
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExtraction yield prediction for the large-scale recovery of cannabinoids†
Hart
Plommer
 ab,
Isaiah O.
Betinol
a,
Tom
Dupree
b,
Markus
Roggen
*b and
Jolene P.
Reid
*a
ab,
Isaiah O.
Betinol
a,
Tom
Dupree
b,
Markus
Roggen
*b and
Jolene P.
Reid
*a
aDepartment of Chemistry, University of British Columbia, Vancouver, British Columbia V6T 1Z1, Canada. E-mail: jreid@chem.ubc.ca
bDelic Labs, 3800 Wesbrook Mall, Vancouver, British Columbia V6S 2L9, Canada. E-mail: markus@deliclabs.com
First published on 28th November 2023
Abstract
The extraction of compounds from natural sources is essential to organic chemistry, from identifying bioactive molecules for potential therapeutics to obtaining complex, chiral molecule building blocks. One industry that is currently leading in innovation of new botanical extraction methods and products is the cannabis industry, although it is still hampered by a lack of efficiency. Similar to chemical syntheses, anticipating the extraction conditions (flow rate, time, pressure, etc.) that will lead to the highest purity or recovery of a target molecule, like cannabinoids, is difficult. Machine learning algorithms have been demonstrated to streamline reaction optimization processes by constraining the parameter space to be physically tested to predicted regions of high performance; however, it is not altogether clear if these techniques extend to the optimization of extractions where the process conditions are even more expensive to evaluate, limiting the data available for assessment. Combining information from several sources could provide access to the requisite data necessary for implementing a data-driven approach to optimization, but little data has been made publicly available. To address this challenge and to evaluate the capabilities of machine learning for optimizing extraction processes, we built a dataset on the carbon dioxide supercritical fluid extraction (CO2 SFE) of cannabis by harmonizing data from various companies. Using this combinatorial dataset and new techniques for maximizing the information obtained from a single large scale experiment, we built robust machine learning models to accurately predict extraction yields. The resulting machine learning models also allow for the prediction of out-of-sample biomass variations, process conditions, and scales.
Introduction
Most optimization problems in organic molecule production focus on applying chemical reactions to facilitate bond constructions through the evaluation of complicated conditions and complex catalyst structures. Traditionally this process has involved the exploration of reaction parameter space to reveal and quantify the variable effects on the experimental outcome (e.g., yield, purity, selectivity).1,2 In practice, this is often achieved by evaluating one parameter at a time, however, such a process is iterative and resource intensive. Indeed, significant research efforts have been dedicated to applying machine learning (ML) algorithms for building statistical models that can quantitatively anticipate how a change in any reaction component alters the experimental outcome.3–8 These predictive models allow reaction conditions to be explored first virtually, allowing bench chemists to narrow down the conditions to be physically tested while increasing the proportion of reactions that lead to good results.9 Likewise, valuable organic molecules can also be located in nature where extraction processes must be optimized to obtain the target compounds in high yields, though anticipating the extraction conditions (flow rate, time, pressure, etc.) that lead to the highest quality or recovery is difficult.10 This overarching issue in extraction optimization is often exasperated by subtle connections across several variables and some of these are not routinely assessed (e.g., leaf size).11 Consequently, it is not clear if the algorithms and techniques typically used to build predictive models to guide the optimization of synthetic reactions can be extended to include extraction processes (Fig. 1).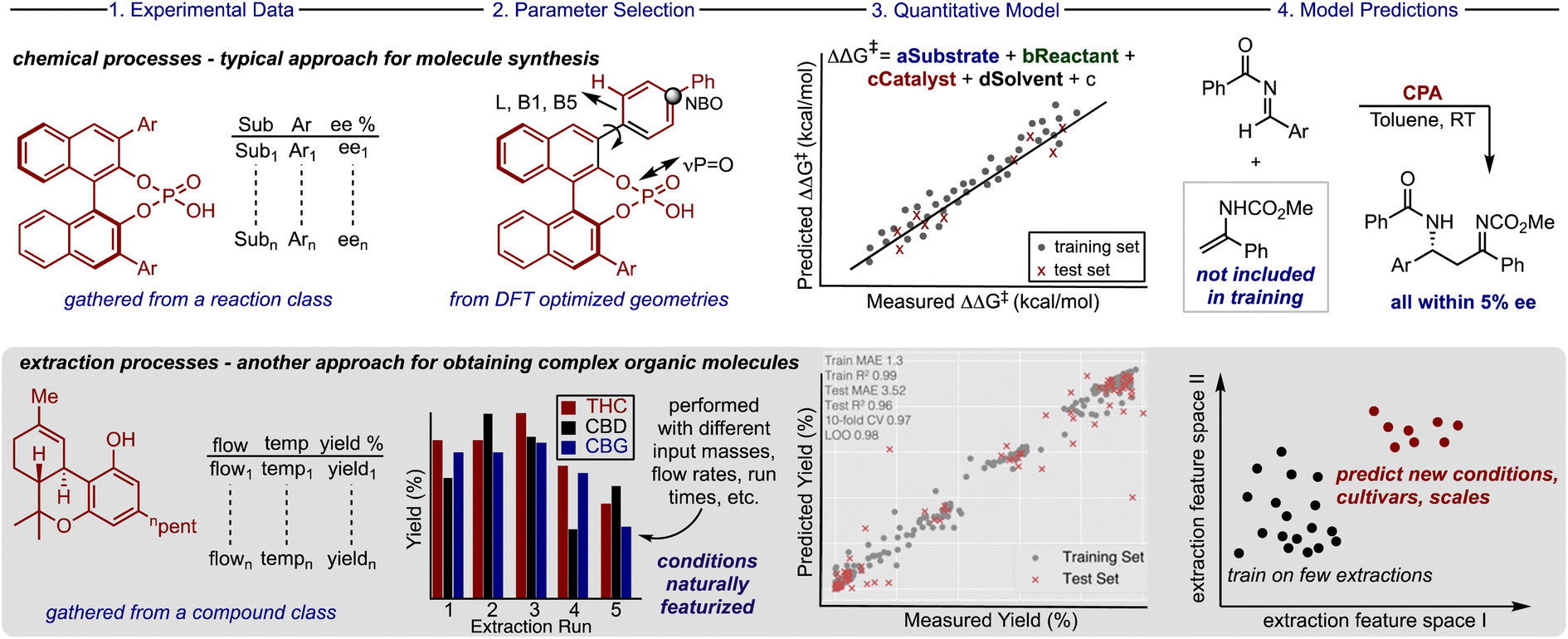 | ||
Fig. 1 Assessing the transferability of the data-driven workflow traditionally employed for optimizing chemical syntheses (top) to extraction (bottom), a different approach for obtaining organic molecules. Ar = aromatic group, NBO = natural bond orbital charge, DFT = density functional theory, CPA = chiral phosphoric acid, L/B1/B5 are 3D sterimol parameters, νP = O is the vibrational frequency of the P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond, THC = tetrahydrocannabinol, CBD = cannabidiol, CBG = cannabigerol. O bond, THC = tetrahydrocannabinol, CBD = cannabidiol, CBG = cannabigerol. | ||
The dichotomy between the two approaches for obtaining important organic molecules is also expressed in the type and amount of data that is used to train powerful ML models. Most efforts in chemical synthesis rely on data sets extracted from the literature,5,12–14 produced by high throughput experimentation,4,15,16 or a combination of the two.17 However, these data sources do not exist or are in limited supply for training robust ML models for extraction processes. One potential explanation is that these separations are generally performed on large scale (input > 1 kg) making the process conditions more expensive to evaluate. Therefore, the effective implementation of these tools will require innovative ways for maximizing the information gained from running a single large-scale experiment.
Considering these challenges and the importance of obtaining complex organic molecules from natural sources, we have evaluated the ability of ML algorithms to predict the impact of extraction conditions on compound recovery. The setting for this study is carbon dioxide supercritical fluid extraction (CO2 SFE), one of the major techniques to recover cannabinoids from cannabis and hemp with high levels of enrichment. Supercritical CO2 extraction stands out in cannabis processing for its efficiency and integrity in separating plant components, ensuring high-quality, safe extracts. This environmentally and economically favorable method aligns with pharmaceutical and food industry standards, preserving the essential properties of cannabis for optimal utilization.18,19
The composition of cannabis is rich with organic molecules including terpenes, phenolics, and cannabinoids, most of which are present in very small quantities (<1 wt%).20,21 Two major exceptions to this are cannabidiolic acid (CBDA) and tetrahydrocannabinolic acid (THCA), typically existing in >10 wt%. These acidic cannabinoids are converted into their neutral counterparts, cannabidiol (CBD) and tetrahydrocannabinol (THC), upon exposure to heat. Owing to their ubiquity, CBD and THC have been the focus of bioactive research in recent years. THC is the primary psychoactive and intoxicating component, while CBD is non-intoxicating but has received interest due to its antiseizure, antianxiety, and analgesic activities.22 In 2018, the FDA approved the first CBD-containing medicine, Epidiolex, for the treatment of epilepsy.23 These factors combined with favourable changes in the legal status of cannabis in many US states and Canada have led to a substantial demand for cannabis goods including cannabinoid concentrates and infused products.
Despite the potential of cannabinoids in pharmaceutical and consumer products, the lab-intensive and time-consuming extraction process impedes applications of these molecules. Consequently, there is an urgent need to develop more effective extraction methods to access these high-value compounds. Herein, we report that a random forest ML model trained on extraction parameters can be applied to predict the yield of cannabinoids obtained from CO2 SFE. To achieve this, we built a unique extraction database gathering results across several industry platforms (3 different instruments and 14 different cultivars) and describing the impact of various process conditions on the yield of a diverse set of cannabinoids. Our workflow includes new techniques for amplifying the information gathered from a single large-scale experiment, while also providing a prediction platform for extraction outcomes of untested conditions, cultivars, and scales (Fig. 1, bottom).
Results and discussion
Building and analysing the extraction database
Harmonizing information from different individual reports is a common technique deployed for acquiring the requisite amount of data to build ML models for organic chemistry applications. Accordingly, to assemble the database, processes executed by various companies involving different cultivars and extractors were combined to increase the number of data points for training. To maximize the data gathered from large scale experiments, which are very expensive to evaluate systematically, we considered the amounts of neutral cannabinoids (THC, CBD, cannabigerol (CBG), and cannabinol (CBN)), and their corresponding acids (THCA, CBDA, and CBGA) afforded from each extraction process. When these extraction components are treated separately, identical sets of process conditions will be present for different molecules which can be distinguished from each other through one-hot encoding. Implementing this approach would permit a single, costly extraction to provide a wealth of information about the impact of the process parameters on the recovery of up to seven different compounds. Because downstream application is variable, not all companies quantify each cannabinoid and, in some cases, it can be advantageous to collect blends of cannabinoid materials; however, implementing this approach is comparable to requiring seven times less experiments for training this extraction prediction model.In compiling the database another important factor to consider was how to measure the efficacy of the extraction process for each of the compounds. Reporting the recovery is the traditional method applied, but for some cannabinoid materials the mass does not simply reflect the original plant composition. As such, for each compound class we calculated the yield which we define as the amount of material obtained from the extraction process as a percentage of the total cannabinoid mass within the input biomass (see ESI for full discussion†). This was viewed as a simple but crucial means to transform the data to a comprehensible scale where the percentages of each cannabinoid obtained would be recorded below 100%. The information is compiled to create a unique database, which we call CannaLit, consisting of 632 data entries covering eight different cannabinoid materials (Fig. 2A).
 | ||
| Fig. 2 Analysis of dataset structures, yields and extractor condition distributions. (A) Individual cannabinoid materials contribute to the various ranges of yield. (B) Several extractor conditions are biased toward certain values of parameter space. | ||
The yield range reported for some cannabinoids is sparse and typically biased towards lower amounts which is expected to be key for accurate predictions. In many cases this value simply reflects the compound's natural abundance, while in other samples, the low number is a result of the complex chemical reactions occurring throughout the extraction process. For example, CBDA and THCA are thermally unstable and eventually converted to CBD and THC at elevated temperatures and times.24–27 Accordingly, it is expected that certain materials are strongly biased towards low yields as they are eventually converted to other compounds. Despite the similarities in the reported range of CBD and THC, the yield distribution dramatically differs. Further inspection shows most CBD recoveries are recorded over 50% while most THC recoveries fall below 50%. This symmetry demonstrates how extraction yields can vary across cultivars even with similar conditions, in this case owing to the fact that CBD is more soluble in CO2 than THC.28,29 Explicitly accounting for different cultivar yields in a single extraction avoids the narrow yield distribution when analysing individual cannabinoids, minimizes biased experiment selection, and could offer improved predictive performances.
The dataset describes the impact of 10 distinctive parameters on the recovery of cannabinoids. Most of the extraction conditions are naturally featurized (e.g., flow rate, temperature, density, input masses etc.) with one-hot encoding required for interpreting categorical data (e.g., cannabinoid and extractor type). Coverage of the continuous process conditions is sparse and is strongly biased toward operating conditions with 330–370 bar and 60–70 °C. This data driven analysis of extraction conditions shows that industrial operators seem to focus on specific process regimes to search for high yield performance (Fig. 2B). To understand the benefit of building and employing a predictive model it is worthwhile to enumerate the accessible process space. Recognising that there are limits in adjusting certain process parameters (e.g., very high flow rates and pressures are not viable options) we calculated the number of all reasonable possible combinations of continuous variables of extraction time, temperature, pressure, and flow rate.
This amounts to 4.4 × 107 possible process conditions to be tried for a single biomass input. These results hint that developing an accurate model for extraction would allow for the prediction of cannabinoid yields for an enormous set of unperformed process conditions.
A comparison of the ML modeling processes between the two approaches for accessing complex organic molecules show inherent differences in terms of data diversity and output range distributions. This assessment is important as it allows further understanding of how performances displayed by ML on predicting the outcome of synthetic reactions could translate to extraction processes. To standardize the comparison, we sought data sets of similar size that were generated in a comparable way of combining different data sources. Considering these constraints, we compared our data set to a portion of another dataset compiled from the synthetic literature, NiCOlit,13 restricted to reactions including a boron coupling partner (677 reactions, a comparable number to our data set of 632 data entries). Fig. 3 shows the yield distribution between the two datasets (i.e., restricted NiCOLit and CannaLit) to be similar, with both showing biases towards lower yields specifically in the range of 0–10%, and towards higher values around 80–100%. In contrast, CannaLit has significantly lower density coverage in the 20–80% range, which could introduce biases that have not yet been studied. Overall, despite similarities in the reported yield distribution, the underlying structure of the reaction data considerably differs. Because extraction processes are primarily unpublished there is no motivation to record only the best results; however, there is a clear tendency to rely on established conditions as additional changes to the set-up are prohibitive. In contrast, literature data focused on chemical syntheses often include a larger sampling of reaction conditions especially early in the optimization campaign when little is known about the impact of the reaction components on the experimental outcome. Therefore, optimization tables are a useful but limited source of negative data due to the reporting biases in the synthetic literature.
 | ||
| Fig. 3 Comparison of the yield distribution between two data sets. One is derived from published literature reports recording synthetic data, while the other is gathered from industry platforms describing the impact of extraction conditions on various cannabinoids. Density is each bins raw count divided by the database size and the bin width. | ||
Training a ML model
The relatively small size of the data set led us to consider random forest (RF) as the ML algorithm for building the predictive model. This algorithm is specifically designed to avoid overfitting by averaging the output of many randomly generated decision trees and recent studies have demonstrated the model's superior performance over a range of other ML techniques.4,30,31 In addition to RF regressors, support vector (SVR), k-nearest neighbour (kNN), and XGBoost regressors were also built as comparisons. Linear regression with various regularizations was also tested as a baseline approach. To identify correlations between all collected process parameters and the recovery of various cannabinoids we applied the ML algorithms to a training set containing 80% of the data entries and evaluated the model performance with the remaining test reactions. Key results are summarized in Table 1, and in our case, tree-based predictors vastly outperformed SVM and linear models. XGBoost and kNN regressors provided the best training set fits as demonstrated by the high R2 and low MAE on comparing predicted and measured yields; however, RF provided training set errors closer to the value calculated for the test set. To limit the possibility for overfitting, the rest of the study focuses on results obtained with the RF model (Fig. 4). Importantly, cross-validation (leave-one-out (LOO) and k-fold) and test set predictions suggest each of the three models to be comparable.| Regressor | Train R2 | Train MAE (%) | Test R2 | Test MAE (%) | 10-fold CV | LOO |
|---|---|---|---|---|---|---|
| a RF model created from only entries with available DFT features. | ||||||
| Random forest | 0.99 | 1.30 | 0.96 | 3.52 | 0.97 | 0.98 |
| XGBoost | 1.00 | 1.09 | 0.96 | 3.30 | 0.98 | 0.98 |
| SVR | 0.52 | 16.15 | 0.47 | 18.02 | 0.46 | 0.49 |
| kNN | 1.00 | 0.02 | 0.98 | 2.78 | 0.97 | 0.97 |
| Linear regression | 0.40 | 21.03 | 0.42 | 21.84 | 0.34 | 0.37 |
| ElasticNet | 0.37 | 23.41 | 0.38 | 24.62 | 0.34 | 0.35 |
| Ridge | 0.40 | 21.03 | 0.42 | 21.84 | 0.35 | 0.37 |
| LASSO | 0.39 | 21.75 | 0.42 | 22.55 | 0.35 | 0.37 |
| NiCOLit13,a | 0.81 | 10.73 | 0.27 | 21.61 | 0.45 | 0.48 |
 | ||
| Fig. 4 RF regression model correlating the yield of various cannabinoids to a set of extractor operating conditions and categorical descriptors. | ||
Despite the clear experimental and yield distribution bias, the predictive performance on CannaLit is significantly better than the one reported for NiCOLit (test R2 = 0.27). In fact, when progressing to this learning task, we obtained some of the highest R2 observed thus far in any yield correlation. One explanation could be that extraction processes are naturally parameterized making the connection between set-up conditions and output relatively straightforward. This is in significant contrast to statistically modelling chemical syntheses where the structure of each reaction component must be described by carefully chosen numerical descriptors. While several reports demonstrate the need for appropriate featurization for high model accuracy,14,32 most reports link the moderate model performance to dataset distribution and size.6,13,33,34 These new findings could encourage the implementation of additional molecular representations to improve statistical model performances even with smaller, biased literature data sets.
To verify the model further we next sought to determine how each variable contributes to the overall result. Gini importance values suggest that the input cannabinoid mass has the greatest effect on the extraction yield, followed by the identity of the cannabinoid encoded as a categorical descriptor which is unsurprising given that this is the only descriptor that differentiates between identical sets of process conditions. Notably, this is congruent with previous reports that demonstrate that recovery of unique cannabinoids is impacted by different process conditions.11 Although identifying the key contributors to model performance confirms appropriate model logic, another important test is to determine the chemical validity of our chosen parameters and ensure the model is not simply learning the structure of the data.35 This is especially noteworthy given that our dataset is comprised of few individual extractions that are repeated. For each extraction, we replaced the features with an array of randomly generated numbers, essentially creating a unique barcode for each extraction but notably with no chemical information. Applying the model architecture to correlate these meaningless reaction barcodes shows the statistical scores to be much worse (R2 = 0.22, Q2 = −0.65, LOO = −0.65, and 10-fold CV = −0.65), suggesting that the regression model relies on chemically meaningful descriptors (see the ESI for more details†). Including the identity of the cannabinoid does improve the performance (R2 = 0.84, Q2 = 0.33, LOO = 0.29, 10-fold CV = 0.25); however, the statistics are still much worse than those with the chemically relevant features especially when comparing performance on cross-validation or external test sets.
Next, we performed tests to determine the extent that our data harmonization strategy improves over standard baseline models. As noted above, different cannabinoids can have different extraction yields based only on its chemical properties and not the extraction conditions. We compared our model to a model trained structural features of each cannabinoid (Morgan fingerprints36 with 2048 bits and radius = 2). Applying a random forest model to this dataset shows little predictive performance (R2 = 0.36, MAE = 21.0, test R2 = 0.38, test MAE = 21.7) and suggests that extraction conditions are key features for predicting recoveries.
Analysis of ML performance on out-of-sample predictions
Previous work in predicting reaction outcomes have focused mainly on using the model to forecast the impact of substrates or catalysts not included in the initial training correlation. Similarly, we chose the prediction of extraction yield for an unseen cannabis cultivar as a task of practical interest. To test this, all experiments including a particular cultivar were held-out; after training on the remainder of the data, the model is applied to predict the recovery of the held-out cultivar. The results from this task showed calculated MAE ranging from 1.3% to 45.4%; however, 13 of 14 strains were predicted with a MAE below 20% and 8 of 14 strains predicted to within 10% on average (Fig. 5). Practically speaking, the poorly predicted cultivar (cultivar 8) was the only set of extractions wherein the reported yield was the combined yield of all cannabinoids and not individually reported yields. Consequently, it is expected that a model that has not been trained on this type of data is unable to adequately predict the combined extraction yields. This cultivar aside, this task demonstrates the model is not dependent on a singular cultivar and can be used to predict new cannabinoid materials given there is some representation in the training data. | ||
| Fig. 5 Mean average error when predicting cultivars left out of training. | ||
On the basis of the key parameters identified in the ML model training and assessment, we surmised that the model could also be applied to predict the recoveries of scaled up procedures, an important goal in process development.
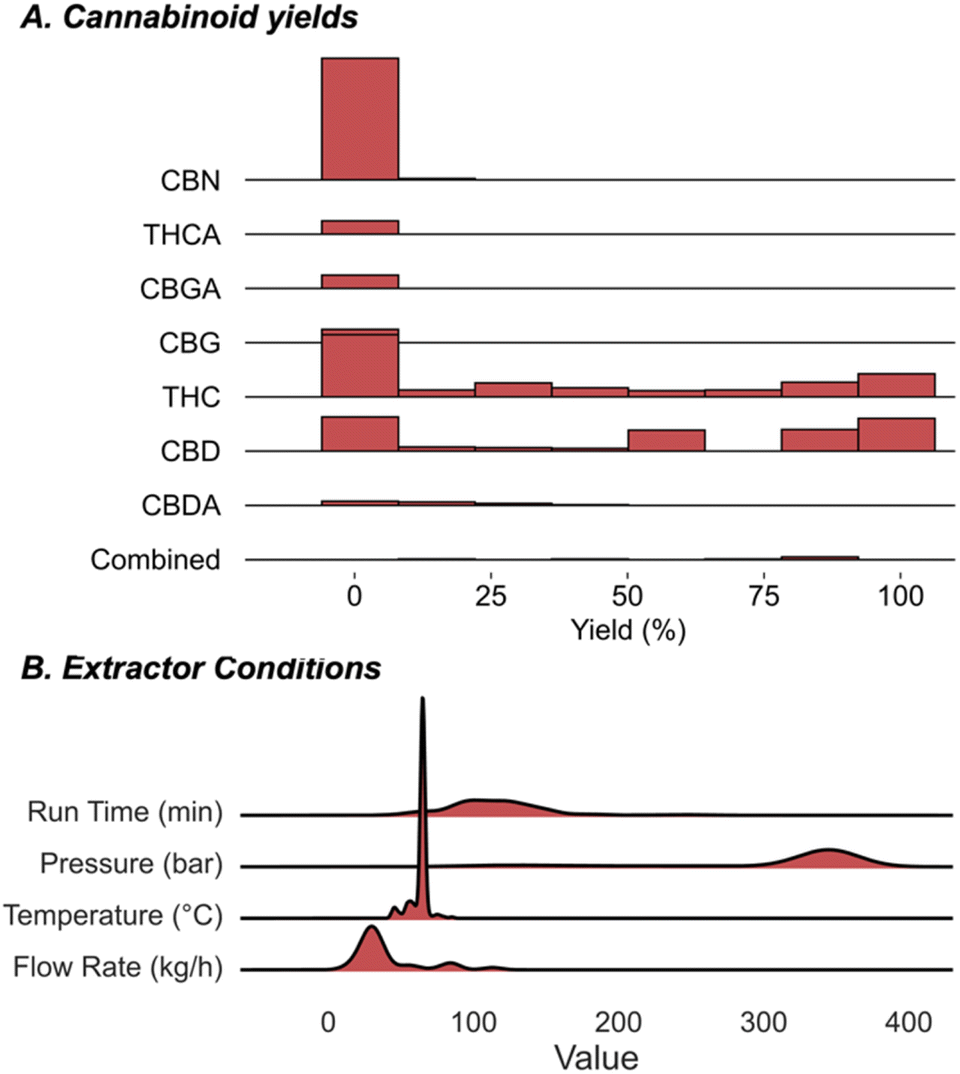
If the optimal extraction conditions are uncertain, scaling up a process can result in significant risk considering the substantial investments in resources, and simply put, a bad outcome would be more costly than running multiple smaller-scale processes to evaluate the process conditions. Consequently, it is unsurprising that a histogram shows that many of the processes are run on smaller scale with a small fraction operated with total input cannabinoid materials >400 g (Fig. 6A). Accordingly, the dataset was split into small- and large-scale processes using 400 g as the threshold for filtering the data into the two bins. The large-scale processes were used as the held-out data while the model was retrained on the remaining data and then deployed to predict recovery of this test set. Fig. 6B shows that the model accurately predicts the recovery for each of the 29 large scale data points with a small MAE of 6.8%. Ultimately, this demonstrates that our model also allows users to make informed decisions about scaling up a procedure and allowing costly resources to be conserved in the process.
 | ||
| Fig. 6 Investigating the impact of extraction scale on the correlation and prediction of yield. (A) Histogram demonstrates the distribution. Extractions with the input mass >400 g are highlighted in red. (B) Retrained RF model utilizing data on extractions with input biomass <400 g and applied to predict larger scale set-ups. | ||
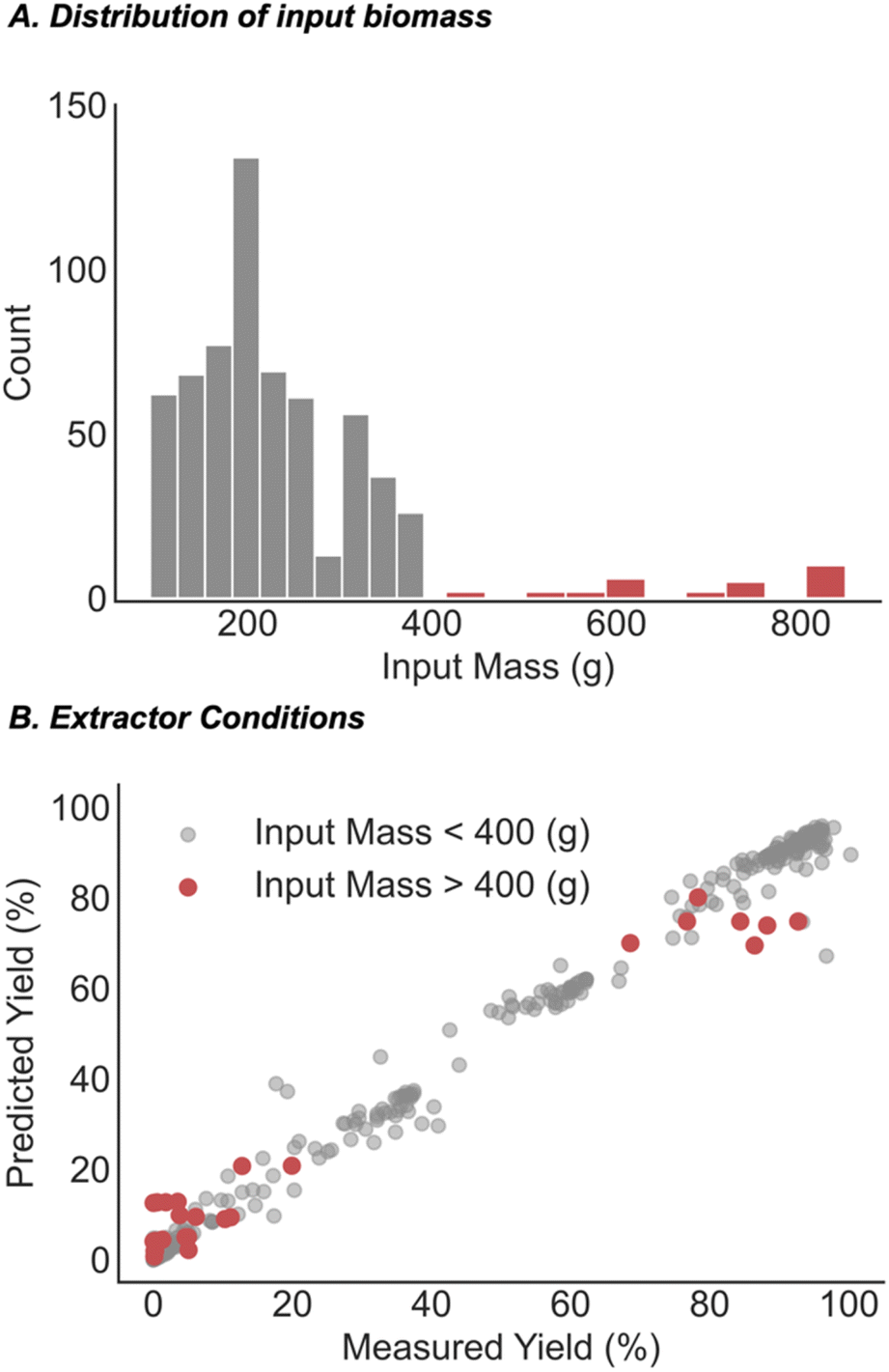
As a final prediction task, we aimed to quantify the improvement that our combinatorial dataset provides. To this end, the most common way of exploring a defined feature space is through design of experiments (DoE) modelling. The data acquired from this process is then used as the basis for model generation and eventual prediction of how changing the set-up conditions will affect the extraction yields. While DoE datasets are highly enabling for specific substrates, they can provide subpar inferences in new areas of feature space (e.g., new cultivars). Despite this, the high cost of running extractions often means that DoE-designed data from one cultivar must be used to predict another. In this regard, a portion of the CannaLit database was generated through DoE to explore how extractor conditions affect cannabinoid extraction yields for cultivar 14 (Fig. 7, blue). We tested how this data would perform in predicting the extraction yield for cultivar 13 (Fig. 7, red) which requires similar conditions to those obtained for cultivar 14 (Fig. 7). While a random forest model trained on only the DoE generated data provides a lower error than a random forest model trained on CannaLit (MAE = 3.5% vs. 11.8%), it is clear that the CannaLit-trained model better captures the trends within the out of sample set. This is exemplified when comparing the range of predicted values where the CannaLit-trained model mirrors the observed range (5.4–22% predicted, 0–20% observed) while the DoE trained dataset only predicts values to be 0% yield (0–1.2% predicted, 0–20% observed).
 | ||
| Fig. 7 Comparing our combinatorial approach with DoE, an industry standard. (A) UMAP visualization of the extractions present in the combinatorial dataset (grey), those obtained from DoE (blue, cultivar 14) and the dataset to be predicted (red, cultivar 13). (B) ML performances when trained on full or DoE only extraction space. | ||
Last, we performed each out-of-sample prediction task with a model trained on a THC-constrained version of CannaLit to demonstrate the benefits of our data harmonization strategy. This model exhibits similar training set statistics to the one trained on full CannaLit, however for each out-of-sample prediction task the model built on full CannaLit outperforms the THC-only model (see ESI for full discussion†). This suggests that harmonizing data does not hinder and may increase model generalizability. Comparisons were not performed with other cannabinoids as the requisite data for out-of-sample comparisons is not available.
Conclusions
Substantial resources are currently expended on accessing complex organic molecules through the implementation of chemical reactions and in cases where the desired compound already exists in a natural source, via extraction. Here we have demonstrated that the machine learning techniques originally deployed for assessing the impact of reaction conditions on product yield can be extended to predicting extraction outputs. This study introduces new techniques for maximizing the information gathered from a single large-scale experiment which was demonstrated to be important for expanding the size of the data set and necessary to increase yield distribution.Evaluation of various machine learning algorithms proved several tree-based models to be accurate for our purposes and we obtained some of the strongest yield correlations observed to date. More broadly, this successful outcome reinforces the utility of a data-driven approach to optimization and the need for carefully constructed databases to achieve adequate prediction power.
Data availability
All models, python scripts, Jupyter notebooks and datasets are available in the ESI.†Author contributions
H. P. generated the database and performed statistical analysis with help from I. O. B. and T. D. M. R. and J. P. R. supervised the research. All authors wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support to J. P. R. was provided by a MITACS Accelerate grant. I. O. B. acknowledges NSERC for a PGSD Research Fellowship.References
- A. McNally, C. K. Prier and D. W. C. MacMillan, Discovery of an α-Amino C–H Arylation Reaction Using the Strategy of Accelerated Serendipity, Science, 2011, 334, 1114–1117 CrossRef PubMed.
- D. W. Robbins and J. F. Hartwig, A Simple, Multidimensional Approach to High-Throughput Discovery of Catalytic Reactions, Science, 2011, 333, 1423–1427 CrossRef PubMed.
- P. Schwaller, A. C. Vaucher, T. Laino and J.-L. Reymond, Prediction of chemical reaction yields using deep learning, Mach. learn.: sci. technol., 2021, 2, 015016 Search PubMed.
- D. T. Ahneman, J. G. Estrada, S. Lin, S. D. Dreher and A. G. Doyle, Predicting reaction performance in C–N cross-coupling using machine learning, Science, 2018, 360, 186–190 CrossRef PubMed.
- J. P. Reid and M. S. Sigman, Holistic prediction of enantioselectivity in asymmetric catalysis, Nature, 2019, 571, 343–348 CrossRef PubMed.
- M. Saebi, B. Nan, J. E. Herr, J. Wahlers, Z. Guo, A. M. Zurański, T. Kogej, P.-O. Norrby, A. G. Doyle, N. V. Chawla and O. Wiest, On the use of real-world datasets for reaction yield prediction, Chem. Sci., 2023, 14, 4997–5005 RSC.
- F. Häse, L. M. Roch, C. Kreisbeck and A. Aspuru-Guzik, Phoenics: A Bayesian Optimizer for Chemistry, ACS Cent. Sci., 2018, 4, 1134–1145 CrossRef PubMed.
- B. J. Shields, J. Stevens, J. Li, M. Parasram, F. Damani, J. I. M. Alvarado, J. M. Janey, R. P. Adams and A. G. Doyle, Bayesian reaction optimization as a tool for chemical synthesis, Nature, 2021, 590, 89–96 CrossRef PubMed.
- I. O. Betinol, Y. Kuang and J. P. Reid, Guiding Target Synthesis with Statistical Modeling Tools: A Case Study in Organocatalysis, Org. Lett., 2022, 24, 1429–1433 CrossRef PubMed.
- P. A. Uwineza and A. Waśkiewicz, Recent Advances in Supercritical Fluid Extraction of Natural Bioactive Compounds from Natural Plant Materials, Molecules, 2020, 25, 3847 CrossRef PubMed.
- S. Rochfort, A. Isbel, V. Ezernieks, A. Elkins, D. Vincent, M. A. Deseo and G. C. Spangenberg, Utilisation of Design of Experiments Approach to Optimise Supercritical Fluid Extraction of Medicinal Cannabis, Sci. Rep., 2020, 10, 9124 CrossRef PubMed.
- A. Shoja, J. Zhai and J. P. Reid, Comprehensive Stereochemical Models for Selectivity Prediction in Diverse Chiral Phosphate-Catalyzed Reaction Space, ACS Catal., 2021, 11, 11897–11905 CrossRef CAS.
- J. Schleinitz, M. Langevin, Y. Smail, B. Wehnert, L. Grimaud and R. Vuilleumier, Machine Learning Yield Prediction from NiCOlit, a Small-Size Literature Data Set of Nickel Catalyzed C–O Couplings, J. Am. Chem. Soc., 2022, 144, 14722–14730 CrossRef CAS PubMed.
- W. Beker, E. P. Gajewska, T. Badowski and B. A. Grzybowski, Prediction of Major Regio-, Site-, and Diastereoisomers in Diels–Alder Reactions by Using Machine-Learning: The Importance of Physically Meaningful Descriptors, Angew. Chem., Int. Ed., 2019, 58, 4515–4519 CrossRef CAS PubMed.
- A. F. Zahrt, J. J. Henle, B. T. Rose, Y. Wang, W. T. Darrow and S. E. Denmark, Prediction of higher-selectivity catalysts by computer-driven workflow and machine learning, Science, 2019, 363, eaau5631 CrossRef CAS PubMed.
- M. K. Nielsen, D. T. Ahneman, O. Riera and A. G. Doyle, Deoxyfluorination with Sulfonyl Fluorides: Navigating Reaction Space with Machine Learning, J. Am. Chem. Soc., 2018, 140, 5004–5008 CrossRef CAS PubMed.
- E. Caldeweyher, M. Elkin, G. Gheibi, M. Johansson, C. Sköld, P.-O. Norrby and J. F. Hartwig, Hybrid Machine Learning Approach to Predict the Site Selectivity of Iridium-Catalyzed Arene Borylation, J. Am. Chem. Soc., 2023, 145, 17367–17376 CrossRef CAS PubMed.
- C. Da Porto, D. Decorti and A. Natolino, Separation of aroma compounds from industrial hemp inflorescences (Cannabis sativa L.) by supercritical CO2 extraction and on-line fractionation, Ind. Crops Prod., 2014, 58, 99–103 CrossRef CAS.
- M. P. Lazarjani, O. Young, L. Kebede and A. Seyfoddin, Processing and extraction methods of medicinal cannabis: a narrative review, J. Cannabis Res., 2021, 3, 32 CrossRef PubMed.
- P. Berman, K. Futoran, G. M. Lewitus, D. Mukha, M. Benami, T. Shlomi and D. Meiri, A new ESI-LC/MS approach for comprehensive metabolic profiling of phytocannabinoids in Cannabis, Sci. Rep., 2018, 8, 14280 CrossRef.
- C. M. Andre, J.-F. Hausman and G. Guerriero, Cannabis sativa: The Plant of the Thousand and One Molecules, Front. Plant Sci., 2016, 7, 19 Search PubMed.
- E. Stockings, D. Zagic, G. Campbell, M. Weier, W. D. Hall, S. Nielsen, G. K. Herkes, M. Farrell and L. Degenhardt, Evidence for cannabis and cannabinoids for epilepsy: a systematic review of controlled and observational evidence, J. Neurol. Neurosurg. Psychiatry, 2018, 89, 741–753 CrossRef.
- A. Mead, The legal status of cannabis (marijuana) and cannabidiol (CBD) under U.S. law, Epilepsy Behav, 2017, 70, 288–291 CrossRef.
- T. Moreno, P. Dyer and S. Tallon, Cannabinoid Decarboxylation: A Comparative Kinetic Study, Ind. Eng. Chem. Res., 2020, 59, 20307–20315 CrossRef CAS.
- S. Qamar, Y. J. M. Torres, H. S. Parekh and J. Robert Falconer, Extraction of medicinal cannabinoids through supercritical carbon dioxide technologies: A review, J. Chromatogr. B, 2021, 1167, 122581 CrossRef CAS PubMed.
- S. Marzorati, D. Friscione, E. Picchi and L. Verotta, Cannabidiol from inflorescences of Cannabis sativa L.: Green extraction and purification processes, Ind. Crops Prod., 2020, 155, 112816 CrossRef CAS.
- W. He, P. J. Foth, M. Roggen, G. M. Sammisand P. Kennepohl, Why Is THCA Decarboxylation Faster than CBDA? an in Silico Perspective, ChemRxiv, 2020, preprint, DOI:10.26434/chemrxiv.12909887.v1.
- H. Perrotin-Brunel, M. C. Kroon, M. J. E. Van Roosmalen, J. Van Spronsen, C. J. Peters and G.-J. Witkamp, Solubility of non-psychoactive cannabinoids in supercritical carbon dioxide and comparison with psychoactive cannabinoids, J. Supercrit. Fluids, 2010, 55, 603–608 CrossRef CAS.
- H. Perrotin-Brunel, P. C. Perez, M. J. E. Van Roosmalen, J. Van Spronsen, G.-J. Witkamp and C. J. Peters, Solubility of Δ9-tetrahydrocannabinol in supercritical carbon dioxide: Experiments and modeling, J. Supercrit. Fluids, 2010, 52, 6–10 CrossRef CAS.
- F. Osisanwo, J. Akinsola, O. Awodele, J. Hinmikaiye, O. Olakanmi, J. Akinjobi and others, Supervised machine learning algorithms: classification and comparison, Int. J. Comput. Trends Technol. IJCTT, 2017, 48, 128–138 CrossRef.
- F. Sandfort, F. Strieth-Kalthoff, M. Kühnemund, C. Beecks and F. Glorius, A Structure-Based Platform for Predicting Chemical Reactivity, Chem, 2020, 6, 1379–1390 CAS.
- G. Skoraczyński, P. Dittwald, B. Miasojedow, S. Szymkuć, E. P. Gajewska, B. A. Grzybowski and A. Gambin, Predicting the outcomes of organic reactions via machine learning: are current descriptors sufficient?, Sci. Rep., 2017, 7, 3582 CrossRef.
- F. Strieth-Kalthoff, F. Sandfort, M. Kühnemund, F. R. Schäfer, H. Kuchen and F. Glorius, Machine Learning for Chemical Reactivity: The Importance of Failed Experiments, Angew. Chem., Int. Ed., 2022, 61, e202204647 CrossRef CAS PubMed.
- W. Beker, R. Roszak, A. Wołos, N. H. Angello, V. Rathore, M. D. Burke and B. A. Grzybowski, Machine Learning May Sometimes Simply Capture Literature Popularity Trends: A Case Study of Heterocyclic Suzuki–Miyaura Coupling, J. Am. Chem. Soc., 2022, 144, 4819–4827 CrossRef CAS.
- K. V. Chuang and M. J. Keiser, Comment on “Predicting reaction performance in C–N cross-coupling using machine learning”, Science, 2018, 362, eaat8603 CrossRef PubMed.
- H. L. Morgan, The Generation of a Unique Machine Description for Chemical Structures-A Technique Developed at Chemical Abstracts Service, J. Chem. Doc., 1965, 5, 107–113 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3dd00176h |
| This journal is © The Royal Society of Chemistry 2024 |