
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hydrogen spillover through hydride transfer: the reaction of ZnO and ZrO2 with strong hydride donors†
Michael
Benz
a,
Osman
Bunjaku
a,
Michal
Nowakowski
b,
Alexander
Allgaier
a,
Indro
Biswas
c,
Joris
van Slageren
a,
Matthias
Bauer
b and
Deven P.
Estes
 *a
*a
aDepartment of Chemistry, University of Stuttgart, Pfaffenwaldring 55, 70569 Stuttgart, Germany. E-mail: deven.estes@itc.uni-stuttgart.de
bDepartment of Chemistry, University of Paderborn, Warburger Straße 100, 33098 Paderborn, Germany
cDepartment of Electrochemical Energy Technology, Institute of Engineering Thermodynamics, German Aerospace Centre (DLR), 70569 Stuttgart, Germany
First published on 19th August 2024
Abstract
Hydrogen spillover, transfer of H2 from a metal surface to a support (often metal oxides), is pivotal for many heterogeneous catalytic processes, including Cu/ZnO and Cu/ZrO2 catalyzed methanol synthesis. Little is known about hydrogen spillover on ZnO or ZrO2, due to the high complexity of the metal–metal oxide interface. Here, we model hydrogen spillover on ZnO and ZrO2 by reacting them with molecular metal hydrides to see how the properties of the hydrides affect hydrogen spillover. While the good H· donors HV(CO)4dppe (1) and CpCr(CO)3H (2) do not react with the metal oxide surfaces, the strong hydride donors iBu2AlH (3), Cp2ZrHCl (4), and [HCu(PPh3)]6 (5) do reduce ZnO and ZrO2 to give defect sites with the same EPR signatures as obtained via hydrogen spillover. We also observe new M–O bonds to the surface using X-ray absorption spectroscopy (XAS). We propose that these metal oxides undergo hydrogen spillover via initial hydride transfer followed by tautomerization of the surface hydride, giving reduced sites and OH bonds. This mechanism is in contrast to the traditional spillover mechanism involving discrete proton- and electron transfer steps. We also observe that ZnO is easier to reduce than ZrO2, explaining the difficulty observing spillover on Cu/ZrO2.
Introduction
Heterogeneous catalysts based on ZnO and ZrO2 are popular in the industrial synthesis of methanol.1,2 In particular, when combined with Cu nanoparticles, as in the Cu/ZnO/Al2O3 and Cu/ZrO2 catalysts for methanol synthesis from either CO or CO2, they show high activity and selectivity for methanol and long catalyst lifetimes.3 However, the activity of such Cu-based catalysts is heavily dependent on the identity of the metal oxide. For example, Cu/SiO2 and Cu/Al2O3 catalysts primarily convert mixtures of CO2 and H2 to CO under the same conditions at which methanol is made using ZnO and ZrO2 as supports.4 This has led researchers to propose that, rather than simply being used as supports for the Cu nanoparticles, the metal oxides play an active role in the catalytically active sites. This phenomenon is known as a strong metal–support interaction (SMSI) and is now widely accepted to occur for many different heterogeneously catalysed reactions, including methanol synthesis,4,5 hydrocracking,6–8 the hydrodeoxygenation of biomass,9–11 and the hydrogen evolution reaction.12–14 In these cases, atoms from the metal oxides play an intimate role in the reaction mechanism by binding to substrates directly, so much so that the reactions are no longer possible without their presence.The mechanisms of SMSIs are not well understood. In the case of methanol synthesis with Cu based catalysts, two mechanisms have been proposed: hydrogen spillover (Fig. 1) and the interfacial mechanism. In the hydrogen spillover mechanism, hydrogen gas adsorbs on the metal surface, which acts as a source of protons and electrons that then ‘spillover’ onto the metal oxide,15,16 reducing it and producing defect sites that can react with CO or CO2 to eventually produce methanol. This has been proven to occur in the Cu/ZnO/Al2O3 catalyst, which forms CuZn alloys on the surface of the Cu particles.17 The Zn atoms of these alloys change back and forth from CuZn in H2-rich atmospheres to Zn-formate species in CO2-rich atmospheres.18
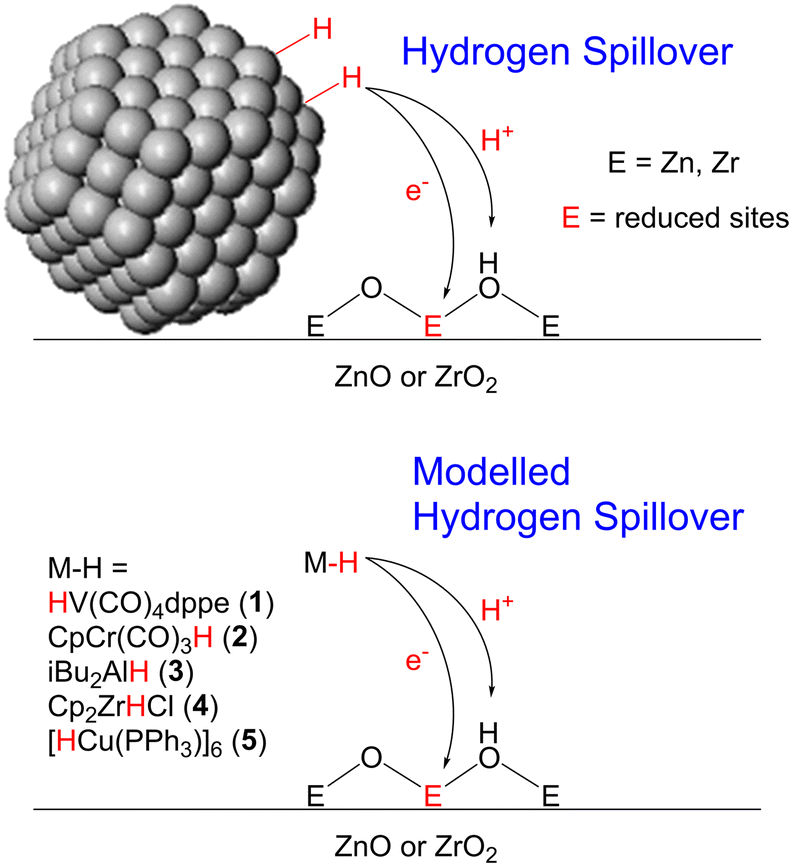 | ||
| Fig. 1 Hydrogen spillover from both nanoparticles and well-defined molecular hydrides on heterogeneous catalyst surfaces. | ||
The mechanism of SMSI in the Cu/ZrO2 case is not as well understood. Indirect evidence for hydrogen spillover such as H/D exchange of Zr–OH species and 1e− reduction of metal dopants has been observed during catalysis.19 However, no direct evidence for the formation of Zr(III) centers during catalysis has been reported. We recently demonstrated that a model oxo-bridged Zr(III) dimer reacts with CO2 to give only CO, rather than formate/methanol, suggesting that even if the sites are present on the surface, they may not contribute to the desired reactivity.20 In contrast, the carbonate, formate, and methoxy intermediates formed during catalysis are bound to the ZrO2 surface.21 Since Lewis acids are known to activate molecular catalysts toward CO2 hydrogenation,22–24 the interface mechanism was proposed in which intermediates bound to Zr Lewis acid sites are activated toward hydride transfer, which occurs from nearby Cu surfaces.21 However, the participation of hydrogen spillover in the catalytic mechanism has not been ruled out.
If indeed hydrogen spillover on these metal oxides is responsible for the production of the catalytically active sites, then the metal surface only serves to reduce the metal oxide. In this case, other metals may result in even more hydrogen spillover and thus a higher proportion of active sites. However, despite hydrogen spillover being known for nearly 60 years,25 our understanding is still limited.16 As a result, we have relatively little information about the mechanisms by which hydrogen spillover might occur on ZnO and ZrO2 or the factors controlling its thermodynamics and kinetics.
Our lack of understanding of the basic chemistry of hydrogen spillover on these oxides is at least partially due to the extremely complex nature of the catalyst surfaces upon which it occurs. In particular, the metal nanoparticles present on the surface have many different potential hydrogen atom positions on the many surface terminations, as well as from defects such as step sites, corner sites, and adatoms that make knowing their basic reactivity very challenging.
One method for investigating hydrogen spillover is to simplify the system by removing the metal nanoparticles from the surface and react pure metal oxide surfaces with well-defined molecular hydrides. Doing so removes the complexity of the metal nanoparticles and hydrogen pressure from the reaction and allows us additionally to vary the reactivity of the metal hydrides at will (pKa,26 bond dissociation free energy (BDFE),27 and hydricity28). We recently demonstrated this strategy by reacting MoO3 with the well-defined hydride HV(CO)4dppe (1),29 which is a strong hydrogen atom donor. This reaction was virtually identical to hydrogen spillover in Pt/MoO3 and mechanistic investigations suggested that the reaction proceeds via sequential proton – and electron transfer steps. We also found that CpCr(CO)3H reacts with CeO2 in a similar way resulting in the same amount of hydrogen spillover as in Pt/CeO2.30 Such a strategy may also yield information about hydrogen spillover on ZnO and ZrO2. Indeed, Mayer and Gamelin have shown that combinations of strong reductants and acids can be used to reduce colloidal nanoparticles of ZnO and TiO2 and that these reduced nanoparticles are good hydrogen atom donors.31–34
Here, we react ZnO and ZrO2 with well-defined molecular metal hydrides to understand how the hydride reactivity affects hydrogen spillover. We found that in contrast to other metal oxides, metal hydrides that are strong hydrogen atom (H·) donors did not reduce ZnO and ZrO2. This lack of reactivity can be rationalized through the metal oxide Pourbaix diagrams. Instead, exposure of the metal oxides to strong hydride (H−) donors resulted in spectral signals characteristic of metal oxide reduction (similar to those observed during hydrogen spillover). The mechanism for this reaction can be understood as initial hydride transfer followed by either surface metal hydride tautomerization to give new OH bonds and inject electrons into the material or via loss of H2 from the surface, leaving behind two reduced sites. We also found that ZrO2 was more difficult to reduce than ZnO, explaining why hydrogen spillover in Cu/ZrO2 is observed less frequently than with ZnO.
Results and discussion
We began the model studies of hydrogen spillover on ZnO and ZrO2 by reacting them with good hydrogen atom donors. We previously showed that the reaction of metal hydrides with weak M–H bonds, such as HV(CO)4dppe (1) or CpCr(CO)3H (2),27,35 with MoO3 and CeO2 leads to hydrogen spillover just as is observed with noble metal nanoparticles.29,30 Therefore, we reacted these two metal hydrides with both ZnO and ZrO2. However, even at elevated (80 °C) temperatures and high metal hydride concentrations (0.1 M), we never observed reduction of either of these metal oxides (by NMR, IR, or EPR).This result was at first puzzling. The V–H and Cr–H bond dissociation free energies of 1 and 2 are 52 and 57 kcal mol−1,35 making them both approximately as reactive as H2, thermodynamically speaking. Since both ZnO and ZrO2 are known to react with H2 at high temperatures,36–40 one would expect 1 to react at some conditions. However, the thermodynamics of the reduction of ZnO and ZrO2 are also dependent on the product that is made during the reduction (Fig. 2). Reduction of metal oxides via PET at low temperatures (near room temperature) generally leads to metal oxyhydroxide products, often termed metal hydrogen bronzes. This is the case for many bulk metal oxides including MoO3,41,42 which we previously observed after reduction with 1.29 It is also possible to reduce metal oxides with loss of H2O, resulting in oxygen vacancies. In this case, the driving force of the reaction is the production of free water in the gas phase (at high temperatures), making these reactions very entropically favorable but often with a high kinetic barrier requiring high temperatures. In the case of ZrO2 and ZnO, it is likely that the second mechanism is thermodynamically favorable while reduction without loss of water does not occur. This is demonstrated nicely by comparison of the redox potentials of ZrO2 and ZnO reduction (both around −0.8 V vs. standard hydrogen electrode (SHE)) obtained from the Pourbaix diagrams,43,44 both of which are more negative than the reduction potential of water to H2. This suggests that the putative oxyhydroxides of these metals are unstable to H2 loss in the presence of water and bulk Zr and Zn oxyhydroxide phases are not stable enough to be shown on their Pourbaix diagrams. However, both metal oxides undergo reduction at >300 °C (ZnO) and >500 °C (ZrO2) under H2 flow in a temperature programmed reduction (TPR) reactor to produce free water and presumably oxygen vacancies,45,46 the water formation being the driving force of the reaction.
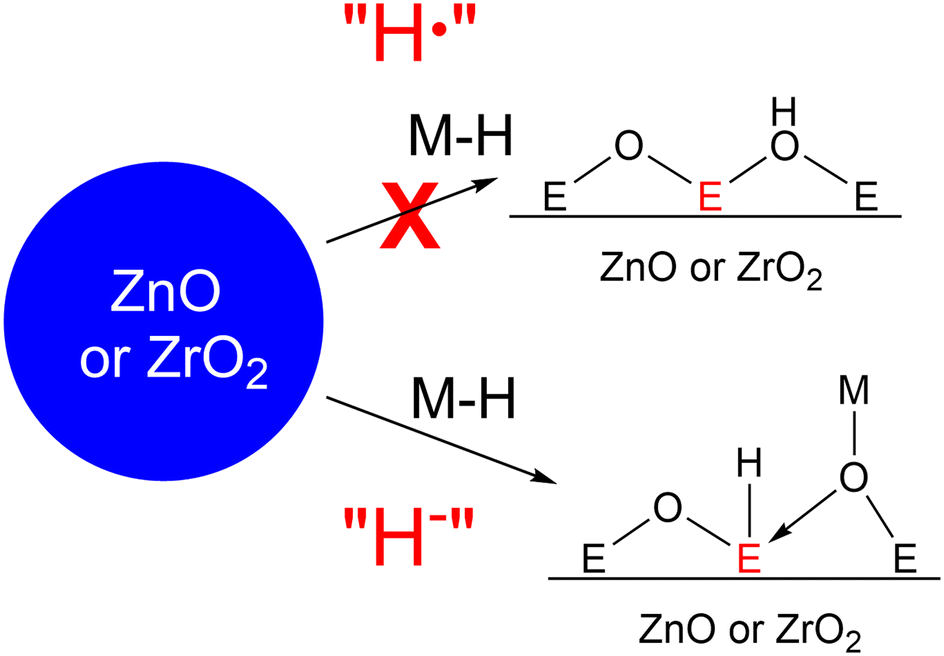 | ||
| Fig. 2 Two potential mechanisms of metal oxide reduction. | ||
Therefore, some additional thermodynamic driving force is necessary for the reduction of ZnO and ZrO2 to occur with molecular metal hydrides. However, at or near room temperature, where we want to perform our well-defined reductions, it is not possible to produce free water. Instead of making free water, we could rather make a new bond to the surface oxygens that compensates for the thermodynamics of the reaction. One well-known reaction on surfaces that produces new M–O bonds is to react hydride donors with metal oxide surfaces (Fig. 3). In this mechanism, a metal hydride donor reacts with bridging oxo ligands of the metal oxide transferring the H− to a metal atom of the surface and making a new M–O bond to the hydride donor.47,48 If the strength of this M–O bond is stronger than a potential OH bond that would be made, this reaction would become thermodynamically favorable.
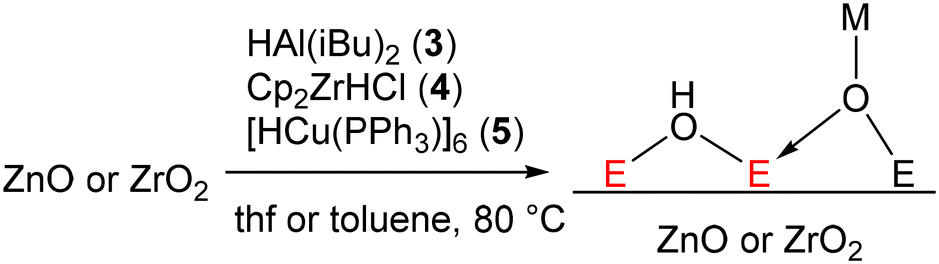 | ||
| Fig. 3 Hydride transfer on metal oxide surfaces. | ||
We attempted this with a variety of hydride donors including diisobutylaluminum hydride (DIBAL, 3), Schwartz's reagent (Cp2ZrHCl, 4), and Stryker's reagent ([HCuPPh3]6, 5) the latter of which somewhat resembles a Cu nanoparticle (which are present in industrial methanol synthesis catalysts). Estimates of Al–O, Zr–O, and Cu–O bond strengths (120–131, ca. 180, and ca. 97 kcal mol−1, respectively)49 suggest that they are indeed much stronger than the weak O–H bonds typical of reduced metal oxide surfaces (typically 50–80 kcal mol−1).50,51 Beginning with ZnO, we reacted a solution of each of these reagents dissolved in toluene with ZnO at 80 °C for 15 h (Fig. 3). This led to a color change from white to grey, indicative of reduction. No reaction was observed at room temperature, indicating that the reactions are likely kinetically limited (i.e. not at equilibrium). We attempted to identify a hydride on the surface, as the characteristic spectroscopic signatures of Zn–H are well known.36–38 We did not observe the signals of Zn–H bonds via NMR or IR (Fig. S1†) for any of the hydride donors used, expected at 0 ppm (ref. 52) and 1710, 1475 cm−1, respectively. However, measurement of X-band EPR spectra of 3-ZnO and 4-ZnO at 77 K (Fig. 4, fitted EPR parameters shown in Table S1†) showed an intense spherically symmetrical signal at g = 1.96, matching the known EPR signal associated with high temperature reduction of the surface with H2.53,54 This signal corresponds to a defect site that is present on the unreduced ZnO in very small quantities (Fig. 4), but which greatly intensifies upon reduction with 3–5. The literature disagrees as to what type of site this represents with suggestions ranging from singly reduced oxygen vacancies (F+ site) to so-called shallow donors with hyperfine interactions to 67Zn and 1H.54,55 However, all sources agree that this site corresponds to a reduction of the ZnO. We also measured XPS spectra of both ZnO and 3-ZnO in order to attempt to learn more about the redox state of the Zn atoms. However, the changes that are typically observed to the Zn 2p3/2 and Zn LMM Auger peaks56,57 were obscured by a general line broadening of the signals in the reduced sample along with the low intensity of the reduced peaks, demonstrated by the low intensity signals due to Al (Fig. S18†). Deng and coworkers also found that the XPS signals due to Zn do not change during H2 reduction of Cu/ZnO/Al2O3 at methanol synthesis conditions (250 °C), similar to our findings.58
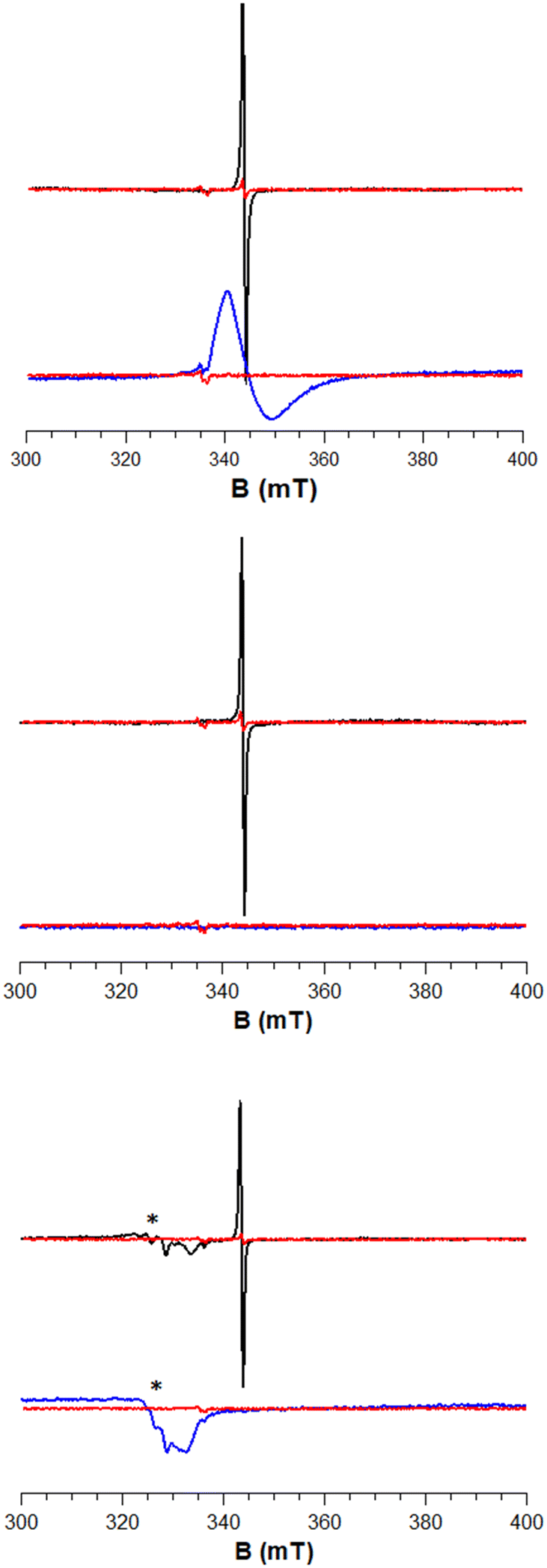 | ||
| Fig. 4 X-band EPR spectra of ZnO (black) and ZrO2 (blue) after reaction with 3 (top), 4 (middle), and 5 (bottom) at 77 K with the spectra of the corresponding unreacted metal oxides in red (* denotes a Cu(II) impurity). | ||
The reaction of ZnO with 5 shows the same characteristic EPR signal for ZnO reduction along with an additional signal with g1 = 2.30, g2 = 2.06, and g3 = 2.03. This new signal shows Cu hyperfine coupling (A = 68 and 300 MHz), meaning that Cu(II) centers are left behind on the surface of the catalyst. We also reacted 5-d6 with ZnO and measured its EPR spectrum. In this spectrum, we see the same EPR signal at g = 1.96 due to the surface reduction but do not see any peaks belonging to Cu(II) species (Fig. S2†). This suggests that the reduced surface sites do not show noticeable hyperfine coupling to the H/D. It also suggests that the Cu(II) peaks are not inherent to the formation of the reduced sites and are likely an impurity in the commercial stryker's reagent that is not present in our self-synthesized 5-d6. In fact, a Cu(II) impurity can be seen in the EPR spectrum of both 5 and 5-d6 (Fig. S2,† the intensity of the Cu(II) signal in our self-synthesized 5-d6 is much lower than that of 5). This impurity is visible by HAADF TEM/EDX as small Cu(II) domains on the surface of the ZnO and ZrO2 as shown in Fig. S5–S8.† However, the XAS analysis demonstrates that the amount of Cu(II) impurity on the surface is negligible compared to the total amount of Cu (vide infra, Fig. S17†). Most of the Cu is dispersed homogeneously across the surface of the metal oxides as shown in the EDX mapping in the ESI.†
We also reacted 3–5 with ZrO2 at 80 °C in a toluene slurry. Similar to ZnO, the reaction of 3–5 with ZrO2 did not show any evidence for Zr–H formation (IR and NMR spectra in Fig. S3†). Unlike ZnO, only 3 led to a new axially symmetric signal in the EPR characteristic of surface reduction (g⊥ = 1.97 and g∥ = 1.91 (overlapping), Fig. 4). A similar EPR spectrum was previously observed for Zr(III) defects in ZrO2 with a distorted tetrahedral coordination geometry.59 Despite not showing signals due to Zr(III) formation, 5-ZrO2 shows the same EPR signals due to Cu(II) as those observed for 5-ZnO (Fig. S4†) suggesting this does indeed come from an impurity and not from reaction of 5 with the surface.
The XANES and EXAFS spectra (Fig. 5 and S9–S17†) also shed additional light on the relationship between the Cu and the metal oxide surface in samples 5-ZnO and 5-ZrO2. Comparison of the absorption edge positions of 5-ZnO, 5-ZrO2 (edge energy = 8978.4 eV for both), and 5 (edge energy = 8978.2 eV) with Cu foil (8977.3 eV) suggests similar oxidation states for 5, 5-ZnO, and 5-ZrO2 (shown in Fig. 5). Simultaneously, the white line intensity at around 8992 eV follows the pattern I5-ZrO2 > I5-ZnO > I5 while the 8979 eV feature follows the trend I5-ZrO2 > I5-ZnO > I5. Comparison of Cu(0) with 5 reveals that 5 is characterized by a broad prepeak. The similar shape to Cu(0) indicates a partial metallic character but its edge position is slightly shifted to higher energy, in accordance with a Cu(I)–H complex. The change in shape around 8992 eV for 5-ZrO2 and 5-ZnO2 could be caused by two factors. Firstly, the residual formation of Cu(II) species upon interaction with the solid support could change the shape of the XANES spectrum. However, this is less likely as the percentage of Cu(II) is likely very small (Fig. S17†). Second, the electron-withdrawing properties of the supports and overlap with ZnO and ZrO2 unoccupied states may modify the available empty 4p levels of Cu, as has been previously observed.60–62 The effect of electron-withdrawing properties of the surfaces on the white line intensity is stronger for ZrO2 than for ZnO (see the longer discussion in the ESI†). This does support a modification of the coordination environment of 5 on the metal oxide surfaces.
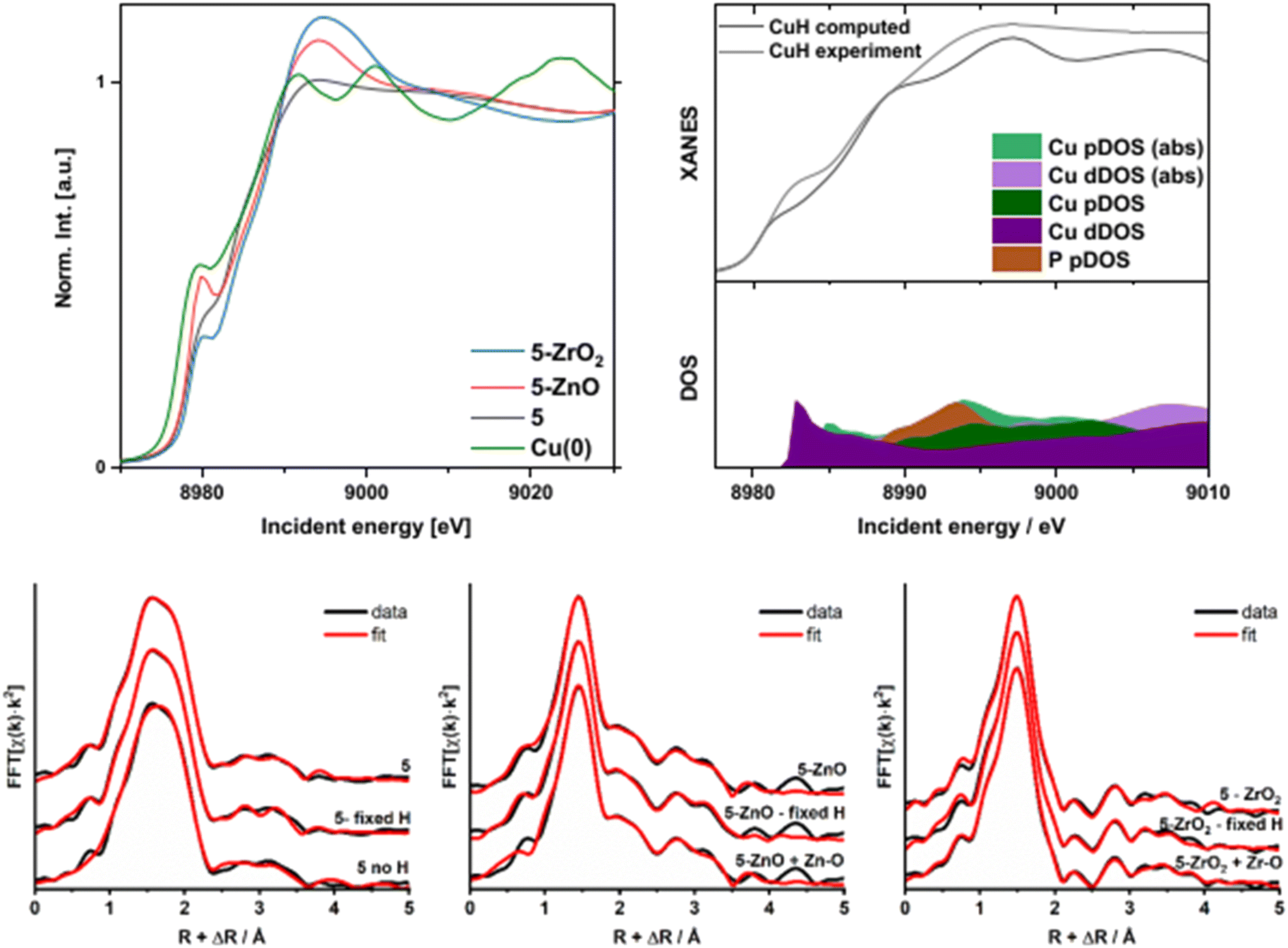 | ||
| Fig. 5 Top left) Cu K-edge XANES and EXAFS spectra of Cu(0), 5, 5-ZnO, and 5-ZrO2. Top right) FMS calculations for 5: comparison of the computed spectrum (top) and relevant density of states functions. (bottom) EXAFS fitting analysis for 5 with various approaches regarding H atoms for: pure 5 (bottom left); 5-ZnO (bottom middle), and 5-ZrO2 (bottom right). | ||
FT-EXAFS also indicates a significant structural difference between 5 and 5-ZnO and 5-ZrO2 (Fig. S6–S8†). The fitting result for pure 5 was obtained as a reference for hydrides reacting with supports. EXAFS (including H atoms) required extremely careful evaluation due to the close-to-zero X-ray scattering factor for hydrogen. However, exclusion of H from the fitting model yields slightly worse fits and, more importantly, unrealistic results (without H atoms in the fit, EXAFS predicts a coordination number for P of ca. 3, which is much higher than the true value of 1). The fit parameters are shown in Table S2.† Since including the H atoms affects the fitting results, we compared the fit with and without H atoms and also used a model with a fixed number of theoretical H atoms. Since the absolute H content modelled from these calculations is somewhat unreliable, we only observe its effect on the larger atoms in the coordination sphere.63–66 For more details and discussion of the EXAFS fitting models, see the ESI.†
If hydride transfer did occur from 5 to ZnO (as in Fig. 3), we would expect a) fewer H− ligands to be present on the Cu and b) a new Cu–O bond to have formed to the surface. Accordingly, we compared the results of EXAFS fitting, shown in Fig. 5 (parameters in Table S3†), for 5-ZnO with either a refined or fixed number of H atoms and also ones containing a potential Cu–O bond. The model for the Cu–O bond of 1.836 Å was taken from the crystal structure of Cu(I)triphenylmethoxide (CCDC identifier WIFQUX).67 The models with both refined and a fixed number of hydrides both gave Debye–Waller factors for 2nd and further shells that were suspiciously high, indicating possible problems with both models, as no extra disorder was anticipated. Additionally, both models required significant anharmonic interactions to be included for convergence. However, inclusion of Cu–O bond resulted in a stable fit with 3 H atoms at 1.568(36) Å, one Cu–O bond at 1.838(12) Å, and Cu–P scatter at 2.155(19) Å (Table S3†). The 2 Cu–Cu scatters from previous attempts became different, one at 2.514(11) Å and the second significantly lengthened to 2.957(52) Å. Such discrepancies would indicate a dramatic structural change upon the interaction of 5 with ZnO and would agree with Fig. 3. Moreover, such a difference is apparent when the FT-EXAFS of 5 and 5-ZnO are directly compared. In this model, we assumed a reaction; thus, the final spectrum would contain contributions from pure 5 and (dominating) from 5-ZnO, thus generating high static disorder, reflected in elevated Debye–Waller factors. This model required no additional H atoms to converge. All things considered, this model reflects the most probable solution among all our attempted fits of this data. Therefore, the XANES and EXAFS data of 5-ZnO support a reaction mechanism involving hydride transfer from 5 to ZnO to produce a new Cu–O bond and reduce the surface.
EXAFS analysis was also conducted for the 5-ZrO2 sample, similar to the 5-ZnO sample, and the results are shown in Fig. 5 (and Table S4†). For varying H atoms and fixed H atoms, the total coordination numbers are almost doubled, which rules out both models. However, once again, the model containing a Cu–O bond gave a satisfactory fit of the experimental data. The 1st coordination shell signal is composed of 4 ± 1 Cu–H scatters at 1.705(21) Å, one Cu–O scatter at 1.823(9) Å and a Cu–P scatter at 2.297(5) Å. Additionally, one Cu–Cu scatter is barely detectable due to the very high Debye–Waller factor at 2.233(11) Å. Since the Cu–Cu scatter was essential for the fit to converge, yet it was impossible to decrease its σ, it can be interpreted as the final stage of hydride decomposition into O[Cu(PPh3)]H subunits, resulting in high static disorder. Since the Cu–H units do not react with ZrO2 to produce reduced surface sites, then this Cu–O bond must form via reaction of the Cu–H with surface Zr–OH bonds. This matches our observation that the OH content of 5-ZrO2 is much lower than bare ZrO2 (Table S12†).
In our previous work on MoO3 and CeO2, our mechanistic investigations suggested that hydrogen spillover in that case proceeds most likely through sequential proton and electron transfer steps (Fig. 6).29 Such a mechanism is corroborated by Boudart and co-workers' observation that strong proton acceptors increase the rate of hydrogen spillover on Pt/WO3.68 This may indeed be the mechanism of hydrogen spillover for many metal oxides. However, for the reactions attempted here with ZnO and ZrO2, this appears not to be the case, since reduction only occurs with hydride donors while no reaction happens with strong proton/hydrogen atom donors (this lack of reactivity with these hydrogen atom donors is likely due to 1 and 2 not being strong enough hydrogen atom donors, and if a stronger hydrogen atom donor were used it would react via proton–electron transfer (PET)). Based on this, we propose that hydrogen spillover on ZnO and ZrO2 can also occur through initial hydride transfer (H−), forming the reduced centers characteristic of hydrogen spillover.
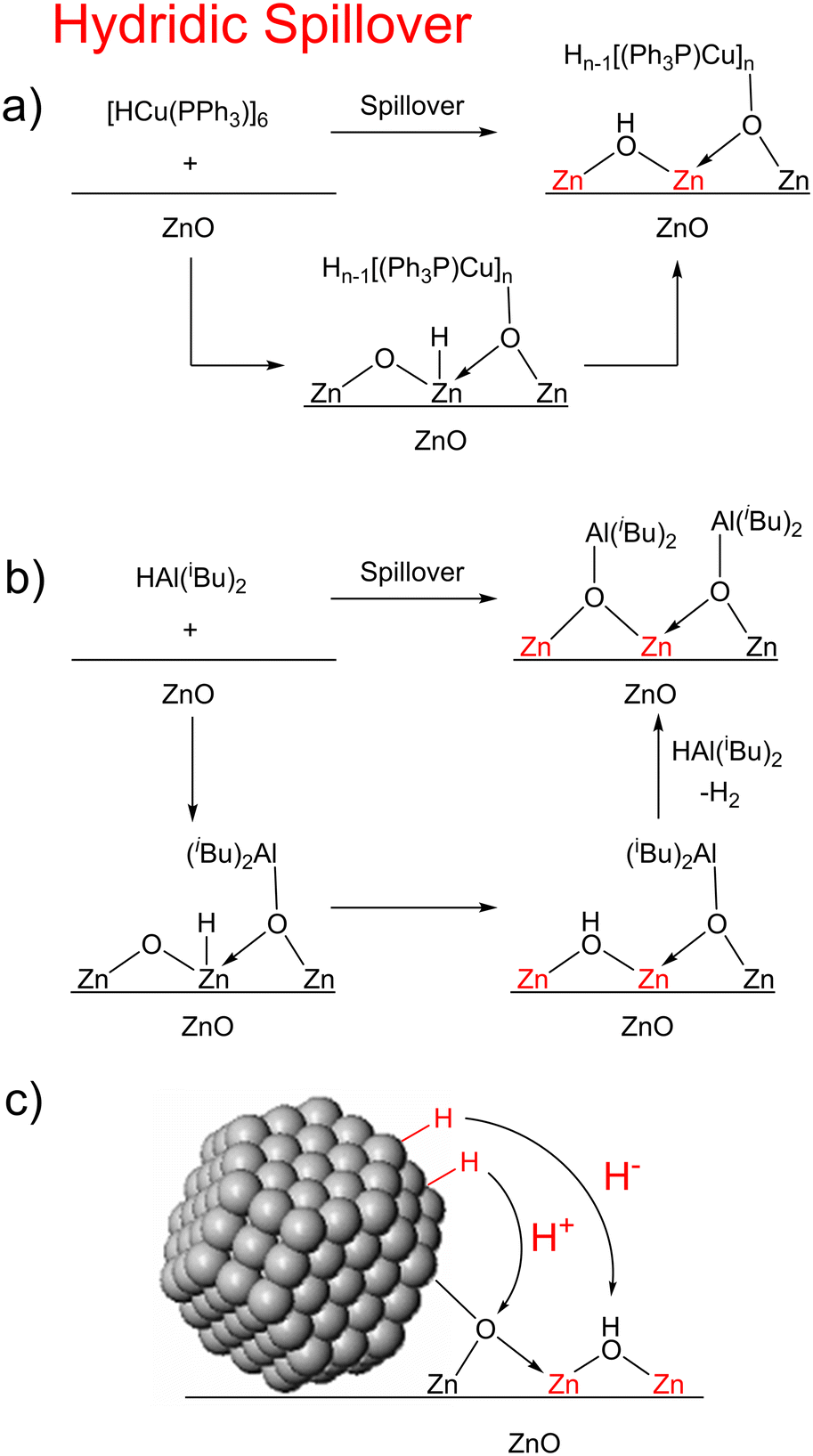 | ||
| Fig. 6 Mechanism of hydridic spillover in 5-ZnO (a), 3-ZnO (b), and for metal nanoparticles on ZnO (c). (red Zn atoms represent reduction of the material). | ||
One question that comes from the investigations above is why we never observe Zn–H or Zr–H species? Indeed, it is likely that the Zn–H's and Zr–H's are intermediates on the pathway to the observed reduced centers. Zn–H can be observed on ZnO surfaces upon treatment of pristine crystalline surfaces and even microcrystalline ZnO with H2 below room temperature, with two types of Zn–H being observed.36,38,52 These two Zn–H species, one terminal one bridging, are thought to originate from heterogeneous dissociation of H2 on surface Zn–O bonds giving new Zn–H and O–H bonds either on Zn terminated surfaces (type 1) or on oxygen vacancies (type 2). However, these hydrides are unstable above room temperature.55,69 There are two possibilities as to the fate of the H atom on the surface. 1) the Zn–H tautomerizes to an OH, acting as a proton electron donor to the surface to make an additional O–H group and transfer its two electrons to the conduction band. This is thought to be the origin of the H2 sensing capability of ZnO. Such a mechanism has also been proposed with ZrO2, where Zr–H species are known,39,40,70,71 but rearrange at elevated temperatures producing additional O–H species and Zr(III) centers.70 To corroborate this, we titrated the OH groups on the surfaces of the bare metal oxides as well as on the reduced surfaces (Table S12†). Treatment of the surfaces with 3 and 4 resulted in a reduction of the number of OH sites on the surface while reaction with 5 increased the number of sites (at least in 5-ZnO where reduction occurs). The increase in OH groups on the surface of 5-ZnO over bare ZnO is consistent with tautomerization of a Zn–H to give new OH groups and reduced surface sites (Fig. 6a). The lower number of OH groups upon treatment with 3 and 4 is likely due to the fact that these hydrides are present in excess and will react with any newly formed OH groups to produce H2 gas (Fig. 6b).
The intermediacy of the metal hydride in this process matches what has been calculated for activation of H2 by ZnO and ZrO2, in which the calculated barrier of heterogeneous activation (producing metal hydrides) is much lower than direct homogeneous activation.72 The tautomerization is most likely not a stepwise deprotonation – electron transfer, since such a process has been calculated to have a very large activation energy. Instead, this would happen simultaneously as a proton-coupled electron transfer (PCET).69 The microscopic reverse of this process has been observed for hydroxy-bridged U(III) dimers, which lose H2 oxidatively to produce an oxo-bridged U(IV) dimer.73 This reaction was shown to proceed via a HO–U(IV)–H intermediate which loses H2via a reverse 1,2-addition type mechanism.74,75
Another possibility is that, upon formation of Zn–H and Zr–H on the surfaces, hydrogen is spontaneously lost from pairs of hydrides to make two reduced centers/inject two electrons into the conduction band. Similar reactions are common for molecular metal hydrides with weak M–H bonds. For example, CpCr(CO)3H (2) is known to reversibly react via a bimolecular pathway forming H2 and two equivalents of CpCr(CO)3 metalloradical, which dimerize rapidly in solution.76,77 Such a mechanism on the surface would require that the metal hydrides be near each other, meaning diffusion of hydrogen atoms across metal oxide surfaces would need to be fast. Calculations suggest that the barrier to hydrogen diffusion through ZnO is higher than the tautomerization of the metal hydrides,78 which suggests that this is not the case, making this mechanism less likely. Additionally, we were not able to observe H2 in the solution during the reaction of hydride donors with the metal oxide surfaces. However, it could be that the amount of H2 formed was too small to measure by NMR in these cases. Therefore, this does not rule out this mechanism, but does make us favor the tautomerization mechanism.
No matter how the reduced sites and OH groups form, the formation of the new M–O bond in the reactions is important for the overall spillover to occur. Depending on the identity of the M fragment that ends up bound to the oxygen atom, a few different things could potentially happen. If the M fragment is highly oxophilic, as is the case for Al and Zr, it is possible that multiple hydride transfers could lead to formation of oxygen vacancies on the surface (along with M–O–M fragments). However, we never observed the formation of the oxo-dimers of our hydride reagents that would indicate formation of oxygen vacancies in any case. For a supported metal catalyst, hydride transfer would form a bond between the metal nanoparticle and a surface oxygen atom (Fig. 6c). From this starting point, the nanoparticle could either transfer another hydride ion (to create an oxygen vacancy with an oxygen atom absorbed on the metal nanoparticle surface) or the M–O bond to the nanoparticle can be broken by proton-transfer, resulting in overall H2 transfer to ZnO.
The mechanism of hydrogen spillover has great consequences for the industrial catalysts for methanol synthesis, Cu/ZnO/Al2O3 and Cu/ZrO2. As discussed above, mechanistic investigations of both catalysts suggest that hydrogen spillover could play a role in the catalytic process. In the case of Cu/ZnO/Al2O3, the reduction of ZnO to form water and CuZn alloy on the metal particle surface is thought to be critical for the catalytic activity, especially for CO2 reduction, where the Zn can be seen switching between the CuZn state and the Zn(O2CH)2 state.18 Therefore, the rate of this transformation is of great importance to the catalytic process.
In the case of Cu/ZrO2, the role of hydrogen spillover is less understood, and has not been directly observed during catalysis. Based on our experiments, this is due to the ZrO2 being more difficult to reduce than the ZnO. Indeed, all hydrides 3–5 result in reduction of ZnO while only 3 is capable of reducing ZrO2. This is most likely either because of a relatively low hydricity (free energy of hydride donation) of DIBAL or the fact that DIBAL is coordinatively unsaturated and can coordinate to the surface oxygen atoms before surface reduction. The Cu nanoparticle may not be capable of reducing ZrO2 under the catalytic conditions, or only minimally. It may indeed be that the Lewis acid sites on ZrO2 as suggested previously, are more important for the synthesis of methanol on Cu/ZrO2 than hydrogen spillover. This does not mean that hydrogen spillover will not play a role on the Cu/ZrO2 catalyst, but does explain why it is more difficult to observe. Our research suggests that nanoparticles that are better hydride donors would also show more hydrogen spillover/surface reduction when supported on ZnO and ZrO2. Therefore, information about the hydricity of metal nanoparticles under H2 would be interesting to see if more hydridic metals do indeed result in greater degrees of reduction.
Conclusions
In conclusion, we reacted both strong hydrogen atom and hydride donors with ZnO and ZrO2 in order to model the hydrogen spillover reaction on these metal oxides. The reduction of ZnO and ZrO2 occurs more readily through the use of hydride donors 3–5 whereas treatment of the surfaces with the strong hydrogen atom donors 1–2 did not result in reduced centers on the surfaces. However, NMR and IR spectroscopy showed no signs of Zn–H and Zr–H after reaction with hydride donors. Instead EPR spectroscopy showed characteristic signals of reduction of ZnO and tetrahedrally coordinated Zr(III) sites on ZrO2 – the same species that form upon hydrogen spillover. The formation of reduced sites on the metal oxide surfaces upon exposure to hydride sources matches the surface science observation that surface hydrides on these metal oxides tautomerize to form surface OH groups above room temperature. This suggests that the surface reduction most likely occurs via a hydride intermediate that then transforms into a new OH group and injects electrons into the material. ZnO is also more readily reduced by hydride donors than ZrO2, which could be the reason for the difficulty in the direct detection of hydrogen spillover in the Cu/ZrO2 heterogeneous catalyst for methanol synthesis. This work is critical to our understanding of hydrogen spillover on supported metal catalysts, particularly ones relevant to renewable energy. It may also be used to inform the choice of metal and metal oxide in future applications where hydrogen spillover sites serve as the catalytically active centers.Data availability
Data for this article, including IR, NMR, EPR, TEM, XPS, and XAS data are available both in the ESI† and at the Data Repository of the University of Stuttgart (DARUS) at https://doi.org/10.18419/darus-4263.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Deutsche Forschungsgemeinschaft (Project no. 444948747 and 358283783 – SFB 1333/2-2022) for funding this project. The authors gratefully acknowledge the core facility SRF AMICA (Stuttgart Research Focus Advanced Materials Innovation and Characterization) at the University of Stuttgart for their support & assistance in this work.Notes and references
- J. Ott, V. Gronemann, F. Pontzen, E. Fiedler, G. Grossmann, D. B. Kersebohm, G. Weiss and C. Witte, Ullmann’s Encyclopedia of Industrial Chemistry, 2012, DOI:10.1002/14356007.a16_465.pub3.
- V. Dieterich, A. Buttler, A. Hanel, H. Spliethoff and S. Fendt, Power-to-liquid via synthesis of methanol, DME or Fischer–Tropsch-fuels: a review, Energy Environ. Sci., 2020, 13, 3207–3252 RSC.
- J. Wambach, A. Baiker and A. Wokaun, CO2 hydrogenation over metal/zirconia catalysts, Phys. Chem. Chem. Phys., 1999, 1, 5071–5080 RSC.
- I. A. Fisher and A. T. Bell, In Situ Infrared Study of Methanol Synthesis from H2/CO over Cu/SiO2and Cu/ZrO2/SiO2, J. Catal., 1998, 178, 153–173 CrossRef CAS.
- M. Zabilskiy, K. Ma, A. Beck and J. A. van Bokhoven, Methanol synthesis over Cu/CeO2–ZrO2 catalysts: the key role of multiple active components, Catal. Sci. Technol., 2021, 11, 349–358 RSC.
- M. Busto, J. M. Grau, J. H. Sepulveda, O. M. Tsendra and C. R. Vera, Hydrocracking of Long Paraffins over Pt–Pd/WO3–ZrO2 in the Presence of Sulfur and Aromatic Impurities, Energy Fuels, 2013, 27, 6962–6972 CrossRef CAS.
- C. Bigey, L. Hilaire and G. Maire, Catalysis on Pd/WO3 and Pd/WO2: Effect of the Modifications of the Surface States Due to Redox Treatments on the Skeletal Rearrangement of Hydrocarbons: Part I. Physical and Chemical Characterizations of Catalysts by BET, TPR, XRD, XAS, and XPS, J. Catal., 1999, 184, 406–420 CrossRef CAS.
- C. Bigey and G. Maire, Catalysis on Pd/WO3 and Pd/WO2: II. Effect of Redox Treatments in Hexanes and Hexenes Re-forming Reactions, J. Catal., 2000, 196, 224–240 CrossRef CAS.
- G. F. Leal, S. Lima, I. Graça, H. Carrer, D. H. Barrett, E. Teixeira-Neto, A. A. S. Curvelo, C. B. Rodella and R. Rinaldi, Design of Nickel Supported on Water-Tolerant Nb2O5 Catalysts for the Hydrotreating of Lignin Streams Obtained from Lignin-First Biorefining, iScience, 2019, 15, 467–488 CrossRef CAS PubMed.
- R. Wojcieszak, A. Jasik, S. Monteverdi, M. Ziolek and M. M. Bettahar, Nickel niobia interaction in non-classical Ni/Nb2O5 catalysts, J. Mol. Catal. A: Chem., 2006, 256, 225–233 CrossRef CAS.
- S. Rong, H. Tan, Z. Pang, Z. Zong, R. Zhao, Z. Li, Z.-N. Chen, N.-N. Zhang, W. Yi and H. Cui, Synergetic effect between Pd clusters and oxygen vacancies in hierarchical Nb2O5 for lignin-derived phenol hydrodeoxygenation into benzene, Renewable Energy, 2022, 187, 271–281 CrossRef CAS.
- D. A. Panayotov and J. T. Yates, Spectroscopic Detection of Hydrogen Atom Spillover from Au Nanoparticles Supported on TiO2: Use of Conduction Band Electrons, J. Phys. Chem. C, 2007, 111, 2959–2964 CrossRef CAS.
- D. A. Panayotov, S. P. Burrows, J. T. Yates and J. R. Morris, Mechanistic Studies of Hydrogen Dissociation and Spillover on Au/TiO2: IR Spectroscopy of Coadsorbed CO and H-Donated Electrons, J. Phys. Chem. C, 2011, 115, 22400–22408 CrossRef CAS.
- T. K. Rahul, M. Mohan and N. Sandhyarani, Enhanced Solar Hydrogen Evolution over In Situ Gold–Platinum Bimetallic Nanoparticle-Loaded Ti3+ Self-Doped Titania Photocatalysts, ACS Sustainable Chem. Eng., 2018, 6, 3049–3059 CrossRef CAS.
- W. C. Conner and J. L. Falconer, Spillover in Heterogeneous Catalysis, Chem. Rev., 1995, 95, 759–788 CrossRef CAS.
- R. Prins, Hydrogen Spillover. Facts and Fiction, Chem. Rev., 2012, 112, 2714–2738 CrossRef CAS PubMed.
- M. Behrens, F. Studt, I. Kasatkin, S. Kühl, M. Hävecker, F. Abild-Pedersen, S. Zander, F. Girgsdies, P. Kurr, B.-L. Kniep, M. Tovar, R. W. Fischer, J. K. Nørskov and R. Schlögl, The Active Site of Methanol Synthesis over Cu/ZnO/Al<sub>2</sub>O<sub>3</sub> Industrial Catalysts, Science, 2012, 336, 893 CrossRef CAS PubMed.
- M. Zabilskiy, V. L. Sushkevich, D. Palagin, M. A. Newton, F. Krumeich and J. A. van Bokhoven, The unique interplay between copper and zinc during catalytic carbon dioxide hydrogenation to methanol, Nat. Commun., 2020, 11, 2409 CrossRef CAS PubMed.
- K.-D. Jung and A. T. Bell, Role of Hydrogen Spillover in Methanol Synthesis over Cu/ZrO2, J. Catal., 2000, 193, 207–223 CrossRef CAS.
- E. J. Wimmer, S. V. Klostermann, M. Ringenberg, J. Kästner and D. P. Estes, Oxo-Bridged Zr Dimers as Well-defined Models of Oxygen Vacancies on ZrO2, Eur. J. Inorg. Chem., 2023, e202200709 CrossRef CAS.
- K. Larmier, W.-C. Liao, S. Tada, E. Lam, R. Verel, A. Bansode, A. Urakawa, A. Comas-Vives and C. Copéret, CO2-to-Methanol Hydrogenation on Zirconia-Supported Copper Nanoparticles: Reaction Intermediates and the Role of the Metal–Support Interface, Angew. Chem., Int. Ed., 2017, 56, 2318–2323 CrossRef CAS PubMed.
- S. Wesselbaum, T. vom Stein, J. Klankermayer and W. Leitner, Hydrogenation of Carbon Dioxide to Methanol by Using a Homogeneous Ruthenium–Phosphine Catalyst, Angew. Chem., Int. Ed., 2012, 51, 7499–7502 CrossRef CAS PubMed.
- K. Thenert, K. Beydoun, J. Wiesenthal, W. Leitner and J. Klankermayer, Ruthenium-Catalyzed Synthesis of Dialkoxymethane Ethers Utilizing Carbon Dioxide and Molecular Hydrogen, Angew. Chem., Int. Ed., 2016, 55, 12266–12269 CrossRef CAS PubMed.
- C. A. Huff and M. S. Sanford, Cascade Catalysis for the Homogeneous Hydrogenation of CO2 to Methanol, J. Am. Chem. Soc., 2011, 133, 18122–18125 CrossRef CAS PubMed.
- S. Khoobiar, Particle to Particle Migration of Hydrogen Atoms on Platinum—Alumina Catalysts from Particle to Neighboring Particles, J. Phys. Chem., 1964, 68, 411–412 CrossRef CAS.
- R. H. Morris, Brønsted–Lowry Acid Strength of Metal Hydride and Dihydrogen Complexes, Chem. Rev., 2016, 116, 8588–8654 CrossRef CAS PubMed.
- M. Tilset and V. D. Parker, Solution homolytic bond dissociation energies of organotransition-metal hydrides, J. Am. Chem. Soc., 1989, 111, 6711–6717 CrossRef CAS.
- E. S. Wiedner, M. B. Chambers, C. L. Pitman, R. M. Bullock, A. J. M. Miller and A. M. Appel, Thermodynamic Hydricity of Transition Metal Hydrides, Chem. Rev., 2016, 116, 8655–8692 CrossRef CAS PubMed.
- A. Almidani, M. Benz, M. Winkler, Y. Ikeda, B. Grabowski, J. van Slageren and D. Estes, EMI Series: The Reaction of HV(CO)4dppe with MoO3: a Well-Defined Model of Hydrogen Spillover, Catal. Sci. Technol., 2021, 11, 7540 RSC.
- O. Bunjaku, J. S. Florenski, J. Wischnat, E. Klemm, O. V. Safonova, J. Van Slageren and D. P. Estes, Understanding the Reducibility of CeO2 Surfaces by Proton-Electron Transfer from CpCr(CO)3H, Inorg. Chem., 2024, 63, 7512 CrossRef CAS PubMed.
- J. N. Schrauben, R. Hayoun, C. N. Valdez, M. Braten, L. Fridley and J. M. Mayer, Titanium and Zinc Oxide Nanoparticles Are Proton-Coupled Electron Transfer Agents, Science, 2012, 336, 1298 CrossRef CAS PubMed.
- C. N. Valdez, M. Braten, A. Soria, D. R. Gamelin and J. M. Mayer, Effect of Protons on the Redox Chemistry of Colloidal Zinc Oxide Nanocrystals, J. Am. Chem. Soc., 2013, 135, 8492–8495 CrossRef CAS PubMed.
- C. N. Valdez, A. M. Schimpf, D. R. Gamelin and J. M. Mayer, Proton-Controlled Reduction of ZnO Nanocrystals: Effects of Molecular Reductants, Cations, and Thermodynamic Limitations, J. Am. Chem. Soc., 2016, 138, 1377–1385 CrossRef CAS PubMed.
- J. Castillo-Lora, R. Mitsuhashi and J. M. Mayer, Revealing the Relative Electronic Landscape of Colloidal ZnO and TiO2 Nanoparticles via Equilibration Studies, J. Phys. Chem. C, 2019, 123, 10262–10271 CrossRef CAS.
- J. Choi, M. E. Pulling, D. M. Smith and J. R. Norton, Unusually Weak Metal−Hydrogen Bonds in HV(CO)4(P−P) and Their Effectiveness as H• Donors, J. Am. Chem. Soc., 2008, 130, 4250–4252 CrossRef CAS PubMed.
- G. Ghiotti, A. Chiorino and F. Boccuzzi, Surface chemistry and electronic effects of H2 (D2) on two different microcrystalline ZnO powders, Surf. Sci., 1993, 287-288, 228–234 CrossRef CAS.
- G. Hussain and N. Sheppard, A Fourier-transform spectral-ratioed reinvestigation of the infrared spectra from H2 and D2 adsorbed on ZnO at room temperature and at 160 °C, J. Chem. Soc., Faraday Trans., 1990, 86, 1615–1617 RSC.
- R. P. Eischens, W. A. Pliskin and M. J. D. Low, The infrared spectrum of hydrogen chemisorbed on zinc oxide, J. Catal., 1962, 1, 180–191 CrossRef CAS.
- A. Trunschke, D. L. Hoang and H. Lieske, In situ FTIR studies of high-temperature adsorption of hydrogen on zirconia, J. Chem. Soc., Faraday Trans., 1995, 91, 4441–4444 RSC.
- J. Kondo, Y. Sakata, K. Domen, K.-i. Maruya and T. Onishi, Infrared study of hydrogen adsorbed on ZrO2, J. Chem. Soc., Faraday Trans., 1990, 86, 397–401 RSC.
- N. Sotani, K. Eda, M. Sadamatu and S. Takagi, Preparation and Characterization of Hydrogen Molybdenum Bronzes, HxMoO3, Bull. Chem. Soc. Jpn., 1989, 62, 903–907 CrossRef CAS.
- X. Sha, L. Chen, A. C. Cooper, G. P. Pez and H. Cheng, Hydrogen Absorption and Diffusion in Bulk α-MoO3, J. Phys. Chem. C, 2009, 113, 11399–11407 CrossRef CAS.
- B. Beverskog and I. Puigdomenech, Revised pourbaix diagrams for zinc at 25–300 °C, Corros. Sci., 1997, 39, 107–114 CrossRef CAS.
- M. H. Kaye and W. T. Thompson, in Uhlig's Corrosion Handbook, ed. R. W. Revie, John Wiley & Sons, Inc., Hoboken, NJ, 3rd edn, 2011 Search PubMed.
- S.-W. Park, O.-S. Joo, K.-D. Jung, H. Kim and S.-H. Han, Development of ZnO/Al2O3 catalyst for reverse-water-gas-shift reaction of CAMERE (carbon dioxide hydrogenation to form methanol via a reverse-water-gas-shift reaction) process, Appl. Catal., A, 2001, 211, 81–90 CrossRef CAS.
- D. L. Hoang and H. Lieske, Effect of hydrogen treatments on ZrO2 and Pt/ZrO2 catalysts, Catal. Lett., 1994, 27, 33–42 CrossRef CAS.
- C. Copéret, D. P. Estes, K. Larmier and K. Searles, Isolated Surface Hydrides: Formation, Structure, and Reactivity, Chem. Rev., 2016, 116, 8463–8505 CrossRef PubMed.
- H.-H. Nguyen, Z. Li, T. Enenkel, J. Hildebrand, M. Bauer, M. Dyballa and D. P. Estes, Probing the Interactions of Immobilized Ruthenium Dihydride Complexes with Metal Oxide Surfaces by MAS NMR: Effects on CO2 Hydrogenation, J. Phys. Chem. C, 2021, 125, 14627–14635 CrossRef CAS.
- D. R. Lide, CRC Handbook of Chemistry and Physics, CRC Press, Boca Raton, FL, 87th edn, 2006 Search PubMed.
- Y. Ikeda, D. P. Estes and B. Grabowski, Comprehensive Understanding of H Adsorption on MoO3 from Systematic Ab Initio Simulations, J. Phys. Chem. C, 2022, 126, 7728–7738 CrossRef CAS.
- R. G. Agarwal, H.-J. Kim and J. M. Mayer, Nanoparticle O–H Bond Dissociation Free Energies from Equilibrium Measurements of Cerium Oxide Colloids, J. Am. Chem. Soc., 2021, 143, 2896–2907 CrossRef CAS PubMed.
- M. Watanabe and T. Ito, Magnetic Resonance Studies of Hydrogen Adsorbed on Zinc Oxide. II. Proton NMR of Chemisorbed Hydrogen and Molecular Hydrogen Adsorbed Ionically, Jpn. J. Appl. Phys., 1980, 19, 1863 CrossRef CAS.
- C. Drouilly, J.-M. Krafft, F. Averseng, S. Casale, D. Bazer-Bachi, C. Chizallet, V. Lecocq, H. Vezin, H. Lauron-Pernot and G. Costentin, ZnO Oxygen Vacancies Formation and Filling Followed by in Situ Photoluminescence and in Situ EPR, J. Phys. Chem. C, 2012, 116, 21297–21307 CrossRef CAS.
- L. S. Vlasenko, Magnetic Resonance Studies of Intrinsic Defects in ZnO: Oxygen Vacancy, Appl. Magn. Reson., 2010, 39, 103–111 CrossRef CAS.
- S. B. Orlinskii, J. Schmidt, P. G. Baranov, D. M. Hofmann, C. de Mello Donegá and A. Meijerink, Probing the Wave Function of Shallow Li and Na Donors in ZnO Nanoparticles, Phys. Rev. Lett., 2004, 92, 047603 CrossRef PubMed.
- S.-K. Jeong, M.-H. Kim, S.-Y. Lee, H. Seo and D.-K. Choi, Dual active layer a-IGZO TFT via homogeneous conductive layer formation by photochemical H-doping, Nanoscale Res. Lett., 2014, 9, 619 CrossRef PubMed.
- P. S. Wehner, G. C. Tustin and B. L. Gustafson, XPS study of the reduction and reoxidation of ZnO-supported palladium, J. Catal., 1984, 88, 246–248 CrossRef CAS.
- W.-L. Dai, Q. Sun, J.-F. Deng, D. Wu and Y.-H. Sun, XPS studies of Cu/ZnO/Al2O3 ultra-fine catalysts derived by a novel gel oxalate co-precipitation for methanol synthesis by CO2+H2, Appl. Surf. Sci., 2001, 177, 172–179 CrossRef CAS.
- C. Gionco, M. C. Paganini, E. Giamello, R. Burgess, C. Di Valentin and G. Pacchioni, Paramagnetic Defects in Polycrystalline Zirconia: An EPR and DFT Study, Chem. Mater., 2013, 25, 2243–2253 CrossRef CAS.
- A. L. Ankudinov, J. J. Rehr, J. J. Low and S. R. Bare, Pt L-edge XANES as a probe of Pt clusters, J. Synchrotron Radiat., 2001, 8, 578–580 CrossRef CAS PubMed.
- K. Sang, J. Zuo, X. Zhang, Q. Wang, W. Chen, G. Qian and X. Duan, Towards a molecular understanding of the electronic metal-support interaction (EMSI) in heterogeneous catalysis, Green Energy Environ., 2023, 8, 619–625 CrossRef CAS.
- M. G. Samant and M. Boudart, Support effects on electronic structure of platinum clusters in Y zeolite, J. Phys. Chem., 1991, 95, 4070–4074 CrossRef CAS.
- S. Mukerjee, J. McBreen, J. J. Reilly, J. R. Johnson, G. Adzic, K. Petrov, M. P. S. Kumar, W. Zhang and S. Srinivasan, In Situ X-Ray Absorption Spectroscopy Studies of Metal Hydride Electrodes, J. Electrochem. Soc., 1995, 142, 2278 CrossRef CAS.
- P. Ngene, A. Longo, L. Mooij, W. Bras and B. Dam, Metal-hydrogen systems with an exceptionally large and tunable thermodynamic destabilization, Nat. Commun., 2017, 8, 1846 CrossRef PubMed.
- B. Lengeler, Lattice Site Location of Hydrogen by Use of Extended X-Ray Absorption Fine Structure, Phys. Rev. Lett., 1984, 53, 74–77 CrossRef CAS.
- J. Purans, A. P. Menushenkov, S. P. Besedin, A. A. Ivanov, V. S. Minkov, I. Pudza, A. Kuzmin, K. V. Klementiev, S. Pascarelli, O. Mathon, A. D. Rosa, T. Irifune and M. I. Eremets, Local electronic structure rearrangements and strong anharmonicity in YH3 under pressures up to 180
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) GPa, Nat. Commun., 2021, 12, 1765 CrossRef CAS PubMed.
GPa, Nat. Commun., 2021, 12, 1765 CrossRef CAS PubMed. - M. Håkansson, C. Lopes and S. Jagner, Copper(I) alkoxides: preparation and structural characterisation of triphenylmethoxocopper(I) and of an octanuclear form of t-butoxocopper(I), Inorg. Chim. Acta, 2000, 304, 178–183 CrossRef.
- R. B. Levy and M. Boudart, The kinetics and mechanism of spillover, J. Catal., 1974, 32, 304–314 CrossRef CAS.
- Y. Wang, B. Meyer, X. Yin, M. Kunat, D. Langenberg, F. Träger, A. Birkner and C. Wöll, Hydrogen Induced Metallicity on the ZnO(10-10) Surface, Phys. Rev. Lett., 2005, 95, 266104 CrossRef CAS PubMed.
- T. Onishi, H. Abe, K.-i. Maruya and K. Domen, I.r. spectra of hydrogen adsorbed on ZrO2, J. Chem. Soc., Chem. Commun., 1985, 617–618, 10.1039/C39850000617.
- J. Kondo, H. Abe, Y. Sakata, K.-i. Maruya, K. Domen and T. Onishi, Infrared studies of adsorbed species of H2, CO and CO2 over ZrO2, J. Chem. Soc., Faraday Trans. 1, 1988, 84, 511–519 RSC.
- J. Kiss, A. Witt, B. Meyer and D. Marx, Methanol synthesis on ZnO(000
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ). I. Hydrogen coverage, charge state of oxygen vacancies, and chemical reactivity, J. Chem. Phys., 2009, 130, 184706 CrossRef PubMed.
). I. Hydrogen coverage, charge state of oxygen vacancies, and chemical reactivity, J. Chem. Phys., 2009, 130, 184706 CrossRef PubMed. - W. W. Lukens, S. M. Beshouri, L. L. Blosch and R. A. Andersen, Oxidative Elimination of H2 from [Cp‘2U(μ-OH)]2 To Form [Cp‘2U(μ-O)]2, Where Cp‘ Is 1,3-(Me3C)2C5H3 or 1,3-(Me3Si)2C5H3, J. Am. Chem. Soc., 1996, 118, 901–902 CrossRef CAS.
- D. P. Estes, G. Siddiqi, F. Allouche, K. V. Kovtunov, O. V. Safonova, A. L. Trigub, I. V. Koptyug and C. Copéret, C–H Activation on Co,O Sites: Isolated Surface Sites versus Molecular Analogs, J. Am. Chem. Soc., 2016, 138, 14987–14997 CrossRef CAS PubMed.
- J. R. Webb, S. A. Burgess, T. R. Cundari and T. B. Gunnoe, Activation of carbon–hydrogen bonds and dihydrogen by 1,2-CH-addition across metal–heteroatom bonds, Dalton Trans., 2013, 42, 16646–16665 RSC.
- J. R. Norton, T. Spataru, D. M. Camaioni, S.-J. Lee, G. Li, J. Choi and J. A. Franz, Kinetics and Mechanism of the Hydrogenation of the CpCr(CO)3•/[CpCr(CO)3]2 Equilibrium to CpCr(CO)3H, Organometallics, 2014, 33, 2496–2502 CrossRef CAS.
- E. O. Fischer, W. Hafner and H. O. Stahl, Über Cyclopentadienyl-metall-carbonyl-wasserstoffe des Chroms, Molybdäns und Wolframs, Z. Anorg. Allg. Chem., 1955, 282, 47–62 CrossRef CAS.
- W. Beyer, U. Breuer, F. Hamelmann, J. Hüpkes, A. Stärk, H. Stiebig and U. Zastrow, Hydrogen diffusion in zinc oxide thin films, MRS Online Proc. Libr., 2009, 1165, 524 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: IR, NMR, EPR, and XAS spectra and fitted simulations as well as experimental detail. See DOI: https://doi.org/10.1039/d4cy00504j |
| This journal is © The Royal Society of Chemistry 2024 |