
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Organo tin precursors for synthesis of zeolite Sn-Beta for alcohol ring opening of epoxides†
Alexander P.
Spanos
a,
Leah
Ford
a,
Jiawei
Guo
 b,
Ryan
Burrows
a,
Ambarish R.
Kulkarni
b and
Nicholas A.
Brunelli
*a
b,
Ryan
Burrows
a,
Ambarish R.
Kulkarni
b and
Nicholas A.
Brunelli
*a
aWilliam G. Lowrie Department of Chemical and Biomolecular Engineering, The Ohio State University, 151 W. Woodruff Ave, Columbus, OH 43210, USA. E-mail: brunelli.2@osu.edu
bDepartment of Chemical Engineering, University of California, Davis, California, 95616 USA
First published on 27th September 2024
Abstract
Lewis acid zeolite Sn-Beta is hydrothermally synthesized using orano tin precursors. NMR experiments indicate that organotin precursors affect the site formed, with methyl tin trichloride increasing the fraction of open defect sites. Catalytic testing showed that the open defect sites are highly active for the epoxide ring opening with methanol.
Synthetic advances have enabled creation of Lewis acid zeolites, such as Sn-Beta,1 that are capable of a substantial range of catalytic chemistry.2,3 The standard synthesis of Sn-Beta – zeolite beta with tin incorporated into the framework – produces different types of catalytic sites including closed, hydrolyzed open, and defect open. As these different types of sites are reported to be highly active in different reactions, it is desirable to create synthetic methods that can produce uniform catalytic sites. Current synthetic work focuses on tuning the structure around these sites using structure directing agents (SDAs) to synthesize different zeolite frameworks. However, this method largely changes the bulk structure of the entire catalyst rather than focusing on tuning the local environment around the active Lewis acid site. Controlling the type of catalytic site and the environment around the sites has the potential to unlock a new paradigm for material design to create highly active and selective catalytic materials. Additional research is needed to create catalysts with uniform catalytic sites.
The typical synthesis of Sn-Beta involves combining water, silica, tetraethylammonium ions as the SDA, hydrofluoric acid, and tin tetrachloride (SnCl4) as a tin source. For hydrothermal synthesis, additional tin(IV) sources have been included in the synthesis, including tin metal, tin acetate, and tin tert-butoxide, as shown in Scheme 1.4,5 These precursors hydrolyze to form tin species that can incorporate into the framework during crystallization to produce catalysts with similar activity for the isomerization of dihydroxyacetone to methyl lactate. An alternative tin source is methyl tin trichloride (Me-SnCl3). Me-SnCl3 has been seen as a possibility to have increased synthetic control of open/closed sites in the material.6,7 Whereas SnCl4 would be able to fully hydrolyze and form four siloxy bonds Sn(–O–Si)4 in the zeolite framework, the organotin bond in Me-SnCl3 would likely not be able to hydrolyze, instead only forming three siloxy bonds at the Sn atom. As discussed below using atomistic simulations, we hypothesize that the methyl group would prevent the formation of a tin-siloxy bond during crystallization and sterically hinder an adjacent silicon atom from being incorporated into the framework, creating an adjacent defect site.
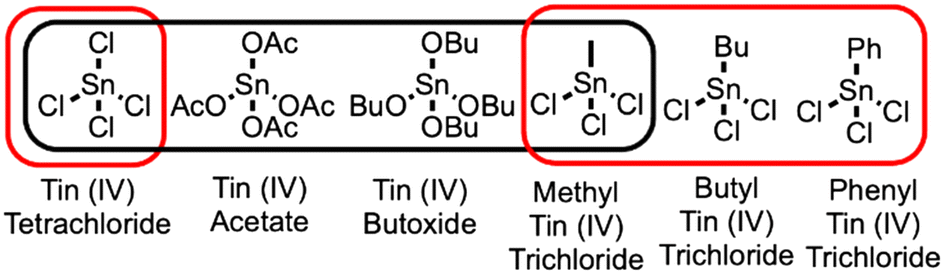 | ||
| Scheme 1 Tin(IV) precursors used in the synthesis of Lewis acid zeolite Sn-Beta. Black box indicates precursors used in previous reports. Red box indicates precursors used in this work, including organo tin(IV) trichlorides. | ||
In this work, we investigate the use of Me-SnCl3 and other organo tin precursors in the hydrothermal synthesis of Sn-Beta. Additional precursors include butyl tin trichloride (Bu-SnCl3) and phenyl tin trichloride (Ph-SnCl3). The resulting materials are characterized using XRD, nitrogen physisorption, DRIFTS, and 31P NMR. The materials are tested for catalytic activity in the alcohol ring opening (ARO) of epoxides with two different epoxides, including epichlorohydrin and 1,2-epoxyhexane. The materials are compared to conventional Sn-Beta synthesized using SnCl4 as the tin source. The observed differences in characterization, further supported by atomistic simulations, are correlated with the differences in catalytic activity to establish synthesis-structure-reactivity relationships for these materials. Overall, our use of organo tin precursors reveals a new method to design the active site in these catalytic materials.
Using methods described in the supporting information, materials consistent with the zeolite Beta framework are successfully synthesized using different tin precursors, including either SnCl4 (to produce Sn-Beta), Me-SnCl3 (Me-Sn-Beta), Bu-SnCl3 (Bu-Sn-Beta), or Ph-SnCl3 (Ph-Sn-Beta). After synthesis, the materials are calcined to remove the SDA and any additional organic content. The materials are characterized using XRD and nitrogen physisorption.
The materials are tested for crystallinity using XRD, as shown in Fig. S1.† These catalysts all show well-defined peaks consistent with the zeolite Beta structure. Since the synthesis of zeolite Beta with tetraethylammonium as the SDA produces a distribution of polymorphs A, B, and C, the XRD data are examined in the region of 2θ of 8°. Interestingly, this 2θ region shows that the broad peak around 8° appears to shift and have different structures for the different samples, as shown in Fig. S2.† Zeolite Beta itself is an intergrowth of different zeolite polymorphs, predominately polymorph A (BEA) and polymorph B (BEB), and the broadness of this peak at 8° is caused by the intergrowth of these two phases. Therefore, the approximate composition of these two frameworks can be determined for different samples by deconvoluting these peaks. Conventional Sn-Beta is typically thought to have a 45![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :55 mixture of A:B.8 Me-Sn-Beta appears to have a similar mixture whereas Ph-Sn-Beta has an enrichment of BEA and Bu-Sn-Beta has an enrichment of BEB. This result is interesting because the difference in bulk framework properties is caused by a small change in the composition of the synthesis gel. A single alteration of the tin precursor, which accounts for less than 1 wt% of the final structure, impacts the overall structure of the zeolite.
:55 mixture of A:B.8 Me-Sn-Beta appears to have a similar mixture whereas Ph-Sn-Beta has an enrichment of BEA and Bu-Sn-Beta has an enrichment of BEB. This result is interesting because the difference in bulk framework properties is caused by a small change in the composition of the synthesis gel. A single alteration of the tin precursor, which accounts for less than 1 wt% of the final structure, impacts the overall structure of the zeolite.
The materials are further characterized using standard methods. Combined with XRD, the nitrogen physisorption data show that the materials are highly crystalline with micropore volumes of ∼0.2 cm3 g−1 (listed in Table S2†; isotherms shown in Fig. S3†). Elemental analysis through ICP-OES shows that these materials all have similar tin wt%, as listed in Table S2;† therefore, the tin incorporation efficiencies did not seem to be greatly affected by the different tin precursors.
These materials are tested for catalytic activity using two different epoxide ring opening reactions (ERO; Scheme 2) with methanol (MeOH) as the nucleophile; one with a small epoxide, epichlorohydrin (ECH; 1a), and one with a large epoxide, 1,2-epoxyhexane (EH; 1b).
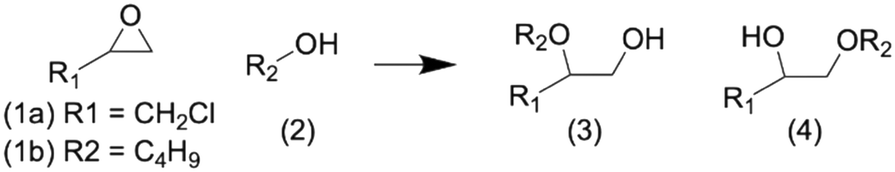 | ||
| Scheme 2 Epoxide (1) ring opening with an alcohol (2) to produce glycol ethers that are either a terminal alcohol (3) or a terminal ether (4). | ||
For ECH ring opening with MeOH, the conversion of ECH over time is plotted for Sn-Beta and Me-Sn-Beta, as shown in Fig. 1. After one hour, Sn-Beta achieves an ECH conversion of 48%. The selectivity for the terminal ether (4) is 97% for all catalysts. As we have done previously,9 the conversion data are fit using a first order model represented by the dashed lines, allowing us to extract a rate constant of k = 0.742 ± 0.009 hr−1. In comparison, Me-Sn-Beta achieves a ECH conversion at one hour of 98%. Fitting the data reveals a reaction rate constant of 3.49 ± 0.05 hr−1, which is an increase in reaction rate by a factor of 4.5 compared to Sn-Beta.
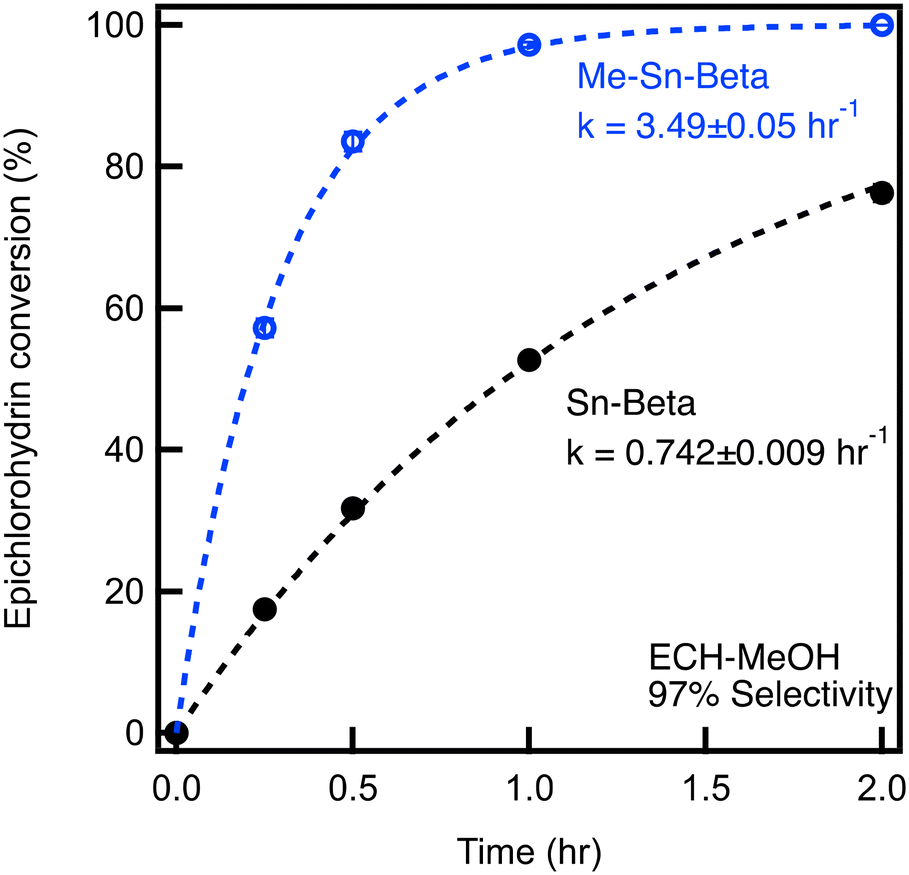 | ||
| Fig. 1 Catalytic testing of materials for epoxide ring opening of 0.4 M epichlorohydrin with methanol at 60 °C with a molar epoxide:Sn of 250:1. | ||
Catalytic testing is also performed using Bu-Sn-Beta and Ph-Sn-Beta. The data for conversion over time are shown in Fig. S4.† Data are fit with a first order model to determine that the reaction rate constants are 3.51 ± 0.04 hr−1 for Bu-Sn-Beta and 1.47 ± 0.03 hr−1 for Ph-Sn-Beta (listed in Table S3†). Interestingly, each material synthesized with an organo tin precursor has a k value greater than the k value for Sn-Beta under identical reaction conditions. The reaction rate constant is plotted for the different orgnao-Sn materials, as shown in Fig. 2. With increasing organo species size, we observe a decrease in the catalytic activity.
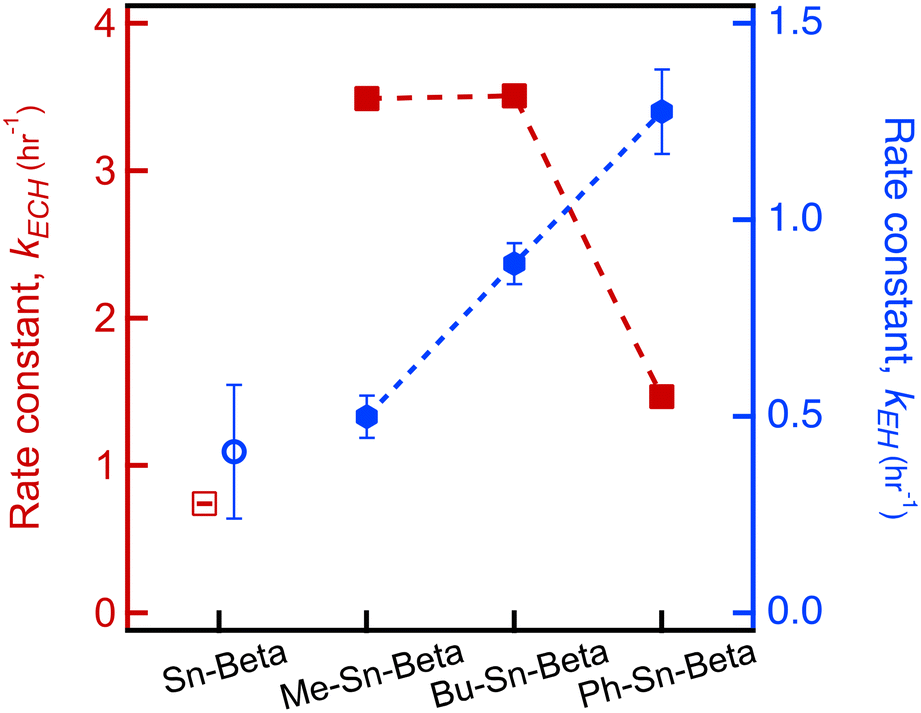 | ||
| Fig. 2 Comparison of the reaction rate constant for zeolite Beta materials made using different organo tin compounds, including Me-Sn-Beta, n-Bu-Sn-Beta, or Ph-Sn-Beta as well as Sn-Beta. The reaction rates are reported for epichlorohydrin ring opening with methanol (left/red axis) and 1,2-epoxyhexane ring opening with methanol (right/blue axis). | ||
The materials are also tested for catalytic activity for ring opening of EH. The conversion over time data are reported in Fig. S5.† Interestingly, the highest conversion in two hours is achieved with Ph-Sn-Beta (conversion of 77%) followed by Bu-Sn-Beta (63%) then Me-Sn-Beta (40%). Whereas the conversion data for ECH can be fit with a first order model over the entire two hour range of data, the conversion data for EH only fit a first order model for the first 0.5 hours. The deviation from first order behavior has been attributed to diffusion limitations caused by the products accumulating in the pores.10–12
The reaction rate constants for the data from the first 0.5 hours are listed in Table S3.† The trend in rate constants is shown in Fig. 2. The trend for ring opening EH is opposite the trend for ECH. Whereas activity decreases as the bulkiness of the organo groups increases when using ECH, the reaction rate constant increases for the materials synthesized with increasing size of the organo species. These results suggest that the catalytic site can be designed to accommodate different epoxides. Yet, it is unclear what is causing the observed differences in catalytic activity.
The structure of the site is investigated using two different forms of spectroscopy, including DRIFTS and NMR. DRIFTS analysis is performed using deuterated acetonitrile as a probe molecule to elucidate specific information on the active Sn sites, as shown in Fig. S6.† The spectra consist of four peaks that are associated with adsorption on silanols (2275 cm−1), the silica surface (2268 cm−1), closed Sn sites (2308 cm−1), and open-defect Sn sites (2316 cm−1). This technique is used to determine the ratio of peaks at 2316 cm−1 and 2308 cm−1, which are believed to correspond to open Sn sites and closed Sn sites, respectively. The materials are dosed with acetonitrile until the appearance of the peak at 2267 cm−1, indicating the presence of weakly physisorbed acetonitrile, which suggests that all other stronger binding sites are saturated. The open:closed ratios are calculated, as reported in Table S2.† Sn-Beta is determined to have an open:closed ratio of 0.22:1. Me-Sn-Beta appears to have a larger fraction of open sites than Sn-Beta since the ratio of open:closed is 0.27:1. Interestingly, the ratio increases for Bu-Sn-Beta to 0.37:1 and for Ph-Sn-Beta to 0.32:1. These results indicate that the increasing size of the tin precursor tends to result in the formation of more open defect sites. This suggests that the organo group on the tin molecule can displace silicon molecules during synthesis, preventing the completion of Si–O–Si bridges and leaving behind a defect site near Sn site. Whereas TGA results indicate that all materials have similar bulk hydrophilicities, DRIFTS results suggest that the different materials could have different hydrophobicity near the Sn site. This could have many catalytic implications as studies implicate the environment around Sn active sites play a crucial role during catalysis.13 Indeed, our recent work has shown that hydrophobic zeolites are more active for epoxide ring opening than hydrophilic zeolites.10
Another method to identify Sn sites in the material involves performing 31P NMR on samples dosed with trimethyl phosphine oxide (TMPO).14 The materials are all degassed and dosed with TMPO to reach a theoretical P:Sn ratio of 0.5:1. After drying, the 31P NMR is recorded for the materials, as shown in Fig. 3. For Sn-Beta, we observe three peaks with the peak at 57.8 and 56.1 ppm associated with closed sites and the peak at 53.4 ppm associated with open defect sites. These assignments are consistent with previous work.14 It is likely that the peak at 56.1 ppm is associated with multiple TMPO molecules binding to the same Sn center. Qualitatively, the NMR results indicate that most sites are closed for Sn-Beta. For Me-Sn-Beta, the open-defect peak (53.4 ppm) is larger than the peaks associated with closed sites (57.8 and 56.1 ppm). The results suggest that the open-defect Sn site is highly active for alcohol ring opening of an epoxide.
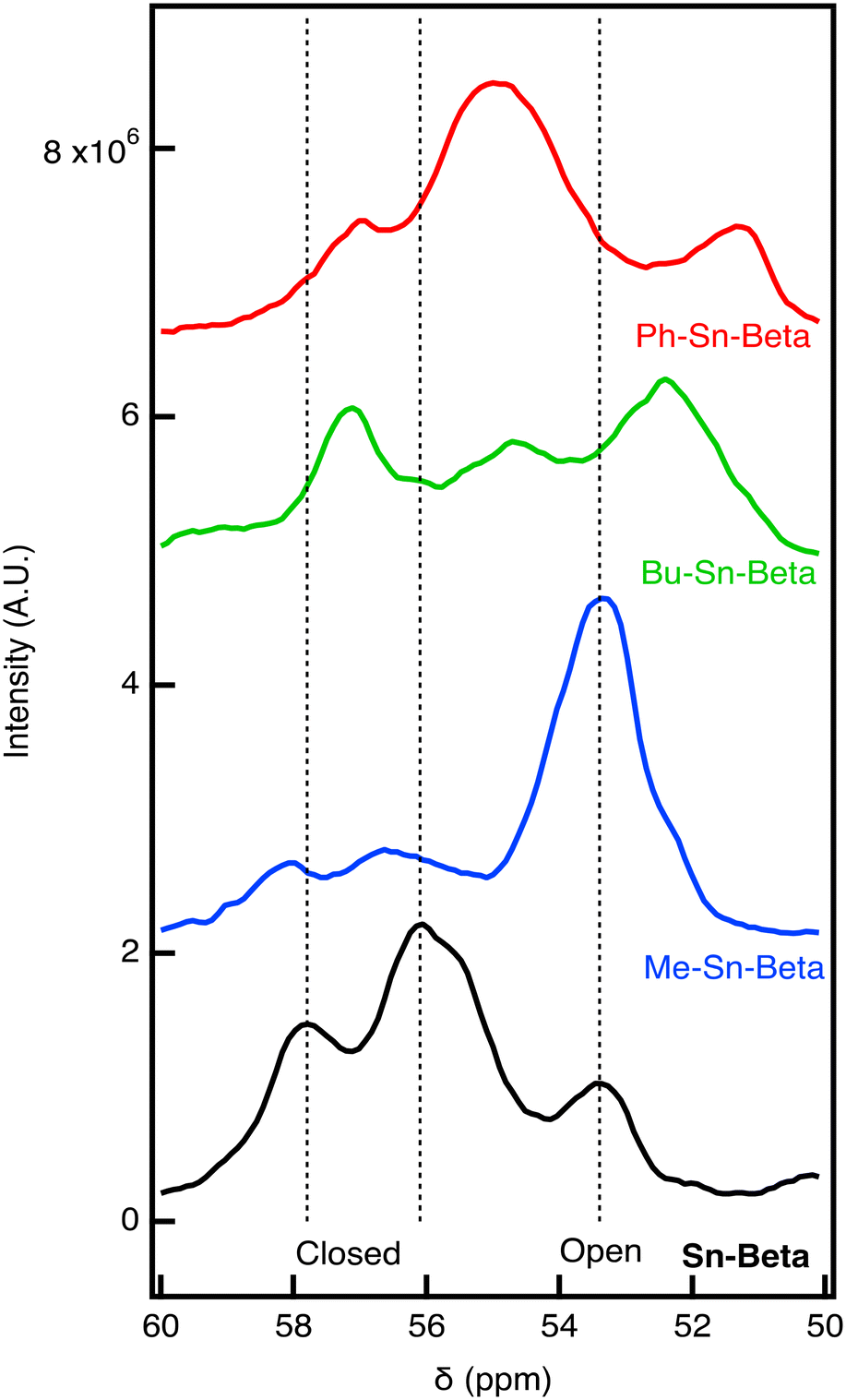 | ||
| Fig. 3
31P NMR of samples dosed with trimethyl phosphine oxide (TMPO) at a ratio of TMPO:Sn of 0.5:1. Vertical dashed lines are drawn at 56.1 and 57.8 ppm that are consistent with previous assignments for closed Sn sites and 53.4 ppm consistent with open Sn sites. | ||
Compared to Sn-Beta and Me-Sn-Beta, we observe distinct chemical shifts for Bu-Sn-Beta and Ph-Sn-Beta. Bu-Sn-Beta has peaks at 57.1, 55, and 52.4 ppm. The peaks for Ph-Sn-Beta are similar (57.1, 55, and 51.3 ppm). The differences in the 31P NMR spectra suggest that distinct sites are present in the material.
With the observed differences in catalytic activity and NMR spectroscopy between the materials, we used atomistic simulations to investigate the impact of the organo-precursor on the local structure close to the Sn atom. Specifically, ab initio molecular dynamics (AIMD) simulations (∼50 ps, 1 fs timestep, T5 cluster models, T = 140 °C, RPBE functional,15 VASP code; additional details in S5†) are used to calculate the excluded volumes (grey volumes in Fig. 4a and b) for Me-Sn and Ph-Sn precursors incorporated within a periodic BEA structure. Using the van der Waals radii as a proxy for steric effects (obtained using the universal force field (UFF) parameters),16 we observe significant differences in the excluded volume for different organo groups. Specifically, compared with Me-Sn, we observed ∼4 times larger excluded volume for Ph-Sn, suggesting the formation of silanol defect site for the latter precursor (Fig. 4c and d, T9 site). These results, which are further decomposed into individual organo-Si interactions in Fig. S7,† suggest that the local structure around an active site can be tuned by choice of the Sn-precursor.
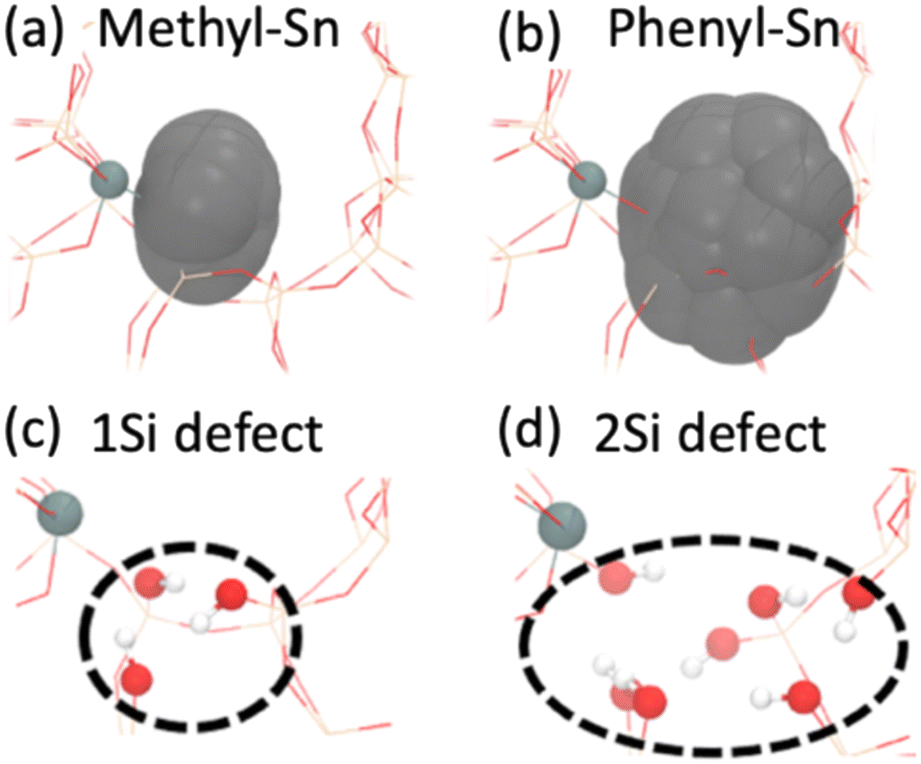 | ||
| Fig. 4 Ab initio molecular dynamics (AIMD) simulations showing the excluded volume (gray) associated with (a) methyl and (b) phenyl of organo tin species. The resulting excluded volume would produce (c) one missing Si atom for Me-SnCl3 and (d) two missing Si atoms for Ph-SnCl3. | ||
A new synthesis procedure using organo tin precursors in the synthesis of Sn-Beta is used to create a catalyst with an enriched amount of open-defect Sn sites. It is proposed that the alkyl group on the Sn atom remains during crystallization, creating a nearby defect in the framework, which could have significant effects on catalytic performance. Whereas characterization methods reveal that the material properties are similar, spectroscopic measurements reveal differences in the local structure. Specifically, DRIFTS measurements with adsorbed acetonitrile show that the alkyl-Sn-Beta materials contain significantly more open Sn sites than conventional Sn-Beta synthesized with tin tetrachloride. Additionally, 31P NMR with adsorbed trimethyl phosphine oxide reveal lower 31P chemical shifts, corresponding to more open sites within these materials. Materials made with organo tin precursors are more active for alcohol ring opening than conventional Sn-Beta, suggesting that open defect sites are highly active for alcohol ring opening of epoxides. Additionally, the size of the organo group on the tin (i.e., Me-Sn-Beta, Bu-Sn-Beta, or Ph-Sn-Beta) impacts the catalytic trends. Bulky organo groups are favorable for EH whereas the opposite is observed for ECH. Overall, this work demonstrates that combining targeted synthesis techniques with powerful characterization methods allows for a deeper understanding of synthesis-structure-reactivity relationships within Lewis acid catalysts.
Data availability
The data supporting this article have been included as part of the ESI.† The VASP setups necessary to reproduce the results included in this work can be found in the ESI.† Initial structure generation is performed using the Multiscale Atomic Zeolite Simulation Environment (MAZE) package, which is an open-source code available at https://github.com/kul-group/MAZE-sim/tree/master/maze. The atomic structures shown in Fig. 4 were prepared using Visual Molecular Dynamics (VMD).Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge grant funding support from NSF CBET (1653587).Notes and references
- A. Corma, L. T. Nemeth, M. Renz and S. Valencia, Nature, 2001, 412, 423–425 CrossRef CAS PubMed.
- M. Moliner, Y. Román-Leshkov and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6164–6168 CrossRef CAS PubMed.
- J. D. Lewis, S. Van de Vyver and Y. Román-Leshkov, Angew. Chem., Int. Ed., 2015, 54, 9835–9838 CrossRef CAS PubMed.
- S. Tolborg, A. Katerinopoulou, D. D. Falcone, I. Sadaba, C. M. Osmundsen, R. J. Davis, E. Taarning, P. Fristrup, M. S. Holm, I. Sádaba, C. M. Osmundsen, R. J. Davis, E. Taarning, P. Fristrup and M. S. Holm, J. Mater. Chem. A, 2014, 2, 20252–20262 RSC.
- M. Kasula, A. P. Spanos, L. Ford and N. A. Brunelli, Ind. Eng. Chem. Res., 2022, 61, 1977–1984 CrossRef CAS.
- R. Bermejo-Deval, R. S. Assary, E. Nikolla, M. Moliner, R. Rodríguez, S.-J. Hwang, A. Palsdottir, D. Silverman, R. F. Lobo, L. A. Curtiss and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 9727–9732 CrossRef CAS PubMed.
- A. Rodríguez-Fernández, J. R. Di Iorio, C. Paris, M. Boronat, A. Corma, Y. Román-Leshkov and M. Moliner, Chem. Sci., 2020, 11, 10225–10235 RSC.
- J. M. Newsam, M. M. J. Treacy, W. T. Koetsier and C. B. De Gruyter, Proc. R. Soc. London, Ser. A, 1988, 420, 375–405 CAS.
- N. Deshpande, A. Parulkar, R. Joshi, B. Diep, A. Kulkarni and N. A. Brunelli, J. Catal., 2019, 370, 46–54 CrossRef CAS.
- A. P. Spanos, A. Parulkar and N. A. Brunelli, J. Catal., 2021, 404, 430–439 CrossRef CAS.
- A. Parulkar, R. Joshi, N. Deshpande and N. A. Brunelli, Appl. Catal., A, 2018, 566, 25–32 CrossRef CAS.
- A. Parulkar, A. P. Spanos, N. Deshpande and N. A. Brunelli, Appl. Catal., A, 2019, 577, 28–34 CrossRef CAS.
- J. C. Vega-Vila and R. Gounder, ACS Catal., 2020, 10, 12197–12211 CrossRef CAS.
- J. D. Lewis, M. Ha, H. Luo, A. Faucher, V. K. Michaelis and Y. Román-Leshkov, ACS Catal., 2018, 8, 3076–3086 CrossRef CAS.
- B. Hammer, L. B. Hansen and J. K. Nørskov, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 7413–7421 CrossRef.
- A. K. Rappe, C. J. Casewit, K. S. Colwell, W. A. Goddard and W. M. Skiff, J. Am. Chem. Soc., 1992, 114, 10024–10035 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cy00837e |
| This journal is © The Royal Society of Chemistry 2024 |