
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Machine learning of stability scores from kinetic data†
Veerupaksh
Singla
 ,
Qiyuan
Zhao
and
Brett M.
Savoie
*
,
Qiyuan
Zhao
and
Brett M.
Savoie
*
Davidson School of Chemical Engineering, Purdue University, West Lafayette, IN 47906, USA. E-mail: bsavoie@purdue.edu
First published on 1st July 2024
Abstract
The absence of computational methods to predict stressor-specific degradation susceptibilities represents a significant and costly challenge to the introduction of new materials into applications. Here, a machine-learning framework is developed that predicts stressor-specific stability scores from computationally generated reaction data. The thermal degradation of alkanes was studied as an exemplary system to demonstrate the approach. The half-lives of ∼32k alkanes were simulated under pyrolysis conditions using 59 model reactions. Using a hinge-loss function, these half-life data were used to train machine learning models to predict a scalar representing the relative stability based only on the molecular graph. These models were successful in transferability case studies using distinct training and testing splits to recapitulate known stability trends with respect to the degree of branching and alkane size. Even the simplest models showed excellent performance in these case studies, demonstrating the relative ease with which thermal stability can be learned. The stability score is also shown to be useful in a design study, where it is used as part of the objective function of a genetic algorithm to guide the search for more stable species. This work provides a framework for converting kinetic reaction data into stability scores that provide actionable design information and opens avenues for exploring more complex chemistries and stressors.
1 Introduction
The design of materials with targeted molecular properties, also called inverse molecular design, has been an enduring challenge in chemistry and materials science.1–5 Several recent developments, including advances in computational resources and simulation throughput, the availability of large datasets amenable to machine learning (ML),6–10 and the proliferation of ML architectures for predicting various material properties,11–16 have broadened the range of properties that are amenable to computational design.17–21 Nevertheless, a major capability gap still exists in predicting the stability of prospective materials with respect to specific stressors (e.g., thermal, chemical, voltaic, radiative, etc.) that might be encountered during use. It would represent a qualitative advance if the stability, degradation, and aging behaviors of prospective chemicals and materials could be directly predicted during the design phase without experimental testing.Despite progress in developing methods to simulate or empirically predict other functional properties, contemporary stability characterization is overwhelmingly done experimentally via empirical make-and-break testing. Although this can reliably characterize a particular material, it is costly in terms of time and material and crucially only delivers information at the end of the design process. Even when such testing is done, generating mechanistic information requires additional analytical monitoring such as differential scanning calorimetry, thermogravimetric analysis, spectroscopy (XRD, XPS, IR, UV-Vis, and NMR), mass spectrometry, cyclic voltammetry, or high-pressure liquid chromatography, amongst others.22–27 To the extent that stability properties are considered during design, it is typically done only indirectly by limiting the scope of materials to established chemistries with known stability or by accounting for a small number of acute susceptibilities that are domain specific.16,21,27–29 Computational contributions at the design stage are primarily through thermodynamic energy estimates. Although this is a reasonable starting point, it provides no information about the kinetics and susceptibility of a material to degradation via specific stressors.30–34 Famously, diamond is thermodynamically unstable relative to graphite, but its thermal decomposition is not kinetically favorable except at temperatures higher than 2000 K.35
Recent advances in reaction prediction create the possibility that first-principles reaction data might provide a basis for estimating the degradation kinetics of prospective chemistries.36,37 Automatic reaction network exploration and transition state characterization algorithms have become more efficient and accurate.38–41 Although such characterization techniques still remain too expensive to be used in high-throughput applications for virtual screening and comparative down selection of prospective materials, they can potentially serve as the basis for generating kinetic data relevant to broad classes of degradation reactions. Critically, material stability is mainly determined by the kinetics of the first irreversible degradation reaction and thus does not necessarily require a full elaboration of a many-step degradation network. Thus, although data scarcity is no longer a fundamental obstacle, the challenge remains in converting this information into a form that generalizes to similar chemistries to avoid the direct simulation of every new material.
Here, we address the absence of inexpensive kinetic measurements of chemical stability by investigating whether ML can be used to turn simulation based half-life data into a scalar stressor-specific stability score (Fig. 1). The thermal stability of alkanes is taken as an exemplary problem and several model architectures are tested to learn a scalar “stability score” that allows for the comparison of the relative thermal stability of alkanes. Although one of the simplest organic classes, the resulting scores compress information from theoretical reaction networks spanning billions of reactions. Remarkably, the relative stability is shown to be easily learned by all ML architectures, suggesting the feasibility of greatly generalizing this approach to other stressors and broader chemical classes. The utility of the derived stability score for materials design is finally demonstrated by performing a case study using a genetic algorithm to discover thermally stable isomers.
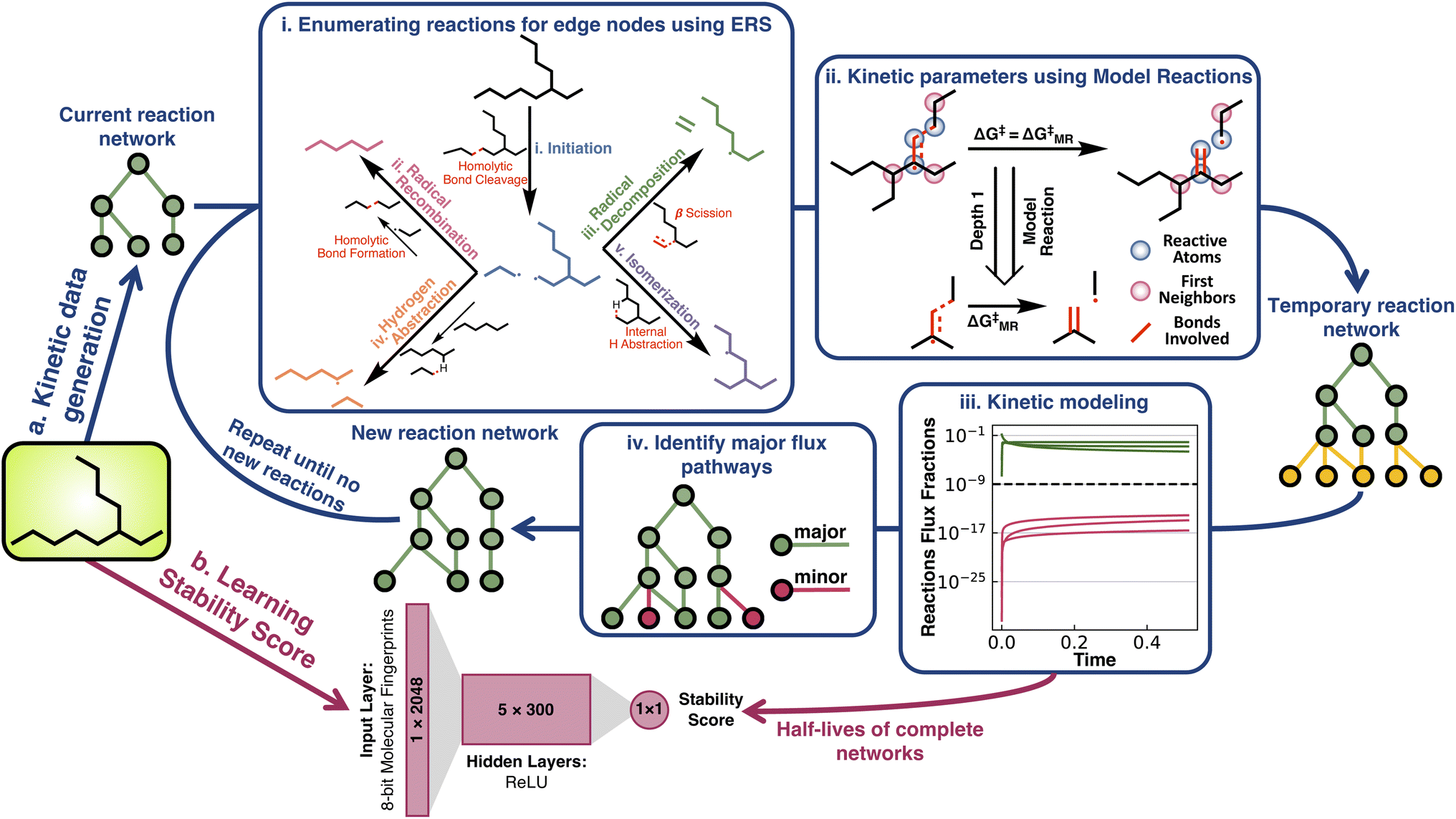 | ||
| Fig. 1 Overview of the methodology for generating material stability scores. (a) Half-life data generation using direct network simulations. (i) The pyrolysis reaction network for each species is expanded using five types of elementary reactions. (ii) Kinetic parameters for all reactions are obtained from transition state simulations. (iii) Kinetic modeling is used to determine if the network is converged with respect to species half-life. (iv) Channels with negligible flux are discarded and the network is expanded until converging with respect to the starting material half-life. (b) MLP architecture used to predict stability scores. After learning the stability score, the expensive top branch can potentially be circumvented to cheaply assess the relative stability of new materials. | ||
2 Results and discussion
2.1 Data generation
Half-life under pyrolysis conditions (700 K and 1 atm) was simulated for all 32![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 421 structural isomers of alkanes with ≤17 carbon atoms (Fig. 1a). Generating these data consisted of assembling an effective reaction network for each species (Fig. 1a(i)), calculating activation energies for the reactions in these networks (Fig. 1a(ii)), and simulating the network kinetics to obtain a half-life for each alkane (Fig. 1a(iii)). See ESI Sections 1.1, 1.2, and 1.3† for complete details on data generation.
421 structural isomers of alkanes with ≤17 carbon atoms (Fig. 1a). Generating these data consisted of assembling an effective reaction network for each species (Fig. 1a(i)), calculating activation energies for the reactions in these networks (Fig. 1a(ii)), and simulating the network kinetics to obtain a half-life for each alkane (Fig. 1a(iii)). See ESI Sections 1.1, 1.2, and 1.3† for complete details on data generation.
The reaction networks for low temperature (i.e., ≤700 K) alkane pyrolysis consist of five elementary reaction steps (Fig. 1a(i)).42 The initiation reaction involves homolytic C–C bond cleavage, which produces two alkyl radicals (C–H bond cleavage is not preferred at such temperatures). Once an alkyl radical has been generated, it can undergo three types of reactions: one bimolecular reaction, hydrogen abstraction (used interchangeably with H-abstraction in the text), and two unimolecular reactions, isomerization and radical decomposition. The hydrogen abstraction reaction occurs between an alkane and an alkyl radical, where an intermolecular hydrogen radical transfer shifts the alkane and alkyl. In contrast, in the isomerization reaction, an intramolecular hydrogen radical transfer within the alkyl radical generates isomeric alkyl radicals. The transition states of isomerization reactions have ring-like structures. Based on whether the hydrogen is abstracted from the 4th, 5th, 6th, or the 7th nearest neighboring carbon to the radical carbon, isomerization reactions have been named 1–4, 1–5, 1–6, or 1–7 isomerization (or iso, used interchangeably through the text). Alkyl radicals can also decompose into alkenes and smaller alkyl radicals through β scission, which is referred to as the radical decomposition reaction. As a termination step, two alkyl radicals can recombine to form stable alkanes via radical recombination. To prevent unlimited network expansion, only radical recombination reactions that produce alkanes with an equal or fewer number of carbon atoms than the starting alkane are considered in the pyrolysis network generation process. Species-specific activation energies for these reactions were calculated from a tractable number of model reactions (MRs) generated from a graph-truncation approach. The reaction graph truncation consists of conserving the reacting atoms and their first bonded neighboring atoms to define the reaction neighborhood. Applying this truncation to all acyclic alkane pyrolysis reactions results in 59 unique MRs. The activation barriers of these MRs were then used in place of larger reactions of the same class whenever they occur in the network (Fig. 1(ii) and ESI Section 1.2 for further details†). The median free energy errors associated with the model reaction approximation were found to be within 3 kcal mol−1 for the 55 model reactions that were benchmarked.
A pruning procedure was used to assemble effective reaction networks and determine accurate half-lives for each alkane (Fig. 1a). The size of the untruncated pyrolysis network grows exponentially with respect to size, making the direct simulation out to terminal products impractical. For example, the reaction network of n-C15H32 is composed of over a billion reactions. However, the downstream reactions from the parent alkane have little impact on the half-life, which allows these networks to be pruned depth-wise based on their simulated flux, while still accurately calculating the half-life (see ESI Section 1.3†). The network pruning approach was benchmarked against complete reaction networks for all structural isomers of alkanes from ethane to decane (i.e., the 149 systems for which complete simulations out to terminal products were practical). Using a minimum relative flux threshold of 10−9, a robust fit was observed (Fig. 2a) with a reduction of up to 108 times in terms of the number of unique reactions required to include in the reaction network, while still obtaining an accurate half-life (Fig. 2b). The growth of unique reactions vs. alkane length also drops to a linear trend compared to the exponential trend for complete networks. The pruned reaction networks exhibit the largest reduction in H-abstraction reactions among the different mechanisms in the full pyrolysis networks (Fig. S2b†).
 | ||
| Fig. 2 Summary of kinetic data generation results. (a) Comparison between the pruned and full network half-lives for all alkane isomers between C2H6 and C10H22 using a maximum relative flux threshold of 10−9. (b) A comparison of the number of unique reactants/reactant pairs between the full (estimated) and kinetically pruned n-alkane reaction networks as a function of n-alkane length. The y-axis is plotted on a log10 scale. | ||
The half-lives of 32421 acyclic alkanes, i.e., all structural isomers up to C17H36, were simulated with Cantera,43 based on their depth-wise pruned networks (see ESI Section 1.3†). Although the absolute accuracy of these half-lives is not at issue in the current study, it is still necessary to establish that qualitatively correct stability trends are reflected in the data for later testing of whether various ML architectures can generalize the implicit stability trends. Experimental kinetic data compiled by Sundaram and Froment for the pyrolysis of ethane, propane, n-butane, and isobutane provide several points of comparison (Fig. 3).44 Additionally, these experimental data and pyrolysis mechanisms were consistent across the literature.45,46 These data were used to replace the quantum chemistry based reaction data in the pyrolysis networks of ethane, propane, n-butane, and isobutane (ESI Section 3†) to resimulate the half-life for comparison with the purely computational results (Fig. 3a). For simple alkanes, both the experimental and simulated half-lives recapitulate established trends that increasing the alkane length (comparing ethane, propane, and n-butane) and branching (comparing n-butane and isobutane) reduce half-lives. Comparisons to experimental kinetic data for more complex alkanes were, however, not possible in the absence of systematic experimental literature. Additionally, sensitivity analysis was performed on both the experimental and computational kinetic data with 1000 simulations done for each reaction network, where random uncertainties from a normal distribution centered at zero and a standard deviation of 3 kcal mol−1 were introduced in all activation barriers. The overlapping shaded region in Fig. 3a implies that the experimental and computational half-lives are within this uncertainty range.
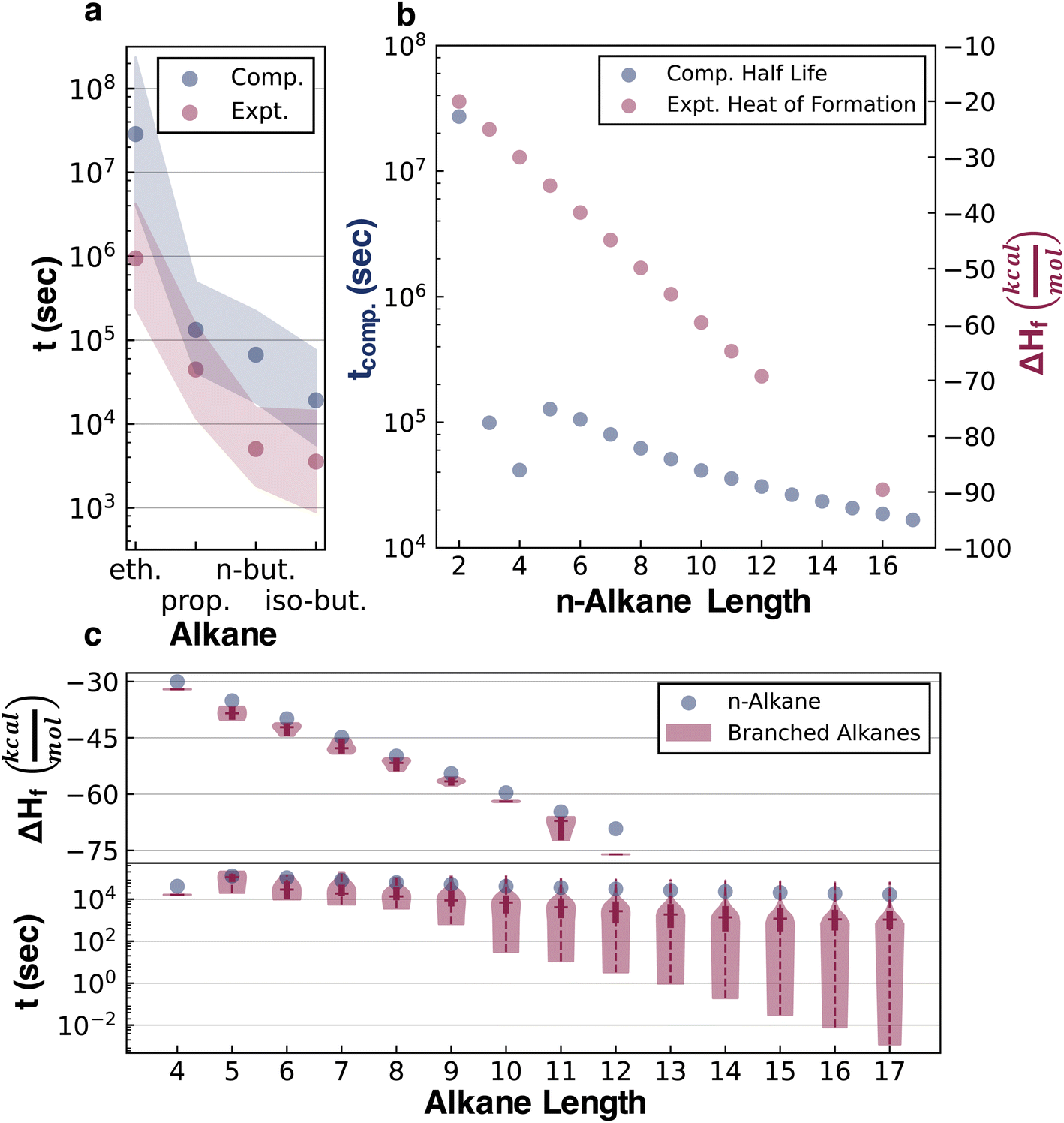 | ||
| Fig. 3 Comparison of computational half-life data with experimental half-life and heat of formation data. For all panels, y-axes with half-lives are on a log10 scale, and the alkane length refers to the number of heavy atoms. (a) Experimental and computational half-lives of ethane, propane, n-butane, and isobutane. The scatter points are the median values from the sensitivity analysis and the shaded regions span the interquartile ranges. (b) Computational half-lives and experimental heats of formation at 298 K of n-alkanes. (c) Comparing n-alkane and branched alkane values: experimental heats of formation at 298 K (upper) and computational pyrolysis half-lives (lower). The horizontal line in the violin plots is the median and the bold vertical line is the interquartile range. | ||
Comparing the half-life data with the experimental heats of formation at 298 K (ΔHf), a frequently used stability surrogate, illustrates the qualitative error of neglecting kinetics in assessing stability. A larger exothermic ΔHf corresponds to higher thermodynamic stability, and a lower half-life corresponds to lower kinetic stability. The thermodynamic data compiled by Pedley, Naylor, and Klein (PNK data)47 show that thermodynamic stability increases with the alkane length, while kinetic stability decreases (Fig. 3b). The thermodynamic trend also misses the half-life discontinuity between n-butane and n-pentane. Comparing ΔHf and the half-life of linear (n-alkanes) and branched alkanes reveals that the thermodynamic trend also qualitatively misrepresents the effect of branching (Fig. 2c). While branching marginally increases thermodynamic stability, it decreases kinetic stability and induces a broad range of half-lives depending on the position and degree of branching. Hence, ΔHf lacks the information required to predict degradation kinetics and gets structure-stability trends qualitatively wrong.
2.2 Model testing
Two neural network models were trained to predict the thermal stability score, a scalar representing the relative rank order of alkane half-lives, using a modified hinge loss function that converts a regression problem to a pairwise classification problem. One model was a simple fully connected multilayer perceptron (labeled MLP in figures and further discussion) trained using 8 bit 2048-length Morgan fingerprints of radius two as inputs with relative stability as a target (the architecture is shown in Fig. 1b). The second architecture was Chemprop,48 a message-passing graph network with stronger input featurization developed for molecular property prediction (full details on model training are provided in ESI Section 1.4†). It is important to clarify here that while the fingerprint-based representation is more prone to saturation for larger molecules compared to the more complex graph-based message-passing representation, stability is strongly influenced by local variability in addition to global variability. This remains true for the alkane studies here as well.Pairwise accuracy was used as a metric to quantify and compare the model predictions. The training and the testing datasets were converted to unique molecule pairs and the pairwise stability trends of the ground truth kinetic data were compared with the pairwise stability trends predicted by models. The accuracy was then measured as the percent of pairs where the model predicts the same stability trend as the ground truth. Several training and testing splits were generated to test the transferability of the stability score under different scenarios (Fig. 4). The consistent accuracy of >90%, even for the relatively simple MLP architecture, illustrates the relative ease of the learning task.
 | ||
| Fig. 4 Stability score performance. (a) Bar plots comparing the performance of the Chemprop and MLP models across the case studies described in the main text. The ratios above the bars represent the train-test split. Accuracy is the percentage of pairwise accuracy (i.e., the percentage of pair-wise ordering relationships based on the half-life that are recapitulated by the stability score). (b) n-alkane stability score prediction comparison between the MLP and Chemprop models. | ||
Using a random training and testing split, the elementary MLP and message-passing Chemprop architectures show comparable performance (Fig. 4a(i)). Although random splitting ensures that the testing set is structurally independent of the training data, the diversity of the training data is sufficient to guarantee that the branching and size distributions of the training and testing data obtained from the random splitting are similar. To carry out a more rigorous test of transferability, four other case studies were performed using distinct train and test splits. In the case study definitions, the backbone refers to the longest chain in the alkane structure. Core branches refer to the number of branches originating from the backbone, whereas total branches refer to all branches and sub-branches from the backbone. Total branches ≤6 put alkanes with at most six total branches in the training set and the rest in the testing set, resulting in a 90:10 train:test split (Fig. 4a(ii)). Core branches ≤4 refer to alkanes with at most four core branches being included in the training set, resulting in a 61:39 train:test split (Fig. 4a(iii)). Backbone ≤10 includes alkanes with a backbone at most ten carbons long as the training set and the rest as the testing set, resulting in a 68:32 train:test split (Fig. 4a(iv)). Length ≤ 10 puts alkanes with at most 16 carbons in the training set and the C17 alkanes in the testing set, resulting in a 47:53 train:test split (Fig. 4a(v)).
The >98% accuracy on splits (iv) and (v) demonstrates the ability of the models to extrapolate to longer alkanes. The >90% accuracy on splits (ii) and (iii) shows that while not as good as extrapolating on length, the models perform reasonably well when extrapolating to highly branched alkanes. Chemprop performs better than the Morgan fingerprint-based MLP model in most of the cases, which is reasonable given that a message-passing architecture is more complex than the MLP fingerprints. However, the fact that the MLP model surpasses 90% accuracy in all the cases, is within 1% of Chemprop accuracy in two instances, and even surpasses Chemprop in one case illustrates that predicting relative stability is a reasonably simple learning problem. The learning curves for all models across all the split types are present in ESI Section 4.†
We also compared the n-alkane stability scores predicted by both the models in Fig. 4b to test their ability to recapitulate the jump in half-life observed between n-butane and n-pentane under pyrolysis conditions. Although the absolute values of the two models cannot be compared, they both predict the stability jump between n-butane and n-pentane (Fig. 3b). This jump, which was unseen during training, demonstrates that the models are learning the relative stability associated with different chemical features rather than just memorizing the training data. These results lend credence to our initial assumption that given consistently generated kinetic data for the stressor-specific degradation reactions, it is possible to learn the inherent relative kinetics, which opens up the avenue to apply this strategy to more complex systems.
2.3 Designing more thermally stable structures
To illustrate a typical usage of the extrapolative ability of the models in material design applications, we implemented a genetic algorithm to discover stable structures using the scores of the MLP and Chemprop models as the corresponding fitness functions. To make the task more challenging, we did not use the models trained on the full dataset; instead, we used the models trained on split (v), alkanes with length ≤16, to design the most stable C17H36 molecule.Three types of operations were applied as part of the genetic search for stable structures: growth, deletion, and insertion of methyl groups (Fig. 5a). Crossovers were not considered because for alkanes they are essentially the same as a couple of generations of mutations. Growth involves adding a methyl group in place of a hydrogen on the alkane, deletion involves removing a primary or secondary carbon atom from the alkane and then generating a smaller alkane by maintaining hybridization using the removal or addition of hydrogens, and insertion involves adding carbon between a C–C bond and balancing the charge with hydrogens.
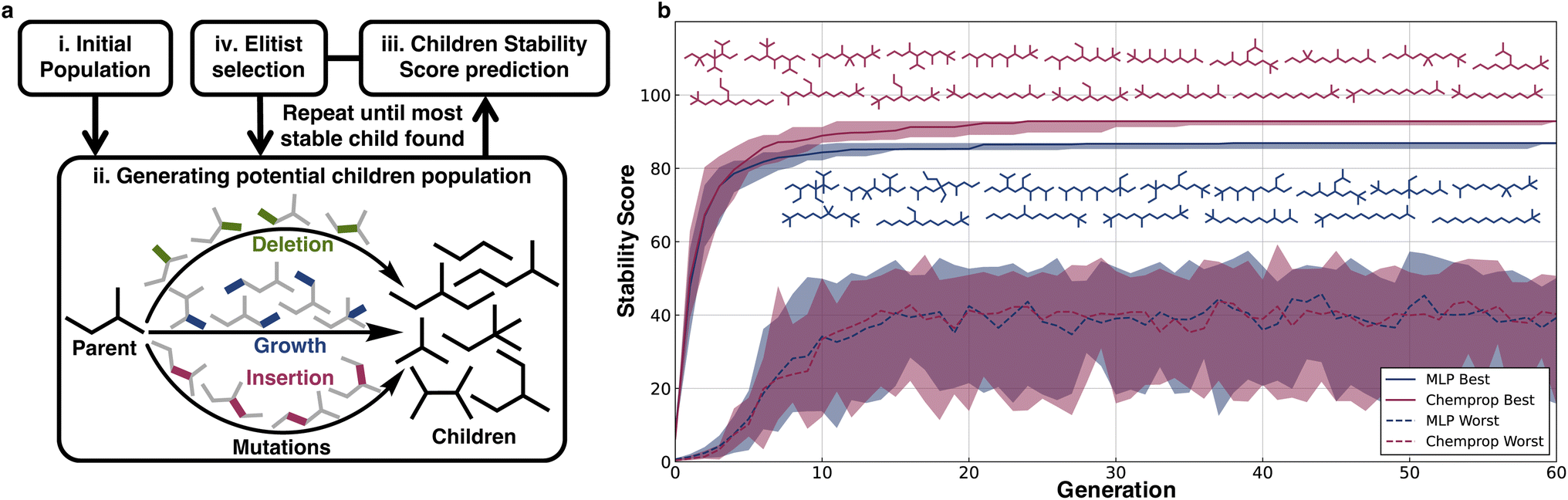 | ||
| Fig. 5 Stability scores can be used in molecular discovery workflows. (a) Genetic algorithm methodology using the stability score as the objective function. (b) Plot of the median highest and lowest stability scores of structures in the Chemprop and MLP populations across 20 runs. The shaded areas are from the zeroth to the hundredth percentile. The alkane structures (read left to right, top to bottom) represent the unique most stable hundredth percentile structures. | ||
The stability scores were used to guide genetic searches to find the most stable C17H36 isomer out of the 24894 possible structures. The genetic searches were initialized with 100 randomly selected C17H36 alkanes selected from the least stable 5000 C17H36 structures, and every generation maintained a population of 100 with the constraint that all molecules in every generation should have 17 C atoms. Elitist selection was used, wherein 20% of the most stable predicted molecules from each generation were propagated to the next generation, and the 50% most stable predicted molecules were used as parents for further mutations to generate children. The remaining 80 molecules to be propagated to the next generation were randomly selected from this group of children using probabilities weighted by the predicted stability scores.
For both the models (MLP and Chemprop), 20 independent genetic runs were conducted and from each run the most stable predicted molecules from every generation were obtained. The median stability of the generated molecules predicted to be the most stable was plotted along with the zeroth and fourth quartile stability scores. The limited variation illustrates that irrespective of starting alkanes, all runs quickly arrive at similar structures (Fig. 5b). Within 30 generations, both models converge with respect to discovering alkanes that maximize their respective scores. The MLP and Chemprop models both show a general trend of predicting less-branched alkanes to be more stable. The searches also consistently converge at late stages on species capped with tert-butyl and isopropyl moieties over species capped with unsubstituted CH3 units. Indeed, a comparison of stability scores for linear alkanes of a given C-value with and without tert-butyl capping shows that those with this termination are slightly preferred as termini, although non-terminal branches in general are disfavored. There are insufficient experimental data from the pyrolysis literature to validate this directly, but it stands as a prediction of the case study.
3 Conclusion
Inexpensive computational methods for predicting stability are scarce despite the central role of degradation susceptibilities in the material translation process. Developments in automated reaction prediction provide access to diverse reaction data that are relevant to predicting such stability behaviors but nevertheless need to be generalized across conserved chemical structures. The current work shows how computationally generated kinetic data can be generalized into scalar stability scores using relatively simple machine learning architectures.There are several avenues for further improving this work. The current work focuses on the thermal stability of alkanes as an entry point for a broader range of degradation susceptibilities that are relevant to materials development. We found that both machine learning architectures were easily able to learn the thermal stability structure–function relationships such that the stability scores could be reliably used in design tasks. Nevertheless, the generalization to other chemistries and stressors is both obvious and necessary. The ease with which the present relationships were learned suggests that the broad data generation performed here so that the scaffolded training:testing case studies could be performed will not be necessary when considering other chemistries and stressors. As such, we do not anticipate fundamental data generation obstacles to extending this approach to redox stability (e.g., by considering reactions involving anions and cations of neutral parents) or other chemistries beyond alkanes. We further showed that the kinetic data contain material stability information and that this relative information can be learned by neural networks and even be extrapolated to unseen data for material design. This means that if properly defined, kinetics can be learned in a way useful to computational material design and virtual screening.
Data availability
The authors declare that the data supporting the findings of this study are available within the paper and its ESI files.† The version of YARP (v2.0) and the reaction conformational sampling package used in this study are available through GitHub under the GNU GPL-3.0 License [https://github.com/Savoie-Research-Group/yarp]. Pretrained models for the stability score and training scripts are available on GitHub [https://github.com/Savoie-Research-Group/papers/tree/main/231101-Stability_Scores].Author contributions
V. S. and B. M. S. conceived and designed the study. V. S. generated and analyzed the data and wrote the paper. Q. Z. provided the model reaction data. B. M. S. oversaw the project and wrote the paper.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The work performed by V. S., Q. Z., and B. M. S. was made possible by the Office of Naval Research (ONR) through support provided by the Energetic Materials Program (MURI grant number: N00014-21-1-2476; Program Manager: Dr Chad Stoltz). B. M. S. also acknowledges partial support for this work from the Purdue Process Safety and Assurance Center.References
- A. Cook, A. P. Johnson, J. Law, M. Mirzazadeh, O. Ravitz and A. Simon, Computer-Aided Synthesis Design: 40 Years On, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 79–107, DOI:10.1002/wcms.61.
- R. Gani, B. Nielsen and A. Fredenslund, A Group Contribution Approach to Computer-Aided Molecular Design, AIChE J., 1991, 37, 1318–1332, DOI:10.1002/aic.690370905.
- C. Kuhn and D. N. Beratan, Inverse Strategies for Molecular Design, J. Phys. Chem., 1996, 100, 10595–10599, DOI:10.1021/jp960518i.
- R. Vaidyanathan and M. El-Halwagi, Computer-Aided Synthesis of Polymers and Blends with Target Properties, Ind. Eng. Chem. Res., 1996, 35, 627–634, DOI:10.1021/ie950072c.
- V. Venkatasubramanian, K. Chan and J. M. Caruthers, Computer-Aided Molecular Design Using Genetic Algorithms, Comput. Chem. Eng., 1994, 18, 833–844, DOI:10.1016/0098-1354(93)E0023-3.
- D. Lowe, Chemical reactions from US patents (1976-Sep2016), figshare, 2017, DOI:10.6084/m9.figshare.5104873.v1.
- J. J. Irwin, K. G. Tang, J. Young, C. Dandarchuluun, B. R. Wong, M. Khurelbaatar, Y. S. Moroz, J. Mayfield and R. A. Sayle, ZINC20—A Free Ultralarge-Scale Chemical Database for Ligand Discovery, J. Chem. Inf. Model., 2020, 60, 6065–6073, DOI:10.1021/acs.jcim.0c00675.
- S. M. Kearnes, M. R. Maser, M. Wleklinski, A. Kast, A. G. Doyle, S. D. Dreher, J. M. Hawkins, K. F. Jensen and C. W. Coley, The Open Reaction Database, J. Am. Chem. Soc., 2021, 143, 18820–18826, DOI:10.1021/jacs.1c09820.
- A. Zakutayev, N. Wunder, M. Schwarting, J. D. Perkins, R. White, K. Munch, W. Tumas and C. Phillips, An Open Experimental Database for Exploring Inorganic Materials, Sci. Data, 2018, 5, 180053, DOI:10.1038/sdata.2018.53.
- K. T. Butler, D. W. Davies, H. Cartwright, O. Isayev and A. Walsh, Machine Learning for Molecular and Materials Science, Nature, 2018, 559, 547–555, DOI:10.1038/s41586-018-0337-2.
- A. C. Mater and M. L. Coote, Deep Learning in Chemistry, J. Chem. Inf. Model., 2019, 59, 2545–2559, DOI:10.1021/acs.jcim.9b00266.
- P. Schwaller, T. Laino, T. Gaudin, P. Bolgar, C. A. Hunter, C. Bekas and A. A. Lee, Molecular Transformer: A Model for Uncertainty-Calibrated Chemical Reaction Prediction, ACS Cent. Sci., 2019, 5, 1572–1583, DOI:10.1021/acscentsci.9b00576.
- J. Shen and C. A. Nicolaou, Molecular Property Prediction: Recent Trends in the Era of Artificial Intelligence, Drug Discovery Today: Technol., 2019, 32–33, 29–36, DOI:10.1016/j.ddtec.2020.05.001.
- J. Wei, X. Chu, X.-Y. Sun, K. Xu, H.-X. Deng, J. Chen, Z. Wei and M. Lei, Machine Learning in Materials Science, InfoMat, 2019, 1, 338–358, DOI:10.1002/inf2.12028.
- O. Wieder, S. Kohlbacher, M. Kuenemann, A. Garon, P. Ducrot, T. Seidel and T. Langer, A Compact Review of Molecular Property Prediction with Graph Neural Networks, Drug Discovery Today: Technol., 2020, 37, 1–12, DOI:10.1016/j.ddtec.2020.11.009.
- R. Gómez-Bombarelli, et al., Design of Efficient Molecular Organic Light-Emitting Diodes by a High-Throughput Virtual Screening and Experimental Approach, Nat. Mater., 2016, 15, 1120–1127, DOI:10.1038/nmat4717.
- C. W. Coley, W. H. Green and K. F. Jensen, Machine Learning in Computer-Aided Synthesis Planning, Acc. Chem. Res., 2018, 51, 1281–1289, DOI:10.1021/acs.accounts.8b00087.
- N. W. A. Gebauer, M. Gastegger, S. S. P. Hessmann, K.-R. Müller and K. T. Schütt, Inverse Design of 3d Molecular Structures with Conditional Generative Neural Networks, Nat. Commun., 2022, 13, 973, DOI:10.1038/s41467-022-28526-y.
- B. Sanchez-Lengeling and A. Aspuru-Guzik, Inverse Molecular Design Using Machine Learning: Generative Models for Matter Engineering, Science, 2018, 361, 360–365, DOI:10.1126/science.aat2663.
- C. Shen, M. Krenn, S. Eppel and A. Aspuru-Guzik, Deep Molecular Dreaming: Inverse Machine Learning for de-Novo Molecular Design and Interpretability with Surjective Representations, Mach. Learn.: Sci. Technol., 2021, 2, 03LT02, DOI:10.1088/2632-2153/ac09d6.
- A. Zunger, Inverse Design in Search of Materials with Target Functionalities, Nat. Rev. Chem, 2018, 2, 1–16, DOI:10.1038/s41570-018-0121.
- G. Ferrer, A. Solé, C. Barreneche, I. Martorell and L. F. Cabeza, Review on the Methodology Used in Thermal Stability Characterization of Phase Change Materials, Renewable Sustainable Energy Rev., 2015, 50, 665–685, DOI:10.1016/j.rser.2015.04.187.
- A. J. Howarth, Y. Liu, P. Li, Z. Li, T. C. Wang, J. T. Hupp and O. K. Farha, Chemical, Thermal and Mechanical Stabilities of Metal–Organic Frameworks, Nat. Rev. Mater., 2016, 1, 1–15, DOI:10.1038/natrevmats.2015.18.
- J. Müller, A. Zhegur, U. Krewer, J. R. Varcoe and D. R. Dekel, Practical Ex-Situ Technique To Measure the Chemical Stability of Anion-Exchange Membranes under Conditions Simulating the Fuel Cell Environment, ACS Mater. Lett., 2020, 2, 168–173, DOI:10.1021/acsmaterialslett.9b00418.
- G. Niu, X. Guo and L. Wang, Review of Recent Progress in Chemical Stability of Perovskite Solar Cells, J. Mater. Chem. A, 2015, 3, 8970–8980, 10.1039/C4TA04994B.
- L. Wu, J. Zhang and W. Watanabe, Physical and Chemical Stability of Drug Nanoparticles, Adv. Drug Delivery Rev., 2011, 63, 456–469, DOI:10.1016/j.addr.2011.02.001.
- D. G. Kwabi, K. Lin, Y. Ji, E. F. Kerr, M.-A. Goulet, D. De Porcellinis, D. P. Tabor, D. A. Pollack, A. Aspuru-Guzik, R. G. Gordon and M. J. Aziz, Alkaline Quinone Flow Battery with Long Lifetime at pH 12, Joule, 2018, 2, 1894–1906, DOI:10.1016/j.joule.2018.07.005.
- J. Hachmann, R. Olivares-Amaya, A. Jinich, A. L. Appleton, M. A. Blood-Forsythe, L. R. Seress, C. Román-Salgado, K. Trepte, S. Atahan-Evrenk, S. Er, S. Shrestha, R. Mondal, A. Sokolov, Z. Bao and A. Aspuru-Guzik, Lead Candidates for High-Performance Organic Photovoltaics from High-Throughput Quantum Chemistry – the Harvard Clean Energy Project, Energy Environ. Sci., 2014, 7, 698–704, 10.1039/C3EE42756K.
- P. Z. Moghadam, T. Islamoglu, S. Goswami, J. Exley, M. Fantham, C. F. Kaminski, R. Q. Snurr, O. K. Farha and D. Fairen-Jimenez, Computer-Aided Discovery of a Metal–Organic Framework with Superior Oxygen Uptake, Nat. Commun., 2018, 9, 1378, DOI:10.1038/s41467-018-03892-8.
- C. J. Bartel, A. Trewartha, Q. Wang, A. Dunn, A. Jain and G. Ceder, A Critical Examination of Compound Stability Predictions from Machine-Learned Formation Energies, npj Comput. Mater., 2020, 6, 1–11, DOI:10.1038/s41524-020-00362-y.
- C. J. Bartel, Review of Computational Approaches to Predict the Thermodynamic Stability of Inorganic Solids, J. Mater. Sci., 2022, 57, 10475–10498, DOI:10.1007/s10853-022-06915-4.
- W. Li, R. Jacobs and D. Morgan, Predicting the Thermodynamic Stability of Perovskite Oxides Using Machine Learning Models, Comput. Mater. Sci., 2018, 150, 454–463, DOI:10.1016/j.commatsci.2018.04.033.
- K. T. Butler, J. M. Frost, J. M. Skelton, K. L. Svane and A. Walsh, Computational Materials Design of Crystalline Solids, Chem. Soc. Rev., 2016, 45, 6138–6146, 10.1039/C5CS00841G.
- Y. Zhang, D. A. Kitchaev, J. Yang, T. Chen, S. T. Dacek, R. A. Sarmiento-Pérez, M. A. L. Marques, H. Peng, G. Ceder, J. P. Perdew and J. Sun, Efficient First-Principles Prediction of Solid Stability: Towards Chemical Accuracy, npj Comput. Mater., 2018, 4, 1–6, DOI:10.1038/s41524-018-0065-z.
- Y. Chen, Polishing of Polycrystalline Diamond Composites, PhD Thesis, The University of Sydney, 2007 Search PubMed.
- Q. Zhao and B. M. Savoie, Simultaneously improving reaction coverage and computational cost in automated reaction prediction tasks, Nat. Comput. Sci., 2021, 1, 479–490 CrossRef PubMed.
- Q. Zhao and B. M. Savoie, Algorithmic Explorations of Unimolecular and Bimolecular Reaction Spaces, Angew. Chem., Int. Ed., 2022, 61, e202210693 CrossRef CAS PubMed.
- C. Shang and Z. P. Liu, Stochastic surface walking method for structure prediction and pathway searching, J. Chem. Theory Comput., 2013, 9, 1838–1845 CrossRef CAS PubMed.
- S. Maeda, T. Taketsugu and K. Morokuma, Exploring transition state structures for intramolecular pathways by the artificial force induced reaction method, J. Comput. Chem., 2014, 35, 166–173 CrossRef CAS PubMed.
- P. M. Zimmerman, Navigating molecular space for reaction mechanisms: an efficient, automated procedure, Mol. Simul., 2015, 41, 43–54 CrossRef CAS.
- T. A. Young, J. J. Silcock, A. J. Sterling and F. Duarte, autodE: Automated Calculation of Reaction Energy Profiles—Application to Organic and Organometallic Reactions, Angew. Chem., Int. Ed., 2021, 60, 4266–4274 CrossRef CAS PubMed.
- I. Safarik and O. P. Strausz, The thermal decomposition of hydro-carbons. Part 1. n-alkanes (C ≥5), Res. Chem. Intermed., 1996, 22, 275–314, DOI:10.1163/156856796X00458.
- D. G. Goodwin, H. K. Moffat, I. Schoegl, R. L. Speth and B. W. Weber, Cantera: An Object-oriented Software Toolkit for Chemical Kinetics, Thermodynamics, and Transport Processes, Version 2.6.0, 2022, https://www.cantera.org Search PubMed.
- K. M. Sundaram and G. F. Froment, Modeling of Thermal Cracking Kinetics. 3. Radical Mechanisms for the Pyrolysis of Simple Paraffins, Olefins, and Their Mixtures, Ind. Eng. Chem. Fundam., 1978, 17, 174–182, DOI:10.1021/i160067a006.
- D. L. Allara and R. Shaw, A Compilation of Kinetic Parameters for the Thermal Degradation of N-alkane Molecules, J. Phys. Chem. Ref. Data, 1980, 9, 523–560, DOI:10.1063/1.555623.
- V. Burklé-Vitzthum, R. Bounaceur, P. M. Marquaire, F. Montel and L. Fusetti, Thermal Evolution of n- and Iso-Alkanes in Oils. Part 1: Pyrolysis Model for a Mixture of 78 Alkanes (C1–C32) Including 13,206 Free Radical Reactions, Org. Geochem., 2011, 42, 439–450, DOI:10.1016/j.orggeochem.2011.03.017.
- J. B. Pedley, R. D. Naylor and S. P. Kirby, in Thermochemical Data of Organic Compounds, ed. Pedley, J. B., Naylor, R. D. and Kirby, S. P., Springer Netherlands, Dordrecht, 1986, pp. 3–6, DOI:10.1007/978-94-009-4099-4_2.
- K. Yang, K. Swanson, W. Jin, C. Coley, P. Eiden, H. Gao, A. Guzman-Perez, T. Hopper, B. Kelley, M. Mathea, A. Palmer, V. Settels, T. Jaakkola, K. Jensen and R. Barzilay, Analyzing Learned Molecular Representations for Property Prediction, J. Chem. Inf. Model., 2019, 59, 3370–3388, DOI:10.1021/acs.jcim.9b00237.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4dd00036f |
| This journal is © The Royal Society of Chemistry 2024 |