
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGotta be SAFE: a new framework for molecular design†
Emmanuel
Noutahi
 *a,
Cristian
Gabellini
a,
Michael
Craig
a,
Jonathan S. C.
Lim
b and
Prudencio
Tossou
a
*a,
Cristian
Gabellini
a,
Michael
Craig
a,
Jonathan S. C.
Lim
b and
Prudencio
Tossou
a
aValence Labs, Montréal, QC, Canada. E-mail: emmanuel@valencelabs.com; cristian@valencelabs.com; michael@valencelabs.com; prudencio@valencelabs.com
bMila & Valence Labs, Montréal, QC, Canada. E-mail: jonathan.lim@u.nus.edu
First published on 21st March 2024
Abstract
Traditional molecular string representations, such as SMILES, often pose challenges for AI-driven molecular design due to their non-sequential depiction of molecular substructures. To address this issue, we introduce Sequential Attachment-based Fragment Embedding (SAFE), a novel line notation for molecules. SAFE reimagines SMILES strings as an unordered sequence of interconnected fragment blocks while maintaining compatibility with existing SMILES parsers. It streamlines complex generative tasks, including scaffold decoration, fragment linking, polymer generation, and scaffold hopping, while facilitating autoregressive generation for fragment-constrained design, thereby eliminating the need for intricate decoding or graph-based models. We demonstrate the effectiveness of SAFE by training an 87-million-parameter GPT-like model on a dataset containing 1.1 billion SAFE line notations. Through targeted experimentation, we show that our SAFE-GPT model exhibits versatile and robust optimization performance. SAFE opens up new avenues for the rapid exploration of chemical space under various constraints, promising breakthroughs in AI-driven molecular design.
1 Introduction
Molecular design, which consists of constructing molecules with desired characteristics, is a critical task in computational drug discovery. It often necessitates the preservation of certain scaffolds or core chemical substructures, which serve as the backbone for the design process, the motivation for preserving these groups and constraints typically stems from their crucial role in the molecule's biological activity. Nevertheless, incorporating such constraints can be challenging when relying on conventional molecular string representations like the Simplified Molecular Input Line Entry System (SMILES).Although SMILES has played a crucial role in chemistry and drug discovery, it is unable to provide a contiguous representation of molecular substructures. This limitation hinders tasks like adding structures to a molecule's scaffold and connecting fragments, limiting its usefulness in improving potential drug candidates, particularly during lead optimization efforts. Addressing these challenges requires the development of an enhanced line notation for molecules, one that can preserve the integrity of molecular scaffolds and fragments while offering flexibility for de novo molecular design.
To this end, we introduce Sequential Attachment-based Fragment Embedding (SAFE),‡ a novel line notation for molecules. In contrast to existing methods, SAFE represents molecules as an unordered sequence of fragment blocks. This re-imagines molecular design tasks, transforming them into simpler sequence completion problems. Moreover, SAFE facilitates autoregressive generation, effectively bypassing the need for intricate decoding schemes or graph-based models (see Fig. 1 and Table 1). Importantly, despite these novel features, SAFE strings are backward compatible with SMILES parsers, promising an easy integration into existing workflows. Our contributions can be summarized as follow:
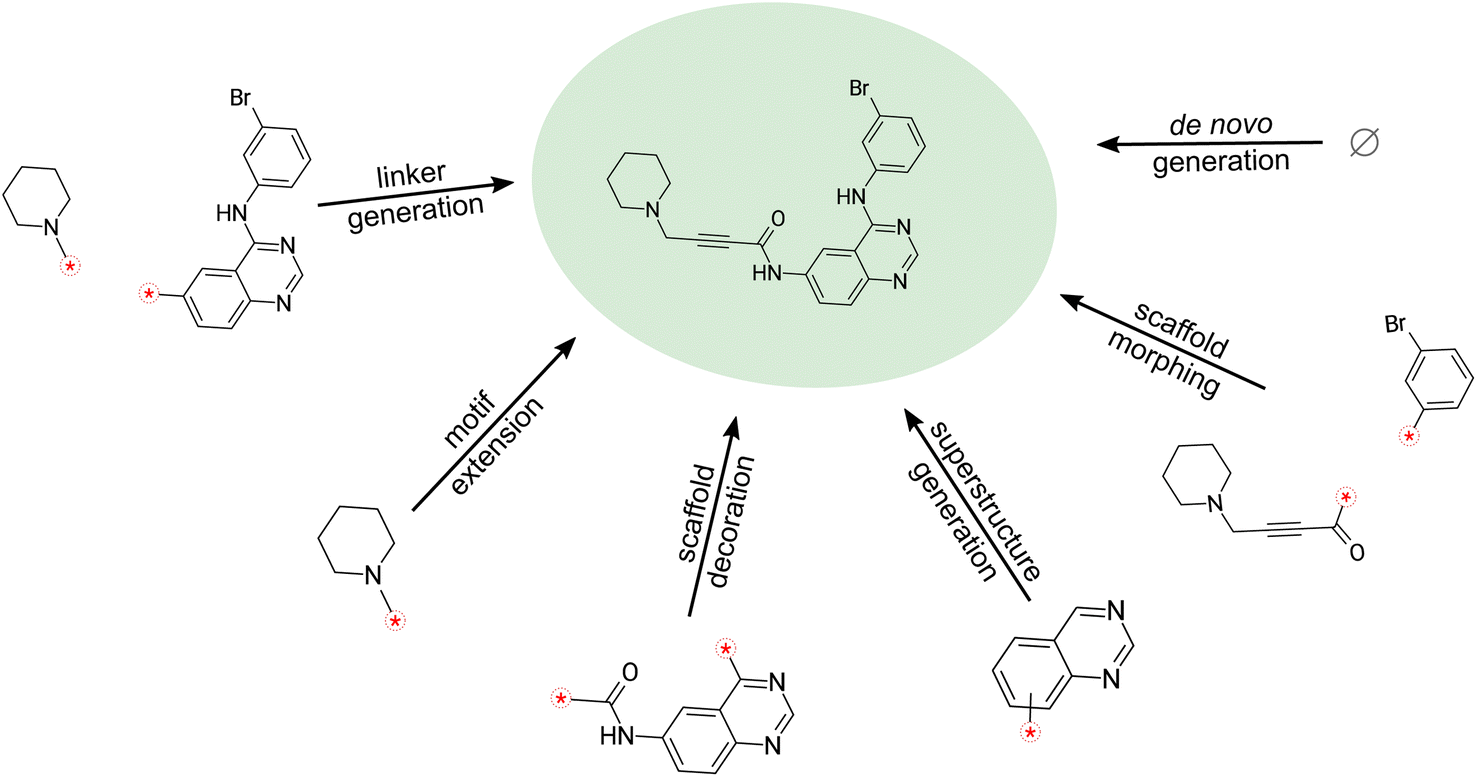 | ||
| Fig. 1 Molecular design tasks that can be performed easily with SAFE. | ||
| Task | SAFE | SMILES | Deep/gen SMILES | SELFIES | Group SELFIES | InChi | Graphs |
|---|---|---|---|---|---|---|---|
| De novo design | ✓ | ✓ | ✓ | ✓ | ✓ | ? | ✓ |
| Linker design | ✓ | ? | ✗ | ✗ | ? | ✗ | ? |
| Motif extension | ✓ | ? | ✗ | ? | ? | ✗ | ✓ |
| Scaffold decoration | ✓ | ? | ✗ | ✗ | ? | ✗ | ✓ |
| Scaffold morphing | ✓ | ✗ | ✗ | ✗ | ? | ✗ | ? |
| Super structure | ✓ | ✗ | ✗ | ✗ | ? | ✗ | ✓ |
• We introduce SAFE, a novel line notation for molecules compatible with SMILES that represents molecules as a sequence of interconnected fragments.
• We introduce SAFE-GPT, an 87.3-million-parameter GPT-like generative model, pretrained on a dataset of 1.1 billion SAFE strings that can be used for diverse downstream tasks. This model is shown to be effective in various molecule generation tasks, capitalizing on SAFE's unique characteristics.
• We propose a new benchmark inspired by real-world drug discovery challenges to assess pure generative models' performance in tasks such as scaffold decoration, linker design, and motif extension.
2 Related works
2.1 Molecular line notation representations
The Simplified Molecular-Input Line-Entry System (SMILES)1 is the most widely adopted molecular line notation in chemoinformatics for its simplicity, compactness, and human readability. In contrast to the International Chemical Identifier (InCHI) that provides global and unique identifier to molecules, SMILES are more suitable for molecular generation tasks. However, SMILES lack robustness to minor changes and struggle with ensuring the validity and integrity of fragments in deep learning-based molecular design. They also underperform in molecular search and substructure matching tasks. To overcome these challenges, alternative notations like Self-Referencing Embedded Strings (SELFIES)2,3 have been developed. SELFIES address the robustness and validity issues in deep generative modeling through a recursive approach, surpassing notations like DeepSmiles4 and GenSMILES,5 but come at the cost of simplicity, interpretability and compactness. None of these notations consistently uphold the integrity of scaffolds and fragments essential for several molecular generation tasks. A recent innovation, Group SELFIES,6 builds on standard SELFIES by introducing functional and chemical group tokens, to improve compactness and chemical motif representation for molecular generative tasks. Yet, neither Group SELFIES nor other line notations facilitate deep generative fragment-based molecule design without extensive, task-specific engineering of training processes and molecule generation steps,7–10 bespoke model architectures,11 or goal-directed optimization frameworks. In Table 1, we contrast the generative capabilities of various molecular line notations, including SAFE.2.2 Deep generative design
To contextualize our work within the domain of deep generative design we refer interested readers to comprehensive reviews provided in ref. 12–14. Herein, we briefly describe sequence-based and graph-based deep generative models. Sequence-based methods, originally focused on character-by-character SMILES generation.15 This approach provided considerable versatility but faced challenges when dealing with fragment-based constraints. Nevertheless, recent advancements have attempted to address this limitation by separately generating scaffolds and side chains,10 introducing transformations derived from matched molecular pairs analysis,16 and employing conditional generation.17,18 In the realm of graph-based methods, our work shares similarities with,19–21 which uses motifs for molecular graphs but encounter difficulties when extending design to scaffold-based generation, linker-design and generating molecules with unseen building blocks. In particular, these methods, while capable of assembling motifs in a tree-like structure, have difficulties creating novel cyclic structures not seen during training.2.3 Constrained molecular design
Notable contributions have emerged in the recent literature on constrained molecular design. Li et al.22 introduced a conditional graph generative model that excels in producing valid molecules while offering the flexibility needed for multi-objective optimization. MolGPT,18 which uses a transformer-decoder architecture for the generation of drug-like molecules, has demonstrated the capacity to conditionally control diverse molecular properties and scaffold designs, highlighting its efficacy in crafting molecules tailored to specific requirements. Furthermore, Multi-Constraint Molecular Generation (MCMG),23 combining conditional transformers, knowledge distillation, and reinforcement learning, has shown the capability to satisfy multiple constraints during the process of molecular generation.2.4 Scaffold-conditioned generation
Under hard scaffold constraints, Lim et al.24 proposed a graph-based model explicitly trained on scaffold and molecule pairs. Under soft scaffold constraints, Li et al.25 have considered the scaffold as part of the input, but their approach does not guarantee its presence in the generated molecules. Arús-Pous et al.11 used an iterative conditional training procedure to perform scaffold decoration with an LSTM trained on SMILES. Their work was extended in ref. 8, where a reaction-driven approach for scaffold decoration was proposed. Finally, Langevin et al.9 proposed a sampling algorithm that can adapt any SMILES-based auto-regressive model to work with scaffolds. However, being trained on SMILES, their models can neither guarantee validity of generated molecules nor the presence of the input scaffold constraint.3 SAFE algorithm
In SMILES, ring structures are marked by using digits to identify the opening and closing ring atom, thus denoting a virtual connection between the corresponding atoms. This rule also contributes to the subjectivity of SMILES representation where multiple different SMILES correspond to the same molecular graph. SAFE (Sequence Attachment-based Fragment Embedding) leverages this rule to discover alternative SMILES strings that enforce an order of SMILES characters in which all SMILES tokens belonging to the same molecular fragment are consistently arranged consecutively (see Fig. 2). As such, SAFE is a molecular line notation that reimagines SMILES as a collection of connected fragments and remains a valid SMILES representation. Furthermore, the arrangement of fragments within a SAFE string has no impact on the underlying molecular graph, ensuring that common data augmentation techniques for generative models, such as randomization, remain applicable.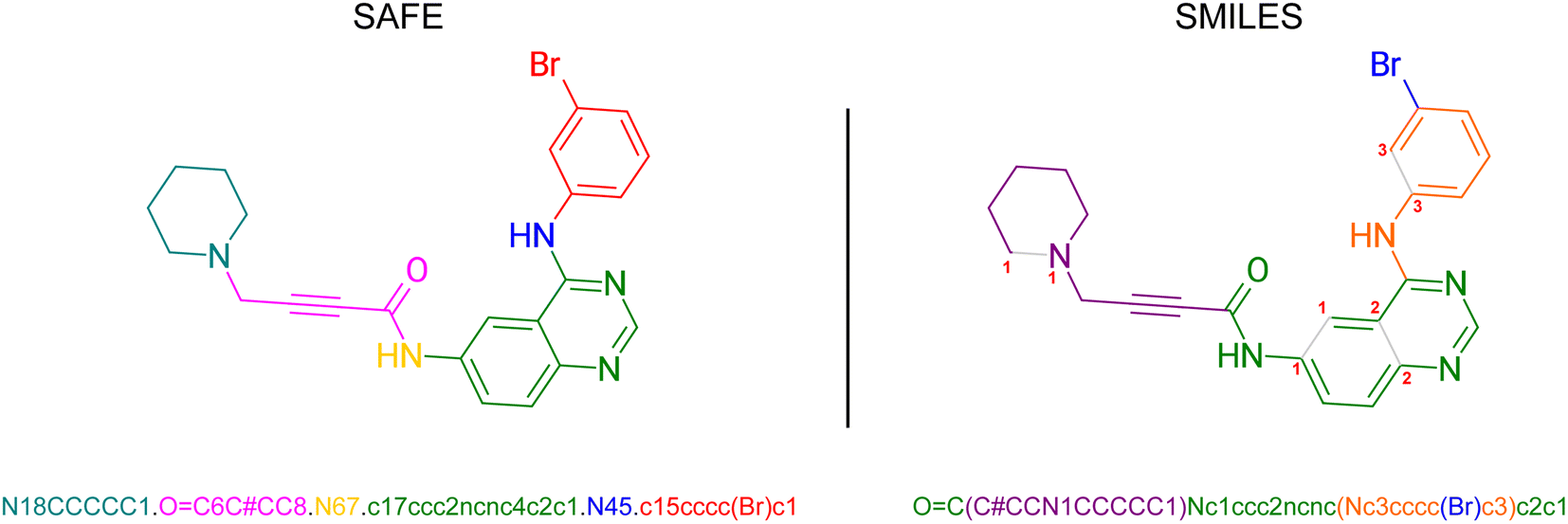 | ||
| Fig. 2 Example of a molecule as a SAFE and SMILES representation. The colored fragments and their corresponding placement in each string show how the ordering of the fragments in the SAFE representation are more easily readable and interpretable than the comparable SMILES string. | ||
3.1 Constructing a SAFE string
The detailed process to convert from SMILES to SAFE is illustrated by Algorithm 1 and Fig. S1.†
It starts by extracting all unique ring digits from the associated molecule and fragmenting it on a desired set of bonds. Our implementation utilizes the BRICS algorithm,26 though other bond-splitting algorithms, such as Hussain-Rea,27 RECAP,28 or custom patterns, are equally valid. These substructures may represent synthetically accessible building blocks that are common in drug-like compounds. The extracted fragments are sorted by size and concatenated, using a dot character (“.”) to mark new fragments in the representation, while preserving their corresponding attachment points. To construct the final SAFE string, we iterate over the numbered attachment points and replace them by novel ring digits to simulate fragment linking. These new ring digits create virtual connections between fragments resulting in a set of linked fragments, as indicated by the dot character. It's worth noting that, similar to canonicalization in SMILES that yields a unique representation from multiple valid forms, we can achieve a similar outcome by enforcing a decoding order not only on SMILES characters within fragments but also on fragment orders within the final SAFE string.
3.2 SAFE facilitates fragment-based design
The inherent sequential block structure of SAFE presents a distinctive advantage for fragment-based design tasks. Traditionally, such endeavors primarily relied on graph-based generative models. However, with a generative model trained on SAFE strings, fragment-based design becomes remarkably straightforward (refer to Fig. 1).Among those, we found the following particularly suitable for SAFE.
4 Experiments
To evaluate the utility of our new molecular line notation, we developed a generative model using a decoder-only transformer architecture. Our aim is to showcase the model's ability, trained on SAFE strings, to generate valid and diverse molecules in de novo scenarios. Additionally, we seek to evaluate its effectiveness in practical, real-world scenarios where tasks like scaffold decoration, scaffold morphing, linker design and goal-directed generation are required.4.1 SAFE-GPT: SAFE generative model
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 warmup steps and an initial lr = 1 × 10−4. We set the batch size to 100 per GPU and used 2 steps of gradient accumulation and gradient checkpointing. The model was trained on 4 Nvidia A100 GPUs, for a maximum of 1000000 steps (7 days).
000 warmup steps and an initial lr = 1 × 10−4. We set the batch size to 100 per GPU and used 2 steps of gradient accumulation and gradient checkpointing. The model was trained on 4 Nvidia A100 GPUs, for a maximum of 1000000 steps (7 days).
4.2 De novo generation results
In de novo design, our objective is to generate entirely novel compounds with desirable profiles. Assessing a model's ability to generate valuable compounds in such a setting, even without an optimization objective is crucial, as some models may encounter problems generating valid or sufficiently diverse and novel compounds. We used classical metrics like molecule validity, uniqueness, and internal diversity33,34 to assess these qualities. Validity measures the percentage of chemically valid structures according to the RDKit's parser, uniqueness is the fraction of non-duplicate molecules, and diversity assesses the internal diversity of generated molecules using the average pairwise Tanimoto distance (ECFP4 representation).
Table 2 showcases a comparison of SAFE-GPT with various generative models across 10000 samples. Despite being trained on a dataset encompassing challenging molecules, SAFE-GPT still demonstrates impressive performance in validity, uniqueness, and diversity. Remarkably, it surpasses other models in uniqueness and diversity, although it has a marginally lower validity score. To determine if this is linked to the complexities in interpreting fragment connectivity, represented by digit pairs—a common challenge also observed in SMILES-based models – we trained a smaller version, SAFE-GPT-20 M, on the MOSES dataset, as well as an alternative model with same architecture that uses Group SELFIES representation (GSELFIES-GPT-20 M). The 100% validity observed for SAFE-GPT-20 M suggests that SAFE-GPT's slightly reduced validity is largely due to its diverse and challenging training dataset. Compared to SAFE-GPT models, GSELFIES-GPT-20 M appears to generate more diverse molecules. However, a closer examination of its outputs (refer to ESI section 2†) reveals a tendency to create large, unstable rings in otherwise “valid” chemical graphs, leading to very low druglikeness and synthetic accessibility.
| Model | Repr. | Valid@10k ↑ | Unique@10k ↑ | Diversity ↑ |
|---|---|---|---|---|
| a SAFE-GPT uses a different training dataset that includes non drug-like and challenging molecules. | ||||
| SAFE-GPT | SAFE | 0.984 | 1 | 0.878 |
| SAFE-GPT-20M | SAFE | 1 | 0.999 | 0.864 |
| GSELFIES-GPT-20M | Group SELFIES | 1 | 0.999 | 0.887 |
| GSELFIES-VAE | Group SELFIES | 1 | 0.999 | 0.859 |
| GMT-SELFIES | SELFIES | 1 | 1 | 0.870 |
| SELFIES-VAE | SELFIES | 1 | 0.999 | 0.858 |
| CharRNN | SMILES | 0.975 | 0.999 | 0.856 |
| VAE | SMILES | 0.977 | 0.998 | 0.856 |
| LatentGAN | SMILES | 0.897 | 0.997 | 0.857 |
| LigGPT | SMILES | 0.900 | 0.999 | 0.871 |
| JT-VAE | GRAPH | 1 | 0.999 | 0.855 |
Hence, we note that SAFE is able to maintain comparable performance in de novo generation in terms of validity, uniqueness and diversity compared to other line notations. Together with Table 1, SAFE has the advantage of enabling new capabilities as exemplified by the fragment-constrained generation tasks while also being able to perform on par with the other methods.
In Fig. S2,† we show a subset of randomly selected molecules generated with SAFE-GPT. This visual representation offers readers an intuitive sense of the quality and reasonableness of the generated molecules. Furthermore, in Fig. S3,† we show the distribution of selected molecular properties for the 10000 generated molecules.
4.3 Performance on fragment-constrained generation
De novo compound generation is only one approach for advancing a drug discovery program. In fact, in many real-world scenarios, generative design involves modifying existing molecules in user-defined ways rather than creating entirely new compounds. This is especially true in later stages of drug discovery, such as hit-to-lead or lead optimization, where well-established structure–activity relationships (SAR) are already in place. Therefore, we examined SAFE's intended capabilities for performing fragment-constrained generative design tasks such as scaffold decoration, scaffold morphing, linker generation, motif extension, and superstructure generation (see Section 3.2). To facilitate this evaluation, we designed a benchmark that involved working with scaffolds and fragments from 10 existing drugs. Further details about the benchmark design can be found in ESI section 4† in the Appendix. Our focus on SAFE-GPT is due to its unique capability to perform these tasks without substantial modifications in the representation, training, or sampling process. In fact attempts at performing those tasks with the Group SELFIES model (GSELFIES-GPT-20 M) mostly resulted in a failure to maintain the fragment constraints. Although we were able to perform the superstructure tasks, the generated samples by the Group SELFIES model exhibit very low uniqueness (6%) and low internal diversity (0.43). Therefore, the aforementioned complexities precludes a straightforward quantitative comparison.Table 3 presents averaged validity, diversity, and uniqueness scores for 1000 molecules sampled in each fragment-constrained design task using SAFE-GPT across all drugs. It displays the average Tanimoto distance between the generated molecules to the original drug molecules, along with the average SA score (Synthetic Accessibility Score),35 which we used the RDKit library36 to generate. We observe that SAFE-GPT maintains full validity for all sampled molecules under constraints, while achieving high internal diversity and novelty compared to the original drugs. Moreover, generated molecules exhibit a low SA score, indicating their ease of synthesis. For a visual inspection of sample molecules from each task using Maribavir as the starting molecule, please refer to Table S2 (ESI section 4†).
| Task | Validity ↑ | Diversity ↑ | Uniqueness ↑ | Distance ↑ | SA score ↓ |
|---|---|---|---|---|---|
| Linker design | 1.000 ± 0.000 | 0.641 ± 0.099 | 0.887 ± 0.191 | 0.712 ± 0.097 | 3.864 ± 0.928 |
| Motif extension | 1.000 ± 0.000 | 0.681 ± 0.089 | 0.923 ± 0.179 | 0.772 ± 0.101 | 3.750 ± 0.651 |
| Scaffold decoration | 1.000 ± 0.000 | 0.571 ± 0.113 | 0.851 ± 0.162 | 0.643 ± 0.137 | 4.017 ± 0.889 |
| Scaffold morphing | 1.000 ± 0.000 | 0.608 ± 0.096 | 0.717 ± 0.219 | 0.688 ± 0.113 | 3.604 ± 0.910 |
| Superstructure | 1.000 ± 0.000 | 0.715 ± 0.059 | 0.929 ± 0.106 | 0.812 ± 0.063 | 3.868 ± 0.919 |
4.4 Goal-directed generative capabilities
To effectively apply generative approaches in live drug discovery projects, it is essential to incorporate goal-directed generation, guiding generation of novel molecules towards specific properties. Therefore, we follow established methodologies37,38 to assess the model's ability for goal-directed generation using simple molecular properties. More precisely, we optimize toward specific values for key molecular properties, including Topological Polar Surface Area (TPSA), Molecular Weight (MW), Calculated LogP (CLOGP), and Quantitative Estimation of Drug-likeness (QED). To achieve this, we use Proximal Policy Optimization (PPO)39 with Adaptive KL Penalty to train a policy for generating molecular samples with the targeted property value. A total of 50 steps was performed with a learning rate of 1 × 10−5 (AdamW optimizer) and a batch size of 100. The reward objective used for this optimization was defined as follows:where prop (mol) represents the calculated molecular property value for a given sample, target signifies the desired target value, and α is set to 0.5.
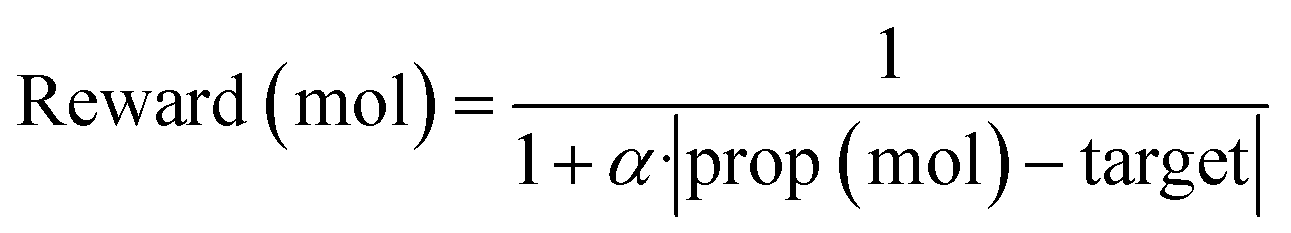
With the methodology described above, we fine-tuned agents for two target values on each molecular property and evaluated their performance by generating 500 samples from each of them. Notably, all generated samples were valid and unique. The property distribution of these samples is visually presented in Fig. 3, where the red line within each plot represents the target value of the molecular property that the agent was optimized towards, and the blue and orange histograms representing the distribution of samples from different agents with distinct goals. The results depicted in Fig. 3 demonstrate that the property distribution of the generated molecules, achieved through goal-conditioned optimization using PPO, is notably centered around the respective target values. This outcome indicates the success of our optimization process in aligning the generated molecules distribution with the desired property targets.
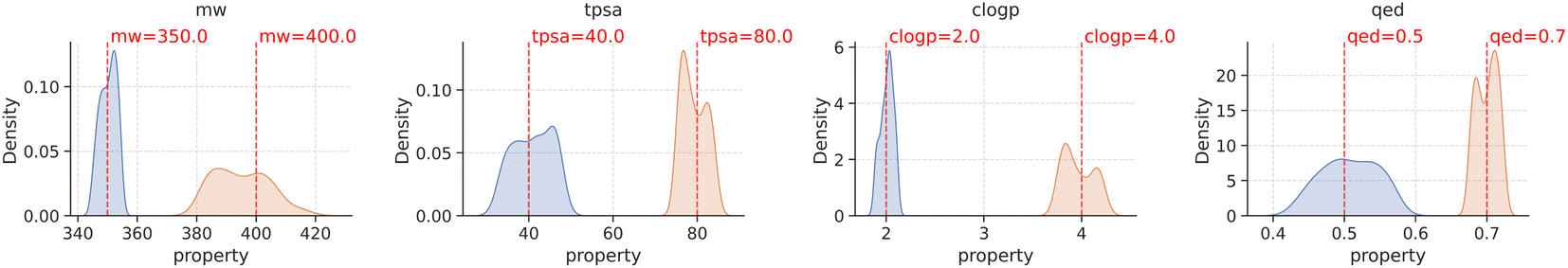 | ||
| Fig. 3 Property distributions of generated molecules, grouped by molecular properties, after goal-conditioned optimization using PPO. The red line in each plot shows the target value the agent was optimized towards. | ||
4.5 Scaffold-constrained optimization of CNS penetration of EGFR inhibitors
In this section, we introduce a novel and challenging optimization task aimed at improving the Central Nervous System (CNS) penetration of EGFR Tyrosine Kinase Inhibitors. This optimization task specifically addresses the challenge of CNS metastases in non-small cell lung cancer, a significant concern in cancer treatment.40 Our objective involves optimizing the CNS-MPO score, a comprehensive metric assessing physico-chemical properties associated with CNS penetration.41 The CNS-MPO score ranges from 0 to 6, with higher scores indicating better desirability, and a score above 4 typically suffices. We introduce additional constraints to our optimization task, requiring that all generated molecules feature a scaffold that has demonstrated activity against EGFR (see Fig. S6†). For an in-depth exploration of this topic, please consult Section 3 in the ESI.†We directly optimize the CNS-MPO score using PPO for 25 steps, and the same training parameters outlined in Section 4.4.
Fig. 4 illustrates the reward distribution obtained by sampling 100 molecules at each optimization iteration. Our findings demonstrate that scaffold-constrained optimization, even when facing challenging metrics, can be efficiently executed with SAFE-GPT using a straightforward optimization algorithm like PPO. As the CNS-MPO policy refines, we observe an expected reduction in the diversity of sampled candidates, while overall validity remains robust. Intriguingly, there's a slight decline in the SA score across iterations, suggesting the presence of synthetically favorable yet optimal compounds within the solution space.
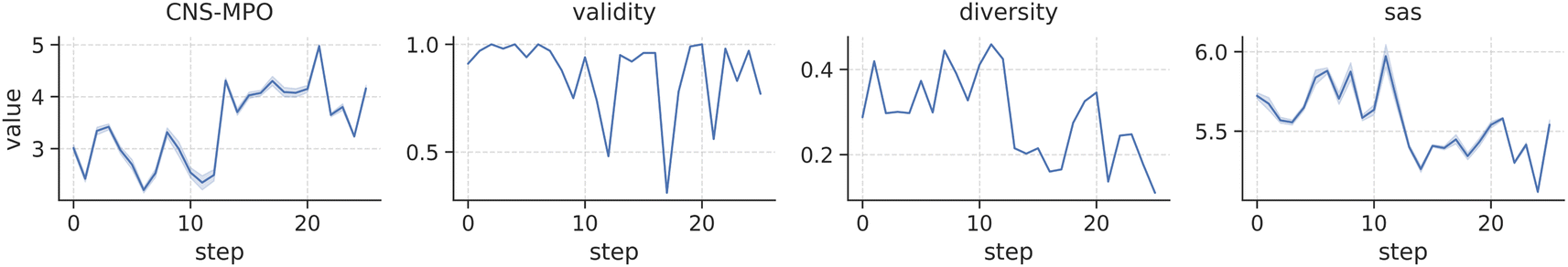 | ||
| Fig. 4 Distribution of CNS-MPO rewards and generative metrics score (validity, internal diversity and SA score) throughout the 25 optimization steps when sampling 100 molecules from the RL agent. | ||
5 Discussion
This work introduces SAFE, a novel molecular line notation that enhances versatility and expressive power in molecular design while retaining compatibility with SMILES parsers. SAFE represents molecules as sequences of interconnected fragments, offering a new paradigm in molecular description. It emerges as a promising alternative to existing molecular line notations, addressing their limitations by striking a balance between simplicity and robustness, thus making it suitable for a wide range of applications.We also present SAFE-GPT, a pioneering generative model with 87.3 million parameters, trained on 1.1 billion diverse SAFE strings. The model's effectiveness in various generative and optimization tasks highlights SAFE's unique attributes. Although we observed slightly lower molecule validity in SAFE-GPT, this can be mostly attributed to the complexity and diversity of its training set. We posit that a better sampling algorithm, potentially enforcing phrasal constraints42 around digit tokens, could address this issue.
The potential for fine-tuning SAFE-GPT on specialized chemical spaces opens avenues for enhancing its utility in targeted tasks. While this work focuses on a benchmark set of 10 drugs for fragment-constrained generation, we plan to extend this to a broader range of drugs, providing a comprehensive evaluation of the model's capabilities in various molecular generation scenarios. In future works, we aim to explore SAFE's performance in multi-property optimization (MPO) scenarios, including the integration of a prediction head into the SAFE-GPT architecture for simultaneous molecular generation and property prediction. Ultimately, we seek to efficiently scale SAFE-GPT to larger models and datasets, laying the groundwork for a new generation of foundational models in drug discovery.
Our work brings significant advancements in molecular representation and generative modeling. We believe that these innovations will continue to drive progress in drug discovery, materials science, and other fields where molecular design plays a pivotal role.
Data availability
All code, data, models, and processing scripts for this work are publicly available at https://github.com/datamol-io/safe/ and have been archived on Zenodo (https://doi.org/10.5281/zenodo.10551924). The SAFE-GPT model can also be accessed on Hugging Face (https://huggingface.co/datamol-io/safe-gpt).Author contributions
E. N. proposed the notation, conceived, and designed the study. E. N., C. G., and J. S. C. L. collected and analyzed the data, and conducted the main and comparative experiments. P. T. assisted in the study design and in analyzing the results, while M. C. helped analyze the results. All authors contributed to writing and revising the manuscript.Conflicts of interest
E. N., C. G., M. C., and P. T. are researchers employed by Valence Labs, a subsidiary of Recursion Pharmaceuticals. J. S. C. L. was an intern at Valence Labs during the study period. All research activities for this project were conducted independently of the commercial interests of Recursion Pharmaceuticals.Acknowledgements
The authors acknowledge the support provided by Valence Labs for this work.References
- D. Weininger, Smiles, a chemical language and information system. 1. introduction to methodology and encoding rules, J. Chem. Inf. Comput. Sci., 1988, 28(1), 31–36 CrossRef CAS.
- M. Krenn, F. Häse, A. K. Nigam, P. Friederich and A. Aspuru-Guzik, Self-referencing embedded strings (selfies): A 100% robust molecular string representation, J. Chem. Inf. Model., 2020, 1(4), 045024 Search PubMed.
- M. Krenn, Q. Ai, S. Barthel, N. Carson, A. Frei, N. C. Frey, P. Friederich, T. Gaudin, A. A. Gayle, K. M. Jablonka and R. F. Lameiro, Selfies and the future of molecular string representations, Patterns, 2022, 3(10), 100588 CrossRef CAS PubMed.
- A. Dalke and N. O'Boyle, Deepsmiles: An adaptation of smiles for use in machine-learning of chemical structures, ChemRxiv, 2018, preprint, DOI:10.26434/chemrxiv.7097960.v1, https://chemrxiv.org/engage/chemrxiv/article-details/60c73ed6567dfe7e5fec388d.
- A. Singh Bhadwal, K. Kumar and N. Kumar, Gensmiles: An enhanced validity conscious representation for inverse design of molecules, Knowl.-Based Systems, 2023, 268, 110429, DOI:10.1016/j.knosys.2023.110429 , https://www.sciencedirect.com/science/article/pii/S095070512300179XISSN 0950-7051..
- A. H. Cheng, A. Cai, S. Miret, G. Malkomes, M. Phielipp and A. Aspuru-Guzik, Group selfies: a robust fragment-based molecular string representation, Digital Discovery, 2023, 2, 748–758, 10.1039/D3DD00012E.
- J. Guo, F. Knuth, C. Margreitter, J. P. Janet, K. Papadopoulos, O. Engkvist and A. Patronov, Link-invent: generative linker design with reinforcement learning, Digital Discovery, 2023, 2(2), 392–408 RSC.
- V. Fialková, J. Zhao, K. Papadopoulos, O. Engkvist, E. Jannik Bjerrum, T. Kogej and A. Patronov, Libinvent: reaction-based generative scaffold decoration for in silico library design, J. Chem. Inf. Model., 2021, 62(9), 2046–2063 CrossRef PubMed.
- M. Langevin, H. Minoux, M. Levesque and M. Bianciotto, Scaffold-constrained molecular generation, J. Chem. Inf. Model., 2020, 60(12), 5637–5646, DOI:10.1021/acs.jcim.0c01015 , PMID: 33301333..
- Z. Liao, L. Xie, H. Mamitsuka and S. Zhu, Sc2mol: a scaffold-based two-step molecule generator with variational autoencoder and transformer, Bioinformatics, 2023, 39(1), btac814 CrossRef CAS PubMed.
- J. Arús-Pous, A. Patronov, E. Jannik Bjerrum, C. Tyrchan, J.-L. Reymond, H. Chen and O. Engkvist, Smiles-based deep generative scaffold decorator for de-novo drug design, J. Cheminf., 2020, 12(1), 1–18 Search PubMed.
- L. David, A. Thakkar, R. Mercado and O. Engkvist, Molecular representations in ai-driven drug discovery: a review and practical guide, J. Cheminf., 2020, 12(1), 1–22 Search PubMed.
- C. Bilodeau, W. Jin, T. Jaakkola, R. Barzilay and K. F. Jensen, Generative models for molecular discovery: Recent advances and challenges, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12(5), e1608, DOI:10.1002/wcms.1608 . https://wires.onlinelibrary.wiley.com/doi/abs/10.1002/wcms.1608.
- Y. Du, T. Fu, J. Sun and S. Liu, Molgensurvey: A systematic survey in machine learning models for molecule design, 2022 Search PubMed.
- R. Gómez-Bombarelli, J. N. Wei, D. Duvenaud, J. M. Hernández-Lobato, B. Sánchez-Lengeling, D. Sheberla, J. Aguilera-Iparraguirre, T. D. Hirzel, R. P. Adams and A. Aspuru-Guzik, Automatic chemical design using a data-driven continuous representation of molecules, ACS Cent. Sci., 2018, 4(2), 268–276 CrossRef PubMed.
- J. He, E. Nittinger, C. Tyrchan, W. Czechtizky, A. Patronov, E. J. Bjerrum and O. Engkvist, Transformer-based molecular optimization beyond matched molecular pairs, J. Cheminf., 2022, 14(1), 18 CAS.
- L. Yang, G. Yang, Z. Bing, T. Yuan, Y. Niu, L. Huang and L. Yang, Transformer-based generative model accelerating the development of novel braf inhibitors, ACS Omega, 2021, 6(49), 33864–33873 CrossRef CAS PubMed.
- V. Bagal, R. Aggarwal, P. K. Vinod and U. D. Priyakumar, Molgpt: molecular generation using a transformer-decoder model, J. Chem. Inf. Model., 2021, 62(9), 2064–2076 CrossRef PubMed.
- W. Jin, R. Barzilay and T. Jaakkola, Junction tree variational autoencoder for molecular graph generation, in Proceedings of the 35th International Conference on Machine Learning, volume 80 of Proceedings of Machine Learning Research, ed. J. Dy and A. Krause, PMLR, 2018, pp. 2323–2332, https://proceedings.mlr.press/v80/jin18a.html Search PubMed.
- W. Jin, R. Barzilay and T. Jaakkola, Multi-objective molecule generation using interpretable substructures, in International conference on machine learning, PMLR, 2020, pp. 4849–4859 Search PubMed.
- K. Maziarz, H. Jackson-Flux, P. Cameron, F. Sirockin, N. Schneider, N. Stiefl, M. Segler, and M. Brockschmidt. Learning to extend molecular scaffolds with structural motifs, arXiv, 2021, preprint, arXiv:2103.03864, DOI:10.48550/arXiv.2103.03864.
- Y. Li, L. Zhang and Z. Liu, Multi-objective de novo drug design with conditional graph generative model, J. Cheminf., 2018, 10, 1–24 Search PubMed.
- J. Wang, C.-Y. Hsieh, M. Wang, X. Wang, Z. Wu, D. Jiang, B. Liao, X. Zhang, B. Yang and Q. He, et al., Multi-constraint molecular generation based on conditional transformer, knowledge distillation and reinforcement learning, Nat. Mach. Intell., 2021, 3(10), 914–922 CrossRef.
- J. Lim, S.-Y. Hwang, S. Moon, S. Kim and W. Y. Kim, Scaffold-based molecular design with a graph generative model, Chem. Sci., 2020, 11(4), 1153–1164, 10.1039/c9sc04503a.
- Y. Li, O. Vinyals, C. Dyer, R. Pascanu and P. Battaglia, Learning deep generative models of graphs, 2018 Search PubMed.
- J. Degen, C. Wegscheid-Gerlach, A. Zaliani and M. Rarey, On the art of compiling and using’drug-like’chemical fragment spaces, ChemMedChem, 2008, 3(10), 1503–1507 CrossRef CAS PubMed.
- J. Hussain and C. Rea, Computationally efficient algorithm to identify matched molecular pairs (mmps) in large data sets, J. Chem. Inf. Model., 2010, 50(3), 339–348 CrossRef CAS PubMed.
- X. Q. Lewell, D. B. Judd, S. P. Watson and M. M. Hann, Recap retrosynthetic combinatorial analysis procedure: a powerful new technique for identifying privileged molecular fragments with useful applications in combinatorial chemistry, J. Chem. Inf. Comput. Sci., 1998, 38(3), 511–522 CrossRef CAS PubMed.
- J. J. Irwin and B. K. Shoichet, Zinc- a free database of commercially available compounds for virtual screening, J. Chem. Inf. Model., 2005, 45(1), 177–182 CrossRef CAS PubMed.
- J. Chambers, M. Davies, A. Gaulton, A. Hersey, S. Velankar, R. Petryszak, J. Hastings, L. Bellis, S. McGlinchey and J. P. Overington, Unichem: a unified chemical structure cross-referencing and identifier tracking system, J. Cheminf., 2013, 5(1), 3 CAS.
- P. Schwaller, T. Laino, T. Gaudin, P. Bolgar, C. A. Hunter, C. Bekas and A. A. Lee, Molecular transformer: a model for uncertainty-calibrated chemical reaction prediction, ACS Cent. Sci., 2019, 5(9), 1572–1583 CrossRef CAS PubMed.
- D. P. Kingma and J. Ba, Adam: A method for stochastic optimization, arXiv, 2014, preprint, arXiv:1412.6980, DOI:10.48550/arXiv.1412.6980.
- D. Polykovskiy, A. Zhebrak, B. Sanchez-Lengeling, S. Golovanov, O. Tatanov, S. Belyaev, R. Kurbanov, A. Artamonov, V. Aladinskiy, M. Veselov, A. Kadurin, S. Johansson, H. Chen, S. Nikolenko, A. Aspuru-Guzik and A. Zhavoronkov, Molecular sets (moses): A benchmarking platform for molecular generation models, Front. Pharmacol., 2020, 11, 565644 CrossRef CAS PubMed.
- K. Huang, T. Fu, W. Gao, Y. Zhao, Y. Roohani, J. Leskovec, C. W. Coley, C. Xiao, J. Sun and M. Zitnik, Therapeutics data commons: Machine learning datasets and tasks for drug discovery and development, Proceedings of Neural Information Processing Systems, NeurIPS Datasets and Benchmarks, 2021 Search PubMed.
- P. Ertl and A. Schuffenhauer, Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions, J. Cheminf., 2009, 1, 1–11 Search PubMed.
- G. Landrum, P. Tosco, B. Kelley, Ric, D. Cosgrove, sriniker, gedeck, R. Vianello, N. Schneider, E. Kawashima, G. Jones, N. Dan, A. Dalke, B. Cole, M. Swain, S. Turk, A. V. AlexanderSavelyev, M. Wójcikowski, I. Take, D. Probst, K. Ujihara, V. F. Scalfani, G. Godin, R. Walker, J. Lehtivarjo, A. Pahl, F. Berenger, Jasondbiggs and strets123, rdkit/rdkit: 2023_09_2 (q3 2023) release, 2023, DOI:10.5281/zenodo.10099869.
- J. Lim, S.-Y. Hwang, S. Moon, S. Kim and W. Y. Kim, Scaffold-based molecular design with a graph generative model, Chem. Sci., 2020, 11(4), 1153–1164 RSC.
- S. Seo, J. Lim and W. Y. Kim, Molecular generative model via retrosynthetically prepared chemical building block assembly, Adv. Sci., 2023, 10(8), 2206674 CAS.
- J. Schulman, F. Wolski, P. Dhariwal, A. Radford, and O. Klimov. Proximal policy optimization algorithms, arXiv, 2017, preprint, arXiv:1707.06347, DOI:10.48550/arXiv.1707.06347.
- M. S. Ahluwalia, K. Becker and B. P. Levy, Epidermal growth factor receptor tyrosine kinase inhibitors for central nervous system metastases from non-small cell lung cancer, Oncologist, 2018, 23(10), 1199–1209 CrossRef CAS PubMed.
- T. T. Wager, X. Hou, P. R. Verhoest and A. Villalobos, Central nervous system multiparameter optimization desirability: application in drug discovery, ACS Chem. Neurosci., 2016, 7(6), 767–775 CrossRef CAS PubMed.
- P. Matt and D. Vilar. Fast lexically constrained decoding with dynamic beam allocation for neural machine translation, arXiv, 2018, preprint, arXiv:1804.06609, DOI:10.18653/v1/N18-1119.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4dd00019f |
| ‡ Code, data and model available at https://github.com/datamol-io/safe/ |
| This journal is © The Royal Society of Chemistry 2024 |