Hydrolysis of amide bonds in dipeptides and nylon 6 over a ZrO2 catalyst†
Received
26th April 2024
, Accepted 28th May 2024
First published on 29th May 2024
Abstract
Hydrolysis of amide bonds is expected as a promising technology, namely in the fields of biorefinery using proteins and chemical recycling of plastics. For the model reaction of glycylglycine hydrolysis, ZrO2 was found to work as an effective catalyst and afforded glycine in up to 97%-C under the optimum conditions, while acidic oxides and basic oxides were inferior to ZrO2. The characterization using N2 physisorption, XRD, NH3-TPD, and CO2-TPD revealed that the weight-basis activity of ZrO2 correlated with its acidic and basic properties. ZrO2 was demonstrated to be applicable to the hydrolysis of amide bonds: dipeptides with acidic or basic side chain into amino acids; small organic amides into both amines and carboxylic acids with their equivalent amounts; and nylon 6 into ε-caprolactam and ε-aminocaproic acid.
1. Introduction
Amide bonds are ubiquitous chemical bonds that are often found in small organic compounds used as solvents (e.g., N,N-dimethylformamide and N,N-dimethylacetamide), pharmaceutical compounds, proteins (in this case, amide bonds are generally called peptide bonds), and synthetic polymers (i.e., polyamides such as nylon and aramid).1–3 In the latter two exemplified categories, their catalytic hydrolysis is an attractive means of producing building blocks, i.e., amino acids from proteins and amines and carboxylic acids from synthetic polymers, from the viewpoints of biorefinery and chemical recycling of plastics.4,5 However, the resonance structure of amides leads to the high stability of C–N bonds and makes the dissociation of amide bonds difficult. The development of catalysts is necessary for the activation of amide bonds to be hydrolyzed. Considering the production of amines and carboxylic acids (or amino acids) via the hydrolysis of amide bonds, strong acids (e.g., HCl and H2SO4) and strong bases (e.g., NaOH), which are typical catalysts for hydrolyzing a variety of chemical bonds, inevitably undergo neutralization by such reaction products to be deactivated; in other words, such strong acids/bases cannot make the hydrolysis of amides catalytic. In this respect, well-designed catalysts need to be devised for the hydrolysis of amide bonds involved in a variety of compounds as described above.
The hydrolysis of peptide bonds involved in various molecules has been achieved by using both homogeneous and heterogeneous catalysts, as summarized in Table S1.† As homogeneous catalysts, Sc(OTf)3 (OTf = trifluoromethanesulfonate)6 and polyoxometalate K15H[Zr(α2-P2W17O61)2] (ref. 7) were reported for the hydrolysis of oligopeptides and dipeptides to produce (co-)monomers in high yields. Cerium hydroxide gel formed via the precipitation of Ce(NH4)2(NO3)6 in a buffer8 and Zr oxide clusters involved in metal–organic frameworks (MOFs) as nodes have also been employed as heterogeneous catalysts for efficiently hydrolyzing peptide bonds in a variety of dipeptides and even proteins (myoglobin and hen egg-white lysozyme) under mild conditions.9–12 This MOF-catalyzed hydrolysis, however, necessitated the careful control of pH by using acid, base, or buffer solution and also could suffer from the restriction of reaction conditions due to the lower thermal stability of MOFs compared to metal oxides; the former disadvantage makes the separation and purification of product(s) complicated, and the latter demerit narrows the substrate scope. Nb2O5 was reported to work as a heterogeneous catalyst for the hydrolysis of aliphatic and aromatic (N-substituted) amides to produce corresponding carboxylic acids in water under reflux conditions (Table S2†).13 As a variant of amide-bond cleavage by solvents, the alcoholysis of various amides to produce esters was achieved by using CeO2,14–17 whose amphoteric surface nature is responsible for a variety of reactions.18–22 Yet, these reaction systems using metal oxides as catalysts continuously released useful co-products (i.e., NH3 or amines) to outside of the reactor or trapped such co-products with an acidic zeolite since the equilibrium of these reactions need to be shifted and also such basic compounds potentially deactivate Lewis acidic metal centers that function as catalytically active sites. Meanwhile, De Vos et al. demonstrated the NH3-assisted depolymerization of nylon 66 by using Nb2O5 in an acetamide solvent to produce N,N′-hexamethylene bis(acetamide), which can be further converted into hexamethylenediamine, in up to 98% yield.23 A homogeneous Zn(OTf)2 catalyst was effective for the esterification of amides possessing a hydroxy group coordinating to the β-carbon atom although it suffered from the narrow substrate scope.24 Mn-containing complexes were also reported as effective catalysts for the esterification of tertiary amides.25,26 For the hydrolysis of nylon 6 (also called polyamide-6) as an example of polyamides, high-temperature water typically at 573 K or above produced its monomer in cyclic form (i.e., ε-caprolactam) and/or linear form (i.e., ε-aminocaproic acid) in up to 92% yield (Table S3†).4,27–34 The use of acids enabled the reaction temperature to decrease to a certain or large extent; some of them including solid acids such as H-beta zeolites and sulfated γ-Al2O3–ZrO2 (ref. 32 and 34) were reported to work as catalysts possibly owing to the production of ε-caprolactam, which possesses neither a carboxy group nor an amino group, instead of ε-aminocaproic acid. As above, catalytic hydrolysis reactions for specific compounds have been reported thus far; however, to the best of our knowledge, there is no report on catalysts that enable hydrolysis of amide bonds in various types of compounds consisting of small organic amides, peptides, and synthetic polymers. In this context, we here demonstrate a widely-applicable catalyst consisting of amphoteric ZrO2, which is also known as an effective catalyst to various reactions due to its acid–base bifunctionality,18,19,35–37 for hydrolytically producing amino acids from peptides, ε-caprolactam and ε-aminocaproic acid from nylon 6, and both amines and carboxylic acids with equivalent amounts from small organic amides.
2. Experimental
2.1. Reagents
All the chemicals listed in Table S4† were purchased from each supplier and used as-received without further purification. Most of the metal oxides tested as catalysts were calcined in an electric furnace in air prior to their use for catalytic reactions (Table S5†); for example, the most intensively studied metal oxide, monoclinic-rich ZrO2 (hereafter denoted as m-ZrO2; RC-100, Daiichi Kigenso Kagaku Kogyo), was calcined in air at 673 K for 3 h. Also, tetragonal-rich ZrO2 (denoted as t-ZrO2) was prepared via the calcination of Zr(OH)4 (Daiichi Kigenso Kagaku Kogyo) in air at 673 K for 3 h. For MgO and CaO, they were heat-treated in a N2 flow (30 mL min−1) at 773 K for 3 h and handled in a glove box filled with Ar in order not to be deactivated by atmospheric CO2.
2.2. Hydrolysis of glycylglycine
Typically, 2.0 mmol of glycylglycine (denoted as Gly-Gly), 0.10 g of catalyst, 5.0 g of H2O, and a stirring bar were charged into a pressure-resistant glass tube (Ace Glass, inner volume 15 mL). The reaction mixture was heated at 353 K in an oil bath for a certain time and then cooled to room temperature in a water bath. The reaction mixture was collected with H2O and filtered with a polytetrafluoroethylene (PTFE) syringe filter (0.2 μm mesh). The unreacted substrate and products were quantified by using a high-performance liquid chromatograph (HPLC; Prominence HPLC System, Shimadzu, refractive index (RI) detector) equipped with an Asahipak NH2P-50 4E column (Shodex, ϕ 4.6 × 250 mm, mobile phase = 20 mM phosphate buffer solution (pH 6)/acetonitrile = 30/70 (vol/vol), 1.0 mL min−1, 313 K) with D-sorbitol as an internal standard. The values of conversion, yield, and balance were calculated by using the following equations. Note that no peaks other than Gly-Gly, glycine, or glycine anhydride were observed in this study.| | 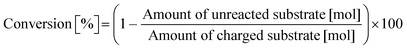 | (1) |
| | 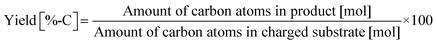 | (2) |
| | 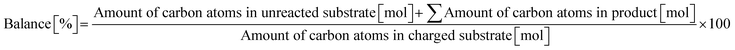 | (3) |
For the reuse test, the spent catalyst was separated from the reaction mixture by centrifugation, washed with H2O, and dried in an oven at 383 K overnight. Afterward, the catalyst was used for the next run without further treatment or after calcination in air at 673 K for 3 h.
2.3. Hydrolysis of various amide compounds
Dipeptides other than Gly-Gly, aliphatic alkylamides (N-ethylpropionamide and N,N-diethylpropionamide), and nylon 6 were employed as substrates via each specific procedure as follows.
Dipeptides.
0.50 mmol of dipeptide, 0.025 g of m-ZrO2, 5.0 g of H2O, and a stirring bar were charged into a pressure-resistant glass tube. The reaction mixture was heated at 373 K in an oil bath for a certain time and then cooled to room temperature in a water bath. The reaction mixture was collected with H2O and filtered with a PTFE syringe filter. The reaction products were quantified by using an HPLC equipped with an Asahipak NH2P-50 4E column (the same as the hydrolysis of Gly-Gly) after the addition of D-sorbitol as an internal standard. The conversion and product yield were calculated by using the following equations.| |  | (4) |
| |  | (5) |
Aliphatic alkylamides.
2.0 mmol of amide, 0.10 g of m-ZrO2, 10 g of H2O, and a stirring bar were charged into a stainless-steel autoclave (HIRO Company, inner volume 190 mL). After sealing, the reactor was purged and pressurized with Ar at 1 MPa at room temperature. The reactor was heated at the designated temperature for a certain time and then cooled to room temperature in a water bath. The reaction mixture was collected with H2O and filtered with a PTFE syringe filter. The reaction products were quantified by a gas chromatograph with a flame-ionization detector (GC-FID; Shimadzu, GC-2014) equipped with an InertCap for Amine capillary column (GL Sciences, ϕ 0.32 mm × 30 m) with tert-butyl alcohol as an internal standard. The conversion and product yield were calculated using eqn (4) and (5). The balance of each moiety (carboxylic acid or amine) was calculated by using the following equation.| | 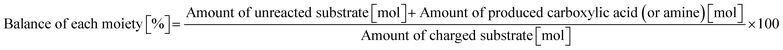 | (6) |
Nylon 6.
0.23 g of nylon 6 (corresponding to 2.0 mmol of monomeric unit, pellets), 0.10 g of m-ZrO2, 10 g of H2O, and a stirring bar were charged into a stainless-steel autoclave. The subsequent procedures were the same as the cases of aliphatic alkylamides (vide supra). The reaction products were collected with H2O. After the filtration through a PTFE syringe filter, the water-soluble products were quantified by using two HPLCs equipped with either an Asahipak NH2P-50 4E column (the same as the hydrolysis of Gly-Gly) or a Gemini NX-C18 column (Phenomenex, ϕ 4.6 × 150 mm, mobile phase = acetonitrile/20 mM phosphate buffer solution (pH 6) = 5/95 (vol/vol), 1.0 mL min−1, 313 K) with both glycerol and tert-butyl alcohol as internal standards. Typical HPLC charts for these two columns are shown in Fig. S1.† The yields of monomers (i.e., ε-caprolactam and ε-aminocaproic acid) were calculated from eqn (7), while the yield of oligomers was estimated with the assumption of their calibration factor from ε-aminocaproic acid in the case of the NH2P-50 4E column and calculated from eqn (8). The value of balance was calculated from eqn (9). We should note that the conversion of nylon 6 could not be estimated since the quantification of unreacted nylon 6 was impossible.| | 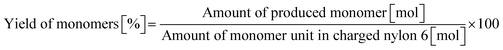 | (7) |
| | 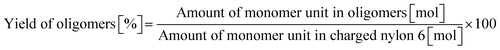 | (8) |
| | 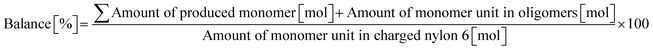 | (9) |
2.4. Characterization of catalysts
N2 physisorption measurement was conducted at 77 K to examine the specific surface area of the catalyst using the Brunauer–Emmett–Teller (BET) equation with an automated instrument (Gemini VII 2360, Micromeritics). X-ray diffraction (XRD) patterns were recorded by using a Rigaku MiniFlex600 diffractometer (Cu Kα (40 kV, 40 mA) radiation). Temperature-programmed desorption with NH3 or CO2 (NH3-TPD or CO2-TPD) was conducted with an automated apparatus BELCAT II (MicrotracMRB) equipped with both a thermal conductivity detector (TCD) and quadrupole mass spectrometer (Q-MS; BELMASS, MicrotracMRB). In each run, 0.30 g of the sample was pretreated in Ar (50 mL min−1) at 673 K for 1 h, exposed to 5.0 vol% of NH3/Ar (50 mL min−1) or 10 vol% of CO2/Ar (50 mL min−1) at 373 K for 30 min, and further treated in Ar (50 mL min−1) at 373 K for 30 min. Afterward, the desorption profile of probe molecules was monitored with both Q-MS and TCD in the temperature range from 373 K to 1273 K with a 10 K min−1 ramp rate in an Ar flow (30 mL min−1). Thermogravimetry-differential thermal analysis (TG-DTA; Rigaku, Thermo Plus EVOII) was conducted to elucidate the presence of organic species deposited on the spent catalysts. The TG-DTA profiles were recorded with ca. 10 mg of each sample in an air flow (30 mL min−1) from room temperature to 1123 K with a 10 K min−1 ramp rate.
3. Results and discussion
3.1. Screening of catalysts for glycylglycine hydrolysis
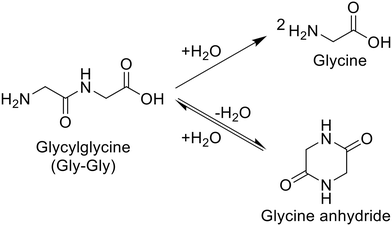
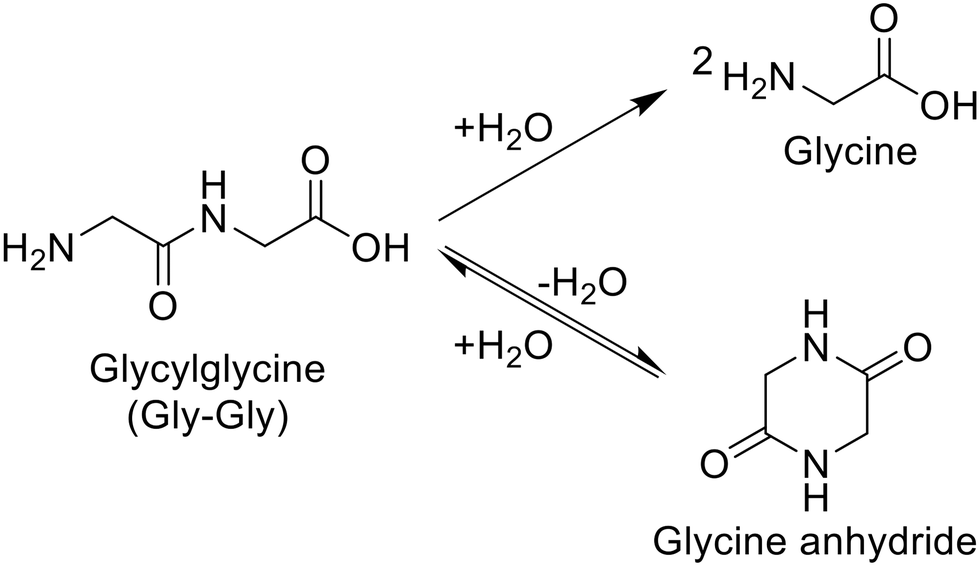
For the hydrolysis of glycylglycine (Gly-Gly), which is a simple dipeptide, a variety of metal oxides, typical solid acids, soluble acid, and soluble base were initially tested in water at 353 K for 4 h, as summarized in Table 1. It should be noted that the reaction conditions employed here were carefully controlled to adjust the conversion of Gly-Gly to the low-medium range due to the desire for fairly comparing the activity of each catalyst. Under the conditions without a catalyst, the hydrolysis of Gly-Gly did not proceed at all (entry 1); this result allowed us to estimate the catalytic activity of each metal oxide precisely by ignoring the contribution of hydrolysis regardless of catalysts opposite to various hydrolysis reactions operated at high temperatures.4,38,39 As seen in Table 1, the hydrolysis of Gly-Gly proceeded to give glycine in the presence of appropriate catalysts, along with the formation of glycine anhydride as a result of the dehydrative cyclization of Gly-Gly. Among the metal oxides tested in this study, m-ZrO2, t-ZrO2, CeO2 (which was reported to be a good catalyst for the alcoholysis of amides),14,15 and CaO provided high values of both Gly-Gly conversion and glycine yield (entries 2, 4, 5, and 6). However, CaO could not be recovered after the reaction due to its dissolution in water. What is worse, the use of half amount of CaO for 8 h converted only 4.8% of Gly-Gly (entry 7), the value of which was 25-fold less than that by 0.10 g of CaO (entry 6), and provided 1.5%-C yield of glycine (the more detailed investigation for the effects of CaO amount and reaction time is available in Table S6 and Fig. S2†). These results indicated that CaO was not an effective catalyst for the hydrolysis of Gly-Gly. The m-ZrO2 and t-ZrO2 catalysts exhibited comparable weight-basis activity to each other and also the highest values (0.25 h−1 and 0.23 h−1) of turnover frequency (TOF) based on the mole of metal atoms in each catalyst and hydrolysis rate (i.e., half of the formation rate of glycine due to the production of two glycine molecules from one Gly-Gly molecule; entries 2 and 4). Their activity is discussed on the basis of their surface properties and structural change in the next section. Furthermore, in stark contrast to CaO (entries 6 and 7), the reaction operated with half the amount of m-ZrO2 for 8 h provided comparable results to those conducted under the original conditions (entries 2 and 3). Some of the metal oxides including Nb2O5 promoted the hydrolysis of Gly-Gly to some extent (entries 8–16). Given that Nb2O5 was demonstrated to achieve the high-yielding production of carboxylic acids from amides under reflux conditions to release co-produced NH3,13 the produced glycine with a basic amino group could act as a catalyst poison toward Nb2O5. The other metal oxides as well as zeolites40 were almost inactive for this reaction (entries 17–26). According to previous papers, beta- and FAU-type zeolites catalyzed the hydrolysis of nylon 6 to produce ε-caprolactam,32,34 while in the current study, they did not hydrolyze Gly-Gly. These contrasting results would be connected to two reasons. The number of basic amino groups in nylon 6 is negligible, and the product (i.e., ε-caprolactam) lacks an amino group; such a situation in the hydrolysis of nylon 6 could allow acid sites of zeolites to avoid their neutralization and maintain their activity, while acid sites readily undergo the neutralization by the amino group of Gly-Gly. Another possible reason is the significant difference in reaction temperatures between the Gly-Gly hydrolysis studied here and previously investigated hydrolysis of nylon 6 (Table S3†). A homogeneous acid, H2SO4, did not promote the hydrolysis of Gly-Gly even when its loading amount was doubled (entries 27 and 28). Gly-Gly underwent hydrolysis in the presence of homogeneous base, NaOH (entry 29), but the glycine yield (24%-C) was lower than that given by m-ZrO2 (40%-C; entry 2). These reaction results stressed ZrO2 to be the appropriate catalyst for hydrolyzing Gly-Gly.
Table 1 Screening of various catalysts for hydrolysis of glycylglycine (Gly-Gly)a
|
|
| Entry |
Catalyst |
S
BET
[m2 g−1] |
Conv. [%] |
Yield [%-C] |
TOFc [h−1] |
Bal.d [%] |
| Glycine |
Glycine anhydride |
|
Reaction conditions: Gly-Gly 2.0 mmol; catalyst 0.10 g; H2O 5.0 g; 353 K; 4 h.
BET specific surface area.
Turnover frequency on the basis of the mole of catalyst and hydrolysis rate (i.e., half of the formation rate of glycine): TOF [h−1] = (formation rate of glycine [mmol h−1])/(2 × (mole of metal atoms in each metal oxide [mmol])). For homogeneous catalysts, the mole of H+ or OH− was used for the calculation instead of the mole of metal atoms.
Balance.
Calcined at 673 K.
Catalyst 0.05 g; 8 h.
Operated in Ar.
2.0 mmol.
|
| 1 |
None |
— |
0 |
0 |
0 |
— |
101 |
| 2 |
m-ZrO2e |
85 |
43 |
40 |
2.8 |
0.25 |
101 |
| 3 |
m-ZrO2e,f |
85 |
45 |
39 |
3.0 |
0.24 |
98 |
| 4 |
t-ZrO2e |
82 |
42 |
38 |
2.9 |
0.23 |
99 |
| 5 |
CeO2 |
72 |
22 |
21 |
0.7 |
0.18 |
99 |
| 6 |
CaOg |
8.7 |
38 |
38 |
0 |
0.11 |
100 |
| 7 |
CaOf,g |
8.7 |
4.8 |
1.5 |
2.4 |
0.004 |
99 |
| 8 |
Sm2O3 |
35 |
16 |
13 |
3.6 |
0.12 |
101 |
| 9 |
Pr6O11 |
46 |
23 |
11 |
5.4 |
0.095 |
94 |
| 10 |
Y2O3 |
34 |
12 |
9.0 |
2.6 |
0.051 |
100 |
| 11 |
Yb2O3 |
28 |
11 |
7.6 |
3.9 |
0.074 |
100 |
| 12 |
Nb2O5 |
48 |
9.9 |
7.3 |
2.5 |
0.049 |
100 |
| 13 |
La2O3 |
31 |
11 |
6.8 |
3.5 |
0.056 |
99 |
| 14 |
ZnO |
9.3 |
10 |
3.3 |
0.7 |
0.014 |
94 |
| 15 |
MgOg |
67 |
3.1 |
0.8 |
0.9 |
0.002 |
99 |
| 16 |
MnO2 |
15 |
1.3 |
0.3 |
0.1 |
0.001 |
99 |
| 17 |
TiO2 |
11 |
0 |
0 |
0.7 |
0 |
101 |
| 18 |
MoO3 |
1.9 |
4.4 |
0 |
2.4 |
0 |
98 |
| 19 |
Fe2O3 |
6.8 |
3.3 |
0 |
0 |
0 |
97 |
| 20 |
α-Al2O3 |
7.7 |
0 |
0 |
0 |
0 |
101 |
| 21 |
SiO2 |
483 |
2.6 |
0 |
0 |
0 |
97 |
| 22 |
SnO2 |
31 |
1.0 |
0 |
0 |
0 |
99 |
| 23 |
SiO2–Al2O3 |
383 |
2.6 |
0 |
0.3 |
0 |
98 |
| 24 |
Beta |
383 |
1.2 |
0 |
0.1 |
0 |
99 |
| 25 |
FAU |
450 |
1.6 |
0 |
0.1 |
0 |
99 |
| 26 |
Hydrotalcite |
59 |
1.4 |
0 |
0.9 |
0 |
100 |
| 27 |
H2SO4 |
— |
2.9 |
0 |
0.1 |
0 |
97 |
| 28 |
H2SO4h |
— |
4.4 |
1.0 |
0.1 |
0.001 |
97 |
| 29 |
NaOH |
— |
23 |
24 |
0 |
0.046 |
101 |
3.2. Relationship between catalytic activity of metal oxides in hydrolysis of glycylglycine and their physicochemical properties
To understand the surface properties that are necessary for the hydrolysis of Gly-Gly, the acidity and basicity of m-ZrO2, t-ZrO2, CeO2, Nb2O5, and MgO were elucidated by NH3- and CO2-TPD measurements (Table 2 and Fig. S3†). The active catalysts (i.e., m-ZrO2, t-ZrO2, and CeO2) were amphoteric, and the higher activity of the two ZrO2 catalysts compared to CeO2 was due to the large amount of acid and base sites (entries 1–3). The comparable activity of the two ZrO2 catalysts can be rationalized by their similar amount of both acid and base sites (entry 1 vs. entry 2). The low activity metal oxides, Nb2O5 and MgO, were acidic and basic, respectively (entries 4 and 5). Yet, considering that MgO was transformed readily into Mg(OH)2 in water at 353 K for 4 h (Fig. S4†), which were the same conditions as the hydrolysis of Gly-Gly (Table 1), the currently acquired reaction result with MgO could actually reflect the activity of the in situ formed Mg(OH)2 phase. The values of TOF on the basis of the hydrolysis rate and amount of acid or base sites are also listed in Table 2. These values stressed the higher activity of the amphoteric metal oxides consisting of m-ZrO2, t-ZrO2, and CeO2, compared to Nb2O5 and MgO. These quantitative data, therefore, demonstrated that acid and base sites cooperatively functioned as active sites for hydrolyzing Gly-Gly. The importance of the amphoteric property of metal oxides for cleaving amide bonds was also demonstrated theoretically and experimentally in a previous report on the alcoholysis of amides over CeO2.15
Table 2 Surface acidity and basicity of metal oxides
| Entry |
Catalyst |
S
BET
[m2 g−1] |
Desorbed amount in TPDb [μmol g−1] |
Conv.c [%] |
Yieldc [%-C] |
TOFd [h−1] |
| NH3 |
CO2 |
Glycine |
Glycine anhydride |
Acid-site basis |
Base-site basis |
|
BET specific surface area.
Total desorbed amount of each probe molecule in the temperature range of 383–773 K in each TPD profile (Fig. S3†).
Results of Gly-Gly hydrolysis (the same as the data in Table 1). Reaction conditions: Gly-Gly 2.0 mmol; catalyst 0.10 g; H2O 5.0 g; 353 K; 4 h.
Turnover frequency calculated from the hydrolysis rate of Gly-Gly (i.e., half of the formation rate of glycine) and amount of acid or base sites: TOF [h−1] = (formation rate of glycine [μmol g−1 h−1])/(2 × (desorbed amount of NH3 (or CO2) [μmol g−1])).
Calcined at 673 K.
Prior to the standard pretreatment, this sample was heat-treated in an Ar flow (30 mL min−1) at 773 K for 3 h.
|
| 1 |
m-ZrO2e |
85 |
275 |
190 |
43 |
40 |
2.8 |
7.3 |
11 |
| 2 |
t-ZrO2e |
82 |
245 |
164 |
42 |
38 |
2.9 |
7.8 |
12 |
| 3 |
CeO2 |
72 |
113 |
94 |
22 |
21 |
0.7 |
9.3 |
11 |
| 4 |
Nb2O5 |
48 |
164 |
1 |
9.9 |
7.3 |
2.5 |
2.2 |
— |
| 5 |
MgOf |
67 |
1 |
94 |
3.1 |
0.8 |
0.9 |
— |
0.4 |
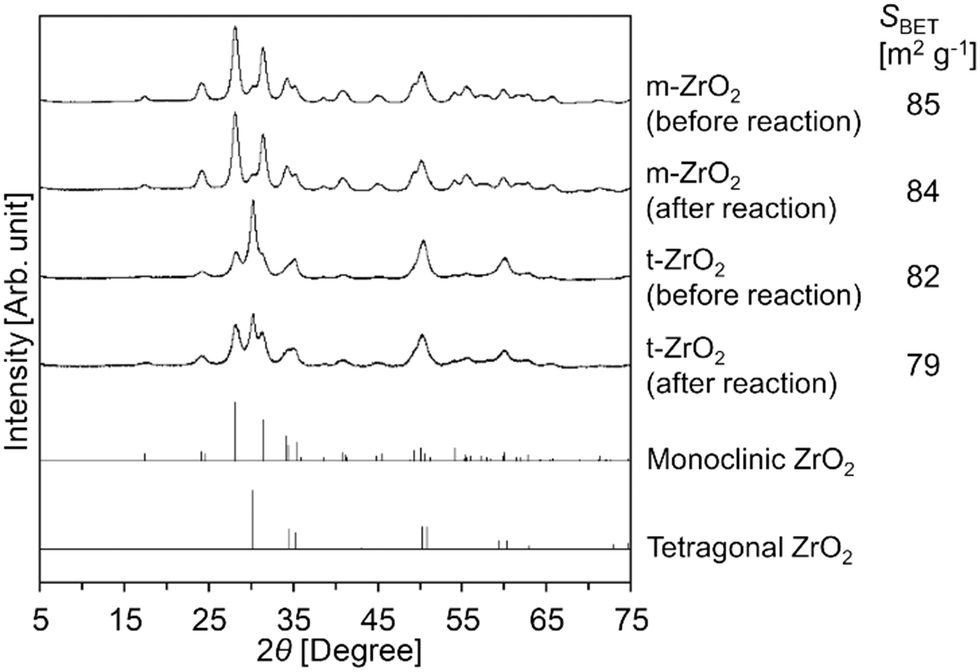
Meanwhile, the XRD patterns of t-ZrO2 before/after the reaction (Fig. 1) indicated that a part of the tetragonal phase underwent phase transition into the monoclinic phase during the hydrolysis reaction. In contrast to t-ZrO2, the XRD pattern of m-ZrO2 remained unchanged after its use for the Gly-Gly hydrolysis (Fig. 1). Although the catalytic activity of m-ZrO2 and t-ZrO2 was similar to each other, m-ZrO2 was determined to be the most suitable catalyst in the later sections due to its higher stability against phase transition under the reaction conditions.
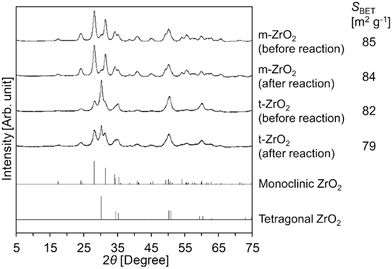 |
| | Fig. 1 XRD patterns of m-ZrO2 and t-ZrO2 before/after the reaction. Reaction conditions: Gly-Gly 2.0 mmol; ZrO2 (calcined at 673 K) 0.10 g; H2O 5.0 g; 353 K; 4 h. References: monoclinic ZrO2 = ICSD card #18190; tetragonal ZrO2 = ICSD card #68781. | |
3.3. Effect of calcination temperature of m-ZrO2 and reaction conditions on glycylglycine hydrolysis
The calcination temperature is well-known to affect the structure, surface properties, and also catalytic activity of various metal oxides,13,20,21,36,41 which motivated us to investigate such effect on the activity of the m-ZrO2 catalyst on the Gly-Gly hydrolysis. The XRD patterns of the m-ZrO2 catalysts calcined at different temperatures (i.e., 673, 773, and 873 K) (Fig. S5†) showed that the tetragonal phase was slightly present in the m-ZrO2 sample calcined at 673 K, but its content gradually decreased upon the increase in the calcination temperature; the highest calcination temperature of 873 K resulted in the formation of monoclinic ZrO2 with negligible content of tetragonal phase. Table 3 lists the specific surface area, acid/base properties, and reaction results of m-ZrO2 calcined at three different temperatures. The conversion of Gly-Gly and yield of glycine steadily decreased by increasing the calcination temperature, while the difference in the conversion rates of Gly-Gly normalized by the specific surface area of each m-ZrO2 was insignificant. Such comparable surface properties of m-ZrO2 could be connected to the similarity of the normalized formation rate. These data have also confirmed the importance of the amphoteric surface property of m-ZrO2 for the hydrolysis of Gly-Gly. In the following reactions, due to the better weight-basis activity of m-ZrO2, the calcination temperature of 673 K was selected.
Table 3 Effect of calcination temperature on the catalytic activity of m-ZrO2
| Entry |
Calcination temp. [K] |
S
BET
[m2 g−1] |
Desorbed amount in TPDb [μmol g−1] |
Conv.c [%] |
Yieldc [%-C] |
Bal.c,d [%] |
v
Gly
[mmol h−1 m−2] |
| NH3 |
CO2 |
Glycine |
Glycine anhydride |
|
BET specific surface area.
Total desorbed amount of each probe molecule in the temperature range of 383–773 K in each TPD profile (Fig. S6†).
Results of Gly-Gly hydrolysis (the data for entry 1 are the same as those of m-ZrO2 in Tables 1 and 2). Reaction conditions: Gly-Gly 2.0 mmol; m-ZrO2 0.10 g; H2O 5.0 g; 353 K; 4 h.
Balance.
Formation rate of glycine on the basis of SBET.
|
| 1 |
673 |
85 |
275 |
190 |
43 |
40 |
2.8 |
101 |
0.048 |
| 2 |
773 |
62 |
216 |
144 |
31 |
28 |
2.5 |
99 |
0.045 |
| 3 |
873 |
45 |
149 |
98 |
27 |
22 |
1.8 |
97 |
0.049 |
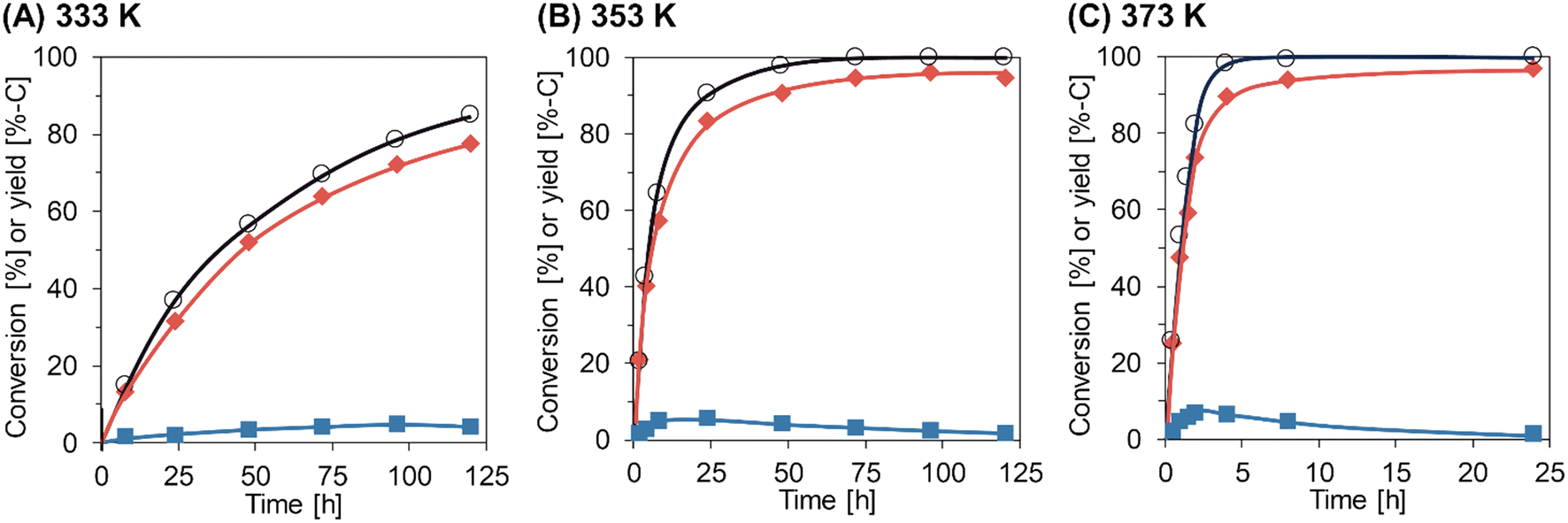
The time courses of Gly-Gly hydrolysis over m-ZrO2 calcined at 673 K were then investigated at different reaction temperatures of 333, 353, and 373 K (Fig. 2 and Table S7†). For all these tests, the material balance was quite high (97–103%), stressing the absence of products other than glycine and glycine anhydride at least within the temperatures investigated here. The reaction at 333 K gradually converted Gly-Gly into glycine and glycine anhydride, but the conversion of Gly-Gly was still 85% even for the reaction time of 120 h. The slope of time course became steeper at higher reaction temperatures. At 373 K, almost all Gly-Gly was converted into glycine (89%-C yield) and glycine anhydride (6.6%-C) for 4 h, and the highest yield of glycine (97%-C) was achieved at 24 h due to the hydrolysis of glycine anhydride. As seen in Fig. 2, the fast hydrolysis of both Gly-Gly and glycine anhydride enabled at high temperatures is the key to the high yield of glycine in a short reaction time.
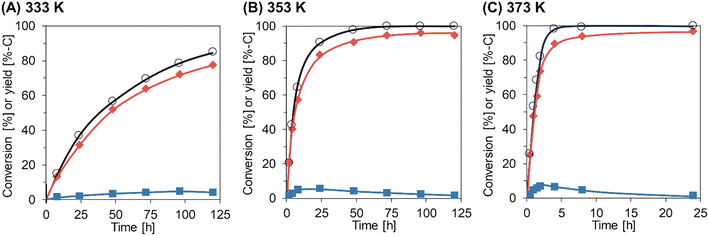 |
| | Fig. 2 Time courses of Gly-Gly hydrolysis over the m-ZrO2 catalyst at different reaction temperatures: (A) 333 K; (B) 353 K; (C) 373 K. Legends: white circles = conversion of Gly-Gly; red diamonds = yield of glycine; blue squares = yield of glycine anhydride. Reaction conditions: Gly-Gly 2.0 mmol; m-ZrO2 (calcined at 673 K) 0.10 g; H2O 5.0 g; 333–373 K; 0.5–120 h. The detailed data are shown in Table S7.† | |
3.4. Reuse test of m-ZrO2 in glycylglycine hydrolysis
Fig. 3A and Table S8† represent the results of the reuse test of the m-ZrO2 catalyst in the hydrolysis of Gly-Gly. In these experiments, the conversion of Gly-Gly was intentionally suppressed within the medium level by controlling the reaction conditions at 353 K for 4 h, which were the same as those for the screening of the catalyst (vide supra), in order to avoid the overestimation of catalyst reusability.42 As above, the fresh m-ZrO2 catalyst under the current conditions afforded the Gly-Gly conversion of 43% and glycine yield of 40%-C. The catalytic activity of m-ZrO2 gradually decreased upon its repeated use. The specific surface area (Fig. 3A) and XRD pattern (Fig. 3B) of m-ZrO2 were unchanged in the three-times reuse tests. These data indicated that the structural change of m-ZrO2 did not occur at least after each run of Gly-Gly hydrolysis. The accumulation of organic species on the catalyst surface was also investigated by TG-DTA (Fig. S7†). Yet, in the TG-DTA profiles for m-ZrO2 after each run, the weight drop with an exothermic peak was not observed clearly. Besides, the catalytic activity could not be recovered after the calcination at 673 K for 3 h (Fig. 3A). These data thus indicated that the deposition of organic species was unlikely to be the reason of catalyst deactivation.
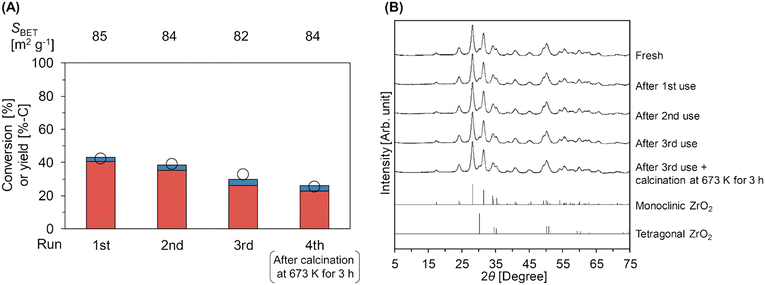 |
| | Fig. 3 (A) Reuse test of the m-ZrO2 catalyst in Gly-Gly hydrolysis and (B) XRD patterns of fresh and spent m-ZrO2 catalysts. Legends for fig. A: white circles = conversion of Gly-Gly; red bars = yield of glycine; blue bars = yield of glycine anhydride. Reaction conditions: Gly-Gly 2.0 mmol; m-ZrO2 (calcined at 673 K) 0.10 g; H2O 5.0 g; 353 K; 4 h. The m-ZrO2 catalyst was reused after washing with water and drying through the 1st to 3rd run, while the 4th run was operated after the calcination of the spent catalyst in air at 673 K for 3 h. The detailed data are shown in Table S8.† References for fig. B: monoclinic ZrO2 = ICSD card #18190; tetragonal ZrO2 = ICSD card #68781. | |
The surface acid/base properties of the catalyst after the 3rd run were examined by TPD measurements (Table S8 and Fig. S8†). The amount of acid sites was unchanged even after the reuse experiments. Meanwhile, the amount of weak base sites that were observed in the temperature range of 383–773 K decreased from 190 μmol g−1 (fresh) to 145 μmol g−1 (after the 3rd run and calcination at 673 K), and instead, the strong base sites (46 μmol g−1) were generated after the 3rd run due to the appearance of new peaks at 853 K and 1033 K in the CO2-TPD profile; such alteration of surface properties of m-ZrO2 could be the reason for its deactivation. According to previous reports on the catalytic alcoholysis of amides over an amphoteric surface of CeO2, a lattice oxygen of CeO2 behaves as a base site and nucleophilically attacks a carbonyl carbon atom of amide to give a tetrahedral intermediate, which is invoked as the rate-determining step of this reaction.15,17 This previously reported insight and similarity of catalytic systems between the previous report on solvolysis and the current study on hydrolysis (i.e., cleavage of amide bonds over an amphoteric surface of metal oxide) state the decrease in the quantity of base sites with appropriate strength to be the most likely reason for the deactivation of the m-ZrO2 catalyst. The development of stable catalysts will be tackled in due course.
3.5. Substrate scope for the hydrolysis of amides over the m-ZrO2 catalyst
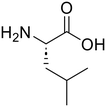
The applicability of the m-ZrO2 catalyst to various types of amide compounds (i.e., dipeptides, small organic amides, and polyamide) was examined under each specific reaction condition (Table 4). A variety of dipeptides which contain bulky, acidic, or basic amino acid residue(s) were employed as substrates with the m-ZrO2 catalyst (entries 1–4). Table 4 lists the highest product yields achieved for each dipeptide, and the time courses for the hydrolysis of dipeptides are also shown in Fig. S9 and Tables S9–S11.† Similar to the hydrolysis of Gly-Gly (entry 1), the hydrolysis of glycyl-L-leucine proceeded well to provide glycine (95%) and L-leucine (95%) under identical reaction conditions at 373 K for 24 h (entry 2). The formation rates of N-terminal and C-terminal amino acids from glycyl-L-leucine (0.12 mmol h−1 and 0.12 mmol h−1, respectively; see Table 5) were clearly lower than those in the Gly-Gly hydrolysis (0.38 mmol h−1), indicating that the presence of a bulky alkyl side chain made the activation of amide bonds difficult and lowered the reaction rate. In the case of glycyl-L-aspartic acid, the hydrolysis operated at 373 K for 72 h produced glycine and L-aspartic acid in 82% and 84% yields, respectively (entry 3 in Table 4). Although glycyl-L-aspartic acid was completely consumed within 24 h, the yields of amino acids could not reach such high values. This gap would be caused by the formation of cyclized products from glycyl-L-aspartic acid, similar to the case of Gly-Gly (Fig. 2); indeed, we observed the formation of poorly soluble white solid species. Yet, such a cyclized product gradually underwent hydrolysis into amino acids to provide their yields over 80% for 72 h. The formation rates of amino acids were significantly lower than those in the hydrolysis of Gly-Gly and glycyl-L-leucine (Table 5). This observed difference pointed to the inhibitory effect of the acidic side chain on the hydrolysis over the m-ZrO2 catalyst, which was consistent with the importance of base sites as discussed in the former parts. In the case of the hydrolysis of L-histidyl-L-leucine, the yields of L-histidine and L-leucine reached 73% and 76%, respectively, for 24 h (entry 4 in Table 4). The formation rates of these amino acids were lower than those in the hydrolysis of Gly-Gly and glycyl-L-leucine while higher than those in the case of glycyl-L-aspartic acid (Table 5). Therefore, in contrast to the acidic side chain in glycyl-L-aspartic acid, the inhibitory effect of the basic side chain was not as large as that of the acidic side chain, which can be again rationalized by the importance of base sites of m-ZrO2. Yet, as seen in the time course for the L-histidyl-L-leucine hydrolysis, the yields of L-histidine and L-leucine plateaued after the reaction time of 24 h (Fig. S9C and Table S11†), suggesting the deactivation of the m-ZrO2 catalyst.
Table 4 Substrate scope for the hydrolysis of amides over the m-ZrO2 catalyst
| Entry |
Substrate |
Temp. [K] |
Time [h] |
Conv. [%] |
Product(s) (yield [%]) |
|
Reaction conditions: Gly-Gly 2.0 mmol; m-ZrO2 (calcined at 673 K) 0.10 g; H2O 5.0 g; 373 K; 24 h.
The same data as Fig. 2C and Table S7.† Yield was calculated on carbon basis.
Reaction conditions: dipeptide 0.50 mmol; m-ZrO2 (calcined at 673 K) 0.025 g; H2O 5.0 g; 373 K; 24 or 72 h.
Reaction conditions: amide 2.0 mmol; m-ZrO2 (calcined at 673 K) 0.10 g; H2O 10 g; 463 K; 120 or 168 h.
Reaction conditions: nylon 6, 0.23 g (corresponding to 2.0 mmol of monomeric unit); m-ZrO2 (calcined at 673 K) 0.10 g; H2O 10 g; 453 or 503 K; 2 or 16 h.
Not determined due to the difficulty of quantification of the unreacted substrate.
|
| 1a,b |
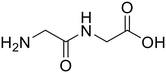 Glycylglycine (Gly-Gly) Glycylglycine (Gly-Gly) |
373 |
24 |
100 |
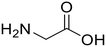 Glycine (97) Glycine (97) |
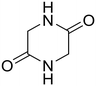 Glycine anhydride (1.6) Glycine anhydride (1.6) |
| 2c |
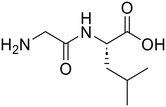 Glycyl-L-leucine (Gly-Leu) Glycyl-L-leucine (Gly-Leu) |
373 |
24 |
100 |
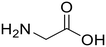 Glycine (95) Glycine (95) |
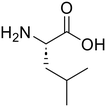 L-Leucine (95)
L-Leucine (95) |
| 3c |
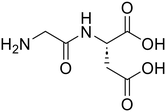 Glycyl-L-aspartic acid (Gly-Asp) Glycyl-L-aspartic acid (Gly-Asp) |
373 |
72 |
100 |
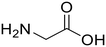 Glycine (82) Glycine (82) |
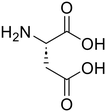 L-Aspartic acid (84)
L-Aspartic acid (84) |
| 4c |
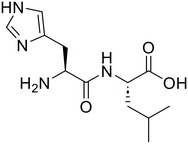 L-Histidyl-L-leucine (His-Leu)
L-Histidyl-L-leucine (His-Leu) |
373 |
24 |
100 |
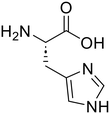 L-Histidine (73)
L-Histidine (73) |
 L-Leucine (76)
L-Leucine (76) |
| 5d |
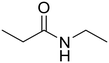 N-Ethylpropionamide
N-Ethylpropionamide |
463 |
120 |
79 |
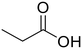 Propionic acid (78) Propionic acid (78) |
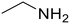 Ethylamine (76) Ethylamine (76) |
| 6d |
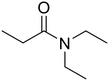 N,N-Diethylpropionamide
N,N-Diethylpropionamide |
463 |
168 |
52 |
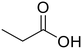 Propionic acid (48) Propionic acid (48) |
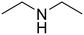 Diethylamine (50) Diethylamine (50) |
| 7e |
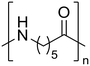 Nylon 6 Nylon 6 |
453 |
16 |
n.d.f |
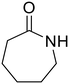 ε-Caprolactam (55) ε-Caprolactam (55) |
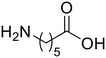 ε-Aminocaproic acid (33) ε-Aminocaproic acid (33) |
| 8e |
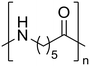 Nylon 6 Nylon 6 |
503 |
2 |
n.d.f |
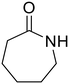 ε-Caprolactam (81) ε-Caprolactam (81) |
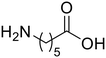 ε-Aminocaproic acid (13) ε-Aminocaproic acid (13) |
Table 5 Comparison of the initial formation rate of amino acids in the hydrolysis of dipeptides over the m-ZrO2 catalysta
| Entry |
Substrate |
v
N-terminal
[mmol h−1] |
v
C-terminal
[mmol h−1] |
|
Reaction conditions: dipeptide 0.50 mmol; m-ZrO2 (calcined at 673 K) 0.025 g; H2O 5.0 g; 373 K; 0.25–4 h.
Formation rate of N-terminal (or C-terminal) amino acid, estimated from the initial slope of each time course where the conversion of dipeptide was below 40% (Fig. S9 and Tables S9–S12†).
Given that the hydrolysis of one Gly-Gly molecule provides two glycine molecules, the halved value of the observed formation rate of glycine (i.e., 0.76 mmol h−1) is shown for both vN-terminal and vC-terminal.
|
| 1 |
Glycylglycine (Gly-Gly) |
0.38c |
0.38c |
| 2 |
Glycyl-L-leucine (Gly-Leu) |
0.12 |
0.12 |
| 3 |
Glycyl-L-aspartic acid (Gly-Asp) |
0.018 |
0.016 |
| 4 |
L-Histidyl-L-leucine (His-Leu) |
0.089 |
0.098 |
As small organic amides, an aliphatic secondary amide (N-ethylpropionamide) and aliphatic tertiary amide (N,N-diethylpropionamide) were tested with the m-ZrO2 catalyst (entries 5 and 6 in Table 4). The time course of N-ethylpropionamide hydrolysis is also shown in Fig. S10 and Table S13.† Although the long reaction times and high reaction temperature were necessary, these amides were hydrolyzed into the equivalent amount of corresponding carboxylic acids and amines. As seen in Tables 4 and S14† as well as Fig. S11,† the hydrolysis of N,N-diethylpropionamide was slower and provided the lower yields of products (i.e., propionic acid and diethylamine) regardless of the longer reaction time, compared to that of N-ethylpropionamide, possibly due to the bulkier structure around the amide bond of the former substrate. The catalytic activity of m-ZrO2 and Nb2O5 was again compared in the hydrolysis of N-ethylpropionamide since the latter metal oxide was previously found as an effective catalyst for the hydrolysis of various amides with the assistance of removal of co-produced NH3.13 As summarized in Table S15,† both catalysts provided the equivalent amount of propionic acid and ethylamine. Meanwhile, the conversion given by m-ZrO2 was higher than that by Nb2O5; considering the slight progress of hydrolysis even in the absence of a catalyst, m-ZrO2 exhibited 9.9-fold higher activity, compared to Nb2O5. These data have thus demonstrated that m-ZrO2 is a suitable catalyst for hydrolyzing small organic amides into corresponding carboxylic acids and amines.
The hydrolysis of nylon 6 was also conducted with the m-ZrO2 catalyst at two different temperatures (entries 7 and 8 in Table 4; time courses are also shown in Fig. S12 and Table S16†); in all cases, two types of monomers, ε-caprolactam and ε-aminocaproic acid, were detected. Note that although a pelletized form of nylon 6 was employed for these tests (Fig. S13A†), all the pellets were melted or dissolved in water at the reaction temperatures of 453 K or higher since no pellet was found after the reaction for 0 h (Fig. S13B†). Yet, it should be noted that the difficulty in collecting the unreacted nylon 6 that adhered to the bottom of the reactor led to the low material balance at the low conversion level. The hydrolysis at 453 K for 16 h afforded ε-caprolactam and ε-aminocaproic acid in 55% and 33% yields, respectively, while the reaction operated at 503 K for 2 h provided a higher yield of ε-caprolactam (81%) in addition to ε-aminocaproic acid (13%). The increase in the molar ratio of ε-caprolactam to ε-aminocaproic acid upon the increase of reaction temperature is due to the shift of reaction equilibrium between these two monomers (the detailed data about the equilibrium level are available in Table S17†). Under identical conditions at 503 K for 2 h without m-ZrO2, only 6.5% yield of ε-caprolactam was produced (Table S16†), stressing the catalytic performance of m-ZrO2. The TOF on the basis of the formation rate of monomers and mole of m-ZrO2 at 503 K was calculated to be 2.2 h−1, where the involvement of hydrolysis in the absence of m-ZrO2 was considered via its subtraction. Compared to previously reported hydrolysis of nylon 6, the hydrolysis of nylon 6 in the presence of m-ZrO2 proceeded at much lower temperatures (453 or 503 K for this study vs. ≥573 K for reported studies27–29,31–34 except for the case operated in a concentrated HCl aq.;30 Table S3†), positing the superiority of the current catalytic hydrolysis using the m-ZrO2 catalyst. Unfortunately, the above-mentioned TOF value for m-ZrO2 cannot be compared fairly to those for the reported systems because of a lack of the insights into the hydrolysis assisted by high-temperature water (or even subcritical/supercritical water) in the absence of a catalyst; such contribution needs to be elucidated and subtracted from the results acquired in the presence of each catalyst to estimate TOF precisely. Altogether, m-ZrO2 has been demonstrated to be applicable to the hydrolysis of amide bonds involved in a variety of compounds.
On the basis of previously reported insights into the solvolysis of amide bonds on amphoteric surfaces15,17 and all the reaction results using the ZrO2 catalyst shown in this work, the plausible reaction mechanism has been illustrated in Fig. 4. The first step is the dissociative adsorption of the water molecule onto the ZrO2 surface to generate both a proton and hydroxide species. An amide molecule is then activated on the ZrO2 surface, where the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O moiety of the amide interacts with the Lewis-acidic Zr species. Toward the activated carbonyl carbon, the Lewis-basic lattice oxygen of ZrO2 attacks nucleophilically to form a tetrahedral intermediate. The amine-side of this intermediate reacts with the water-derived proton to be desorbed from the catalyst surface as an amine product. Meanwhile, the surface-remaining intermediate undergoes nucleophilic attack by the water-derived hydroxide species to be a carboxylic-acid product.
O moiety of the amide interacts with the Lewis-acidic Zr species. Toward the activated carbonyl carbon, the Lewis-basic lattice oxygen of ZrO2 attacks nucleophilically to form a tetrahedral intermediate. The amine-side of this intermediate reacts with the water-derived proton to be desorbed from the catalyst surface as an amine product. Meanwhile, the surface-remaining intermediate undergoes nucleophilic attack by the water-derived hydroxide species to be a carboxylic-acid product.
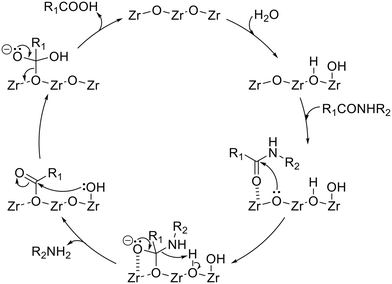 |
| | Fig. 4 Plausible reaction mechanism of amide hydrolysis over the ZrO2 catalyst. | |
4. Conclusions
ZrO2 was demonstrated to behave as an effective catalyst for hydrolyzing amide bonds in various compounds. Among the metal oxides as well as typical acids and bases tested in this study, monoclinic-rich ZrO2 (m-ZrO2) and tetragonal-rich ZrO2 (t-ZrO2) exhibited good catalytic activity for the hydrolysis of glycylglycine (Gly-Gly). The temperature-programmed desorption measurement using either NH3 or CO2 as a probe molecule for both active and inactive catalysts as well as control reactions using homogeneous acid (H2SO4) and base (NaOH) indicated that the presence of both acid and base sites on the same surfaces makes ZrO2 a good catalyst for the hydrolysis of Gly-Gly. Under the optimized reaction conditions, as high as 97%-C yield of glycine was achieved with the m-ZrO2 catalyst. This catalyst gradually underwent deactivation upon its reuse, and such deactivation was caused by the decrease of base sites probably due to the alteration of the local surface structure of m-ZrO2. This m-ZrO2 catalyst exhibited a broad substrate scope: the hydrolysis of dipeptides possessing bulky, acidic, and/or basic side chain(s) into amino acids; that of small organic amides into amines and carboxylic acids; and that of nylon 6 into ε-caprolactam and ε-aminocaproic acid. Although the reusability of the m-ZrO2 catalyst needs to be improved, such wide applicability could pave the way to the utilization of proteins as chemical feedstocks as well as chemical recycling of polyamides.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported financially by the following grants: International Leading Research (23K20034) and a Grant-in-Aid for Scientific Research (C) (23K04494) from the Japan Society for the Promotion of Science (JSPS).
References
- C. A. G. N. Montalbetti and V. Falque, Tetrahedron, 2005, 61, 10827–10852 CrossRef CAS.
- V. R. Pattabiraman and J. W. Bode, Nature, 2011, 480, 471–479 CrossRef CAS PubMed.
- K. Mashima, Y. Nishii and H. Nagae, Chem. Rec., 2020, 20, 332–343 CrossRef CAS PubMed.
- A.-J. Minor, R. Goldhahn, L. Rihko-Struckmann and K. Sundmacher, Chem. Eng. J., 2023, 474, 145333 CrossRef CAS.
- W. Stuyck, K. Janssens, M. Denayer, F. De Schouwer, R. Coeck, K. V. Bernaerts, J. Vekeman, F. De Proft and D. E. De Vos, Green Chem., 2022, 24, 6923–6930 RSC.
- J. Ni, Y. Sohma and M. Kanai, Chem. Commun., 2017, 53, 3311–3314 RSC.
- G. Absillis and T. N. Parac-Vogt, Inorg. Chem., 2012, 51, 9902–9910 CrossRef CAS PubMed.
- T. Takarada, M. Yashiro and M. Komiyama, Chem. – Eur. J., 2000, 6, 3906–3913 CrossRef CAS PubMed.
- H. G. T. Ly, G. Fu, A. Kondinski, B. Bueken, D. De Vos and T. N. Parac-Vogt, J. Am. Chem. Soc., 2018, 140, 6325–6335 CrossRef CAS PubMed.
- A. Loosen, F. de Azambuja, S. Smolders, J. Moons, C. Simms, D. De Vos and T. N. Parac-Vogt, Chem. Sci., 2020, 11, 6662–6669 RSC.
- H. G. T. Ly, G. Fu, F. de Azambuja, D. De Vos and T. N. Parac-Vogt, ACS Appl. Nano Mater., 2020, 3, 8931–8938 CrossRef CAS.
- S. Wang, H. G. T. Ly, M. Wahiduzzaman, C. Simms, I. Dovgaliuk, A. Tissot, G. Maurin, T. N. Parac-Vogt and C. Serre, Nat. Commun., 2022, 13, 1284 CrossRef CAS PubMed.
- S. M. A. H. Siddiki, M. N. Rashed, A. S. Touchy, M. A. R. Jamil, Y. Jing, T. Toyao, Z. Maeno and K. Shimizu, Catal. Sci. Technol., 2021, 11, 1949–1960 RSC.
- S. M. A. H. Siddiki, A. S. Touchy, M. Tamura and K. Shimizu, RSC Adv., 2014, 4, 35803–35807 RSC.
- T. Kamachi, S. M. A. H. Siddiki, Y. Morita, M. N. Rashed, K. Kon, T. Toyao, K. Shimizu and K. Yoshizawa, Catal. Today, 2018, 303, 256–262 CrossRef CAS.
- T. Toyao, M. N. Rashed, Y. Morita, T. Kamachi, S. M. A. H. Siddiki, M. A. Ali, A. S. Touchy, K. Kon, Z. Maeno, K. Yoshizawa and K. Shimizu, ChemCatChem, 2019, 11, 449–456 CrossRef CAS.
- M. N. Rashed, S. M. A. H. Siddiki, A. S. Touchy, M. A. R. Jamil, S. S. Poly, T. Toyao, Z. Maeno and K. Shimizu, Chem. – Eur. J., 2019, 25, 10594–10605 CrossRef CAS PubMed.
- M. G. Cutrufello, I. Ferino, R. Monaci, E. Rombi and V. Solinas, Top. Catal., 2002, 19, 225–240 CrossRef CAS.
- G. Li, S. Dissanayake, S. L. Suib and D. E. Resasco, Appl. Catal., B, 2020, 267, 118373 CrossRef CAS.
- K. Onodera, Y. Nakaji, M. Yabushita, Y. Nakagawa and K. Tomishige, Appl. Catal., A, 2023, 663, 119321 CrossRef CAS.
- R. Fujii, M. Yabushita, D. Asada, M. Tamura, Y. Nakagawa, A. Takahashi, A. Nakayama and K. Tomishige, ACS Catal., 2023, 13, 1562–1573 CrossRef CAS.
- S. Mihara, M. Yabushita, Y. Nakagawa and K. Tomishige, ChemSusChem, 2024, 17, e202301436 CrossRef CAS PubMed.
- R. Coeck and D. E. De Vos, Chem. Commun., 2024, 60, 1444–1447 RSC.
- Y. Kita, Y. Nishii, T. Higuchi and K. Mashima, Angew. Chem., Int. Ed., 2012, 51, 5723–5726 CrossRef CAS PubMed.
- H. Nagae, T. Hirai, D. Kato, S. Soma, S. Akebi and K. Mashima, Chem. Sci., 2019, 10, 2860–2868 RSC.
- T. Hirai, D. Kato, B. K. Mai, S. Katayama, S. Akiyama, H. Nagae, F. Himo and K. Mashima, Chem. – Eur. J., 2020, 26, 10735–10742 CrossRef CAS PubMed.
- M. Braun, A. B. Levy and S. Sifniades, Polym.-Plast. Technol. Eng., 1999, 38, 471–484 CrossRef CAS.
- T. Iwaya, M. Sasaki and M. Goto, Polym. Degrad. Stab., 2006, 91, 1989–1995 CrossRef CAS.
- J. Chen, Z. Li, L. Jin, P. Ni, G. Liu, H. He, J. Zhang, J. Dong and R. Ruan, J. Mater. Cycles Waste Manage., 2010, 12, 321–325 CrossRef CAS.
- X.-X. Yuan, Q. Zhou, X.-Y. Li, P. Yang, K.-K. Yang and Y.-Z. Wang, Polym. Degrad. Stab., 2014, 109, 171–174 CrossRef CAS.
- W. Wang, L. Meng and Y. Huang, Polym. Degrad. Stab., 2014, 110, 312–317 CrossRef CAS.
- W. Wang, L. Meng, K. Leng and Y. Huang, Polym. Degrad. Stab., 2017, 136, 112–120 CrossRef CAS.
- H. Hu, Q. Xu, L. Sun, R. Zhu, T. Gao, Y. He, B. Ma, J. Yu and X. Wang, ACS Appl. Polym. Mater., 2023, 5, 751–763 CrossRef CAS.
- W. Wang, L. Meng, J. Yu, F. Xie and Y. Huang, J. Anal. Appl. Pyrolysis, 2017, 125, 218–226 CrossRef CAS.
- K. Tomishige, T. Sakaihori, Y. Ikeda and K. Fujimoto, Catal. Lett., 1999, 58, 225–229 CrossRef CAS.
- K. Tomishige, Y. Ikeda, T. Sakaihori and K. Fujimoto, J. Catal., 2000, 192, 355–362 CrossRef CAS.
- A. Masudi and O. Muraza, Energy Fuels, 2018, 32, 2840–2854 CrossRef CAS.
- H. Kobayashi, M. Yabushita, T. Komanoya, K. Hara, I. Fujita and A. Fukuoka, ACS Catal., 2013, 3, 581–587 CrossRef CAS.
- M. Yabushita, H. Kobayashi, K. Hara and A. Fukuoka, Catal. Sci. Technol., 2014, 4, 2312–2317 RSC.
- L. Vilcocq, P. C. Castilho, F. Carvalheiro and L. C. Duarte, ChemSusChem, 2014, 7, 1010–1019 CrossRef CAS PubMed.
- K. Pokrovski, K. T. Jung and A. T. Bell, Langmuir, 2001, 17, 4297–4303 CrossRef CAS.
- S. L. Scott, ACS Catal., 2018, 8, 8597–8599 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Yoshinao
Nakagawa
*a,
Yoshinao
Nakagawa
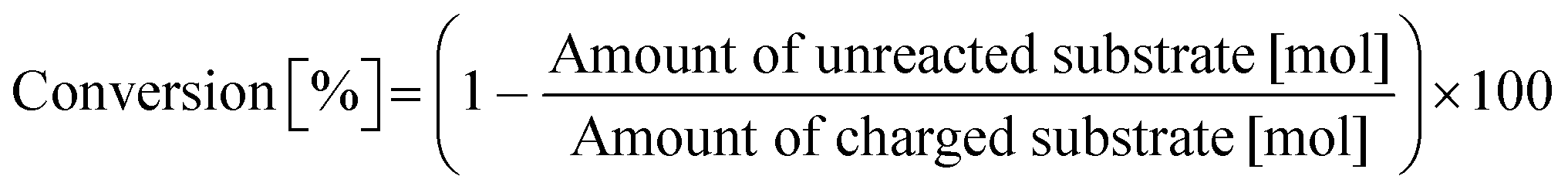


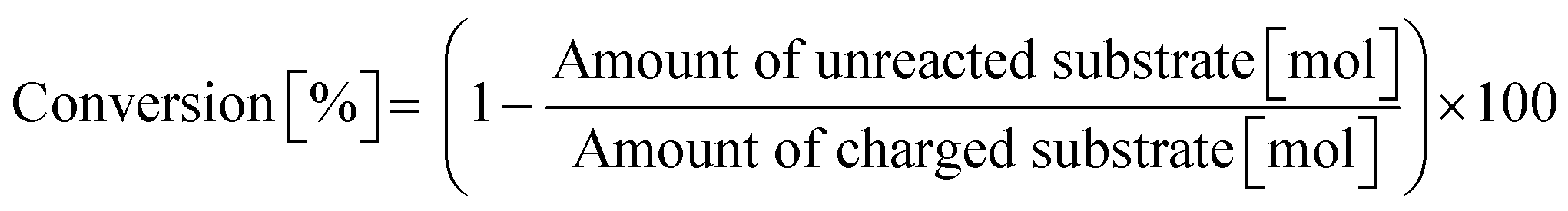
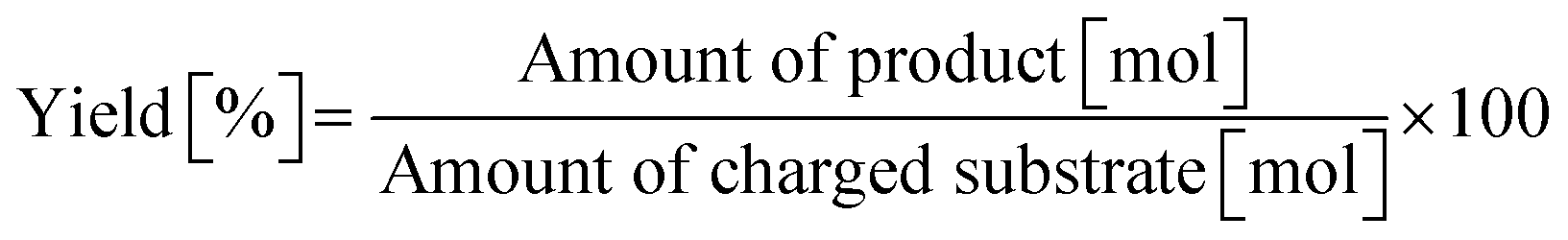
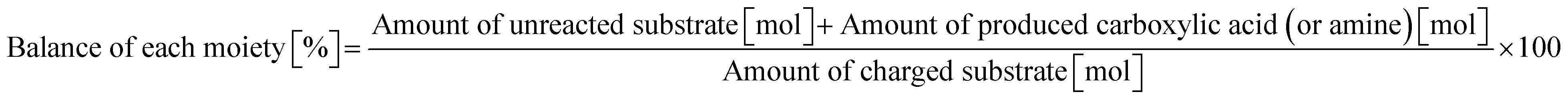

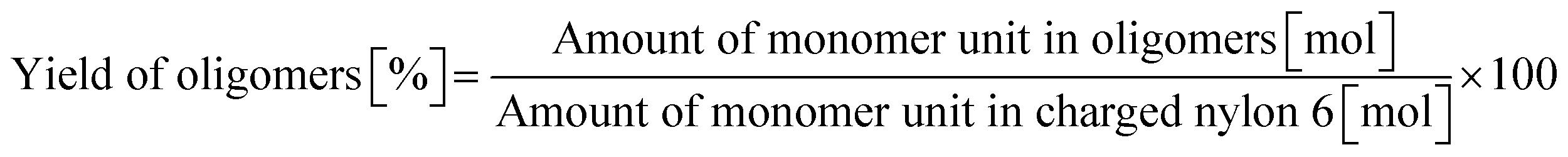
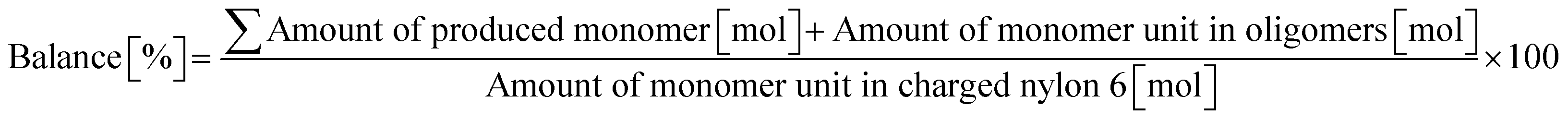

 Glycylglycine (Gly-Gly)
Glycylglycine (Gly-Gly)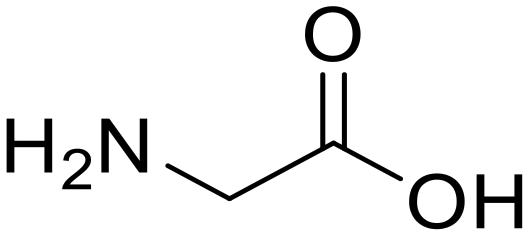 Glycine (97)
Glycine (97) Glycine anhydride (1.6)
Glycine anhydride (1.6)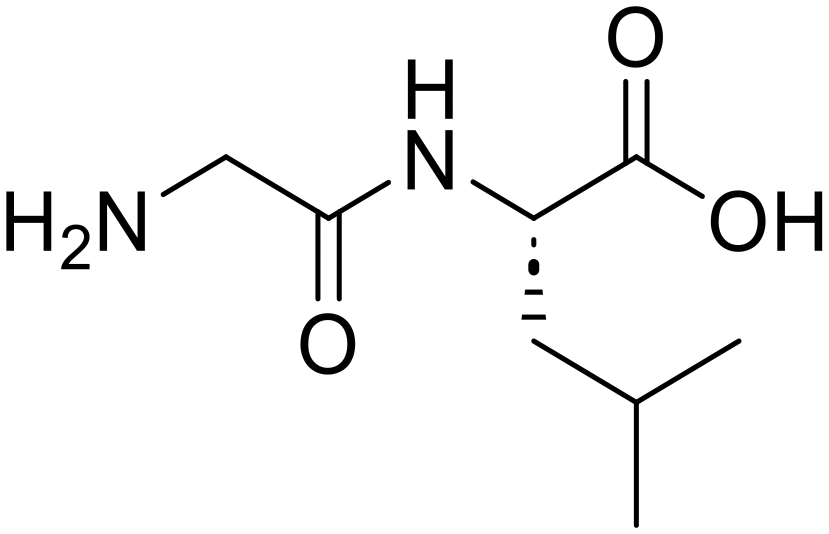 Glycyl-L-leucine (Gly-Leu)
Glycyl-L-leucine (Gly-Leu)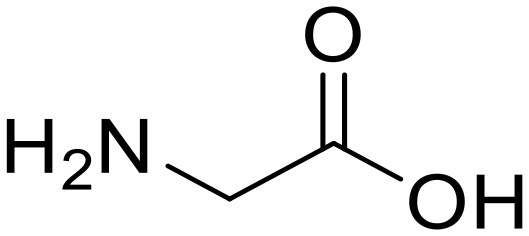 Glycine (95)
Glycine (95) L-Leucine (95)
L-Leucine (95)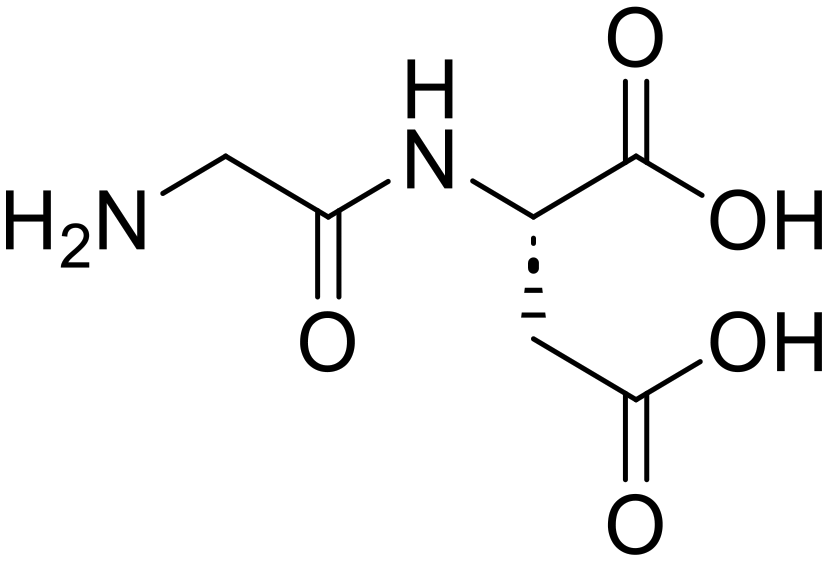 Glycyl-L-aspartic acid (Gly-Asp)
Glycyl-L-aspartic acid (Gly-Asp)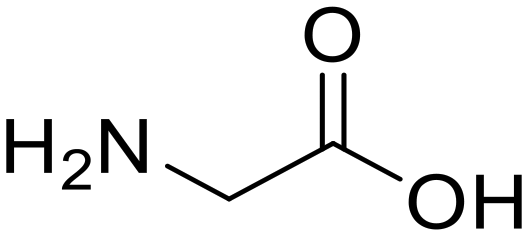 Glycine (82)
Glycine (82)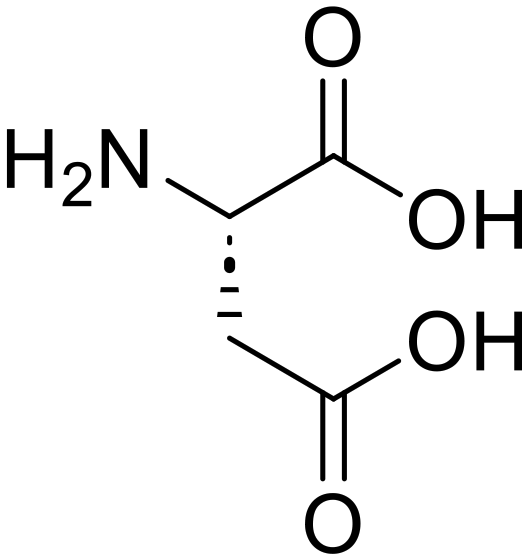 L-Aspartic acid (84)
L-Aspartic acid (84)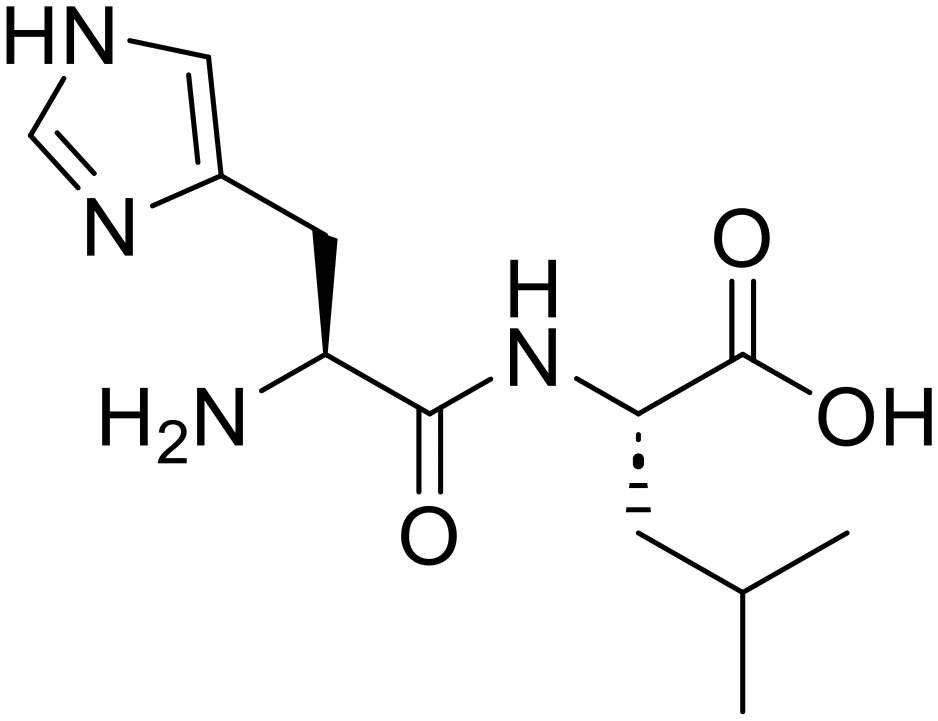 L-Histidyl-L-leucine (His-Leu)
L-Histidyl-L-leucine (His-Leu) L-Histidine (73)
L-Histidine (73) L-Leucine (76)
L-Leucine (76)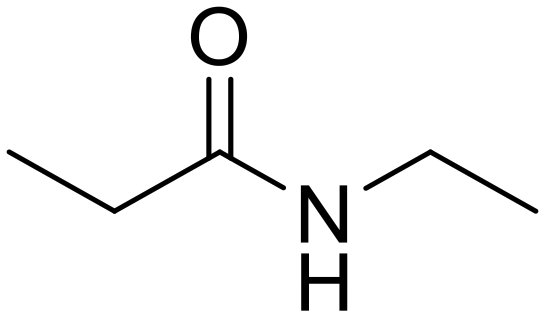 N-Ethylpropionamide
N-Ethylpropionamide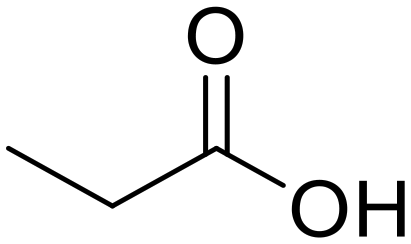 Propionic acid (78)
Propionic acid (78)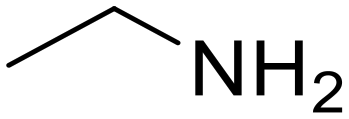 Ethylamine (76)
Ethylamine (76)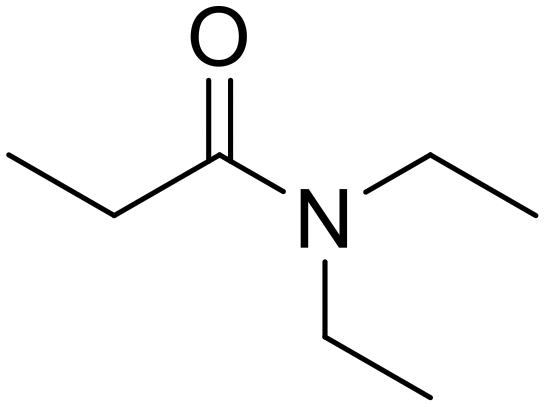 N,N-Diethylpropionamide
N,N-Diethylpropionamide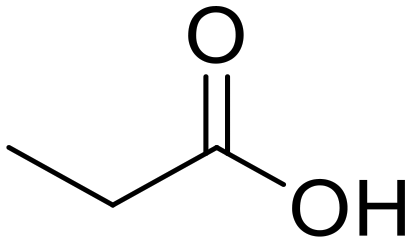 Propionic acid (48)
Propionic acid (48)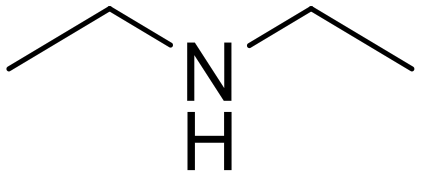 Diethylamine (50)
Diethylamine (50)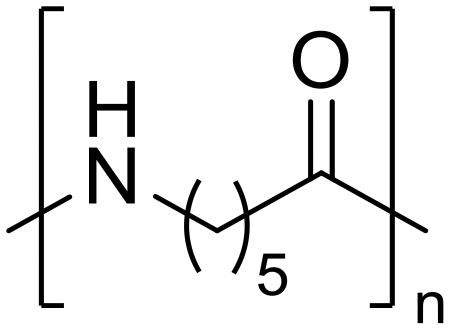 Nylon 6
Nylon 6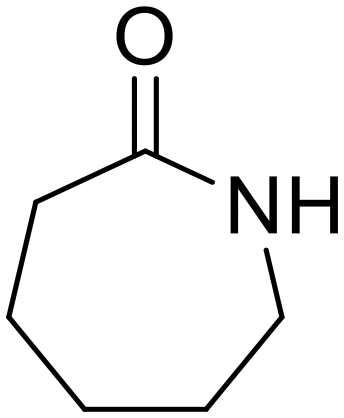 ε-Caprolactam (55)
ε-Caprolactam (55)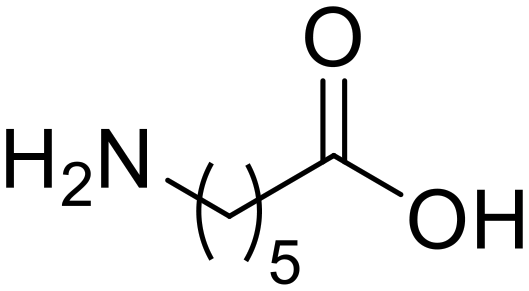 ε-Aminocaproic acid (33)
ε-Aminocaproic acid (33)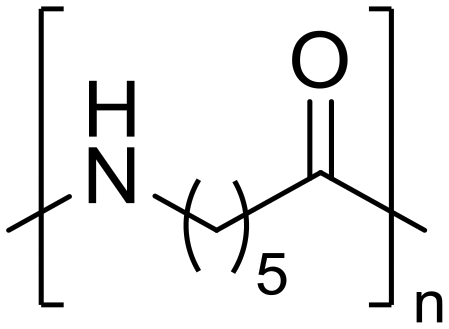 Nylon 6
Nylon 6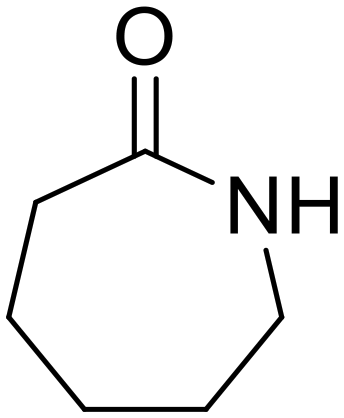 ε-Caprolactam (81)
ε-Caprolactam (81) ε-Aminocaproic acid (13)
ε-Aminocaproic acid (13)