
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUltrasmall RuO2/CoFe2O4 nanoparticles with robust interfacial interactions for the enhanced acidic oxygen evolution reaction†
Ming
Wei
ab,
Liuhua
Mu
 bc,
Zhiwei
Liu
b,
Feng
Gao
b,
Guangjian
Song
ab,
Qiankang
Si
b,
Mao
Zhang
a,
Fangfang
Dai
b,
Min
Zhang
b,
Rui
Ding
b,
Li
Yang
*a,
Zhonggui
Gao
*a and
Sanzhao
Song
*b
bc,
Zhiwei
Liu
b,
Feng
Gao
b,
Guangjian
Song
ab,
Qiankang
Si
b,
Mao
Zhang
a,
Fangfang
Dai
b,
Min
Zhang
b,
Rui
Ding
b,
Li
Yang
*a,
Zhonggui
Gao
*a and
Sanzhao
Song
*b
aCollege of Physics and Technology, Guangxi Normal University, Guilin, 541004, China
bWenzhou Institute, University of Chinese Academy of Sciences, Wenzhou, Zhejiang 325001, China. E-mail: songsanzhao@ucas.ac.cn
cSchool of Physical Science, University of Chinese Academy of Sciences, Beijing 100049, China
First published on 21st October 2024
Abstract
The acidic stability of RuO2-based electrocatalysts remains a critical hurdle for proton exchange membrane electrolyzers due to ruthenium leaching. Here, we report an ultrasmall RuO2/CoFe2O4 (RFC) catalyst with robust interfacial interactions, synthesized via an adsorption–pyrolysis method. The RFC catalyst demonstrates an exceptionally low overpotential of 191 mV and outstanding stability, retaining its performance for over 100 hours in 0.5 M H2SO4. Experimental analyses indicate that the robust interfacial interactions between RuO2 and CoFe2O4 facilitate efficient charge transfer, significantly enhancing the performance of the oxygen evolution reaction (OER). After the stability test, XRD, Raman, and TEM characterization confirmed that the RFC catalyst maintains its crystal structure and morphology, indicating excellent durability. These findings highlight the potential of RFC catalysts for sustainable hydrogen production and provide a novel approach to the design of advanced electrocatalysts through strategic interfacial engineering, paving the way for improved stability and performance in acidic OER applications.
1. Introduction
The increasing global demand for clean and renewable energy has made the development of efficient and stable electrocatalysts for water splitting, which is a crucial technology for sustainable hydrogen production.1–5 In water electrolysis, the oxygen evolution reaction (OER) at the anode is a critical step, yet its reaction kinetics are hindered by the complex four-electron transfer mechanism and high overpotential requirements.6,7 This challenge is exacerbated under acidic conditions, where OER catalysts must exhibit excellent performance and long-term stability in highly corrosive electrolytes.8,9 Proton exchange membrane water electrolyzers (PEMWEs) are considered ideal for large-scale hydrogen production due to their high energy efficiency and compact design.10 However, developing highly efficient OER electrocatalysts that can operate stably in acidic environments remains a core challenge.11RuO2 is widely recognized as one of the most promising catalysts for the OER due to its excellent electronic properties and catalytic activity in acidic media.12 However, its limited stability, particularly under long-term acidic operating conditions, remains a major challenge.13 Based on previous studies, the instability of RuO2 under acidic conditions is mainly attributed to two factors: the oxidative release of lattice oxygen and the leaching of surface Ru. The former occurs due to the over-oxidation of Ru into soluble RuO4 species and the involvement of lattice oxygen in the OER, while the latter results from the de-metallization of surface Ru.14 To address these challenges, researchers have employed various strategies to modify RuO2, including doping with other metals, controlling the catalyst's nanostructure, and utilizing interface engineering techniques.15–17
Recently, interface engineering has gained significant attention due to its potential to enhance the activity and stability of catalysts. By designing and optimizing the interface structure of catalysts, charge transfer can be effectively promoted and interfacial interactions can be enhanced, leading to improved catalytic performance.17–20 Additionally, ultrasmall nanoparticles offer unique advantages in enhancing catalytic reaction efficiency due to their high surface area and abundant active sites.21–23 Therefore, combining the advantage of ultrasmall nanoparticles and interface engineering will present an ideal strategy for enhancing the performance of RuO2-based catalysts.
CoFe2O4 has been recognized for its promising OER performance, attributed to its stable structure and adaptable electronic properties.24 Liu et al. reported a novel SrCo0.4Fe0.6O3/CoFe2O4 nanocomposite that exhibited outstanding OER activity in alkaline media, with a low overpotential of 294 mV at a current density of 10 mA cm−2, and demonstrated stability for 110 hours at an applied potential of 1.56 V (vs. RHE). The exceptional performance and stability were attributed to the strong coupling effect between CoFe2O4 nanoparticles and the graphene substrate, underscoring the potential of CoFe2O4 in enhancing OER performance through interfacial engineering.25
Similarly, Deng et al. reported a CoOx/RuOx nano-heterostructure on carbon cloth (CoOx/RuOx–CC) with remarkable OER activity in acidic media, achieving an overpotential of 180 mV at 10 mA cm−2 and stable operation for 100 hours at 100 mA cm−2. The enhanced performance was attributed to efficient electron transfer from Co to Ru, which minimized ion leaching and over-oxidation of Ru, while optimizing the Ru–O bond structure to lower the energy barrier for the OER process.26
Building on these findings, the design of CoFe2O4 and RuO2 composites via interfacial engineering emerges as a promising strategy to further enhance OER performance. Although the application of CoFe2O4 in the acidic OER remains underexplored, its potential is highly intriguing. Specifically, combining CoFe2O4 with RuO2 through interfacial engineering offers a compelling opportunity to improve both catalytic activity and stability under acidic conditions, presenting a promising avenue for future research.
In this study, we designed and synthesized an ultrasmall RuO2/CoFe2O4 (RFC) catalyst with strong interfacial interactions. The RFC catalyst was prepared using an adsorption–pyrolysis method, resulting in significant structural optimization and performance enhancement. Specifically, the strong interfacial interactions and effective charge transfer between RuO2 and CoFe2O4 significantly enhanced the OER activity of the catalyst, while the ultrasmall nanoparticles provided a larger surface area and more active sites. Experimental results demonstrated that the RFC catalyst exhibited excellent catalytic performance in 0.5 M H2SO4 solution, with an overpotential of only 191 mV at a current density of 10 mA cm−2. Moreover, the RFC catalyst maintained its structure and catalytic performance during a long-term stability test of up to 100 hours, indicating high durability and stability. Overall, this study not only successfully designed and prepared a high-performance RFC catalyst through the synergistic utilization of interface engineering and ultrasmall nanoparticles, but also provided new insights into the design of efficient and stable OER catalysts.
2. Experimental
2.1 Syntheses of RFC catalysts
The RFC catalysts were synthesized using an adsorption–pyrolysis method. Initially, a certain amount of lignin was added to 300 mL of ultrapure water and stirred for 20 minutes. CoCl2·6H2O and FeCl3 were dissolved in 100 mL of ultrapure water at specific ion concentrations (Table S1†) and stirred for 20 minutes. Similarly, RuCl3·H2O was dissolved in 100 mL of ultrapure water and stirred for 20 minutes. Subsequently, the mixed solution of CoCl2 and FeCl3 was poured into the lignin solution and stirred for 30 minutes, followed by the addition of the RuCl3 solution, and the resulting mixture was stirred for 12 hours to obtain solutions with different concentrations. The precipitates from the solution were collected using vacuum filtration. The dried samples were then subjected to calcination at 500 °C for 2 hours in air to obtain the final catalysts.2.2 Syntheses of CoFe2O4 (CFO) catalysts
A certain amount of lignin was added to 300 mL of ultrapure water and stirred for 20 minutes. CoCl2·6H2O and FeCl3 were then dissolved together in 200 mL of ultrapure water and stirred for 20 minutes. This mixture was added to the lignin dispersion and stirred continuously for 12 hours. The concentrations of Co2+ and Fe3+ in the solution were adjusted to 0.4 g L−1 and 0.5 g L−1, respectively. The resulting precipitates were separated by vacuum filtration, dried, and subjected to calcination at 500 °C for 2 hours in air to produce the catalysts.2.3 Syntheses of RuO2 catalysts
Lignin was stirred in 300 mL of ultrapure water for 20 minutes to create a dispersion. RuCl3·H2O was dissolved in 200 mL of ultrapure water, also with 20 minutes of stirring. The RuCl3 solution was then introduced to the lignin dispersion and stirred for 12 hours. The Ru3+ concentration in the solution was maintained at 0.3 g L−1. The precipitates obtained were collected through vacuum filtration, dried, and calcined at 500 °C for 2 hours in air to form the final RuO2 catalysts.3. Results and discussion
3.1 Characterization of the catalysts
To confirm the crystallographic phases and structural properties of the catalysts, we conducted X-ray diffraction (XRD) and Raman spectroscopy analyses. As shown in Fig. 1a, the diffraction peaks of CFO and RuO2 correspond well with the standard PDF patterns PDF#22-1086 and PDF#43-1027, respectively. The diffraction peaks for the RuO2/Fe2O3 (RF), RuO2/CoFe2O4 (RFC), and RuO2/Co3O4 (RC) catalysts were in alignment with the phase of RuO2 (PDF # 43-1027).27 For the RF catalyst, the peaks corresponded to the phase of Fe2O3 (PDF # 33-0664), predominantly exhibiting the (012), (104), and (110) crystal planes.28 The RC catalyst showed diffraction peaks matching the Co3O4 phase (PDF # 74-2120), primarily displaying the (111), (220), and (311) crystal planes.29 For the RFC catalyst, the diffraction peaks corresponding to the (101) plane of RuO2 and the (311) plane of CoFe2O4 exhibit slight shifts, which could be attributed to interfacial interactions between RuO2 and CoFe2O4 in the catalyst. Additionally, due to the limited resolution of the instrument and the broader diffraction peaks of the RuO2 phase, other diffraction peaks of CoFe2O4 in the RFC catalyst are less distinct. The broader peaks suggest the presence of lattice strain or lower crystallinity in the RFC catalyst.30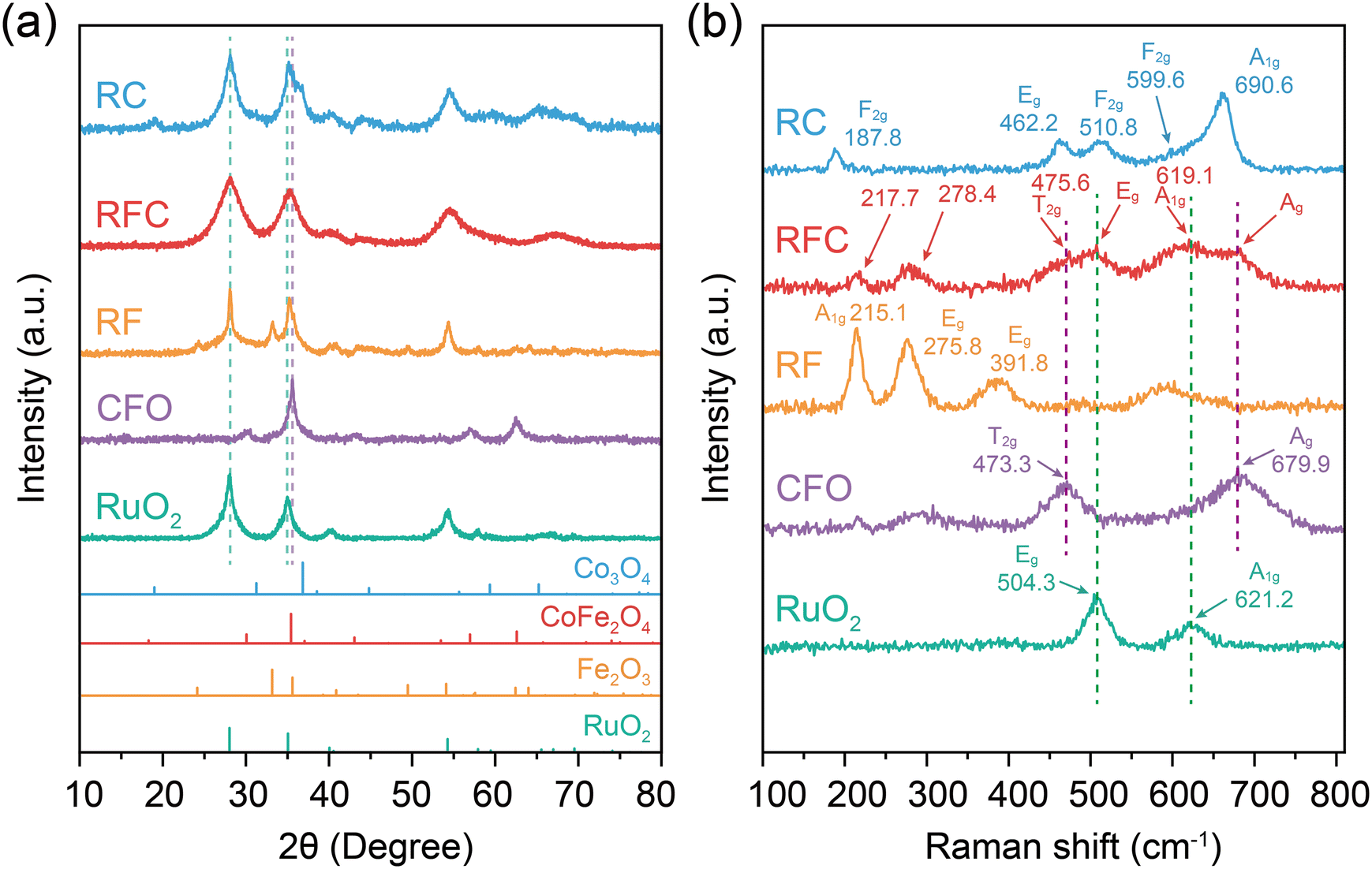 | ||
| Fig. 1 Structural characterization of catalysts. (a) XRD spectra of the catalysts. (b) Raman spectra of the catalysts. | ||
The Raman spectra provide further evidence of interfacial interactions between RuO2 and CoFe2O4 in the RFC catalyst. As shown in Fig. 1b, the pure CoFe2O4 (CFO) sample exhibits characteristic vibrational modes typical of spinel-type oxides, such as the T2g mode at 473.3 cm−1 and the A1g mode at 679.9 cm−1.31 In contrast, the pure RuO2 sample displays modes at 504.3 cm−1 and 621.2 cm−1, corresponding to the E9 and A1g vibrational modes of RuO2, respectively.32 The RF catalyst exhibited two A1g vibrational modes (215.1 and 490.9 cm−1) and three Eg modes (275.8, 391.8, and 602.1 cm−1), corresponding to the α-Fe2O3 phase. The RC catalyst displayed vibrational modes characteristic of Co3O4, including an A1g mode at 660.6 cm−1, an Eg mode at 462.2 cm−1, and three F2g modes at 599.6 cm−1, 510.8 cm−1, and 187.8 cm−1. The Raman modes corresponding to both RuO2 and CFO are detected in the RFC composite, showing slight peak shifts and variations in intensity. Specifically, the A1g mode of RuO2 shifts from 621.2 cm−1 in pure RuO2 to 619.1 cm−1 in the RFC composite, while the T2g mode of CFO shifts from 473.3 cm−1 to 475.6 cm−1. These shifts in the vibrational modes indicate structural modifications at the interface, which are likely attributed to strain or charge redistribution between RuO2 and CoFe2O4, suggesting strong interfacial interactions between the two components.
To observe the detailed morphology and microstructure of the catalysts, transmission electron microscopy (TEM) and scanning electron microscopy (SEM) were used for analysis. Fig. 2(a)–(c) show the TEM images of the RF, RFC, and RC catalysts, respectively. TEM images show that all three catalysts consist of interconnected nanoparticles with different sizes, specifically RF: 16 nm (Fig. 2a), RFC: 3 nm (Fig. 2b), RC: 11 nm (Fig. 2c), CFO: 11 nm (Fig. S2a†) and RuO2: 15 nm (Fig. S2b†). It is evident that compared to other synthesized catalysts, the RFC catalyst exhibits the smallest particle sizes, indicating that the introduction of Co has significantly influenced the morphology of the RFC nanoparticles. The SEM images provided in Fig. S1† further illustrate the morphology of the RF, RFC, and RC catalysts. The RF and RC catalysts display relatively smooth surfaces, while the RFC catalyst exhibits a rougher surface. This rough surface morphology in RFC is attributed to its smaller nanoparticle size, consistent with the TEM results.
 | ||
| Fig. 2 Morphology and elemental characterization of the catalysts. TEM images of (a) RF, (b) RFC and (c) RC, with insets showing the particle size distribution histograms. HRTEM images of (d) RF, (e) RFC and (f) RC. EDX elemental mapping images of (g) RF, (h) RFC and (i) RC. | ||
Smaller particle sizes often correspond to a larger specific surface area. Brunauer–Emmett–Teller (BET) analysis indicates that the RFC catalyst exhibits a surface area of 108.9 m2 g−1, which is higher than those of the RF (63.2 m2 g−1) and RC (88.2 m2 g−1) catalysts (Fig. S3†). This increased surface area provides more active sites for the OER, contributing to the improved catalytic performance. The detailed microstructure and crystallographic planes of the catalysts were elucidated using high-resolution TEM (HRTEM). For the RF catalyst (Fig. 2d), the lattice fringe spacings of 3.18 Å and 2.70 Å correspond to the (110) plane of RuO2 and the (104) plane of α-Fe2O3, respectively.33 In contrast, the RC catalyst (Fig. 2f) exhibits lattice spacings of 2.55 Å and 3.18 Å, corresponding to the (110) and (101) planes of RuO2, while spacings of 2.86 Å and 2.43 Å match the (220) and (311) planes of Co3O4.34 Furthermore, the HRTEM images of the RFC catalyst reveal a well-defined interfacial structure, indicating interactions between CoFe2O4 and RuO2 (Fig. 2e). The lattice fringe spacing of the (311) plane in pure CoFe2O4 (2.53 Å) is slightly expanded to 2.54 Å within the RFC composite, suggesting the presence of interfacial strain (Fig. 2e and S2c†). This lattice expansion, consistent with the XRD results, provides compelling evidence of interfacial interactions between RuO2 and CoFe2O4.
Elemental composition and distribution of the catalysts were analyzed using energy-dispersive X-ray (EDX) spectroscopy. The analysis revealed that the RF catalyst contains Ru, Fe, and O elements, each uniformly distributed (Fig. 2g). In the RFC catalyst, Ru, Fe, and Co elements were found, with an even distribution (Fig. 2h). Similarly, the RC catalyst consists of Ru, Co, and O elements, all evenly distributed (Fig. 2i). Additionally, the Ru content in various RFC samples was measured using ICP-MS, as shown in Table S3.† The Ru contents in the RFC catalyst were found to be 14.5 wt% and 23.2 at%, which are lower than those of RF (15.7 wt% and 25.0 at%) and RC (19.4 wt% and 29.2 at%), as detailed in Tables S2 and S3.†
The surface composition and chemical states of the catalysts were analyzed using X-ray photoelectron spectroscopy (XPS). All spectra were calibrated against the C 1s peak at a binding energy of 284.8 eV. The wide-scan spectra revealed the presence of Ru, Fe, Co, and O in the RFC catalyst, with no detectable interference from other impurities (Fig. S4†). As depicted in Fig. 3a, the Fe 2p spectrum of the CFO catalyst exhibits characteristic peaks at binding energies of 710.5 eV for Fe 2p3/2 and 724.0 eV for Fe 2p1/2, with satellite peaks at 718.6 eV and 731.7 eV corresponding to Fe 2p3/2. These peaks confirm that the Fe in the CFO sample is in the Fe3+ oxidation state.35 In comparison, the Fe 2p peaks in the RFC catalyst show no shift relative to the CFO catalyst, indicating that the Fe in the RFC sample is also in the Fe3+ oxidation state.
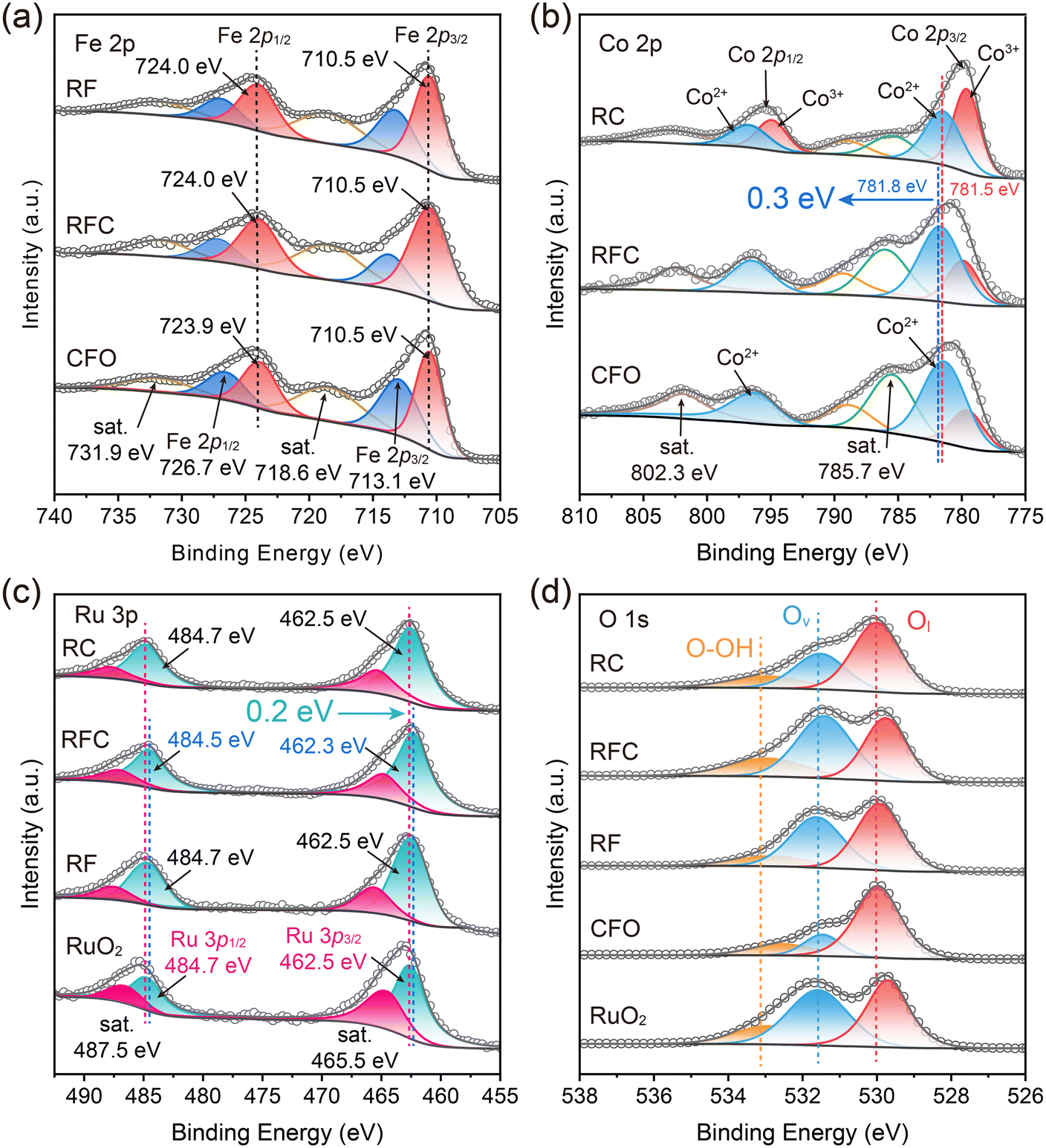 | ||
| Fig. 3 Electronic characterization of the catalysts. (a) Fe 2p, (b) Co 2p, (c) Ru 3p and (d) O 1s XPS spectra of the catalysts. | ||
The Co 2p spectra of the RC catalyst can be divided into four peaks: Co 2p3/2 and Co 2p1/2 (Fig. 3b). The binding energies of 779.6 eV and 794.9 eV are attributed to Co3+ at Co 2p3/2 and Co 2p1/2 sites, respectively, while the peaks at 781.4 eV and 796.7 eV are identified as Co2+. The left-side peaks represent satellites of Co 2p3/2 and Co 2p1/2.36
For the CFO catalyst, the peaks at 781.5 eV and 796.4 eV correspond to the 2p3/2 and 2p1/2 states of Co2+ in tetrahedral and octahedral sites, respectively. These peaks are accompanied by satellite peaks around 785.7 eV. Additionally, the peak at 779.7 eV is assigned to the 2p3/2 state of Co3+ in the octahedral site.37 Compared to the CFO catalyst, the Co 2p3/2 peaks of the RFC catalyst exhibit a positive shift in the binding energy by 0.3 eV, suggesting an elevation in the oxidation state of Co in the RFC catalyst. In addition, the Ru 3p spectra were measured to further investigate the oxidation state of Ru (Fig. 3c). For the RuO2 catalyst, the characteristic Ru 3p spectra exhibit spin–orbit doublet peaks at 462.5 eV and 484.7 eV, corresponding to Ru 3p3/2 and Ru 3p1/2, along with satellite peaks at 465.5 eV and 487.5 eV, respectively. In comparison, the Ru 3p peaks of the RFC catalyst are slightly shifted to lower binding energies (462.3 eV and 484.5 eV), indicating an electron-rich environment around the Ru sites. This negative shift suggests that a significant number of electrons are injected into the Ru sites, leading to a partial reduction of the Ru oxidation state in the RFC catalyst. The O 1s spectrum of the RFC catalyst, as shown in Fig. 3d, is deconvoluted into three distinct peaks: Ol at 529.8 eV (lattice oxygen), Ov at 531.4 eV (oxygen vacancies), and O–OH at 532.9 eV (hydroxyl groups). The proportion of Ov in RFC (45%) is higher than those in RF (41%) and RC (30%), indicating a greater number of oxygen vacancies in RFC (Table S4†). This increased ratio of oxygen vacancies enhances the oxidation kinetics, thereby improving the OER performance.
Overall, the XPS analysis demonstrates substantial electronic interactions at the CoFe2O4 and RuO2 interface in the RFC catalyst. The positive shift in the Co 2p3/2 binding energy indicates an increased oxidation state of Co, while the negative shift in the Ru 3p binding energy suggests a reduced oxidation state of Ru due to electron transfer to the Ru sites. These binding energy shifts highlight significant interfacial charge transfer between RuO2 and CoFe2O4, indicating strong interfacial interactions and charge redistribution at the interface. This redistribution likely enhances the overall catalytic performance by facilitating charge transfer. Furthermore, the increased presence of oxygen vacancies in RFC is expected to accelerate oxidation kinetics, thereby further improving the OER activity of the RFC catalyst.
3.2 Electrochemical performance
The electrocatalytic performance of the catalysts was assessed using a standard three-electrode system in 0.5 M H2SO4, with all potentials referenced to the reversible hydrogen electrode (RHE). The linear sweep voltammetry (LSV) curve for the RFC catalyst (Fig. 4a) reveals an overpotential of only 191 mV at a current density of 10 mA cm−2, which is notably lower than the overpotentials observed for RC (215 mV), RF (244 mV), RuO2 (225 mV) and CFO (501 mV). This suggests the superior catalytic activity of the RFC catalyst. Additionally, the RFC catalyst prepared with 0.5 g L−1 Co2+ demonstrated optimal oxygen evolution reaction (OER) performance, as depicted in Fig. S5.† The Tafel slope analysis (Fig. 4b) indicates that the RFC catalyst has a Tafel slope of 74.8 mV dec−1, which is lower than those of RC (104.5 mV dec−1), RF (149.4 mV dec−1), RuO2 (115.1 mV dec−1) and CFO (296.4 mV dec−1), reflecting faster reaction kinetics. Electrochemical impedance spectroscopy (EIS) results (Fig. S6†) show that the RFC catalyst exhibits the lowest charge-transfer resistance, implying more efficient charge transfer and enhanced reaction kinetics in acidic media. A comparison of overpotentials at 10 mA cm−2 and Tafel slopes (Fig. 4c) further highlights the superior performance of the RFC catalyst. Fig. S7† presents a comparison of the overpotential at 10 mA cm−2 and the average particle size of the various catalysts, highlighting the crucial role of the ultrasmall particle size of RFC in enhancing its catalytic performance. The reduced particle size of RFC significantly increases the catalyst's surface area, providing more active sites for the oxygen evolution reaction (OER), which in turn leads to improved catalytic activity. Furthermore, the electrochemical double-layer capacitance (Cdl) was estimated in the non-faradaic region using cyclic voltammetry (CV). Among all tested catalysts, RFC exhibited the highest Cdl value of 22.7 mF cm−2, which is markedly higher than those of RC (15.6 mF cm−2), RF (10.3 mF cm−2), RuO2 (13.5 mF cm−2), and CFO (7.6 mF cm−2). This suggests that RFC possesses a larger electrochemically active surface area, providing a greater number of active sites for the OER (Fig. 4d and S8†). The increased surface area of RFC, combined with its ultrasmall particle size, plays a vital role in boosting its catalytic performance. Additionally, linear sweep voltammetry (LSV) curves normalized to the electrochemically active surface area (ECSA) further confirm this finding. As shown in Fig. S9,† the RFC catalyst achieves a higher current density than the other catalysts, underscoring its superior intrinsic activity and overall effectiveness for the OER. To evaluate the effect of Ru mass loading on the electrochemical performance, we normalized the linear sweep voltammetry (LSV) curves to the Ru mass, as illustrated in Fig. S10.† The RFC-0.5 sample shows the highest mass activity, underscoring the effectiveness of the RuO2 and CoFe2O4 combination in the RFC structure. Even at lower Ru loading levels (Table S5†), RFC-0.5 maintains its superior intrinsic activity compared to other samples. This improvement is likely attributed to more efficient charge transfer and catalytic reaction pathways facilitated by the robust interfacial interactions between the two components. In addition to its high intrinsic activity, the RFC catalyst demonstrates excellent long-term stability. As shown in Fig. 4e, the stability test at a constant current density of 10 mA cm−2 indicates that RFC maintains a stable potential for over 100 hours, significantly outperforming RuO2 and CFO, which exhibit noticeable potential drift. Moreover, Fig. 4f illustrates that the LSV curves of RFC, after 500 continuous CV cycles, show only a minor potential shift of 8 mV at 100 mA cm−2, indicating minimal performance degradation. To assess the catalyst's durability under industrially relevant conditions, stability tests were also conducted at a higher current density of 200 mA cm−2, as shown in Fig. S11.† The RFC catalyst maintained a stable potential of approximately 1.75 V vs. RHE over 3000 minutes of continuous operation, further highlighting its robustness and suitability for large-scale water electrolysis systems. As a result, the comparisons of various Ru-based catalysts for the OER, as summarized in Table S6,† reveal that the RFC catalyst shows superior acidic OER catalytic performance compared to the RF and RC catalysts, as well as many of the recently reported Ru-based electrocatalysts.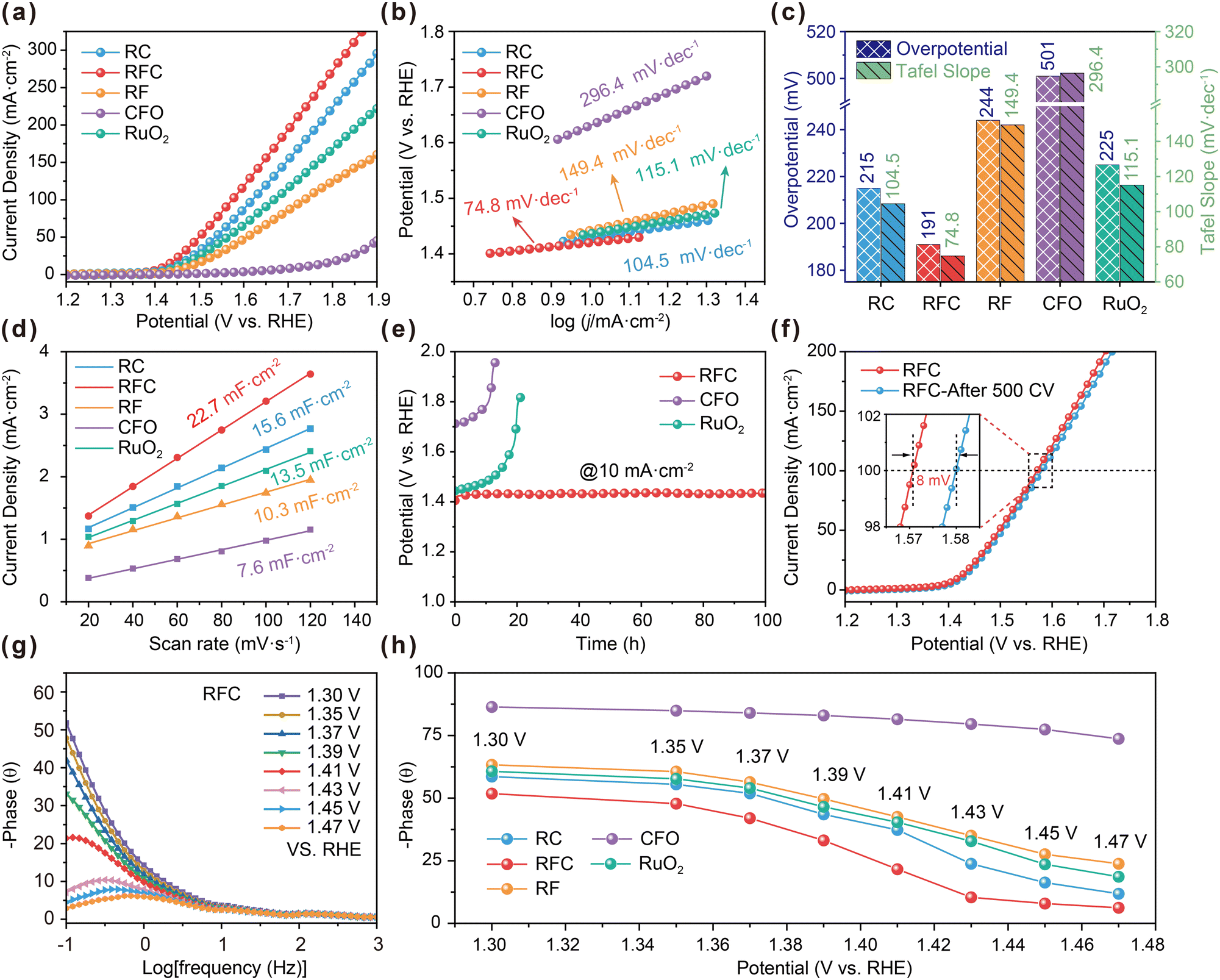 | ||
| Fig. 4 OER performance of the catalysts in 0.5 M H2SO4 solution. (a) LSV curves of the catalysts. (b) Tafel plots derived from the LSV curves. (c) Comparison of the overpotentials at 10 mA cm−2 and Tafel slopes of the catalysts. (d) Cdl plots of the catalysts. (e) Stability test of the catalysts at 10 mA cm−2. (f) The LSV curves of RFC before and after 500 CV cycles. (g) Bode phase plots of RFC. (h) Response of the phase angle to the applied potential of the catalysts. | ||
The in situ impedance Bode phase plots for the RC, RFC, RF, RuO2 and CFO catalysts (Fig. 4g) elucidate the key electrochemical characteristics and charge transfer processes during the OER in a 0.5 M H2SO4 solution. The RFC catalyst exhibits a notable decrease in the phase angle in the low-frequency region (−1 to 0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) log [Hz]) as the potential increases, indicating lower charge transfer resistance and more efficient charge transfer processes compared to the RF and RC catalysts (Fig. 4h and S12†). In the mid-frequency range (0 to 1log [Hz]), the RFC catalyst demonstrates significant phase angle variations, suggesting higher charge transfer efficiency and lower interfacial impedance.38–40 Additionally, at high frequencies (1 to 3log [Hz]), the RFC catalyst shows lower phase angles, indicating higher double-layer capacitance and improved interfacial electrochemical performance. These observations suggest that the RFC catalyst outperforms the RF, RC, RuO2 and CFO catalysts in terms of charge transfer efficiency and interfacial electrochemical properties, likely due to increased oxygen vacancies and stronger interfacial interactions in the RFC catalyst.
log [Hz]) as the potential increases, indicating lower charge transfer resistance and more efficient charge transfer processes compared to the RF and RC catalysts (Fig. 4h and S12†). In the mid-frequency range (0 to 1log [Hz]), the RFC catalyst demonstrates significant phase angle variations, suggesting higher charge transfer efficiency and lower interfacial impedance.38–40 Additionally, at high frequencies (1 to 3log [Hz]), the RFC catalyst shows lower phase angles, indicating higher double-layer capacitance and improved interfacial electrochemical performance. These observations suggest that the RFC catalyst outperforms the RF, RC, RuO2 and CFO catalysts in terms of charge transfer efficiency and interfacial electrochemical properties, likely due to increased oxygen vacancies and stronger interfacial interactions in the RFC catalyst.
Following the constant current test to assess stability, detailed characterization was performed using transmission electron microscopy (TEM) and X-ray diffraction (XRD). As shown in Fig. S13,† the TEM analysis revealed that the RFC catalyst retained its nanoparticle morphology without significant changes in particle size or distribution, indicating high morphological stability. The uniformly distributed nanoparticles demonstrate the catalyst's physical stability under prolonged electrochemical conditions. The XRD analysis in Fig. S14(a)† shows that the diffraction peaks remained consistent with those observed before the reaction, indicating that the crystal structure and crystallinity of the RFC catalyst were maintained during long-term stability testing. Additionally, Raman spectroscopy, as shown in Fig. S14(b),† further supports the structural stability, with A1g and Eg vibrational modes retaining their peak shapes and positions, suggesting preserved structural integrity. These comprehensive characterization results demonstrate the RFC catalyst's exceptional morphological and structural stability after the stability test. The robust interfacial interactions within the RFC catalyst contribute to its superior electrochemical stability. The stability of the nanoscale structure and the abundance of oxygen vacancies play crucial roles in sustaining the catalyst's high performance. Consequently, the RFC catalyst shows great promise for practical applications in proton exchange membrane water electrolysis (PEMWE), due to its long-term stability and catalytic efficiency under acidic OER conditions.
3.3 Acidic OER performances in PEMWE
To evaluate the practical applications of the RFC catalysts, we employed a two-electrode water electrolysis cell with a proton exchange membrane (PEM). The RFC catalysts served as the anode for the acidic oxygen evolution reaction (OER), while commercial 20% Pt/C was used as the cathode, as depicted in Fig. 5a. The linear sweep voltammetry (LSV) results presented in Fig. 5b indicate that the RFC catalyst exhibits superior catalytic activity, achieving a cell voltage of 1.60 V to deliver a current density of 10 mA cm−2, which is lower than those of the RF (1.71 V) and RC (1.63 V) catalysts. This demonstrates the enhanced performance of the RFC catalyst under acidic conditions. The long-term stability of the RFC catalyst was further evaluated via chronopotentiometry at 10 mA cm−2, as shown in Fig. 5c. The RFC catalyst maintained excellent stability throughout the test, with only a minor voltage increase of 30 mV over 50 hours. This notable electrochemical stability underscores the potential of RFC catalysts for practical applications in proton exchange membrane water electrolysis (PEMWE), offering both high efficiency and durability in acidic environments.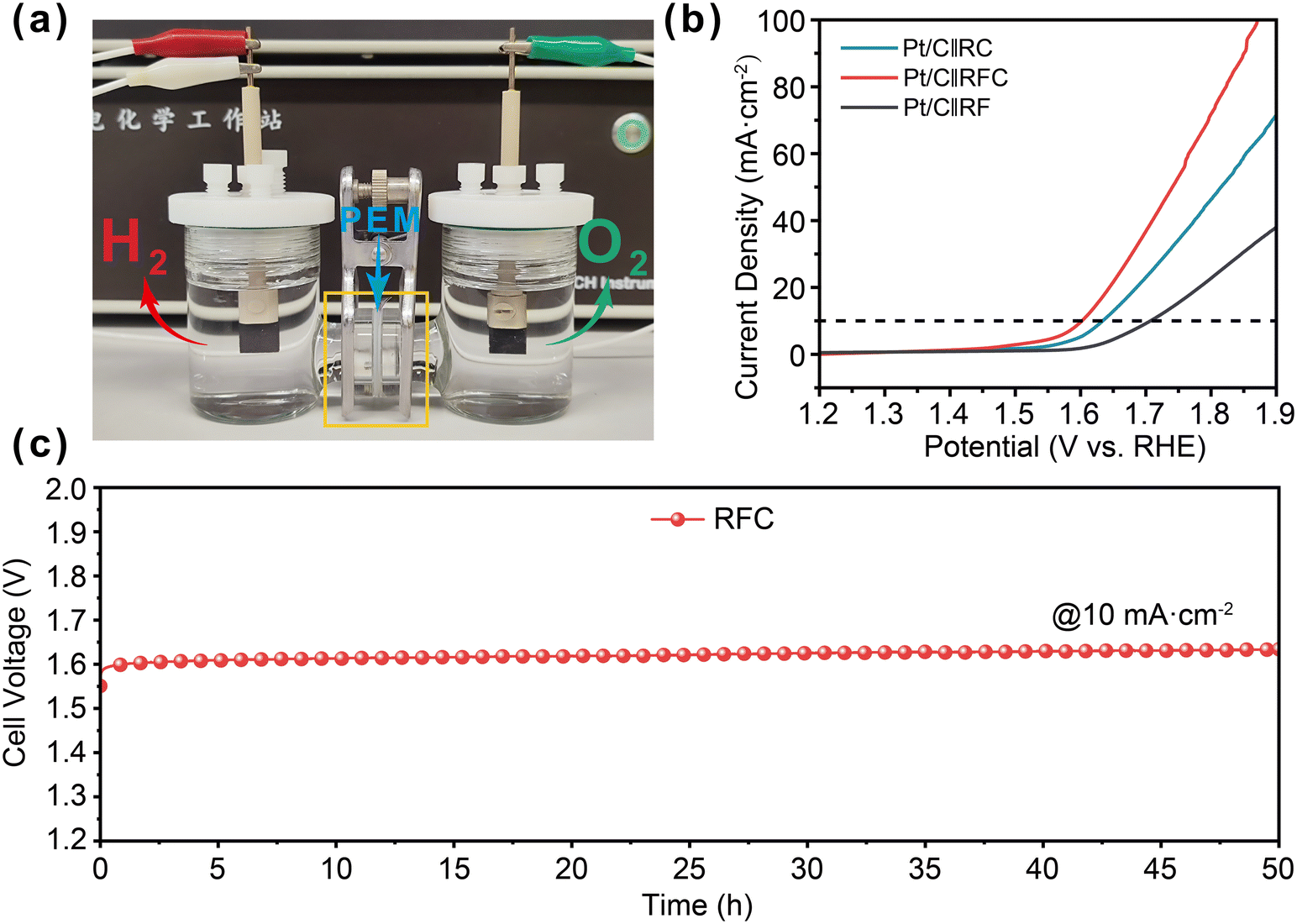 | ||
| Fig. 5 Acidic OER performance of RFC in PEMWE. a) Photograph of the two-electrode system with a PEM for water electrolysis. b) LSV curves of the catalysts in the two-electrode system. c) Long-term stability test of Pt/C‖RFC. | ||
4. Conclusions
In this study, we successfully fabricated ultrasmall RuO2/CoFe2O4 (RFC) catalysts through an adsorption–pyrolysis approach, achieving an average size of 3 nm. These ultrasmall dimensions led to a substantial increase in the surface area and intensified interfacial interactions, which are crucial determinants for their enhanced OER performance. Detailed characterization confirmed the robust structure and exceptional catalytic activity of the RFC catalysts. Electrochemical measurements demonstrated a low overpotential of 191 mV at 10 mA cm−2, significantly outperforming RF and RC catalysts. The RFC catalysts also exhibited exceptional stability in 0.5 M H2SO4, maintaining their performance over prolonged operation. The enhanced performance of the RFC catalysts is attributed to their ultrasmall size, which provides a larger specific surface area, and the strong interfacial interactions between RuO2 and CoFe2O4, facilitating effective charge transfer. Additionally, the higher proportion of oxygen vacancies in the RFC catalysts enhances oxidation kinetics, further improving their OER performance. These findings highlight the potential of RFC catalysts for sustainable hydrogen production and provide a novel approach to designing advanced electrocatalysts via strategic interfacial engineering, underscoring their promising performance for practical applications in proton exchange membrane water electrolysis (PEMWE).Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Ming Wei contributed to the methodology, experiments, formal analysis, and writing – original draft. Sanzhao Song, Liuhua Mu, Zhonggui Gao, and Li Yang provided supervision, project administration, and funding acquisition. Guangjian Song, Qiankang Si, and Fangfang Dai were responsible for the conceptualization and methodology of the study. Ming Wei, Zhiwei Liu, Feng Gao, Min Zhang, Rui Ding, and Mao Zhang conducted the investigation and data curation. All authors participated in the validation, visualization, and writing – review & editing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Guangxi Natural Science Foundation (No. 2022GXNSFAA035487), the Wenzhou Institute, University of Chinese Academy of Sciences (No. WIUCASQD2022021, WIUCASQD2021014), the Postdoctoral Fellowship Program of CPSF under Grant Number GZC20232610, and the Guangxi Natural Science Foundation (No. 2020GXNSFAA297241).References
- H. Mosallaei, H. Hadadzadeh, A. A. Ensafi, K. Z. Mousaabadi, M. Weil, A. Foelske and M. Sauer, Int. J. Hydrogen Energy, 2023, 48, 1813–1830 CrossRef CAS.
- J. Shan, C. Guo, Y. Zhu, S. Chen, L. Song, M. Jaroniec, Y. Zheng and S.-Z. Qiao, Chem, 2019, 5, 445–459 CAS.
- Z.-Y. Wu, F.-Y. Chen, B. Li, S.-W. Yu, Y. Z. Finfrock, D. M. Meira, Q.-Q. Yan, P. Zhu, M.-X. Chen, T.-W. Song, Z. Yin, H.-W. Liang, S. Zhang, G. Wang and H. Wang, Nat. Mater., 2023, 22, 100–108 CrossRef CAS.
- S. Chu, Y. Cui and N. Liu, Nat. Mater., 2017, 16, 16–22 CrossRef.
- H. Sun, X. Xu, H. Kim, Z. Shao and W. Jung, InfoMat, 2024, 6, e12494 CrossRef CAS.
- Y. Qin, T. Yu, S. Deng, X.-Y. Zhou, D. Lin, Q. Zhang, Z. Jin, D. Zhang, Y.-B. He, H.-J. Qiu, L. He, F. Kang, K. Li and T.-Y. Zhang, Nat. Commun., 2022, 13, 3784 CrossRef CAS.
- L. Zhang, H. Jang, H. Liu, M. G. Kim, D. Yang, S. Liu, X. Liu and J. Cho, Angew. Chem., Int. Ed., 2021, 60, 18821–18829 CrossRef CAS PubMed.
- F. Zeng, C. Mebrahtu, L. Liao, A. K. Beine and R. Palkovits, J. Energy Chem., 2022, 69, 301–329 CrossRef CAS.
- M. Guo, R. Deng, C. Wang and Q. Zhang, J. Energy Chem., 2023, 78, 537–553 CrossRef CAS.
- H. Sun, X. Xu, H. Kim, W. Jung, W. Zhou and Z. Shao, Energy Environ. Mater., 2023, 6, e12441 CrossRef CAS.
- R. Fan, Y. Zhang, J. Lv, G. Han, Y. Chai and B. Dong, Small, 2023, 20, 2304636 CrossRef.
- Y. Wang, X. Lei, B. Zhang, B. Bai, P. Das, T. Azam, J. Xiao and Z. Wu, Angew. Chem., Int. Ed., 2024, 63, e202316903 CrossRef CAS.
- Q. Qin, T. Wang, Z. Li, G. Zhang, H. Jang, L. Hou, Y. Wang, M. Gyu Kim, S. Liu and X. Liu, J. Energy Chem., 2024, 88, 94–102 CrossRef CAS.
- H. Yan, Z. Jiang, B. Deng, Y. Wang and Z. Jiang, Adv. Energy Mater., 2023, 13, 2300152 CrossRef CAS.
- X. Cao, J. Huo, L. Li, J. Qu, Y. Zhao, W. Chen, C. Liu, H. Liu and G. Wang, Adv. Energy Mater., 2022, 12, 2202119 CrossRef CAS.
- L. Hou, X. Gu, X. Cui, J. Tang, Z. Li, X. Liu and J. Cho, EES Catal., 2023, 1, 619–644 RSC.
- K. Du, L. Zhang, J. Shan, J. Guo, J. Mao, C.-C. Yang, C.-H. Wang, Z. Hu and T. Ling, Nat. Commun., 2022, 13, 5448 CrossRef CAS.
- N. Wang, S. Ning, X. Yu, D. Chen, Z. Li, J. Xu, H. Meng, D. Zhao, L. Li, Q. Liu, B. Lu and S. Chen, Appl. Catal., B, 2022, 302, 120838 CrossRef CAS.
- S. Niu, S. Wei, D. Yue, Y. Cai, Z. Ma, K. Liu, Y. Huang, H. Wang, Q. Li and T.-N. Ye, Catal. Sci. Technol., 2023, 13, 7076–7084 RSC.
- S. Sun, H. Jiang, Z. Chen, Q. Chen, M. Ma, L. Zhen, B. Song and C. Xu, Angew. Chem., Int. Ed., 2022, 61, e202202519 CrossRef CAS PubMed.
- S. P. Schwaminger, R. Surya, S. Filser, A. Wimmer, F. Weigl, P. Fraga-García and S. Berensmeier, Sci. Rep., 2017, 7, 12609 CrossRef PubMed.
- H. Xu, Z. Jin, Y. Zhang, X. Lin, G. Xie, X. Liu and H.-J. Qiu, Chem. Sci., 2023, 14, 771–790 RSC.
- Z. Li, B. Li, Y. Hu, S. Wang and C. Yu, Mater. Adv., 2022, 3, 779–809 RSC.
- H. Wang, Q. Hu, J. Qiu, R. Guo and X. Liu, Catal. Sci. Technol., 2023, 13, 6102–6125 RSC.
- H. Liu, Y. Wang, P. Tan, J. Pan and C. D'Agostino, Small, 2024, 23, 2308948 CrossRef.
- L. Deng, S. Liu, D. Liu, Y. Chang, L. Li, C. Li, Y. Sun, F. Hu, H. Chen, H. Pan and S. Peng, Small, 2023, 19, 2302238 CrossRef CAS.
- Z. Guan, Q. Chen, L. Liu, C. Xia, L. Cao and B. Dong, Nanoscale, 2024, 16, 10325–10332 RSC.
- A. Lassoued, B. Dkhil, A. Gadri and S. Ammar, Results Phys., 2017, 7, 3007–3015 CrossRef.
- M. Wang, S. Li, J. Kang, Y. Tang, J. Wang, Z. Xu and J. Liu, Chem. Eng. J., 2023, 451, 138611 CrossRef CAS.
- P. E. Karthik, L. Sangaletti, M. Ferroni and I. Alessandri, Catal. Sci. Technol., 2024, 14, 2268–2274 RSC.
- Y. Hao, G. Du, Y. Fan, L. Jia, D. Han, W. Zhao, Q. Su and B. Xu, Appl. Surf. Sci., 2023, 614, 156237 CrossRef CAS.
- Y.-N. Zhou, N. Yu, Q.-X. Lv, B. Liu, B. Dong and Y.-M. Chai, J. Mater. Chem. A, 2022, 10, 16193–16203 RSC.
- T. Dong, W. Yi, T. Deng, T. Qin, X. Chu, H. Yang, L. Zheng, S. J. Yoo, J. Kim, Z. Wang, Y. Wang, W. Zhang and W. Zheng, Energy Environ. Mater., 2023, 6, e12262 CrossRef CAS.
- K. Shah, R. Dai, M. Mateen, Z. Hassan, Z. Zhuang, C. Liu, M. Israr, W. Cheong, B. Hu, R. Tu, C. Zhang, X. Chen, Q. Peng, C. Chen and Y. Li, Angew. Chem., Int. Ed., 2022, 61, e202114951 CrossRef CAS.
- G. Song, M. Wei, J. Zhou, L. Mu and S. Song, ACS Catal., 2024, 14, 846–856 CrossRef CAS.
- G. Song, L. Mu, P. Qiao, L. Yang, M. Zhang, F. Dai, L. Dai, H. Wu, Z. Gao, G. Shi and S. Song, Results Eng., 2023, 20, 101606 CrossRef CAS.
- E. M. Davis, A. Bergmann, H. Kuhlenbeck and B. Roldan Cuenya, J. Am. Chem. Soc., 2024, 146, 13770–13782 CrossRef CAS PubMed.
- Y. Hao, S.-F. Hung, W.-J. Zeng, Y. Wang, C. Zhang, C.-H. Kuo, L. Wang, S. Zhao, Y. Zhang, H.-Y. Chen and S. Peng, J. Am. Chem. Soc., 2023, 145, 23659–23669 CrossRef CAS PubMed.
- N. O. Laschuk, E. B. Easton and O. V. Zenkina, RSC Adv., 2021, 11, 27925–27936 RSC.
- Y. Qi, Y. Zhang, L. Yang, Y. Zhao, Y. Zhu, H. Jiang and C. Li, Nat. Commun., 2022, 13, 4602 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cy00719k |
| This journal is © The Royal Society of Chemistry 2024 |