
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Restructuring of Ag catalysts for methanol to formaldehyde conversion studied using in situ X-ray ptychography and electron microscopy†
Srashtasrita
Das
 a,
Maik
Kahnt
b,
Youri
van Valen
c,
Tina
Bergh
c,
Sara
Blomberg
de,
Mikhail
Lyubomirskiy
f,
Christian G.
Schroer
f,
Hilde J.
Venvik
*c and
Thomas L.
Sheppard‡
*a
a,
Maik
Kahnt
b,
Youri
van Valen
c,
Tina
Bergh
c,
Sara
Blomberg
de,
Mikhail
Lyubomirskiy
f,
Christian G.
Schroer
f,
Hilde J.
Venvik
*c and
Thomas L.
Sheppard‡
*a
aInstitute for Chemical Technology and Polymer Chemistry, Karlsruhe Institute of Technology, 76131 Karlsruhe, Germany. E-mail: thomas.sheppard@kit.edu
bMAX IV Laboratory, Lund University, 22100 Lund, Sweden
cDepartment of Chemical Engineering, Norwegian University of Science and Technology, NO-7491 Trondheim, Norway. E-mail: hilde.j.venvik@ntnu.no
dNanoLund, Lund University, Box 118, 211 00 Lund, Sweden
eDepartment of Chemical Engineering, Lund University, Box 118, 221 00 Lund, Sweden
fCenter for X-ray and Nano Science, Deutsches Elektronen-Synchrotron, 22607 Hamburg, Germany
First published on 26th August 2024
Abstract
Dynamic restructuring of silver catalysts during the industrial conversion of methanol to formaldehyde leads to surface faceting and pinhole formation. Subsequent sintering under reaction conditions, followed by increased pressure drop and decreased catalyst activity requires catalyst bed replacement after several months of operation. This necessitates a comprehensive understanding of the bulk catalyst restructuring under exposure to different gas environments. In this work, Ag restructuring was studied at elevated temperatures under different reactive and inert gas environments. Bubble formation within catalysts of 5–8 μm thickness was visualized in real-time using in situ X-ray ptychography. Stepwise heating up to 650 °C in combination with imaging was used to determine the effect of temperature on silver restructuring. Dynamic changes within the catalyst were further quantified in terms of relative changes in mass on selected regions at a constant temperature of 500 °C. Quantitative assessment of dynamic changes in the catalyst resulting from bubble growth and movement revealed the influence of temperature, time, and gas environment on the degree of restructuring. Post-mortem scanning electron microscopy with energy-dispersive X-ray spectroscopy mapping confirmed the redistribution of material as a consequence of bubble rupture and collapse. The formation of pores and cavities under different gas environments was additionally confirmed using a fixed bed reactor, and subsequent examination using focused-ion beam milling, providing detailed analysis of the surface structure. This study demonstrates the unique advantage of correlative hard X-ray and electron microscopy for quantitative morphological studies of industrial catalysts.
1. Introduction
The industrial catalytic conversion of methanol to formaldehyde is important due to the broad usage of formaldehyde in various sectors such as automotive, pharmaceuticals, construction, and cosmetics.1,2 High-temperature methanol conversion over unsupported, polycrystalline silver (Ag) at temperatures around 600–720 °C and under atmospheric pressures accounts for around 50% of industrial formaldehyde production.1,3 Under reaction conditions, the Ag catalyst experiences surface faceting, formation of dynamic pinhole-like surface pores, and subsequent sintering. This results in increased pressure drop over time, leading to a decrease in catalyst activity and replacement of the catalyst bed typically after 6–12 months of industrial operation.1,4 Despite the morphological issues accompanying the Ag catalyst, its non-toxic nature, high catalytic activity, and low costs have led to its widespread usage.The overall catalytic reaction process over Ag involves a feed mixture of methanol vapor, steam, and air. Two different reaction pathways have been proposed for the conversion of methanol to formaldehyde: 1) endothermic dehydrogenation (eqn (1)) and 2) exothermic partial oxidation (eqn (2)). An overview of the proposed reaction network has been discussed using eqn (1)–(9).5 It has been well-established that the presence of oxygen is essential for both formaldehyde production routes (eqn (1) and (2)), either as adsorbed O(a) species or in the gas phase, respectively.3,6–8 Oxygen also plays a key role in facilitating side reactions such as eqn (3) and eqn (5)–(8). Primary non-selective industrial byproducts in the reactor off-gas include H2, H2O and CO2 (eqn (1)–(9)).1,5,9 Hence, to mitigate the formation of carbon byproducts and improve the selectivity of formaldehyde, it is essential to minimize side-reactions. This refers both to homogeneous gas phase reactions, as well as surface oxygen routes to CO2 formation, both of which may be relevant.
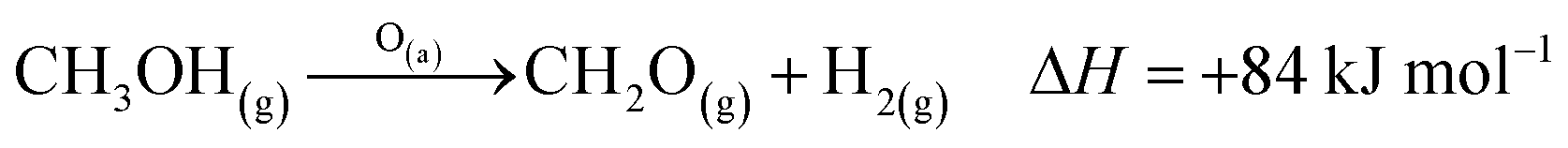 | (1) |
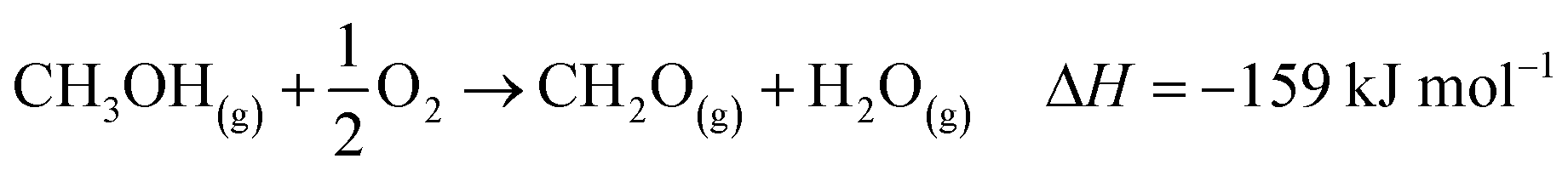 | (2) |
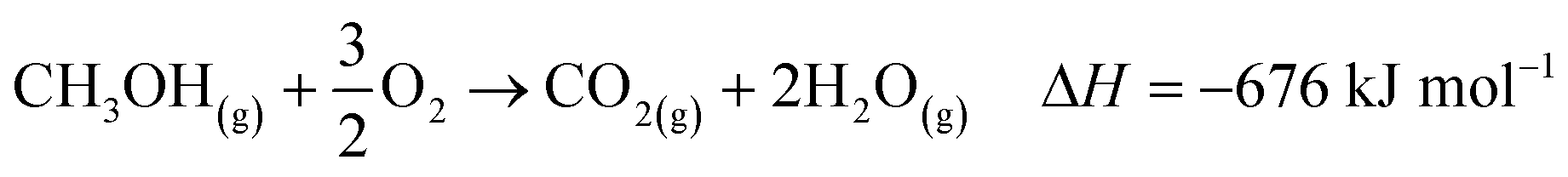 | (3) |
| CH3OH(g) → CO(g) + 2H2(g) ΔH = +91 kJ mol−1 | (4) |
| CH2O(g) + O2 → CO2(g) + H2O(g) ΔH = −519 kJ mol−1 | (5) |
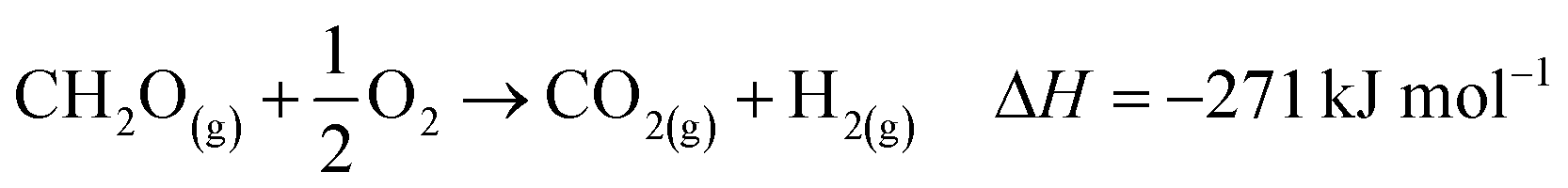 | (6) |
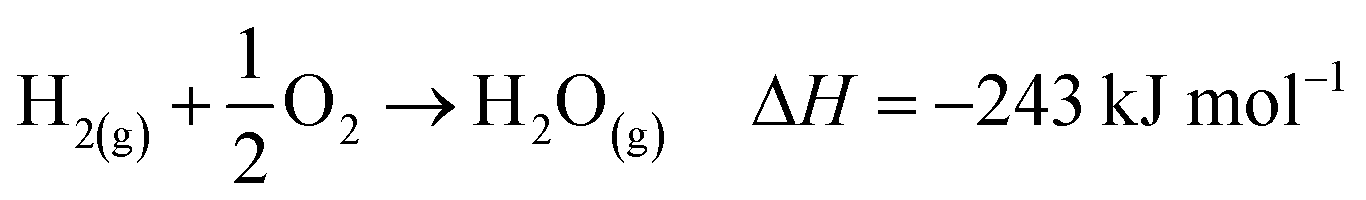 | (7) |
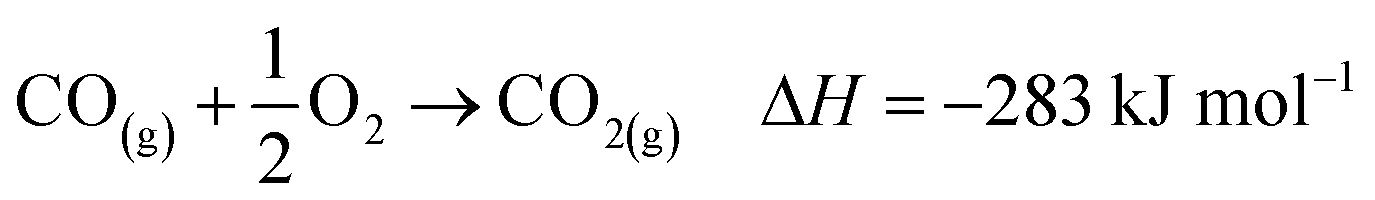 | (8) |
| CH2O(g) → CO(g) + H2(g) ΔH = +12 kJ mol−1 | (9) |
While the morphological changes in Ag have been intensively studied in the literature using surface studies, the relationship between the bulk catalyst restructuring and the different gas environments during methanol conversion has not been thoroughly investigated. Recent reactor studies and post-mortem scanning electron microscopy (SEM) characterization revealed the influence of high reaction temperatures on the Ag morphology, with different extents of faceting and pinhole formation under oxygen-containing atmospheres.5 However, electron microscopy still may not properly represent changes in the bulk catalyst, since it is primarily restricted to the surface (SEM) or requires preparation of nm scale subvolumes (e.g. for transmission electron microscopy (TEM)). In situ electron microscopy, such as ESEM, likewise can investigate only surface restructuring under model reaction conditions, and cannot reveal the full structural changes experienced by the bulk of the catalyst.11,18 In contrast, the high-penetration depth of X-rays offers the advantage of spatially-resolved non-invasive studies of technical samples, which can be readily combined with in situ measurements. This fosters an in-depth investigation of physicochemical changes without being restricted to the surface.20,21 Recent efforts in this context have led to the development of in situ X-ray microscopy cells for use at synchrotron radiation sources, many of which use micro-electromechanical systems (MEMS) based TEM chips,22–25 to perform experiments under industrially relevant conditions. Of currently available X-ray imaging methods, X-ray ptychography (also known as scanning coherent diffraction imaging) has consistently shown potential for high-resolution physicochemical studies of extended samples, including with in situ measurements.26,27 X-ray ptychography is sensitive to the local electron density of the sample,28 allowing for imaging of even weakly absorbing materials with enhanced contrast, which is particularly useful in studying porous systems.29,30
In this study, we utilize in situ 2D X-ray ptychography and correlative post-mortem electron microscopy to study the restructuring of Ag catalysts treated under various model gas environments. In situ morphological changes in the Ag catalyst were observed under industrially relevant conditions (ambient pressure and elevated temperatures up to 650 °C) using a MEMS-based nanoreactor (Fig. 1). A spatially-resolved study was facilitated using correlative in situ hard X-ray ptychography as a function of time and temperature to probe the initiation and progress of catalyst restructuring. Post-mortem SEM with energy-dispersive X-ray spectroscopy (EDX) mapping was employed to understand the observed redistribution of material after exposure of catalysts to reaction environments. Here, real-time study of Ag catalysts at technically relevant length scales, supported by visual and quantitative determination of these dynamic changes in the catalyst under a controlled environment was possible exclusively due to X-ray microscopy with synchrotron radiation. Additional catalyst studies in a laboratory-scale tubular reactor were performed to correlate the Ag restructuring observed in the nanoreactor to a regular reaction environment, thereby excluding other factors such as X-ray beam-induced changes or limited and unrepresentative sample volumes. This approach complements established methods, such as electron microscopy, and can be further extended to monitor industrial catalysts under reaction conditions.
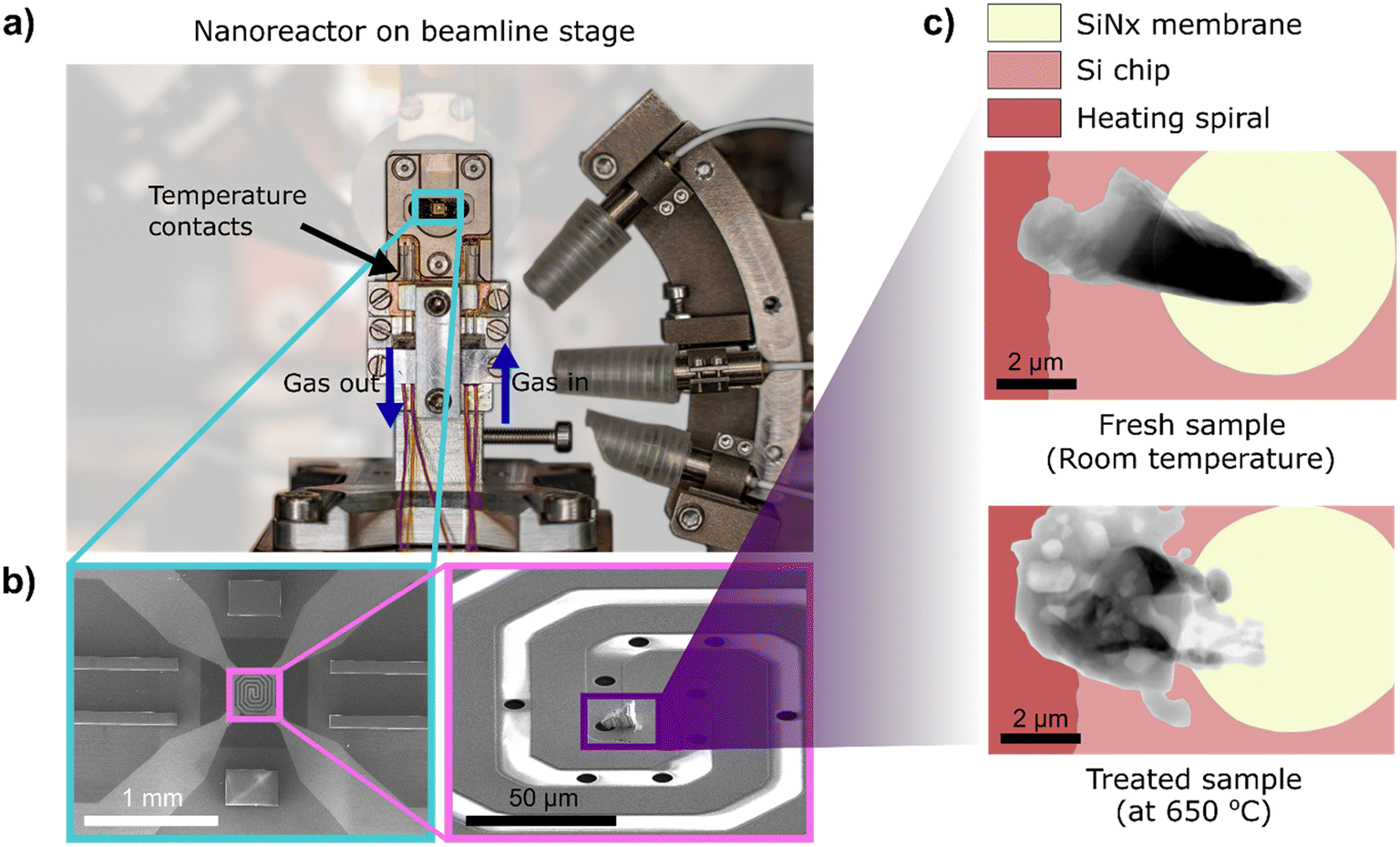 | ||
| Fig. 1 In situ nanoreactor a) implemented for use at synchrotron radiation sources, showing b) heating spiral (left) and zoomed-in view of the sample mounting region (right). c) Ptychographic projections (grayscale) of a fresh sample (top) mounted on a MEMS-based TEM chip, and a treated sample (bottom) illustrate changes in Ag catalyst morphology after exposure to elevated temperatures under different gas environments at atmospheric pressure in the nanoreactor. | ||
2. Experimental
2.1. Sample preparation
All samples used in this study were supplied by K. A. Rasmussen AS (Hamar, Norway). High purity (>99.99%) crystalline Ag particles with diameters in the range of 0.15 to 0.30 mm were used for in situ X-ray ptychography studies. Three irregularly shaped specimens studied at the P06 beamline, PETRA III, were prepared using a Helios G4 focused ion beam scanning electron microscope (FIB-SEM, Thermo Fisher Scientific, US). A particle edge with thickness ≤5 μm was fastened to the lift-out needle, and subsequently mounted covering the circular silicon nitride window (as shown in Fig. 1b and c) of the MEMS chips (DENSSolutions B.V., Delft, Netherlands) by the ion-beam-induced deposition of Pt. For the experiment at the NanoMAX beamline, MAX IV Laboratory, FIB milling using a Ga-ion source Nova NanoLab 600 DualBeam FIB/SEM (Thermo Fisher Scientific, US), and subsequent lift-out using an Omniprobe micromanipulator, was performed to mount specimens. A selected region of interest was cut out of the Ag particle edge. The Ag specimens were then transferred and fastened to the MEMS chips. With this technique, two irregularly-shaped specimens of 5–8 μm were prepared. A cuboid-shaped lamella was also prepared by milling a specimen out of the bulk Ag particles. For all irregularly-shaped samples, care was taken not to expose the bulk specimen to the FIB so that the original morphology and structure of the Ag particles were retained.2.2. In situ catalyst studies using X-ray ptychography
X-ray ptychography was performed on the samples mounted on MEMS-based TEM chips. Two different X-ray ptychography experiments were performed for in situ studies (protocol details in Table 1): 1) at the nanoprobe endstation PtyNAMi of the P06 beamline of PETRA III (Deutsches-Elektronen Synchrotron DESY, Hamburg, Germany) at an incident photon energy of 10.3 keV using an EIGER X 4 M detector (DECTRIS AG, Baden, Switzerland),31–33 and 2) at the diffraction endstation of the NanoMAX beamline34,35 at the MAX IV Laboratory (Lund University, Lund, Sweden),36 at an incident photon energy of 10 keV using an EIGER2 X 4 M detector (DECTRIS AG, Baden, Switzerland).37 Beamline-respective X-ray scanning parameters were used to record high-resolution ptychographic projections with a scan duration of 4–5 min per projection (see ESI,† section 3).| Catalyst behavior | Sequential gas flow composition (vol%) | Maximum temp. (°C) | Catalyst shape | Sample IDc | ||
|---|---|---|---|---|---|---|
| a Two similar samples were tested under enclosed ambient air conditions. b Initial gas flow of pure He. The top chip membrane was found to be broken at the end of the temperature cycle resulting in unknown gas mixture of He/Air. c Samples are named according to synchrotron P: PETRA III, M: MAX IV, followed by the first gas environment they were exposed to. | ||||||
| Restructuring study (P06 beamline) | He | 650 | Irregular | PHe | ||
| Aira (enclosed) | 650 | Irregular | PA1, PA2 | |||
| Dynamic study (NanoMAX beamline) | Air (enclosed) | Air (enclosed) | — | 500 | Irregular | MA1 |
| He/airb | O2/He = 11.6/88.4 | O2/H2/He = 11.6/3.8/84.6 | 500 | Lamella | MA2 | |
| He | O2/H2/He = 11.6/3.8/84.6 | H2/He = 3.8/94.2 | 500 | Irregular | MHe | |
The commercially available Climate nanoreactor (DENSSolutions B.V., Delft, Netherlands, compatible with JEOL electron microscopes) was adapted through an in-house developed beamline interface for facilitating experiments at synchrotron radiation sources (ESI,† section 1–2). This nanoreactor was used to study the mounted Ag catalyst samples under exposure to different temperatures and various gas mixtures comprising O2, H2, and He (Table 1). All experiments were carried out at atmospheric pressure. A total gas flow rate of 0.5 mL min−1 was maintained in the nanoreactor setup for all conditions using mass flow controllers (Bronkhorst High-Tech B.V., Ruurlo, Netherlands) integrated into the in-house gas dosing system. The sample is heated using the commercial heating control unit equipped with a closed-loop feedback control to adjust for the differences in temperature that might occur during the experiment or due to the gas flow. Temperature simulations of a similar setup have been shown previously in literature.23 The Ag samples were step-wise heated up to 650 °C at the P06 beamline (to study catalyst restructuring), and up to 500 °C at the NanoMAX beamline (to study the dynamic behavior of catalysts at elevated temperatures), using the heating control unit with ptychographic projections recorded at each temperature setpoint (see ESI,† Fig. S14 and S15 for temperature cycles of all samples measured at both beamlines).
2.3. Tubular reactor setup
750 mg of electrolytic Ag particles (0.25–0.50 mm) were placed in a quartz reactor (inner diameter: 11 mm). A thermocouple was placed in a quartz duct through the center of the catalytic bed. The gas atmosphere was introduced at ambient temperature, adjusting the flows to maintain approximately 80 ms contact time. The furnace was heated to and kept at 650 °C for 48 h. The catalyst was then cooled to ambient temperature.5,38 The respective treatments have been outlined in Table 2.2.4. Post-mortem electron microscopy
High-resolution field emission secondary electron SEM images of specimens after the in situ catalytic studies performed at 650 °C were acquired using the Everhart–Thornley detector on the Nova Nano SEM 450 (Thermo Fisher Scientific, US) at the DESY NanoLab. SEM–EDX elemental mapping was performed using an X-Max 150 EDS silicon drift detector (Oxford Instruments, UK). For samples treated up to 500 °C at the NanoMAX beamline, a post-mortem SEM study was performed using a FlexSEM 1000 VP-SEM (HITACHI Ltd., Japan). The SEM–EDX mapping for these samples was performed using the XFlash 6–30 (Bruker, US) at the MAX IV Laboratory. Post-mortem analysis of specimens treated in the quartz reactor was performed using a Helios G4 FIB-SEM (Thermo Fisher Scientific, Netherlands) for cross-sectioning and imaging of the particles. During the cross-sectioning, a Pt protection layer was deposited on the top surface, before the milling was carried out, using an ion beam acceleration voltage of 30 kV and a current of 2.4 nA. Secondary electron SEM images were acquired using an acceleration voltage of 5 kV.3. Results and discussion
3.1. In situ X-ray ptychography to study structural changes
To investigate the catalyst restructuring as a function of treatment time, specimens measured at PETRA III under He (PHe), and under synthetic air (PA1, PA2) were examined via X-ray ptychography during each temperature cycle (Table 1). This comprised stepwise heating up to 650 °C, a constant temperature condition of 650 °C, and subsequent cooling to room temperature. Supporting videos for all samples show accompanying changes under experimental conditions (see ESI† videos). The ptychographic projections were first visually assessed to determine the onset of bubble formation (Fig. 2). Catalyst restructuring was found to occur at 200 °C under ambient air. Since this was mostly in regions closer to the deposited Pt, it is difficult to conclude whether this portrays changes in Ag structure or in the carbon content (which can vary between 20–70 at% (ref. 39)) of the deposited Pt under oxidizing conditions. However, bubble growth and propagation were vividly observed at ca. 250 °C for PA1 and PA2 (Fig. 2b and c), followed by accelerated growth at elevated temperatures. Surprisingly, catalyst restructuring was found to occur in PHe under non-reactive He environment. This is presumably due to the dissolution of He and diffusion through the Ag metal. Previous studies have shown that He can penetrate as small clusters into the interstitial sites of the Ag lattice.40 Increase in temperature of the Ag metal results in a higher diffusion constant of He.41 This increased He diffusivity could result in a higher concentration of He in solid solution in Ag during the experiment. Consequently, larger gas bubbles were observed in the Ag specimens as a function of temperature (Fig. 2a). A delayed onset of restructuring was observed under He environment compared to oxidizing environment (Fig. 2). The deposited Pt region of PHe was not in the imaged field of view, hence only the direction of propagation of such gas bubbles was noted for PHe. However, an increase in temperature above 350 °C resulted in accelerated bubble formation within PHe, again at notably higher temperature than under oxidizing conditions.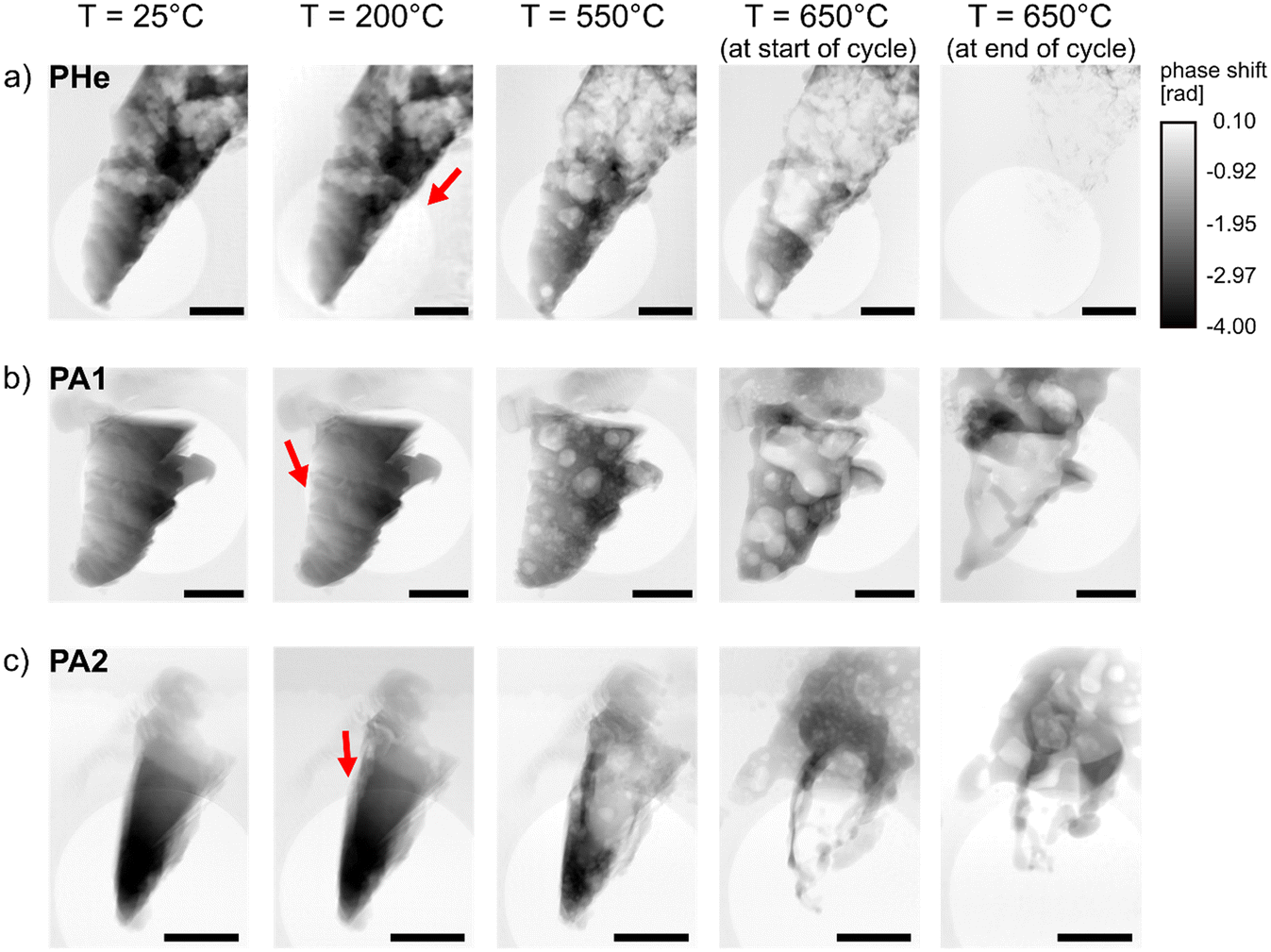 | ||
| Fig. 2 In situ X-ray ptychography projections of a) PHe, b) PA1, and c) PA2 as a function of applied temperature for T = 25, 200, 550, and 650 °C (showing start and end of the constant temperature cycle) under their respective gas environment. The red arrows indicate both bubble formation and the direction of bubble propagation. All scale bars = 2.4 μm. | ||
Surface pinholes were previously observed in literature at temperatures above 400 °C and near defect sites, with the initial formation of hill-like nodules on the catalyst surface.10–12 While in situ ESEM studies had earlier shown widespread formation of surface pinholes around 600 °C, in situ X-ray ptychography provides a deeper understanding of rapid bubble formation and coalescence occurring well below 400 °C by providing depth information on the entire catalyst cross-section. Further temperature increase showed an increase in porosity in all samples (PA1, PA2, and PHe), resulting in catalysts which were completely filled with bubbles upon heating from 500 °C to 550 °C. The formation of larger bubbles resulting from the rapid development, movement, and coalescence of tiny bubbles was observed. This shows that the high mobility of Ag above the Tammann temperature facilitates the propagation of gas bubbles, thereby contributing to the redistribution of material. This phenomenon is clearly not limited to the catalyst surface.
It is important to note that the surface pinholes and the gas bubbles at 650 °C are comparable in size to the prepared specimen. The resultant hollow catalyst is then susceptible to structural collapse due to the sintering of Ag at such high temperatures. A constant temperature condition was maintained at 650 °C for approximately 2 hours to understand the influence of treatment time on the Ag catalyst. All samples showed signs of severe structural collapse (Fig. 2 and SEM images in ESI,† Fig. S7). The key influence of temperature revealed here on the structural collapse can be attributed to the synergistic effect of increased porosity and sintering of Ag catalyst, which is known to occur at such high temperatures of 600–700 °C.1 The movement of these bubbles in the catalyst material, visualized as a function of temperature in Fig. 2 and ESI,† section 4.1, reveals rapid material transfer within the catalyst. This is accompanied by the movement of material away from the scanning area due to surface explosion of bubbles and redistribution of material to different parts of the sample holder at temperatures above 550 °C (also, SEM images of ESI,† Fig. S7).
The observed restructuring of Ag was then quantitatively assessed to relate the movement of bubbles to the catalyst structure as a function of temperature. To study the influence of time and temperature as key control parameters, the sample region overlapping with the circular silicon nitride window was selected as the region of interest (ROI) for all chips (ESI,† Fig. S12) to obtain a uniform background. The relative change in mass, measured as the change in integrated phase shift from X-ray ptychography projections in the selected ROIs, was then monitored as a function of time and temperature (Fig. 3a–c).
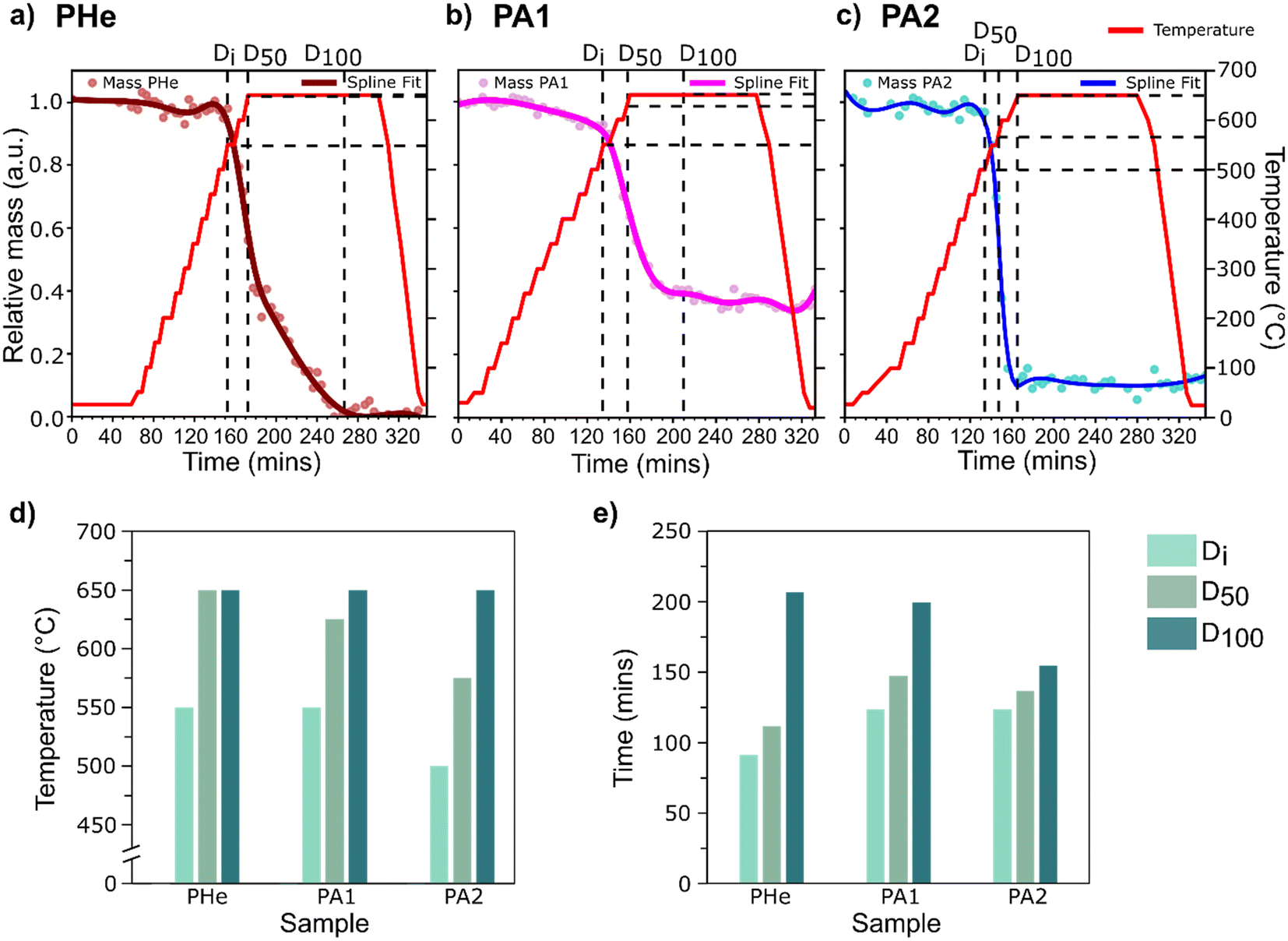 | ||
| Fig. 3 Relative change in mass of samples a) PHe, b) PA1, and c) PA2 over time, including temperature program during step-wise heating and cooling. The extent of catalyst restructuring is described using three different events, Di, D50, and D100, and their corresponding d) temperature and e) treatment time for each sample. Time values were offset to account for the different absolute time points of the first heating ramps. | ||
For the quantitative mass change study, the following three events were studied:
1) The catalyst restructuring indicating a resultant initial mass change or ‘Di’,
2) 50% of the maximum attainable catalyst restructuring or ‘D50’,
3) The maximum attainable catalyst restructuring at 650 °C or ‘D100’.
The restructuring extents, Di and D50, under ambient air conditions (PA2) were attained at temperatures at least 50 °C below that under He (PHe), indicating more extensive restructuring in the presence of O2. Although D100 by definition indicates the maximum structural change experienced by three samples at 650 °C (Fig. 3d), the relative time required to achieve this is significantly different (Fig. 3e). Overall, the maximum restructuring was achieved relatively faster and at a lower temperature in air compared to He. The difference in relative mass observed at 650 °C between samples treated under similar gas environments (PA1 and PA2) can be attributed to the potential influence of the initial material thickness (which was not uniform for each sample) or the minor differences in heating ramp rate.
The observed first derivative plot shows a concave-up profile for all samples (ESI,† Fig. S14 and S15). The steeper derivative curve for PA2 shows a faster restructuring rate compared to PA1. This difference in the rate of movement of material can be attributed to different catalyst thicknesses, heterogeneity in the initial distribution of defects, or varying morphology between the two specimens. The inflection point for all samples coincides around the D50, with a diminishing rate of change after the inflection point. A comparatively gradual mass change in PHe was observed where a change in slope after the inflection point reflects delayed catalyst restructuring, and hence the longer time required to reach D100. Therefore, although the morphological changes are more aggressive at higher temperatures, the plateaued catalyst restructuring is reflective of the limited scope for mass change due to the absence of sufficient catalyst material to further restructure after reaching 650 °C.
Post-mortem SEM–EDX maps (ESI,† section 5) showed the presence of Ag redistributed as fragments/large chunks from surface explosions of bubbles and the homogeneous layers of Ag debris in all directions around the specimens. The constant unidirectional gas flow did not seem to influence the direction of redeposition of Ag material on the MEMS-based chips, implying a high pressure event during bubble rupture. This also correlates to the formation of Ag debris on the catalyst surface previously visualized in literature.12 The SEM–EDX maps revealed the distribution of Pt over the entire catalyst surface (ESI,† Fig. S17-S20). The ion-beam induced deposition of Pt was originally carried out at one selected point on the chip to fasten the sample, which contradicts this observed post-mortem Pt distribution on the catalyst. Previous studies have shown that the deposited Pt undergoes noticeable changes in structure under high temperatures,24 due to the organometallic ligands present in the compound. This might also contribute to the changes observed in the sample region adjacent to the deposited Pt already at 200 °C, a temperature lower than those reported in literature.
SEM EDX maps showed Ga distribution across the entire sample, even though Ga exposure occurred only on one edge of the Ag particle, unlike the FIB-milled lamella. Literature studies have shown that the FIB-milling is often accompanied by the Ga implantation on the substrate and its redeposition during material sputtering.39 Additionally, the ion-beam induced deposition of Pt at 30 kV also results in Ga implantation in the exposed sample region, with an estimated composition of Pt/C/Ga/O = 45/45/5/5 at%.39,42 However, the high mobility of the Ag under operating conditions resulted in a redistribution of the Pt and Ga within the entire bulk catalyst. Since Pt can be reactive under H2/O2, catalytic activity data (not included here) measured from samples prepared and mounted via FIB must be considered with caution.
The formation of dynamic surface pinholes, as known from literature, was confirmed here to result from the movement of bubbles within the catalyst material and their eventual explosion at the catalyst surface (see ESI† video). The observed onset of bubble formation at regions closer to the deposited Pt can be attributed to increased FIB-induced sample damage, Pt deposition, and likely Ga implantation into the specimens, thereby introducing additional defects or activation promoters to the catalyst structure. Despite the notable difference in the delayed onset of catalyst restructuring under He environment compared to oxidizing environment, the effect of temperature was found to be a dominating factor under all gas environments. It is important to note that the formation of gas bubbles in Ag catalysts might be crucial to industrial formaldehyde production. The constant formation, mobility, and explosion of bubbles allow constant material movement and evolution of active adsorbed sub-surface Oγ species, facilitating reaction between adsorbed Oγ species and methanol.
3.2. In situ X-ray ptychography to study dynamic bubble formation and growth
It was established above that an increase in temperature beyond 500 °C has a dramatic effect on bubble formation and Ag restructuring. Hence, the dynamic behavior of the catalysts was studied while avoiding significant catalyst restructuring. This was done by monitoring bubble formation and growth in the specimens measured at MAX IV under He (MHe), and under synthetic air (MA1 and MA2) during temperature ramps up to a maximum of 500 °C, with exposure of each sample to a series of different gas environments (Table 1). Resultant mass change was quantified to estimate of effect of different environments on catalyst restructuring (Fig. 4 and ESI,† section 4.2). The difference in catalyst restructuring was again visualized as a function of treatment time, as shown in supporting videos for samples MHe, MA1, and MA2 (see ESI† videos). | ||
| Fig. 4 In situ X-ray ptychography projections of samples a) MA1, b) MA2, and c) MHe. The top row for each sample shows the fresh sample. The subsequent rows for each sample exhibit morphological changes at the start and end of each constant temperature condition of 500 °C, under their respective environments. The red arrows indicate bubble formation and the direction of bubble propagation. All scale bars = 2 μm. | ||
Using MHe, due to a wider imaging field of view, a delayed onset of bubble formation could be confirmed at around 250 °C under the He environment (Fig. 4c, cycle 1), compared to air in MA1 (Fig. 4a, cycle 1). Also, significantly fewer gas bubbles were observed in MHe in the first temperature cycle. Subsequently in a H2/O2 mixture (O2/H2/He = 11.6/3.8/84.6 vol%) (Fig. 4c, cycle 2), bubble formation occurred at 450 °C during the second temperature heating ramp, with rapid catalyst restructuring at 500 °C. In addition to O2 modifying the surface, the exothermic formation of water can lead to localized heat generation which could in principle contribute to the relatively higher rate of catalyst restructuring. On the other hand, the nanoreactor design includes a fast feedback control which can compensate for changes in temperature which may occur due to exothermic processes. During the third MHe temperature heating ramp (H2/He = 3.8/96.2 vol%) (Fig. 4c, cycle 3), less bubble formation was observed around 450 °C, and slower bubble growth or propagation. This could indicate relatively low solubility of H2. The absence of O2 limits the possibility of catalyst restructuring and potential formation of hydroxyl species. For the sample MA2 pretreated in He/Air, larger dimension bubbles were formed already after the first temperature cycle (Fig. 4b, cycle 1). Since the initial gas condition involved pure He, the broken top chip membrane during the experiment resulted in loss of inert environment (Table 1). This led to reduced extent of material restructuring compared to MA1, with relatively fewer but larger gas bubbles. Such large gas bubbles were prevalent in samples exposed to ambient air (i.e., MA1 Fig. 4a, cycle 1) which showed highly accelerated bubble formation and growth in comparison to inert environment. The second temperature heating ramp of MA2 under O2/He = 11.6/88.4 vol% showed rapid bubble formation around 450 °C (Fig. 4b, cycle 2), resulting in a highly porous catalyst structure. In contrast, during the third temperature heating ramp under O2/H2/He = 11.6/3.8/84.6 vol% (Fig. 4b, cycle 3), relatively less sample restructuring was seen, despite the presence of O2. Reduced catalyst restructuring observed during the third temperature heating ramp, despite a change in gas environment in both MHe and MA2, indicates a potential threshold in catalyst restructuring at a specific temperature. Hence, the influence of O2 on restructuring is evident, however the effect of H2 requires further study. This also indicates catalyst restructuring to be a temperature- and time-dependent phenomenon. Since the catalyst restructuring and pinhole/pore formation correlate with formaldehyde formation,10 the reaction under adiabatic conditions is likely kinetically controlled.
Quantitative assessment was carried out using selected ROIs (ESI,† Fig. S13) for all three samples. Estimation of the Pearson coefficient for bivariate relationships shows a negative correlation between the mass change and the measurement time for MA1, MA2, and MHe (Fig. 5a–c). It is evident from calculated statistical metrics such as high absolute r-value and low P-value, with high R-squared values denoting an excellent linear fit for the initial temperature cycles (ESI,† Table S3). Here, a strong correlation of relative mass change with treatment time within the first/initial constant temperature conditions for all samples, followed by a weak correlation in the final constant temperature condition (indicated by decreased absolute r-value) for all three samples, shows the significant effect of both gas environment and treatment time on material movement. This is because the material experienced almost complete restructuring during the first/initial temperature cycles in air. Hence, during the final temperature cycle, treatment time had less or no influence on the relative mass changes. No changes in catalyst structure confirm the previous hypothesis that bubble formation within the Ag catalyst is likely to be time-dependent at a specific temperature. It is important to note that the relative mass values shown in the plots do not always begin at 1.0 (Fig. 5). This is prominent in MA1 where notable catalyst restructuring and material movement in air within the selected ROI occurred before the first constant temperature condition (see ESI,† Fig. S15a for a complete overview of the temperature steps while heating up to 500 °C and the corresponding change in mass observed at each temperature). This reflects the change in catalyst structure and corresponding material redistribution occurring during heating, which precedes the constant-temperature observations. Hence, the specimens examined at 500 °C are no longer regarded as in the fresh state, and this again confirms the associated bubble formation and material movement at lower temperatures, particularly on exposure to oxidizing environment.
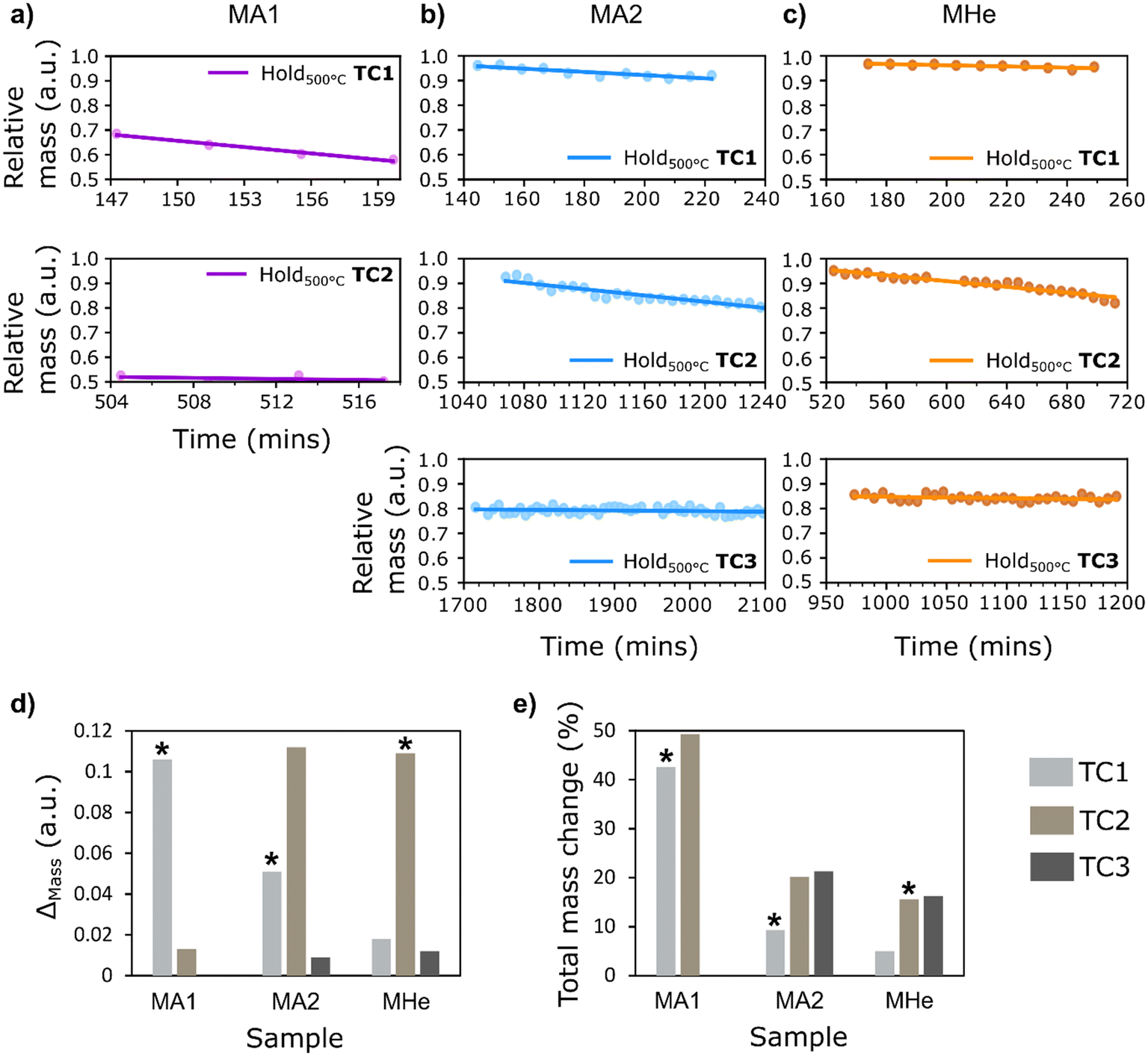 | ||
| Fig. 5 Relative change in mass in the selected ROIs plotted as a function of time to study the catalyst behavior at a constant temperature of 500 °C for three samples – a) MA1, b) MA2, and c) MHe. d) Bar chart showing change in mass (ESI,† Table S4) during each temperature cycle (denoted as TC) for all three samples. e) The corresponding percentage change in mass with respect to the initial mass, showing estimated percentage decrease in the observed ROI after each period of holding at a constant temperature. (* denotes the cycle with the first exposure of sample to O2). | ||
For MA1, the first temperature ramp in air resulted in significant material restructuring at lower temperatures, well below the hold temperature of 500 °C (also see ESI,† Fig. S15a for a detailed overview of the corresponding mass changes). As observed in Fig. 4, MA1 experienced maximum catalyst restructuring and hollow gas bubble formation in air compared to other samples within the same time frame. Hence, the temperature cycles resulted in a total mass change of 49.3% in MA1 (Fig. 5e). The changes occurring in MA2 under sequential exposure to He/Air, O2/He = 11.6/88.4 vol%, and O2/H2/He = 11.6/3.8/84.6 vol% show comparatively less change in mass, with a total reduction of 21.3% after the final constant temperature condition. A net reduction in mass by 9.3% and 20.2% after the first and second holding time periods, respectively, was observed in MA2 under exposure to their respective mixtures. Sample MHe, initially treated under He in the first temperature cycle followed by O2/H2/He = 11.6/3.8/84.6 vol% and H2/He = 3.8/96.2 vol% in the subsequent cycles, was found to have the lowest total change in mass by roughly 16.3% (also evident in visual changes noted in Fig. 4c). This is due to the significantly less catalyst restructuring during the first step under He exposure (a net decrease of 5%), followed by reduced restructuring in the subsequent steps. The gas environment, temperature, and time are the key factors in the first temperature cycle. However, the relatively lower mass change in the final temperature cycle in all samples shows that over a prolonged experiment duration the treatment time becomes a restricting factor at a specific temperature.
While differences in material behavior were clearly distinguishable under inert vs. oxidizing conditions, the pretreatment of the sample under these gas environments in the first temperature cycle does not provide conclusive evidence regarding the effect of gas mixtures of O2, H2, and He on the bubble evolution rates in the successive temperature cycles. Millar et al. and Lervold et al. showed that a higher formation of pinholes was found when O2 was introduced to the CH3OH feed.5,10,38 This is supported by the current study that formation of tiny bubbles and catalyst restructuring is accelerated in the O2/H2 rich environment. In future studies, quantitative assessment of the influence of various gas environments at different temperatures will benefit from measurements on samples of identical size and shape, without prior exposure to other gas conditions. This would allow better control experiments and further quantification of the dynamic changes resulting from bubble formation and growth. Furthermore, the employment of faster measurement techniques, such as multi-beam ptychography,43–45 could improve time resolution and therefore capture the rapid formation of gas bubbles to accurately study such dynamic effects within the catalyst. Simultaneous gas-phase analysis of the reaction products could help future studies to accurately correlate the structural changes within the catalyst to its activity and selectivity. However, this requires sensitive gas analysis instrumentation due to the small sample volume mounted inside the nanoreactor and consequently a low product concentration.
3.3. Ex situ SEM of catalysts treated in a tubular reactor
To validate the catalyst restructuring under different gas environments, and free from the influence of FIB-introduced defects or other measurement conditions, catalyst testing was performed in a fixed-bed quartz reactor. Ex situ FIB-SEM studies of the catalysts (Fig. 6) show that varying degrees of morphological changes occurred after exposure to different atmospheres at elevated temperatures (650 °C). The fresh Ag particles display a few surface pinholes and some cavities below the surface (Fig. 6a). Overall, the fresh particles are crystalline and heterogeneous with respect to the size and occurrence of such cavities. For the samples exposed to N2, H2/N2, or He, changes to the surface relative to the fresh Ag cannot be distinguished on the scale accessed here. The sample exposed to N2 exhibits a small number of sub-surface pores, similar to the fresh material (Fig. 6b). However, when looking at the He treated sample, a larger number of cavities of varying sizes (mostly Ø < 1 μm) are found below the surface (Fig. 6c). Exposure to a H2 containing atmosphere (H2/N2 = 2/98 vol%) did not result in significant changes below the surface (Fig. 6d). The two elongated cavities seen below the surface resemble imperfections that occur in fresh Ag samples. Hence, neither for N2 nor H2/N2 can the sub-surface cavities observed after exposure be unequivocally attributed to an effect of the gases. The outlier to this is the He exposed sample, which showed substantially more sub-surface cavities due to apparent He dissolution.40,41 This observation is important for studies where He is used as a carrier or inert gas, and it is consistent with the bubble formation observed in specimens tested in the nanoreactor under non-reactive and oxygen-free gas environments. Both the effect of He on the formation of sub-surface cavities and the underlying mechanism should be clarified in the future. | ||
| Fig. 6 SEM image (top) and FIB-milled cross-sectioned SEM image (bottom) of Ag catalysts, a) fresh, and after 48 h of heat treatment under different gas environments: b) N2, c) He, d) H2/N2 = 2/98 vol%, e) synthetic air and f) H2/O2/N2 = 2/6/92 vol%. The Pt protection layer was deposited on the top surface before FIB milling (pink dashed line). The pores on the catalyst surface (yellow circles) and the cavities inside the catalyst exposed after FIB-milling (green circles) have been indicated. | ||
After exposure to synthetic air (O2/N2 = 21/79 vol%), more pronounced structural changes were observed. The surface was smoothened, with terraces and pinholes, and a large amount of sub-surface cavities formed as a result of O2 dissolution into Ag (Fig. 6e). The specific cross-section that was milled displayed a relatively large region without cavities that is pointing out of the surface and likely is a result of so-called Ag spitting or nodule formation (i.e., relocation of Ag resulting from dissolution and enhanced mobility). The presence of both H2 and O2 (H2/O2/N2 = 2/6/92 vol%) resulted in surface smoothing and terracing, as well as a large number of pinholes visible on the surface (Fig. 6f). Below the surface, the catalyst shows a large number of pores/cavities, some considerably larger (Ø ∼ 5 μm) than those observed in the sample exposed to air. The formation of such large pinholes in bulk Ag is similar to those observed in the nanoreactor under synthetic air or oxidizing conditions which explains the eventual structural collapse of 5–8 μm sized Ag specimens tested at the P06 or NanoMAX beamline. While the formation of pinholes in both air and H2 oxidation samples is documented in the literature,5,14 the significant increase in pinholes and cavity density for the H2 oxidation catalyst is interesting in relation to methanol oxidation. In the presence of adsorbed O, surface and subsurface hydroxyl species can be formed. This results in hydrostatic pressure build-up inside the Ag catalyst, and the release of this stress leads to the formation of surface pinholes.12,18
4. Conclusion
The Ag-catalyzed partial oxidation of methanol experiences catalyst restructuring with formation of surface pinholes under operating conditions. In this study, dissolution of gases and induced catalyst restructuring were visualized with depth information using in situ X-ray ptychography of 5–8 μm thick Ag catalysts. The gas-filled bubbles formed were eventually found to rupture on the catalyst surface, resulting in dynamic surface pinholes/pores under different gas environments. Redeposition of material after surface explosion of these bubbles was observed on various parts of the sample using post-mortem SEM. The Ag catalyst was found to restructure in non-reactive environments with the formation of pores or cavities within the bulk catalyst. The specimen exposed to N2 showed very few pores and was similar in appearance to the fresh catalyst. The relatively higher number of cavities and bubble formation during heating under He environment revealed the notable phenomenon of He dissolution into the Ag catalyst. The presence of restructuring under non-reactive gas environments (likely due to gas dissolution) can be influenced and accelerated by the presence of contaminants or existing defects in the Ag catalyst. Such defects may be introduced during the fabrication process, for example. More extensive surface faceting and catalyst restructuring were observed in the presence of O2/H2. This is explained by reaction of O2 with the Ag surface O2, which is known to increase the surface pinhole formation. The relative movement of mass, which was quantified using in situ ptychography within selected regions of interest, shows that treatment time and temperature are the key driving factors for gas dissolution and catalyst restructuring, with different dynamics depending on the gaseous environment. It is worth noting that no noticeable beam damage was observed during consecutive scans at regular intervals. Increased temperature led to severe catalyst restructuring due to increased bubble formation and higher Ag mobility. Furthermore, prolonged treatment time at constant elevated temperature showed time-dependent catalyst restructuring. This study confirms that the Ag catalyst develops dynamic pore structures at elevated temperatures and particularly in the presence of O2 or He. Supporting evidence on catalyst restructuring was also obtained using post-mortem FIB-SEM on catalysts treated in a lab-scale tubular reactor. This study acts as a benchmark for the identification of crucial experimental parameters influencing the restructuring of the Ag catalyst, while extending the application of hard X-ray microscopy under realistic conditions at technically relevant length scales. The current reactor cell limits the thickness of measured specimens to a few μm, although in principle sample throughput can be improved by placing multiple samples on the same chip simultaneously. Faster scanning techniques can in future be exploited for kinetic studies on the same catalyst system. The new insights into the formation of surface pinholes resulting from the dissolution of O, H, and He within the catalyst help deepen the understanding of the material behavior under realistic conditions while forming the basis for future multimodal and multiscale morphological, kinetic, and chemical studies.Data availability
Data is available at KITOpen, the open access repository of Karlsruhe Institute of Technology. The 2D reconstructions of Ag catalysts studied using in situ X-ray ptychography at P06 beamline and NanoMAX beamline are available at https://doi.org/10.35097/ebe9hsrmkpcwq5fv and https://doi.org/10.35097/prxrp7rx8rgfhdxu, respectively. Further data supporting this article have been included as part of the ESI.†Author contributions
Conceptualization: MK, HJV, CGS, TLS; data curation: SD, MK, YvV, TB; formal analysis: SD, MK, YvV, TB; funding acquisition: HJV, TLS; investigation: SD, MK, YvV, TB, SB, HJV, TLS; methodology: SD, MK, ML, CGS, TLS; project administration: CGS, HJV, TLS; resources: SD, MK, SB, CGS, HJV, TLS; software: SD, MK, ML, CGS; supervision: CGS, HJV, TLS; validation: SD, MK, YvV, TB; visualization: SD, MK, TB; writing – original draft: SD, MK, YvV, TB, TLS; writing – review & editing: all authors.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was funded by the German Federal Ministry of Education and Research (Bundesministerium für Bildung und Forschung, BMBF), project COSMIC 05K19VK4. The silver particles were provided by K. A. Rasmussen AS, Norway. The authors acknowledge the support from the Research Council of Norway to iCSI (industrial Catalysis Science and Innovation, no. 237922) and NorFab (Norwegian Micro- and Nano-Fabrication Facility, no. 295864). We also acknowledge Lund University for instrument access at Lund NanoLab and thank Ofentse Makgae for helping with the preparation of samples used during the NanoMAX beamtime. We thank Jan-Torben Roeh and Anite Ehnes from DESY for their assistance in developing the nanoreactor. The authors are grateful to PETRA III, DESY (Hamburg, Germany), a member of the Helmholtz Association, for the provision of beamtime within the scope of a long-term proposal with ID II-20190762. We would like to thank Sina Roeper, Sebastian Weber, and Shweta Sharma for their help during these beamtimes. We acknowledge the support of the P06 group of PETRA III provided during the experiment setup. We acknowledge MAX IV Laboratory for time on the NanoMAX Beamline under Proposal 20200394. Research conducted at MAX IV, a Swedish national user facility, is supported by the Swedish Research Council under contract 2018-07152, the Swedish Governmental Agency for Innovation Systems under contract 2018-04969, and Formas under contract 2019-02496. We thank Arno Jeromin, Satish Kulkarni, and Thomas Keller from the DESY NanoLab for helping with the acquisition of SEM and SEM–EDX images.References
- G. J. Millar and M. Collins, Industrial Production of Formaldehyde Using Polycrystalline Silver Catalyst, Ind. Eng. Chem. Res., 2017, 56(33), 9247–9265, DOI:10.1021/acs.iecr.7b02388.
- A. W. Franz, H. Kronemayer, D. Pfeiffer, R. D. Pilz, G. Reuss, W. Disteldorf, A. O. Gamer and A. Hilt, Formaldehyde, in Ullmann's Encyclopedia of Industrial Chemistry, John Wiley & Sons, Ltd, 2016, pp. 1–34, DOI:10.1002/14356007.a11_619.pub2.
- M. Qian, M. A. Liauw and G. Emig, Formaldehyde Synthesis from Methanol over Silver Catalysts, Appl. Catal., A, 2003, 238(2), 211–222, DOI:10.1016/S0926-860X(02)00340-X.
- K. V. Raun, L. F. Lundegaard, J. Chevallier, P. Beato, C. C. Appel, K. Nielsen, M. Thorhauge, A. D. Jensen and M. Høj, Deactivation Behavior of an Iron-Molybdate Catalyst during Selective Oxidation of Methanol to Formaldehyde, Catal. Sci. Technol., 2018, 8(18), 4626–4637, 10.1039/C8CY01109E.
- S. Lervold, K. Arnesen, N. Beck, R. Lødeng, J. Yang, K. Bingen, J. Skjelstad and H. J. Venvik, Morphology and Activity of Electrolytic Silver Catalyst for Partial Oxidation of Methanol to Formaldehyde Under Different Exposures and Oxidation Reactions, Top. Catal., 2019, 62(7–11), 699–711, DOI:10.1007/s11244-019-01159-0.
- G. I. N. Waterhouse, G. A. Bowmaker and J. B. Metson, Mechanism and Active Sites for the Partial Oxidation of Methanol to Formaldehyde over an Electrolytic Silver Catalyst, Appl. Catal., A, 2004, 265(1), 85–101, DOI:10.1016/j.apcata.2004.01.016.
- A. J. Nagy, G. Mestl, D. Herein, G. Weinberg, E. Kitzelmann and R. Schlögl, The Correlation of Subsurface Oxygen Diffusion with Variations of Silver Morphology in the Silver–Oxygen System, J. Catal., 1999, 182(2), 417–429, DOI:10.1006/jcat.1998.2388.
- C.-B. Wang, G. Deo and I. E. Wachs, Interaction of Polycrystalline Silver with Oxygen, Water, Carbon Dioxide, Ethylene, and Methanol: In Situ Raman and Catalytic Studies, J. Phys. Chem. B, 1999, 103(27), 5645–5656, DOI:10.1021/jp984363l.
- G. Halbritter, W. Muehlthaler, H. Sperber, H. Diem, C. Dudeck and G. Lehmann, Manufacture of Formaldehyde, US Pat., US4072717A, 1978 Search PubMed.
- G. J. Millar, M. L. Nelson and P. J. R. Uwins, In Situ Imaging of Catalytic Etching on Silver during Methanol Oxidation Conditions by Environmental Scanning Electron Microscopy, J. Catal., 1997, 169(1), 143–156, DOI:10.1006/jcat.1997.1653.
- P. J. R. Uwins, G. J. Millar and M. L. Nelson, Dynamic Imaging of Structural Changes in Silver Catalysts by Environmental Scanning Electron Microscopy, Microsc. Res. Tech., 1997, 36(5), 382–389, DOI:10.1002/(SICI)1097-0029(19970301)36:5<382::AID-JEMT8>3.0.CO;2-N.
- G. J. Millar, M. L. Nelson and P. J. R. Uwins, Insitu Observation of Structural Changes in Polycrystalline Silver Catalysts by Environmental Scanning Electron Microscopy, J. Chem. Soc., Faraday Trans., 1998, 94(14), 2015–2023, 10.1039/A802674B.
- X. Bao, M. Muhler, B. Pettinger, Y. Uchida, G. Lehmpfuhl, R. Schlögl and G. Ertl, The Effect of Water on the Formation of Strongly Bound Oxygen on Silver Surfaces, Catal. Lett., 1995, 32(1), 171–183, DOI:10.1007/BF00806112.
- L. Lefferts, J. G. van Ommen and J. R. H. Ross, An X-Ray Photoelectron Spectroscopy Study of the Influence of Hydrogen on the Oxygen–Silver Interaction, J. Chem. Soc., Faraday Trans. 1, 1987, 83(10), 3161–3165, 10.1039/F19878303161.
- T. Pu, A. Setiawan, A. C. Foucher, M. Guo, J.-M. Jehng, M. Zhu, M. E. Ford, E. A. Stach, S. Rangarajan and I. E. Wachs, Revealing the Nature of Active Oxygen Species and Reaction Mechanism of Ethylene Epoxidation by Supported Ag/α-Al2O3 Catalysts, ACS Catal., 2024, 14(1), 406–417, DOI:10.1021/acscatal.3c04361.
- C. I. Carlisle, T. Fujimoto, W. S. Sim and D. A. King, Atomic Imaging of the Transition between Oxygen Chemisorption and Oxide Film Growth on Ag111, Surf. Sci., 2000, 470(1), 15–31, DOI:10.1016/S0039-6028(00)00831-1.
- G. J. Millar, J. B. Metson, G. A. Bowmaker and R. P. Cooney, In Situ Raman Studies of the Selective Oxidation of Methanol to Formaldehyde and Ethene to Ethylene Oxide on a Polycrystalline Silver Catalyst, J. Chem. Soc., Faraday Trans., 1995, 91(22), 4149–4159, 10.1039/FT9959104149.
- G. J. Millar, M. L. Nelson and P. J. R. Uwins, A Combined Environmental Scanning Electron Microscopy and Raman Microscopy Study of Methanol Oxidation on Silver(I) Oxide, Catal. Lett., 1997, 43(1), 97–105, DOI:10.1023/A:1018938522320.
- G. I. N. Waterhouse, G. A. Bowmaker and J. B. Metson, Influence of Catalyst Morphology on the Performance of Electrolytic Silver Catalysts for the Partial Oxidation of Methanol to Formaldehyde, Appl. Catal., A, 2004, 266(2), 257–273, DOI:10.1016/j.apcata.2004.02.015.
- S. Das, R. Pashminehazar, S. Sharma, S. Weber and T. L. Sheppard, New Dimensions in Catalysis Research with Hard X-Ray Tomography, Chem. Ing. Tech., 2022, 94(11), 1591–1610, DOI:10.1002/cite.202200082.
- L. Mino, E. Borfecchia, J. Segura-Ruiz, C. Giannini, G. Martinez-Criado and C. Lamberti, Materials Characterization by Synchrotron X-Ray Microprobes and Nanoprobes, Rev. Mod. Phys., 2018, 90(2), 25007, DOI:10.1103/RevModPhys.90.025007.
- G. T. van de Kerkhof, J. M. Walker, S. Agrawal, S. M. Clarke, M. H. Sk, D. J. Craske, R. Lindsay, M. Dowhyj, O. Ayomide, M. E. Schuster and J. E. Parker, An in Situ Liquid Environment for Synchrotron Hard X-Ray Nanoprobe Microscopy, Mater. High Temp., 2023, 40(4), 371–375, DOI:10.1080/09603409.2023.2213579.
- J. E. Parker, M. Gomez-Gonzalez, Y. Van Lishout, H. Islam, D. Duran Martin, D. Ozkaya, P. D. Quinn and M. E. Schuster, A Cell Design for Correlative Hard X-Ray Nanoprobe and Electron Microscopy Studies of Catalysts under in Situ Conditions, J. Synchrotron Radiat., 2022, 29(2), 431–438, DOI:10.1107/S1600577521013576.
- Y. Fam, T. L. Sheppard, J. Becher, D. Scherhaufer, H. Lambach, S. Kulkarni, T. F. Keller, A. Wittstock, F. Wittwer, M. Seyrich, D. Brueckner, M. Kahnt, X. Yang, A. Schropp, A. Stierle, C. G. Schroer and J.-D. Grunwaldt, A Versatile Nanoreactor for Complementary in Situ X-Ray and Electron Microscopy Studies in Catalysis and Materials Science, J. Synchrotron Radiat., 2019, 26(5), 1769–1781, DOI:10.1107/S160057751900660X.
- S. Weber, A. Diaz, M. Holler, A. Schropp, M. Lyubomirskiy, K. L. Abel, M. Kahnt, A. Jeromin, S. Kulkarni, T. F. Keller, R. Gläser and T. L. Sheppard, Evolution of Hierarchically Porous Nickel Alumina Catalysts Studied by X-Ray Ptychography, Adv. Sci., 2022, 9(8), 2105432, DOI:10.1002/advs.202105432.
- A. P. Hitchcock, Soft X-Ray Spectromicroscopy and Ptychography, J. Electron Spectrosc. Relat. Phenom., 2015, 200, 49–63, DOI:10.1016/j.elspec.2015.05.013.
- F. Pfeiffer, X-Ray Ptychography, Nat. Photonics, 2018, 12(1), 9–17, DOI:10.1038/s41566-017-0072-5.
- A. Diaz, P. Trtik, M. Guizar-Sicairos, A. Menzel, P. Thibault and O. Bunk, Quantitative X-Ray Phase Nanotomography, Phys. Rev. B, 2012, 85(2), 020104, DOI:10.1103/PhysRevB.85.020104.
- J. C. da Silva, K. Mader, M. Holler, D. Haberthür, A. Diaz, M. Guizar-Sicairos, W.-C. Cheng, Y. Shu, J. Raabe, A. Menzel and J. A. van Bokhoven, Assessment of the 3 D Pore Structure and Individual Components of Preshaped Catalyst Bodies by X-Ray Imaging, ChemCatChem, 2015, 7(3), 413–416, DOI:10.1002/cctc.201402925.
- J. Ihli, L. Bloch, F. Krumeich, K. Wakonig, M. Holler, M. Guizar-Sicairos, T. Weber, J. C. da Silva and J. A. van Bokhoven, Hierarchical Structure of NiMo Hydrodesulfurization Catalysts Determined by Ptychographic X-Ray Computed Tomography, Angew. Chem., Int. Ed., 2020, 59(39), 17266–17271, DOI:10.1002/anie.202008030.
- C. G. Schroer, M. Seyrich, M. Kahnt, S. Botta, R. Döhrmann, G. Falkenberg, J. Garrevoet, M. Lyubomirskiy, M. Scholz, A. Schropp and F. Wittwer, PtyNAMi: Ptychographic Nano-Analytical Microscope at PETRA III: Interferometrically Tracking Positions for 3D x-Ray Scanning Microscopy Using a Ball-Lens Retroreflector, in Proc. SPIE, 2017, vol. 10389, p. 103890E. DOI:10.1117/12.2273710.
- A. Schropp, S. Achilles, S. Patjens, F. Seiboth, M. E. Stückelberger, Z. Jiang, J. H. Pikul and C. G. Schroer, 3D Scanning Coherent X-Ray Microscopy at PtyNAMi, in Developments in X-Ray Tomography XIV, ed. B. Müller and G. Wang, SPIE, 2022, vol. 12242, p. 1224208. DOI:10.1117/12.2632619.
- A. Schropp, R. Döhrmann, S. Botta, D. Brückner, M. Kahnt, M. Lyubomirskiy, C. Ossig, M. Scholz, M. Seyrich, M. E. Stuckelberger, P. Wiljes, F. Wittwer, J. Garrevoet, G. Falkenberg, Y. Fam, T. L. Sheppard, J.-D. Grunwaldt and C. G. Schroer, PtyNAMi: Ptychographic Nano-Analytical Microscope, J. Appl. Crystallogr., 2020, 53(4), 957–971, DOI:10.1107/S1600576720008420.
- U. Johansson, D. Carbone, S. Kalbfleisch, A. Björling, M. Kahnt, S. Sala, T. Stankevic, M. Liebi, A. Rodriguez Fernandez, B. Bring, D. Paterson, K. Thånell, P. Bell, D. Erb, C. Weninger, Z. Matej, L. Roslund, K. Åhnberg, B. Norsk Jensen, H. Tarawneh, A. Mikkelsen and U. Vogt, NanoMAX: The Hard X-Ray Nanoprobe Beamline at the MAX IV Laboratory, J. Synchrotron Radiat., 2021, 28(6), 1935–1947, DOI:10.1107/S1600577521008213.
- D. Carbone, S. Kalbfleisch, U. Johansson, A. Björling, M. Kahnt, S. Sala, T. Stankevic, A. Rodriguez-Fernandez, B. Bring, Z. Matej, P. Bell, D. Erb, V. Hardion, C. Weninger, H. Al-Sallami, J. Lidon-Simon, S. Carlson, A. Jerrebo, B. Norsk Jensen, A. Bjermo, K. Åhnberg and L. Roslund, Design and Performance of a Dedicated Coherent X-Ray Scanning Diffraction Instrument at Beamline NanoMAX of MAX IV, J. Synchrotron Radiat., 2022, 29(3), 876–887, DOI:10.1107/S1600577522001333.
- A. Robert, Y. Cerenius, P. F. Tavares, A. Hultin Stigenberg, O. Karis, A.-C. Lloyd Whelan, C. Runéus and M. Thunnissen, MAX IV Laboratory, Eur. Phys. J. Plus, 2023, 138(6), 495, DOI:10.1140/epjp/s13360-023-04018-w.
- T. Donath, D. Šišak Jung, M. Burian, V. Radicci, P. Zambon, A. N. Fitch, C. Dejoie, B. Zhang, M. Ruat, M. Hanfland, C. M. Kewish, G. A. van Riessen, D. Naumenko, H. Amenitsch, G. Bourenkov, G. Bricogne, A. Chari and C. Schulze-Briese, EIGER2 Hybrid-Photon-Counting X-Ray Detectors for Advanced Synchrotron Diffraction Experiments, J. Synchrotron Radiat., 2023, 30(4), 723–738, DOI:10.1107/S160057752300454X.
- S. Lervold, R. Lødeng, J. Yang, J. Skjelstad, K. Bingen and H. J. Venvik, Partial Oxidation of Methanol to Formaldehyde in an Annular Reactor, Chem. Eng. J., 2021, 423, 130141, DOI:10.1016/j.cej.2021.130141.
- I. Utke, P. Hoffmann and J. Melngailis, Gas-Assisted Focused Electron Beam and Ion Beam Processing and Fabrication, J. Vac. Sci. Technol., B: Microelectron. Nanometer Struct.--Process., Meas., Phenom., 2008, 26(4), 1197–1276, DOI:10.1116/1.2955728.
- W. D. Wilson, C. L. Bisson and M. I. Baskes, Self-Trapping of Helium in Metals, Phys. Rev. B: Condens. Matter Mater. Phys., 1981, 24(10), 5616–5624, DOI:10.1103/PhysRevB.24.5616.
- V. Sciani and P. Jung, Diffusion of Helium in FCC Metals, Radiat. Eff., 1983, 78(1–4), 87–99, DOI:10.1080/00337578308207362.
- R. M. Langford, T.-X. Wang and D. Ozkaya, Reducing the Resistivity of Electron and Ion Beam Assisted Deposited Pt, Microelectron. Eng., 2007, 84(5), 784–788, DOI:10.1016/j.mee.2007.01.055.
- F. Wittwer, M. Lyubomirskiy, F. Koch, M. Kahnt, M. Seyrich, J. Garrevoet, C. David and C. G. Schroer, Upscaling of Multi-Beam x-Ray Ptychography for Efficient x-Ray Microscopy with High Resolution and Large Field of View, Appl. Phys. Lett., 2021, 118(17), 171102, DOI:10.1063/5.0045571.
- M. Lyubomirskiy, F. Wittwer, M. Kahnt, F. Koch, A. Kubec, K. V. Falch, J. Garrevoet, M. Seyrich, C. David and C. G. Schroer, Multi-Beam X-Ray Ptychography Using Coded Probes for Rapid Non-Destructive High Resolution Imaging of Extended Samples, Sci. Rep., 2022, 12(1), 6203, DOI:10.1038/s41598-022-09466-5.
- T. Li, M. Kahnt, T. L. Sheppard, R. Yang, K. V. Falch, R. Zvagelsky, P. Villanueva-Perez, M. Wegener and M. Lyubomirskiy, X-Ray Multibeam Ptychography at up to 20 KeV: Nano-Lithography Enhances X-Ray Nano-Imaging, Adv. Sci., 2024, 11(30), 2310075, DOI:10.1002/advs.202310075.
Footnotes |
| † Electronic supplementary information (ESI) available: Videos demonstrating the rapid restructuring effect for three samples exposed to 650 °C in the nanoreactor setup at the P06 beamline of PETRA III are included to facilitate visualization of influence of temperature under He or ambient air conditions. These are namely: PA1_scan005.avi, PA2_scan001.avi, and PHe_scan004.avi. Videos demonstrating the gas bubble formation and growth within three samples exposed to 500 °C under different gas mixtures in the nanoreactor setup mounted at the NanoMAX beamline of the MAX IV Laboratory are included, namely MA1_chip6.avi, MA2_chip9.avi, and MHe_chip8.avi. These videos help visualize bubble formation and consequent surface explosion on the left panel, and bubble propagation using difference maps of adjacent scans on the right panel of each video for all three samples. See DOI: https://doi.org/10.1039/d4cy00770k |
| ‡ Current address: Institute of Chemical Technology, Leipzig University, Linnéstrasse 3, 04103 Leipzig, Germany. |
| This journal is © The Royal Society of Chemistry 2024 |