
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Innovations in nanocomposite photocatalysts for CO2 to CH3OH conversion
Shuang
Deng
a,
Nannan
Wang
 *a,
Yanqiu
Zhu
b and
Kunyapat
Thummavichai
*c
*a,
Yanqiu
Zhu
b and
Kunyapat
Thummavichai
*c
aState Key Laboratory of Featured Metal Materials and Life-cycle Safety for Composite Structures, School of Resources, Environment and Materials, Guangxi University, Nanning, 530004, Guangxi, China. E-mail: wangnannan@gxu.edu.cn
bCollege of Engineering, Mathematics and Physical Sciences, University of Exeter, Exeter, EX4 4QF, UK
cDepartment of Mathematics, Physics and Electrical Engineering, Faculty of Engineering and Environment, Northumbria University, Newcastle-upon-Tyne, NE1 8ST, UK. E-mail: kunyapat.thummavichai@northumbria.ac.uk
First published on 10th September 2024
Abstract
Nowadays, the excessive use of fossil fuels has led to a global energy shortage and exacerbated the greenhouse effect. One of the main contributors to the greenhouse effect is the excessive emission of carbon dioxide (CO2). Therefore, to achieve sustainable development and harmonious symbiosis between humans and nature, reducing CO2 emission has become a global concern. Converting CO2 into value-added products can effectively address this issue. At present, the photocatalytic reduction of CO2 using synthetic nanocomposites is a common method. Among the products of photocatalytic CO2 reduction, methanol (CH3OH) shows promise as a new clean fuel and basic chemical raw material, with potential to reduce energy consumption. This paper reviews the research progress of photocatalytic reduction of CO2 to CH3OH in recent years. The difficulties and challenges in the photocatalytic process, along with potential solutions, are briefly discussed. Finally, the future prospects of this research are outlined.
1. Introduction
In the course of human development, science and technology have not only brought convenience to human life, but also enriched the living environment. However, for the entire ecological environment, science and technology act as a “double-edged sword”, presenting a series of negative effects. During the rapid pace of global industrialization, the excessive use of fossil fuels has led to significant greenhouse gas emissions, resulting in the greenhouse effect and global climate warming. Carbon dioxide (CO2) is one of the most common greenhouse gases. Under normal circumstances, CO2 is absorbed by plants through photosynthesis to produce oxygen, essential for human survival. However, due to the indiscriminate exploitation of forest resources, CO2 emissions have increased year by year, failing to meet the requirements for sustainable human development. In order to solve this problem, the most effective approach is to convert CO2 into chemical raw materials needed for human development, thereby achieving its recycling. Through continuous exploration, researchers have identified several effective methods to reduce the concentration of CO2 in the atmosphere, such as thermal–chemical methods, biological methods, electrochemical methods and photocatalysis methods.1 Simulating plant photosynthesis to convert CO2 into hydrocarbons through photocatalysis is considered a more viable approach for achieving clean energy and sustainable development. Compared to other methods, this method is not only simple and convenient, but also avoids secondary pollution. Additionally, the final product can be used as green renewable energy, thereby alleviating the global energy shortage problem.Because the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond energy of CO2 is high, the activation energy required to disconnect this bond is substantial, posing a significant challenge to the photocatalytic CO2 reduction process. To design and synthesize efficient photocatalysts, researchers commonly use nanocomposites for photocatalytic CO2 reduction. The final products are mainly divided into two categories: C1 products and C2 and C2+ products. Among them, C1 products include CO, CH4, CH3OH, HCOOH, etc. C2 and C2+ products include C2H4, C2H6, C3H6, C2H5OH, etc.2 Methanol (CH3OH) is the simplest saturated monohydric alcohol. It is not only an important basic organic chemical raw material but also a new type of clean energy fuel. CH3OH is widely used for the synthesis of various organic compounds such as aldehydes, olefins, esters, etc. Additionally, it can be incorporated into gasoline as an alternative fuel and used as a fuel cell raw material. Currently, the industry primarily synthesizes methanol through pressurized catalytic hydrogenation of carbon monoxide and carbon dioxide. Although this technology is relatively mature, it involves a cumbersome process flow and large energy consumption. In contrast, the photocatalytic reduction of CO2 to CH3OH can be achieved at atmospheric pressure, offering a promising method for reducing energy consumption.
O bond energy of CO2 is high, the activation energy required to disconnect this bond is substantial, posing a significant challenge to the photocatalytic CO2 reduction process. To design and synthesize efficient photocatalysts, researchers commonly use nanocomposites for photocatalytic CO2 reduction. The final products are mainly divided into two categories: C1 products and C2 and C2+ products. Among them, C1 products include CO, CH4, CH3OH, HCOOH, etc. C2 and C2+ products include C2H4, C2H6, C3H6, C2H5OH, etc.2 Methanol (CH3OH) is the simplest saturated monohydric alcohol. It is not only an important basic organic chemical raw material but also a new type of clean energy fuel. CH3OH is widely used for the synthesis of various organic compounds such as aldehydes, olefins, esters, etc. Additionally, it can be incorporated into gasoline as an alternative fuel and used as a fuel cell raw material. Currently, the industry primarily synthesizes methanol through pressurized catalytic hydrogenation of carbon monoxide and carbon dioxide. Although this technology is relatively mature, it involves a cumbersome process flow and large energy consumption. In contrast, the photocatalytic reduction of CO2 to CH3OH can be achieved at atmospheric pressure, offering a promising method for reducing energy consumption.
There have been reviews on the application of different photocatalysts in photocatalytic CO2 reduction reactions. However, very few reviews focus on the preparation of CH3OH. This paper aims to summarize the research on the photocatalytic reduction of CO2 to CH3OH over the past five years, briefly discussing the reaction mechanism and the opportunities and challenges faced in this field.
2. Theoretical basis of photocatalytic reduction of CO2 to CH3OH
A catalyst is a substance that can accelerate a chemical reaction without itself participating in the reaction. Photocatalysts are a general term for chemicals that can play a catalytic role under the excitation of photons. Through continuous research, various types of photocatalysts have been developed, with semiconductor photocatalysts, particularly nanoscale titanium dioxide (TiO2), being the most widely studied and applied. The principle of photocatalytic reactions can be explained by the band theory of semiconductors.3 Unlike metals and insulators, semiconductors have a band gap between the valence band (VB) and the conduction band (CB). When they are illuminated by light with photon energy equal to or higher than the bandgap energy, electrons in the VB can be excited into the CB, creating corresponding holes in the VB and forming electron–hole pairs. The photocatalytic reaction process in semiconductors can be simply described as follows: (i) semiconductors absorb light to form electron–hole pairs, which then (ii) migrate and recombine. These processes allow redox reactions to occur on the surface of semiconductors through the adsorption of reactants and the desorption of products.Fig. 1 illustrates the photocatalytic process of semiconductors, where electron–hole pair recombination can occur either within the material or on its surface. So far, the biggest bottleneck in photocatalytic reactions is low photocatalytic efficiency, primarily due to the rapid recombination of photogenerated electrons and holes.4 Improving energy conversion efficiency involved expanding the range of light absorption to achieve more efficient photocatalytic reactions. However, developing a photocatalyst with high light-trapping capacity and effective charge separation, as well as abundant CO2 adsorption active sites, remains a significant challenge.5
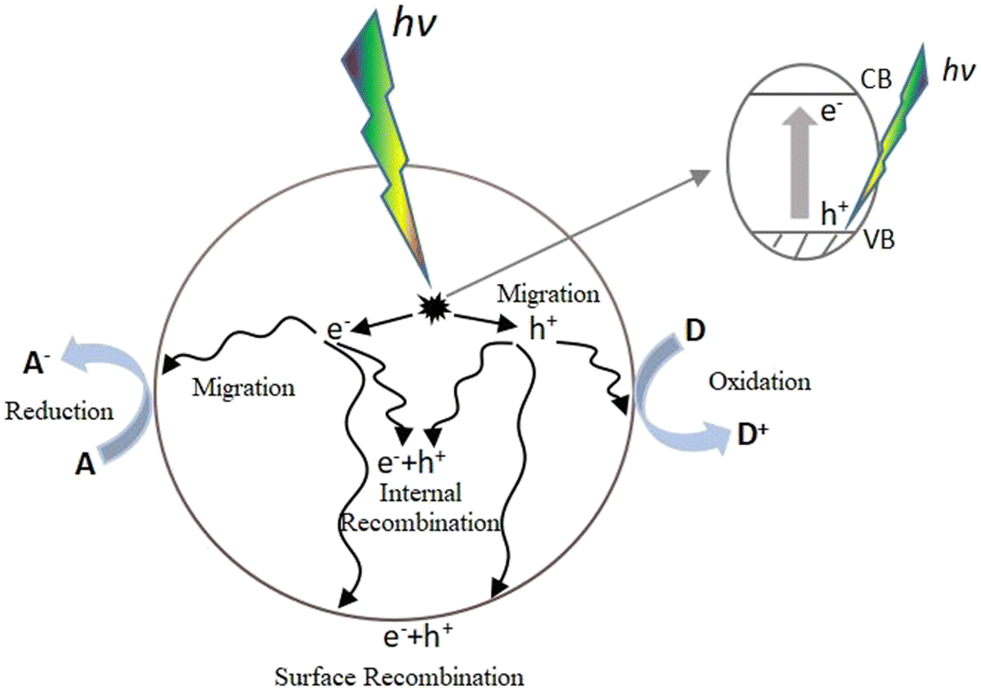 | ||
| Fig. 1 Photocatalytic process of a semiconductor. | ||
CO2 is a symmetrical linear molecule, and because CO (750 kJ mol−1) has a much higher bond energy than C–C (336 kJ mol−1), C–O (327 kJ mol−1), and C–H (411 kJ mol−1) bonds,6 photocatalytic conversion of CO2 usually requires a large amount of energy input to break CO bonds. At the same time, CO2 is optically inert under visible or ultraviolet radiation with a wavelength of 200–900 nm. Therefore, it is necessary to use a photocatalyst with a suitable band structure to enable photoexcitation and generate photoelectron–hole pairs to complete the reduction process of CO2.7 As the highest oxidation state of carbon, CO2 can be reduced to a variety of products by obtaining different numbers of electrons and protons. The presence of by-products depletes valuable photogenerated electron–hole pairs, thereby reducing the formation of target products. Consequently, obtaining high yields of target products is a primary goal in photocatalytic CO2 reduction reactions.8 In the process of photocatalytic reduction of CO2 to produce CH3OH, the following chemical reaction formula is followed:6
| CO2 + 2H+ + 2e− → CO + H2O E1 = −0.53 V | (1) |
| CO2 + 6H+ + 6e− → CH3OH + H2O E2 = −0.38 V | (2) |
The adsorption of CO2 on the surface of the photocatalyst is a key step in facilitating the reaction. The adsorption structures of CO2 on the surface of the photocatalyst mainly include oxygen, carbon and mixed coordination.8 Different CO2 adsorption structures will form different reaction pathways, resulting in different reaction products and intermediates. In the process of photocatalytic reduction of CO2 to CH3OH, there is often a competitive reaction with another product, CH4. When CO2 molecules obtain photogenerated electrons and H+, they will be converted into ·COOH free radicals. Through a series of dehydration hydrogenation reactions, the product CH3OH can be obtained. If CH3OH is weakly bound to the photocatalyst surface, the desorption of CH3OH can be carried out more easily. However, if CH3OH binds strongly to the photocatalyst surface, it becomes difficult for CH3OH to be released on the surface of the photocatalyst, leading to further hydrodeoxidation reaction, and finally generating CH4. This results in a decrease in the yield of CH3OH. Therefore, synthesizing suitable photocatalysts and optimising the experimental conditions of photocatalysis are crucial strategies to improve the yield of CH3OH.
3. Reduction of CO2 to CH3OH by different photocatalytic systems
Photocatalysis is a technology that emerged in the 1970s, based on the redox ability of photocatalysts under light conditions to achieve the purification of pollutants, as well as the synthesis and transformation of the substances. The typical natural photocatalyst is chlorophyll, which promotes the conversion of CO2 and H2O in the air into oxygen and carbohydrates during plant photosynthesis. Photocatalytic reduction of CO2 aims to use light energy to convert CO2 into high value-added chemicals or fuels by simulating this natural process. The photocatalytic systems used for photocatalytic reduction of CO2 to CH3OH can be roughly divided into the following categories: layered bimetallic hydroxide-based (LDH) photocatalysts, graphite carbon nitride-based (g-C3N4) photocatalysts, graphene oxide-based (GO) photocatalysts, mixed metal compound photocatalysts, and single metal-doped photocatalysts. And from Fig. 2, it is easy to find that in the last 5 years, the most studies on mixed metal compound photocatalysts for photocatalytic reduction of CO2 to C2H5OH have been carried out.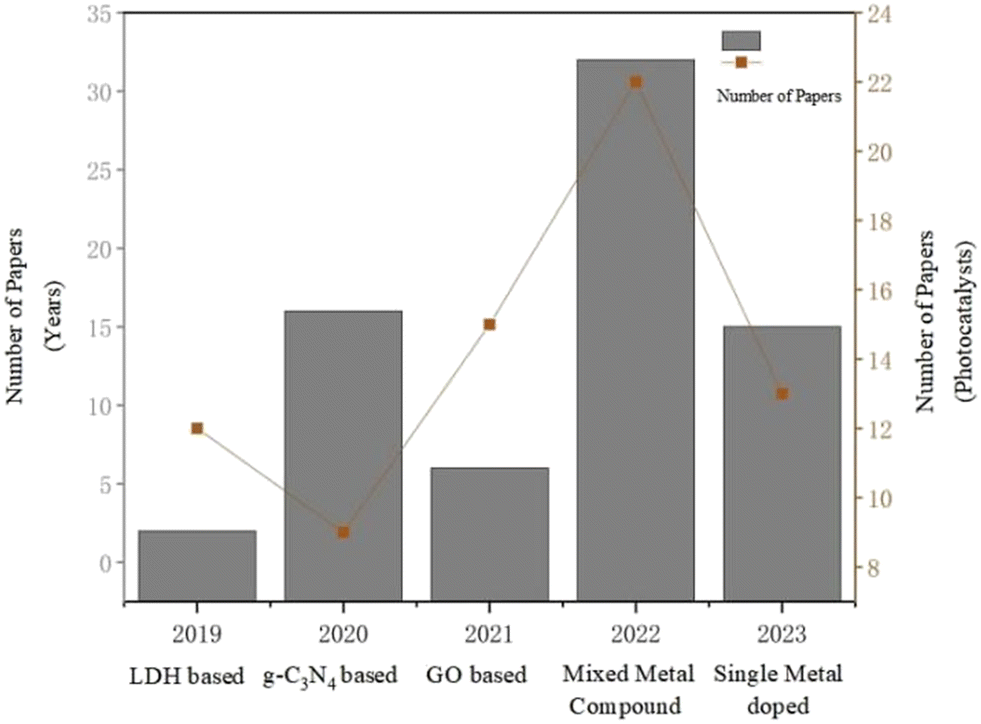 | ||
| Fig. 2 The number of published papers on the photocatalytic reduction of CO2 to CH3OH over different years, as well as the number of papers related to various types of photocatalysts in the past five years (data from Scholar Search). | ||
3.1 Layered double hydroxide (LDH)-based photocatalyst
LDH is a two-dimensional (2D) nanomaterial with the general formula [M2+1−xM3+x(OH)2]q+(Xn−)q/n·yH2O, where typical M2+ ions include Mg2+, Co2+, Ni2+, Cu2+, and Zn2+, and M3+ = Al3+, Cr3+, Fe3+, In3+, Ga3+, etc.9 In addition, M1+ and M4+ can also combine to form LDH, but currently, this is limited to combination of Li+, Ti4+, Zr4+ and Sn4+.10 Photocatalytic materials with various properties can be obtained by adjusting the types and proportions of cations in the LDH layer. Furthermore, the photocatalytic performance can also be adjusted by changing the type of interlayer anion. Common interlayer anions are CO32−, OH−, SO42−, NO3−, F−, Cl−, Br−, etc.As early as 2009, LDH was mainly used as a heterogeneous catalyst for base-catalyzed reactions or redox transformations due to its unique structure. However, in 2009, Silva et al.11 considered the presence of two different metals in the LDH structure and the controllable ratio between them, hypothesizing that LDHs could function as “doped semiconductors”. Silva et al. then prepared a series of LDHs with different Zn/metal atomic ratios (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 to 4:25) by the co-precipitation method, aiming to create an efficient photocatalyst for visible light oxygen production in water. The results showed that all the prepared materials exhibited good photocatalytic activity, with the Zn–Cr containing materials displaying the highest photocatalytic activity. Using monochromatic light, the apparent quantum yield of oxygen generation at 410 nm was as high as 60.9%.
:2 to 4:25) by the co-precipitation method, aiming to create an efficient photocatalyst for visible light oxygen production in water. The results showed that all the prepared materials exhibited good photocatalytic activity, with the Zn–Cr containing materials displaying the highest photocatalytic activity. Using monochromatic light, the apparent quantum yield of oxygen generation at 410 nm was as high as 60.9%.
In 2012, Teramura et al.12 proposed that Mg–Al hydrotalcite anionic clays could catalyze the aldol reaction of carbonyl compounds in aqueous solutions due to the high water resistance of the surface basic groups. They suggested that if CO2 is first adsorbed on the surface of the photocatalyst and then activated under light exposure, most LDH could act as a photocatalyst in water. Therefore, they synthesized LDH by co-precipitation and hydrothermal methods. The photocatalytic reduction of CO2 was then carried out in a closed circulation system. The results showed that the surface base sites of LDH with high water resistance not only act as CO2 adsorbents, but also as active sites for CO2 activation under light irradiation. This confirms the potential for photocatalytic CO2 conversion on various LDHs such as NiAl-LDH, ZnAl-LDH, NiGa-LDH, ZnLn-LDH, MgAl-LDH, etc., greatly expanding the photocatalytic application of LDH materials.
In recent years, LDHs have shown great potential in the design and manufacture of nanomaterials for photocatalytic reduction of CO2 due to their versatility in composition, morphology and structure, as well as their unique structural properties. However, over the past five years, there have been relatively few studies on the use of LDH materials in photocatalytic reduction to produce CH3OH. It has been found that the photocatalytic activity of single LDH materials is limited by the rapid recombination of photogenerated carriers and their weak CO2 adsorption and activation abilities, making them difficult to apply directly to photocatalytic CO2 reduction. Therefore, further modifications are often necessary for these materials to achieve higher photocatalytic efficiency. Ziarati et al.13 designed a 3D yolk@shell structure TiO2/LDH containing oxygen vacancies through morphology control (Fig. 3). This structure is special in that it has a specific void space between the core and the shell, providing high light reflection potential and reduced diffusion resistance, as well as performance adjustability, which offers various opportunities for photocatalysis. In addition, the interior pore space can be used as a nanoreactor and enable efficient diffusion of solvents, substrates, and products through the porous surface. This structure reveals the high efficiency of photoreduction of CO2 into a solar fuel in the absence of a precious metal co-catalyst. Its experiments showed that the generation of CH3OH was almost selective before 2 h, up to 251 μmol gcat−1 h−1. To improve the efficiency of photocatalysis, many methods have been tried, such as atom doping,14 defect engineering,15 component control,16 surface modification,17 and heterojunction construction.18 And the precise regulation of the intrinsic internal electric field of the photocatalyst through the construction of a heterojunction structure seems to be the most direct method for coordinating the separation of photogenerated carriers. Jiang et al.19 prepared CuO quantum dot/ultra-thin CoAl-LDH (CuO/CoAl-u) direct Z-type 0D/2D heterojunctions by a simple electrostatic self-assembly process for photocatalytic CO2 reduction. The results showed that the 4.5% CuO/CoAl-u heterojunction exhibited excellent photocatalytic performance, with a CH3OH yield of 283.26 μmol g−1 h−1, which is higher than the original CoAl-u. This enhanced photocatalytic performance is due to the outstanding light absorption properties of CuO/CoAl-u, the efficient separation of photoinduced carriers, and the high reduction capacity.
 | ||
| Fig. 3 Overall flowchart for the fabrication of the 3D Y@S TiO2−x/LDH architecture (steps I–III); corresponding (a–f) FESEM, (g–i) TEM, and (j–l) HRTEM images of the 3D Y@S TiO2−x/LDH sites.13 | ||
Overall, although a single LDH material may have limitations in the efficiency of photocatalytic CO2 reduction, its unique layered structure and the versatility of interlayer anions and cations offer significant potential for developing new photocatalysts. The efficiency of converting CO2 to CH3OH can already be greatly enhanced through various modification methods. However, these structural modifications increase the material's complexity, making it more challenging to understand the photocatalytic mechanism. Employing advanced technologies to achieve precise micro-level regulation of the material structure could help address this issue.
3.2 Graphitic carbon nitride (g-C3N)-based photocatalyst
g-C3N4 is a covalent solid mainly composed of carbon and nitrogen, forming a planar two-dimensional sheet structure similar to graphene. It is composed of two basic units, the triazine ring (C3N3) and the 3-s-triazine ring (C6N7), with the two-dimensional nanosheets bound by van der Waals forces. As a metal-free polymer N-type semiconductor, g-C3N4 possesses unique electrical, optical, structural and physicochemical properties. Unlike many other semiconductors, g-C3N4 offers a wide range of prospects for the development of novel nanostructured g-C3N4 photocatalysts due to its unique polymerization structure, shape, size, composition, thickness, and pore size and distribution.20,21 Back in 2009, Wang et al.22 discovered that g-C3N4 can produce hydrogen from water under visible light irradiation in the presence of a donor. This discovery opened a new chapter for g-C3N4-based photocatalysts and since then, they have garnered increasing global attention. The photocatalytic performance of g-C3N4 has been greatly enhanced through various modifications. In recent years, many important breakthroughs have been made in the synthesis and application of g-C3N4-based photocatalysts.It has been reported that the selectivity to certain products can be influenced by changing the energy of CB electrons. Zero-dimensional (0D) semiconductor quantum dots have attracted significant attention due to their unique advantages such as large surface area, high atomic utilization, and short effective charge transfer length. One of the most important characteristics of 0D semiconductor quantum dots is their tunable band structures, which can be used to control the evolution of specific products through quantum confinement effects. Inspired by this, Li et al.23 designed and prepared a g-C3N4/CdSe quantum dot (p-CNCS) photocatalyst with different CdSe particle sizes for highly selective and highly active photocatalytic CO2 reduction. The synthesized CdSe quantum dots had a statistical diameter ranging from 1.2 to 4.1 nm. Based on the quantum confinement effect, the energy of CB electrons was adjusted to an appropriate value, inhibiting the formation of H2 and promoting the formation of CH3OH, thereby enhancing product selectivity. The final results showed that when the size of CdSe quantum dots was 2.2 nm, the selectivity to CH3OH reached 73% with a formation rate of 186.4 μmol g−1 h−1.
Although g-C3N4 exhibits excellent photocatalytic activity, its pure form suffers from rapid recombination of photogenerated electron–hole pairs, which significantly hampers its photocatalytic efficiency. To address these issues, the combination of g-C3N4 with heterojunction semiconductors can promote electron–hole separation, effectively preventing charge recombination. Liang et al.24 designed a hollow structure g-C3N4@CeO2 photocatalyst with oxygen-rich vacancies by combining the advantages of the heterostructure, oxygen-rich vacancies and hollow structure. Its unique hollow structure enabled light to be reflected and utilized multiple times within its inner cavity, ultimately improving the utilization efficiency of light. As the result, this novel photocatalyst demonstrated high CO2 reduction capacity, with a CH3OH yield of 5.2 μmol g−1 when the CeO2 mass fraction was 49.7 wt%. Similarly, in order to improve the photocatalytic performance of g-C3N4, Li et al.25 considered the strong adsorption capacity of MgO, an alkali metal oxide, for CO2, as well as the use of precious metals (such as Au, Ag, Pt and Pd) as electron traps to improve the separation efficiency of photogenerated electron–hole pairs. They prepared a series of MgO-modified g-C3N4 composites by calcination and then loaded the Au cocatalyst onto the composite using the NaBH4 reduction method. Under simulated solar illumination, the CO2 reduction was studied by using the ternary photocatalyst. The results showed that the enhanced photocatalytic activity of the photocatalyst is due to the synergistic action between the components. Among them, the g-C3N4 photocatalyst co-modified with 3% MgO had the best photocatalytic performance, and CH3OH could reach 47.2 μmol g−1 when the reaction time was 3 h.
The photocatalytic reduction of CO2 to CH3OH is a process involving multi-electron transfer. During the electron transfer process, it is easy to produce other products requiring fewer electrons and protons, such as CO, HCHO, HCOOH, etc. To achieve the selective production of CH3OH by photocatalytic CO2 reduction with high activity, Ma et al.26 prepared a CN supported cobalt sulfide (CS) reduced photocatalyst, which can target the activation of H2O and induce the selective production of CH3OH. The results showed that the selectivity to CH3OH (87.2%) was 2.3 times higher than that of CN (38.6%), and the CH3OH yield increased from 22.0 to 97.3 μmol g−1 h−1. As potential photocatalysts for CO2 reduction, metal semiconductor oxide photocatalysts have been receiving significant attention. However, these photocatalysts often suffer from metal leaching caused by photocorrosion, which can lead to environmental pollution. To address this issue, Ding et al.27 developed a metal-free core–shell photocatalyst composed of graphite carbon nitride (g-C3N4, CN) covalently attached to a melamine–resorcinol–formaldehyde (MRF) microsphere polymer. Under reaction conditions of 80 °C and 0.5 MPa, the CH3OH yield was 0.99 μmol h−1. This yield was approximately 20 and 10 times that of its components CN and MRF, respectively. The external quantum efficiency ranged from 5.5 to 1.7% at wavelengths of 380 to 550 nm.
The development of g-C3N4 in photocatalytic CO2 reduction is severely limited by the fast quenching rate of its photogenic carrier and its poor adsorption capacity of CO2. The number of photogenerated electrons received by CO2 is highly dependent on the close contact between the two. Therefore, one of the ideal ways to improve the CO2 conversion rate is to increase the adsorption capacity of g-C3N4 for CO2. Wang et al.28 synthesized a new type of g-C3N4/UiO-66 (Zr/Ce) nanosheet using an in situ method. As shown in Fig. 4, the g-C3N4 and UiO-66 interfaces are connected by N–Zr/Ce–O bonds. This two-dimensional structure tightly connected by chemical bonds accelerates the transport of electrons and significantly inhibit the quenching of photogenerated carriers. In Fig. 5b, UiO-66 (Zr/Ce) nanosheets form close contact with g-C3N4. The TEM results (Fig. 5d) also show that UiO-66 (Zr/Ce) and g-C3N4 have been successfully combined, with the edges of the UiO66 (Zr/Ce) nanosheets being almost transparent, indicating an ultra-thin thickness. The results of high-angle toroidal dark-field scanning transmission electron microscopy (HAADF-STEM) and the distribution of EDS components indicate that C, N, O, Zr and Ce are evenly distributed (Fig. 5e–j). It can be clearly found that Zr, Ce and O are mainly concentrated in the same region, and no obvious signals of Zr, Ce and O are found elsewhere. This shows that Zr, Ce, and O originate from UiO-66 (Zr/Ce), rather than the single atoms of Zr and Ce scattered on g-C3N4. In summary, individual atoms (Zr and Ce) successfully come together in UiO-66 (Zr/Ce) to form metal nodes through the connection of organic ligands. After that, CO2 photocatalytic reduction experiments were carried out in an acetonitrile–water mixed system without adding a sacrificial agent. As shown in Fig. 6, the product yield of CO2 photocatalytic reduction by g-C3N4/UiO-66 (Zr/Ce) was much higher than that by g-C3N4 and UiO-66 (Zr/Ce), and compared with photocatalysts of other systems, g-C3N4/UiO-66 (Zr/Ce) showed excellent performance in photocatalytic reduction of CO2 to produce CH3OH and C2H5OH. In particular, after many cycles of experiments, the photocatalyst still showed good photocatalytic performance, indicating that it has excellent chemical stability.
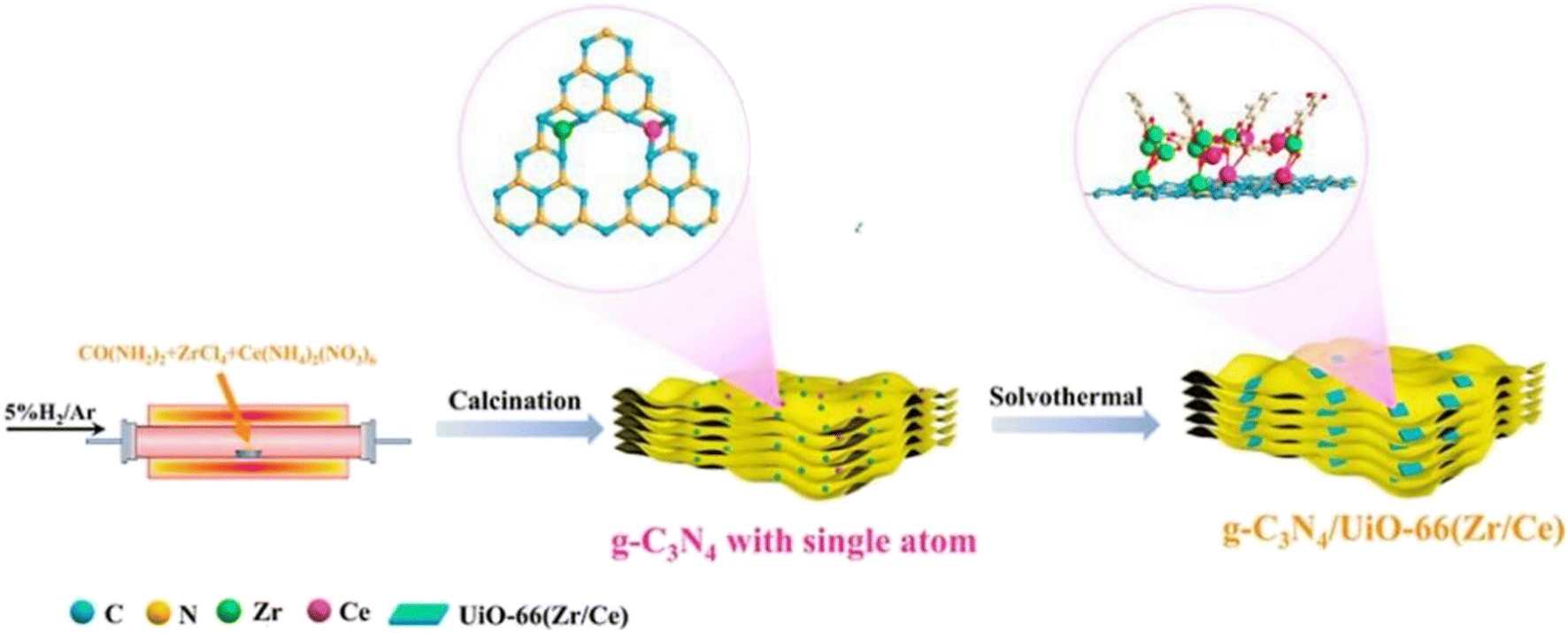 | ||
| Fig. 4 Synthesis process of the g-C3N4/UiO-66 (Zr/Ce) photocatalyst.28 | ||
 | ||
| Fig. 5 SEM images of (a) g-C3N4 and (b) g-C3N4/UiO-66 (Zr/Ce); TEM images of (c) g-C3N4 and (d) g-C3N4/UiO-66 (Zr/Ce); (e–j) EDS mapping of g-C3N4/UiO-66 (Zr/Ce).28 | ||
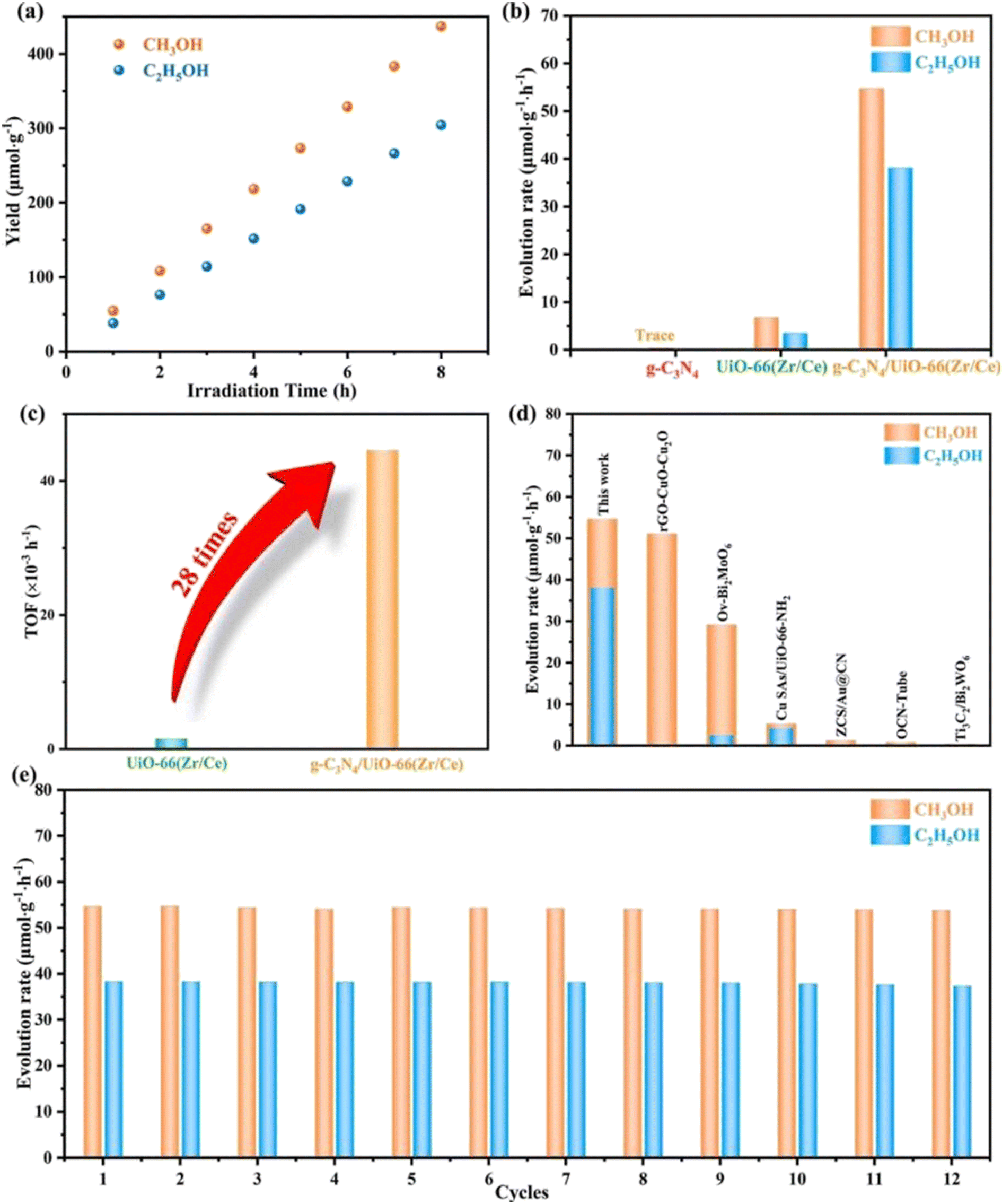 | ||
| Fig. 6 (a) Production of CH3OH and C2H5OH on g-C3N4/UiO-66 (Zr/Ce). (b) Evolution rate of CH3OH and C2H5OH on UiO-66 (Zr/Ce), g-C3N4, and g-C3N4/UiO-66 (Zr/Ce). (c) TOF of UiO-66 (Zr/Ce) and g-C3N4/UiO-66 (Zr/Ce). (d) Comparison of the yields of CH3OH and C2H5OH with other photocatalysts. (e) Catalytic cyclic tests of g-C3N4/UiO-66 (Zr/Ce).28 | ||
In general, the photocatalytic performance of the modified g-C3N4 is greatly improved. Currently, commonly used modification methods can be categorized into surface modification, morphology control, doping, and hybrid formation, among others. In addition to the development of conventional binary systems, g-C3N4-based photocatalysts with ternary or multicomponent systems have also emerged in recent years. Table 1 shows the reduction of CO2 to CH3OH by different g-C3N4-based photocatalysts in recent years. It can be seen from the table that whether in a binary system or ternary system, people are more inclined to choose metal compounds to modify g-C3N4. This is because the surface of metal compounds often has abundant active sites, so their catalytic performance is very prominent. In addition, after the semiconductor is excited by light, because the work function of the metal is higher than that of the semiconductor material, the electrons migrate to the loaded metal and are trapped, which can inhibit the recombination of electron–hole pairs, thereby improving the efficiency of the photocatalytic reaction.
| Photocatalysts | Reaction conditions | CH3OH yield | Ref. |
|---|---|---|---|
| g-C3N4/CdSe | Visible light (λ > 420 nm) | 186.4 μmol g−1 h−1 | 23 |
| g-C3N4@CeO2 | Monochromatic light (525 nm)/4 h | 5.2 μmol g−1 | 24 |
| MgO/Au@g-C3N4 | Xenon lamp (300 W)/3 h | 47.2 μmol g−1 | 25 |
| CoS/g-C3N4 | Xenon lamp (300 W) | 97.3 μmol g−1 h−1 | 26 |
| g-C3N4–melamine–resorcinol | Xenon lamp with a 420 nm cut-off filter/5 h | 0.99 μmol h−1 | 27 |
| Formaldehyde | |||
| g-C3N4/UiO-66 (Zr/Ce) | Xenon lamp (300 W) | 54.71 μmol g−1 | 28 |
| ZnV O/RGO/g-C N | Xenon lamp (35 W) | 3488 μmol g−1 | 29 |
| g-C3N4/Sn2S3-DETA | Visible light (λ > 420 nm) | 1.35 μmol g−1 h−1 | 30 |
| Zn0.2Cd0.8S/g-C3N4 | Xenon lamp (300 W) | 11.5 ± 0.3 μmol g−1 h−1 | 31 |
| Br/g-C3N4 | Xenon lamp (300 W)/1 h | 0.6 μmol g−1 | 32 |
| Cu/P/g-C3N4/TiO2 | 20 W LED light | 859 μmol gcat−1 | 33 |
| Ag/UiO-66@g-C3N4 | UV light (λ < 380 nm)/2 h | 17.76 μmol g−1 h−1 | 34 |
3.3 Graphene oxide (GO)-based photocatalyst
GO is a very important derivative of graphene-based materials, which is generally prepared by stripping graphite with acid. As a two-dimensional layered material, its structure extends to tens of microns in transverse dimensions. Although it lacks the highly conjugated structure of graphene, the oxidation process makes the base surface and edge of GO modified by a variety of oxygen-containing functional groups.35 This modification provides numerous active sites and a larger specific surface area for the surface of graphene oxide materials. Additionally, GO materials can modulate their electrical conductivity and band gap by regulating the type and number of oxygen-containing functional groups they contain, which greatly expands their range of applications. In photocatalytic systems, GO can be used as both a cocatalyst and a photocatalyst. As a semiconductor, it has a low degree of oxidation and an inherently wide bandgap. Furthermore, its bandgap width can be adjusted by changing its C/O ratio and the type and number of oxygen-containing functional groups it contains.36Unlike graphite, GO is hydrophilic due to its surface oxygen-containing groups, which enhances its dispersion in water. Given that GO is a polymer-like graphite semiconductor composed solely of carbon, oxygen, and hydrogen, with a large surface area, Yeh et al.37 suggested that these structural characteristics should enhance GO's effectiveness as a photocatalyst for H2 generation from water. In 2010, they synthesized a GO semiconductor photocatalyst using an improved Hummers method, achieving an apparent band gap ranging from 2.4 to 4.3 eV. They found that under ultraviolet or visible light irradiation, the GO photocatalyst could stably catalyze H2 production from a 20 vol% methanol solution and pure water. This study demonstrated for the first time the potential of graphene-based materials as a medium for water decomposition by sunlight. GO-based photocatalytic composites have significant advantages in the field of photocatalysis due to their unique layered structure. In recent years, the research on GO-based photocatalysts in the field of photocatalytic CO2 reduction has mainly used metal oxides and GO to form composite materials, or used GO as a cocatalyst to improve the photocatalytic performance of photocatalytic materials. For example, Shi et al.38 synthesized a series of In2O3–GO composite photocatalysts modified by GO by a precipitation method. The effect of different contents of graphene oxide on the activity of CO2 hydrogenation to CH3OH by the In2O3–GO photocatalyst was studied. The results showed that GO can promote the transformation of cubic In2O3 (c-In2O3) to hexagonal In2O3 (h-In2O3) during CO2 hydrogenation and form a homojunction between h-In2O3(110) and c-In2O3(440), which strengthens the interaction between the two phases. Thus, the reduction of In2O3 and the formation of oxygen vacancies on the surface are promoted, which is greatly conducive to the formation of methanol. When the content of GO reaches 8%, the space time yield (STY) of methanol can be as high as 0.93 gMEOH h−1 g−1, and the selectivity can reach more than 76%. Zhang et al.39 prepared 2D/2D nanostructures composed of BiVO4 and layered GO by a hydrothermal method and realized the efficient effect of CO2 photoreduction to CH3OH. The optimized 10-CGO/BiVO4 composite catalyzed CO2 reduction to produce CH3OH under light, which was 537.78 μmol g−1 h−1, about 6.47 times that of BiVO4 nanosheets. In addition, multi-component GO based photocatalysts39–43 have also been constructed by doping and composite methods. Due to their unique 2D or 3D structure, they have shown excellent performance in selectively capturing CO2 and driving CO2 reduction.
In general, GO materials have significant advantages in the field of photocatalysis due to their huge specific surface area, zero band gap width, 2D-layered structure and other characteristics.44,45 However, at present, there are few studies on low-cost, mass-produced GO materials, which has limited their application scope to some extent, so there is an urgent need to conduct in-depth research on this class of materials. However, to solve this problem, the integration of resources appears to be very critical, to build a more reasonable industrial ecological chain system and to avoid the waste of resources in order to be more conducive to in-depth research.
3.4 Mixed metal compound photocatalyst
Metal oxides are a common class of compounds in daily life. Because of their excellent stability and good electrical conductivity, they have been widely studied as effective photocatalysts. In the process of photocatalytic CO2 reduction, the most important thing is the separation and transfer of electron–hole pairs. Therefore, the band gap width of the photocatalyst and the potential corresponding to the conduction band and valence band are the key factors determining the photocatalytic efficiency.46 Due to the wide variety of metal oxides, their band gap width and redox potential are adjustable in a wide range, which makes them have broad application prospects in the field of photocatalysis. Metal oxides have two electronic configurations: d0 (transition metal) and d10 (main group metal). Among them, the transition metals that can form metal oxides mainly include Ti4+, Zn2+, Zr4+, Ta5+, Nb5+, W6+ and Mo6+, and the main group metals mainly include In3+, Ga3+, Ge4+, Sn4+ and Sb5+.47TiO2 is a widely used semiconductor material in various fields, including hydrogen production, carbon dioxide conversion, organic pollutant degradation, and water purification, where it plays a highly significant role. This is because TiO2-based materials have good light absorption performance and stability under UV-visible light irradiation.46 As early as 1972, Fujishima et al.48 found for the first time that TiO2 catalyzed the H2O redox reaction under light irradiation. This research has opened the prelude to the study of photocatalytic materials. However, TiO2, due to its wide band gap (3.2 eV), has a very low utilization of visible light in the UV-visible region and can only absorb a small part of ultraviolet light. This limits the wide application of TiO2 in the field of high efficiency photocatalysis. As research progresses, a variety of metal compound photocatalytic materials have been developed. Some of the most common include ZnO, CdS, Fe2O3, WO3, ZnS, BiOBr, Ag3PO4, etc.49 However, simple photocatalytic materials often have some shortcomings such as low photoresponse, high recombination rate of photogenerated electron pairs and low photocatalytic efficiency. In addition, photocatalytic systems in which metal compounds and organic compounds are constructed into composites have appeared,50,51 but those that can reduce CO2 to CH3OH are rare. In order to improve the photocatalytic efficiency, at present, people often use a variety of metal compound materials to construct heterojunctions, so as to promote the separation of photogenerated electron–hole pairs, accelerate charge transfer, and finally achieve efficient photocatalytic reactions.
Because of the differences in the extranuclear electron structure, different metal compounds have their own unique band gap structure. In general, two or more materials whose bandgap positions can be combined in a “misaligned” manner can be combined to form a heterojunction structure, which facilitates the transport of electron–hole pairs. There are three types of common heterojunctions, namely type-II heterojunctions, Z heterojunctions and S heterojunctions. The modes of electron–hole transport between different types of heterojunctions are different. But ultimately, it is for the same purpose, that is, to improve the separation of photogenerated electron–hole pairs, so as to improve the photocatalytic efficiency. At present, coupling two or more semiconductors into heterojunctions is considered as a promising method to obtain highly photocatalytically active photocatalysts. Wang et al.52 synthesized BiVO4/Bi4Ti3O12 heterojunction composite materials by an in situ hydrothermal method. This novel composite material has excellent photocatalytic ability and can reduce CO2 and H2O to CH3OH and CO. Among them, the CH3OH and CO yields of the BiVO4/10% Bi4Ti3O12 sample were the highest (Fig. 7a), up to 16.6 and 13.29 μmol g−1 h−1, which were 12.39 and 5.68 times higher than pure BiVO4, and 9.88 and 2.80 times higher than pure Bi4Ti3O12, respectively. In addition, the cyclic experiment results in Fig. 7b show that the photocatalytic stability of BiVO4/10% Bi4Ti3O12 is good. In Fig. 8c and d, BiVO4 has successfully and uniformly grown on the surface of Bi4Ti3O12. There was no apparent reunion. Afterwards, XPS analysis was performed (Fig. 9), which verified the electron transfer mechanism of the BiVO4/10% Bi4Ti3O12 sample by comparing the binding energies, i.e., the electrons migrated from BiVO4 to Bi4Ti3O12, and a heterojunction was formed between BiVO4 and Bi4Ti3O12. This provides effective evidence to prove its photocatalytic ability.
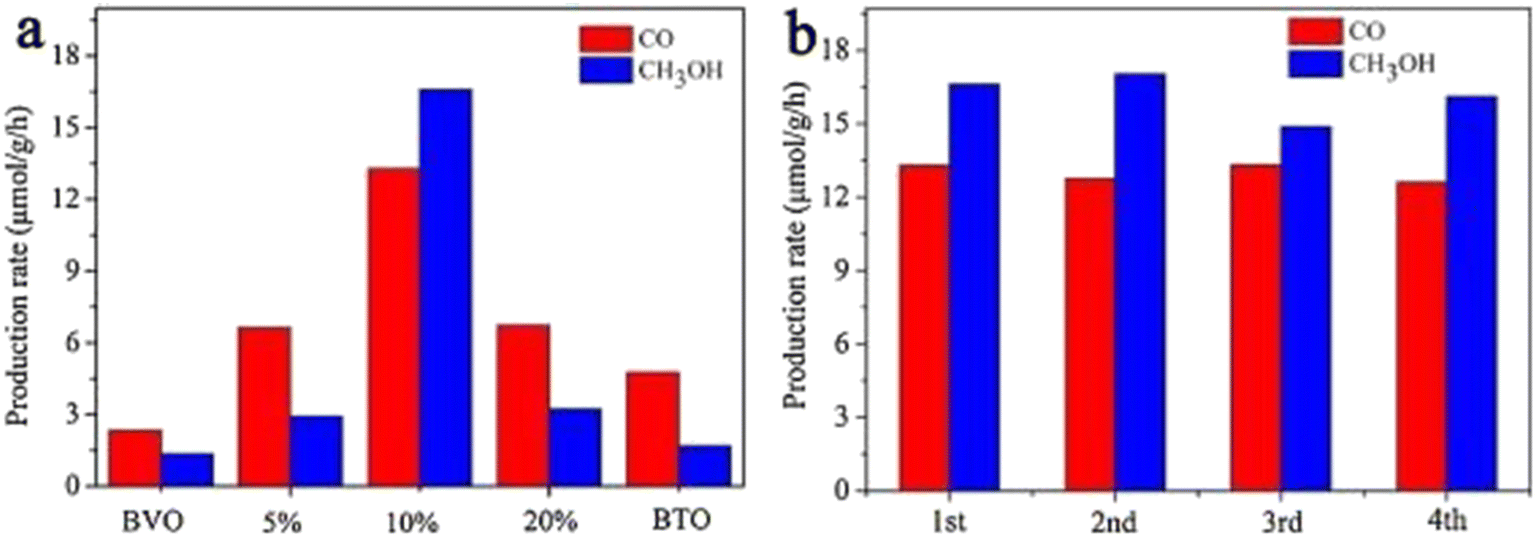 | ||
| Fig. 7 (a) CH3OH and CO evolutions on BiVO4/Bi4Ti3O12 with different Bi4Ti3O12 contents under a 300 W Xe lamp and (b) photocatalytic activity stability of the BiVO4/10% Bi4Ti3O12 sample within 12 h with 3 h per circulation.52 | ||
 | ||
| Fig. 8 SEM images of (a) BiVO4, (b) Bi4Ti3O12, and (c and d) BiVO4/10% Bi4Ti3O12.52 | ||
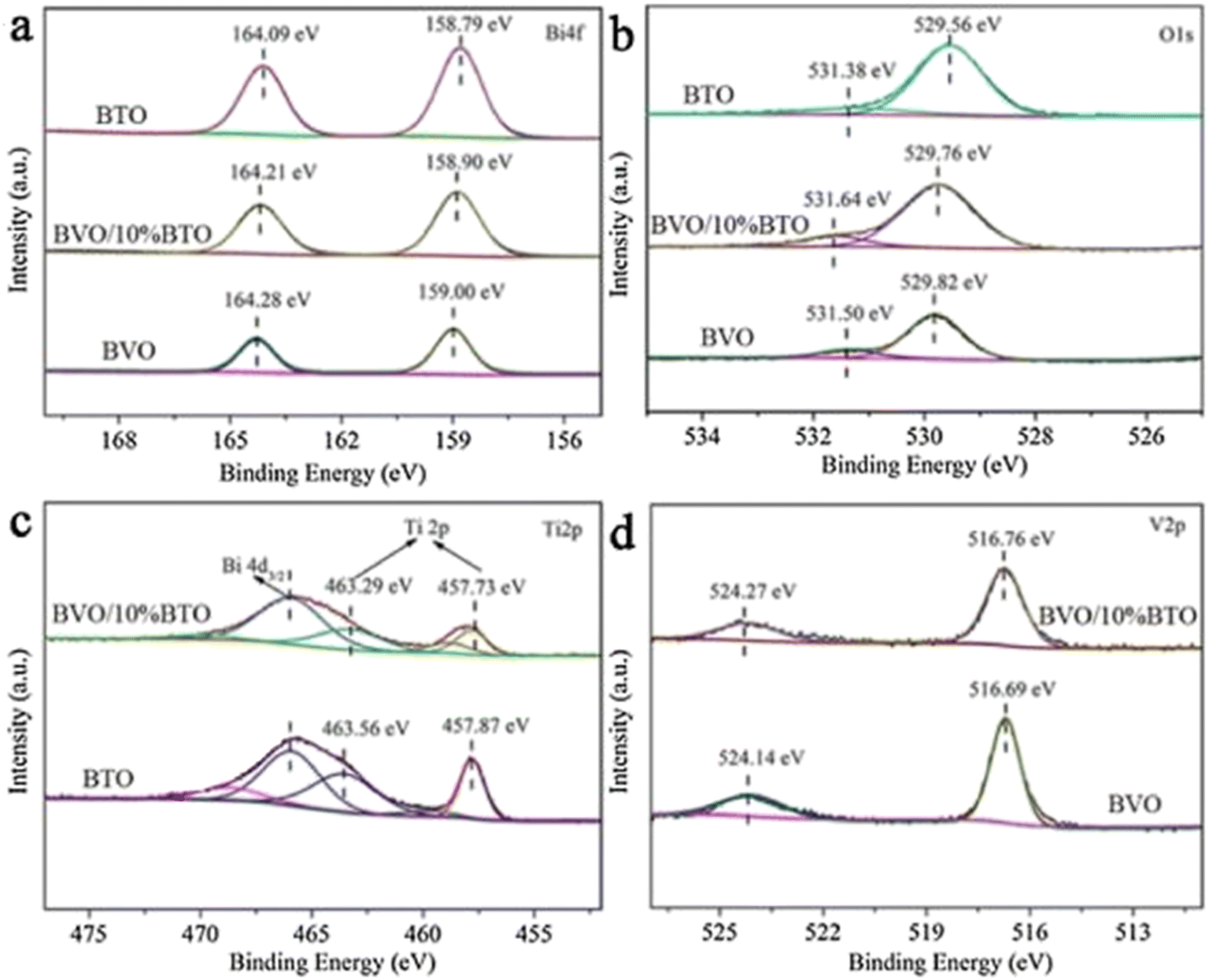 | ||
| Fig. 9 The Bi 4f (a), O 1s (b), Ti 2p (c) and V 2p (d) spectra of BiVO4, BiVO4/10% Bi4Ti3O12 and Bi4Ti3O12.52 | ||
Interface engineering often has outstanding performance in improving the efficiency of photocatalysis. This is because abundant catalytically active sites can be generated on the surface of the material through interface modification, thereby enhancing the adsorption capacity of the material for CO2, while shortening the charge transfer distance, thus ensuring efficient electron–hole pair transport. Wang et al.53 developed porous CuO ultra-thin nanoribbons with well-controlled oxygen vacancies (Vo) by a three-step method using wet chemical methods combined with rapid calcination strategies (Fig. 10). The ultrathin band structure of CuO, along with micropores and slight lattice perturbations, was confirmed via TEM and HAADF-STEM, indicating the presence of point defects. Oxygen vacancies (Vo) were further investigated using various techniques, including low-loss EELS, XPS, and EPR. These defects enhanced the selective adsorption and enrichment of gas molecules on the photocatalyst surface during CO2 reduction. The photocatalytic reaction achieved an optimal CH3OH generation rate of 12.3 μmol g−1 h−1 with a selectivity of 62.5%. Isotope labeling confirmed CO2 as the carbon source, and cycling experiments demonstrated the material's good photostability (Fig. 11). Li et al.54 developed V–Bi19Br3S27, a nanowire photocatalyst with abundant Br and S double vacancies and surface Bi–O bonds, through alkali etching. Compared with the synthetic Bi19Br3S27 nanowires, the treated nanowires V–Bi19Br3S27 retained their original nanowire morphology (Fig. 12). EDS spectra analysis detected low-density oxygen in the structure. FT-XANES and EXAFS results revealed the formation of double vacancies, and EXAFS wavelet transform analysis confirmed the Bi–O bond formation in V–Bi19Br3S27. Ultrafast transient absorption (TA) spectroscopy showed that both Bi19Br3S27 and V–Bi19Br3S27 nanowires have transient absorption peaks at 1150 nm, indicating NIR light excitation capability. The electron decay kinetics revealed that the V–Bi19Br3S27 nanowires had a delay time 22 times longer than the Bi19Br3S27 nanowires, suggesting that vacancy formation and oxygen doping enhanced the photogenerated electron lifetime. The resulting V–Bi19Br3S27 nanowires produced 1.6 μmol g−1 CH3OH in the photocatalytic reduction of CO2 under near-infrared irradiation without any co-catalyst and sacrificial agent, which was 2.3 times higher than that of the Bi19Br3S27 nanowires (Fig. 13).
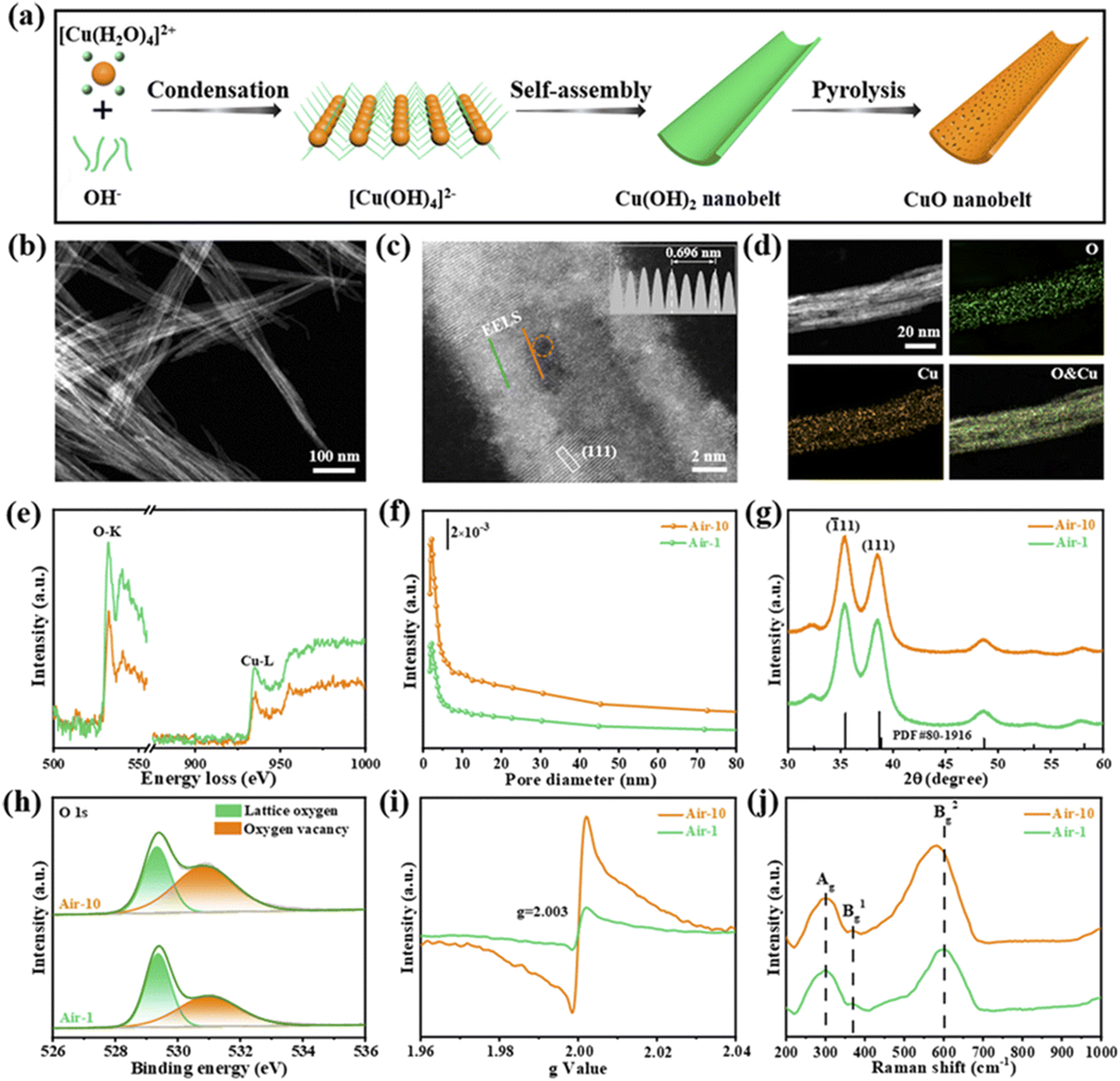 | ||
| Fig. 10 (a) Schematic illustration of the synthetic process of a CuO nanobelt. (b) TEM image, (c) HAADF-STEM image (inset: intensity profiles recorded from the grey rectangular area), and (d) EDX elemental mappings of air-10. (e) EELS spectra for the O-K and Cu-L edges (the solid orange and green lines are from the corresponding scan lines in (c)). (f) The pore-size distribution curves, (g) XRD patterns, (h) O 1s XPS spectra, (i) EPR spectra, and (j) Raman spectra of air-1 and air-10.53 | ||
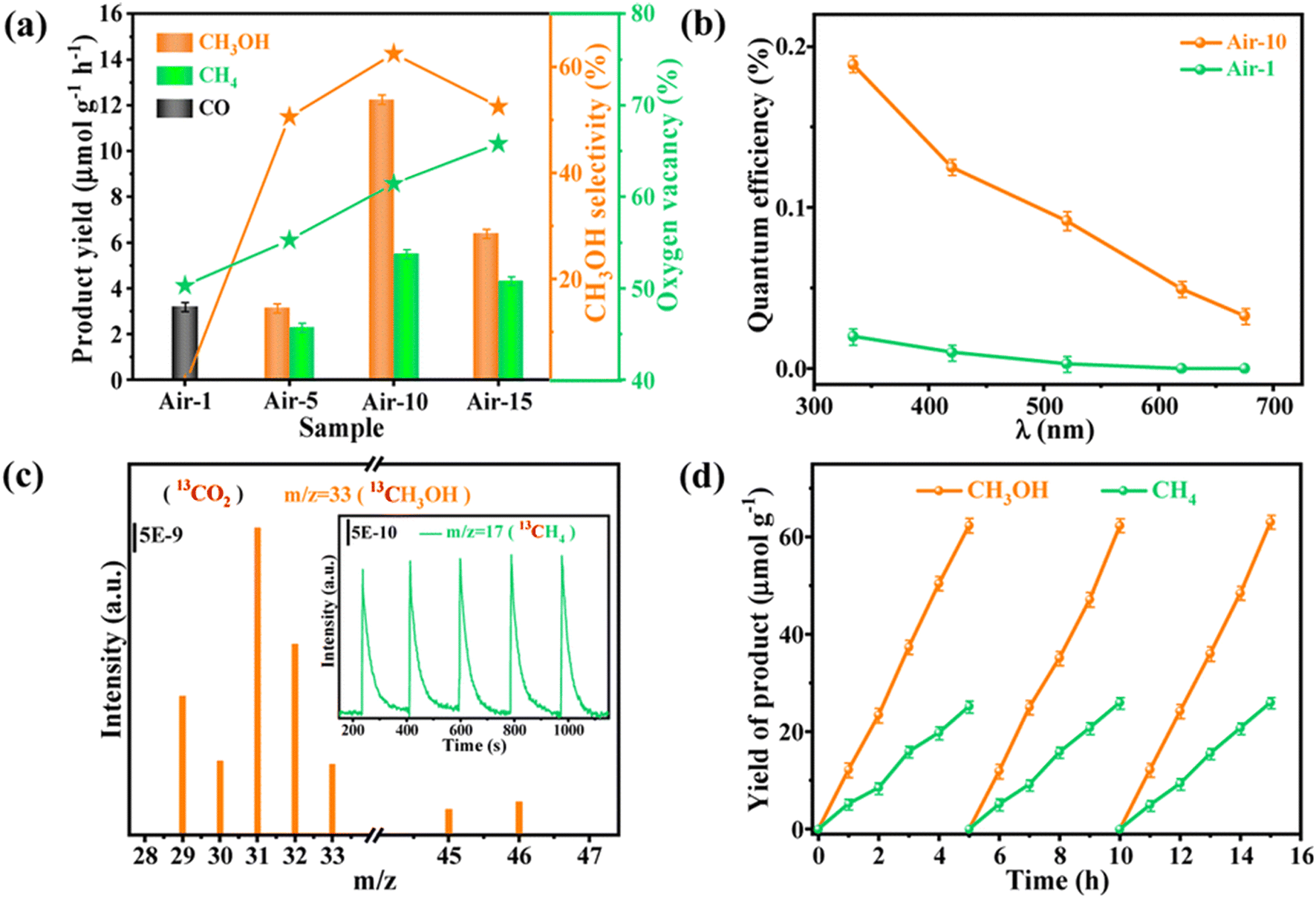 | ||
| Fig. 11 (a) Product yields, CH3OH selectivity and oxygen vacancy content of different photocatalysts. (b) QE values of air-1 and air-10, respectively. (c) GC-MS spectra of 13CH3OH and 13CH4 generated from 13CO2 photoreduction of air-10. (d) Photocatalytic stability of air-10.53 | ||
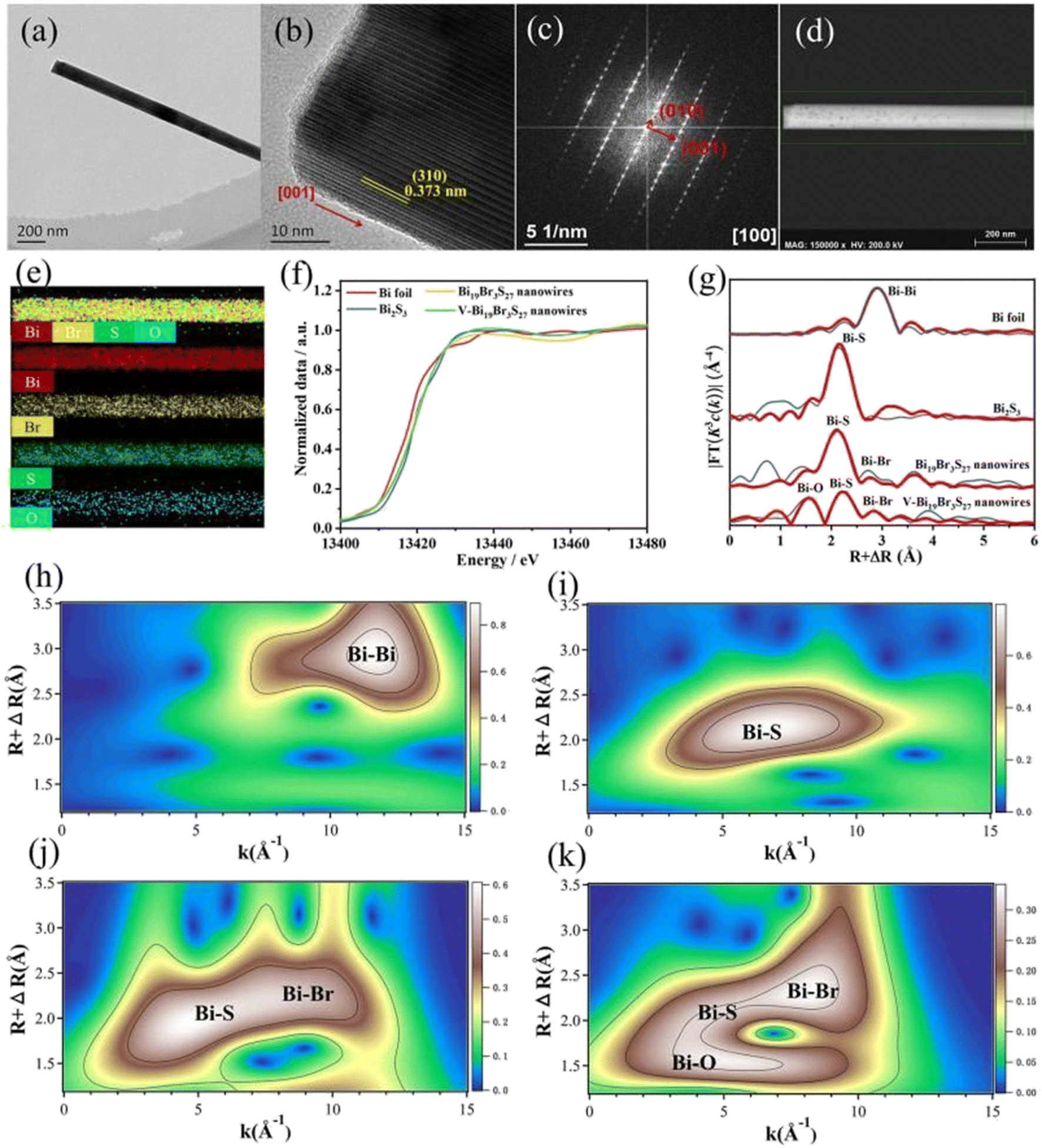 | ||
| Fig. 12 (a) TEM image, (b) HRTEM image, (c) SAED image, and (d and e) EDS mapping images of a V–Bi19Br3S27 nanowire. (f) Bi LIII-edge XANES spectra of Bi19Br3S27, V–Bi19Br3S27 nanowires and bismuth references. (g) Corresponding Fourier transforms R space fitting results. Red: observed data; blue: fitted data. (h–k) Wavelet transforms for k2-weighted EXAFS signals of Bi, Bi2S3, Bi19Br3S27, and V–Bi19Br3S27, respectively.54 | ||
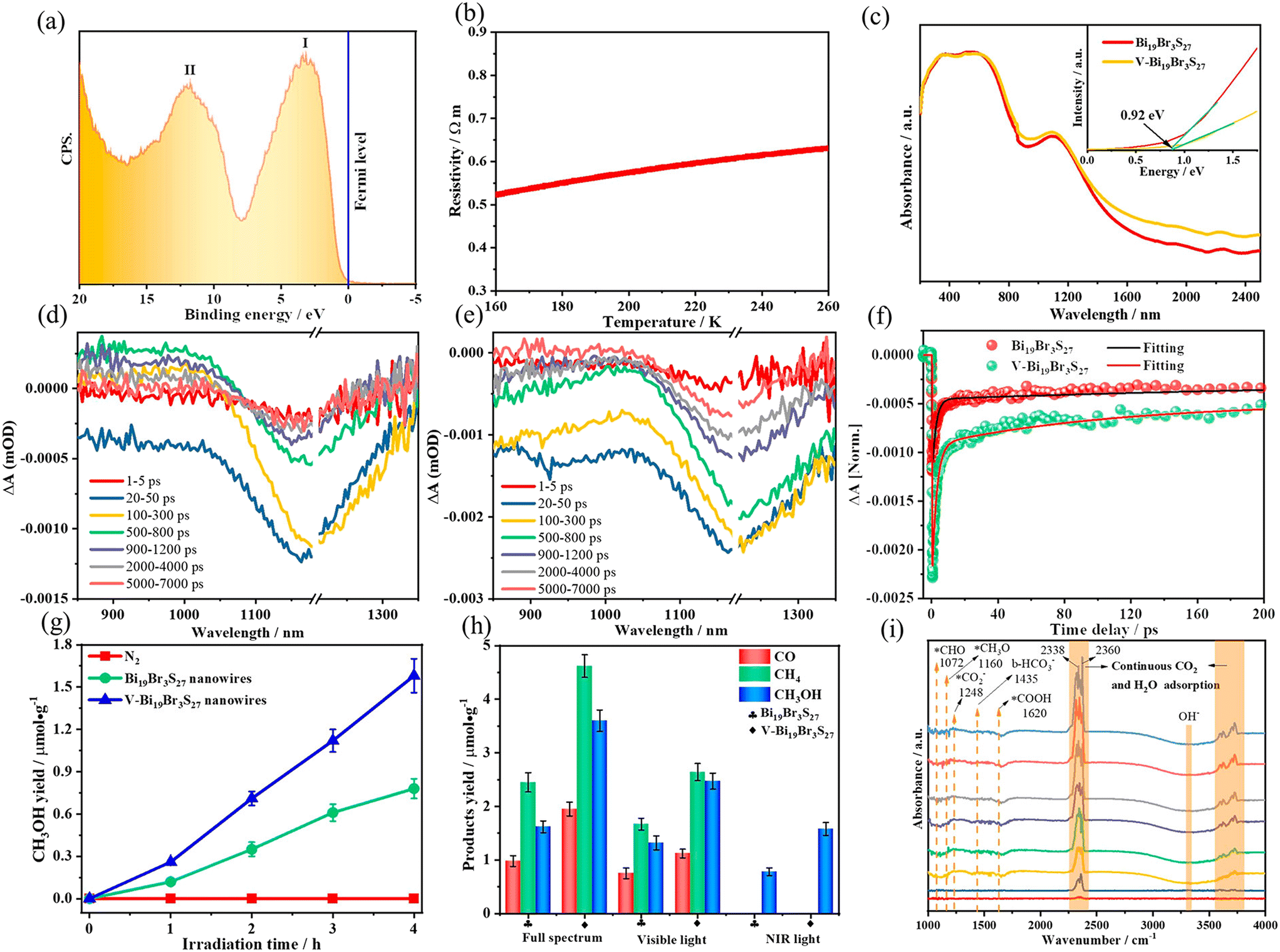 | ||
| Fig. 13 (a) Valence band XPS spectrum and (b) temperature dependence of resistivities of V–Bi19Br3S27 nanowires. (c) Optical absorption spectra and the corresponding optical bandgaps of Bi19Br3S27 and V–Bi19Br3S27 nanowires. Transient absorption (TA) spectra of (d) Bi19Br3S27 and (e) V–Bi19Br3S27 nanowires. (f) Corresponding TA kinetics. Photocatalytic CH3OH evolution under (g) NIR and (h) full-spectrum light irradiation for Bi19Br3S27 and V–Bi19Br3S27 nanowires. (i) In situ FT-IR spectra showing coadsorption of CO2 and H2O on V–Bi19Br3S27 nanowires.54 | ||
At present, there are numerous research studies on mixed metal compounds in the field of photocatalysis. In this paper, we summarized the recent research results on the reduction of CO2 to CH3OH by photocatalysts with different mixed metal compounds. As shown in Table 2, we find that most of these photocatalysts are composed of transition metal compounds, which can be divided into polymetallic oxides, polymetallic sulfides, and binary or multi-metal oxide composites. In the past few decades, a lot of research has been done on various photocatalytic materials, such as TiO2, Cds, Bi2O3, CeO2, etc. However, due to the single composition and structure of these photocatalysts, the overall photoconversion efficiency and product selectivity are often not ideal due to the rapid recombination of photogenerated holes in the process of photocatalytic CO2 reduction. In order to improve the photocatalytic efficiency, it has been found that mixed metal compounds synthesized by hybridization,55–58 etching,54 surface reconstruction,59,60 defect control,53,61,62 and heterojunction construction52,63 often have better photocatalytic properties than single metal compounds. This is similar to the study of alloys in order to improve the defects of pure metals in certain properties. However, it is not enough to focus only on the composition of the photocatalyst. In order to increase the specific surface area and obtain more CO2 conversion active sites, it is necessary to pay attention to the homogeneity of the composite during the preparation process, as well as the dispersion of metal vacancies and oxygen vacancies on the surface of the material during the surface reconstruction and defect control process.
| Photocatalysts | Reaction conditions | CH3OH yield (μmol g−1 h−1) | Ref. |
|---|---|---|---|
| BiVO4/Bi4Ti3O12 | Commercial xenon lamp (300 W) | 16.6 | 52 |
| CuO | Xenon lamp (300 W) | 12.3 | 53 |
| Bi19Br3S27 | Xenon lamp (300 W) | 1.6 | 54 |
| M0.33WO3 | Xenon lamp (300 W) | 15.48 | 55 |
| (Air as a carbon source) | |||
| ZnO/Fe2O3 | Xenon lamp (300 W) | 178.3 | 64, 65 |
| Mo2C/TiO2 | UV-visible LED lamp (5 mW cm−2) | 11.8 | 63 |
| CuO | Visible LED lamp (25 °C) | 3.7 | 59 |
| Mg(OH)2/CuO/Cu2O | Halogen lamp (20 W, 300–900 nm) | 6 | 66 |
| CeO2 | Xenon lamp (300 W) | 0.702 | 61 |
| VO2/ZnV2O4 | HID xenon lamp (35 W) | 202 | 67 |
| CeO2/ZnIn2S4 | Xenon lamp (300 W) | 0.542 | 56 |
| CoO/Co/TiO2 | Xenon lamp (300 W, 120 °C) | 39.6 | 68 |
| MoS2/Mn0.2Cd0.8S | Xenon lamp (300 W, 24 h) | 1017.7 | 57 |
| NiSe2/WSe2 | Xenon lamp (300 W) | 3.80 | 69 |
| Cu2O/TiO2 | UV light (6 h) | 21.0–70.6 | 70 |
| NiMoO4 | LED light (20 W) | 3365 | 71 |
| Ce-MOF/Bi2MoO6 | Xenon lamp (300 W, 6 h) | 40.59 | 72 |
| Bi12SiO20/BiO2−x | Xenon lamp (300 W) | 31.16 | 73 |
| LiNbO3 | UV pen lamp | 35 | 58 |
| Bi2O2SiO3 | Xenon lamp (300 W, 4 h) | 12.78 | 60 |
| CuCo2O4/CeO2 | Xenon lamp (300 W, 9 h) | 1320 | 74 |
| WO3−x | Monochromatic visible light (675 nm) | 17 | 62 |
3.5 Single metal-doped photocatalyst
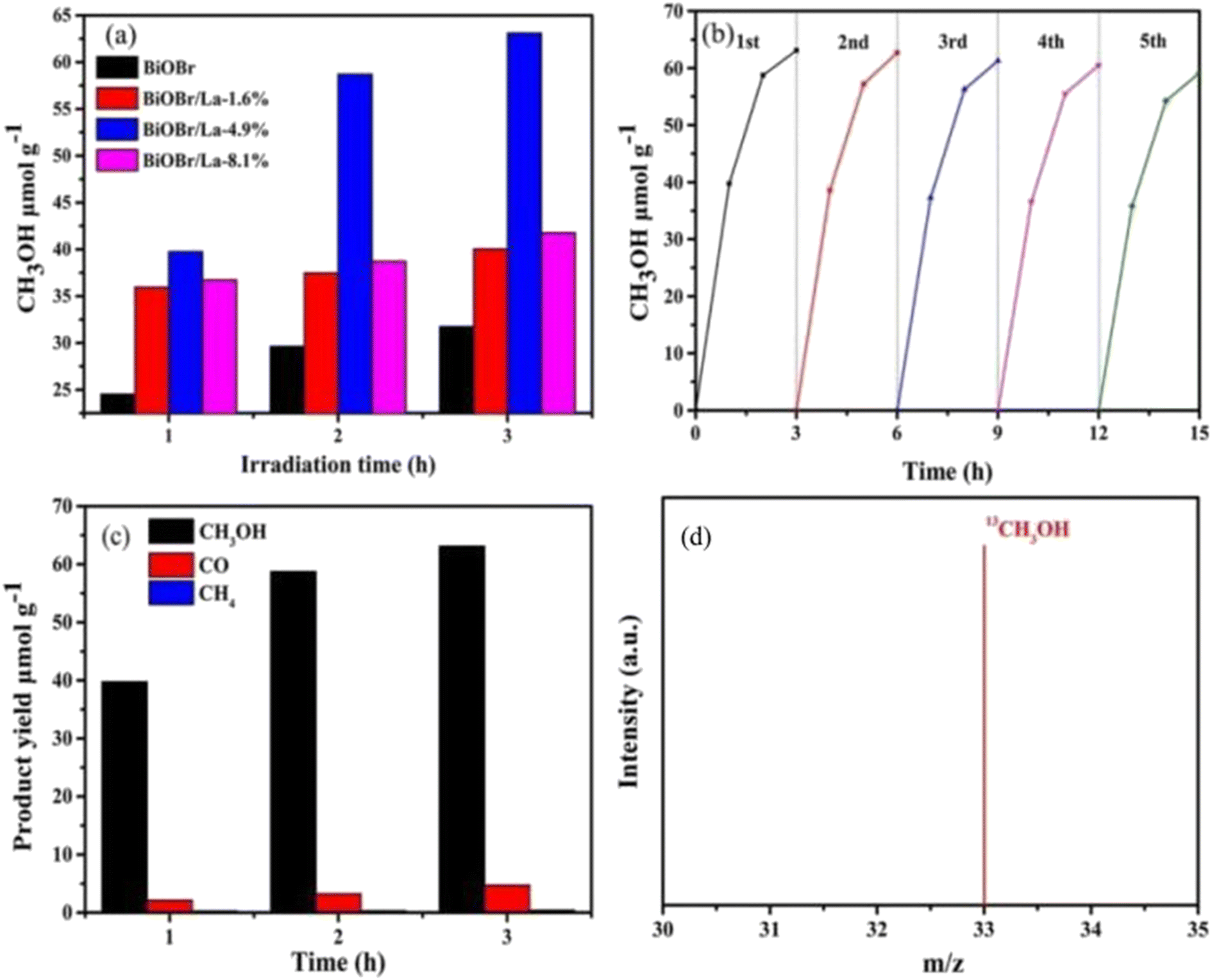
Single metal-doped photocatalysts mainly refer to the single metal as an impurity atom to modify the structure of metal compounds to form composite materials with efficient photocatalytic performance. Among them, the single metal is divided into two categories, one refers to precious metals, often as doping elements of precious metals which are: gold (Au), silver (Ag), platinum (Pt), palladium (Pd) and so on; the other category refers to non-precious metals, mainly including iron (Fe), cobalt (Co), nickel (Ni), copper (Cu) and so on. It has been reported that the successful doping of single Pd, Pt, Cd and Hg atoms into gold nanoparticles can not only enhance the stability of nanoparticles, but also adjust the catalytic performance of nanoparticles.75 As for the application of single atom doping in the field of photocatalysis, as early as 2014, Xing et al.76 stably synthesized dispersed metal atoms (Pt, Pd, Rh or Ru) on TiO2 for the first time in order to solve the problem of the aggregation of precious metals as co-catalysts in the supported process or photocatalytic reaction. The dispersed metal atom-based photocatalyst has excellent hydrogen evolution stability, and its photocatalytic activity is 6–13 times higher than that of metal clusters supported on TiO2 by traditional methods.At present, various types of single metal doped photocatalytic materials are explored for photocatalytic CO2 reduction. Ma et al.77 synthesized Pr1–N6/CN photocatalysts using a “two-step calcination-induced metal vaporization strategy” (Fig. 14a). Then, using the same strategy, carbon nitride (CN)-loaded Pr monoatomic oxygen coordination materials were designed by adding oxygen-containing species. The engineered oxygen coordination configuration of the Pr monoatom (Pr1–N4O2/CN) was successfully obtained and its ultra-high loading of Pr atoms is highly dispersed on the surface of the CN substrate. This photocatalyst utilizes oxygen atoms to modulate the electronic structure of a single Pr atom, which can be directed to induce the enrichment of photogenerated electrons at the Pr monoatomic site under light thus significantly promoting the adsorption and activation of CO2. Photocatalytic CO2 reduction results show that, as shown in Fig. 15a, the CH3OH yield is significantly increased after the introduction of atomically dispersed Pr1–N6 and Pr1–N4O2 active sites into CN. In addition, its CH3OH formation rate is higher than the CN loading of different kinds of metal monoatoms and other metal elements (Fig. 14e and f). However, in addition to CH3OH, other by-products such as H2, CO, CH4 and C2H4 appear during CO2 reduction (Fig. 3c). Nevertheless, it can be observed that the CH3OH selectivity on Pr1–N4O2/CN is very high, reaching more than 90%. The experimental results of repeated use of Pr1–N4O2/CN for 5 cycles (∼20 h) to determine the durability of Pr1–N4O2/CN showed that the yield and selectivity of Pr1–N4O2/CN to CH3OH were very stable, and there was no obvious deactivation. This indicates that Pr1–N4O2/CN has good durability for the synthesis of CH3OH by CO2 photoreduction. In addition, Jiao et al.78 developed novel flower-like La-doped BiOBr nanosheets, the synthesis process of which is schematically shown in Fig. 16. From the TEM imaging analysis, it can be seen that the structure is composed of nanospheres of nanosheets with uniform elemental distribution (Fig. 17). The results of its photoelectrochemical test analysis (Fig. 18) showed that due to the doping of La3+, the photogenerated electrons located in the conduction band of BiOBr can be transferred to La3+, which prolongs the lifetime and reduces the compounding rate, thus improving the photocatalytic performance. From the photocatalytic results (Fig. 19), we can find that the yield of CH3OH products for BiOBr/La-4.9% is much higher than that for the other groups after 3 h of irradiation. At the end of 5 cycles of experiments, the yield of CH3OH products can reach more than 90% of the first experiment, which indicates that BiOBr/La-4.9% has good chemical stability in the CO2 reduction process. Comparison of the amount of each product shows that BiOBr/La-4.9% has a high selectivity for the generation of CH3OH. The GC-MS analysis in Fig. 19d is the reduction product of 13CO2 photoreduction, where the strong signal in the MS spectrum at m/z = 33 is 13CH3OH. This result proves that CO2 is the only carbon source of CH3OH reduction products.
 | ||
| Fig. 14 Synthesis process and structural information of the as-fabricated photocatalysts. (a) Schematic of the synthesis process of Pr1–N6/CN and Pr1–N4O2/CN; (b) XRD patterns; high-resolution XPS spectra of (c) C 1s and (d) N 1s over CN, O/CN, Pr1–N6/CN, and Pr1–N4O2/CN; (e) and (f) AC-HAADF-STEM images, and (g) corresponding EDX mapping images of the as-prepared Pr1–N4O2/CN.77 | ||
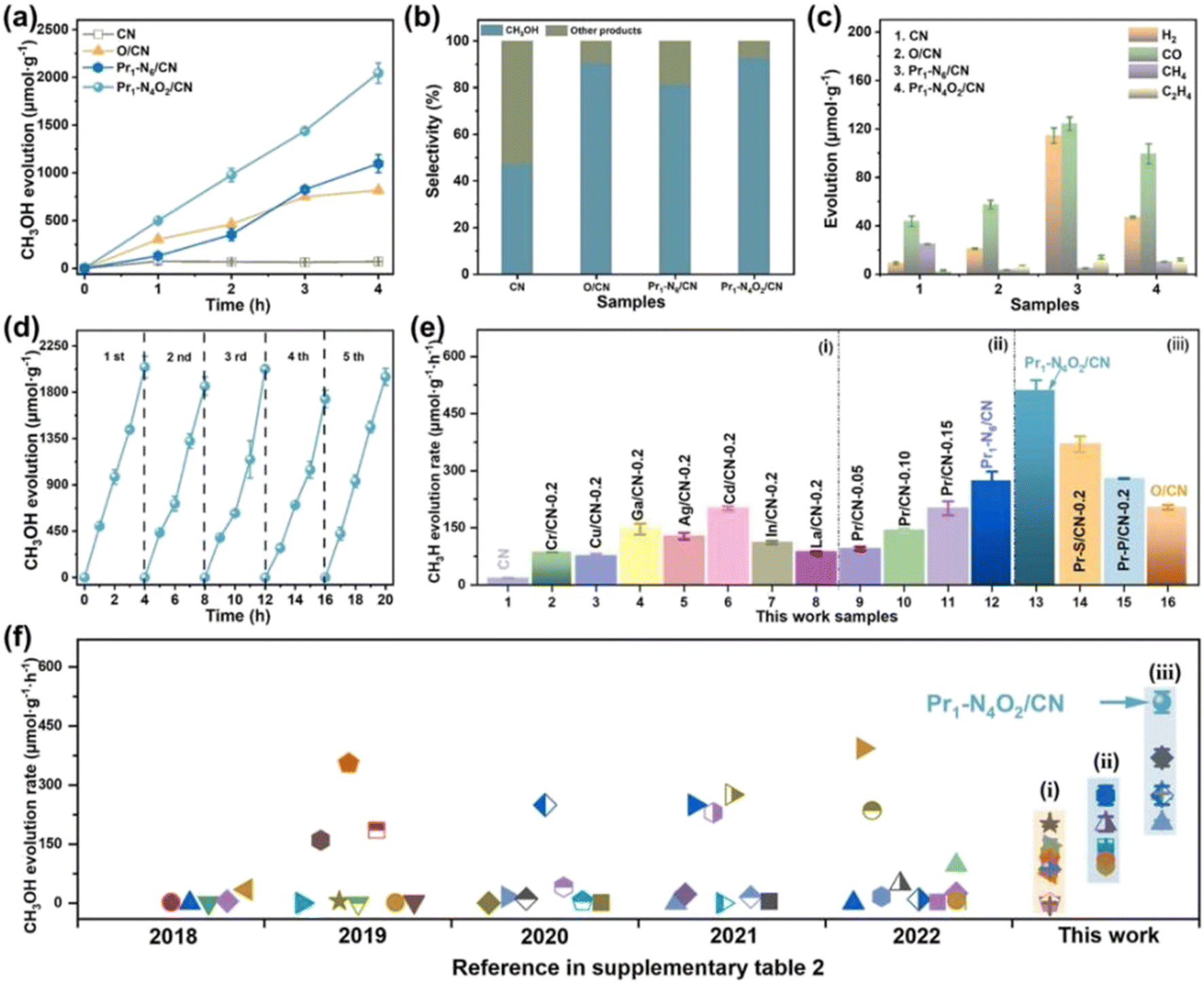 | ||
| Fig. 15 Photocatalytic reduction CO2 performance of the as-prepared photocatalysts. (a) CH3OH production with time for CN, O/CN, Pr1–N6/CN, and Pr1–N4O2/CN; (b) the selectivity to CH3OH and other products; (c) H2, CO, CH4, and C2H4 production in 4 h; (d) CH3OH production corresponding to cycle experiments (∼20 h) for Pr1–N4O2/CN; (e) CH3OH production of the reference photocatalysts synthesized in this work, such as (i) CN supported different kinds of metal elements, (ii) CN supported Pr single-atom with different density, and (iii) CN supported Pr single-atom with different coordination configurations; and (f) CH3OH evolution rate of various photocatalysts during photocatalytic reduction of CO2 with H2O under similar reaction conditions over the past five years.77 | ||
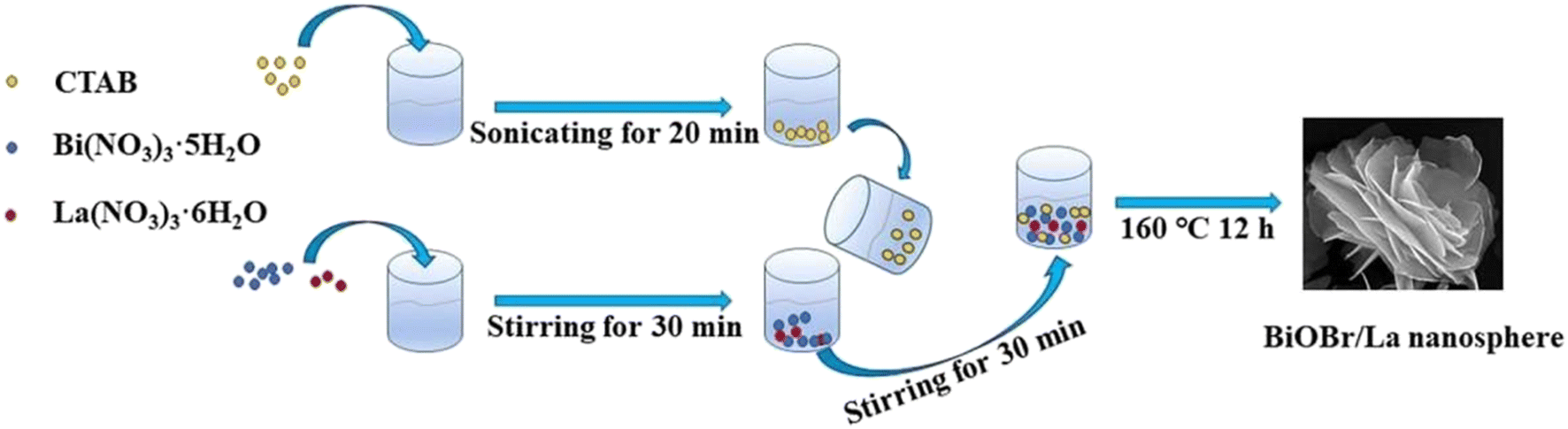 | ||
| Fig. 16 Process diagram of BiOBr/La synthesis.78 | ||
 | ||
| Fig. 17 TEM images of BiOBr (a and b), TEM images of BiOBr/La-4.9% (c and d), HRTEM images of BiOBr (e) and BiOBr/La-4.9% (f). EDX elements mapping images of Bi (g), Br (h), O (i), and La (j) in BiOBr/La-4.9%.78 | ||
 | ||
| Fig. 18 Transient photocurrent curves (a), PL spectra (b), and EIS curves (c) for BiOBr and BiOBr/La catalysts.78 | ||
 | ||
| Fig. 19 (a) The yields of CH3OH for BiOBr and BiOBr/La catalysts, (b) cycling performance for BiOBr/La-4.9%, (c) products from CO2 reduction on BiOBr/La-4.9% and (d) MS spectrum of CH3OH produced by 13CO2 reduction.78 | ||
In Table 3, we summarize the results of some studies on the reduction of CO2 to CH3OH by single metal-doped photocatalysts. This kind of photocatalytic material mainly improves the photocatalytic performance of the original matrix material by loading metal atoms on the surface of the matrix material, thereby forming an active site on the surface of the matrix material and promoting charge transfer. At present, the methods for preparing such materials include pyrolysis,77 solvent/hydrothermal,78–81 acid-assisted,82,83 sol–gel,84,85etc. The most commonly used method is the solvent/hydrothermal method, which is convenient and simple, and is also a common method for synthesizing other photocatalytic materials. From Table 3, we can also find that most of the single metal atoms used for doping belong to transition metals, because transition metals often contain d orbital electrons or empty d orbitals, which makes its d electron layer easy to lose electrons or seize electrons to form variable valence metal ions, which is conducive to the transfer of electron–hole pairs during photocatalytic reactions. Although single-metal doping has shown remarkable effect in improving the photocatalytic performance of the substrate materials, the transition metals used in it are often costly, and there are still problems of catalyst poisoning caused by improper use. Therefore, the development of green low-cost single metal-doped photocatalytic materials is worthy of further exploration by researchers.
| Photocatalysts | Reaction conditions | CH3OH yield | Ref. |
|---|---|---|---|
| Pr1–N4O2/CN | Xenon lamp (300 W) | 511.1 μmol g−1 h−1 | 77 |
| La/BiOBr | Xenon lamp (300 W) | 22.77 μmol g−1 h−1 | 78 |
| Pt/ZnO | Continuous visible light (9 h) | 668 μmol g−1 | 84 |
| Co/TiO2/rGO | Xenon lamp (500 W) | 936 μmol gcat−1 | 86 |
| Co/ZnO/rGO | High pressure xenon lamp (400 W) | 30.1 μmol g−1 | 85 |
| Pt/ZnO–ZnS | Xenon lamp (300 W) | 81.1 μmol g−1 h−1 | 87 |
| Ag NPs/ACFs | Xenon lamp (300 W) | 13.9 μmol g−1 h−1 | 82 |
| Ag–Al2O3 | UVC (200–280 nm) | 36.3 ppm | 79 |
| Cu/g-C3N4 | UV-visible xenon lamp (300 W) | 25.0 μmol g−1 | 80 |
| Pr3+/La1−xPrxMn0.6Ni0.4O3−δ | Visible light (300 °C) | 3.97 mmol g−1 | 88 |
| Ti/WO3 | — | 16.8 μmol g−1 h−1 | 83 |
| Cu/SnS2 | Visible light (λ ≥ 420 nm) | 0.99 μmol g−1 h−1 | 81 |
3.6 Characteristics of different photocatalyst systems
Here, we can easily find a rich variety of photocatalytic systems used for photocatalytic reduction of CO2 to CH3OH, and different photocatalytic systems have their own characteristics. In Table 4, we briefly summarize the characteristics of the five photocatalytic systems mentioned in the paper, and it can be seen that no photocatalyst is perfect at present, and all of them have their own shortcomings. In the future, when designing photocatalytic materials, not only their photocatalytic performance should be considered, but also the service life, atomic utilization, recyclability, etc. of the materials should be taken into account from the perspective of environmental protection. A single material as a photocatalyst has obviously exposed its shortcomings, so, combined with the characteristics of various materials, the development of new composite materials to achieve “1 + 1 > 2” performance improvement is a good choice.| Photocatalysts | Traits | Limitations |
|---|---|---|
| LDH-based | Unique layered structure, interlayer cations can be selective | Rapid recombination of photogenerated charge carriers, low CO2 adsorption activity, agglomerate easily |
| g-C3N4-based | Planar two-dimensional sheet structure, unique physical and chemical properties, safe and pollution-free | Small specific surface area, low visible light absorption power, rapid recombination of photogenerated charge carriers |
| GO-based | Single atomic layer structure, high stability, excellent electrical conductivity, multifunctional groups | High cost, difficult to synthesize, research immaturity |
| Mixed metal compound | Good electrical conductivity, appropriate bandgap width | Uniformity of materials, the dispersion of elements on the surface of materials is difficult to control |
| Single metal-doped | Highly active site, high selectivity, stability, easy separation | High surface energy, atoms easily aggregate |
4. Challenges and future prospects
In the context of global energy shortages, the production of solar fuels through the photocatalytic reduction of CO2 presents a promising strategy to address environmental challenges and ensure future energy security. Methanol (CH3OH), as a new type of clean energy fuel, has the potential to replace some fossil fuels and offers advantages in reducing energy consumption. Therefore, using photocatalytic processes to convert CO2 into CH3OH holds great promise for addressing future energy needs.Recent years have seen noted innovations in the development of nanocomposite photocatalysts designed to enhance the efficiency of CO2 reduction to CH3OH. A key advancement has been made in layered double hydroxide (LDH) photocatalysts, where the synergistic integration of metal cations within a single-layered structure improves charge separation and optimizes electronic properties, leading to more efficient CO2 conversion. Similarly, significant improvements in graphitic carbon nitride (g-C3N4) photocatalysts have been achieved through structural modifications and element doping, which extend light absorption into the visible spectrum and enhance charge separation, thereby boosting the overall photocatalytic performance. Innovations in graphene oxide (GO)-based photocatalysts have also been noteworthy; their high surface area and exceptional electron conductivity have driven the development of GO-based composites. These composites combine GO with other materials, effectively reducing electron–hole recombination and increasing CO2 adsorption, resulting in higher CH3OH yields. Collectively, these advancements, along with progress in mixed metal compounds and single metal-doped photocatalysts, underscore the potential for developing more effective and scalable solutions for CO2 reduction, as they improve light absorption, enhance charge separation, and facilitate multi-electron transfer processes.
However, despite these advancements, challenges remain in the photocatalytic reduction process. These include competitive by-product reactions, limited light absorption, the recombination of photogenerated electron–hole pairs, low photocatalyst stability, and the need for improved CO2 capture. To address these challenges, researchers have focused on enhancing photocatalyst performance by synthesizing binary or multivariate composites, metal doping, and ligand displacement. Moving forward, it will be crucial to explore ways to further improve the selectivity to and yield of CH3OH in photocatalytic reactions by optimizing the process. Additionally, achieving large-scale mass production of CH3OH through photocatalytic CO2 reduction remains a significant challenge that will require substantial attention and effort from the research community for sustainable long-term development.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project is supported by The Leverhulme Trust Early Career Fellowship funding under award number ECF-2021-657.References
- Y. Chen, G. Jia and Y. Hu, et al., Two-dimensional nanomaterials for photocatalytic CO2 reduction to solar fuels, Sustainable Energy Fuels, 2017, 1, 1875–1898 RSC.
- H. Lin, S. Luo and H. Zhang, et al., Toward solar-driven carbon recycling, Joule, 2022, 6(2), 294–314 CrossRef CAS.
- Y. A. Ye, B. Rtga and A. Lfh, A review of metal oxide-based Z-scheme heterojunction photocatalysts: Actualities and developments, Mater. Today Energy, 2021, 21, 100829 CrossRef.
- Q. Xu, L. Zhang and B. Cheng, et al., S-Scheme Heterojunction Photocatalyst, Chem, 2020, 6(7), 1543–1559 CAS.
- A. Mn, B. Vs and C. Tian, et al., Recent advances in visible-light-driven carbon dioxide reduction by metal-organic frameworks, Sci. Total Environ., 2020, 762, 144101 Search PubMed.
- Z. Fu, Q. Yang, Z. Liu, F. Chen, F. Yao, T. Xie, Y. Zhong, D. Wang, J. Li, X. Li and G. Zeng, Photocatalytic conversion of carbon dioxide: From products to design the catalysts, J. CO2 Util., 2019, 34, 63–73 CrossRef CAS.
- K. Li, B. Peng and T. Peng, Recent Advances in Heterogeneous Photocatalytic CO2 Conversion to Solar Fuels, ACS Catal., 2016, 7485–7527 CrossRef CAS.
- J. Fu, K. Jiang and X. Qiu, et al., Product selectivity of photocatalytic CO2 reduction reactions, Mater. Today, 2019, 32, 222–243 CrossRef.
- X. Bian, S. Zhang and Y. Zhao, et al., Layered double hydroxide-based photocatalytic materials toward renewable solar fuels production, InfoMat, 2021, 3(7), 719–738 CrossRef CAS.
- Z. Bi, R. Guo and X. Hu, et al., Research progress on photocatalytic reduction of CO2 based on LDH materials, Nanoscale, 2022, 14(9), 3367–3386 RSC.
- C. G. Silva, Y. Bouizi and V. Fornés, et al., Layered double hydroxides as highly efficient photocatalysts for visible light oxygen generation from water, J. Am. Chem. Soc., 2009, 131(38), 13833–13839 CrossRef PubMed.
- K. Teramura, S. Iguchi and Y. Mizuno, et al., Photocatalytic conversion of CO2 in water over layered double hydroxides, Angew. Chem., Int. Ed., 2012, 51(32), 8008–8011 CrossRef CAS PubMed.
- A. Ziarati, A. Badiei and R. Grillo, et al., 3D Yolk@ Shell TiO2–x/LDH architecture: tailored structure for visible light CO2 conversion, ACS Appl. Mater. Interfaces, 2019, 11(6), 5903–5910 CrossRef CAS PubMed.
- L. Zhu, C. Qin, Y. Wang and J. Cao, Single-atom Pt supported on non-metal doped WS2 for photocatalytic CO2 reduction: A first-principles study, Appl. Surf. Sci., 2023, 626, 157252 CrossRef CAS.
- J.-C. Wang, N. Li, A. M. Idris, J. Wang, X. Du, Z. Pan and Z. Li, Surface Defect Engineering of CsPbBr3 Nanocrystals for High Efficient Photocatalytic CO2 Reduction, Sol. RRL, 2021, 5(7), 2100154 CrossRef CAS.
- P. M. Stanley, K. Hemmer, M. Hegelmann, A. Schulz, M. Park, M. Elsner, M. Cokoja and J. Warnan, Topology- and wavelength-governed CO2 reduction photocatalysis in molecular catalyst-metal-organic framework assemblies, Chem. Sci., 2022, 13(41), 12164–12174 RSC.
- X. Zhang, K. Hu, X. Zhang, W. Ali, Z. Li, Y. Qu, H. Wang, Q. Zhang and L. Jing, Surface co-modification with highly-dispersed Mn & Cu oxides of g-C3N4 nanosheets for efficiently photocatalytic reduction of CO2 to CO and CH4, Appl. Surf. Sci., 2019, 492, 125–134 CrossRef CAS.
- D. Chen, Z. Wang, J. Zhang, O. Ruzimuradov, S. Mamatkulov, K. Dai and J. Low, Modulation of internal electric field in S-scheme heterojunction towards efficient photocatalytic CO2 conversion, Mater. Today Phys., 2024, 40, 101315 CrossRef CAS.
- Y. Jiang, J. Guo and X. Li, et al., Direct Z-scheme 0D/2D heterojunction of CuO quantum Dots/ultrathin CoAl-LDH for boosting charge separation and photocatalytic CO2 reduction, Sol. Energy, 2022, 231, 705–715 CrossRef CAS.
- J. Wen, J. Xie and X. Chen, et al., A review on g-C3N4-based photocatalysts, Appl. Surf. Sci., 2017, 391, 72–123 CrossRef CAS.
- Z. Sun, H. Wang and Z. Wu, et al., g-C3N4 based composite photocatalysts for photocatalytic CO2 reduction, Catal. Today, 2018, 300, 160–172 CrossRef CAS.
- X. Wang, K. Maeda and A. Thomas, et al., A metal-free polymeric photocatalyst for hydrogen production from water under visible light, Nat. Mater., 2009, 8(1), 76–80 CrossRef CAS PubMed.
- A. Li, T. Wang and C. Li, et al., Adjusting the reduction potential of electrons by quantum confinement for selective photoreduction of CO2 to methanol, Angew. Chem., Int. Ed., 2019, 58(12), 3804–3808 CrossRef CAS PubMed.
- M. Liang, T. Borjigin and Y. Zhang, et al., Controlled assemble of hollow heterostructured g-C3N4@ CeO2 with rich oxygen vacancies for enhanced photocatalytic CO2 reduction, Appl. Catal., B, 2019, 243, 566–575 CrossRef CAS.
- N. Li, M. Huang and J. Zhou, et al., MgO and Au nanoparticle Co-modified g-C3N4 photocatalysts for enhanced photoreduction of CO2 with H2O, Chin. J. Catal., 2021, 42(5), 781–794 CrossRef CAS.
- M. Ma, Z. Huang and R. Wang, et al., Targeted H2O activation to manipulate the selective photocatalytic reduction of CO2 to CH3OH over carbon nitride-supported cobalt sulfide, Green Chem., 2022, 24(22), 8791–8799 RSC.
- J. Ding, Q. Tang and Y. Fu, et al., Core–Shell Covalently Linked Graphitic Carbon Nitride–Melamine–Resorcinol–Formaldehyde Microsphere Polymers for Efficient Photocatalytic CO2 Reduction to Methanol, J. Am. Chem. Soc., 2022, 144(22), 9576–9585 CrossRef CAS PubMed.
- W. Wang, S. Song and P. Wang, et al., Chemical Bonding of g-C3N4/UiO-66 (Zr/Ce) from Zr and Ce Single Atoms for Efficient Photocatalytic Reduction of CO2 under Visible Light, ACS Catal., 2023, 13(7), 4597–4610 CrossRef CAS.
- A. Bafaqeer, M. Tahir and A. Ali Khan, et al., Indirect Z-scheme assembly of 2D ZnV2O6/RGO/g-C3N4 nanosheets with RGO/pCN as solid-state electron mediators toward visible-light-enhanced CO2 reduction, Ind. Eng. Chem. Res., 2019, 58(20), 8612–8624 CrossRef CAS.
- Y. Huo, J. Zhang and K. Dai, et al., All-solid-state artificial Z-scheme porous g-C3N4/Sn2S3-DETA heterostructure photocatalyst with enhanced performance in photocatalytic CO2 reduction, Appl. Catal., B, 2019, 241, 528–538 CrossRef CAS.
- H. Guo, J. Ding and S. Wan, et al., Highly efficient CH3OH production over Zn0.2Cd0.8S decorated g-C3N4 heterostructures for the photoreduction of CO2, Appl. Surf. Sci., 2020, 528, 146943 CrossRef CAS.
- M. S. Akple, S. P. Chimmikuttanda and G. K. S. Takyi, et al., Fabrication and density functional theory calculations of bromine doped carbon nitride nanosheets with enhanced photocatalytic reduction of CO2 into solar fuels, Biointerface Res. Appl. Chem., 2021, 11, 14602–14619 CAS.
- M. H. Foghani, O. Tavakoli and M. J. Parnian, et al., Enhanced visible light photocatalytic CO2 reduction over direct Z-scheme heterojunction Cu/P co-doped g-C3N4@ TiO2 photocatalyst, Chem. Pap., 2022, 76(6), 3459–3469 CrossRef CAS.
- H. Guo, T. Zhang and W. Ma, et al., Construction of sandwich-like Ag/UiO-66@ g-C3N4 Z-scheme ternary heterojunction for photocatalytic CO2 conversion to CH3OH and CO, Fuel, 2023, 344, 127911 CrossRef CAS.
- H. C. Hsu, I. Shown and H. Y. Wei, et al., Graphene oxide as a promising photocatalyst for CO2 to methanol conversion, Nanoscale, 2013, 5(1), 262–268 RSC.
- Y. Kuang, J. Shang and T. Zhu, Photoactivated graphene oxide to enhance photocatalytic reduction of CO2, ACS Appl. Mater. Interfaces, 2019, 12(3), 3580–3591 CrossRef PubMed.
- T. F. Yeh, J. M. Syu and C. Cheng, et al., Graphite oxide as a photocatalyst for hydrogen production from water, Adv. Funct. Mater., 2010, 20(14), 2255–2262 CrossRef CAS.
- Y. Shi, W. Su and L. Kong, et al., The homojunction formed by h-In2O3 (1 1 0) and c-In2O3 (4 4 0) promotes carbon dioxide hydrogenation to methanol on graphene oxide modified In2O3, J. Colloid Interface Sci., 2022, 623, 1048–1062 CrossRef CAS.
- Y. Zhang, L. Zheng and J. Jia, et al., Construction of 2D-coal-based graphene/2D-bismuth vanadate compound for effective photocatalytic CO2 reduction to CH3OH, Colloids Surf., A, 2022, 639, 128321 CrossRef CAS.
- A. Bafaqeer, M. Tahir and A. Ali Khan, et al., Indirect Z-scheme assembly of 2D ZnV2O6/RGO/g-C3N4 nanosheets with RGO/pCN as solid-state electron mediators toward visible-light-enhanced CO2 reduction, Ind. Eng. Chem. Res., 2019, 58(20), 8612–8624 CrossRef CAS.
- M. Mgolombane, O. M. Bankole and E. E. Ferg, et al., Construction of Co-doped TiO2/rGO nanocomposites for high-performance photoreduction of CO2 with H2O: Comparison of theoretical binding energies and exploration of surface chemistry, Mater. Chem. Phys., 2021, 268, 124733 CrossRef CAS.
- A. Nosrati, S. Javanshir and F. Feyzi, et al., Effective CO2 Capture and Selective Photocatalytic Conversion into CH3OH by Hierarchical Nanostructured GO–TiO2– Ag2O and GO–TiO2–Ag2O–Arg, ACS Omega, 2023, 8, 3981–3991 CrossRef CAS PubMed.
- Y. Xia, B. Cheng and J. Fan, et al., Near-infrared absorbing 2D/3D ZnIn2S4/N-doped graphene photocatalyst for highly efficient CO2 capture and photocatalytic reduction, Sci. China Mater., 2020, 63(4), 552 CrossRef CAS.
- P. Kumar, H. P. Mungse and S. Cordier, et al., Hexamolybdenum clusters supported on graphene oxide: Visible-light induced photocatalytic reduction of carbon dioxide into methanol, Carbon, 2015, 94, 91–100 CrossRef CAS.
- W. Zhang, F. Dong and W. Zhang, Capture of atmospheric CO2 into (BiO)2CO3/graphene or graphene oxide nanocomposites with enhanced photocatalytic performance, Appl. Surf. Sci., 2015, 358, 75–83 CrossRef CAS.
- Y. Y. Lee, H. S. Jung and Y. T. Kang, A review: Effect of nanostructures on photocatalytic CO2 conversion over metal oxides and compound semiconductors, J. CO2 Util., 2017, 20, 163–177 CrossRef CAS.
- M. Aggarwal, N. P. Shetti and S. Basu, et al., Two-dimensional ultrathin metal-based nanosheets for photocatalytic CO2 conversion to solar fuels, J. Environ. Manage., 2022, 313, 114916 CrossRef CAS PubMed.
- A. Fujishima and K. Honda, Electrochemical photolysis of water at a semiconductor electrode, Nature, 1972, 238(5358), 37–38 CrossRef CAS PubMed.
- C. Ray and T. Pal, Retracted Article: Recent advances of metal–metal oxide nanocomposites and their tailored nanostructures in numerous catalytic applications, J. Mater. Chem. A, 2017, 5(20), 9465–9487 RSC.
- Z.-W. Huang, K.-Q. Hu, L. Mei, D.-G. Wang, J.-Y. Wang, W.-S. Wu, Z.-F. Chai and W.-Q. Shi, Encapsulation of Polymetallic Oxygen Clusters in a Mesoporous/Microporous Thorium-Based Porphyrin Metal–Organic Framework for Enhanced Photocatalytic CO2 Reduction, Inorg. Chem., 2022, 61(8), 3368–3373 CrossRef CAS PubMed.
- Z.-W. Huang, K.-Q. Hu, X.-B. Li, Z.-N. Bin, Q.-Y. Wu, Z.-H. Zhang, Z.-J. Guo, W.-S. Wu, Z.-F. Chai and L. Mei, et al., Thermally Induced Orderly Alignment of Porphyrin Photoactive Motifs in Metal–Organic Frameworks for Boosting Photocatalytic CO2 Reduction, J. Am. Chem. Soc., 2023, 145(32), 18148–18159 CrossRef CAS PubMed.
- X. Wang, Y. Wang and M. Gao, et al., BiVO4/Bi4Ti3O12 heterojunction enabling efficient photocatalytic reduction of CO2 with H2O to CH3OH and CO, Appl. Catal., B, 2020, 270, 118876 CrossRef CAS.
- Q. Wang, Y. Zhou and K. Zhang, et al., Defect-enrichment in porous interface of ultrathin CuO nanobelts realizes a novel CO2 photoreduction pathway, J. Mater. Chem. A, 2023, 11, 8776–8782 RSC.
- J. Li, W. Pan and Q. Liu, et al., Interfacial engineering of Bi19Br3S27 nanowires promotes metallic photocatalytic CO2 reduction activity under near-infrared light irradiation, J. Am. Chem. Soc., 2021, 143(17), 6551–6559 CrossRef CAS PubMed.
- X. Wu, Y. Li and G. Zhang, et al., Photocatalytic CO2 conversion of M0.33WO3 directly from the air with high selectivity: insight into full spectrum-induced reaction mechanism, J. Am. Chem. Soc., 2019, 141(13), 5267–5274 CrossRef CAS PubMed.
- C. Yang, Q. Li and Y. Xia, et al., Enhanced visible-light photocatalytic CO2 reduction performance of Znln2S4 microspheres by using CeO2 as cocatalyst, Appl. Surf. Sci., 2019, 464, 388–395 CrossRef CAS.
- C. Luo, T. Yang and Q. Huang, et al., CuMoxW (1-x) O4 Solid Solution Display Visible Light Photoreduction of CO2 to CH3OH Coupling with Oxidation of Amine to Imine, Nanomaterials, 2020, 10(7), 1303 CrossRef CAS PubMed.
- E. Luévano-Hipólito and L. M. Torres-Martínez, CO2 photoreduction with H2O to C1 and C2 products over perovskite films of alkaline niobates ANbO3 (A= Li, Na, K), Fuel, 2022, 320, 123934 CrossRef.
- M. A. Ávila-López, E. Luévano-Hipólito and L. M. Torres-Martínez, CO2 adsorption and its visible-light-driven reduction using CuO synthesized by an eco-friendly sonochemical method, J. Photochem. Photobiol., A, 2019, 382, 111933 CrossRef.
- K. Wang, Y. Du and Y. Li, et al., Atomic-level insight of sulfidation-engineered Aurivillius-related Bi2O2SiO3 nanosheets enabling visible light low-concentration CO2 conversion, Carbon Energy, 2023, 5(2), e264 CrossRef CAS.
- A. Hezam, K. Namratha and Q. A. Drmosh, et al., CeO2 nanostructures enriched with oxygen vacancies for photocatalytic CO2 reduction, ACS Appl. Nano Mater., 2019, 3(1), 138–148 CrossRef.
- W. Zhao, M. Ding and P. Yang, et al., Pit-embellished low-valent metal active sites customize CO2 photoreduction to methanol, EES Catal., 2023, 1, 36–44 RSC.
- J. Albo and G. García, Enhanced visible-light photoreduction of CO2 to methanol over Mo2C/TiO2 surfaces in an optofluidic microreactor, React. Chem. Eng., 2021, 6(2), 304–312 RSC.
- J. Lu, Z. Zhang and L. Cheng, et al., MoS2-wrapped Mn0.2Cd0.8S nanospheres towards efficient photocatalytic H2 generation and CO2 reduction, New J. Chem., 2020, 44(32), 13728–13737 RSC.
- K. Zheng, Y. Wu and J. Zhu, et al., Room-temperature photooxidation of CH4 to CH3OH with nearly 100% selectivity over hetero-ZnO/Fe2O3 porous nanosheets, J. Am. Chem. Soc., 2022, 144(27), 12357–12366 CrossRef CAS PubMed.
- M. Flores-Flores, E. Luévano-Hipólito and L. M. Torres-Martínez, et al., CO2 adsorption and photocatalytic reduction over Mg (OH)2/CuO/Cu2O under UV-Visible light to solar fuels, Mater. Chem. Phys., 2019, 227, 90–97 CrossRef CAS.
- M. Tahir, Hierarchical 3D VO2/ZnV2O4 microspheres as an excellent visible light photocatalyst for CO2 reduction to solar fuels, Appl. Surf. Sci., 2019, 467, 1170–1180 CrossRef.
- Z. H. He, C. S. Jiang and K. Wang, et al., Photothermal CO2 hydrogenation to methanol over a CoO/Co/TiO2 catalyst in aqueous media under atmospheric pressure, Catal. Today, 2020, 356, 579–588 CrossRef CAS.
- Z. Luo, Y. Li and F. Guo, et al., Carbon dioxide conversion with high-performance photocatalysis into methanol on NiSe2/WSe2, Energies, 2020, 13(17), 4330 CrossRef CAS.
- S. P. Cheng, L. W. Wei and H. P. Wang, Photocatalytic reduction of CO2 to methanol by Cu2O/TiO2 heterojunctions, Sustainability, 2021, 14(1), 374 CrossRef.
- M. A. Ávila-López, E. Luévano-Hipólito and L. M. Torres-Martínez, Optimizing the CO2 reduction to produce CH3OH using flexible NiMoO4 coatings as a photocatalyst, J. Alloys Compd., 2022, 918, 165549 CrossRef.
- W. Cai, X. Yu and Y. Cao, et al., Electron-coupled enhanced interfacial interaction of Ce-MOF/Bi2MoO6 heterostructure for boosted photoreduction CO2, J. Environ. Chem. Eng., 2022, 10(3), 107461 CrossRef CAS.
- Y. Liu, X. Chu and A. Shi, et al., Construction of 2D Bismuth Silicate Heterojunctions from Natural Mineral toward Cost-Effective Photocatalytic Reduction of CO2, Ind. Eng. Chem. Res., 2022, 61(34), 12294–12306 CrossRef CAS.
- K. A. Alzahrani, A. A. Ismail and N. Alahmadi, CuCo2O4/CeO2 S-scheme photocatalyst for promoted CO2 photoreduction to CH3OH, J. Mol. Liq., 2023, 376, 121509 CrossRef CAS.
- S. Wang, H. Abroshan and C. Liu, et al., Shuttling single metal atom into and out of a metal nanoparticle, Nat. Commun., 2017, 8(1), 848 CrossRef PubMed.
- J. Xing, J. F. Chen and Y. H. Li, et al., Stable isolated metal atoms as active sites for photocatalytic hydrogen evolution, Chem. – Eur. J., 2014, 20(8), 2138–2144 CrossRef CAS PubMed.
- M. Ma, Z. Huang and L. Li, et al., Modulating photogenerated electron density of Pr single-atom sites by coordination environment engineering for boosting photoreduction of CO2 to CH3OH, Appl. Catal., B, 2023, 330, 122626 CrossRef CAS.
- W. Jiao, Y. Xie and F. He, et al., A visible light-response flower-like La-doped BiOBr nanosheets with enhanced performance for photoreducing CO2 to CH3OH, Chem. Eng. J., 2021, 418, 129286 CrossRef CAS.
- H. J. Yoon, J. H. Yang and S. J. Park, et al., Thermal CO oxidation and photocatalytic CO2 reduction over bare and M-Al2O3 (M= Co, Ni, Cu, Rh, Pd, Ag, Ir, Pt, and Au) cotton-like nanosheets, Nanomaterials, 2021, 11(5), 1278 CrossRef CAS PubMed.
- H. Du, X. Gao and Q. Ma, et al., Cu/PCN Metal-Semiconductor Heterojunction by Thermal Reduction for Photoreaction of CO2-Aerated H2O to CH3OH and C2H5OH, ACS Omega, 2022, 7(19), 16817–16826 CrossRef CAS PubMed.
- T. Di, T. Cao and H. Liu, et al., Cu doped SnS2 nanosheets with superior visible-light photocatalytic CO2 reduction performance, Phys. Chem. Chem. Phys., 2023, 25, 5196–5202 RSC.
- R. Wang, J. Ding and Q. Zhong, et al., Plasmonic Ag nanoparticles decorated acid-aching carbon fibers for enhanced photocatalytic reduction of CO2 into CH3OH under visible-light irradiation, Catal. Lett., 2021, 1–10 Search PubMed.
- P. Ling, J. Zhu and Z. Wang, et al., Ultrathin Ti-doped WO3 nanosheets realizing selective photoreduction of CO2 to CH3OH, Nanoscale, 2022, 14(38), 14023–14028 RSC.
- S. M. Albukhari and A. A. Ismail, Highly dispersed Pt nanoparticle-doped mesoporous ZnO photocatalysts for promoting photoconversion of CO2 to methanol, ACS Omega, 2021, 6(36), 23378–23388 CrossRef CAS PubMed.
- M. Mgolombane, S. Majodina and O. M. Bankole, et al., Influence of surface modification of zinc oxide–based nanomaterials on the photocatalytic reduction of carbon dioxide, Mater. Today Chem., 2021, 20, 100446 CrossRef CAS.
- M. Mgolombane, O. M. Bankole and E. E. Ferg, et al., Construction of Co-doped TiO2/rGO nanocomposites for high-performance photoreduction of CO2 with H2O: Comparison of theoretical binding energies and exploration of surface chemistry, Mater. Chem. Phys., 2021, 268, 124733 CrossRef CAS.
- R. M. Mohamed, I. A. Mkhalid and M. Alhaddad, et al., Enhanced CO2 photocatalytic conversion into CH3OH over visible-light-driven Pt nanoparticle-decorated mesoporous ZnO–ZnS S-scheme heterostructures, Ceram. Int., 2021, 47(19), 26779–26788 CrossRef CAS.
- R. Han, L. Chen and B. Xing, et al., Pr3+-doped La1-xPrxMn0.6Ni0.4O3-δ as efficient artificial photosynthesis catalysts for solar methanol, Catal. Commun., 2022, 165, 106440 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |