
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Directed evolution of C-methyltransferase PsmD for enantioselective pyrroloindole derivative production†
Diana A.
Amariei
a,
Julia
Tenhaef
 b,
Thomas
Classen
b,
Benoit
David
c,
Tobias M.
Rosch
b,
Holger
Gohlke
cd,
Stephan
Noack
b and
Jörg
Pietruszka
*ab
b,
Thomas
Classen
b,
Benoit
David
c,
Tobias M.
Rosch
b,
Holger
Gohlke
cd,
Stephan
Noack
b and
Jörg
Pietruszka
*ab
aInstitute of Bioorganic Chemistry & Bioeconomy Science Center (BioSC), Heinrich Heine University Düsseldorf in Forschungszentrum Jülich, Jülich, Germany. E-mail: j.pietruszka@fz-juelich.de
bInstitute of Bio- and Geosciences (IBG-1: Biotechnology), Forschungszentrum Jülich, Jülich, Germany
cInstitute of Bio- and Geosciences (IBG-4: Bioinformatics), Forschungszentrum Jülich, Jülich, Germany
dInstitute for Pharmaceutical and Medicinal Chemistry & Bioeconomy Science Center (BioSC), Heinrich Heine University Düsseldorf, Düsseldorf, Germany
First published on 5th September 2024
Abstract
The natural product physostigmine is known for its capacity to inhibit acetylcholinesterase (AChE). The pyrroloindole-based scaffold of physostigmine is prevalent among various compounds demonstrating AChE inhibition, suggesting that its structural diversification holds promise as a strategy for the development of novel AChE inhibitors. The C-methyltransferase PsmD is involved in the biosynthesis of physostigmine. While the two described variants from Streptomyces griseofuscus and Streptomyces albulus display an extended substrate range, their specificity hinders the efficient methylation of substrate derivatives. In order to improve the activity of PsmD towards voluminous non-natural substrates, we employed an iterative saturation mutagenesis strategy, which led to an increase in the available space in the catalytic site, while maintaining stereoselectivity. To aid our efforts and provide an efficient platform for the evolution of pyrroloindole-forming enzymes, we developed a modular automated process for the expression, enzymatic reaction and activity screening of the obtained mutant libraries, using an integrated robotic system. In this way, we identified multiple mutants, which led to increased specific activity towards our target substrates. Our results enabled the identification of amino acid position 166 as a key site for the modulation of substrate specificity. We immobilized the best mutant W166C, and used it for the preparative synthesis of an AChE inhibitor, in the presence of a SAM cofactor recycling system.
Introduction
The pyrroloindole scaffold is present in multiple compounds known to exhibit acetylcholinesterase (AChE) inhibition, some of which are approved drugs against Alzheimer's disease, glaucoma or cholinergic poisoning.1–7 Physostigmine is one of the most well-known compounds in this class and several of its analogs were also found to exhibit AChE and butyrylcholinesterase (BChE) inhibition (Fig. 1).8 Other drugs and natural products containing the 1,2,3,3a,8,8a-hexahydropyrrolo[2,3-b]indole (pyrroloindole) motif show a variety of therapeutic effects, ranging from analgesics to antibiotics or tumor suppressants.9–11 The structural diversification of this scaffold could provide a path to new drugs, and the possibility to improve the pharmacological properties of the existing ones. For this reason, multiple chemical routes have been developed for the synthesis of these compounds. While in some cases the methylation of the indole leads to a racemic mixture, which then needs to be separated, asymmetric synthesis methods were also developed using chiral catalysts to introduce the stereogenic center earlier in the sequence.12–17 An enzymatic route towards these compounds can be attractive in particular due to the late-stage enantioselective introduction of the stereogenic center on the indole ring.18 The biosynthetic pathway of physostigmine has been elucidated and it revealed that a C-methylation step on a tryptophan metabolite is responsible for the chirality of the natural product (Scheme 1).19 The responsible methyltransferase, PsmD, has been characterized and a more stable homolog has been identified and used in preparative enantioselective methylation in the 3-position.8,20 In our previous work, we obtained the crystal structure of PsmD from S. griseofuscus (PsmD_Sg), mapped the catalytic site and identified the amino acid residues that are essential for the enzymatic activity.20 We also assessed the activity of PsmD towards other derivatives of the natural substrate. Based on this information, in the work presented here, we attempted to evolve the enzyme in order to increase activity towards a larger variety of substrates while maintaining stereoselectivity. Most enzymes, in particular those belonging to secondary metabolism are not performant enough for preparative approaches and less so for economically viable industrial processes.21,22 Enzyme engineering is now a firmly established procedure for the improvement of catalytic properties.23 There are multiple paths such as rational and computational design, semi-rational design or directed evolution.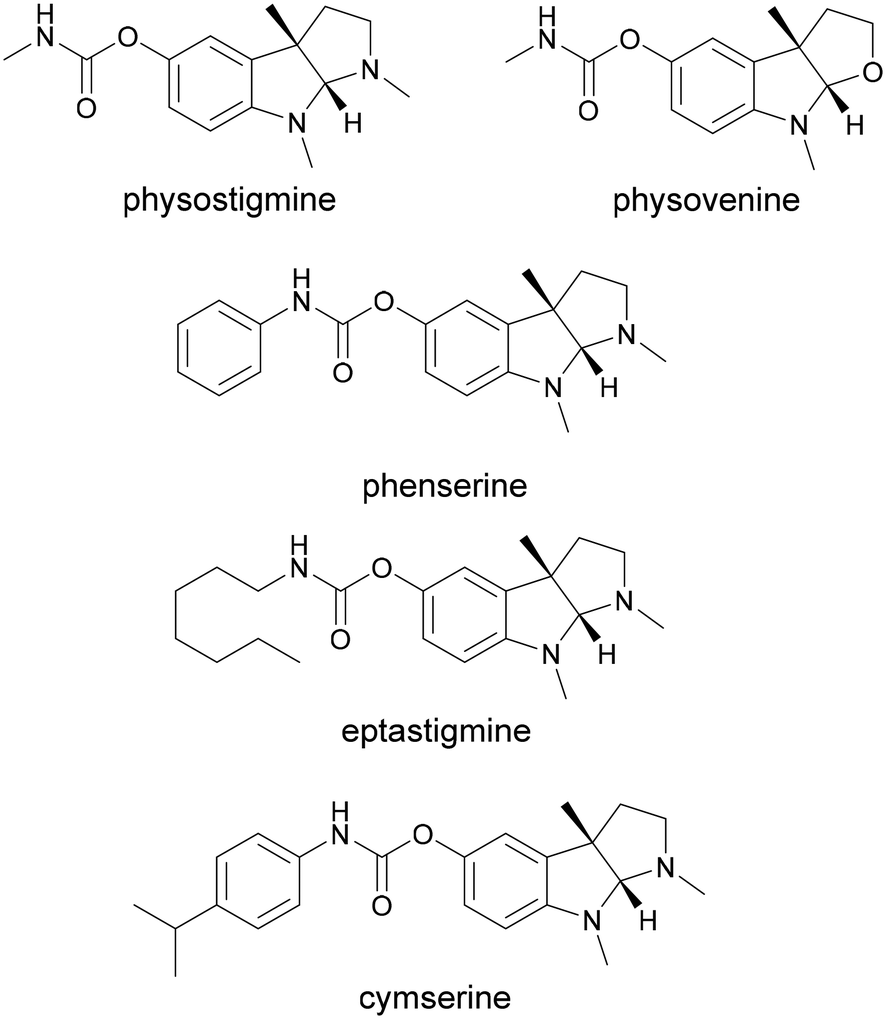 | ||
| Fig. 1 Known biologically active compounds sharing the physostigmine scaffold. | ||
 | ||
| Scheme 1 Reaction catalyzed by PsmD, the natural substrate (1a) and product (1b). | ||
While the rational approach is more accessible experimentally, it requires detailed structural and mechanistic knowledge in order to accurately predict the optimal mutations. Directed evolution eliminates bias and greatly expands the possibility of favorable outcomes but requires extensive library generation and screening efforts. In many cases, a middle ground is the preferred choice, having a semi-rational approach while still maintaining some degree of randomness. This generates focused mutant libraries, significantly reducing the screening efforts.24–27 Small molecule methyltransferases have attracted attention as synthetic options for complex methylated compounds, due to their chemo-, regio- and stereoselectivity.28–32 Several of these enzymes have found their way into the synthesis and diversification of clinically relevant natural products.33–40 The engineering of natural product methyltransferases can be challenging, due to the low reaction rates, limited availability of substrates and the currently limited number of identified and characterized enzymes in this class.29 The necessity of the SAM cofactor in stoichiometric amounts is also a significant barrier to extensive screening efforts due to its price and chemical lability.
Several SAM recycling systems have been recently developed offering much-needed solutions to the cofactor issue when it comes to preparative and industrial applications.41–45 Nevertheless, the recent advances in this line of study have produced several very successful examples of methyltransferase engineering in the last years.46–52 A 235-fold improvement in the total turnover number was achieved for the methylation of α-keto acids by engineering the C-methyltransferase SgvM, while the engineering of human NMT produced up to 118-fold increase in activity for the N-methylation of pyrazoles.53,54 This indicates the potential for the evolution of small molecule methyltransferases, albeit the practical implementation is still in its early stage. In this work, we modified the C-methyltransferase PsmD from S. albulus (PsmD_Sa, the organism of origin also recently annotated as Streptomyces noursei) in order to expand its substrate scope, while maintaining stereoselectivity.55 We used a semi-rational design approach by performing saturation mutagenesis on selected residues in the catalytic site and we developed an automated screening strategy for the obtained mutant libraries. In this way, we could identify variants displaying increased activity for larger substrates, and use them in combination with a cofactor recycling system for the preparative synthesis of physostigmine analogs.
Results and discussion
Mutagenesis targets
We chose two model substrates for our screening efforts. Although multiple physostigmine analogues present structural diversity on the carbamate moiety, diversification of the amide was scarcely explored.56 For this reason, we chose compound 2a as a substrate, containing a t-butyl residue which adds considerable extra volume to the molecule and was found to exhibit significant AChE and BChE inhibitory effects.8 Conversely, we also explored the use of 3a as a substrate for mutant libraries, with the aim of obtaining a precursor of the AChE inhibitor phenserine, which is not produced naturally (Fig. 2A).57 After site-directed mutagenesis to alanine or phenylalanine to assess the importance of the amino acids lining the catalytic pocket, we chose positions that can sterically hinder the binding of larger substrates, but do not affect the catalytic process (Table S2†). Our previous computational simulations revealed two possible positions of the substrate in the catalytic pocket.20 | ||
| Fig. 2 A. Substrates used for the mutant library screening. B. Scheme of the catalytic mechanism and the positions of the chosen amino acids relative to the substrate in the catalytic site. C. Catalytic site of PsmD with SAM (blue) and the docked substrate (yellow). The Glu-His-Tyr catalytic triad is highlighted in green. D. Catalytic site of PsmD with SAM (blue) and the docked substrate (yellow). The residues chosen for saturation mutagenesis are highlighted in red. | ||
Due to the loss of activity upon replacement of the Glu-His-Tyr catalytic triad, we expected the pose in which the indole amine is oriented towards Y128 (Fig. 2C and D) to be productive for methylation.20 We chose two voluminous residues on each side of the docked substrate as saturation mutagenesis positions: W33 and W166, which are part of a previously observed Trp cluster lining the back of the catalytic pocket, and Y197 and Y15 from the lid region (Fig. 2B–D).20 The aromatic residues are not highly conserved in the sequences of similar methyltransferases, suggesting the evolutionary variability of these positions (Fig. S7†). Due to the high metabolic price for incorporating tryptophan in protein sequences, the presence of four tryptophans (W33, W166, W171 and W182) in this region of the catalytic pocket is likely important for the substrate specificity (Fig. S13†).58 A “compression” motion within the catalytic site of methyltransferases was suggested to promote the enzymatic SN2-type methylation, by reducing the energy barrier to the transition state.59–62 This offers a plausible explanation for the two observed conformations (open and closed) of PsmD. It was found that the closure of the lid influences the conformation of the cofactor and reduces the available space. In this context, we aimed to reduce the steric constraints in the catalytic pocket as much as possible, while maintaining a compact space to enable methylation.
Mutant library production
The mutant libraries were generated using the 22c trick, which allows for a reduction of codon redundancies while maintaining full variance of amino acids, consequently reducing the screening resources.63 As such, degenerated primer mixtures were used to introduce mutations to all 20 amino acids in each position. 66 colonies were selected from each library to probably represent all 20 amino acids. E. coli BL21 Gold(DE3) cells were transformed with the obtained plasmid mixtures and the obtained agar plates were used directly in the automated process.Process design
The library screening process involved multiple process steps and was divided into three main modules comprising the protein expression, the enzymatic reaction and the activity assay (Fig. 3). To increase reproducibility and consistency across multiple libraries, we used an automated approach for all modules. This also enabled easy adaptation and extension of the process for further enzymes or different assay conditions. The automated sequence started with colony picking, followed by two consecutive cultivation steps during which the enzymes were expressed using autoinduction. The liquid handling, plate transport, centrifugation, mixing and incubation steps were performed using the integrated AutoBioTech laboratory platform.64 The membrane permeability of the PsmD substrates and products allowed us to perform the enzymatic reactions using whole-cell systems. To screen for activity, a modified version of our previously described indole assay was used, which utilizes p-dimethylaminobenzaldehyde (DMAB) and H2SO4 to form colored products with the indole ring of the substrate (Fig. 4).65 The absorbance at 580 nm was measured and heatmaps were created in order to select the positions with the highest substrate consumption. The system allowed the processing of up to four plates in parallel and provided an almost complete removal of human experimental input. | ||
| Fig. 3 Operation scheme of the automated screening process using the AutoBioTech platform. | ||
 | ||
| Fig. 4 Scheme of the colorimetric assay used for the detection of the substrate indole and example of a library plate after the assay. EV refers to the “empty vector” negative control. | ||
Library screening using substrate 2a
After identification of the mutants, the hits of each round were isolated and their specific activity was determined. Interestingly, position W166 turned out to be the most versatile, leading to the best hits for both 2a and 3a. Replacing the tryptophan in this position with a cysteine led to a three-fold increase in the specific activity towards 2a, compared to the wild type (Fig. 5A). The best hits containing new mutations in each library were used as parents for subsequent mutagenesis rounds, in the hope of identifying cooperative effects of multiple mutations. | ||
| Fig. 5 A. Hits from every round of mutagenesis and screening using 2a as a substrate and their activity relative to WT PsmD. B. Specific activities of all the hits from the screening experiments for the natural substrate 1a (blue) and the selected substrate 2a (orange) as well as the enantiomeric excess for reactions with 2a (diamond shape). The specific activities were determined using the commercial MTase-Glo™ assay. In the case of W33C and W166P/W33L, the enantiomeric excess could not be determined. | ||
Unfortunately, this was not the case, as further mutagenesis rounds did not improve the performance beyond mutant W166C. Most mutants did however keep an increased activity towards this substrate, when compared to the wild type. As we expected, increasing the space in the catalytic site drives an improvement in the acceptance of the bulkier 2a containing the t-butyl amide, at the expense of the activity towards the natural substrate (Fig. 5B). Indeed, the degree of freedom added to the natural substrate within the modified catalytic site can hinder its binding in a productive position for methylation. The reduced activity of the single mutants in the positions Y15, Y197 and W33 suggests that further optimization in these positions is not beneficial for the tested substrates. All active mutants maintained the stereoselectivity of the transformation to 2b (Fig. 5B). The structural explanation for the importance of position W166 was found by docking the respective substrates into the homology model of PsmD_Sa. We used the homolog PsmD_Sg, for which X-ray structures are available (PDB ID: 7ZKH, 7ZGT, 7ZKG), as a template for the homology model generation with MODELLER.66 Two protein forms were identified upon crystallization, determined by the movement of an N-terminal “lid”.
Our previous mechanistic analysis suggests that the closed conformation is active, therefore it was selected for the generation of the PsmD_Sa homology model and the docking experiments. An extensive probing of the active site of WT PsmD_Sa and the W166C variant was performed using molecular docking of the substrate 2a. Several thousands of poses were obtained for each enzyme-substrate combination, which were then subjected to principal component analysis, to reveal the most probable productive configuration of substrate 2a in the catalytic site. The data was then analyzed in regards to binding energy and distance between the cofactor and the methylation site. Our previous mechanistic study of PsmD revealed the productive binding mode of the natural substrate 1a (Fig. 2C and D). This overlaps with one of the pose clusters obtained after docking substrate 2a in both variants (cluster 6, Fig. S2 and S3†). However, the cluster occupation differs significantly between WT PsmD and mutant W166C (Fig. 6). The productive binding mode was markedly more likely to occur among the docking results of W166C than WT PsmD. This is in accord with the observed activity difference between the two variants, confirming our choice of pose as the most probable active configuration of the substrate 2a in the catalytic site of PsmD.
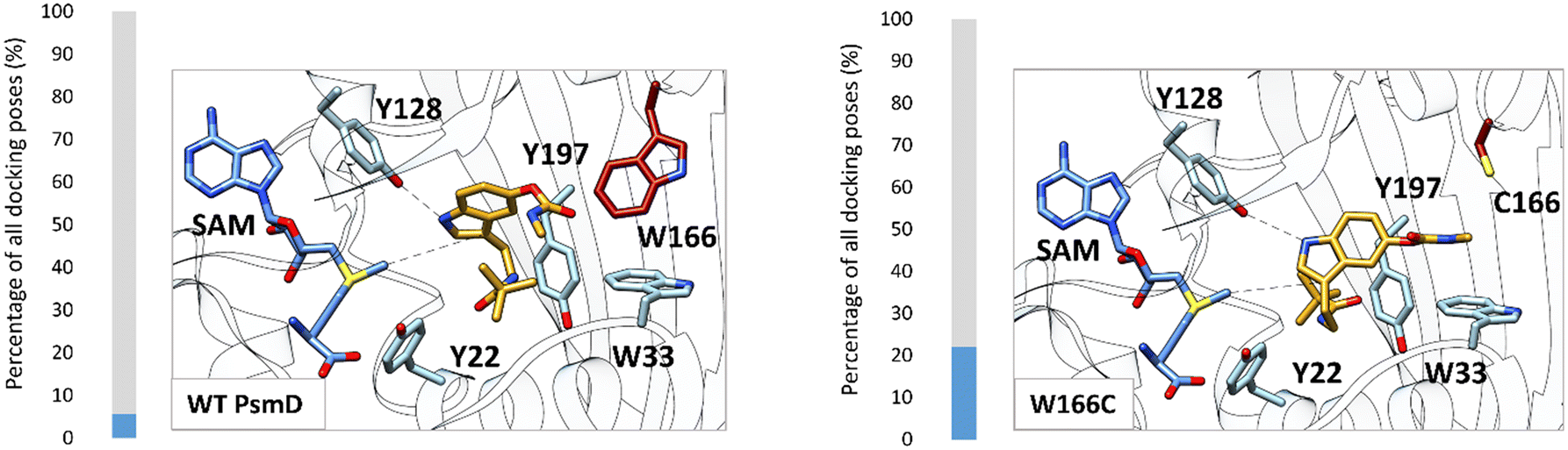 | ||
| Fig. 6 Comparison of the active pose of docked substrate 2a in the catalytic pocket of WT PsmD and the mutant W166C. The proportion of the active binding mode in all the obtained docking poses is represented as percentages (blue). 1999 poses were analyzed for each enzyme with substrate 2a. The displayed poses were selected as corresponding to activity based on the similarity with the previously determined active pose of the natural substrate and the preservation of the experimentally observed stereoselectivity (Fig. 2C).20 The correlation of the pose clusters with the calculated binding energy and distance from the cofactor supported the choice (full analysis in the ESI,† Fig. S2 and S3). The residue in position 166 is highlighted in red. | ||
The comparison between the docked substrate 2a in WT PsmD_Sa and mutant W166C shows that a gap forms between the residues in the positions 166 and 33 (Fig. 6). This allows for a more relaxed substrate conformation within the catalytic site, accommodating the carbamate and supporting the interaction with the catalytic Y128 for the activation of the indole ring. Our previous molecular dynamics simulation study on PsmD_Sg suggested that the W33–W166 interaction could be involved in the exit of the product after methylation, opening a channel between the connected α-helices.20 Replacing residue W166 could also presumably affect the dynamics of the cavity opening, influencing the overall reaction rate.
Preparative enzymatic methylation of 2a
The best mutant, W166C, was used to obtain the product 2b in a preparative manner, starting with 50 mg substrate, using a halide methyl transferase (CtHMT) for the recycling of the SAM cofactor. The enzymes containing His-tags were immobilized on Ni-NTA resin and incubated in the presence of MeI, SAH and substrate 2a (Fig. 7). This set-up was successfully used before for the preparative enzymatic methylation using StspM1 using the HMT-based cofactor recycling.34 In this system, immobilized mutant W166C lead to product 2b with 65% conversion after 20 h and 60% yield after purification by column chromatography. | ||
| Fig. 7 Scheme of the procedure for the preparative enzymatic methylation of 2a using the immobilized W166C mutant and the HMT-based cofactor recycling system. | ||
Library generation and screening using substrate 3a
We chose to perform a limited screening for substrate 3a due to its low solubility in the reaction medium. As such, we attempted a trial screen of three libraries: the randomized positions 33 and 166, as well as the combination between the two. We adapted the colorimetric assay by diluting the reaction sample with isopropanol to a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, to address the substrate's poor solubility in water. This improved the detection of the substrate and allowed us to compare the absorbance at 580 nm within the wells of the reaction plates. Nevertheless, the assay sensitivity decreased substantially compared to the screening of compound 2a, which could affect the detection of the poorer-performing mutants. However, we could identify hits in one of the three libraries, which upon further testing, performed better than the wild type in reaction with compound 3a. Saturation mutagenesis in position 33 did not lead to any detectable hit. Interestingly, similarly to our previous screening with compound 2a, the mutation of W166, this time to proline, provided a significant improvement in activity towards substrate 3a (Fig. 8A). Starting with almost no activity of the wild type towards substrate 3a, W166P increased the specific activity 28-fold. Although the activity towards compound 3a needs further improvement for an efficient biocatalytic production of the phenserine precursor 3b, we found that position 166 plays a key role, and further modifications could be considered for its surroundings. The double mutant W166P/W33L, although identified as hit in the high-throughput assay, displayed no detectable activity towards substrate 3a when isolated.
:1 ratio, to address the substrate's poor solubility in water. This improved the detection of the substrate and allowed us to compare the absorbance at 580 nm within the wells of the reaction plates. Nevertheless, the assay sensitivity decreased substantially compared to the screening of compound 2a, which could affect the detection of the poorer-performing mutants. However, we could identify hits in one of the three libraries, which upon further testing, performed better than the wild type in reaction with compound 3a. Saturation mutagenesis in position 33 did not lead to any detectable hit. Interestingly, similarly to our previous screening with compound 2a, the mutation of W166, this time to proline, provided a significant improvement in activity towards substrate 3a (Fig. 8A). Starting with almost no activity of the wild type towards substrate 3a, W166P increased the specific activity 28-fold. Although the activity towards compound 3a needs further improvement for an efficient biocatalytic production of the phenserine precursor 3b, we found that position 166 plays a key role, and further modifications could be considered for its surroundings. The double mutant W166P/W33L, although identified as hit in the high-throughput assay, displayed no detectable activity towards substrate 3a when isolated.
 | ||
| Fig. 8 A. Results of saturation mutagenesis screening rounds using 3a as substrate. B. Specific activity of hit mutants towards 1a (blue, left axis) and 3a (green, right axis), compared to WT PsmD_Sa. C. Comparison of the docked active pose of substrate 3a in the catalytic pocket of WT PsmD and mutant W166P. The proportion of the active binding mode in all the obtained docking poses is represented as percentages (blue). 1861 poses were analyzed for WT PsmD with substrate 3a and 1791 poses for W166P. The displayed poses were selected as corresponding to activity based on the similarity with the previously determined active pose of the natural substrate.20 The correlation of the poses with the calculated binding energy and distance from the cofactor supported the choice (full analysis in the ESI;† Fig. S4 and S5). The residue in position 166 is highlighted in red. | ||
Given the uncertain positioning of the substrate in the catalytic pocket, we explored the idea that the hits identified in both screenings could also catalyse the reaction with the respective complementary substrate. To quantify their specific activity, we isolated and tested all the hits with both substrates using the MTase-Glo™ assay, which quantifies SAH production by converting it to ATP and measuring the luminescence in a luciferase reaction.67 Interestingly, two hits from the substrate 2a screening also show improved activity towards substrate 3a. Y197H and the triple mutant W166C/Y197F/W33M increased the activity towards compound 3a by 5- and 18-fold compared to the wild type (Fig. 8B).
An extensive docking approach was used for docking substrate 3a into WT PsmD_Sa and the W166P variant, following the method described earlier. Molecular docking of 3a into the W166P homology model (Fig. 8C) reveals a pose similar to that obtained for substrate 2a in the W166C variant (Fig. 6), as well as the active pose of 1a in WT (Fig. 2C and D). The mutation in position 166 promotes a relaxed conformation of the substrate, with the carbamate sidechain extending into the gap between W33 and P166. The π-stacking of the phenyl ring of the substrate with W33 could additionally stabilize the enzyme-substrate complex. When testing variants W166C and W166P with the other respective substrates, the results showed that the beneficial effects of the mutations are largely specific to the screened substrate. While variant W166C provided a 3-fold increase in activity towards substrate 3a, compared to the WT, variant W166P led to a loss in activity towards substrate 2a (Fig. S14†). The observed effect of mutations to residue W166 indicates its importance in substrate binding and enzyme selectivity. Further engineering approaches for other substrates substituted on the amide or carbamate side could benefit from addressing this position. The further evolution potential of this enzyme type might be worth exploring in relation to this site.
Conclusions
The use of an automated screening strategy allowed for the identification of multiple PsmD mutants displaying activity towards substrates with expanded non-polar functionalities, known to display AChE inhibition. Although methyltransferase engineering instances are relatively rare in literature, expanding the portfolio of successful examples highlights the potential of engineering this class of enzymes in order to provide valuable routes in the synthesis of complex natural products and pharmaceuticals.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
DAA – conceptualization, data curation, formal analysis, investigation, methodology, validation, visualisation, writing – original draft. JT – data curation, formal analysis, investigation, methodology. TC – formal analysis, methodology. BD – formal analysis, methodology. TMR – methodology. HG, SN – project administration, supervision, resources. JP – conceptualization, funding acquisition, project administration, supervision, resources. All authors contributed to writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the German Federal Ministry of Education of Research (BMBF, “Modellregion, BioRevierPlus: BioökonomieREVIER Innovationscluster Biotechnologie & Kunststofftechnik-BioTech”, grant number 031B1134A), as well as the Heinrich Heine University Düsseldorf and the Forschungszentrum Jülich GmbH for their ongoing support.Notes and references
- K. L. Davis, R. C. Mohs, J. R. Tinklenberg, A. Pfefferbaum, L. E. Hollister and B. S. Koppel, Science, 1978, 201, 272–274 CrossRef CAS PubMed.
- D. J. Triggle, J. M. Mitchell and R. Filler, CNS Drug Rev., 1998, 4, 87–136 CrossRef CAS.
- A. Galli, G. Renzi, E. Grazzini, R. Bartolini, P. Aiello-Malmberg and A. Bartolini, Biochem. Pharmacol., 1982, 31, 1233–1238 CrossRef CAS PubMed.
- A. Kadir, N. Andreasen, O. Almkvist, A. Wall, A. Forsberg, H. Engler, G. Hagman, M. Lärksäter, B. Winblad, H. Zetterberg, K. Blennow, B. Långström and A. Nordberg, Ann. Neurol., 2008, 63, 621–631 CrossRef CAS PubMed.
- Q. Yu, C. Liu, M. Brzostowska, L. Chrisey, A. Brossi, N. Greig, J. R. Atack, T. T. Soncrant, S. I. Rapoport and H. Radunz, Helv. Chim. Acta, 1991, 74, 761–766 CrossRef CAS.
- N. Canal and B. P. Imbimbo, Clin. Pharmacol. Ther., 1996, 60, 218–228 CrossRef CAS PubMed.
- M. A. Kamal, A. A. Al-Jafari, Q.-S. Yu and N. H. Greig, Biochim. Biophys. Acta, Gen. Subj., 2006, 1760, 200–206 CrossRef CAS PubMed.
- P. Schneider, B. Henßen, B. Paschold, B. P. Chapple, M. Schatton, F. P. Seebeck, T. Classen and J. Pietruszka, Angew. Chem., Int. Ed., 2021, 60, 23412–23418 ( Angew. Chem. , 2021 , 133 , 23600–23606 ) CrossRef CAS PubMed.
- T. A. Amador, L. Verotta, D. S. Nunes and E. Elisabetsky, Planta Med., 2000, 66, 770–772 CrossRef CAS PubMed.
- M. A. Schallenberger, T. Newhouse, P. S. Baran and F. E. Romesberg, J. Antibiot., 2010, 63, 685–687 CrossRef CAS PubMed.
- E. Viziteu, C. Grandmougin, H. Goldschmidt, A. Seckinger, D. Hose, B. Klein and J. Moreaux, Br. J. Cancer, 2016, 114, 519–523 CrossRef CAS PubMed.
- Q.-s. Yu, X.-F. Pei, H. W. Holloway, N. H. Greig and A. Brossi, J. Med. Chem., 1997, 40, 2895–2901 CrossRef CAS PubMed.
- J. C. Yi, C. Liu, L. X. Dai and S. L. You, Chem. – Asian J., 2017, 12, 2975–2979 CrossRef CAS PubMed.
- T. Matsuura, L. E. Overman and D. J. Poon, J. Am. Chem. Soc., 1998, 120, 6500–6503 CrossRef CAS.
- T. Bui, S. Syed and C. F. Barbas, J. Am. Chem. Soc., 2009, 131, 8758–8759 CrossRef CAS PubMed.
- A. Pinto, Y. Jia, L. Neuville and J. Zhu, Chem. – Eur. J., 2007, 13, 961–967 CrossRef CAS PubMed.
- A. Huang, J. J. Kodanko and L. E. Overman, J. Am. Chem. Soc., 2004, 126, 14043–14053 CrossRef CAS PubMed.
- C. Sun, W. Tian, Z. Lin and X. Qu, Nat. Prod. Rep., 2022, 39, 1721–1765 RSC.
- J. Liu, T. Ng, Z. Rui, O. Ad and W. Zhang, Angew. Chem., Int. Ed., 2014, 53, 136–139 ( Angew. Chem. , 2014 , 126 , 140–143 ) CrossRef CAS PubMed.
- D. A. Amariei, N. Pozhydaieva, B. David, P. Schneider, T. Classen, H. Gohlke, O. H. Weiergräber and J. Pietruszka, ACS Catal., 2022, 12, 14130–14139 CrossRef CAS.
- S. E. O'Connor, Annu. Rev. Genet., 2015, 49, 71–94 CrossRef PubMed.
- A. Bar-Even and D. Salah Tawfik, Curr. Opin. Biotechnol., 2013, 24, 310–319 CrossRef CAS PubMed.
- C. Zeymer and D. Hilvert, Annu. Rev. Biochem., 2018, 87, 131–157 CrossRef CAS PubMed.
- M. T. Reetz and J. D. Carballeira, Nat. Protoc., 2007, 2, 891–903 CrossRef CAS PubMed.
- R. M. P. Siloto and R. J. Weselake, Biocatal. Agric. Biotechnol., 2012, 1, 181–189 CrossRef CAS.
- Y. Wang, P. Xue, M. Cao, T. Yu, S. T. Lane and H. Zhao, Chem. Rev., 2021, 121, 12384–12444 CrossRef CAS PubMed.
- N. J. Turner, Nat. Chem. Biol., 2009, 5, 567–573 CrossRef CAS PubMed.
- H. L. Schubert, R. M. Blumenthal and X. Cheng, Trends Biochem. Sci., 2003, 28, 329–335 CrossRef CAS PubMed.
- M. R. Bennett, S. A. Shepherd, V. A. Cronin and J. Micklefield, Curr. Opin. Chem. Biol., 2017, 37, 97–106 CrossRef CAS PubMed.
- D. Aynetdinova, M. C. Callens, H. B. Hicks, C. Y. X. Poh, B. D. A. Shennan, A. M. Boyd, Z. H. Lim, J. A. Leitch and D. J. Dixon, Chem. Soc. Rev., 2021, 50, 5517–5563 RSC.
- H. Schönherr and T. Cernak, Angew. Chem., Int. Ed., 2013, 52, 12256–12267 ( Angew. Chem. , 2013 , 125 , 12480–12492 ) CrossRef PubMed.
- E. Abdelraheem, B. Thair, R. F. Varela, E. Jockmann, D. Popadić, H. C. Hailes, J. M. Ward, A. M. Iribarren, E. S. Lewkowicz, J. N. Andexer, P. L. Hagedoorn and U. Hanefeld, ChemBioChem, 2022, 23, e202200212 CrossRef CAS PubMed.
- B. J. C. Law, A.-W. Struck, M. R. Bennett, B. Wilkinson and J. Micklefield, Chem. Sci., 2015, 6, 2885–2892 RSC.
- M. Haase, B. David, B. Paschold, T. Classen, P. Schneider, N. Pozhydaieva, H. Gohlke and J. Pietruszka, ACS Catal., 2024, 14, 227–236 CrossRef CAS PubMed.
- A. Gutmann, M. Schiller, M. Gruber-Khadjawi and B. Nidetzky, Org. Biomol. Chem., 2017, 15, 7917–7924 RSC.
- E. Abdelraheem, E. Jockmann, J. Li, S. Günther, J. N. Andexer, P. L. Hagedoorn and U. Hanefeld, ChemCatChem, 2023, 16, e202301217 CrossRef.
- R. Roddan, F. Subrizi, J. Broomfield, J. M. Ward, N. H. Keep and H. C. Hailes, Org. Lett., 2021, 23, 6342–6347 CrossRef CAS PubMed.
- J. Fricke, A. Sherwood, R. Kargbo, A. Orry, F. Blei, A. Naschberger, B. Rupp and D. Hoffmeister, ChemBioChem, 2019, 20, 2824–2829 CrossRef CAS PubMed.
- H. Stecher, M. Tengg, B. J. Ueberbacher, P. Remler, H. Schwab, H. Griengl and M. Gruber-Khadjawi, Angew. Chem., Int. Ed., 2009, 48, 9546–9548 ( Angew. Chem. , 2009 , 121 , 9710–9712 ) CrossRef CAS PubMed.
- E. Jockmann, F. Subrizi, M. K. F. Mohr, E. M. Carter, P. M. Hebecker, D. Popadić, H. C. Hailes and J. N. Andexer, ChemCatChem, 2023, 15, e202300930 CrossRef CAS.
- S. Mordhorst, J. Siegrist, M. Müller, M. Richter and J. N. Andexer, Angew. Chem., Int. Ed., 2017, 56, 4037–4041 ( Angew. Chem. , 2017 , 129 , 4095–4099 ) CrossRef CAS PubMed.
- C. Liao and F. P. Seebeck, Nat. Catal., 2019, 2, 696–701 CrossRef CAS.
- S. Mordhorst and J. N. Andexer, Nat. Prod. Rep., 2020, 37, 1316–1333 RSC.
- X. Wen, F. Leisinger, V. Leopold and F. P. Seebeck, Angew. Chem., Int. Ed., 2022, 61, e202208746 ( Angew. Chem. , 2022 , 134 , e202208746 ) CrossRef CAS PubMed.
- D. Popadić, D. Mhaindarkar, M. H. N. Dang Thai, H. C. Hailes, S. Mordhorst and J. N. Andexer, RSC Chem. Biol., 2021, 2, 883–891 RSC.
- G.-Y. Yang, G.-W. Zheng, B.-B. Zeng, J.-H. Xu and Q. Chen, Mol. Catal., 2023, 550, 113533 CrossRef CAS.
- X. Wang, C. Wang, L. Duan, L. Zhang, H. Liu, Y.-m. Xu, Q. Liu, T. Mao, W. Zhang, M. Chen, M. Lin, A. A. L. Gunatilaka, Y. Xu and I. Molnár, J. Am. Chem. Soc., 2019, 141, 4355–4364 CrossRef CAS PubMed.
- B. Aberle, D. Kowalczyk, S. Massini, A. N. Egler-Kemmerer, S. Gergel, S. C. Hammer and B. Hauer, Angew. Chem., Int. Ed., 2023, 62, e202301601 ( Angew. Chem. , 2023 , 135 , e202301601 ) CrossRef CAS PubMed.
- A. Kunzendorf, B. Zirpel, L. Milke, J. P. Ley and U. T. Bornscheuer, ChemCatChem, 2023, 15, e202300951 CrossRef CAS.
- Q. Tang, C. W. Grathwol, A. S. Aslan-Üzel, S. Wu, A. Link, I. V. Pavlidis, C. P. S. Badenhorst and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2021, 60, 1524–1527 ( Angew. Chem. , 2021 , 133 , 1547–1551 ) CrossRef PubMed.
- K. H. Schulke, J. S. Frose, A. Klein, M. Garcia-Borras and S. C. Hammer, ChemBioChem, 2024, e202400079 CrossRef PubMed.
- C. Y. Gao, G. Y. Yang, X. W. Ding, J. H. Xu, X. Cheng, G. W. Zheng and Q. Chen, Angew. Chem., Int. Ed., 2024, 63, e202401235 ( Angew. Chem. , 2024 , 136 , e202401235 ) CrossRef CAS PubMed.
- S. Ju, K. P. Kuzelka, R. Guo, B. Krohn-Hansen, J. Wu, S. K. Nair and Y. Yang, Nat. Commun., 2023, 14, 5704 CrossRef CAS PubMed.
- L. L. Bengel, B. Aberle, A. N. Egler-Kemmerer, S. Kienzle, B. Hauer and S. C. Hammer, Angew. Chem., Int. Ed., 2021, 60, 5554–5560 ( Angew. Chem. , 2021 , 133 , 5614–5620 ) CrossRef CAS PubMed.
- W. Butdee, S. Muangham, D. Chonudomkul and K. Duangmal, Int. J. Syst. Evol. Microbiol., 2023, 73, 005639 CAS.
- Q.-s. Yu, H. W. Holloway, J. L. Flippen-Anderson, B. Hoffman, A. Brossi and N. H. Greig, J. Med. Chem., 2001, 44, 4062–4071 CrossRef CAS PubMed.
- J. Klein, Expert Opin. Invest. Drugs, 2007, 16, 1087–1097 CrossRef CAS PubMed.
- S. Barik, Int. J. Mol. Sci., 2020, 21, 8776 CrossRef CAS PubMed.
- K. Świderek, I. Tuñón, I. H. Williams and V. Moliner, J. Am. Chem. Soc., 2018, 140, 4327–4334 CrossRef PubMed.
- R. J. Boyd, C. K. Kim, Z. Shi, N. Weinberg and S. Wolfe, J. Am. Chem. Soc., 1993, 115, 10147–10152 CrossRef CAS.
- J. Zhang, H. J. Kulik, T. J. Martinez and J. P. Klinman, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 7954–7959 CrossRef CAS PubMed.
- J. Zhang and J. P. Klinman, J. Am. Chem. Soc., 2016, 138, 9158–9165 CrossRef CAS PubMed.
- S. Kille, C. G. Acevedo-Rocha, L. P. Parra, Z.-G. Zhang, D. J. Opperman, M. T. Reetz and J. P. Acevedo, ACS Synth. Biol., 2013, 2, 83–92 CrossRef CAS PubMed.
- T. M. Rosch, J. Tenhaef, T. Stoltmann, T. Redeker, D. Kosters, N. Hollmann, K. Krumbach, W. Wiechert, M. Bott, S. Matamouros, J. Marienhagen and S. Noack, ACS Synth. Biol., 2024, 13, 2227–2237 CrossRef CAS PubMed.
- D. A. Amariei, M. Haase, M. K. T. Klischan, M. Wäscher and J. Pietruszka, ChemCatChem, 2024, e202400052 CrossRef CAS.
- B. Webb and A. Sali, Curr. Protoc. Bioinf., 2016, 54, 5.6 Search PubMed.
- K. Hsiao, H. Zegzouti and S. A. Goueli, Epigenomics, 2016, 8, 321–339 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cy00657g |
| This journal is © The Royal Society of Chemistry 2024 |