Table S4,† the N weight fraction in the solidified LiCl–KCl salt from the experiment at 500 °C was 0.783 wt%, still larger than that at 400 °C (0.559 wt%). The results indicated that, although the NH3 absorption was weakened, the nitrogen fixation ability of Li was stronger at a higher temperature. Nevertheless, it should be noted that the steady values for NH3 synthesis at 400 °C and 500 °C were relatively close, indicating it suitable to operate at relatively low temperatures, especially considering the development trend toward milder and greener conditions for NH3 synthesis.
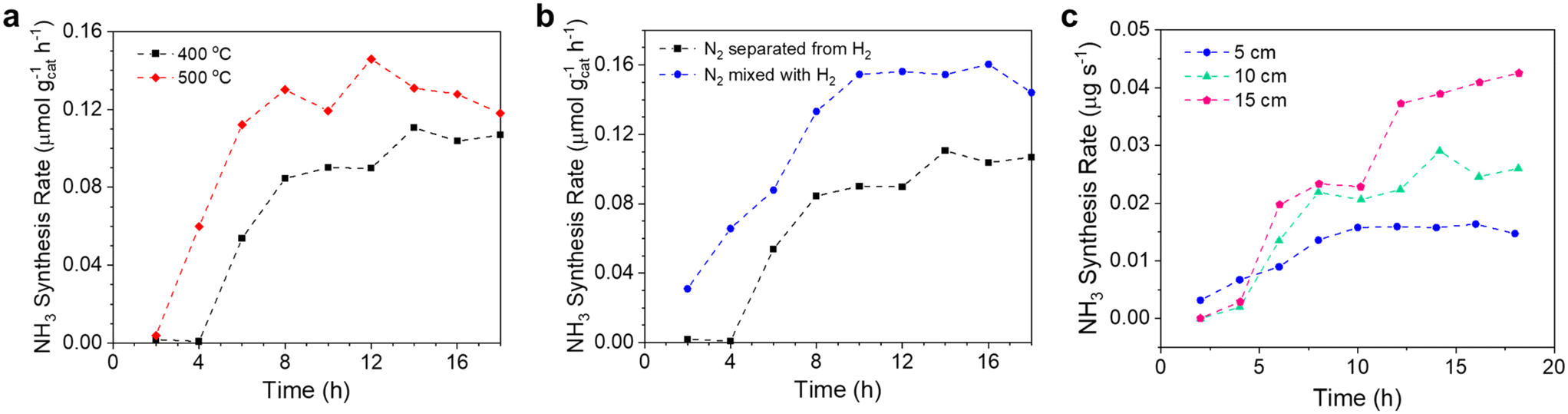 | ||
| Fig. 6 (a) The NH3 synthesis rate in liquid catalytic systems comprising Li–Zn alloy (height: 5 cm) and molten LiCl–KCl (height: 5 cm) at 400 and 500 °C. N2 (5 cm3 min−1) and H2 (15 cm3 min−1) were fed to the alloy and the salt, respectively. (b) The NH3 synthesis rate in liquid catalytic systems comprising Li–Zn alloy (height: 5 cm) and molten LiCl–KCl (height: 5 cm) at 400 °C. In one experiment as denoted by black squares, N2 (5 cm3 min−1) and H2 (15 cm3 min−1) were fed to the alloy and salt, respectively. In the other experiment as denoted by the blue spheres, the mixture of N2 (5 cm3 min−1) and H2 (15 cm3 min−1) was fed to the liquid alloy. (c) The NH3 synthesis rate in liquid catalytic systems comprising Li–Zn (5 cm) and LiCl–KCl of different heights (5, 10, and 15 cm). The mixture of N2 (5 cm3 min−1) and H2 (15 cm3 min−1) was directly introduced to the liquid alloy. | ||
3.5. Summary of major reactions in the liquid catalytic system and the outlook
Improving the understanding of the molten catalytic system provides a guideline for designing better molten catalytic systems. The lithium-based loop presented in the previous work is over-simplified.40 Based on the afore-mentioned experimental and simulation results, here we have proposed a more complete picture of the major reactions taking place in the molten catalytic system for NH3 synthesis from N2 and H2, as illustrated in Fig. 7. The ammonia synthesis process involves multiple reaction steps especially in the molten salt. It has taken into account the stepwise reactions of N3− hydrogenation (reactions (4a)–(4c)), the absorption of NH3 by N3− (reaction (7)) and H− (reaction (8)), and N2 fixation by H− (reaction (10)). Reactions (6), (7), (8), and (10) lead to the accumulation of the relatively stable intermediate species (i.e., NH2−, NH2−) in the molten salt, corresponding to the limited NH3 yield, as discussed in section 3.2.3.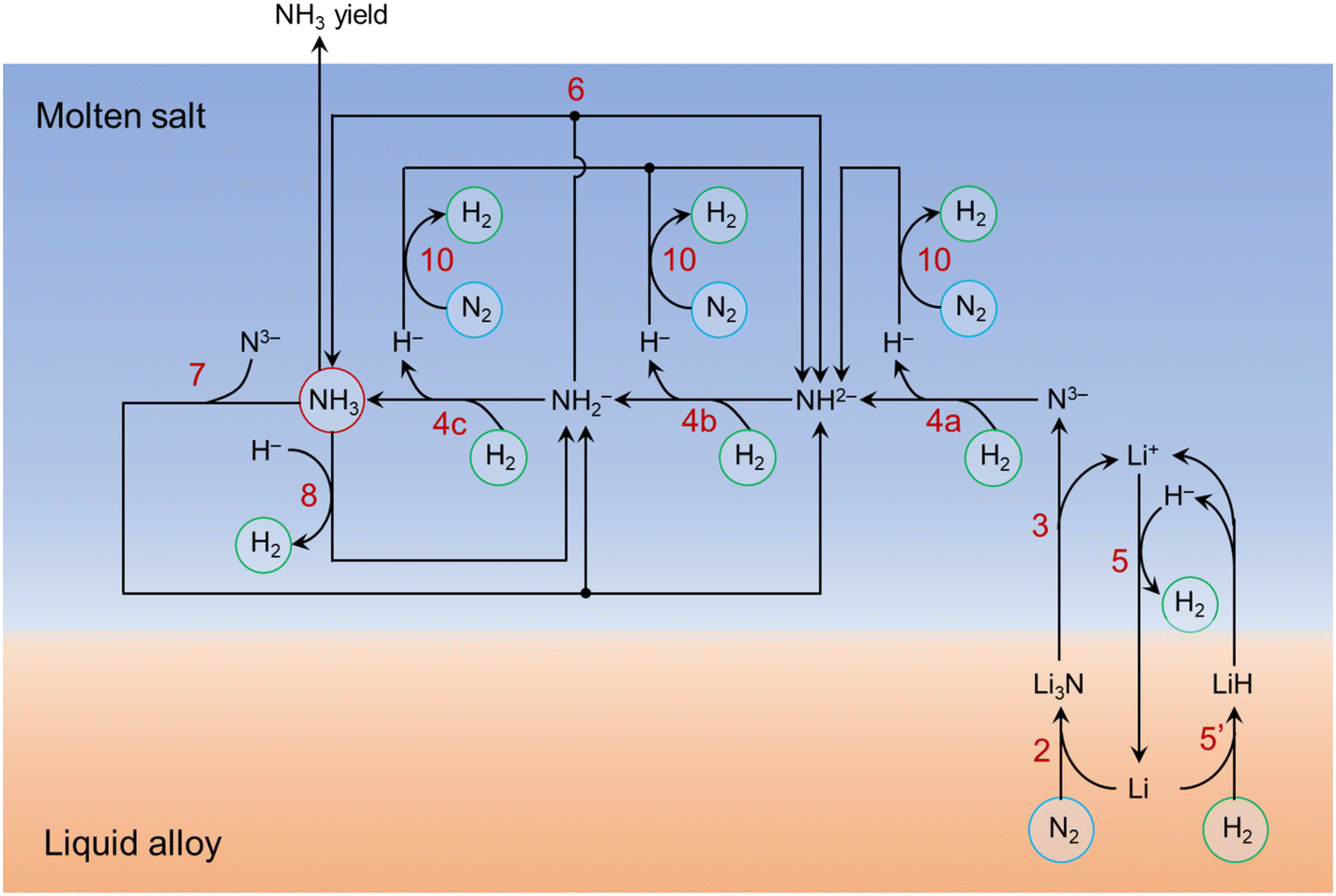 | ||
| Fig. 7 A holistic illustration of the major reactions taking place in the molten catalytic system comprising Li–Zn alloy and LiCl–KCl salt. The gaseous molecules (N2, H2, NH3) are circled to increase clarity. The numbers in red refer to the reactions listed in the main text. The effect of HCl is not considered here. Reaction (5′) is the reverse of reaction (5). | ||
To evaluate the catalytic performance of the molten catalytic system for ammonia synthesis from N2 and H2, we have compared the activity levels of the molten catalytic systems studied in this work with several relevant catalysts reported in the literature, as summarized in Table S5.† The highest activity level obtained was 0.137 μmol gcat−1 h−1 in the molten catalytic system comprising 70 mol% Li–Zn (5 cm) and eutectic LiCl–KCl (15 cm) at 400 °C and 1 bar. This value was greater than that obtained in pure liquid sodium29 but smaller than those of solid catalysts such as the Zn/LiH system,59 La–TM–Si systems (TM![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Co, Fe, Mn),28 and Cs-promoted Ru/MgO.82 Overall, the molten catalytic system has a significant ammonia yield rate but a low calculated activity level. The reason is actually clear. The molten catalytic system uses a heavy bubble column (21.55–39.15 g) with large-size bubbles at a limited flow rate (5–20 cm3 min−1), and the effective gas/liquid interfacial areas for nitrogen fixation and ammonia synthesis are small; in comparison, the solid catalysts reported in the literature28,59,82 use much less material (0.03–0.1 g) with large surface areas and high gas flows.
Co, Fe, Mn),28 and Cs-promoted Ru/MgO.82 Overall, the molten catalytic system has a significant ammonia yield rate but a low calculated activity level. The reason is actually clear. The molten catalytic system uses a heavy bubble column (21.55–39.15 g) with large-size bubbles at a limited flow rate (5–20 cm3 min−1), and the effective gas/liquid interfacial areas for nitrogen fixation and ammonia synthesis are small; in comparison, the solid catalysts reported in the literature28,59,82 use much less material (0.03–0.1 g) with large surface areas and high gas flows.
Apparently, there are opportunities for improving the molten catalytic system for ammonia synthesis significantly. For example, the material selection for the liquid alloy and molten salt is not restricted to Li–Zn and LiCl–KCl. It is necessary to investigate other reactive alloys and ionic salts. It is also critical to advance the understanding of the high atomic mobility and the complex atomic structure in the melts of various compositions with in situ characterization techniques83 and artificial intelligence methods,84 which might considerably improve the catalytic performance of the system. The dynamic interface between the alloy and the salt is also of great interest. In addition, it will be necessary to carry out scale-up studies and develop pressurized reactors that allow the generation of a significant amount of very tiny gas bubbles to increase the area of the gas/liquid reaction interface. Besides, as indicated by Fig. 4i, the introduction of a suitable amount of HCl into the molten salt can improve the ammonia synthesis rate significantly due to rapid reactions of HCl with N3−, NH2−, and NH2−. HCl readily disturbs the accumulation of the relatively stable NH2− and NH2− in the molten salt. Moreover, the liquid alloy–salt catalytic system can incorporate electrolysis to facilitate the ammonia synthesis and remove the oxide impurities if formed by the reaction between the reactive metal and any water or oxygen present in the feeding gas. These exciting research opportunities are worthy of thorough investigations in the future work.
4. Conclusions
The liquid alloy–salt catalytic system is an integrated, synergistic, and multi-phase (i.e., liquid alloy, molten salt, gas bubbles) system that offers unique chemistry and reaction pathways for ammonia synthesis under relatively mild conditions. Through combined experiments and simulations, we have studied the mechanism of nitrogen fixation and ammonia synthesis in the catalytic system comprising liquid Li–Zn and molten LiCl–KCl. The 70 mol% Li–Zn liquid alloy is effective for N2 dissociation and fixation, and Li is identified to be the reactive species. Li3N floats from the self-cleaning liquid alloy to the highly ionic molten salt, circumventing the scaling relation exerted on Li. When feeding H2 to the molten salt, the relationship between the Li3N concentration in the molten salt and the NH3 synthesis rate exhibits a volcano plot. In the liquid catalytic system, Li3N is first formed in the alloy without LiH. The NH3 generated from Li3N and H2 in the molten salt might be absorbed by the excessive Li3N, limiting the NH3 synthesis rate. As the reactions proceeds, LiH accumulates and absorbs NH3 as well. These findings provide guidelines for the optimization of the experiments. Different strategies have been investigated for improving the NH3 synthesis rate, including (i) feeding HCl to the salt, (ii) changing the temperature, (iii) flowing the mixture of N2 and H2 to the alloy, and (iv) increasing the height of the molten salt column. In summary, this work has demonstrated the effectiveness of the catalytic system based on liquid Li–Zn and molten LiCl–KCl for NH3 synthesis at relatively low temperatures (400–500 °C) and ambient pressure, and will motivate further research and development on decentralized ammonia synthesis with complex melts.Author contributions
X. G. conceived the study and supervised the project. X. M., Z. T., and B. X. performed the experiments, characterized the materials, and analyzed the data under X. G.'s guidance. J. L. and B. Y. designed and performed the AIMD simulations, and analyzed the computational results. P. Y. performed the STEM and EELS experiments and analyzed the results under K. C.'s guidance. X. W. assisted in the material characterization and the technical discussion. X. M., J. L., Z. T., B. X., B. Y., and X. G. wrote the manuscript with feedback from all authors.Conflicts of interest
The following may be considered as potential conflict of interest: X. G., Z. T., and X. M. have patents (CN112266002A, CN112250088A) issued to ShanghaiTech University.Acknowledgements
This material is based on the work supported by ShanghaiTech University. Part of the characterization experiments was performed at the Analytical Instrumentation Center (SPST-AIC10112914) and the Center for High-resolution Electron Microscopy (CħEM, EM02161943) at ShanghaiTech University. The authors thank Ms. Jiayin Zhou for the kind help with the SEM, EDS, and Raman characterization, and Mr. Xu Zhang for generously sharing experiences in the HCl generation experiments.References
- D. R. MacFarlane, P. V. Cherepanov, J. Choi, B. H. R. Suryanto, R. Y. Hodgetts, J. M. Bakker, F. M. Ferrero Vallana and A. N. Simonov, Joule, 2020, 4, 1186–1205 CrossRef CAS.
- N. Lazouski, Z. J. Schiffer, K. Williams and K. Manthiram, Joule, 2019, 3, 1127–1139 CrossRef CAS.
- C. Smith and L. Torrente-Murciano, Chem Catal., 2021, 1, 1163–1172 CrossRef CAS.
- S. F. Yin, B. Q. Xu, X. P. Zhou and C. T. Au, Appl. Catal., A, 2004, 277, 1–9 CrossRef CAS.
- Q. Wang, J. Guo and P. Chen, J. Energy Chem., 2019, 36, 25–36 CrossRef.
- H. Liu, Chin. J. Catal., 2014, 35, 1619–1640 CrossRef CAS.
- A. Buttler and H. Spliethoff, Renewable Sustainable Energy Rev., 2018, 82, 2440–2454 CrossRef CAS.
- C. Zhou, J. Ni, H. Chen and X. Guan, Sustainable Energy Fuels, 2021, 5, 4355–4367 RSC.
- J. W. Erisman, M. A. Sutton, J. Galloway, Z. Klimont and W. Winiwarter, Nat. Geosci., 2008, 1, 636–639 CrossRef CAS.
- C. Smith, A. K. Hill and L. Torrente-Murciano, Energy Environ. Sci., 2020, 13, 331–344 RSC.
- United Nations Sustainable Development Goals, https://sdgs.un.org/goals, (accessed 5/29/2023).
- M. Kitano, Y. Inoue, Y. Yamazaki, F. Hayashi, S. Kanbara, S. Matsuishi, T. Yokoyama, S.-W. Kim, M. Hara and H. Hosono, Nat. Chem., 2012, 4, 934–940 CrossRef CAS PubMed.
- M. Kitano, S. Kanbara, Y. Inoue, N. Kuganathan, P. V. Sushko, T. Yokoyama, M. Hara and H. Hosono, Nat. Commun., 2015, 6, 6731 CrossRef CAS PubMed.
- M. Kitano, Y. Inoue, H. Ishikawa, K. Yamagata, T. Nakao, T. Tada, S. Matsuishi, T. Yokoyama, M. Hara and H. Hosono, Chem. Sci., 2016, 7, 4036–4043 RSC.
- R. Kojima and K.-I. Aika, Chem. Lett., 2000, 29, 514–515 CrossRef.
- P. Wang, F. Chang, W. Gao, J. Guo, G. Wu, T. He and P. Chen, Nat. Chem., 2017, 9, 64–70 CrossRef CAS PubMed.
- P. Mehta, P. Barboun, F. A. Herrera, J. Kim, P. Rumbach, D. B. Go, J. C. Hicks and W. F. Schneider, Nat. Catal., 2018, 1, 269–275 CrossRef.
- T. Wang and F. Abild-Pedersen, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2106527118 CrossRef CAS PubMed.
- X. Peng, H. X. Liu, Y. Zhang, Z. Q. Huang, L. Yang, Y. Jiang, X. Wang, L. Zheng, C. Chang, C. T. Au, L. Jiang and J. Li, Chem. Sci., 2021, 12, 7125–7137 RSC.
- A. Vojvodic and J. K. Nørskov, Natl. Sci. Rev., 2015, 2, 140–143 CrossRef CAS.
- C. Mao, J. Wang, Y. Zou, G. Qi, J. Y. Yang Loh, T. Zhang, M. Xia, J. Xu, F. Deng, M. Ghoussoub, N. P. Kherani, L. Wang, H. Shang, M. Li, J. Li, X. Liu, Z. Ai, G. A. Ozin, J. Zhao and L. Zhang, J. Am. Chem. Soc., 2020, 142, 17403–17412 CrossRef CAS PubMed.
- Y. Gong, H. Li, C. Li, X. Yang, J. Wang and H. Hosono, Chem. Mater., 2022, 34, 1677–1685 CrossRef CAS.
- Z. Wang, Y. Gong, M. L. Evans, Y. Yan, S. Wang, N. Miao, R. Zheng, G.-M. Rignanese and J. Wang, J. Am. Chem. Soc., 2023, 145, 26412–26424 CrossRef CAS PubMed.
- S. Feng, Y. Yao, J.-C. Charlier, G.-M. Rignanese and J. Wang, Chem. Mater., 2023, 35, 9019–9028 CrossRef CAS.
- Y. Gong, J. Wu, M. Kitano, J. Wang, T.-N. Ye, J. Li, Y. Kobayashi, K. Kishida, H. Abe, Y. Niwa, H. Yang, T. Tada and H. Hosono, Nat. Catal., 2018, 1, 178–185 CrossRef CAS.
- Y. Gong, H. Li, J. Wu, X. Song, X. Yang, X. Bao, X. Han, M. Kitano, J. Wang and H. Hosono, J. Am. Chem. Soc., 2022, 144, 8683–8692 CrossRef CAS PubMed.
- Y. Gong, H. Li, C. Li, X. Bao, H. Hosono and J. Wang, J. Adv. Ceram., 2022, 11, 1499–1529 CrossRef CAS.
- H. Li, Y. Gong, H. Yang, X. Yang, K. Li, J. Wang and H. Hosono, ChemSusChem, 2023, 16, e202301016 CrossRef CAS PubMed.
- F. Kawamura and T. Taniguchi, Sci. Rep., 2017, 7, 11578 CrossRef PubMed.
- F. Kawamura, M. Morishita, K. Omae, M. Yoshimura, Y. Mori and T. Sasaki, J. Mater. Sci.: Mater. Electron., 2005, 16, 29–34 CrossRef CAS.
- D. Roy, A. Navarro-Vazquez and P. V. R. Schleyer, J. Am. Chem. Soc., 2009, 131, 13045–13053 CrossRef CAS PubMed.
- J. M. McEnaney, A. R. Singh, J. A. Schwalbe, J. Kibsgaard, J. C. Lin, M. Cargnello, T. F. Jaramillo and J. K. Nørskov, Energy Environ. Sci., 2017, 10, 1621–1630 RSC.
- Z. Tang and X. Guan, J. Sustain. Metall., 2021, 7, 203–214 CrossRef.
- O. Takeda, M. Li, T. Toma, K. Sugiyama, M. Hoshi and Y. Sato, J. Electrochem. Soc., 2014, 161, D820–D823 CrossRef CAS.
- S. Yamaguchi, T. Ichikawa, Y. Wang, Y. Nakagawa, S. Isobe, Y. Kojima and H. Miyaoka, ACS Omega, 2017, 2, 1081–1088 CrossRef CAS PubMed.
- K. Shinzato, K. Tagawa, K. Tsunematsu, H. Gi, P. K. Singh, T. Ichikawa and H. Miyaoka, ACS Appl. Energy Mater., 2022, 5, 4765–4773 CrossRef CAS.
- T. Yamaguchi, K. Shinzato, K. Yamamoto, Y. Wang, Y. Nakagawa, S. Isobe, T. Ichikawa, H. Miyaoka and T. Ichikawa, Int. J. Hydrogen Energy, 2020, 45, 6806–6812 CrossRef CAS.
- R. Michalsky, A. M. Avram, B. A. Peterson, P. H. Pfromm and A. A. Peterson, Chem. Sci., 2015, 6, 3965–3974 RSC.
- K. Tagawa, H. Gi, K. Shinzato, H. Miyaoka and T. Ichikawa, J. Phys. Chem. C, 2022, 126, 2403–2409 CrossRef CAS.
- Z. Tang, X. Meng, Y. Shi and X. Guan, ChemSusChem, 2021, 14, 4697–4707 CrossRef CAS PubMed.
- K. Wang, K. Jiang, B. Chung, T. Ouchi, P. J. Burke, D. A. Boysen, D. J. Bradwell, H. Kim, U. Muecke and D. R. Sadoway, Nature, 2014, 514, 348–350 CrossRef CAS PubMed.
- Y. Hu, G. Z. Chen, L. Zhuang, Z. Wang and X. Jin, Cell Rep. Phys. Sci., 2021, 2, 100425 CrossRef CAS.
- Z. J. Schiffer, A. M. Limaye and K. Manthiram, Joule, 2021, 5, 135–148 CrossRef CAS.
- H. Okamoto, J. Phase Equilib. Diffus., 2012, 33, 345 CrossRef CAS.
- Z. Moser, F. Sommer, J. J. Lee and B. Predel, Thermochim. Acta, 1989, 142, 117–125 CrossRef CAS.
- T. Goto, T. Iwaki and Y. Ito, Electrochim. Acta, 2005, 50, 1283–1288 CrossRef CAS.
- T. Goto, K. Toyoura, H. Tsujimura and Y. Ito, Mater. Sci. Eng., A, 2004, 380, 41–45 CrossRef.
- I. J. McPherson, T. Sudmeier, J. P. Fellowes, I. Wilkinson, T. Hughes and S. C. E. Tsang, Angew. Chem., Int. Ed., 2019, 58, 17433–17441 CrossRef CAS PubMed.
- A. Jain, H. Miyaoka and T. Ichikawa, Int. J. Hydrogen Energy, 2016, 41, 5969–5978 CrossRef CAS.
- Y. Wang, Y. Zhang and C. Wolverton, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 88, 024119 CrossRef.
- X. Gu, J. Dong and C. Lai, Eng. Rep., 2020, 3, e12339 CrossRef.
- T. R. Hogness, J. Am. Chem. Soc., 1921, 43, 1621–1628 CrossRef CAS.
- H. W. Davison, NASA Technical Note D-4650, 1968.
- H. Ito, Y. Hasegawa and Y. Ito, J. Chem. Eng. Data, 2001, 46, 1203–1205 CrossRef CAS.
- NIST X-ray Photoelectron Spectroscopy Database, NIST Standard Reference Database Number 20, National Institute of Standards and Technology, Gaithersburg MD, 20899 (2000), DOI: DOI:10.18434/T4T88K, (retrieved on 2023/5/29).
- Q. Yang, J. Liu, C. Zhou, J. Ni, E. I. Vovk, Y. Yang, B. Yang and X. Guan, Mater. Today Chem., 2022, 25, 100949 CrossRef CAS.
- Q. Yang, C. Zhou, J. Ni and X. Guan, Sustainable Energy Fuels, 2020, 4, 2768–2774 RSC.
- Y. Sun, Y. Li, J. Sun, Y. Li, A. Pei and Y. Cui, Energy Storage Mater., 2017, 6, 119–124 CrossRef.
- R. Wang, W. Gao, S. Feng, Y. Guan, Q. Wang, J. Guo and P. Chen, ChemSusChem, 2023, 16, e202300813 CrossRef CAS PubMed.
- M. S. Kim, Z. Zhang, J. Wang, S. T. Oyakhire, S. C. Kim, Z. Yu, Y. Chen, D. T. Boyle, Y. Ye, Z. Huang, W. Zhang, R. Xu, P. Sayavong, S. F. Bent, J. Qin, Z. Bao and Y. Cui, ACS Nano, 2023, 17, 3168–3180 CrossRef CAS PubMed.
- G. O. Hartley, L. Jin, B. J. Bergner, D. S. Jolly, G. J. Rees, S. Zekoll, Z. Ning, A. T. R. Pateman, C. Holc, P. Adamson and P. G. Bruce, Chem. Mater., 2019, 31, 9993–10001 CrossRef CAS.
- Y. Kojima, T. Ichikawa and H. Fujii, in Encyclopedia of Electrochemical Power Sources, ed. J. Garche, Elsevier, Amsterdam, 2009, pp. 473–483 Search PubMed.
- S. Suzuki, T. Shodai and J. Yamaki, J. Phys. Chem. Solids, 1998, 59, 331–336 CrossRef CAS.
- F. Wang, S. W. Kim, L. Wu, D. Su, Y. Zhu and J. Graetz, Microsc. Microanal., 2013, 19, 1490–1491 CrossRef.
- Y. Chen, H. Liu, N. Ha, S. Licht, S. Gu and W. Li, Nat. Catal., 2020, 3, 1055–1061 CrossRef CAS.
- Z. Zhang, Y. Zhao, B. Sun, J. Xu, Q. Jin, H. Lu, N. Lyu, Z.-M. Dang and Y. Jin, ACS Appl. Mater. Interfaces, 2022, 14, 19419–19425 CrossRef CAS PubMed.
- C. Ruffman, K. G. Steenbergen, A. L. Garden and N. Gaston, Chem. Sci., 2024, 15, 185–194 RSC.
- E. Sanville, S. D. Kenny, R. Smith and G. Henkelman, J. Comput. Chem., 2007, 28, 899–908 CrossRef CAS PubMed.
- D. C. Frost, C. A. McDowell and C. E. H. Bawn, Proc. Math. Phys. Eng. Sci., 1956, 236, 278–284 CAS.
- H. M. Jin, J. Z. Luo and P. Wu, Appl. Phys. Lett., 2007, 90, 084101 CrossRef.
- T. Murakami, T. Nishikiori, T. Nohira and Y. Ito, J. Electrochem. Soc., 2005, 152, D75 CrossRef CAS.
- T. Zhang, H. Miyaoka, H. Miyaoka, T. Ichikawa and Y. Kojima, ACS Appl. Energy Mater., 2018, 1, 232–242 CrossRef CAS.
- N. Serizawa, H. Miyashiro, K. Takei, T. Ikezumi, T. Nishikiori and Y. Ito, J. Electrochem. Soc., 2012, 159, E87 CrossRef CAS.
- M. Che, Catal. Today, 2013, 218–219, 162–171 CrossRef CAS.
- Z. Li, J. Zhang, S. Wang, L. Jiang, M. Latroche, J. Du and F. Cuevas, Phys. Chem. Chem. Phys., 2015, 17, 21927–21934 RSC.
- Y. Kojima and Y. Kawai, J. Alloys Compd., 2005, 395, 236–239 CrossRef CAS.
- B. Paik and A. Wolczyk, J. Phys. Chem. C, 2019, 123, 1619–1625 CrossRef CAS.
- N. Serizawa, K. Takei, T. Nishikiori, Y. Katayama and Y. Ito, Electrochemistry, 2018, 86, 88–91 CrossRef CAS.
- N. Serizawa, H. Miyashiro, K. Takei, T. Ikezumi, T. Nishikiori and Y. Ito, J. Electrochem. Soc., 2012, 159, E87–E91 CrossRef CAS.
- H. Miyaoka, H. Fujii, H. Yamamoto, S. Hino, H. Nakanishi, T. Ichikawa and Y. Kojima, Int. J. Hydrogen Energy, 2012, 37, 16025–16030 CrossRef CAS.
- L. Wang, M. Z. Quadir and K.-F. Aguey-Zinsou, Int. J. Hydrogen Energy, 2016, 41, 18088–18094 CrossRef CAS.
- Q. Wang, J. Pan, J. Guo, H. A. Hansen, H. Xie, L. Jiang, L. Hua, H. Li, Y. Guan, P. Wang, W. Gao, L. Liu, H. Cao, Z. Xiong, T. Vegge and P. Chen, Nat. Catal., 2021, 4, 959–967 CrossRef CAS.
- L. Chen, Z. Song, S. Zhang, C.-K. Chang, Y.-C. Chuang, X. Peng, C. Dun, J. J. Urban, J. Guo, J.-L. Chen, D. Prendergast, M. Salmeron, G. A. Somorjai and J. Su, Science, 2023, 381, 857–861 CrossRef CAS PubMed.
- S. S. Fatima, K. Zuraiqi, A. Zavabeti, V. Krishnamurthi, K. Kalantar-Zadeh, K. Chiang and T. Daeneke, Nat. Catal., 2023, 6, 1131–1139 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cy00202d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |