
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLigand coordination controlled by monomer binding: a hint from DFT for stereoselective lactide polymerization†
Massimo Christian
D'Alterio
 a,
Serena
Moccia
a,
Yolanda
Rusconi
b,
Claudio
De Rosa
a and
Giovanni
Talarico
*ab
a,
Serena
Moccia
a,
Yolanda
Rusconi
b,
Claudio
De Rosa
a and
Giovanni
Talarico
*ab
aDepartment of Chemical Sciences, Università Degli Studi Di Napoli Federico II, Via Cintia, 80126 Napoli, Italy. E-mail: talarico@unina.it
bScuola Superiore Meridionale, Largo San Marcellino 10, 80138 Napoli, Italy
First published on 9th August 2024
Abstract
Stereoselective ring opening polymerization (ROP) of racemic lactide (rac-LA) is a challenging goal because a rationale connecting the catalyst structure and polymer microstructure (as has been established for α-olefin polymerization) is still missing. In this work, we reveal the origin of the stereoselective preference for D and L-lactide with two enantiopure salen–Al complexes, which have so far been claimed as the most efficient in enantiomorphic site control, using Density Functional Theory calculations. We introduce active site reorganization and monomer/chain switching throughout the reaction pathway, unconventional aspects necessitating careful consideration when confronting the intricacies associated with chiral catalyst recognition. We show how the catalytic pocket easily rearranges in the reaction path establishing a novel concept of the ligand coordination controlled by monomer binding. The resulting final picture of PLA stereoselectivity is much more complex than that of α-olefin polymerization catalysis, and a “complete” prediction by brute-force is (currently) hard, but the principles evolving should – even in their incomplete form – be useful in the design of new selective catalysts.
1. Introduction
The aliphatic polyester poly(lactide) (PLA) has gained increasing popularity as an environmentally friendly substitute for conventional olefin-based plastics, owing to its distinctive attributes of biorenewability, biocompatibility, and biodegradability.1–4 The PLA life-cycle assessment has shown a consistent reduction in non-renewable-energy compared with petrochemical-derived polymers such as polypropylene and can be recycled and degraded in its end-of-life fate.1The lactide (LA) monomer has two chiral centers and, as a consequence, three diastereoisomers, namely D-LA (RR), L-LA (SS) and meso-LA (RS). The homopolymerization of a single enantiomer by ring opening polymerization (ROP) results in an isotactic polymer whereas the stereocontrolled ROP of rac- and meso-lactide utilizing metal complexes as initiators may lead to a wider range of polymer microstructures (Scheme 1).5,6 Analogous to α-olefin polymerization transition metal (TM) catalysis,7–10 the ROP stereochemistry can be dictated by either the enantiomorphic site control (ESC) mechanism, where catalyst chirality dictates homochiral preference, or the chain-end control (CEC) mechanism, where the preference for homochirality or heterochirality depends on the last inserted monomer unit.11 Indeed, the stereochemistry influence on the physical and mechanical properties of PLA materials has been carefully assessed, and manipulating the microstructure becomes pivotal for tailoring PLA properties with the aim to replace polyolefins in target applications.12 However, simple relationships between ligand catalysts/PLA microstructures are not yet achieved13–20 diversely from the α-olefin counterpart where the polymer microstructure can be finely tuned by suitable ligand catalyst modification a priori.21
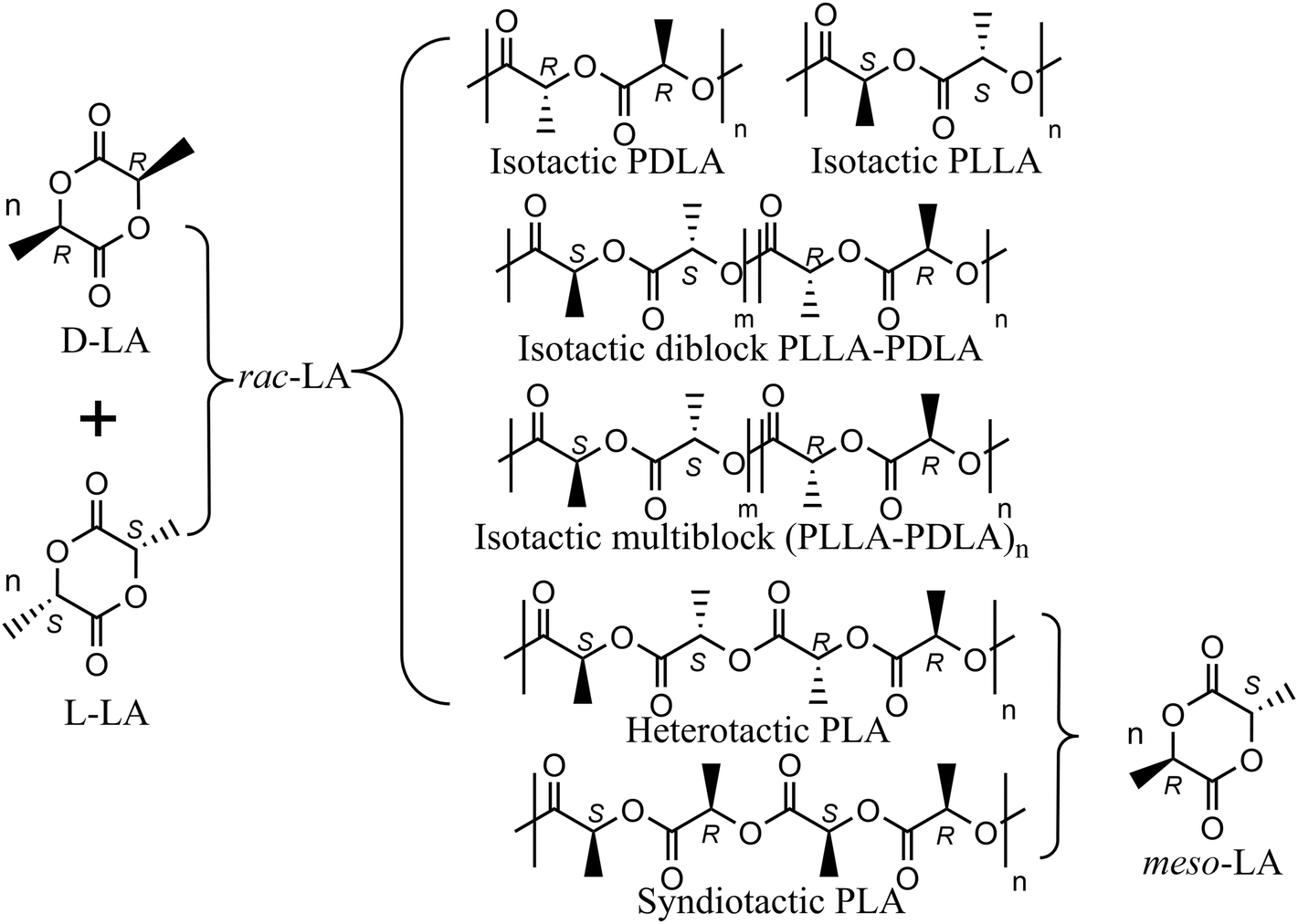 | ||
| Scheme 1 Ordered microstructures from stereoselective ROP of rac-LA and meso-LA. | ||
In a seminal study of Spassky et al.,22 the enantiomerically pure binaphthyl–salen aluminum complex (system (R)-1, Scheme 2) was employed for the synthesis of a block copolymer poly(D-lactide)–poly(L-lactide) [(PDLA–PLLA)n] using rac-LA monomer feed. Indeed, system (R)-1 revealed, for the first time, a clear ESC preference toward D-LA rather than L-LA (kRR/kSS = 20, Scheme 2), with the formation of a stereocopolymer due to the incorporation of L-LA at a high conversion rate, when the reaction mixture is poor in D-LA. Relevant insights have been later reported by Coates et al. by using rac-1 for rac-LA polymerization still obtaining isotactic-enriched PLA with a high melting temperature (Tm = 179–191 °C) even at low conversion.23 The formation of a stereoblock microstructure at low conversion was explained by assuming a polymer chain exchange among the two enantiomeric forms of rac-1 catalysts.23 Interestingly, Feijen et al.24 demonstrated that the enantiopure aluminum complexes incorporating a Jacobsen ligand ((R,R)-2A, Scheme 2) also exhibited significant isoselectivity in rac-LA ROP by ESC but with a clear preference toward L-LA (kSS/kRR = 25, Scheme 2). As mentioned for system (R)-1, the progressive increase in the relative concentration of the D-LA isomer within the monomer pool, ascribed to the initial L-LA transformation into PLLA, results into a stereosequence gradient, transitioning from extended isotactic L-lactyl units to extended isotactic D-lactyl units. Finally, the enantioselectivity dramatically drops when the tBu moieties in the ortho position are substituted with H atoms (Scheme 2), as recently reported by Wang and coworkers (kSS/kRR = 5, Scheme 2 for ((R,R)-2B).25
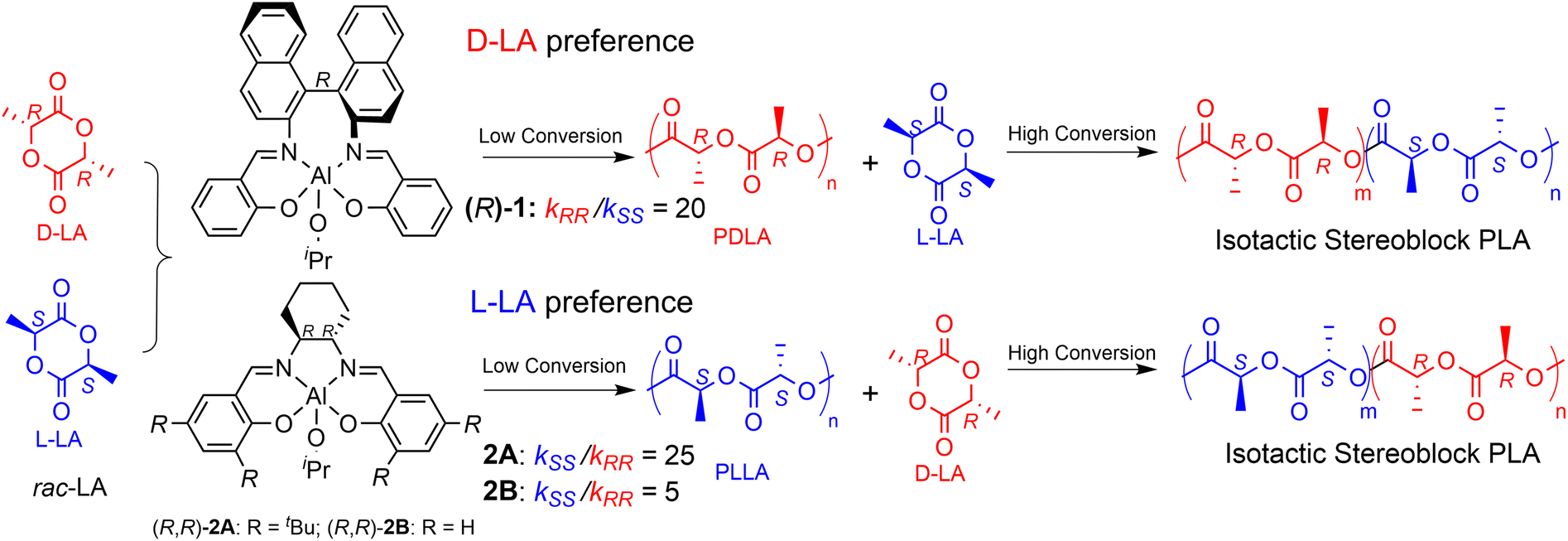 | ||
| Scheme 2 Stereoselective ROP behavior of (R)-1 and (R,R)-2 toward rac-LA. | ||
Inspired by these results, the enantiomeric resolution and stereoselective polymerization of rac-LA have become a challenge in the last two decades with the final target to achieve high levels of isoselectivity combined with an enhanced productivity.26–30
However, factors governing ESC and CEC for ROP mechanisms, as well as the interplay between those two stereocontrol mechanisms remain obscure.31,32 In our recent communication,33 we anticipated that the origin of the stereoselectivity in rac-LA ROP achieved by the enantiopure system (R)-1 is different from the classical ES reported so far. Novel mechanistic steps have been used to trace the experimental stereoselectivity as summarized in Scheme 3. This (remarkable) complexity is due to several factors: 1) multiple ligand wrapping modes at the aluminum center; 2) variations of the wrapping mode configurations during the ROP catalytic cycle; 3) rate limiting step (RLS) energetics dependent on the monomer (D and L) LA as well as on the chirality of the last inserted unit in the growing chain.34 This complicated scenario makes the prediction of the stereoselective ROP of rac-LA a challenging game for chemists31 and justifies the lack of ligand structure/polymer microstructure relationships as consolidated in stereoselective TM catalyzed olefin polymerization.7,10,21 Going into more detail, we found that the ligand wrapping mode for (R)-1 may assume fac–mer 1 (fm1), fac–mer 2 (fm2) and fac–fac (ff) configurations and they may interconvert going from the nucleophilic addition (TS1) to the ring opening (TS2) steps.
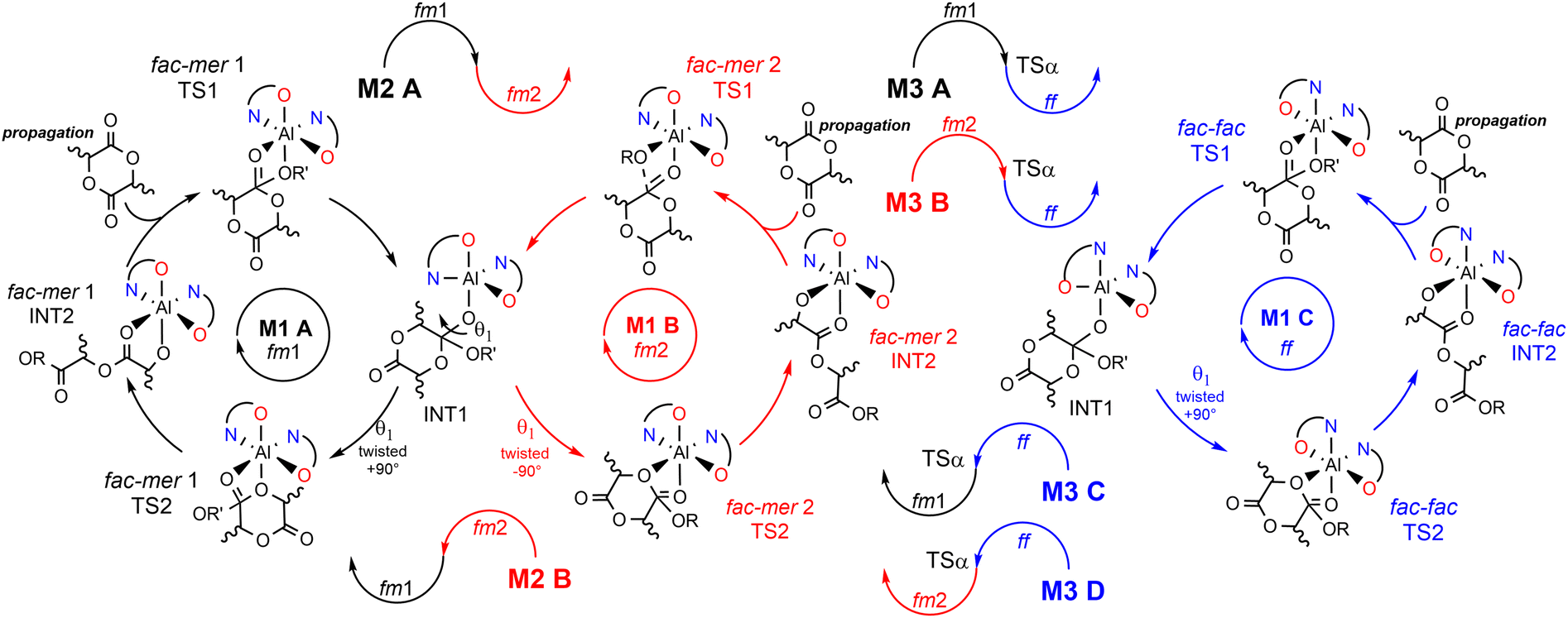 | ||
| Scheme 3 Stereoselective ROP of rac-LA at an octahedral aluminum center following the mechanistic paths depending on the wrapping mode. M1-A (TS1 and TS2 at fm1) is reported in black; M1-B (TS1 and TS2 at fm2) in red and M1-C (TS1 and TS2 at ff) in blue. Additional mechanisms (M2-A and M2-B) where TS1 and TS2 show fm1 and fm2 configurations are reported in half black–half red and half red–half black, respectively, sharing the same INT1. | ||
We identified several reaction paths under the assumption that for the first mechanism (M1), both TS1 and TS2 occur with the same wrapping mode (fm1, fm2 or ff, M1-A, M1-B, M1-C, respectively). For the second mechanism (M2), TS1 occurs at fm1 and TS2 at fm2 and vice versa (M2-A and M2-B, respectively). The third mechanism, (M3), based on the interconversion among fac-mer and fac-fac, is also possible but it requires an additional TS that involves active site reorganization via the modification of the OL–Al–OL valence angle (defined as α) (see later).
The aim of this work is to furnish, by using computational methods rooted into Density Functional Theory (DFT), a comprehensive analysis of the ESC mechanism(s) for the stereoselective ROP of rac-LA promoted by (R)-1 and (R,R)-2A to understand the key factors leading to asymmetric kinetic resolution polymerization (AKRP) processes35 for D and L-LA. We will elucidate how the two enantiopure salen–Al complexes, despite the ligand/metal similarity, act inversely in the AKRP of rac-LA (Scheme 2). The structural features of the ligands, the mechanistic ROP intricacies and the role of the chirality of the growing polymer chain will be analyzed. We will also highlight the microstructure differences derived from our DFT calculations for the so-called stereoblock PLAs promoted by enantiopure (R)-1 and (R,R)-2A complexes. Finally, a novel concept for the stereoselective ROP of rac-LA will be defined, aiming to establish ligand catalyst/PLA microstructure relationships.
2. Methods
DFT results were obtained using the B3LYP/6-311G(d,p)/SVP level of theory for geometry optimization and the energies were refined by single point calculations with B3LYP-D3BJ(CPCM)/6-311G(d,p). This approach was already tested in the stereoselective ROP33,36,37 of LA and regioselective methyl glycolide38 as well as conventional olefin polymerization showing a good match with the experimental results.39–41 Details are reported in the ESI.†3. Results
3.1 Stereoselective rac-LA ROP promoted by (R)-1
The simplified energetic paths explaining the ROP origin of the stereoselectivity toward RR-LA promoted by (R)-1 is reported in Fig. 1. We report the minimum energy pathways (MEPs) of the isotactic enchainment for RR- and SS-LA insertions with the active species in the preferred ligand coordination modes (fm for RR-LA and ff for SS-LA, respectively) extracted from a combinatorial analysis of all the elements of chirality occurring in this reaction, also including the prochirality of the monomer (Scheme S1†). In the context of the classical M1-A mechanism in which the catalyst is in fm1 wrapping mode, DFT calculations did not reveal any preference for RR- versus SS-LA insertion promoted by (R)-1. In fact, the calculated Gibbs energy values (ΔG) of the RLS for RR-LA propagation are very close to those for SS-LA (16.4 and 16.2 kcal mol−1, Fig. 1). The experimental preference for RR-LA is matched only by assuming novel mechanisms as M2-A (TS1 fm1 and TS2 fm2) and/or M3-A (ligand re-organization going from fm TS1 to ff TS2).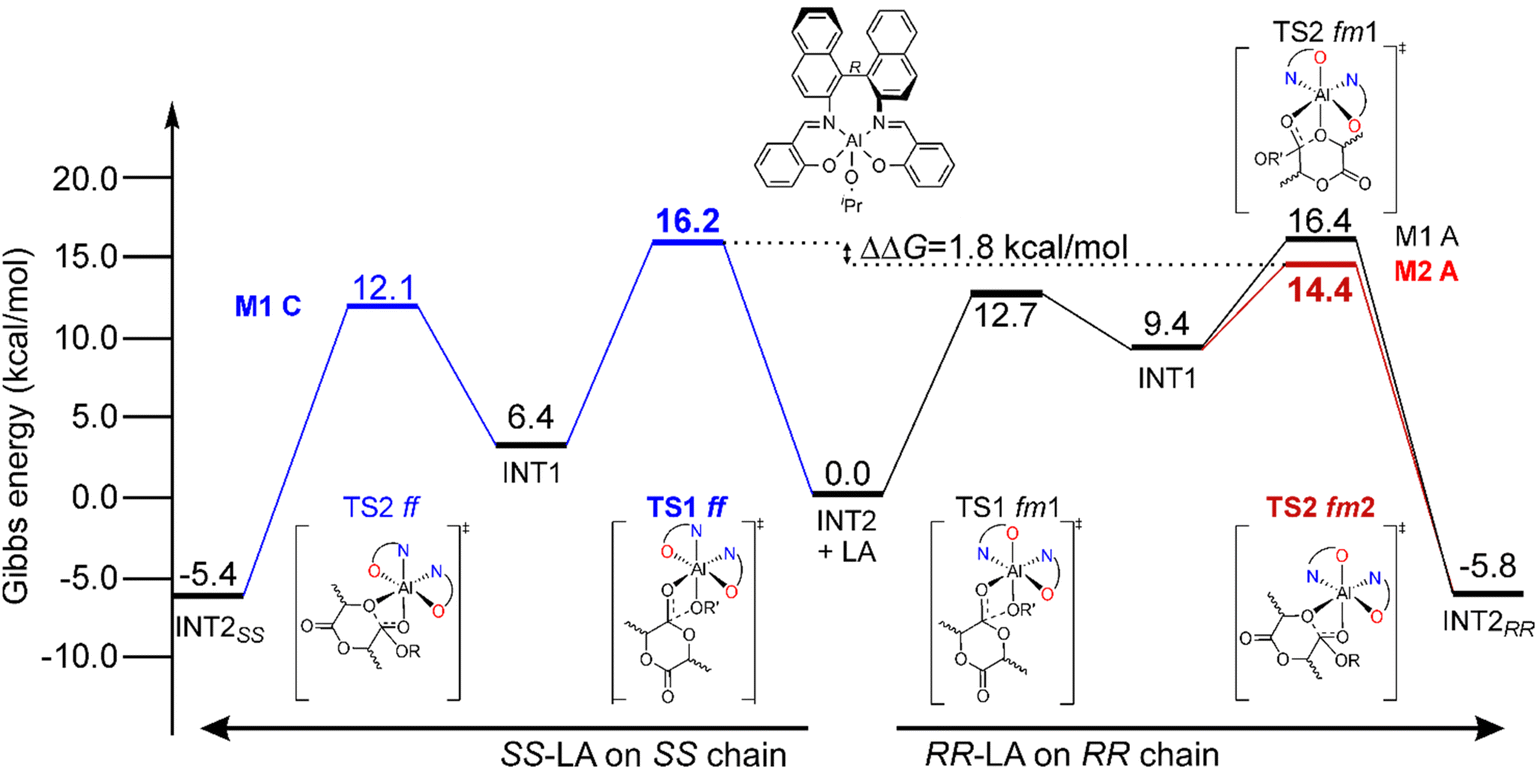 | ||
| Fig. 1 Preferred Gibbs energy paths for RR-LA (right) and SS-LA (left) propagations promoted by (R)-1 following the M1–M2 mechanisms. The RLSs for the reaction paths (see text) are reported in bold. | ||
In Scheme 3, we draw the (easy) interconversion between fm1 and fm2 wrapping modes going from TS1 to TS2. In detail, this interconversion is possible through a barrierless rotation at INT1 around the dihedral angle indicated as θ1 and involves a single bond (Scheme 3). This mechanism is particularly noteworthy since it enables the catalysts to find the lowest energy paths among the various accessible on the potential energy surface, particularly when the ring opening step (TS2) shows remarkable differences in the Gibbs energies depending on the ligand wrapping mode (Fig. 1). The DFT geometries of these two TSs are reported in Fig. 2 and the main steric interactions leading to the TS2 energetic preference for fm2 (Fig. 2A) vs. fm1 (Fig. 2B) configurations are indicated with arrows.
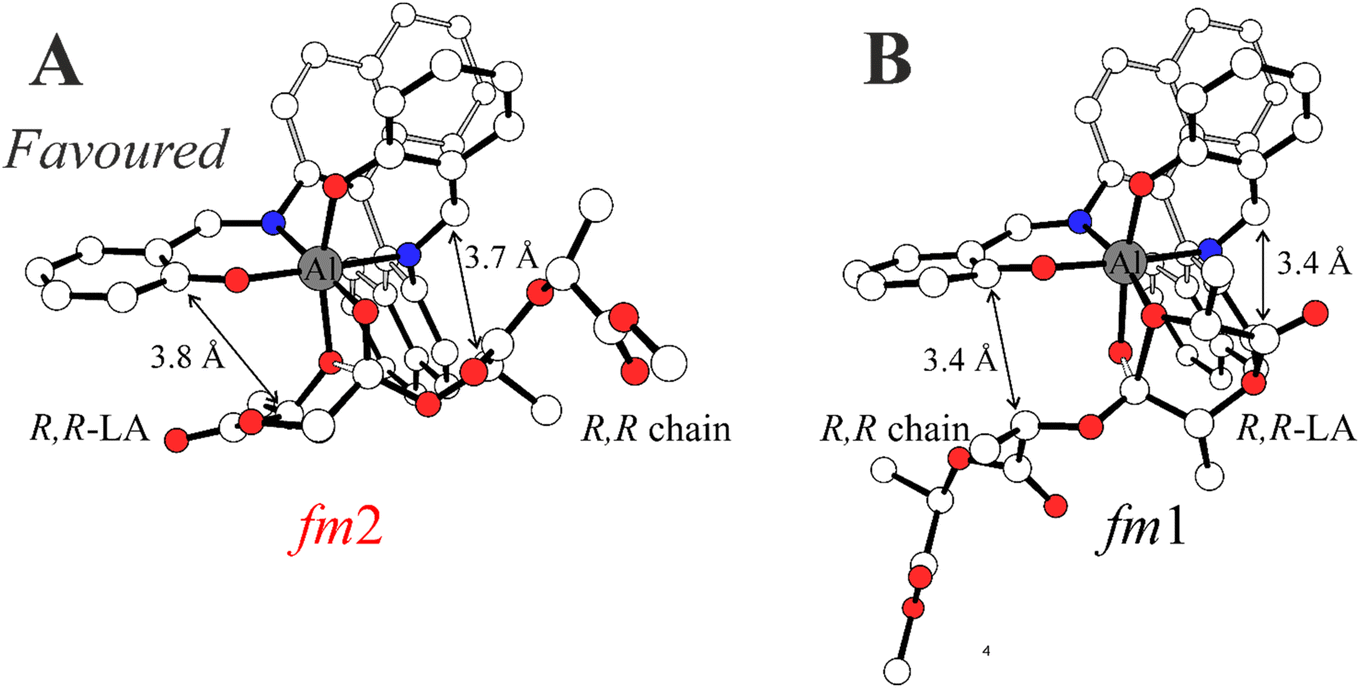 | ||
| Fig. 2 Optimized DFT geometries for the RLS of RR propagation promoted by (R)-1 corresponding to the ring opening step (TS2). The energetic preference of the fm2 wrapping mode (A) with respect to fm1 (B) is due to lower steric interactions reported with arrows. Oxygen and nitrogen atoms in red and blue. Distances in Å and H atoms omitted for clarity. | ||
The steric maps obtained with a molecular descriptor (% VBur)42–44 reported in Fig. 3 may help to visualize the (R)-1 ligand features leading to the energetic differences reported in Fig. 2. Indeed, the key aspect of the Spassky ligand is the binaphthyl linker (see the red zone in Fig. 3) that is closer to the monomer moiety in TS2 having an fm1 configuration (Fig. 3B) than to the more flexible polymer chain in fm2 (Fig. 3A). Indeed, at fm2, the couple RR-chain and RR-LA monomer finds a better accommodation rather than at fm1, where the monomer points toward the hindered area of the NE quadrant. On the other side, at fm2, the monomer lies in the free SW quadrant and the chain avoids clashing, positioning itself on the z-axis out of the plane. In the previous considerations, the chirality of the chain also plays an important role in the orientation assumed during the TSs. Indeed, in its conformational minimum, the RR-chain at the C2–O2–C3–C4 dihedral angle is G+ (≈+60°), whereas the SS-chain at the same dihedral angle is G− (≈−60°).
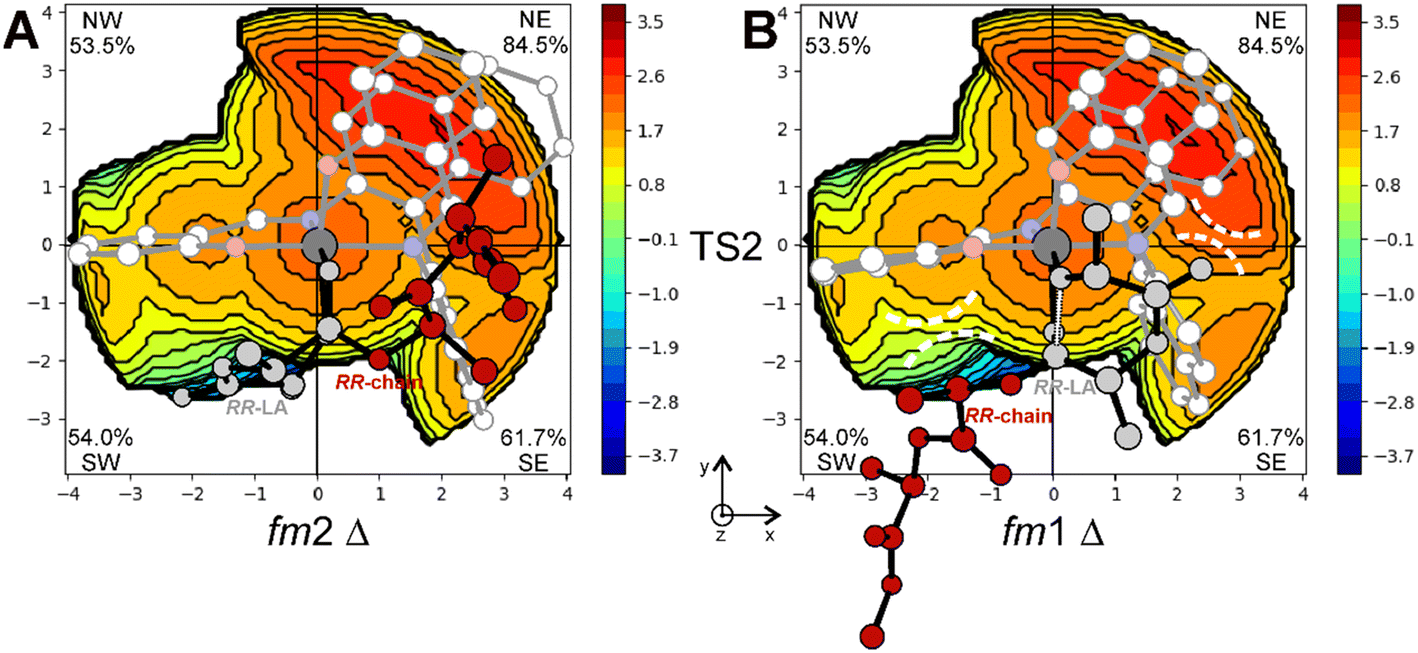 | ||
| Fig. 3 Steric maps obtained by the % VBur analysis explaining the energetic preference for fm2 (A) vs. fm1 (B) wrapping modes in the ring opening step (TS2) of RR-LA polymerization promoted by the (R)-1 system. The main steric interactions are reported with dashed lines. | ||
The interconversion among fac–mer and fac–fac is also possible but it requires the energetic evaluation of an additional TS that involves active site reorganization via the modification of the OL–Al–OL valence angle (defined as α) from ≈90 to ≈180° (Fig. 4). In Fig. 4A, we report the preferred Gibbs energy path for the mechanism moving from TS1 (in the preferred fm1) to TS2 (ff) as a function of α for (RR)-LA polymerization promoted by (R)-1 and we located the TS for such ligand interconversion at α = 141°, with a barrier of 14.6 kcal mol−1 very close to the RLS found for M2-A (14.4 kcal mol−1, see Fig. 1). These computations suggest that fm–ff transformation has to be considered in the stereoselectivity computation, increasing the number of mechanistic paths to be computed. However, we noted that such a feature is strictly related to the catalyst flexibility. In our computational screening looking for a simple method to evaluate the ligand wrapping mode energetics, we found that the use of a bidentate ligand such as acetylacetonate (acac−) to simulate the monomer and the growing chain binding appears promising. In fact, the computations with acac− (Fig. 4B) to mimic the polylactide growing chain avoided the complications derived from the several degrees of freedom of the chain (Fig. 4A), still showing similar energetic profile and geometrical features of TSα.
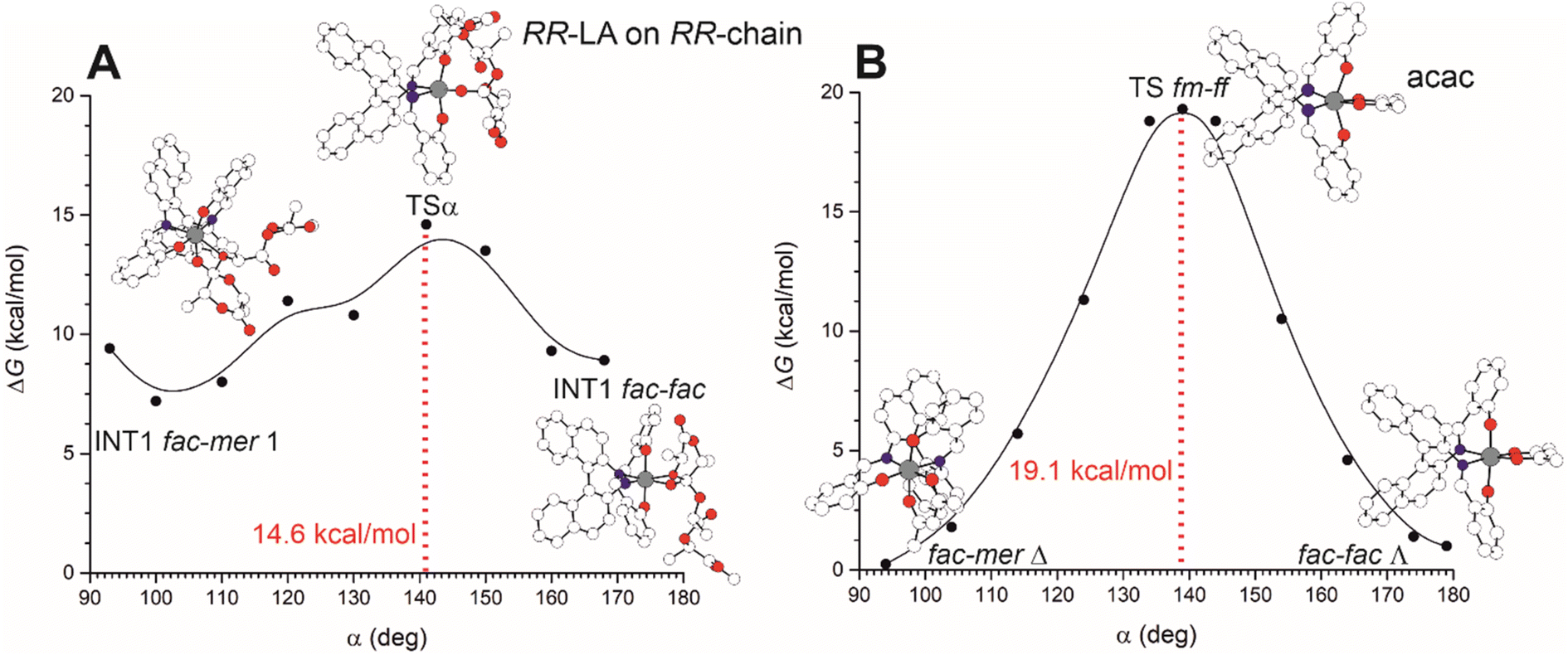 | ||
| Fig. 4 Free energy profiles of the transformation from fac–mer Δ to fac–fac Λ for (R)-1 as a function of the OL–Al–OL valence bond. The re-organization path moving from TS1 (fm1) to TS2 (ff) as a function of α for RR-LA polymerization is reported in A. The analogous path by using the bidentate acac− is reported in B. Aluminum, oxygen and nitrogen atoms in gray, red and blue. Hydrogen atoms have been omitted for clarity. | ||
These findings achieved for (R)-1 need to be better discussed and inserted in a more general framework for a comparison with system (R,R)-2. Four different diastereoisomers are derived from the various wrapping modes of the salen ligand around the Al metal center (Scheme 4). These geometries arise from the combination of fac and mer with Λ and Δ isomerism in the octahedral geometries adopted by the ligand in the ROP TSs (Scheme 3). The mer–mer isomer was ruled out since the chain and the monomer would be in trans of each other, forming an angle of ≈180° (not feasible with the ROP mechanism). Complexes with ff or fm configurations are inherently chiral and the chirality of the bridge (indicated as X in Scheme 4) “forces” the catalyst to adopt a specific octahedral wrapping mode.
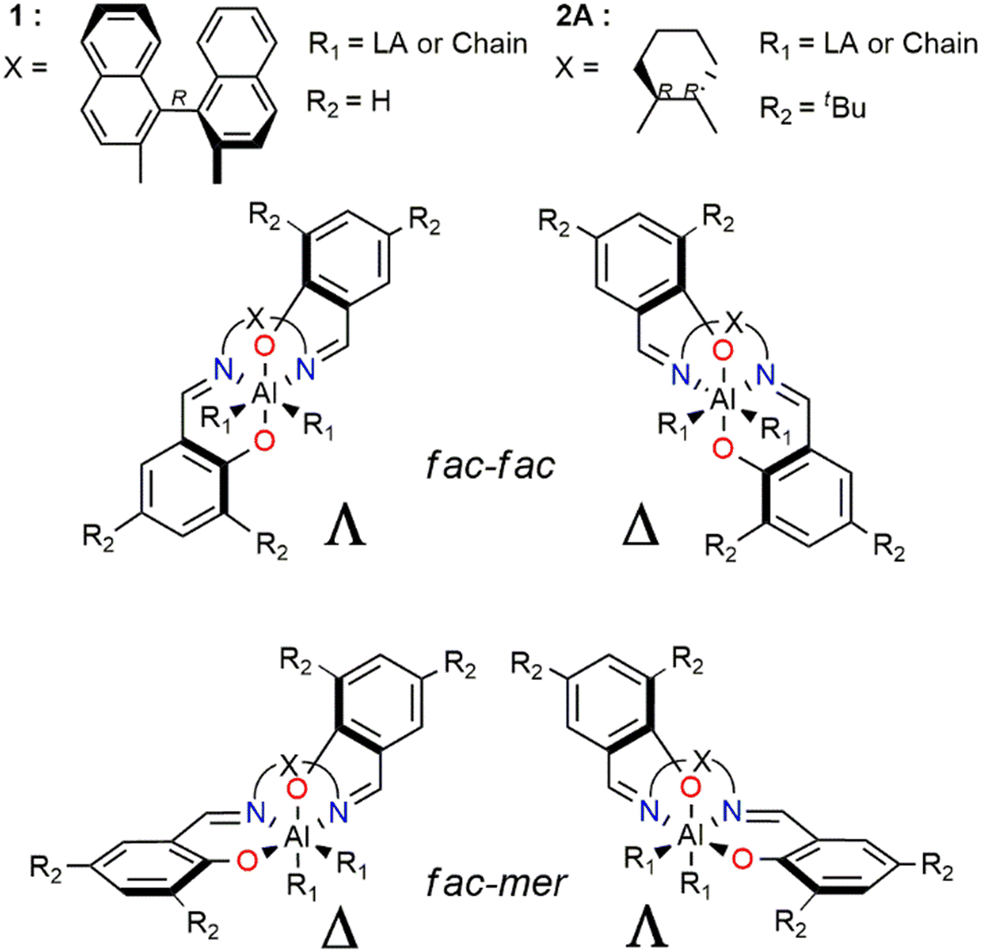 | ||
| Scheme 4 Illustrations of the combination of fac and mer with Λ and Δ isomerism for octahedral chiral salen-type catalysts. | ||
It is worth to note that (R)-1, bearing X = (R)-binaphthyl as the bridge, presents two stable diastereoisomers, fm Δ and ff Λ which differ only by 1 kcal mol−1 (Table 1) due to the flexible binaphthyl linker, constituted by four carbon atoms.
| Diastereoisomers | Catalyst | |
|---|---|---|
| (R)-1 | (R,R)-2A | |
| fac–mer Δ | 0.0 | 0.0 |
| fac–mer Λ | 29.2 | 8.5 |
| fac–fac Δ | 40.2 | 35.9 |
| fac–fac Λ | 1.0 | 13.7 |
This explains why the SS-LA monomer dictates the ff configuration of the catalyst (Fig. 1) with a Λ octahedral chirality that is preferred to the fm with a Δ octahedral chirality. Indeed, the SS-LA polymerization is more easily accommodated in the ff Λ wrapping mode (Fig. 5A) than in the fm Δ wrapping mode (Fig. 5B) with an energetic preference of 4.0 kcal mol−1 (Table S1†). In Fig. 5A, where the favored situation is depicted, the monomer is located in the southern region of the map and the chain lies parallel to the z-axis. Steric interactions between the ligand and one of the methyl groups of SS-LA are present, explaining the energetic difference in TS1 with RR-LA (16.2 vs. 12.7 kcal mol−1, Fig. 1). However, the situation is additionally disadvantaged with fm1 (Fig. 5B), by the fact that the chain is directed toward the hindered SE zone of the map.
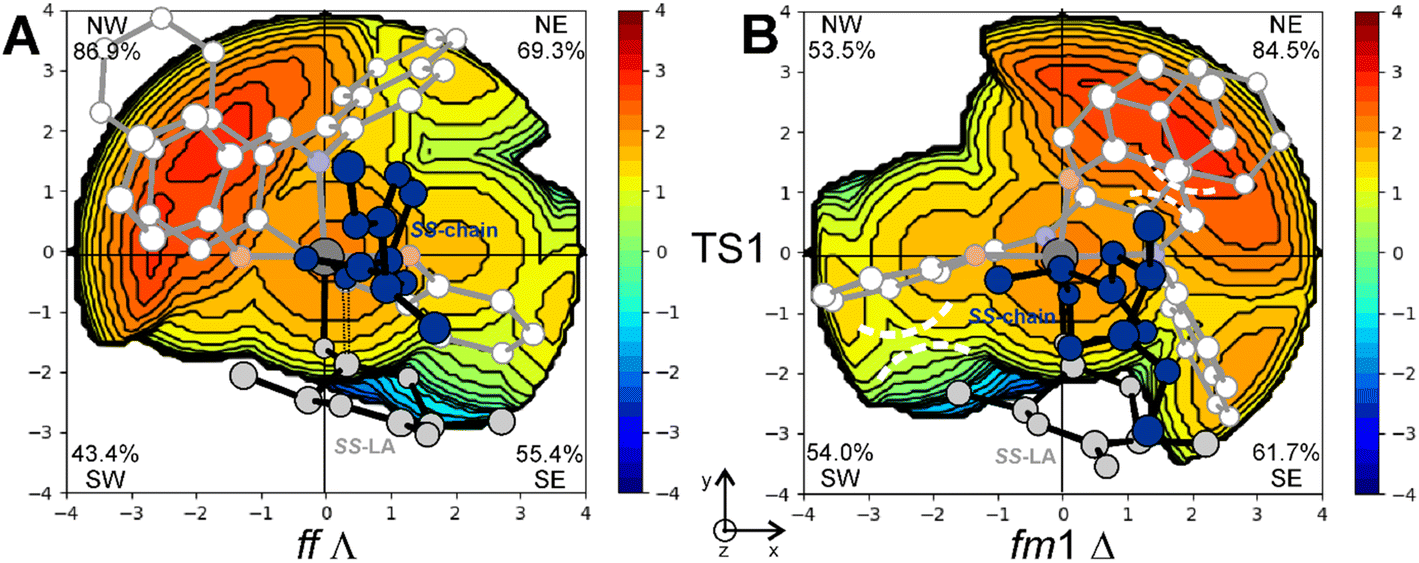 | ||
| Fig. 5 Steric maps obtained by the % VBur analysis explaining the wrapping mode energetic preference for ff (A) vs. fm1 (B) configurations in the nucleophilic addition step (TS1) of SS-LA polymerization promoted by the (R)-1 system. Main steric interactions are reported with dashed lines. | ||
It is worth to remark that the ff wrapping mode is precluded for (R,R)-2A having X = (R,R)-cyclohexyl bridge, which is far less flexible being constituted by two carbon atoms. Indeed, in this case, only the fac–mer Δ isomer is energetically feasible (Table 1). This important aspect that we will take into account in the following sections sheds a new light on the understanding of chiral catalyst recognition toward rac-LA.
3.2 Stereoselective rac-LA ROP promoted by (R,R)-2A and (R,R)-2B
System (R,R)-2A experimentally shows a clear preference toward the SS-LA monomer in the ROP of rac-LA (Scheme 2), although the ff wrapping mode that leads the SS-LA propagation for (R)-1 is too high in energy (Table 1 and ESI†). This implies that the AKRP origin of (R,R)-2A should be traced within the fm Δ wrapping mode. The simplified MEP of rac-LA isotactic enchainment extracted from a combinatorial analysis of all the elements of chirality (Tables S2 and S3†) is summarized in Fig. 6. The energetic preference for SS-LA (Fig. 6-left) vs. RR-LA (Fig. 6-right) propagation is sorted out by our DFT results, and the calculated stereoselectivity (ΔΔG of 2.2 kcal mol−1, Fig. 6) matches with the experimental results (Scheme 2). However, the preferred SS-LA energetic enchainment shows a fm2 wrapping mode (M1-B mechanism) and the RLS for this enchainment (TS1, 11.3 kcal mol−1) is preferred to the analogous having fm1 configuration (18.8 kcal mol−1, Fig. S1 and Table S2†). The preference for fm2 is also maintained for the low-lying RR-LA enchainment (Table S3†), although the free energies are upshifted (13.5 kcal mol−1), consistent with the experimental preference. The calculated enantioselectivity (ΔΔG among the two RLSs at fm2) is similar to that of catalyst (R)-1, (≈2 kcal mol−1) and analogously, we found the switching of fm configurations going from TS1 to TS2 for RR-LA propagation (comparing the M2-B mechanism in Fig. 6 with M2-A in Fig. 3). Such a mechanism helps in a better refinement of the MEP, the TS2 at fm2 being 2.4 kcal mol−1 higher than the TS2 at fm1 so a much higher enantioselectivity (3.6 kcal mol−1) would have been predicted.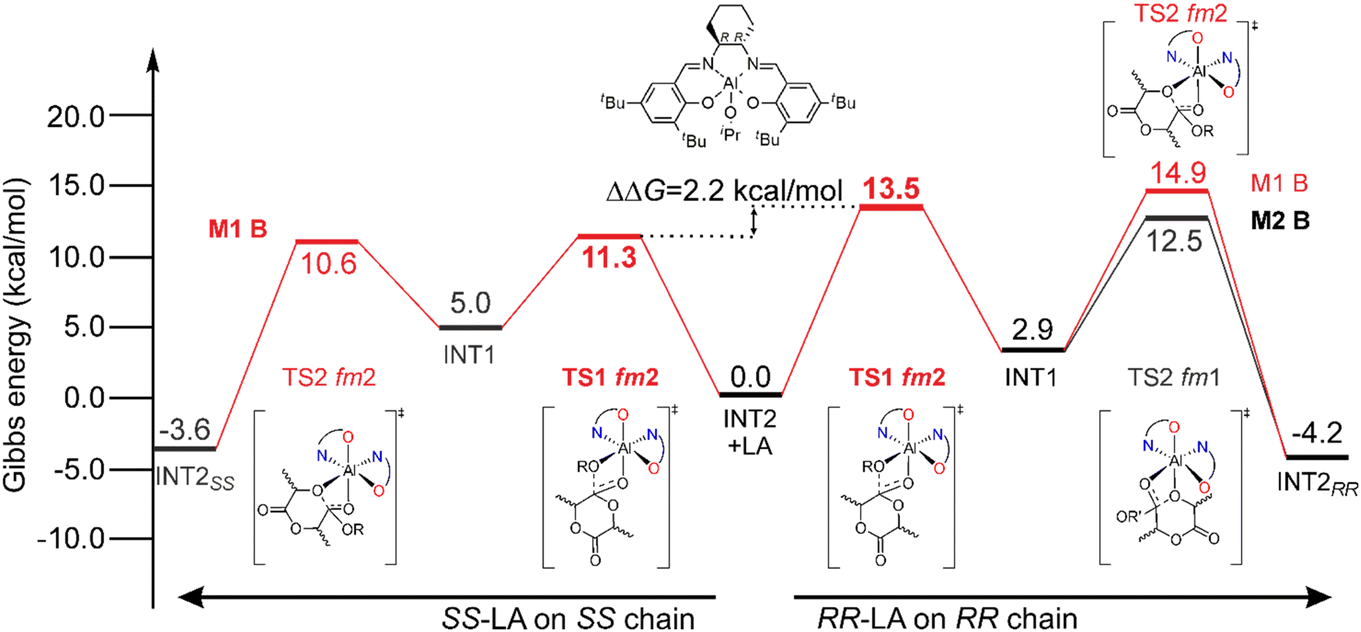 | ||
| Fig. 6 Preferred Gibbs energy paths for SS-LA (left) and RR-LA (right) propagations promoted by the (R,R)-2A system following the M1–M2 mechanisms. The RLSs for the reaction paths (see text) are reported in bold. | ||
Once a reasonable agreement with the experimental data is achieved, the origin of the stereoselectivity can be rationalized. In particular, the high steric hindrance exerted by bulky tBu groups in the ortho position of the (R,R)-2A system is crucial for inferring a stereoselective outcome to this reaction. This feature can be visualized by the % VBur steric maps reported in Fig. 7. Indeed, TS1 for SS propagation accommodates both the monomer and the growing chain in the least hindered area of the ligand (Fig. 7A). For RR propagation, the orientation of the RR-chain points towards the SW occupied by tBu groups (Fig. 7B), justifying the energetic difference between the two RLSs determining the enantioselectivity. As a matter of fact, the conformation of the chain with the steric hindrance exerted by the ligand pushes the disfavored axial positioning of the lactide methyl substituents of the nucleophilic addition TS (Fig. S2†). Lastly, the % VBur steric maps also explain the energetic reason for the switching of the monomer and growing chain positions in the ring opening step (TS2) with fm1 (Fig. 7C) having less unfavorable interactions with respect to fm2 conformations (Fig. 7D).
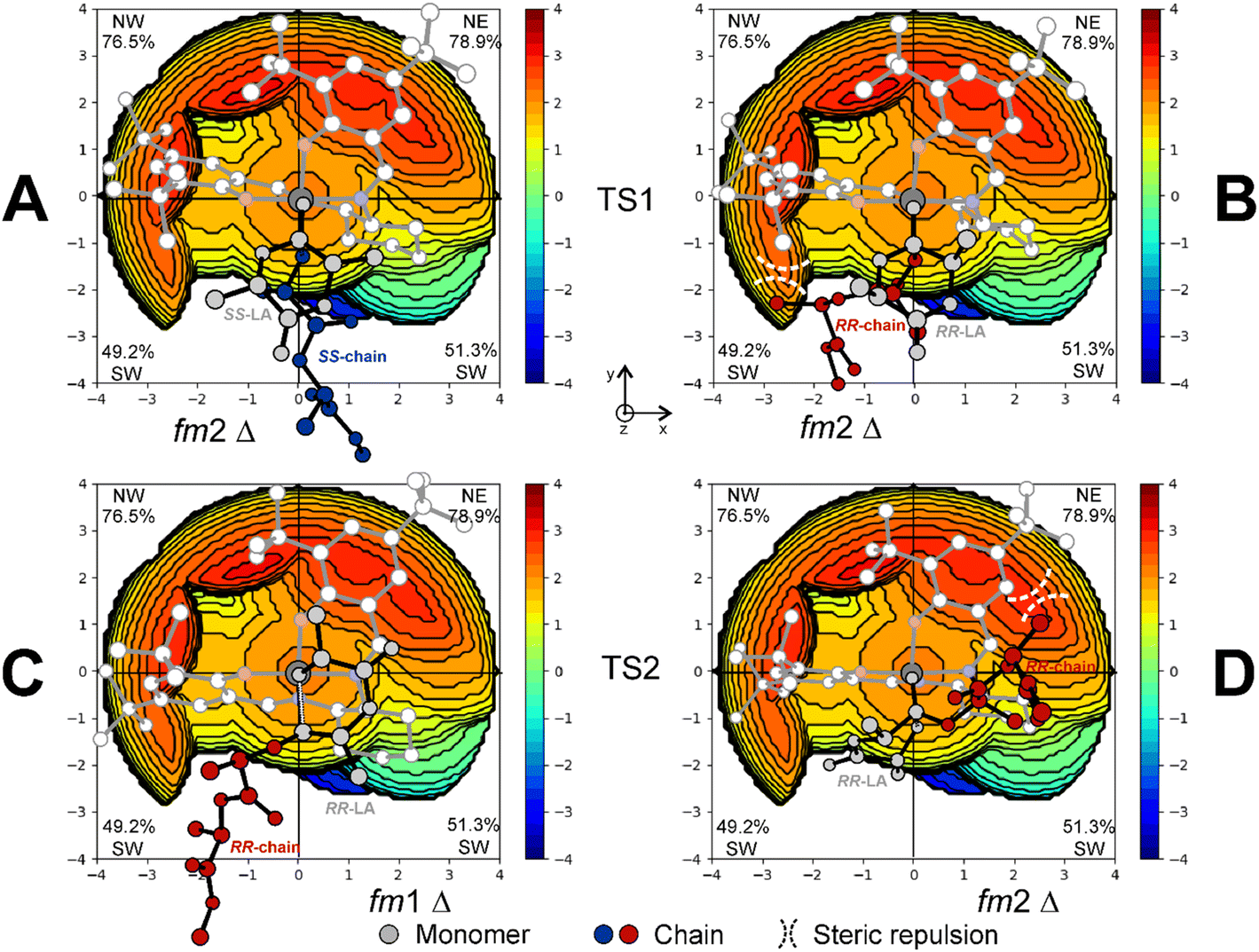 | ||
| Fig. 7 % VBur steric maps of the RLS corresponding to TS1 for SS-LA (A) and RR-LA (B) propagations with catalyst (R,R)-2A. Steric interactions leading to the TS2 preference for fm1 (C) vs. fm2 (D) for RR-LA propagation are reported with dashed lines. The z-axis is perpendicular to the plane containing the map. | ||
The importance of tBu groups in the ortho position has been further confirmed by computing the stereoselectivity of the (R,R)-2B system (replacing tBu with H atoms) in the rac-LA polymerization. The calculated enantioselectivity of (R,R)-2B decreases to 0.5 kcal mol−1 (RLS structures are reported in Fig. S3†), in good agreement with recent experimental data by Wang et al. who claimed a poor stereoselectivity in the ROP of rac-LA toward SS-LA promoted by (R,R)-2B.25 The % VBur steric maps of the RLS for SS-LA (Fig. 8A) and RR-LA (Fig. 8B) obtained with (R,R)-2B show that the occupancies of NW and SW quadrants drop respectively to 57.1% and 43.9% from 76.5% and 49.2% calculated for (R,R)-2A (Fig. 7A and B).
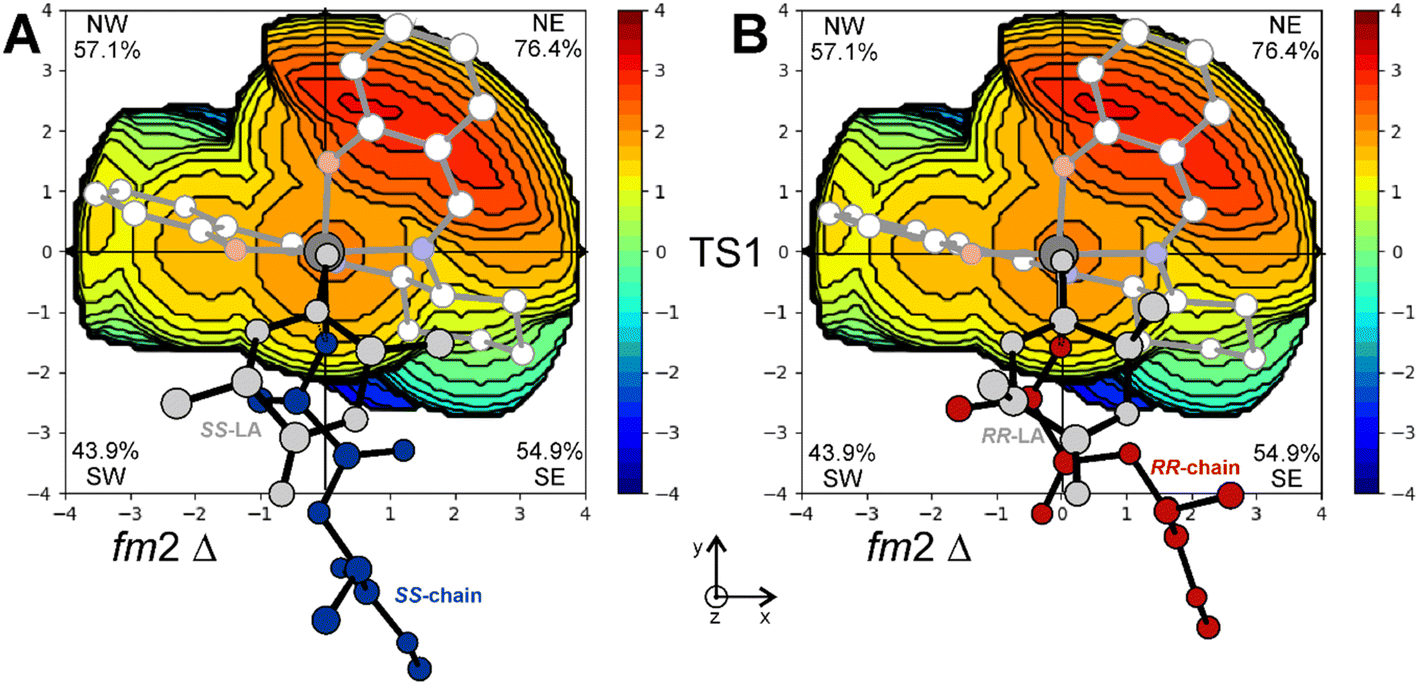 | ||
| Fig. 8 % VBur steric maps of the RLS corresponding to TS1 for SS-LA (A) and RR-LA (B) propagation with catalyst (R,R)-2B. | ||
The DFT energetic plots in Fig. 1 and 6, combined with steric maps in Fig. 3 and 5, rationalized the origin of ESC control in the rac-LA ROP promoted by (R)-1 (RR preference) and by (R,R)-2A (SS preference) including the intermediate situation of (R,R)-2B. The multiple mechanistic paths we proposed are the only way to explain the experimental data (Table S4†). However, additional insights are disclosed by computing the activation Gibbs energies for all enchainments, including the RR and SS insertions on SS- and RR-chains, respectively. The main results summarized in Table 2 (complete lists reported in Tables S5 and S6†) suggest that the occasional insertion of SS-LA is corrected by the ESC of (R)-1 being the heterotactic enchainment intermediate between the isotactic ones (Table 2-left and Fig. S4†). Occasional RR insertion at (R,R)-2A are, instead, favoured by the last inserted RR-chain (Table 2-right and Fig. S5†). The additional CEC interplay we found might explain the isotactic stereosequences reported by Nomura et al.29 in the rac-LA ROP promoted by an achiral salen catalyst (X = Et; R2 = tBu, Scheme 4) as well as the subtle microstructure differences of PLA stereoblocks obtained by (R)-1 and (R,R)-2A.
| System (R)-1 | System (R,R)-2A | ||||
|---|---|---|---|---|---|
| Assembly mode | ΔG‡RLS (kcal mol−1) | ΔΔG (kcal mol−1) | Assembly mode | ΔG‡RLS (kcal mol−1) | ΔΔG (kcal mol−1) |
| RR-LA + RR chain | 14.4 | 0.0 | SS-LA + SS chain | 11.3 | 0.0 |
| RR-LA + SS chain | 14.9 | 0.5 | RR-LA + RR chain | 13.5 | 2.2 |
| SS-LA + RR chain | 15.2 | 0.8 | RR-LA + SS chain | 13.9 | 2.6 |
| SS-LA + SS chain | 16.2 | 1.8 | SS-LA + RR chain | 14.5 | 3.2 |
4. Conclusions
This computational study revealed the origin of the stereoselectivity in the rac-LA ROP promoted by two enantiopure salen–Al systems which have so far been claimed as the most stereoselective catalysts through ESC control.5,6 Despite belonging to the same ligand class, (R)-1 and (R,R)-2A preferentially select opposite enantiomers in the polymerization of rac-LA. We succeeded in explaining the experimental stereoselectivity, and our analysis provided additional insights. First, the presence of a chiral flexible bridge, such as (R)-binaphthyl in (R)-1, makes both ff and fm octahedral wrapping modes feasible, with their energetic preference dictated by the monomer's chirality. This feature, combined with multiple possible reaction pathways due to the easy switching of the monomer and chain positions, explains (R)-1's preference for forming PDLA from rac-LA.In contrast, with a less flexible chiral bridge, such as (R,R)-cyclohexyl in (R,R)-2A, access to ff chiral octahedral wrapping modes is precluded. However, multiple monomer-controlled reaction paths remain. The stereoselective outcome, in this case, is ensured by a fixed chiral octahedral environment enforced by the steric hindrance of the tBu groups in the ortho position of the salen ligand, leading to the preferred formation of PLLA from rac-LA. This feature was further confirmed by modeling the ROP of rac-LA promoted by (R,R)-2B, which has H atoms in the ortho position, resulting in a lower AKRP.25
Finally, the DFT calculations of the overall microstructures revealed the interplay between ESC and CEC, opening the way to rationalize the CEC promoted by achiral salen systems.29 We believe that the insights from this work can be useful for both the theoretical and experimental communities in the rational development of ROP stereoselective catalysts,45 thereby bridging the gap with TM-catalyzed olefin polymerization.46
Data availability
Data for this article including computational details, Scheme S1, Fig. S1–S5, Tables S1–S6,† and Cartesian coordinates (xyz) of the MEP structures mentioned in the text are available free of charge at https://doi.org/10.1039/d4cy00937a.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Italian Ministry of University and Research (PRIN 2022, CUP E53D23008360006). This study was carried out within the MICS (Made in Italy-Circular and Sustainable) Extended Partnership funded from the European Union Next-Generation EU (Piano Nazionale di Ripresa e Resilienza (PNRR)–Missione 4 Componente 2, Investimento 1.3 – D.D. 1551.11-10-2022, PE00000004).References
- F. Vidal, E. R. van der Marel, R. W. F. Kerr, C. McElroy, N. Schroeder, C. Mitchell, G. Rosetto, T. T. D. Chen, R. M. Bailey, C. Hepburn, C. Redgwell and C. K. Williams, Nature, 2024, 626, 45–57 CrossRef CAS PubMed.
- C. Shi, E. C. Quinn, W. T. Diment and E. Y. X. Chen, Chem. Rev., 2024, 124, 4393–4478 CrossRef CAS PubMed.
- S. Lambert and M. Wagner, Chem. Soc. Rev., 2017, 46, 6855–6871 RSC.
- E. Castro-Aguirre, F. Iñiguez-Franco, H. Samsudin, X. Fang and R. Auras, Adv. Drug Delivery Rev., 2016, 107, 333–366 CrossRef CAS PubMed.
- C. M. Thomas, Chem. Soc. Rev., 2010, 39, 165–173 RSC.
- M. J. Stanford and A. P. Dove, Chem. Soc. Rev., 2010, 39, 486–494 RSC.
- L. Resconi, L. Cavallo, A. Fait and F. Piemontesi, Chem. Rev., 2000, 100, 1253–1346 CrossRef CAS PubMed.
- V. Busico, R. Cipullo, R. Pellecchia, S. Ronca, G. Roviello and G. Talarico, Proc. Natl. Acad. Sci., 2006, 103, 15321–15326 CrossRef CAS PubMed.
- N. Bahri-Laleh, A. Hanifpour, S. A. Mirmohammadi, A. Poater, M. Nekoomanesh-Haghighi, G. Talarico and L. Cavallo, Prog. Polym. Sci., 2018, 84, 89–114 CrossRef CAS.
- L. Cavallo, Polymer, 2024, 291, 126589 CrossRef CAS.
- M. J. L. Tschan, R. M. Gauvin and C. M. Thomas, Chem. Soc. Rev., 2021, 50, 13587–13608 RSC.
- H. Tsuji, Adv. Drug Delivery Rev., 2016, 107, 97–135 CrossRef CAS PubMed.
- X. Li, Z. Jia, X. Pan and J. Wu, Chem. – Asian J., 2019, 14, 662–669 CrossRef CAS PubMed.
- N. Yuntawattana, T. M. McGuire, C. B. Durr, A. Buchard and C. K. Williams, Catal. Sci. Technol., 2020, 10, 7226–7239 RSC.
- X. Pang, R. Duan, X. Li, C. Hu, X. Wang and X. Chen, Macromolecules, 2018, 51, 906–913 CrossRef CAS.
- P. Daneshmand, A. van der Est and F. Schaper, ACS Catal., 2017, 7, 6289–6301 CrossRef CAS.
- R. Hador, A. Botta, V. Venditto, S. Lipstman, I. Goldberg and M. Kol, Angew. Chem., Int. Ed., 2019, 58, 14679–14685 CrossRef CAS.
- W. Rao, C. Cai, J. Tang, Y. Wei, C. Gao, L. Yu and J. Ding, Chin. J. Chem., 2021, 39, 1965–1974 CrossRef CAS.
- W.-H. Rao, L. Yu and J.-D. Ding, Chin. J. Polym. Sci., 2023, 41, 745–759 CrossRef CAS.
- X. Wang, Y. Huang, X. Xie, Y. Liu, Z. Huo, M. Lin, H. Xin and R. Tong, Nat. Commun., 2023, 14, 3647 CrossRef CAS PubMed.
- C. De Rosa, R. Di Girolamo, A. B. Muñoz-García, M. Pavone and G. Talarico, Macromolecules, 2020, 53, 2959–2964 CrossRef CAS.
- N. Spassky, M. Wisniewski, C. Pluta and A. Le Borgne, Macromol. Chem. Phys., 1996, 197, 2627–2637 CrossRef CAS.
- T. M. Ovitt and G. W. Coates, J. Am. Chem. Soc., 2002, 124, 1316–1326 CrossRef CAS PubMed.
- Z. Zhong, P. J. Dijkstra and J. Feijen, J. Am. Chem. Soc., 2003, 125, 11291–11298 CrossRef CAS PubMed.
- Z. Peng, H. Ahmed, G. Xu, X. Guo, R. Yang, H. Sun and Q. Wang, Polym. Chem., 2023, 14, 2174–2180 RSC.
- P. McKeown, M. G. Davidson, G. Kociok-Köhn and M. D. Jones, Chem. Commun., 2016, 52, 10431–10434 RSC.
- S. M. Kirk, G. Kociok-Köhn and M. D. Jones, Organometallics, 2016, 35, 3837–3843 CrossRef CAS.
- K. Press, I. Goldberg and M. Kol, Angew. Chem., Int. Ed., 2015, 54, 14858–14861 CrossRef CAS PubMed.
- N. Nomura, R. Ishii, Y. Yamamoto and T. Kondo, Chem. – Eur. J., 2007, 13, 4433–4451 CrossRef CAS PubMed.
- P. Hormnirun, E. L. Marshall, V. C. Gibson, R. I. Pugh and A. J. P. White, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15343–15348 CrossRef CAS PubMed.
- S. M. Guillaume, E. Kirillov, Y. Sarazin and J.-F. Carpentier, Chem. – Eur. J., 2015, 21, 7988–8003 CrossRef CAS PubMed.
- M. H. Chisholm, J. C. Gallucci, K. T. Quisenberry and Z. Zhou, Inorg. Chem., 2008, 47, 2613–2624 CrossRef CAS PubMed.
- M. C. D'Alterio, C. De Rosa and G. Talarico, ACS Catal., 2020, 10, 2221–2225 CrossRef.
- M. C. D'Alterio, C. De Rosa and G. Talarico, Chem. Commun., 2021, 57, 1611–1614 RSC.
- X. Guo, G. Xu, R. Yang and Q. Wang, J. Am. Chem. Soc., 2024, 146, 9084–9095 CrossRef CAS PubMed.
- A. Grillo, Y. Rusconi, M. C. D'Alterio, C. De Rosa, G. Talarico and A. Poater, Int. J. Mol. Sci., 2024, 25, 1647 CrossRef CAS PubMed.
- Y. Rusconi, M. C. D'Alterio, A. Grillo, A. Poater, C. De Rosa and G. Talarico, Polymer, 2024, 292, 126639 CrossRef CAS.
- Y. Rusconi, M. C. D'Alterio, C. De Rosa, Y. Lu, S. M. Severson, G. W. Coates and G. Talarico, ACS Catal., 2024, 14, 318–323 CrossRef CAS PubMed.
- E. Romano, V. Barone, P. H. M. Budzelaar, C. De Rosa and G. Talarico, Chem. – Asian J., 2024, 19, e202400155 CrossRef CAS PubMed.
- O. D'Anania, E. Romano, V. Barone and G. Talarico, J. Comput. Chem., 2024, 45, 1483–1492 CrossRef PubMed.
- L. Falivene, V. Barone and G. Talarico, Mol. Catal., 2018, 452, 138–144 CrossRef CAS.
- L. Falivene, Z. Cao, A. Petta, L. Serra, A. Poater, R. Oliva, V. Scarano and L. Cavallo, Nat. Chem., 2019, 11, 872–879 CrossRef CAS PubMed.
- S. Escayola, N. Bahri-Laleh and A. Poater, Chem. Soc. Rev., 2024, 53, 853–882 RSC.
- L. Falivene, L. Cavallo and G. Talarico, ACS Catal., 2015, 5, 6815–6822 CrossRef CAS.
- X. Tang, A. H. Westlie, L. Caporaso, L. Cavallo, L. Falivene and E. Y.-X. Chen, Angew. Chem., Int. Ed., 2020, 59, 7881–7890 CrossRef CAS PubMed.
- J. W. Han, F. Hollmann, R. Luque, I. K. Song, G. Talarico, T. Tatsumi and N. Yan, Mol. Catal., 2022, 522, 112233 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cy00937a |
| This journal is © The Royal Society of Chemistry 2024 |