
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Acrylamides from 1,2-dichloroethane via palladium-catalyzed carbonylation†
Ren-Rui
Xu
ab,
Chang-Sheng
Kuai
 bc and
Xiao-Feng
Wu
*abc
bc and
Xiao-Feng
Wu
*abc
aDalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, 116023 Dalian, Liaoning, China. E-mail: xwu2020@dicp.ac.cn
bLeibniz-Institut für Katalyse e.V., Albert-Einstein-Straße 29a, 18059 Rostock, Germany. E-mail: Xiao-Feng.Wu@catalysis.de
cUniversity of Chinese Academy of Sciences, Beijing, China
First published on 9th October 2024
Abstract
1,2-Dichloroethane, a widely produced chemical raw material, has attracted considerable attention due to its versatility in various applications. Herein, we report an efficient strategy for the synthesis of acrylamides via palladium-catalyzed carbonylation, using 1,2-dichloroethane and amines as starting materials. The catalytic system demonstrated remarkable tolerance towards a broad range of amines, producing the corresponding acrylamides in good to excellent yields. Remarkably, a variety of aromatic and alkyl amines were completely converted within 40 minutes, delivering the target products with an impressive 99% yield. Additionally, the strategy's versatility was confirmed by its successful application in the synthesis of functionalized acrylates. Importantly, the reaction was scaled up to 1 mmol scale with consistent yields, underscoring its potential for practical use in organic synthesis.
Introduction
1,2-Dichloroethane, an indispensable chemical raw material, plays a central role in industrial production. Particularly, it has been crucial in the preparation of vinyl chloride,1,2 which has directly propelled the development of polyvinyl chloride (PVC) in the field of new materials.3–6 Given its significance in the industry, the production processes and output of 1,2-dichloroethane have been continuously optimized and improved. As a valuable raw material in organic synthesis, the accessibility and cost-effectiveness of 1,2-dichloroethane make it an ideal choice for the preparation of high-value-added compounds, a field that merits further exploration and development.Acrylamides, which are very important compounds in organic chemistry, have structures that are the main ones of various natural products and bioactive molecules and are widely used in fields such as food,7,8 medicine,9 and materials.10 In the past decade, acrylamides have been widely used in the research and development of targeted covalent drugs due to their electrophilicity.11–16 These drug molecules achieve precise regulation of specific biological processes through covalently binding to target proteins.17 Furthermore, acrylamides are pivotal polymer monomers, serving as a key component in numerous significant processes and offering unique benefits in the investigation of enhanced material performance.18 In recent years, the production of polyacrylamide has reached several hundred tons, representing a significant increase in output compared to previous years.19 Due to its excellent performance, it is currently employed extensively, particularly in the context of oil extraction.20 Acrylamides were traditionally mainly synthesized by the acylation of acryloyl chlorides and their derivatives with amines as nitrogen sources (Scheme 1A).21,22 In addition, under the catalysis of samarium diiodide, a one-step synthesis of acrylamides from azide and ester compounds was achieved (Scheme 1B).23 The synthesis of N-phenylacrylamide via the coupling reaction of aryl iodide with acrylamide was achieved using CuTC as a catalyst, eliminating the need for additional ligands (Scheme 1C).24 The synthesis of acrylamides through deprotonation of 3-substituted-N-phenylpropanamide was also reported, involving leaving groups such as –Br,25 –SCH3,26 –OCH3,27 –Ts (ref. 28) in these works (Scheme 1D). Nevertheless, the study of synthesizing acrylamides remains a subject of ongoing interest.
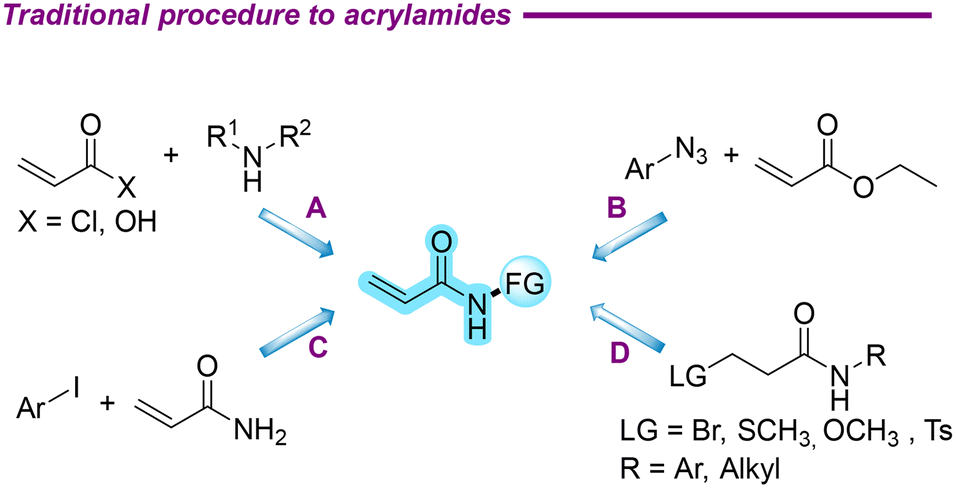 | ||
| Scheme 1 Traditional procedure to obtain acrylamides. | ||
Carbon monoxide (CO) is a highly significant C1 unit in organic chemistry, exhibiting optimal economic efficiency and extensive applicability. By adjusting the pressure of carbon monoxide, it is possible to achieve reactions that would otherwise be difficult to carry out with some carbon monoxide substitutes.29,30 Over the past five decades, remarkable progress has been made in the synthesis of high-value carbonyl compounds, such as aldehydes,31 carboxylic acids,32 carboxylic esters,33 amides,34 and others,35–37 through carbonylation. Given the increasing importance of acrylamides, there have been numerous reports in recent years on their synthesis via transition-metal-catalyzed carbonylation reactions. Beller et al. reported a modular and diverse synthesis of acrylamides using palladium-catalyzed hydroaminocarbonylation of acetylene (Scheme 2A).38 Notably, bioactive compound derivatives such as ibrutinib and osimertinib can be synthesized through this strategy. Acrylamides have also been synthesized via carbonylation reactions using vinyl halides, including vinyl chloride and vinyl iodide, as starting materials (Scheme 2B).39,40 In industrial settings, vinyl chloride is primarily produced by the high-temperature cracking of 1,2-dichloroethane, typically at 500–550 °C and 25–35 bar.1,2 Research has also explored the electrochemical removal of HCl from 1,2-dichloroethane to generate vinyl chloride.41 Additionally, the synthesis of vinyl chloride from acetylene has been well-documented over the past decade.42–44 We propose the direct synthesis of acrylamides via carbonylation using 1,2-dichloroethane as the substrate under palladium-catalyzed conditions (Scheme 2C).
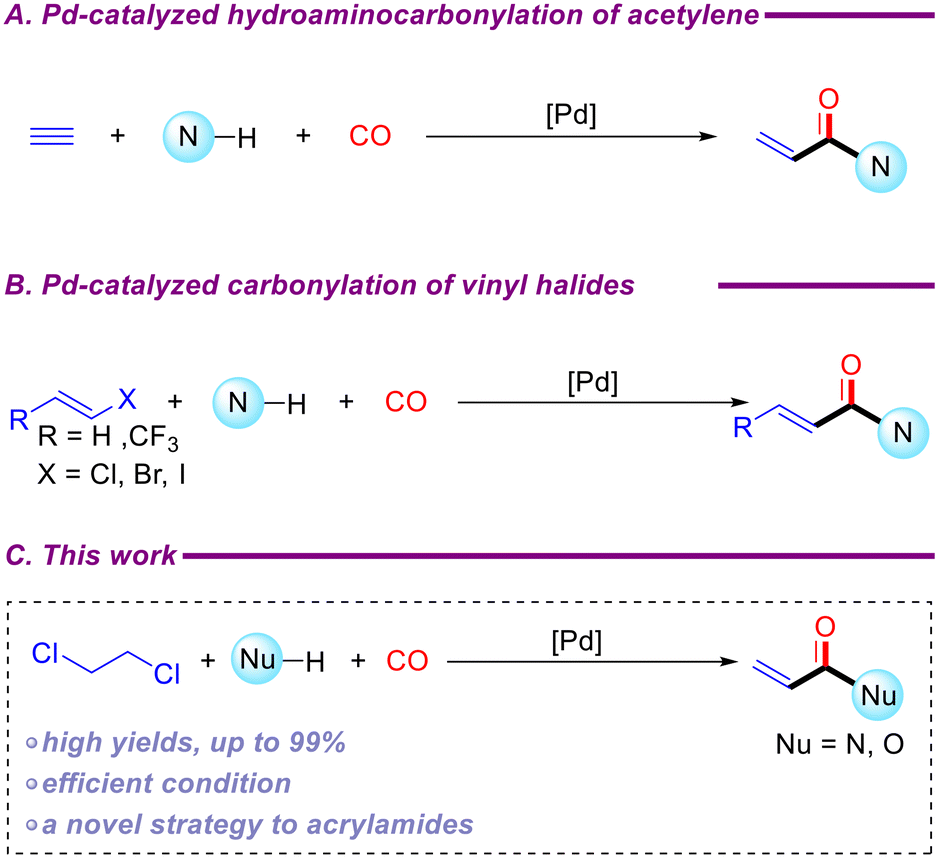 | ||
| Scheme 2 Previous carbonylation reactions for synthesizing acrylamides and this work. | ||
Results and discussion
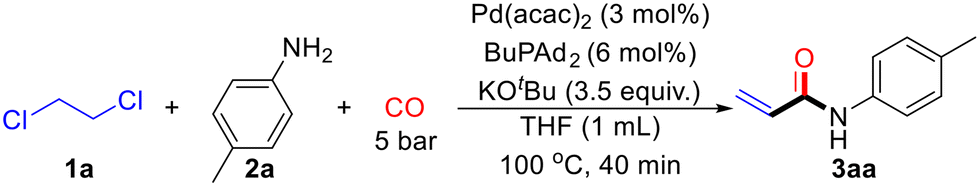
To investigate an efficient catalytic reaction system, 1,2-dichloroethane (1a) and p-toluidine (2a) were selected as model substrates. The reaction was performed with 1 mL of 1,2-dichloroethane (1a), 0.2 mmol of p-toluidine (2a), Pd(acac)2 (5 mol%), BuPAd2 (10 mol%), and 2.5 equivalents of KOtBu under 10 bar of CO at 100 °C for 20 hours. To our delight, the desired product 3aa was obtained in a 79% yield (Table 1, entry 1). Next, a series of bidentate and monodentate phosphine ligands were evaluated. The reaction was first tested with monodentate phosphine ligands, including PPh3, PCy3, and TFP, but resulted in low yields of 3aa (Table 1, entries 2–4). However, when using bidentate phosphine ligands such as DPPP, Xantphos, BINAP, and DPPF, the target product 3aa was successfully detected (Table 1, entries 5–8). Increasing the amount of KOtBu to 3.5 equivalents led to a significant increase in the yield of 3aa, reaching 91% (Table 1, entry 9). The reduction of 1,2-dichloroethane was also crucial. When the solvent was switched to THF and the amount of 1,2-dichloroethane was reduced to 10 equivalents, the yield of 3aa remained at 91% (Table 1, entry 10). Further optimization of reaction time, temperature, and CO pressure was conducted (Table 1, entries 11–12). Under optimized conditions, with 10 equivalents of 1,2-dichloroethane (1a), 0.2 mmol of p-toluidine (2a), Pd(acac)2 (3 mol%), BuPAd2 (6 mol%), and 3.5 equivalents of KOtBu under 5 bar of CO at 100 °C for 40 minutes, the desired product 3aa was obtained in an excellent 99% yield (Table 1, entry 12). However, further decrease the pressure of CO to atmospheric pressure led to low yield of the desired product.|
|
||||
|---|---|---|---|---|
| Entry | Ligand | Solvent | Time | Yieldb (%) |
| a Reaction conditions: 1a (1 mL), 2a (0.2 mmol), CO (10 bar), Pd(acac)2 (5 mol%), BuPAd2 (10 mol%), KOtBu (2.5 equiv.) at 100 °C for 20 hours. b Yields were determined by GC-FID analysis using n-dodecane as internal standard. c Yield of isolated product. ND = not detection. d KOtBu (3.5 equiv.). e 1a (2 mmol, 10 equiv.). f CO (5 bar). g Pd(acac)2 (3 mol%), BuPAd2 (6 mol%). | ||||
| 1 | BuPAd2 | w/o | 20 h | 79 |
| 2 | PPh3 | w/o | 20 h | 11 |
| 3 | PCy3 | w/o | 20 h | 17 |
| 4 | TFP | w/o | 20 h | 14 |
| 5 | DPPP | w/o | 20 h | ND |
| 6 | Xantphos | w/o | 20 h | ND |
| 7 | BINAP | w/o | 20 h | ND |
| 8 | DPPF | w/o | 20 h | ND |
| 9d | BuPAd2 | w/o | 20 h | 91 |
| 10e | BuPAd2 | THF | 20 h | 91 |
| 11e,f | BuPAd2 | THF | 40 min | 99 |
| 12e,f,g | BuPAd2 | THF | 40 min | 99 (96c) |
Subsequently, we explored the scope of various amine compounds under optimal reaction conditions (Scheme 3). Aryl substrates bearing electron-donating groups, such as methyl, methoxy, and trifluoromethoxy, were well-tolerated, and the corresponding acrylamides were obtained in good to excellent yields ranging from 77% to 99% (3ab–3af). Substrates with halogen substituents, such as fluoro and chloro, also gave the desired products in good yields (3ag, 3ah). However, aryl substrates with electron-withdrawing groups performed less satisfactorily, yielding 3ai and 3aj in 28% and 68%, respectively. We also investigated the impact of biphenyl and 2-nitro-9H-fluorene moieties, both of which produced the corresponding products in 92% yields (3ak, 3al). 1-Naphthylamine and 2-naphthylamine were suitable for this transformation, yielding 3am and 3an in 69% and 67%, respectively. Furthermore, the catalytic system demonstrated good tolerance for heterocyclic amines, including 3,4-methylenedioxyaniline, 8-aminoquinoline, and 3-aminoquinoline, with yields ranging from 66% to 91% (3ao–3aq). In addition, a variety of alkyl amines reacted smoothly and efficiently with 1,2-dichloroethane. Primary alkyl amines such as benzylamine, n-butylamine, cyclobutylamine, cyclododecylamine, and amantadine were highly compatible, yielding products in 64% to 99% (3ar–3av). The system also converted secondary amines, such as butylamine and hexamethyleneimine, producing the target compounds in 83% and 77% yields (3aw, 3ax). Notably, alkyl amines containing electron-withdrawing groups or carbon–carbon triple bonds also reacted successfully, yielding products in 68% and 41% (3ay, 3az). Furthermore, a heteroaryl-substituted alkylamine was also compatible with standard conditions, for example, 2-(aminomethyl)pyrazine (4aa). 4-Methoxybenzyl alcohol was smoothly converted to the corresponding acrylate 5aa with a yield of 53% under prolonged reaction conditions. Cyclohexanol was used as a substrate, and a small amount of the target product (5ab) was detected by GC-MS. (4-(Trifluoromethyl) phenyl) methanol cannot be converted into the corresponding acrylate in the catalytic system. Unfortunately, phenols and thiophenols were incompatible with this catalytic system and could not be smoothly transformed under standard or prolonged reaction conditions. It is worth to mention that the reactions failed when 1,2-dichloropropane, 1,1,2-trichloroethane, and 1,1,2,2-tetrachloroethane were tested instead of DCE under our standard conditions.
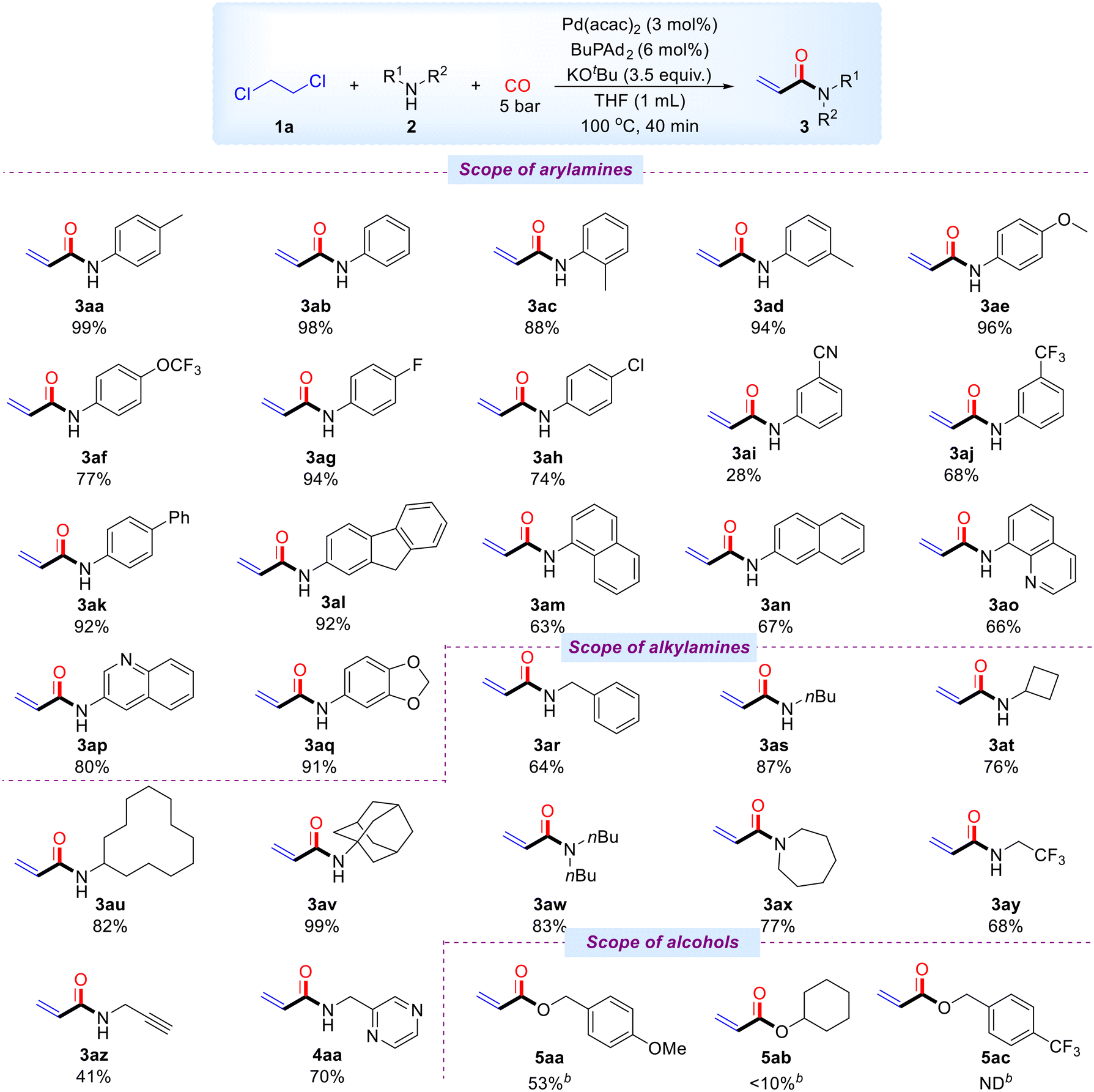 | ||
| Scheme 3 Substrate scopea aReaction conditions: 1a (2 mmol, 10 equiv.), 2 (0.2 mmol), CO (5 bar), Pd(acac)2 (3 mol%), BuPAd2 (6 mol%), KOtBu (3.5 equiv.), THF (1 mL) at 100 °C for 40 minutes, isolated yields. b1a (1 mL), 4-methoxybenzylalcohol (0.2 mmol), CO (10 bar), Pd2(dba)3 (3.6 mg, 2 mol%), BuPAd2 (2.9 mg, 4 mol%), K2CO3 (3.5 equiv., 96.8 mg) at 100 °C for 20 hours, isolated yield. | ||
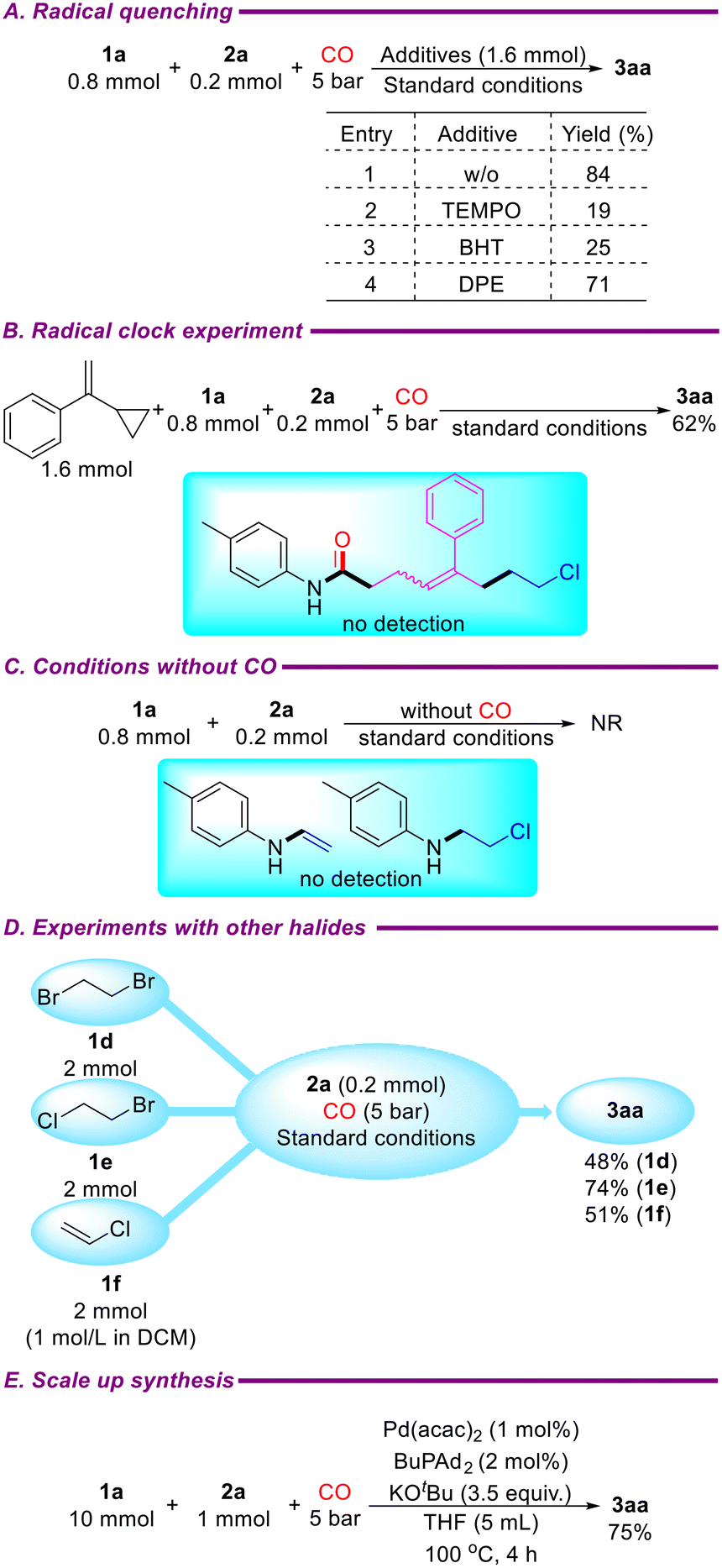
To further explore the reaction mechanism of the catalytic system, several control experiments were conducted. First, the amount of 1,2-dichloroethane was reduced to 0.8 mmol, and radical scavengers TEMPO (1.6 mmol), BHT (1.6 mmol), or DPE (1.6 mmol) were added under standard conditions, resulting in the target product 3aa with yields of 19%, 25%, and 71%, respectively (Scheme 4A). Next, a radical clock experiment was performed by adding (1-cyclopropylvinyl)benzene (1.6 mmol) under standard conditions, yielding 3aa in 67% yield, with no radical-captured products detected (Scheme 4B). These results indicate that the reaction does not proceed via a radical pathway. In another control experiment without carbon monoxide, neither 4-methyl-N-vinylaniline nor N-(2-chloroethyl)-4-methylaniline was detected, and p-toluidine (2a) remained unreacted (Scheme 4C). The catalytic system also exhibited good tolerance for 1,2-dibromoethane and 1-bromo-2-chloroethane, yielding 3aa in 48% and 51%, respectively, under standard conditions. Additionally, vinyl chloride was converted into 3aa with a yield of 51% under the same conditions (Scheme 4C). Based on these results and previous reports,39,40 it is likely that the catalytic system involves the elimination of 1,2-dichloroethane to form C(sp2)–Cl bonds, followed by oxidative addition with Pd(0) to initiate the reaction. Finally, the reaction was successfully scaled up to 1 mmol using a 1 mol% catalyst and a 4-hour reaction time, delivering the target product 3aa in 75% yield (Scheme 4D).
 | ||
| Scheme 4 Control experiments and scale-up reaction. | ||
Conclusions
In summary, we have developed a novel and efficient method for preparing acrylamides using 1,2-dichloroethane and amines as substrates. This catalytic system demonstrated broad substrate versatility, allowing amines to be smoothly converted into the corresponding acrylamides with good to excellent yields, reaching up to 99%. The reaction was successfully scaled up to a 1 mmol scale. Furthermore, this strategy is also applicable to the preparation of acrylates.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the financial supports from National Key R&D Program of China (2023YFA1507500) and DICP.Notes and references
- I. Mochida, T. Tsunawaki, C. Sotowa, Y. Korai and K. Higuchi, Ind. Eng. Chem. Res., 1996, 35, 3803–3807 CrossRef.
- C. Li, G. Hu, W. Zhong, W. He, W. Du and F. Qian, Ind. Eng. Chem. Res., 2013, 52, 17501–17516 CrossRef.
- J. A. Witjes, G. Del Popolo, M. Marberger, O. Jonsson, H. P. Kaps and C. R. Chapple, J. Urol., 2009, 182, 2794–2798 CrossRef PubMed.
- C. Hou, T. Li, T. Zhao, J. Lv, W. Zhang and Y. Ma, J. Reinf. Plast. Compos., 2012, 31, 1526–1531 CrossRef.
- X. Colom, J. Cañavate, F. Carrillo and M. Lis, J. Compos. Mater., 2014, 48, 1061–1069 CrossRef.
- A. T. Mohamed, Meas., 2016, 89, 28–33 CrossRef.
- C. Khongsiri, A. Ratsamisomsi, P. Wilairat and W. Tiyapongpattana, ACS Food Sci. Technol., 2024, 4, 1834–1843 CrossRef CAS.
- B. Nivine, A. Hadiya, P. Montserrat, S. Franscesc and H. Amira, Food Sci. Nutr., 2024, 12, 1046–1055 CrossRef PubMed.
- D. Yamane, R. Tetsukawa, N. Zenmyo, K. Tabata, Y. Yoshida, N. Matsunaga, N. Shindo and A. Ojida, J. Med. Chem., 2023, 66, 9130–9146 CrossRef CAS PubMed.
- Z. Liu, Q. Feng, Z. Xu and S. Yang, Molecules, 2024, 29, 2589 CrossRef CAS PubMed.
- A. Osoegawa, T. Karashima, Y. Takumi, T. Sato, M. Abe, T. Hashimoto and K. Sugio, J. Thorac. Dis., 2023, 15, 5566–5573 CrossRef PubMed.
- Y. Song, K. Zhou, D. Zou, J. Zhou, J. Hu, H. Yang, H. Zhang, J. Ji, W. Xu, J. Jin, F. Lv, R. Feng, S. Gao, H. Guo, L. Zhou, R. Elstrom, J. Huang, W. Novotny, R. Wei and J. Zhu, Clin. Cancer Res., 2020, 26, 4216–4224 CrossRef CAS PubMed.
- P. Hillmen, J. R. Brown, B. F. Eichhorst, N. Lamanna, S. MO'Brien, L. Qiu, T. Salmi, J. Hilger, K. Wu, A. Cohen, J. Huang and C. S. Tam, Future Oncol., 2020, 16, 517–523 CrossRef CAS PubMed.
- F. Skoulidis, B. T. Li, G. K. Dy, T. J. Price, G. S. Falchook, J. Wolf, A. Italiano, M. Schuler, H. Borghaei, F. Barlesi, T. Kato, A. Curioni-Fontecedro, A. Sacher, A. Spira, S. S. Ramalingam, T. Takahashi, B. Besse, A. Anderson, A. Ang, Q. Tran, O. Mather, H. Henary, G. Ngarmchamnanrith, G. Friberg, V. Velcheti and R. Govindan, N. Engl. J. Med., 2021, 384, 2371–2381 CrossRef CAS PubMed.
- D. A. Martin, J. Telliez, S. Pleasic-Williams, Y. Zhang, B. Tierney, M. Blatnik, J. D. Gale, C. Banfield, Y. Zhou, A. Lejeune, S. H. Zwillich, E. Stevens, N. Tiwari, E. Kieras and A. Karanam, J. Clin. Pharmacol., 2024, 64, 67–79 CrossRef CAS PubMed.
- B. King, X. Zhang, W. G. Harcha, J. C. Szepietowski, J. Shapiro, C. Lynde, N. A. Mesinkovska, S. H. Zwillich, L. Napatalung, D. Wajsbrot, R. Fayyad, A. Freyman, D. Mitra, V. Purohit, R. Sinclair and R. Wolk, Immunotherapy, 2023, 15, 1093–1103 CrossRef CAS.
- D. Chen, D. Guo, Z. Yan and Y. Zaho, Med. Chem. Commun., 2018, 9, 244–253 RSC.
- D. Taeymans, J. Wood, P. Ashby, I. Blank, A. Studer, R. H. Stadler, P. GONDÉ, T. Whitmore, P. Eijck, S. Lalljie, H. Lingnertt, M. Lindblom, R. Matissek, D. Müller, D. Tallmadge, J. O'brien, S. Thompson, D. Silvani and T. Whitmore, Crit. Rev. Food Sci. Nutr., 2004, 44, 323–347 CrossRef CAS PubMed.
- T. Zhao, J. Xing, W. Pu, Z. Dong, C. Yuan, G. Peng, F. Jin and J. Xia, Polym. Compos., 2018, 39, 368–376 CrossRef CAS.
- S. Liang, Y. Liu, S. Hu, A. Shen, Q. Yu, H. Yan and M. Bai, Energies, 2019, 12, 562 CrossRef CAS.
- Q. L. Luo, L. Lv, Y. Li, J. P. Tan, W. Nan and Q. Hui, Eur. J. Org. Chem., 2011, 6916–6922 CrossRef CAS.
- L. Zhang, S. Bai and L. Zheng, Organometallics, 2023, 42, 2262–2268 CAS.
- X. Wang, J. Chem. Res., 2006, 37, 484–485 Search PubMed.
- Z. J. Quan, H. D. Xia, Z. Zhang, Y. X. Da and X. C. Wang, Appl. Organomet. Chem., 2014, 28, 81–85 CrossRef CAS.
- F. Pandolfi, I. Chiarotto, L. Mattiello, R. Petrucci and M. Feroci, ChemistrySelect, 2019, 4, 12871–12874 CrossRef CAS.
- K. Yang, Y. Li, Z. Ma, L. Tang, Y. Yin, H. Zhang, Z. Li and X. Sun, Eur. J. Org. Chem., 2019, 2019, 5812–5814 CrossRef CAS.
- R. Zhao, B. L. Zeng, W. Q. Jia, H. Y. Zhao, L. Y. Shen, X. J. Wang and X. D. Pan, RSC Adv., 2020, 10, 34938–34942 RSC.
- E. C. Wang, K. S. Huan, G. W. Li, J. R. Lin and M. K. Hsu, J. Chin. Chem. Soc., 2001, 48, 83–90 CrossRef CAS.
- L. C. Wang, B. Chen and X. F. Wu, Angew. Chem., Int. Ed., 2022, 61, e202203797 CrossRef CAS PubMed.
- Y. Wang, H. Yang, Y. Zheng, M. Hu, J. Zhu, Z. P. Bao, Y. Zhao and X. Wu, Nat. Catal., 2024 DOI:10.1038/s41929-024-01204-6.
- C. Zhou, J. Hu, Y. Wang, C. Yao, P. Chakraborty, H. Li, C. Guan, M. H. Huang and K. W. Huang, Org. Chem. Front., 2019, 6, 721–724 RSC.
- A. R. Elman, Tetrahedron Lett., 2013, 54, 5527–5531 CrossRef.
- L. C. Wang, B. Chen, Y. Zhang and X. F. Wu, Angew. Chem., Int. Ed., 2022, 61, e202207970 CrossRef.
- R. R. Xu, D. Wen, X. Qi and X. F. Wu, Org. Biomol. Chem., 2022, 20, 2605–2608 RSC.
- R. R. Xu, W. Wang, X. Qi and X. F. Wu, Org. Chem. Front., 2022, 9, 1417–1421 RSC.
- R. R. Xu, X. Bao, Y. W. Huo, R. G. Miao, D. Wen, W. Dai, X. Qi and X. F. Wu, Org. Lett., 2022, 24, 6477–6482 CrossRef PubMed.
- F. P. Wu, Y. Yang, D. P. Fuentes and X. F. Wu, Chem, 2022, 8, 1982–1992 Search PubMed.
- M. Beller, Z. Cao, Q. Wang and H. Neumann, Angew. Chem., Int. Ed., 2024, e202410597 Search PubMed.
- J. Eriksson, O. Åberg and B. Långström, Eur. J. Org. Chem., 2007, 455–461 CrossRef CAS.
- Y. Li, Y. Jing, Y. R. Shi, H. Li, M. G. Yang, Y. L. Kou and Q. W. Fan, Eur. J. Org. Chem., 2024, 27, e202301255 CrossRef.
- Y. Liang, F. Lin, Y. Adeli, R. Jin and N. Jiao, Angew. Chem., Int. Ed., 2019, 58, 4566–4570 CrossRef PubMed.
- X. Wang, B. Dai, Y. Wang and F. Yu, ChemCatChem, 2014, 6, 2339–2344 CrossRef.
- J. Zhao, Y. Bolin Wang, S. Yue, Y. Di, H. Zhai, G. He, H. Sheng, Y. Lai, L. Guo Zhu and X. Li, J. Catal., 2018, 365, 153–162 CrossRef.
- S. K. Kaiser, R. Lin, F. Krumeich, O. V. Safonova and J. Pérez-Ramírez, Angew. Chem., Int. Ed., 2019, 58, 12297–12304 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: General comments, general procedure, analytic data, and NMR spectra. See DOI: https://doi.org/10.1039/d4cy01117a |
| This journal is © The Royal Society of Chemistry 2024 |