
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Vibrational frequencies utilized for the assessment of exchange–correlation functionals in the description of metal–adsorbate systems: C2H2 and C2H4 on transition-metal surfaces†
Ray
Miyazaki‡
 *ab,
Somayeh
Faraji‡
a,
Sergey V.
Levchenko
c,
Lucas
Foppa
*a and
Matthias
Scheffler
a
*ab,
Somayeh
Faraji‡
a,
Sergey V.
Levchenko
c,
Lucas
Foppa
*a and
Matthias
Scheffler
a
aThe NOMAD Laboratory at the Fritz Haber Institute of the Max Planck Society, Faradayweg 4-6, Berlin 14195, Germany. E-mail: foppa@fhi-berlin.mpg.de
bInstitute for Catalysis, Hokkaido University, N21 W10 Kita-ku, Sapporo, Hokkaido 001-0021, Japan. E-mail: ray_miyazaki@cat.hokudai.ac.jp
cCenter for Energy Science and Technology, Skolkovo Institute of Science and Technology, Moscow 121205, Russia
First published on 15th October 2024
Abstract
Describing the interaction between reactive species and surfaces is crucial for designing catalyst materials. Density-functional approximation is able to quantitatively model such interaction, but its accuracy strongly depends on the choice of exchange–correlation (XC) functional approximation. In this work, we assess the performance of XC functionals for describing the interaction of C2H2 and C2H4 with the (111) surfaces of Cu, Pt, Pd, and Rh by particularly focusing on RPBE and mBEEF functionals. We study the geometry and the vibrational frequencies associated with the adsorbed molecules as well as the adsorption energies and the reaction enthalpy of semi-hydrogenation of C2H2 in the gas phase. Crucially, experimental values for vibrational frequencies of molecules adsorbed on metal surfaces are available for more systems compared to physical quantities typically used to benchmark XC functionals, such as adsorption energies. Thus, vibrational frequencies can be utilized as a reference to assess the reliability of the exchange–correlation functionals. We find that the mean percentage errors (MPEs) of RPBE and mBEEF with respect to reported experimental values of vibrational frequencies are 0.64% and −3.88%, respectively (36 data points). For adsorption enthalpy, RPBE and mBEEF provide MPEs of 27.61% and −59.81%, respectively, with respect to reported experimental values (7 data points). Therefore, the performance of RPBE is superior to that of mBEEF for the considered systems.
1. Introduction
Transition metals are widely used as heterogeneous catalysts.1–7 Thus, investigating the interaction of adsorbates with metal surfaces and their impact on the reaction mechanisms is important for the design of novel catalysts. Electronic-structure calculations can provide the needed detailed description.8 In particular, density functional theory (DFT)9 in the Kohn–Sham10 framework is a widely applied theoretical approach for catalytic systems.11 However, in practice, we are bound to use approximated exchange–correlation (XC) functionals, and the accuracy of such density functional approximation (DFA) strongly depends on the XC functional. Consequently, many studies have been carried out (see ref. 12–15) to assess the performance and accuracy of XC functionals for, e.g., bulk band structures, structural properties, and surface properties of transition metals. According to benchmark studies on the adsorption of small molecules on metal surfaces, the alternative revision of the Perdew–Burke–Ernzerhof XC functional (RPBE)16 provides an accurate description of the most stable adsorption sites and adsorption energies.13,14 Recently, a benchmark study by Kabalan et al.17 on the performance of XC functionals, specifically for transition metals, showed that meta-generalized gradient XC functional approximation based on Bayesian error estimation (mBEEF)18 provides good agreement with experimental data, such as surface energy and work function.17 Ref. 17 focused on pristine surfaces without adsorbates, and RPBE was not considered in the context of surface properties because its error for experimental bulk properties is larger than that of the other tested XC functionals.Ethylene (C2H4) is one of the most important chemical building blocks for polymer production.19–21 Ethylene is typically obtained by naphtha steam cracking. In this process, acetylene (C2H2) is formed as a by-product. However, the presence of acetylene is undesirable, as it deactivates the catalyst for C2H4 polymerization, which is one of the key processes where ethylene is utilized. Thus, C2H2 has to be removed before the polymerization process. This can be achieved by the catalytic selective hydrogenation of C2H2 to C2H4. The catalyst selectively hydrogenates C2H2 while suppressing the hydrogenation of C2H4 to ethane (C2H6). Pd and Pd-based alloys have been used as catalyst materials in this process.6,7,22,23 Many DFA studies investigated the selective hydrogenation of acetylene.24–28 In order to design new catalyst, the accurate description of both acetylene and ethylene adsorption is necessary.11 All the benchmark studies proclaim the challenge of finding an optimal functional that could simultaneously capture both covalent and non-covalent interactions of adsorbates on metal surfaces, with no clear preference for a particular functional. Thus, the accuracy of the XC functional for these specific target systems still needs to be scrutinized. However, reliable experimental and/or higher-level theoretical reference data for assessing the performance of the XC functional for adsorption of molecules on surfaces are limited.
In this study, we assess the performance of RPBE and mBEEF by focusing on the vibrational frequencies associated to C2H4 and C2H2 molecules adsorbed on transition-metal (TM) surfaces. The vibrational frequencies of adsorbed molecules reflect the nature and strength of the molecules' interaction with the TM surfaces. Additionally, reliable and accurate experimental values for vibrational frequencies of molecules adsorbed on TM surfaces are available. The aim of this study is not to find the best XC functional for describing the adsorption of C2H4 and C2H2 molecules on TM surfaces among a wide range of functionals. Instead, we aim at demonstrating how the performance of XC functionals for describing surfaces and catalysis can be assessed via the experimental vibrational frequencies. The RPBE and mBEEF are widely used XC functionals and they provide good accuracy for the surface properties and adsorption energies of molecules on TM surfaces. Thus, these two XC functionals are adopted to showcase the concept. We would like to stress that the properties of adsorbed molecules, such as bond elongation, have been directly correlated to catalytic activity.29 Additionally, the accuracy of XC functionals for the vibrational frequency is important in the context of calculating energy curvatures around equilibrium and transition states and the calculated properties of adsorbed molecules can be considered as potentially relevant parameters in artificial intelligence (AI) analysis that aim to identify intricate correlations between material properties and catalytic performance. These calculated parameters can be offered along with experimental parameters related to other underlying processes in the AI analysis.30 The accuracy and reliability of the data employed in AI is crucial. Thus, proposing the proper way for assessment of the performance of XC functionals is also an urgent task for data-centric heterogeneous catalyst design.
We study adsorbed C2H2 and C2H4 on the surfaces of Pd, Pt, Cu, and Rh. These are transition metals widely used as catalysts for the selective hydrogenation of C2H2, and the vibrational frequencies of C2H2 and C2H4 on these metals have been determined experimentally.31–39 We also assess the performance of the functionals based on adsorption energies of the molecules on Pt, for which experimental values are available. However, the available reference data are scarcer than in the case of the vibrational frequencies. Additionally, we investigate the performance for the reaction enthalpy of semi-hydrogenation of C2H2 to C2H4 in the gas phase. To demonstrate further application of our concept, we also investigate the C2H4 adsorption on Pt(111) by using other XC functionals (PBEsol40 and TPSS41) and RPBE with the Tkatchenko–Scheffler van der Waals (vdW) interaction correction42 (RPBE + TS).
2. Computational details
2.1 DFA calculations
All the calculations were performed within the DFA framework as implemented in the all-electron full-potential electronic-structure package FHI-aims43 (version 210226). RPBE is a generalized gradient approximation (GGA) functional, which only differs from the original PBE XC functional by the mathematical form of the exchange energy enhancement factor (see ref. 16). The functional was mainly proposed to improve the chemisorption energetics of atoms and molecules on TM surfaces. The mBEEF is a meta-GGA, which combines machine learning with Bayesian statistics to find the exchange model parameters (see ref. 18). It was designed to give reasonably accurate predictions of material properties, such as cohesive energies and lattice constants. The mBEEF calculations are done by interfacing FHI-aims with the LibXC44 library. In all the calculations, scalar relativity is included via the atomic zero-order regular approximation (ZORA).45 The calculations are done by employing a tight basis set and a converged Γ-centered k-grid of 20 × 20 × 20 and 12/n × 12/n × 1 (n is the supercell size) for the bulk and surfaces, respectively. The Broyden–Fletcher–Goldfarb–Shanno (BFGS) algorithm46 with a force convergence criterion of 10 meV Å−1 along all directions is employed for the geometry optimizations. A dipole correction is applied along the surface normal (non-periodic direction, see the following section for more details) to eliminate any potential electrostatic dipole that could result from a non-symmetric slab relaxation and molecule adsorption.2.2 Surface models
The (111) surface is the most stable facet of the TMs adopted in this study (i.e., Cu, Pt, Pd, Rh), which have the face-centered cubic (fcc, space group F![[m with combining macron]](https://www.rsc.org/images/entities/i_char_006d_0304.gif) 3m) bulk structure,47 and is the focus of experimental studies. To model the surfaces, firstly, the lattice parameters of the bulk structures were optimized by mBEEF or RPBE. Then, the equilibrium lattice parameters were used to model the (111) surfaces by a two-dimensional slab with the periodic boundary conditions. Unless otherwise stated, the surfaces are modeled by 2 × 2 and 3 × 3 surface supercells to account for the adsorption coverages observed in experiments. Each slab is separated by at least a 20 Å vacuum perpendicular to the (111) surface (z-direction) to decrease the interaction between slab periodic images so that it can be ignored. Each slab consists of seven atom layers with the three bottom layers kept fixed at their ideal bulk positions during the geometry optimization. The atomic simulation environment (ASE) Python package48 is used to create surface slab models from the fully optimized bulk geometries.
3m) bulk structure,47 and is the focus of experimental studies. To model the surfaces, firstly, the lattice parameters of the bulk structures were optimized by mBEEF or RPBE. Then, the equilibrium lattice parameters were used to model the (111) surfaces by a two-dimensional slab with the periodic boundary conditions. Unless otherwise stated, the surfaces are modeled by 2 × 2 and 3 × 3 surface supercells to account for the adsorption coverages observed in experiments. Each slab is separated by at least a 20 Å vacuum perpendicular to the (111) surface (z-direction) to decrease the interaction between slab periodic images so that it can be ignored. Each slab consists of seven atom layers with the three bottom layers kept fixed at their ideal bulk positions during the geometry optimization. The atomic simulation environment (ASE) Python package48 is used to create surface slab models from the fully optimized bulk geometries.
2.3 Vibrational frequencies
Vibrational modes and frequencies of the C2H2 and C2H4 molecules in the gas phase and adsorbed on TM surfaces are computed by FHI-aims and Phonopy49 using a finite-differences approach to calculate second derivatives of forces. The absolute displacement of each atom for the finite difference calculations is 0.01 Å. The forces are converged within 1 meV Å−1 during the self-consistency cycle. To exclude the atomic displacements of the fixed layers in the slab models, we employ the modified version of the Phonopy–FHI-aims interface.502.4 Adsorption energies
The adsorption energy (Eads) is calculated using the following equation.| Eads = Emol+slab − Eslab − Emol | (1) |
The experimental adsorption energies (heat of adsorption) cited in the present study were measured by microcalorimetry. Thus, they should be compared to calculated adsorption enthalpy (Hads).
| Hads = Hmol+slab − Hslab − Hmol | (2) |
The adsorption of C2H4/Pt(111) can be dissociative, resulting in adsorbed ethylidyne and hydrogen:
| C2H4 + Pt(111) → (CCH3 + H)/Pt(111) | (3) |
3. Results and discussion
3.1 Geometry analysis
As a first step, we performed geometry optimizations for C2H2 and C2H4 in the gas phase and adsorbed on the TM surfaces, and the results were compared with the corresponding experimental data. The geometry optimizations are evaluated at 0 K without zero-point vibrational corrections. As shown in Table S1 in the ESI,† the calculated geometry parameters in the gas phase are in good agreement with the experimental results: the mean absolute errors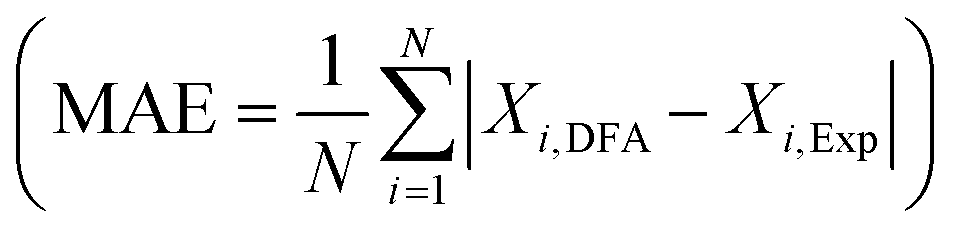 in calculated bond lengths are 8 × 10−3 and 6 × 10−3 Å for the mBEEF and RPBE functionals, respectively. For bond angles, the MAE for both functionals is 0.23°.
in calculated bond lengths are 8 × 10−3 and 6 × 10−3 Å for the mBEEF and RPBE functionals, respectively. For bond angles, the MAE for both functionals is 0.23°.
As described in the computational details, the slab models of TM surfaces were prepared from bulk structures obtained by minimizing the total energy at T = 0 K as listed in Table S2 of the ESI.† The results of bulk calculations show that both functionals accurately represent the experimental lattice constant of each metal. The mean absolute percentage errors  of RPBE and mBEEF are 1.342% and 0.589%, respectively. Although RPBE provides larger error in comparison with mBEEF, the difference is only 0.753% (the MAE of mBEEF and RPBE are 0.022 Å and 0.052 Å, respectively).
of RPBE and mBEEF are 1.342% and 0.589%, respectively. Although RPBE provides larger error in comparison with mBEEF, the difference is only 0.753% (the MAE of mBEEF and RPBE are 0.022 Å and 0.052 Å, respectively).
Thus, both RPBE and mBEEF provide accurate results for lattice constants of the metals in this study. We also investigated the thermal effects on the lattice constants by using the quasi-harmonic approximation (QHA) implemented in Phonopy.49,52 As shown in Table S2,† at the experimental temperature for the measurements (291–298 K),53,54 the change in the lattice constants induced by the thermal effects is rather small (the maximum difference from the value without the thermal effect is 0.067 Å).
Several different adsorption configurations of the molecules on the surfaces are studied. Fig. 1a illustrates the possible adsorption structures for C2H2 on a fcc(111) metal surface. According to theoretical and experimental literature, the preferred C2H2 adsorption site on Pd(111),55–57 Pt(111),58 and Rh(111)59 surfaces is the threefold fcc hollow site (h-fcc in Fig. 1a) with di-σ/π bonding (i.e., σ-bonding to two metal atoms along the C–C axis direction, and π-bonding to the remaining metal atom). However, the favorite site on Cu(111) is the bridge site (b-per in Fig. 1a) with the C–C axis perpendicular to the Cu–Cu bond.60 We investigated all the shown adsorption sites and found that the preferred sites obtained from both functionals are in agreement with previous theoretical and experimental studies cited above. In addition to the most stable configuration, we also looked for metastable adsorption configurations.
 | ||
| Fig. 1 Different adsorption sites for (a) C2H2 and (b) C2H4 on a fcc(111) metal surface. The CCH3 + H in (b) shows the adsorption structure of C2H4 in dissociative mode. Here, CCH3 and H adsorb at neighbouring h-fcc sites. Metal, carbon, and hydrogen atoms are shown as purple, black, and white spheres, respectively. | ||
According to available literature, the b-par (known as di-σ bonding) and top (known as π bonding) are the configurations for C2H2 on Pd(111) and Pt(111), which can be present at finite temperature and/or coverage. Based on our RPBE results shown in Table 1 and Table S6 of the ESI,† we obtain the following order of stability (starting from the most stable site with lowest adsorption energy): h-fcc, b-par, and top. This order is in agreement with other studies.59,61–65 However, we find that geometry optimization with mBEEF misses some configurations that can be obtained by RPBE, such as the top configurations on Pd(111) and Cu(111).
| System | Site | θ (ML) | mBEEF | RPBE | Exp. | Other DFA studies |
|---|---|---|---|---|---|---|
| C2H2/Pt(111) | h-fcc | 1/4 | −2.67 | −1.89 | −2.37 (PW91),59 −2.26 (PW91)66 | |
| 1/9 | −2.70 | −1.83 | −2.11 (BEEF-vdW)67 | |||
| 1/16 | −2.87 | −2.01 | −2.18 (173 K)68 | |||
| [−2.64 (173 K)] | [−1.79 (173 K)] | |||||
| b-par | 1/4 | −2.09 | −1.47 | −1.99 (PW91)59 | ||
| Top | 1/4 | −0.68 | −0.23 | −0.73 (PW91)59 | ||
| C2H4/Pt(111) | Top | 1/4 | −0.80 | −0.28 | −0.41 ± 0.1 (112 K)69 | −0.76 (PW91),61 −0.55 (PW)62 |
| [−0.69 (112 K)] | [−0.18 (112 K)] | |||||
| [−0.70 (110 K)] | [−0.19 (110 K)] | |||||
| −0.22–−0.39 (110 K)70 | ||||||
| b-par | 1/4 | −1.36 | −0.74 | −0.39–−0.74 (110 K)70 | −1.21 (PW91),61 −1.26 (PW)62 | |
| [−1.23 (110 K)] | [−0.62 (110 K)] | −0.74 (100 K)71 | ||||
| [−1.23 (100 K)] | [−0.62 (100 K)] | |||||
| CCH3 + H | 1/9 | −1.78 | −1.12 | −1.36 (300 K)72 | −1.65 (PBE),15 −1.74 (PBE),72 −1.20 (RPBE),15 −1.75 (optPBE),72 −1.83 (optPBE-vdW),15 −1.34 (BEEF-vdW),15 −1.43 (BEEF),72 −1.68 (MS2),15 −2.21 (SCAN),15 −2.33 (SCAN + rVV10),15 −2.21 (HSE06),15 −1.44 (PW91)61 | |
| [−1.40 (300 K)] | [−0.81 (300 K)] | −1.24 (300 K)73 |
Finally, the adsorption of C2H4 was investigated. C2H4 can also adsorb dissociatively on TM surfaces: ethylidyne (CCH3) and H. For the adsorption of C2H4/Pt(111), all the possible adsorption sites shown in Fig. 1b were examined. By employing both functionals, we find that the b-par site with a di-σ bonding is the global minimum for the adsorption of the C2H4 molecule for both functionals, which is in agreement with other theoretical and experimental studies.31,66,71,74
3.2 Vibrational frequencies
The vibrational frequencies of C2H2 and C2H4 both in the gas phase and adsorbed on the TM surfaces are computed with mBEEF and RPBE functionals. For the molecules in the gas phase, the mBEEF functional tends to overestimate the vibrational frequencies, and the MPE of mBEEF (3.04%) is larger than that of RPBE (0.12%). In particular, the C–C and C–H stretching modes calculated by mBEEF show a large deviation from the experimental results. More details of the obtained results are discussed in Sections S3 and S4 of the ESI.†For the molecules adsorbed on each metal, we used the adsorption site reported for the corresponding experiments. Except for ref. 38 and 39 where Fourier-transform reflection-adsorption infrared (FT-RAIR) spectroscopy was employed, the other experimental vibrational frequencies used in our study are obtained by high-resolution electron energy loss spectroscopy (HREELS). The calculated vibrational frequencies are summarized in Table S5 of the ESI.† Although experimental studies of vibrational spectroscopy report the configuration of the adsorbed molecule on the surface and the chamber exposure, the coverages are rarely mentioned. Therefore, in this study, the effect of the coverage on the calculated vibrational frequencies is investigated by comparing the results for two coverages: 1/4 and 1/9 monolayer (ML). These selected coverages are close to the C2H2 and C2H4 gas exposures reported in the experimental references (see Table S4 of the ESI†). As can be seen in Fig. 2, the obtained results are altogether only little affected by the change of coverage in the considered range. The MAE of mBEEF at both coverages is 66 cm−1 and that of RPBE slightly increases from 42 cm−1 to 43 cm−1 with increasing coverage. Therefore, in the following, we only discuss the results of 1/9 ML.
 | ||
Fig. 2 Deviations of calculated vibrational frequencies for C2H2 and C2H4 molecules adsorbed on fcc(111) TM surfaces from corresponding experimental values. ν, δ and γ denote stretching mode, in-plane bending mode, and out-of-plane bending mode, respectively. The blue squares and pink triangles indicate the deviations obtained with mBEEF and RPBE XC functionals, respectively (see Table S5 of the ESI† for the values of vibrations and experimental references). The mean percentage error is calculated by using the following formula: 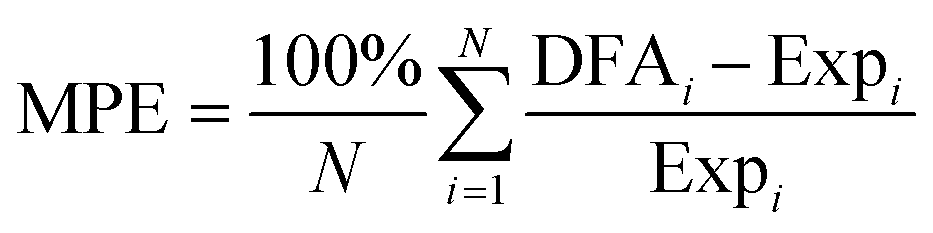 . Some vibrations are absent either because of experimental method limitations or because only the most intense modes were listed in the references. . Some vibrations are absent either because of experimental method limitations or because only the most intense modes were listed in the references. | ||
According to Table S5,† the values of all the experimental vibrational frequencies assigned to the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C stretching mode for chemisorbed C2H2 display a large shift from 1974 cm−1 (in the gas phase) to 1260–1402 cm−1 (adsorbed on TM surfaces). This frequency shift is a consequence of the partial rehybridization of the CC triple bond from sp to sp2 or sp3 hybrid orbitals due to the formation of the covalent bond between carbon atoms of C2H2 and the metal surfaces. Our mBEEF and RPBE results also show such a large shift from 2082 and 2001 to 1295–1353 and 1238–1303 cm−1, respectively, and the range of the shift depends on the adsorption configurations. For other modes, the shift in vibrational frequencies relative to the free molecules occurs as well, but it is not as strong as for the νCC mode. Similar results are obtained for the νCC mode of C2H4 adsorbed on Pt(111) and Pd(111). For this molecule, the absolute changes of the frequencies are 573 and 520 cm−1 at Pt(111) and Pd(111), respectively. Both mBEEF and RPBE XC functionals reproduce this large drop in frequency. The absolute changes obtained with mBEEF are 674 and 595 cm−1 at Pt(111) and Pd(111), respectively. The results from RPBE give absolute changes of 651 and 544 cm−1 at Pt(111) and Pd(111), respectively. Thus, the absolute changes of the frequencies obtained from RPBE are closer to the experimental changes. Therefore, according to our results, the adsorbate–substrate bonding is described more accurately by RPBE.
C stretching mode for chemisorbed C2H2 display a large shift from 1974 cm−1 (in the gas phase) to 1260–1402 cm−1 (adsorbed on TM surfaces). This frequency shift is a consequence of the partial rehybridization of the CC triple bond from sp to sp2 or sp3 hybrid orbitals due to the formation of the covalent bond between carbon atoms of C2H2 and the metal surfaces. Our mBEEF and RPBE results also show such a large shift from 2082 and 2001 to 1295–1353 and 1238–1303 cm−1, respectively, and the range of the shift depends on the adsorption configurations. For other modes, the shift in vibrational frequencies relative to the free molecules occurs as well, but it is not as strong as for the νCC mode. Similar results are obtained for the νCC mode of C2H4 adsorbed on Pt(111) and Pd(111). For this molecule, the absolute changes of the frequencies are 573 and 520 cm−1 at Pt(111) and Pd(111), respectively. Both mBEEF and RPBE XC functionals reproduce this large drop in frequency. The absolute changes obtained with mBEEF are 674 and 595 cm−1 at Pt(111) and Pd(111), respectively. The results from RPBE give absolute changes of 651 and 544 cm−1 at Pt(111) and Pd(111), respectively. Thus, the absolute changes of the frequencies obtained from RPBE are closer to the experimental changes. Therefore, according to our results, the adsorbate–substrate bonding is described more accurately by RPBE.
Comparing the calculated vibrational frequencies with the corresponding experimental values illustrated in Fig. 2 shows that the calculated stretching modes display the largest deviations, and the mBEEF functional tends to overestimate the frequencies. These trends are similar to the results for free molecules as discussed in Section S4 in the ESI.† For each adsorbed system, the RMSE for both functionals is also calculated. As shown in Fig. 2, the RMSE of RPBE is smaller than that of mBEEF in all the TM systems studied here. For the adsorbed C2H2 at all the four fcc(111) surfaces, the values of RMSE obtained with mBEEF and RPBE are 98 and 70 cm−1, respectively (see Fig. 2a–d). For the case of C2H4 over the Pt and Pd surfaces (see Fig. 2e and f), the RMSEs of mBEEF and RPBE are 80 and 41 cm−1, respectively. Altogether, the overall RMSEs for the studied systems (i.e., 36 vibrational modes listed in Table S5†) are 118 cm−1 and 61 cm−1 for mBEEF and RPBE XC functionals, respectively. The highest achieved resolution of HREELS reported before 2010, when the reference values adopted in this study were measured, was 60–80 cm−1.31,75 Thus, the RPBE RMSE of 61 cm−1 is within the range of the reported HREELS resolution.
3.3 Adsorption energies of C2H2 and C2H4
Let us now examine the performance of the two XC functionals for adsorption enthalpy by comparing with the experimental heat of adsorption measured by microcalorimetry. The Hads values of C2H2 and C2H4 on Pt(111) are computed and summarized in Table 1 along with the available experimental data and those from other studies. Ideally, the performance of XCs for the heats of adsorption should be assessed for multiple systems, and the results should be compared with the performance for the vibrational frequencies. However, we only did it for Pt because the measured data of the heat of adsorption are not available for other systems to the best of our knowledge.For C2H2 adsorbed at h-fcc site on Pt(111), the absolute adsorption energy differences (|HDFT − HExp.|) obtained from mBEEF and RPBE with the experimental value (−2.18 eV)68 are 0.46 and 0.39 eV, respectively. Thus, the adsorption enthalpy obtained with both functionals deviates from experiment, but the RPBE results are closer to experiment. Our RPBE results are also closer to those obtained by BEEF-vdW67 and PW91.59,66 According to the experimental and theoretical studies summarized in Table 1, for the adsorption of C2H4/Pt(111) without dissociation, the molecule can adsorb at top and b-par sites. The RPBE adsorption enthalpy is closer to the experimental values than mBEEF adsorption enthalpy. The absolute difference of adsorption enthalpy obtained with RPBE and mBEEF for the top configuration in comparison with the experimental value of −0.41 eV (ref. 69) is 0.23 and 0.28 eV, respectively. For the molecular adsorption of C2H4/Pt(111) on the b-par site, the obtained result from RPBE is in the range of the reported experimental values, while the value obtained from the mBEEF functional is largely overestimated (see Table 1). In the case of the dissociative adsorption of C2H4/Pt(111) to form ethylidyne (CCH3) and hydrogen, the energy differences in the adsorption energies calculated with RPBE and mBEEF and available experimental results (−1.36 eV)72 are 0.55 and 0.04 eV, respectively. Thus, for the dissociative adsorption of C2H4/Pt(111), Hads calculated by mBEEF is closer to the experimental value than that calculated by RPBE. Table 1 also summarizes other theoretical values reported in the literature: RPBE, BEEF-vdW, BEEF, and PW91 show good agreement with the reported experimental values. In order to analyze the general performance of the two functionals, we plot in Fig. 3 the computed values of Hadsversus the experimental ones. As can be seen in the figure, RPBE with RMSE of 0.32 eV for adsorption enthalpy displays a better agreement with the experimental results. mBEEF tends to overestimate Hads. This systematic tendency towards the overestimation of the adsorption enthalpy is confirmed by the obtained MPE values, also shown in Fig. 3: MPE = −59.81% (mBEEF) and 27.61% (RPBE). The measurement error for the heat of adsorption cited in the present work is 0.085–0.175 eV. Thus, the RMSEs for Hads of the RPBE and mBEEF XC functionals are larger than the experimental error.
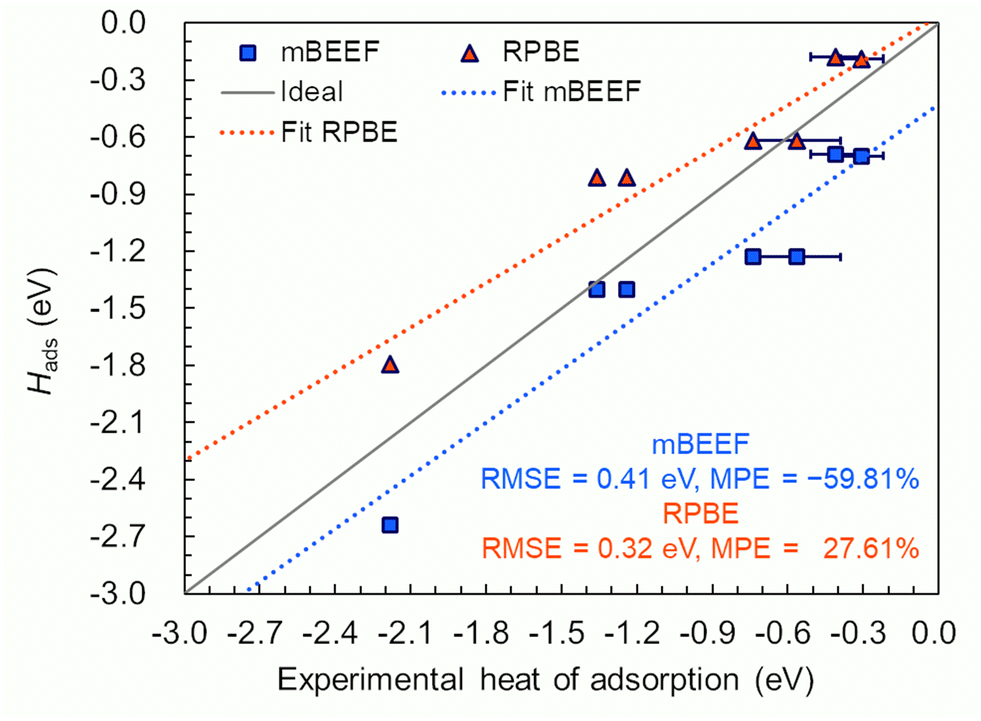 | ||
Fig. 3 Calculated adsorption enthalpy (Hads) at temperatures listed in Table 1 of acetylene and ethylene on the Pt(111) surface versus corresponding experimental values. Dotted lines show least-squares-fit for the mBEEF and RPBE results. The gray line shows the ideal match between theory and experiment. In cases when a range of experimental adsorption energies were given, the average value along with the error bars are included in the plot. The mean percentage error is calculated by using the following formula:  . . | ||
Finally, we also assessed the performance of two additional XC functionals and the influence of vdW corrections. In particular, we calculated vibrational frequencies and adsorption enthalpies for the C2H4/Pt(111) system with PBEsol and TPSS. The details of this analysis are discussed in Section S7 in the ESI.† TPSS is reported as the reliable functional for the bulk and surface properties of metals,17 and PBEsol is also a widely used functional for solid systems. Additionally, for the metal–adsorbate systems, the importance of adding the vdW correction to the GGA functionals has been discussed.76–78 Thus, we also assess the performance of the RPBE functional with the Tkatchenko–Scheffler (TS) vdW correction. As shown in Fig. S3a and Table S7,† PBEsol shows the lowest RMSE for the vibrational frequency (30 cm−1). TPSS shows a moderate performance among the adopted XC functionals (RMSE = 61 cm−1), and there is no significant effect of the vdW correction on vibrational frequency in RPBE + TS (RMSE increased by 2 cm−1). RPBE shows the second best RMSE for the vibrational frequencies (42 cm−1), but mBEEF shows the highest RMSE among the considered XC functionals (83 cm−1). On the other hand, as shown in Fig. S3b,† RPBE + TS overestimates the adsorption enthalpy by 0.47 eV. This overestimation is at least partly due to overestimation of long-range vdW interaction with metal surfaces by pairwise dispersion interaction models.79 Additionally, PBEsol shows the highest error among the considered functionals (0.94 eV) while TPSS shows a moderate performance (error = 0.35 eV). For the adsorption enthalpy, RPBE shows the best performance (error = 0.05 eV), but mBEEF shows the second worst performance (error = 0.66 eV). The reaction enthalpy of the semi-hydrogenation of C2H2 to C2H4 in the gas phase was also calculated with all considered functionals. Interestingly, the accuracy trend across functionals is the same as the one for the adsorption enthalpy (Fig. S3c†).
Because RPBE shows high performance for all considered properties (assuming that the long-range vdW correction weakly depends on the specific functional among the considered ones), our study shows that RPBE is the most reliable functional for the considered system. On the other hand, PBEsol shows good accuracy for the vibrational frequency, but its accuracy for the adsorption and reaction enthalpy is lowest among the functionals. These results suggest that, if possible, the reliability of XC functionals should be assessed by investigating the accuracy for several physical quantities. However, in practice, we are limited by the scarcity of reliable experimental data, such as vibrational frequencies, for the metal–adsorbate systems.
We note that modelling the full catalytic progression by DFA and statistical mechanics is impractical, since catalysis is governed by an intricate interplay of several underlying processes, such as the surface reactions, the material restructuring under reaction conditions, and the transport of reactants and products. However, DFA can be used to obtain possibly relevant descriptive parameters correlated with atomistic processes. These parameters can be combined with experimental data in order to model catalysis via artificial intelligence (AI).30 Indeed, we have recently considered such an AI approach for identifying the key descriptive parameters correlated to the experimental performance, out of many offered candidate descriptive parameters obtained from theory and/or experiment.30,80–82 In analogy to genes in biology, these key parameters might be called “material genes” of catalysis,80 as they correlate with the processes triggering, favouring or hindering the catalytic performance without providing the full understanding of the underlying processes.
Conclusions
In this study, we have evaluated the performance of mBEEF and RPBE functionals by mainly focusing on the vibrational frequencies of C2H2 and C2H4 both in the gas phase and adsorbed on TM surfaces. The experimentally measured frequencies are available for multiple systems such as the gas-phase molecules and the molecules adsorbed on Pt, Pd, Rh, and Cu surfaces (in total 36 data points). According to our vibrational frequency analysis for the molecules in the gas phase, the RMSEs obtained with RPBE are smaller than those obtained with mBEEF by 79 cm−1 for C2H2 and 58 cm−1 for C2H4. For the adsorbed molecules, we studied different adsorption structures at each TM surface. Our results show that the mBEEF functional misses some metastable adsorption structures. Moreover, we find that for vibrational frequencies, mBEEF, despite being in relatively good agreement with experimental data (RMSE = 118 cm−1), performs worse than RPBE (RMSE = 61 cm−1) for the considered systems. The accuracy of the functionals is also evaluated by studying the adsorption energy of the molecules on the Pt(111) surfaces. This study also shows that RPBE is more accurate than mBEEF. RMSEs of 0.41 eV and 0.32 eV are obtained for adsorption enthalpy with mBEEF and RPBE, respectively.To demonstrate further application of our approach, the adsorption enthalpy and vibrational frequencies of the C2H4/Pt(111) system were also investigated with TPSS, PBEsol, and RPBE with the Tkatchenko–Scheffler pairwise long-range vdW correction. The performance of these functionals is worse than that of RPBE. Our results indicate that RPBE shows high reliability in describing the interaction between the metal surfaces and C2H2 or C2H4. Our study has demonstrated that the measured vibrational frequencies can be utilized for assessing the accuracy of the XC functionals. Such assessment is particularly important for the application of the DFA data to building an AI model for heterogeneous catalysis.
Data availability
Part of the data for this article is available at NOMAD (upload ID: oiPDzxLcTlClT6CAeiMsKg).Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the discussion with our colleagues from the Max Planck-Cardiff Centre on the Fundamentals of Heterogeneous Catalysis (FUNCAT). This work received funding from the European Union's Horizon 2020 Research and Innovation Programme (grant agreement No. 951786), the NOMAD CoE. The work was partially supported by the Max-Planck Network on BigData-Driven Materials Science (BiGmax). We gratefully acknowledge the scientific support and HPC resources provided by the Erlangen National High Performance Computing Center (NHR@FAU) of the Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU) under the NHR project ID c100ba. NHR funding is provided by federal and Bavarian state authorities. NHR@FAU hardware is partially funded by the German Research Foundation (DFG) – 440719683. A part of the computation was also performed at the Research Center for Computational Science, Okazaki, Japan (project: 24-IMS-C002), and the Center for Computational Materials Science, Institute for Materials Research, Tohoku University is acknowledged for the use of MASAMUNE-IMR (project No. 202403-SCKXX-0026). R. M. acknowledges the support by the Joint Usage/Research Center for Catalysis (Proposal 22AY0053), the MEXT project “Integrating Research Consortium on Chemical Science”, and JSPS KAKENHI Grant Number JP24K17554. S. F. acknowledges the Alexander von Humboldt Foundation for financial support. S. V. L. is grateful for the support by RSCF grant 24-13-00317. Open Access funding provided by the Max Planck Society.Notes and references
- M. Niu, Y. Wang, W. Li, J. Jiang and Z. Jin, Catal. Commun., 2013, 38, 77–81 CrossRef CAS.
- K. K. Tanabe, M. S. Ferrandon, N. A. Siladke, S. J. Kraft, G. Zhang, J. Niklas, O. G. Poluektov, S. J. Lopykinski, E. E. Bunel, T. R. Krause, J. T. Miller, A. S. Hock and S. T. Nguyen, Angew. Chem., Int. Ed., 2014, 53, 12055–12058 CrossRef CAS PubMed.
- O. B. Ayodele, R. Cai, J. Wang, Y. Ziouani, Z. Liang, M. C. Spadaro, K. Kovnir, J. Arbiol, J. Akola, R. E. Palmer and Y. V. Kolen'ko, ACS Catal., 2020, 10, 451–457 CrossRef CAS.
- Z. Chen, C. Cai and T. Wang, J. Phys. Chem. C, 2022, 126, 3037–3042 CrossRef CAS.
- M. Bowker, S. DeBeer, N. F. Dummer, G. J. Hutchings, M. Scheffler, F. Schüth, S. H. Taylor and H. Tüysüz, Angew. Chem., Int. Ed., 2022, 61, e202209016 CrossRef CAS PubMed.
- K. S. Kley, J. De Bellis and F. Schüth, Catal. Sci. Technol., 2023, 13, 119–131 RSC.
- I.-T. Trotuş, T. Zimmermann and F. Schüth, Chem. Rev., 2014, 114, 1761–1782 CrossRef PubMed.
- F. Studt, F. Abild-Pedersen, T. Bligaard, R. Z. Sørensen, C. H. Christensen and J. K. Nørskov, Science, 2008, 320, 1320–1322 CrossRef CAS PubMed.
- P. Hohenberg and W. Kohn, Phys. Rev. A, 1964, 136, B864–B871 CrossRef.
- W. Kohn and L. J. Sham, Phys. Rev. A, 1965, 140, A1133–A1138 CrossRef.
- J. K. Nørskov, F. Abild-Pedersen, F. Studt and T. Bligaard, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 937–943 CrossRef PubMed.
- C. J. Cramer and D. G. Truhlar, Phys. Chem. Chem. Phys., 2009, 11, 10757–10816 RSC.
- B.-T. Teng, X.-D. Wen, M. Fan, F.-M. Wu and Y. Zhang, Phys. Chem. Chem. Phys., 2014, 16, 18563–18569 RSC.
- J. Wellendorff, T. L. Silbaugh, D. Garcia-Pintos, J. K. Nørskov, T. Bligaard, F. Studt and C. T. Campbell, Surf. Sci., 2015, 640, 36–44 CrossRef CAS.
- S. Mallikarjun Sharada, R. K. B. Karlsson, Y. Maimaiti, J. Voss and T. Bligaard, Phys. Rev. B, 2019, 100, 035439 CrossRef.
- B. Hammer, L. B. Hansen and J. K. Nørskov, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 7413–7421 CrossRef.
- L. Kabalan, I. Kowalec, C. R. A. Catlow and A. J. Logsdail, Phys. Chem. Chem. Phys., 2021, 23, 14649–14661 RSC.
- J. Wellendorff, K. T. Lundgaard, K. W. Jacobsen and T. Bligaard, J. Chem. Phys., 2014, 140, 144107 CrossRef PubMed.
- E. Espí, A. Salmerón, A. Fontecha, Y. García and A. I. Real, J. Plast. Film Sheeting, 2006, 22, 85–102 CrossRef.
- H. Yue, Y. Zhao, X. Ma and J. Gong, Chem. Soc. Rev., 2012, 41, 4218–4244 RSC.
- J. Wu, Z. Wu, Y. Wei, H. Ding, W. Huang, X. Gui, W. Shi, Y. Shen, K. Tao and X. Xie, ACS Appl. Mater. Interfaces, 2020, 12, 19069–19079 CrossRef CAS PubMed.
- K. Xie, K. Xu, M. Liu, X. Song, S. Xu and H. Si, Materials Today Catalysis, 2023, 3, 100029 CrossRef.
- X. Cao, B. W.-L. Jang, J. Hu, L. Wang and S. Zhang, Molecules, 2023, 28, 2572 CrossRef CAS PubMed.
- C. S. Spanjers, J. T. Held, M. J. Jones, D. D. Stanley, R. S. Sim, M. J. Janik and R. M. Rioux, J. Catal., 2014, 316, 164–173 CrossRef CAS.
- L. Ye, X. Duan, S. Wu, T.-S. Wu, Y. Zhao, A. W. Robertson, H.-L. Chou, J. Zheng, T. Ayvalı, S. Day, C. Tang, Y.-L. Soo, Y. Yuan and S. C. E. Tsang, Nat. Commun., 2019, 10, 914 CrossRef PubMed.
- Y. Cao, H. Zhang, S. Ji, Z. Sui, Z. Jiang, D. Wang, F. Zaera, X. Zhou, X. Duan and Y. Li, Angew. Chem., Int. Ed., 2020, 59, 11647–11652 CrossRef CAS PubMed.
- Y. Wang, W. Zheng, B. Wang, L. Ling and R. Zhang, Chem. Eng. Sci., 2021, 229, 116131 CrossRef CAS.
- W. Zheng, Y. Wang, B. Wang, M. Fan, L. Ling and R. Zhang, J. Phys. Chem. C, 2021, 125, 15251–15261 CrossRef CAS.
- A. Mazheika, Y. G. Wang, R. Valero, F. Vines, F. Illas, L. M. Ghiringhelli, S. V. Levchenko and M. Scheffler, Nat. Commun., 2022, 13, 419 CrossRef CAS PubMed.
- R. Miyazaki, K. S. Belthle, H. Tüysüz, L. Foppa and M. Scheffler, J. Am. Chem. Soc., 2024, 146, 5433–5444 CrossRef CAS PubMed.
- H. Ibach and S. Lehwald, J. Vac. Sci. Technol., 1978, 15, 407–415 CrossRef CAS.
- P. Y. Timbrell, A. J. Gellman, R. M. Lambert and R. F. Willis, Surf. Sci., 1988, 206, 339–347 CrossRef CAS.
- J. A. Gates and L. L. Kesmodel, Surf. Sci., 1983, 124, 68–86 CrossRef CAS.
- C. M. Mate, C. T. Kao, B. E. Bent and G. A. Somorjai, Surf. Sci., 1988, 197, 183–207 CrossRef CAS.
- B. J. Bandy, M. A. Chesters, M. E. Pemble, G. S. McDougall and N. Sheppard, Surf. Sci., 1984, 139, 87–97 CrossRef CAS.
- M. A. Chesters and E. M. McCash, J. Electron Spectrosc. Relat. Phenom., 1987, 44, 99–108 CrossRef CAS.
- H. Steininger, H. Ibach and S. Lehwald, Surf. Sci., 1982, 117, 685–698 CrossRef CAS.
- D. Stacchiola, F. Calaza, T. Zheng and W. T. Tysoe, J. Mol. Catal. A: Chem., 2005, 228, 35–45 CrossRef CAS.
- M. Sock, A. Eichler, S. Surnev, J. N. Andersen, B. Klötzer, K. Hayek, M. G. Ramsey and F. P. Netzer, Surf. Sci., 2003, 545, 122–136 CrossRef CAS.
- J. P. Perdew, A. Ruzsinszky, G. I. Csonka, O. A. Vydrov, G. E. Scuseria, L. A. Constantin, X. Zhou and K. Burke, Phys. Rev. Lett., 2008, 100, 136406 CrossRef PubMed.
- J. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed.
- A. Tkatchenko and M. Scheffler, Phys. Rev. Lett., 2009, 102, 073005 CrossRef PubMed.
- V. Blum, R. Gehrke, F. Hanke, P. Havu, V. Havu, X. Ren, K. Reuter and M. Scheffler, Comput. Phys. Commun., 2009, 180, 2175–2196 CrossRef CAS.
- S. Lehtola, C. Steigemann, M. J. T. Oliveira and M. A. L. Marques, SoftwareX, 2018, 7, 1–5 CrossRef.
- E. van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1994, 101, 9783–9792 CrossRef CAS.
- J. Nocedal and S. J. Wright, Numerical Optimization, Springer, New York, NY, 2th edn, 2006 Search PubMed.
- A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner, G. Ceder and K. A. Persson, APL Mater., 2013, 1, 011002 CrossRef.
- A. Hjorth Larsen, J. Jørgen Mortensen, J. Blomqvist, I. E. Castelli, R. Christensen, M. Dułak, J. Friis, M. N. Groves, B. Hammer, C. Hargus, E. D. Hermes, P. C. Jennings, P. Bjerre Jensen, J. Kermode, J. R. Kitchin, E. Leonhard Kolsbjerg, J. Kubal, K. Kaasbjerg, S. Lysgaard, J. Bergmann Maronsson, T. Maxson, T. Olsen, L. Pastewka, A. Peterson, C. Rostgaard, J. Schiøtz, O. Schütt, M. Strange, K. S. Thygesen, T. Vegge, L. Vilhelmsen, M. Walter, Z. Zeng and K. W. Jacobsen, J. Phys.: Condens. Matter, 2017, 29, 273002 CrossRef PubMed.
- A. Togo and I. Tanaka, Scr. Mater., 2015, 108, 1–5 CrossRef CAS.
- https://github.com/fidanyan/phonopy .
- I. Kowalec, L. Kabalan, C. R. A. Catlow and A. J. Logsdail, Phys. Chem. Chem. Phys., 2022, 24, 9360–9373 RSC.
- A. Togo, L. Chaput, I. Tanaka and G. Hug, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 174301 CrossRef.
- K. Hellwege, A. Hellwege, B. Eisenmann and H. Schaefer, Structure data of elements and intermetallic phases, Springer, 1971 Search PubMed.
- G. Grube, A. Schneider and U. Esch, Heraeus Festschrift, 1951 Search PubMed.
- L. Lie-Quan and C. Pei-Lin, J. Phys.: Condens. Matter, 1996, 8, 3313 CrossRef.
- J. C. Dunphy, M. Rose, S. Behler, D. F. Ogletree, M. Salmeron and P. Sautet, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, R12705–R12708 CrossRef CAS.
- C. Matsumoto, Y. Kim, T. Okawa, Y. Sainoo and M. Kawai, Surf. Sci., 2005, 587, 19–24 CrossRef CAS.
- T. Okada, Y. Kim, M. Trenary and M. Kawai, J. Phys. Chem. C, 2012, 116, 18372–18381 CrossRef CAS.
- J. W. Medlin and M. D. Allendorf, J. Phys. Chem. B, 2003, 107, 217–223 CrossRef CAS.
- S. Bao, K. M. Schindler, P. Hofmann, V. Fritzsche, A. M. Bradshaw and D. P. Woodruff, Surf. Sci., 1993, 291, 295–308 CrossRef CAS.
- R. M. Watwe, R. D. Cortright, M. Mavrikakis, J. K. Nørskov and J. A. Dumesic, J. Chem. Phys., 2001, 114, 4663–4668 CrossRef CAS.
- Q. Ge and D. A. King, J. Chem. Phys., 1999, 110, 4699–4702 CrossRef CAS.
- R. M. Watwe, B. E. Spiewak, R. D. Cortright and J. A. Dumesic, J. Catal., 1998, 180, 184–193 CrossRef CAS.
- Y.-T. Wong and R. Hoffmann, J. Chem. Soc., Faraday Trans., 1990, 86, 4083–4094 RSC.
- H. A. Aleksandrov, L. V. Moskaleva, Z. J. Zhao, D. Basaran, Z. X. Chen, D. H. Mei and N. Rösch, J. Catal., 2012, 285, 187–195 CrossRef CAS.
- F. Mittendorfer, C. Thomazeau, P. Raybaud and H. Toulhoat, J. Phys. Chem. B, 2003, 107, 12287–12295 CrossRef CAS.
- Z.-J. Zhao, J. Zhao, X. Chang, S. Zha, L. Zeng and J. Gong, AIChE J., 2019, 65, 1059–1066 CrossRef CAS.
- B. E. Spiewak, R. D. Cortright and J. A. Dumesic, J. Catal., 1998, 176, 405–414 CrossRef CAS.
- J. Kubota, S. Ichihara, J. N. Kondo, K. Domen and C. Hirose, Surf. Sci., 1996, 357-358, 634–638 CrossRef CAS.
- M. Salmeron and G. A. Somorjai, J. Phys. Chem., 1982, 86, 341–350 CrossRef CAS.
- J. M. Essen, J. Haubrich, C. Becker and K. Wandelt, Surf. Sci., 2007, 601, 3472–3480 CrossRef CAS.
- S. Gautier, S. N. Steinmann, C. Michel, P. Fleurat-Lessard and P. Sautet, Phys. Chem. Chem. Phys., 2015, 17, 28921–28930 RSC.
- W. A. Brown, R. Kose and D. A. King, Chem. Rev., 1998, 98, 797–832 CrossRef CAS PubMed.
- E. A. Carter and B. E. Koel, Surf. Sci., 1990, 226, 339–357 CrossRef CAS.
- O. L. Krivanek, N. Dellby, J. A. Hachtel, J. C. Idrobo, M. T. Hotz, B. Plotkin-Swing, N. J. Bacon, A. L. Bleloch, G. J. Corbin, M. V. Hoffman, C. E. Meyer and T. C. Lovejoy, Ultramicroscopy, 2019, 203, 60–67 CrossRef CAS PubMed.
- R. B. Araujo, G. L. S. Rodrigues, E. C. Dos Santos and L. G. M. Pettersson, Nat. Commun., 2022, 13, 6853 CrossRef CAS PubMed.
- J. B. Davis, F. Baletto and R. L. Johnston, J. Phys. Chem. A, 2015, 119, 9703–9709 CrossRef CAS PubMed.
- E. O. Bartaquim, R. C. Bezerra, A. F. B. Bittencourt and J. L. F. Da Silva, Phys. Chem. Chem. Phys., 2022, 24, 20294–20302 RSC.
- W. Liu, A. Tkatchenko and M. Scheffler, Acc. Chem. Res., 2014, 47, 3369–3377 CrossRef CAS PubMed.
- L. Foppa, L. M. Ghiringhelli, F. Girgsdies, M. Hashagen, P. Kube, M. Hävecker, S. J. Carey, A. Tarasov, P. Kraus, F. Rosowski, R. Schlögl, A. Trunschke and M. Scheffler, MRS Bull., 2021, 46, 1016–1026 CrossRef CAS PubMed.
- L. Foppa, F. Rüther, M. Geske, G. Koch, F. Girgsdies, P. Kube, S. J. Carey, M. Hävecker, O. Timpe, A. V. Tarasov, M. Scheffler, F. Rosowski, R. Schlögl and A. Trunschke, J. Am. Chem. Soc., 2023, 145, 3427–3442 CrossRef CAS PubMed.
- L. Foppa, C. Sutton, L. M. Ghiringhelli, S. De, P. Löser, S. A. Schunk, A. Schäfer and M. Scheffler, ACS Catal., 2022, 12, 2223–2232 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Geometry analysis for the C2H2 and C2H4 molecules in the gas phase, bulk lattice constants of each metal, vibrational frequencies of the molecules in the gas phase and adsorbed on the metal surfaces, adsorption energies on Pd(111), and comparison with other XC functionals for the C2H4/Pt(111) system. See DOI: https://doi.org/10.1039/d4cy00685b |
| ‡ These authors contributed equally: Ray Miyazaki and Somayeh Faraji. |
| This journal is © The Royal Society of Chemistry 2024 |