
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
DrugPose: benchmarking 3D generative methods for early stage drug discovery
Zygimantas
Jocys
 *,
Joanna
Grundy
and
Katayoun
Farrahi
*,
Joanna
Grundy
and
Katayoun
Farrahi
University of Southampton, UK. E-mail: z.jocys@soton.ac.uk
First published on 14th June 2024
Abstract
Molecule generation in 3D space has gained attention in the past few years. These models typically have a hypothesis that they need to satisfy (i.e. shape) or they are designed to fit into a protein pocket. However, there's been limited evaluation of the 3D poses they produce. In the previous work, the generated molecules are redocked and the generated poses are disregarded. Moreover, many of the generated molecules are not synthesisable and druglike. To tackle these challenges we propose DrugPose, a novel benchmark framework, that utilises Simbind to evaluate the generated molecules based on their coherence with the initial hypothesis formed from available data (e.g., active compounds and protein structures) and their adherence to the laws of physics. Moreover, it offers enhanced insights into synthesizability by directly cross-referencing with a commercial database and utilising the Ghose filter for assessing drug-likeness. Considering current generative methods, the percentage of generated molecules with the intended binding mode ranges from 4.7% to 15.9%, with commercial accessibility spanning 23.6% to 38.8% and fully satisfying the Ghose filter between 10% and 40%. These results highlight the need for further research to develop more reliable and transparent methodologies for 3D molecule generation.
1 Introduction
Drug discovery is a complex, lengthy and expensive process,1 which allows medicine to be brought to the patients who need it. Generative machine learning methods offer a promising way to speed up the early stages of drug discovery by exploring synthetically accessible molecules in an efficient and cost-effective way.2 However, current generative models that use simplified molecular-input line-entry system (SMILES),3 a representation of the molecule as a string, or graph-based representations without 3D information,4 have limited ability to generate diverse and novel compounds with specific properties relevant to drug discovery.5 In contrast, 3D representations have been widely used in non-machine learning methods for drug discovery, such as docking6 and rapid overlay of chemical structures (ROCS) used for pharmacophore screening,7 as they can capture the more nuanced features of molecules important for predicting biological activity. More recent 3D methods, such as LigDream,8 Pocket2mol,9 and SQUID10 allow the generation of novel molecules in 3D space.One of the very successful applications is using machine learning for docking. Models like Equibind,11 Equidock,12 DiffDock13 and recently DockGen are some examples.14 Moreover, PoseBusters15 were introduced showing that even though these models claim to have superior performance, it often is because the splits between training and testing sets are not strict enough allowing for the model to overfit over certain pockets and chemical structures.
While there is growing interest in using generative models as discussed in multiple reviews,16–18 the lack of benchmarks has made it difficult to draw conclusions about the generalisability of these methods. There have been some case studies where SMILES19–22 or graph-based methods without 3D information23–26 were claimed to be successful in discovering novel compounds, but further analysis has revealed questionable performance. For instance, a recent study used a SMILES-based generative model to design DDR1 inhibitors in just 21 days (ref. 27) and pre-clinically validated them in 25 days. However, upon closer inspection,5 it was found that the molecule highlighted in the paper bore a striking similarity to a number of previously published DDR1 inhibitors, including the marketed drug ‘ponatinib’. In early-stage drug discovery, the chemical matter needs to be novel so that it can be patented later on. Moreover, similar observations have been noted with AlphaFold in relation to matching experimental data. According to Terwilliger et al.,28 while some AlphaFold predictions align closely with experimental maps, most show discrepancies on a global scale in terms of distortion and domain orientation, and on a local scale in backbone and side-chain conformations,28 showing the need for sophisticated evaluation frameworks for advanced ML models.
Existing methods lack transparency, as the underlying causal reasoning is not adequately assessed. By causal reasoning, we mean the process wherein each design decision for a de novo molecule aims to deliberately facilitate specific interactions and binding in the desired manner or improve certain properties. A recent research article introduced a benchmark29 to evaluate the docking capabilities of generated molecules. The study revealed that graph-based (without 3D information) methods struggle to effectively learn the generation of molecules that form a low energy binding pose.
A benchmark should evaluate:
• Is the binding mode consistent with the input molecule?
• Can the molecule be synthesisable?
• Are the generated molecules druglike?
In this paper, we extend the work of Cieplinski et al.29 and PoseCheck30 to evaluate the performance of generative models in early-stage drug discovery. Ciepinski et al. used a Vinardo score: weighted scoring system over ligand-protein steric interactions, hydrophobic interactions, and non-directional hydrogen bonds to assess binding. We extend the work by checking if the binding mode meets the hypothesis space and that the model is able to create the desired interactions, contrary to using the energy score. Moreover, while PoseCheck evaluates the quality of the binding pose, we evaluate if the generated molecule binds as designed. In addition, we evaluate synthesisability by assessing if a similar compound exists in the Enamine REAL Database, which allows us to evaluate if the molecule can readily be made by the external provider. QED is only a proxy model to evaluate the synthesizability and thus our methodology is a more realistic representation of what needs to be satisfied early on. Moreover, to evaluate druglikeness, we propose not to use the classic QED score, but by using the Ghose filter's criteria. This binary decision will prevent averaging of results masking very not druglike molecules, which would be undetected when averaging the QED score.
Moreover, we argue that the same framework can be applied to both ligand-based and structure-based drug discovery. Why is this feasible? In structure-based drug discovery, it is essential to effectively leverage knowledge about the pocket to make decisions that ensure the model, by adding new fragments, which creates desired interactions. It is problematic if adding a fragment causes the molecule to change the binding mode. The same principle holds in ligand-based drug discovery, which is currently underexplored. When working with many active ligands and attempting to modify the seed molecule or create similar molecules, the binding mode should remain stable unless a change is specifically intended.
Thus, a good practice would be to take a molecule from a known PDB, generate it based on the active ligand's pose, redock it, and check if the pose remains similar. If the desired behavior is not observed, it suggests that the model is not making deliberate changes but is instead performing random exploration. Therefore, the same workflow should be used, though different performance outcomes should be expected.
The following contributions have been made in this paper:
• A benchmark system is designed to evaluate the usefulness of current 3D generative models for early drug discovery, incorporating a new binding mode similarity metric, Simbind, a more rigorous druglikeness measure, and a new synthetic accessibility metric.
• An evaluation of existing 3D generative models is performed and presented:
• The generated poses cannot be reproduced even once most of the time.
• The generated molecules cannot be found in Enamine REAL Space.
• Most of the generated molecules are not drug like.
2 Related work
2.1 Early stage drug discovery
Traditionally, three core techniques are used to identify drug candidates: high-throughput screening (HTS),31 fragment-based screening,32 and DNA-encoded libraries (DELs).33 Prebuilt molecule libraries are required for HTS and fragment-based screening, whereas DELs allow for the construction of custom libraries.Once a hit has been identified, the process moves on to lead optimization, where decisions must be made to improve the molecule, given chemical constraints. Given the lengthy and expensive nature of the process and the vast chemical space to explore, each decision must be made with the intent to deliberately improve the required properties within the synthetic constraints imposed by chemical space. Thus, the generative machine learning models must have different requirements for different stages (hit identification, hit-to-lead and lead optimization). In hit identification, the primary goal is to identify early-stage chemical matter. During this step, depending on the technology used, new chemical matter is discovered that could be optimized. In the hit-to-lead phase, the goal is to validate hits and demonstrate their potential for optimization, ensuring they exhibit favorable structure–activity relationships (SAR). At this stage, only analogs that can be easily synthesized or acquired are considered. During lead optimization, our focus is on refining compound properties based on specific requirements. This involves making small changes that are constrained by the available synthetic routes.
2.2 Generative methods
The success of a generative method depends on an expressive representation. Some of these methods represent molecules as string sequences (such as SMILES,38 SELFIES39 and DeepSMILES40). Models based on generative adversarial networks (GANs) and long short-term memory (LSTM) networks have been reported to be effective. For example, MolGAN, a GAN-based model, has demonstrated its ability to generate valid molecules.41 Similarly, GENTRL, which utilizes LSTMs, has been claimed to generate active molecules.27 Others use graph representations (without 3D information), such as JT-VAE,4 and Reinvent42 which have shown some promise. Graph neural networks (GNNs) without 3D information were used with DNA encoded libraries (DEL)43 to screen iteratively, with an overall hit rate of ≈30% at 30 μM and discovery of potent compounds (IC50 < 10 nM) for every target. However, they have limited applicability, as the full library is screened, and GNNs have been shown to have significant limitations.44 In general, it's recognized that there are instances where two different molecules can be depicted with the same graph representation. For example, this occurs with decalin and bicyclopentyl, despite their distinct chemical structures.2.3 3D generative methods
Among the first methods described is LigDream,8 where molecules are generated from a shape descriptor and the 3D conformation is fed to a convolutional neural network (CNN). The resultant SMILES string is captioned with a LSTM network. However, few of the molecules could be correctly decoded. This work has been extended to LIGANN,45 adding a shape generation step, captioned with LigDream. However this model has the same limitations as LigDream. An electron density-based GPT for optimization and suggestion of a host–guest binder learning model has been proposed.46 It was trained on electron density data and effectively generates three-dimensional representations and optimizes host–guest molecular systems using a variational autoencoder, achieving over 98% accuracy in predicting SMILES formats. Moreover, molecule generation using flow networks has been explored in AR47 and Pocket2Mol9 models. Additionally, FLAG,48 an autoregressive substructure-based method, has also been developed. Despite an improved SA score, generative models still yield non-synthesisable molecules,49 because the score is optimised to resemble synthetically accessible molecules,50 rather than actually being synthetically accessible. Synthetic accessibility (SA) is estimating the ease of synthesizing molecules, using a hybrid approach that combines historical synthetic knowledge learned from a PubChem subset enhanced with standard rules to create a composite SA score. To be suitable for molecule library design or optimization tasks, synthesisability and interpretability must be ensured.2.4 Benchmark methods
GuacaMol34 and MOSES35 are evaluation frameworks that have been heavily used in the field as benchmark frameworks, however, these frameworks focus too much on property improvement, such as uniqueness, novelty drug-likeness (QED), synthetic accessibility, etc. All these properties are computationally calculable. A success for these benchmark frameworks is when you have molecules with an initial distribution of properties and you are able to generate a new set of molecules with another distribution of properties. This process is irrelevant for hit identification and is only partially relevant for the lead optimization stage.51 However, it must have additional considerations to be useful for lead optimisation, such as available synthetic route, interpretability, and maintenance of the binding mode, to name a few.More benchmarks have been released, as shown in Table 1, such as the sample efficiency matters,37 where the number of steps needed to optimize the molecule is evaluated, and the therapeutics data commons,36 where they aggregated the existing datasets to evaluate machine learning models for a vast range of drug discovery related tasks. However, none of these frameworks are aligned with the goals of early-stage drug discovery:
• Find novel chemical matter that shows desired activity with a high likelihood of the desired profile.
• Optimize chemical matter through accessible synthetic space.
Multiple evaluation methods have been proposed for evaluating generative methods, as mentioned above. However, these benchmarks optimize activity, drug-likeness, and synthetic accessibility without considering the available synthetic space, which is limited by the available reagents and reactions. Current methods optimize synthetic accessibility (SA),52 which means that they are trying to make the molecules look synthesisable, but the actual synthetic routes are not computed during the generation.
Two major approaches have been adopted regarding synthesisability.
2.5 Score based generation
Current methods in machine learning (ML) for assessing synthetic accessibility of molecules typically involve training the ML model to score molecules, enabling the model to differentiate between synthetically accessible and inaccessible molecules. The score based generation was used in Pocket2mol,9 Flag48 and many others.2.6 Rule-based exploration
Another approach involves restricting the generative process by specifying the permissible reagents and reactions, as demonstrated in a study by atomwise.53 They utilized the Combinatorial Synthesis Library Variational AutoEncoder (CSLVAE)53 to generate molecules using only the available blocks and reactions.Furthermore, many papers use the QED score, a continuous measure for assessing a molecule's drug-likeness. However, comparing these scalar values does not reveal how frequently the model is incorrect. When scoring and averaging a large group of molecules, the knowledge of the quantity of generated molecules with abnormal scores is obscured. Therefore, the Ghose filter provides a more insightful approach for evaluating drug-likeness by offering a discrete output: either drug-like or not drug-like. In the drug discovery process, the critical distinction lies not in the degree of drug-likeness but rather in whether the molecule is classified as drug-like or not. In the case of Pocket2mol, it is evident that numerous molecules are not drug-like, contrary to what the scalar value of the QED in the paper might suggest. We observed numerous molecules consisting of two to three atoms generated by Pocket2mol from the PDBBind dataset, resulting in structures that are not druglike.
3 Methodology
Our evaluation methodology is designed to address the following problems:• Problem 1 molecule alignment before comparison. We propose to align on the protein and see if there exists a low-energy conformer similar to the original crystal structure. A prevalent method involves aligning molecules according to their pharmacophores, which presents challenges when the number of pharmacophores greatly varies without a clear solution. Our approach resolves this issue by aligning molecules based on the protein structure, essentially comparing binding modes.
• Problem 2 sensitivity to interactions. The binding mode is different if the molecule is rotated. Models that take into account only the atom overlap would fail to distinguish that the binding mode has changed, as the interactions will be different.
• Problem 3 druglikeness. The utilization of quantitative estimate of drug-likeness (QED) for assessing the drug-likeness of molecules does not indicate the proportion of generated molecules that do not possess drug-like characteristics.
• Problem 4 synthetic accessibility. Generated molecules can have a high SA score, but still be hard to synthesise.
3.1 Simbind: a new score for evaluating binding mode similarity
There are two main situations (with some variants in between) in early drug discovery with respect to available data for the chemistry part: the protein structure is available or some active compounds are available. For both cases, once the molecule is generated we need to make sure that a low-energy molecule protein complex exists and is aligned with the initial hypothesis (input to the generative model). In both cases, we need to ensure that a low-energy complex exists with a similar binding mode, as the initial hypothesis.Measuring the shift in mass of the centre has been used to evaluate the binding mode similarity, however if a molecule is flipped over by 180°, this could result in a negligible shift in mass, but a very different binding mode.
To account for this, we introduce a new binding mode similarity score, Simbind. Simbind works as follows:
1. A molecule protein complex is taken from PDBBind,54 where the molecule (A) from the crystal structure is used as a seed molecule for the generative model.
2. Each of the generated molecules is docked to the protein within a binding box with dimensions of 25 × 25 × 25 Å. Thus, for each generated molecule, we have a set of poses, where each pose B will have a number of atoms NB with position pj.
3. Consider each docked pose, where D represents the matrix containing the pairwise Euclidean distance between atoms pi and pj, where pi and pj denote the atom positions of A and the generated molecule B, where i = 1, …, NA and j = 1, …, NB. NA and NB are the number of atoms in A and generated molecule B respectively:
| Dij = ‖pi − pj‖ | (1) |
4. The minimum distance for each atom of A to any atom of B is therefore the minimum along each row of D, so posi indicates the index in B.
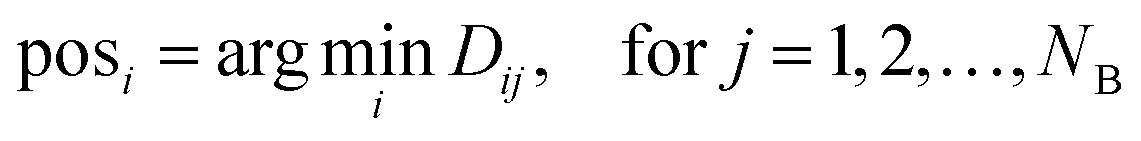 | (2) |
5. Fposi and Fposj are forces applied on atoms i and j, respectively. We check if the smallest distance is shorter than hyperparameter d and we want to check if the force difference between Fposi and Fposj, which denotes the force applied on the atom i and j in the pocket for molecules A and B, respectively, is smaller than the force at posj scaled by hyperparameter β. This way, we determine whether there is an atom from molecule B near posi of molecule A, and whether the forces between the two atoms fall within expected ranges for Gauss (van der Waals), repulsion, hydrophobic, and non-directional hydrogen bonding forces. The force vector for each atom is extracted from SMINA output as denoted in eqn (3). We perform this step because different atoms can interact in a similar manner. Thus, we are assessing if the force falls within a certain range.
| Fposi = [FGauss,FRepulsion,FHydrophobic,FNon-directional HB] | (3) |
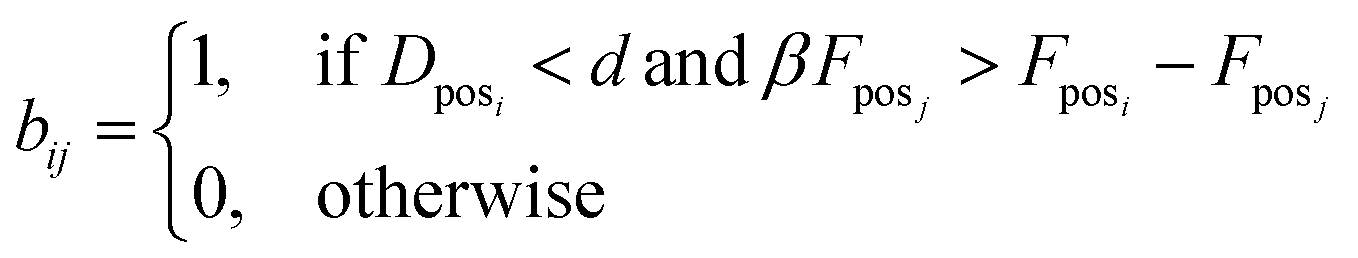 | (4) |
6. The score is computed by adding up all the atoms that are interacting in a similar manner divided by the total number of atoms of the seed molecule NA. We chose to divide by NA, because if there is a single atom in a position close to any of the atoms of the molecules, the binding mode similarity would be 100%, thus it is necessary for the molecule to occupy a similar shape in space.
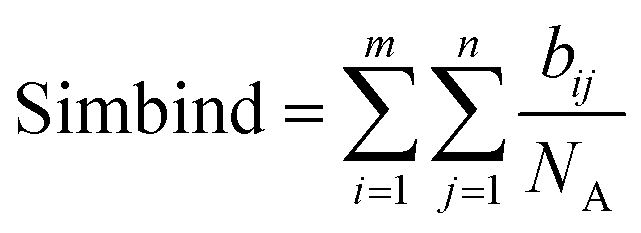 | (5) |
To demonstrate the superiority of Simbind over other methods, we conducted two experiments:
1. Extract a molecule from a PDB, redock the same molecule to the same protein as shown in Fig. 2, and compute the similarities between the poses.
2. Extract a molecule from a PDB, generate a new molecule using LIGANN, redock the generated molecule to the same protein as shown in Fig. 3, and compute the similarities between the poses.
3.2 Comparing different models
We evaluate 3D generative models based on the three criteria as introduced earlier: binding mode similarity, synthetic accessibility, and Ghose filter (druglikeness). Each model is evaluated in the following way:• We select 100 protein-data complexes and extract the ligands and proteins (Appendix A).
• For the ligand-based method, we use the crystallized ligand as the seed compound and we generate 30 compounds for each crystal structure.
• For structure based models, we use the protein structure and define the bounding box by computing the center of the ligand and selecting the area around the center of the mass of the crystallised ligand.
3.3 Ghose filter (druglikeness)
The Pocket2mol model generated many simple molecules, such as single system rings or two carbon atoms as a molecule, which is not a complex enough binder to be a selective and potent compound as a starting point of a drug discovery program. At best, it is a fragment of the compound. Currently, QED is commonly used as a continuous metric between zero and one to assess the druglikeness as it is the most convenient way to train the model and optimise for it. QED55 is a geometric mean of the individual functions: molecular weight (MW), octanol–water partition coefficient (ALOGP), number of hydrogen bond donors (HBD), number of hydrogen bond acceptors (HBA), molecular polar surface area (PSA), number of rotatable bonds (ROTB), number of aromatic rings (AROM) and number of structural alerts (ALERTS). However, when a model outputs very simple molecules, such as a dicarbon compound, the quantitative estimate of the drug-likeness (QED)55 score provides a scalar value, which is difficult to interpret when averaging over all molecules. This averaging will obscure the presence of low-scoring molecules. However, in reality, this molecule is completely unacceptable as a starting point, as the molecule is too small.Thus, we propose to assess the molecule with the Ghose filter56 in a classification manner, even if it is optimised for QED by an external scoring model, to learn how many molecules are within the desired property range.
All the Ghose filter metrics should be satisfied for the molecule to be druglike:
• Log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P values constrained within the range −0.4 to 5.6.
P values constrained within the range −0.4 to 5.6.
• Molecular masses required to fall within 180 to 480 atomic mass units.
• Total atom counts limited to between 20 and 70.
• Refractivity values restricted to the interval 40 to 130.
3.4 Evaluating binding mode similarity across the generated compounds
For each generated compound, we prepare the molecule and protein for docking and we dock the compounds to generate 10 poses for each generated molecule. We used SMINA57 software with the bounding box dimensions of [25,25,25]. For each docked generated molecule, we compute a Simbind score and we consider two cases:• BMStotal: for each generated molecule, we check if there exists a complex with a similar binding mode.
• BMScoarse: for each seed molecule, we check if there exists at least one low energy binding mode.
Thereafter, we are summing over a matrix M of size m × n, where m is the number of generated molecules and n is the number of docked poses. The function 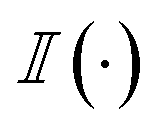 represents the indicator function, which returns 1 if the condition inside the parentheses is true and 0 otherwise. BMSij denotes the element at the ith row and jth column of matrix BMS. The hyperparameter Smin determines the cutoff threshold at which we conclude that molecules share a similar binding pose.
represents the indicator function, which returns 1 if the condition inside the parentheses is true and 0 otherwise. BMSij denotes the element at the ith row and jth column of matrix BMS. The hyperparameter Smin determines the cutoff threshold at which we conclude that molecules share a similar binding pose.
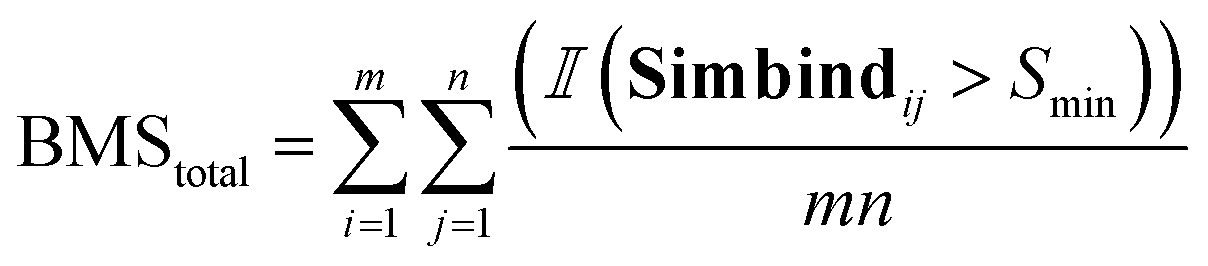 | (6) |
For the coarse score, we check if there was at least one docked pose that binds similarly to the crystal structure.
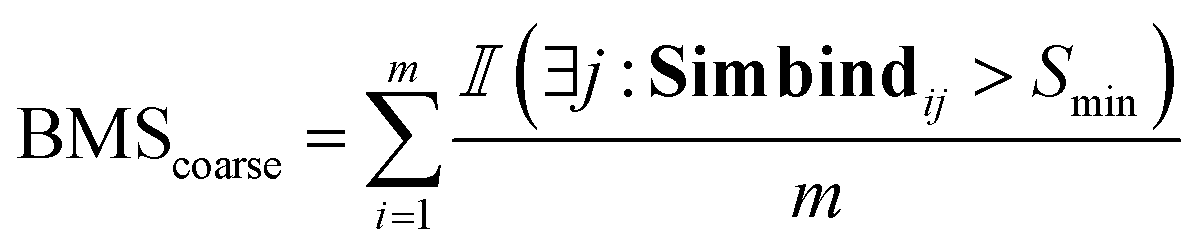 | (7) |
3.5 Commercial availability of the compound
Often to assess synthesisability, a synthetic accessibility (SA) score is computed based on a combination of fragment contributions and a complexity penalty. Fragment contributions have been calculated based on the analysis of one million representative molecules from PubChem. The molecular complexity score takes into account the presence of non-standard structural features, such as large rings, non-standard ring fusions, stereocomplexity and molecule size. However, it is not that helpful in the early stage of the program, as the compound should be readily available and synthesisable by outside vendors or available within your own library, reactions and fragments. Thus, in this case, we propose to evaluate if the compound is available within the Enamine REAL Database, which constitutes 30 billion compounds via an API. For the commercially available score, we get the number of molecules, that are an exact match in the database.4 Results
4.1 Binding similarity score
Following the outlined methodology, we begin by exclusively redocking the same seed molecule into the same pocket of tyrosine kinase C-src with PDBID 1 A1E. We also redock a single generated molecule by Ligdream from the seed molecule, aiming to illustrate the limitations of commonly used shape similarity algorithms Shape Tanimoto using the rdkit implementation58 and PMapper.59 First, the shape of each molecule is encoded onto a grid and these grids are then compared using the Shape Tanimoto. PMapper is a Python module to generate 3D pharmacophore signatures and fingerprints. Signatures uniquely encode 3D pharmacophores with hashes.In Fig. 2, the docked pose (b) is a perfect example where the molecule is flipped 180° degrees and has a completely different binding mode. Thus, even if the volume is similar, the interactions between the molecule and the protein are different. With our proposed Simbind score, it gets a 64% similarity, contrary to shape Tanimoto. However, for pose (f), the Simbind score is greater in comparison to the position (b), which follows the original binding mode with mild differences to the crystal pose. We can see that PMapper is actually giving very sensible scores, when looking at the positions a, b and f. The model is able to disambiguate the closest molecule (position f) with the seed molecule, with the score of 0.67 in comparison to pose 2, where the score is 64.51%
| Position | Shape Tanimoto | PMapper | Simbind | |||
|---|---|---|---|---|---|---|
| Crystal | LigDream | Crystal | LigDream | Crystal | LigDream | |
| a | 0.00 | 0.79 | 0.00 | −1.00 | 100.00% | 30.00% |
| b | 0.56 | 0.81 | 1.59 | −1.00 | 64.51% | 35.00% |
| c | 0.92 | 0.79 | 2.09 | −1.00 | 3.22% | 15.00% |
| d | 1.00 | 0.80 | 1.77 | −1.00 | 0.00% | 15.00% |
| e | 0.65 | 0.82 | 2.14 | −1.00 | 48.38% | 27.00% |
| f | 0.51 | 0.79 | 0.67 | −1.00 | 70.96% | 22.00% |
4.2 Model evaluation
Using the proposed Simbind, druglikeness, and synthesisability scores described in the Methodology section, we evaluate the following models using the methodology described in the previous section and how well they perform in a practical scenario. Thus, key results are presented in Table 3 with key metrics for three molecular docking models: LigDream, SQUID, and Pocket2mol. The Coarse Binding Mode Similarity (BMS) is highlighted, with LigDream exhibiting the highest value at 45%, followed by SQUID at 16.7%, and Pocket2mol at 18.3%. In terms of total BMS, LigDream leads with 15.9%, while SQUID and Pocket2mol have lower values of 4.7% and 7.4%, respectively. Druglikeness, another crucial parameter, shows that LigDream possesses a value of 37.8%, SQUID stands at 46.5%, and Pocket2mol trails with 10.36%. Lastly, the commercial availability of these models is indicated, with LigDream at 32.4%, SQUID at 23.6%, and Pocket2mol at 38.8%. These metrics provide insights into the performance and commercial accessibility of each model, assisting researchers and practitioners in choosing an appropriate molecular docking tool based on their specific requirements.| Model | Coarse BMS | Total BMS | Druglikeness | Commercial availability |
|---|---|---|---|---|
| LigDream | 45% | 15.9% | 37.8% | 32.4% |
| SQUID | 16.7% | 4.7% | 46.5% | 23.6% |
| Pocket2mol | 18.3% | 7.4% | 10.36% | 38.8% |
4.3 Assessment of the models
The best model was LigDream where the model was able to generate molecules of which 15.9% could have at least one similar binding mode to the seed compound. Only for 45%, the model was able to generate at least one molecule that could have a similar binding mode. However, 37.8% of the molecules are druglike and 32.4% can be purchased on Enamine.From the molecules that SQUID generated, only 4.7% were binding similarly and for only 16.7% of seed molecules potential similar binders were generated. It means that for 83.3% of the generated molecules not a single docked pose could satisfy the initial hypothesis. It has the best druglikeness compared to LigDream and Pocket2mol. However, this model output the least amount of synthetically accessible compounds with 23.6% available commercially (i.e. can be purchased).
We evaluate the model with the structure based evaluation as described in Fig. 1. The metrics for this model were slightly better than those of SQUID with respect to binding mode similarity with 16.7% for BMScoarse and 7.4% for BMStotal, however the task is slightly different. Nonetheless, the model had the lowest number of molecules that passed the Ghose filter with 10.36% of molecules, with 38.8% of molecules accessible commercially.
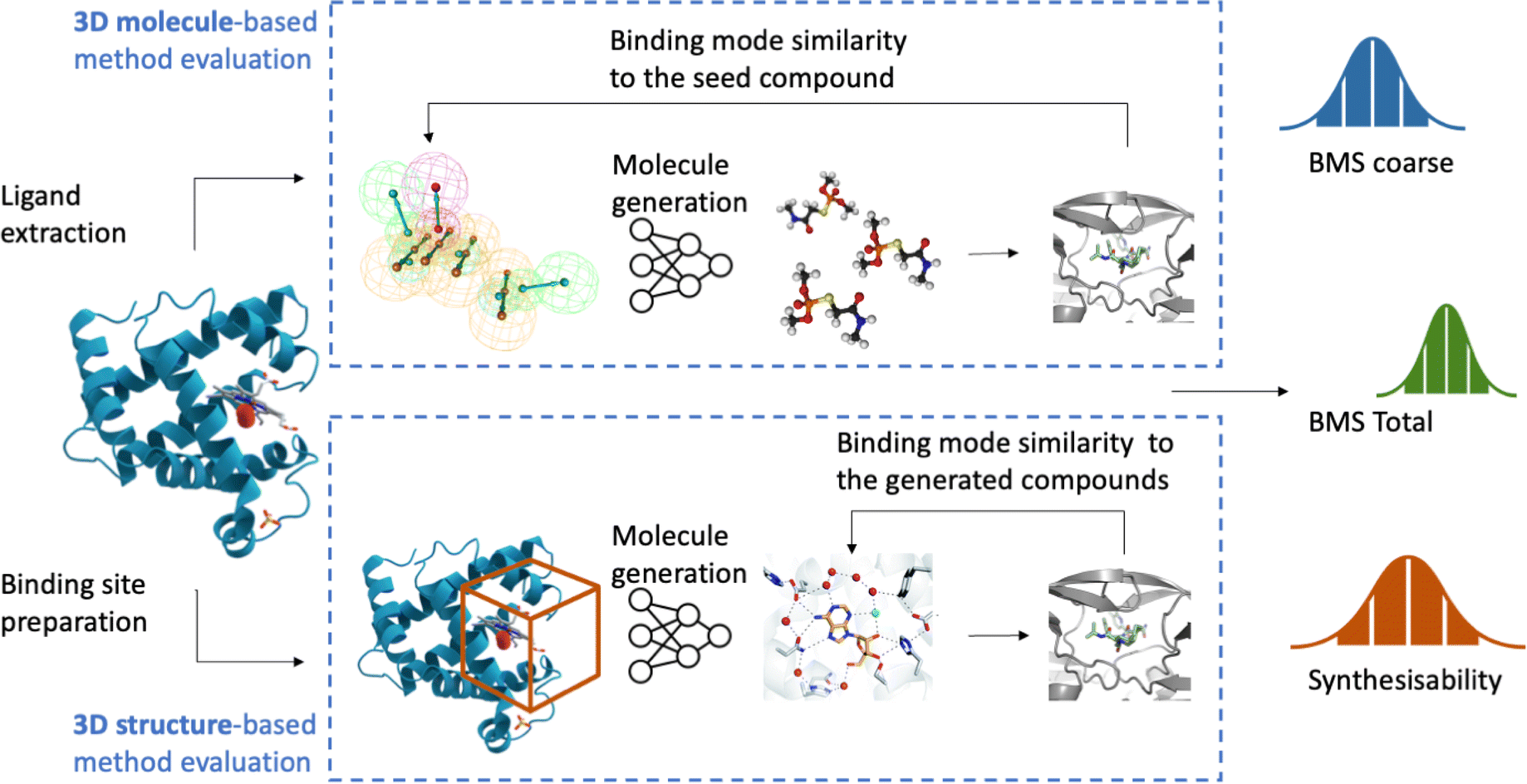 | ||
| Fig. 1 Using PDB input (active ligand or binding site), the model generates molecules, which are redocked into the binding site to assess binding similarity across 100 inputs. The evaluation yields BMS coarse, BMS total, and a synthesisability score. | ||
 | ||
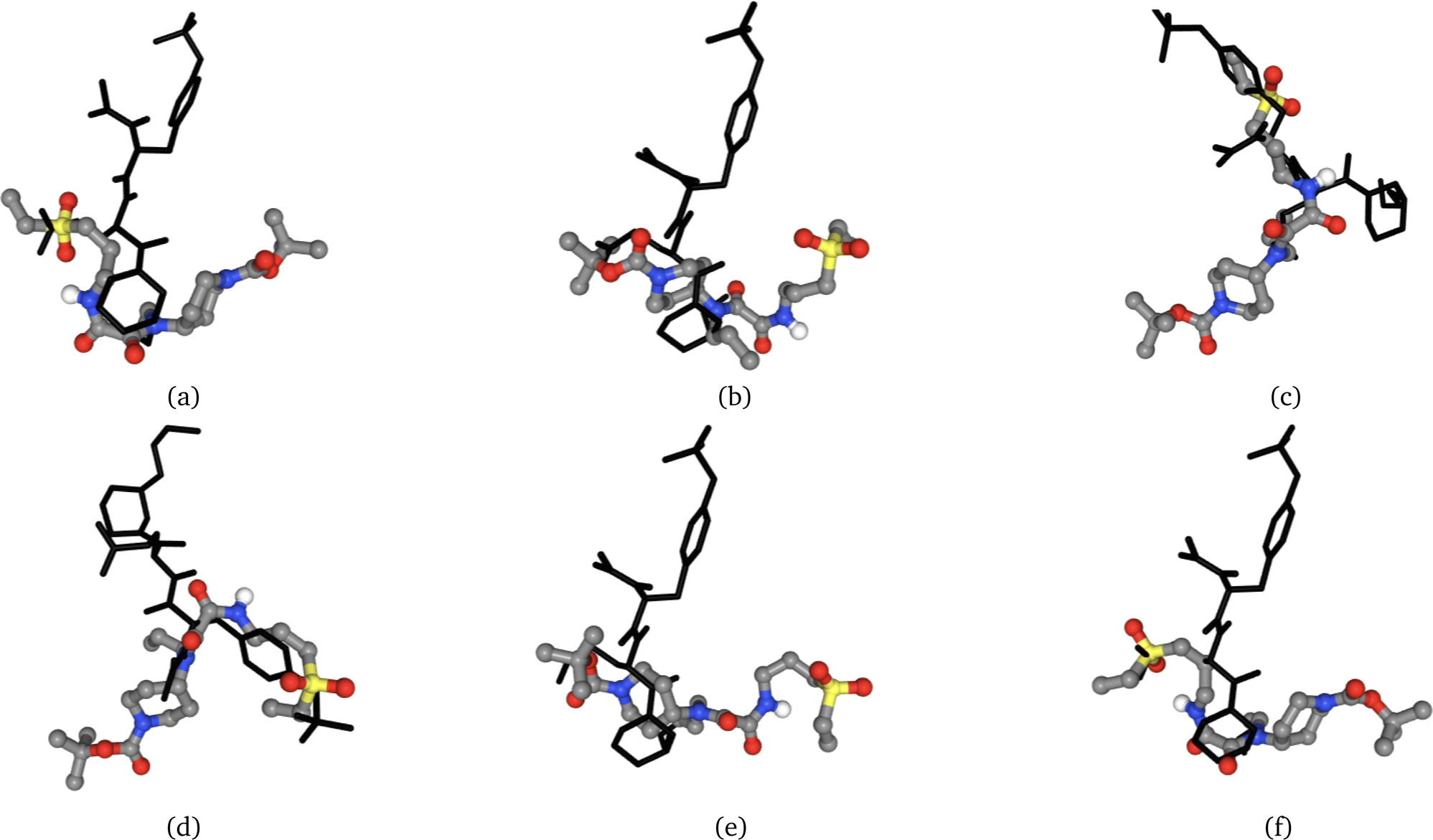
| Fig. 2 Molecule extracted from the crystal structure (coloured) in position (a) and the same molecule is redocked (black) to the same protein 5 times (b–f). We can see that pose (b) occupies the same volume, however the molecule is flipped by 180°, leading to a completely different binding mode. On the other hand, pose (f) is the closest to the crystallized molecule and poses (c) to (e) have completely different binding modes. | ||
 | ||
| Fig. 3 Molecule from the crystal structure (coloured) was used as a seed with the LigDream model, and the generated molecules(black) have been docked and imposed on the seed molecule. As we can see the generated molecules bind with a completely different binding mode in comparison to the crystallized molecule. | ||
5 Discussion
5.1 Generative methods are unable to generate molecules that bind with the desired binding mode
For early drug discovery programs, we need models that would generate molecules, that can be made (synthesised) in a cost and time effective manner, and are novel, druglike and diverse. Current models fail to reliably satisfy these requirements. First of all, from our results, it can be observed that current models do not consistently generate molecules that could bind with a desired binding mode. By “could”, we mean that the molecule would be in one of the possible binding modes identified by the docking software. The models are trained to satisfy the conditions of a molecule by taking a certain shape and generating a molecule that satisfies the shape, however no tests are done to evaluate if the molecule can be redocked to the similar pose. From the results, we see that LigDream had the best BMS scores; at the same time, the generated molecules have the greatest molecular weight.For the model to be useful, the BMScoarse score should be 100%, meaning from 30 generated molecules there should be at least one docked pose that satisfies the initial hypothesis and should bind similarly to the seed compound. Moreover, in order to make our ML model valuable in early stage drug discovery, we set a stringent criterion, requiring a minimum BMStotal of 70%. This threshold ensures that 7 out of 10 generated molecules exhibit the expected binding pose, while also being synthesisable and demonstrating satisfactory druglikeness.
5.2 Druglikeness and QED
Druglikeness is an important property, when talking about drug discovery and development. Most machine learning metrics are using QED scores, which is a continuous value score that can be assigned to any chemical structure. Most of the papers declare that they improve the score. Nevertheless, transitioning from QED to the Ghose filter implies a more discrete approach in determining whether the molecule exhibits drug-like properties. In this paper, we show that even if molecules seem to be druglike statistically from the results reported in their papers, in reality many molecules fall outside of druglikeness once the criteria were changed to percentage of molecules satisfying the Ghose filter, where there are only: 37.8% druglike molecules for Ligdream, 46.5% for SQUID and 10.36% for Pocket2Mol. The low druglikeness for Pocket2mol is due to the generation of many molecules that are of a very low molecular weight. As can be seen in Fig. 4, most of the generated molecules are below the threshold of druglike space.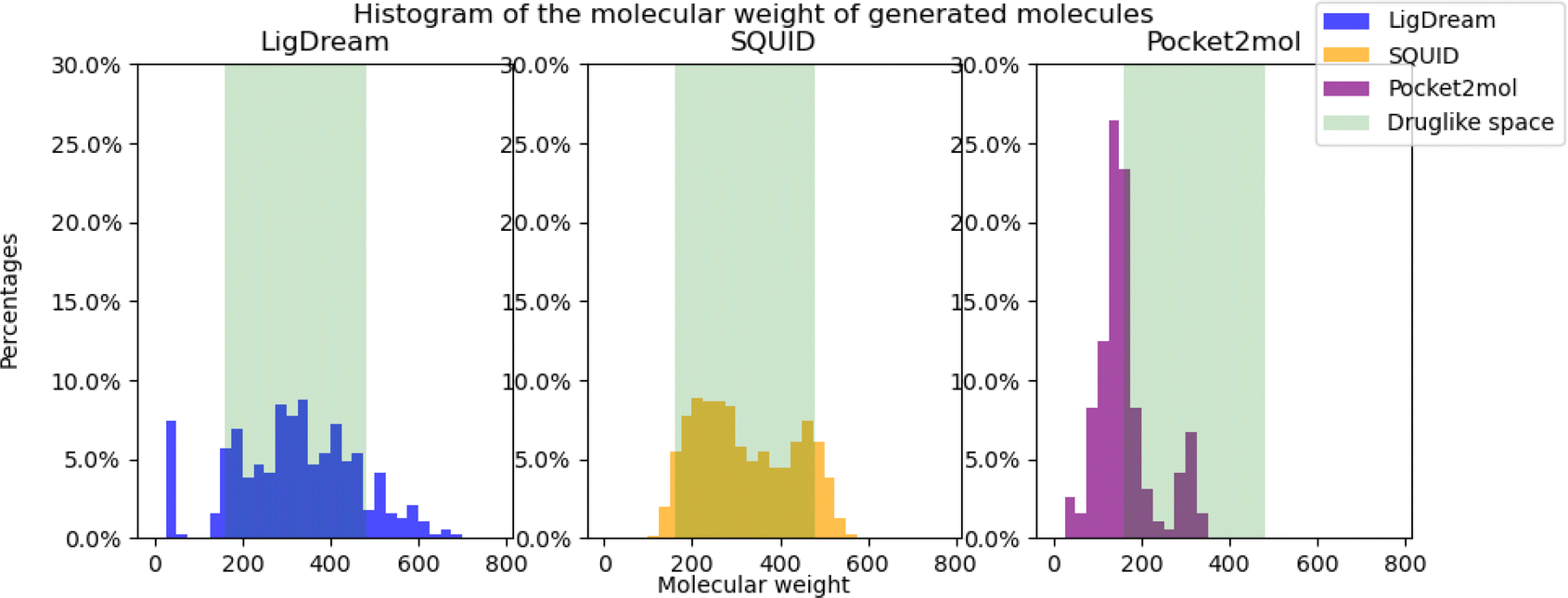 | ||
| Fig. 4 The figure shows the molecular weight of the generated molecules for each model (LigDream, SQUID, and Pocket2mol) and the druglike space according to the Ghose filter. Most of the molecules from LigDream and SQUID are in the druglike space, while most of the molecules generated by Pocket2mol are outside of the druglike space. | ||
5.3 Synthesisability and the SA score
Skoraczynski et al.60 investigated whether synthetic accessibility scores can reflect the complexity of retrosynthesis planning. Their results show that higher RAscore61 and SAscore values correlate with lower complexity, particularly in terms of the number of nodes. For early stage drug discovery, it would be a worthwhile task to find the retrosynthetical path of the generated molecule. However, as the current predictive models to identify 30% (very high estimate) of active compounds,43 7 out of 10 compounds will be inactive, and thus the acquisition of the compound should not be the bottleneck for the model to provide value in real world drug discovery scenarios. In this paper, we show that current 3D generative models do not generate many commercially available molecules, casting some doubt on their usefulness in a drug discovery scenario. Moreover, synthesizability is important because it supports transitioning generative predictions from in silico to an experimental laboratory setting.6 Conclusions and future work
It is an advancement, overcoming limitations of previous evaluation methods. We propose a new binding mode similarity score, Simbind, which effectively assesses if two molecules bind to a protein in a similar manner. The evaluation of different models reveals that the current generative models have limitations in generating molecules with the desired binding mode. The models show varying levels of druglikeness and synthesisability, with LigDream performing the best in terms of binding mode similarity, Pocket2mol with commercial availability due to the generation of simple molecules and SQUID generating the most druglike molecules. However, none of the models fully satisfy the criteria for a useful drug discovery tool.Future work should focus on improving the models themselves, particularly in terms of synthesisability, 3D information, and causal reasoning. Additionally, benchmark improvements are needed to evaluate models in different scenarios and incorporate factors such as cost and time for synthesis. These advancements will contribute to the development of more reliable and effective deep learning methods for drug discovery, enabling researchers to generate molecules with the desired binding mode and druglike properties.
Data and software availability
The Simbind and BMS code, along with the data, can be found on Github at this link: https://github.com/zygiauzas/DrugPose.The codes for LigDream, SQUID and Pocket2mol are available in the following repositories:
– LigDream: https://github.com/playmolecule/ligdream.
– SQUID: https://github.com/keiradams/SQUID.
– Pocket2mol: https://github.com/pengxingang/Pocket2Mol.
All the evaluations that were performed on the PDBbind dataset are available here: http://www.pdbbind.org.cn/.
The PDBs utilized for evaluation are listed in Table 4 of Appendix A.
The synthesis evaluation was done using this API: https://github.com/matteoferla/Python_SmallWorld_API.
Conflicts of interest
There are no conflicts to declare.Appendix A
In Table 4, we show the PDBs used for model evaluation.| 4rdn | 4mo4 | 3s0b | 3fwv | 1qin |
|---|---|---|---|---|
| 4b5d | 3sus | 3b26 | 2v25 | 6g3a |
| 4oc2 | 4f5y | 1bai | 2avo | 1ppi |
| 5c28 | 3vf7 | 3ckz | 2wky | 1fkf |
| 5cxa | 5nbw | 5yjm | 4zb8 | 3eax |
| 1hdq | 6c0s | 3nxq | 1e2k | 3dx3 |
| 5ot8 | 3ibi | 6ezq | 5kqx | 3ebp |
| 3l4w | 1gx8 | 2ygf | 2qbs | 4oc5 |
| 5dnu | 3bxh | 2xxx | 3sut | 4yrd |
| 1yc4 | 5g1a | 1jmf | 4gih | 2vwo |
| 5z99 | 3s0e | 4hp0 | 3d83 | 4y79 |
| 5orj | 3ibn | 3cd5 | 1m7y | 1e2l |
| 3dx4 | 4lzs | 3kgu | 5tya | 4urz |
| 5tp0 | 4f9u | 4xaq | 4q7v | 3w5n |
| 6g98 | 3tcg | 5wcm | 2bok | 5otz |
| 2v7a | 1bdq | 4mc1 | 5nkd | 4q87 |
| 1rpj | 5kr2 | 1zsf | 4jpx | 1ew8 |
| 4zb6 | 5g2g | 6mu3 | 2qnq | 1dhj |
| 3imc | 4ij1 | 1fkh | 4clj | 1k1y |
| 4y8x | 4ly9 | 4dzy | 3su1 | 4acc |
References
- O. J. Wouters, M. McKee and J. Luyten, JAMA, 2020, 323, 844–853 CrossRef PubMed.
- J. Meyers, B. Fabian and N. Brown, Drug Discovery Today, 2021, 26, 2707–2715 CrossRef CAS PubMed.
- L. Yang, G. Yang, Z. Bing, Y. Tian, Y. Niu, L. Huang and L. Yang, ACS Omega, 2021, 6, 33864–33873 CrossRef CAS PubMed.
- W. Jin, R. Barzilay and T. S. Jaakkola, Proceedings of the 35th International Conference on Machine Learning, 2018, 80, 2323 Search PubMed.
- W. P. Walters and R. Barzilay, Acc. Chem. Res., 2021, 54, 263–270 CrossRef CAS PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
- P. C. D. Hawkins, A. G. Skillman, G. L. Warren, B. A. Ellingson and M. T. Stahl, J. Med. Chem., 2007, 50, 1312–1313 CrossRef PubMed.
- M. Skalic, J. Jiménez, D. Sabbadin and G. De Fabritiis, J. Chem. Inf. Model., 2019, 59, 1205–1214 CrossRef CAS PubMed.
- X. Peng, S. Luo, J. Guan, Q. Xie, J. Peng and J. Ma, Proceedings of the 39th International Conference on Machine Learning, 2022, pp. 17644–17655 Search PubMed.
- K. Adams and C. W. Coley, The Eleventh International Conference on Learning Representations, 2023 Search PubMed.
- H. Stärk, O.-E. Ganea, L. Pattanaik, R. Barzilay and T. Jaakkola, EquiBind: Geometric Deep Learning for Drug Binding Structure Prediction, 2022 Search PubMed.
- O.-E. Ganea, X. Huang, C. Bunne, Y. Bian, R. Barzilay, T. Jaakkola and A. Krause, Independent SE(3)-Equivariant Models for End-to-End Rigid Protein Docking, 2022 Search PubMed.
- G. Corso, H. Stärk, B. Jing, R. Barzilay and T. Jaakkola, DiffDock: Diffusion Steps, Twists, and Turns for Molecular Docking, 2023 Search PubMed.
- G. Corso, A. Deng, B. Fry, N. Polizzi, R. Barzilay and T. Jaakkola, Deep Confident Steps to New Pockets: Strategies for Docking Generalization, 2024 Search PubMed.
- M. Buttenschoen, G. M. Morris and C. M. Deane, PoseBusters: AI-based docking methods fail to generate physically valid poses or generalise to novel sequences, 2023 Search PubMed.
- C. Pang, J. Qiao, X. Zeng, Q. Zou and L. Wei, J. Chem. Inf. Model., 2024, 64, 2174–2194 CrossRef CAS PubMed.
- W. Xie, F. Wang, Y. Li, L. Lai and J. Pei, J. Chem. Inf. Model., 2022, 62, 2269–2279 CrossRef CAS PubMed.
- D. M. Anstine and O. Isayev, J. Am. Chem. Soc., 2023, 145, 8736–8750 CrossRef CAS PubMed.
- O. Prykhodko, S. V. Johansson, P.-C. Kotsias, J. Arús-Pous, E. J. Bjerrum, O. Engkvist and H. Chen, J. Cheminf., 2019, 11, 74 Search PubMed.
- G. L. Guimaraes, B. Sanchez-Lengeling, C. Outeiral, P. L. C. Farias and A. Aspuru-Guzik, Objective-Reinforced Generative Adversarial Networks (ORGAN) for Sequence Generation Models, 2018 Search PubMed.
- J. Lim, S. Ryu, J. W. Kim and W. Y. Kim, Molecular generative model based on conditional variational autoencoder for de novo molecular design, 2018 Search PubMed.
- F. Grisoni, M. Moret, R. Lingwood and G. Schneider, J. Chem. Inf. Model., 2020, 60, 1175–1183 CrossRef CAS PubMed.
- C. Zang and F. Wang, Proceedings of the 26th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining, 2020 Search PubMed.
- B. Samanta, A. De, G. Jana, P. K. Chattaraj, N. Ganguly and M. Gomez-Rodriguez, NeVAE: A Deep Generative Model for Molecular Graphs, 2019 Search PubMed.
- Q. Liu, M. Allamanis, M. Brockschmidt and A. L. Gaunt, Constrained Graph Variational Autoencoders for Molecule Design, 2019 Search PubMed.
- C. Shi, M. Xu, Z. Zhu, W. Zhang, M. Zhang and J. Tang, GraphAF: a Flow-based Autoregressive Model for Molecular Graph Generation, 2020 Search PubMed.
- A. Zhavoronkov, Y. A. Ivanenkov and A. Aliper, et al. , Nat. Biotechnol., 2019, 37, 1038–1040 CrossRef CAS PubMed.
- T. C. Terwilliger, D. Liebschner, T. I. Croll, C. J. Williams, A. J. McCoy, B. K. Poon, P. V. Afonine, R. D. Oeffner, J. S. Richardson, R. J. Read and P. D. Adams, Nat. Methods, 2024, 21, 110–116 CrossRef CAS PubMed.
- T. Ciepliński, T. Danel, S. Podlewska and S. Jastrzebski, J. Chem. Inf. Model., 2023, 63, 3238–3247 CrossRef PubMed.
- C. Harris, K. Didi, A. R. Jamasb, C. K. Joshi, S. V. Mathis, P. Lio and T. Blundell, Benchmarking Generated Poses: How Rational is Structure-based Drug Design with Generative Models?, 2023 Search PubMed.
- J. Inglese, C. Shamu and R. K. Guy, Nat. Chem. Biol., 2007, 3, 438–441 CrossRef CAS PubMed.
- C. Murray and D. Rees, Nat. Chem., 2009, 1, 187–192 CrossRef CAS PubMed.
- A. Gironda-Martínez, E. J. Donckele, F. Samain and D. Neri, ACS Pharmacol. Transl. Sci., 2021, 4, 1265–1279 CrossRef PubMed.
- N. Brown, M. Fiscato, M. H. Segler and A. C. Vaucher, J. Chem. Inf. Model., 2019, 59, 1096–1108 CrossRef CAS PubMed.
- D. Polykovskiy, A. Zhebrak, B. Sánchez-Lengeling, S. Golovanov, O. Tatanov, S. Belyaev, R. Kurbanov, A. Artamonov, V. Aladinskiy, M. Veselov, A. Kadurin, S. I. Nikolenko, A. Aspuru-Guzik and A. Zhavoronkov, Front. Pharmacol., 2020, 11, 565644 CrossRef CAS PubMed.
- K. Huang, T. Fu, W. Gao, Y. Zhao, Y. Roohani, J. Leskovec, C. W. Coley, C. Xiao, J. Sun and M. Zitnik, arXiv, 2021, preprint, arXiv:2102.09548v2, DOI:10.48550/arXiv.2102.09548.
- W. Gao, T. Fu, J. Sun and C. W. Coley, Sample Efficiency Matters: A Benchmark for Practical Molecular Optimization, 2022 Search PubMed.
- V. Bagal, R. Aggarwal, P. K. Vinod and U. D. Priyakumar, J. Chem. Inf. Model., 2022, 62, 2064–2076 CrossRef CAS PubMed.
- M. Krenn, Q. Ai, S. Barthel, N. Carson, A. Frei, N. C. Frey, P. Friederich, T. Gaudin, A. A. Gayle, K. M. Jablonka, R. F. Lameiro, D. Lemm, A. Lo, S. M. Moosavi, J. M. Nápoles-Duarte, A. Nigam, R. Pollice, K. Rajan, U. Schatzschneider, P. Schwaller, M. Skreta, B. Smit, F. Strieth-Kalthoff, C. Sun, G. Tom, G. Falk von Rudorff, A. Wang, A. D. White, A. Young, R. Yu and A. Aspuru-Guzik, Patterns, 2022, 3, 100588 CrossRef CAS PubMed.
- N. O'Boyle and A. Dalke, ChemRxiv, 2018, preprint, DOI:10.26434/chemrxiv.7097960.v1.
- N. D. Cao and T. Kipf, MolGAN: An implicit generative model for small molecular graphs, 2022 Search PubMed.
- T. Blaschke, J. Arús-Pous, H. Chen, C. Margreitter, C. Tyrchan, O. Engkvist, K. Papadopoulos and A. Patronov, ChemRxiv, 2020, preprint, DOI:10.26434/chemrxiv.12058026.v2.
- K. McCloskey, E. A. Sigel, S. Kearnes, L. Xue, X. Tian, D. Moccia, D. Gikunju, S. Bazzaz, B. Chan, M. A. Clark, J. W. Cuozzo, M.-A. Guié, J. P. Guilinger, C. Huguet, C. D. Hupp, A. D. Keefe, C. J. Mulhern, Y. Zhang and P. Riley, J. Med. Chem., 2020, 63, 8857–8866 CrossRef CAS PubMed.
- R. Sato, A Survey on The Expressive Power of Graph Neural Networks, 2020 Search PubMed.
- M. Skalic, D. Sabbadin, B. Sattarov, S. Sciabola and G. De Fabritiis, Mol. Pharmaceutics, 2019, 16, 4282–4291 CrossRef CAS PubMed.
- J. Parrilla-Gutiérrez, J. Granda and J. Ayme, et al. , Nat. Comput. Sci., 2024, 4, 200–209 CrossRef PubMed.
- S. Luo, J. Guan, J. Ma and J. Peng, Adv. Neural Inf. Process. Syst., 2021, 34 Search PubMed.
- Z. ZHANG, Y. Min, S. Zheng and Q. Liu, The Eleventh International Conference on Learning Representations, 2023 Search PubMed.
- W. Gao and C. W. Coley, J. Chem. Inf. Model., 2020, 60, 5714–5723 CrossRef CAS PubMed.
- C. Bilodeau, W. Jin, T. Jaakkola, R. Barzilay and K. Jensen, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2022, 12, e1608 CrossRef.
- W. L. Jorgensen, Acc. Chem. Res., 2009, 42, 724–733 CrossRef CAS PubMed.
- P. Ertl and A. Schuffenhauer, J. Cheminf., 2009, 1, 8 Search PubMed.
- A. Pedawi, P. Gniewek, C. Chang, B. M. Anderson and H. van den Bedem, Adv. Neural Inf. Process. Syst., 2022, 35 Search PubMed.
- R. Wang, X. Fang, Y. Lu, C. Y. Yang and S. Wang, J. Med. Chem., 2004, 47, 2977–2980 CrossRef CAS PubMed.
- G. R. Bickerton, G. V. Paolini, J. Besnard, S. Muresan and A. L. Hopkins, Nat. Chem., 2012, 4, 90–98 CrossRef CAS PubMed.
- A. K. Ghose, V. N. Viswanadhan and J. J. Wendoloski, J. Comb. Chem., 1999, 1, 55 CrossRef CAS PubMed.
- D. R. Koes, M. P. Baumgartner and C. J. Camacho, J. Chem. Inf. Model., 2013, 53, 1893–1904 CrossRef CAS PubMed.
- G. Landrum, 2006, http://www.rdkit.org.
- A. Kutlushina, A. Khakimova, T. Madzhidov and P. Polishchuk, Molecules, 2018, 23, 3094 CrossRef PubMed.
- G. Skoraczyński, M. Kitlas, B. Miasojedow and A. Gambin, J. Cheminf., 2023, 15, 6 Search PubMed.
- A. Thakkar, V. Chadimová, E. J. Bjerrum, O. Engkvist and J.-L. Reymond, Chem. Sci., 2021, 12, 3339–3349 RSC.
| This journal is © The Royal Society of Chemistry 2024 |